
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel ion-imprinted amidoxime-functionalized UHMWPE fiber based on radiation-induced crosslinking for selective adsorption of uranium†
Junxuan Aoab,
Hongjun Zhangc,
Xiao Xua,
Fujia Yaod,
Lin Maab,
Lan Zhanga,
Bangjiao Yec,
Qingnuan Li*a,
Lu Xu*a and
Hongjuan Ma*a
aShanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201800, China. E-mail: liqingnuan@sinap.ac.cn; xulu@sinap.ac.cn; mahongjuan@sinap.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cHefei National Laboratory for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei 230026, China
dJining University, Qufu 273155, China
First published on 10th September 2019
Abstract
A novel uranium-imprinted adsorbent (AO-Imp fiber) was prepared by radiation-induced crosslinking of amidoxime-functionalized ultra-high molecular weight polyethylene fiber (AO fiber). The porous structure was characterized by scanning electron microscopy (SEM) and positron annihilation lifetime (PAL) spectroscopy after ion imprinting. This ion-imprinted fiber exhibited enhanced adsorption selectivity for uranium in the form of both UO22− and [UO2(CO3)3]4− in batch experiments. Compared with AO fiber, the adsorption capacity of the AO-Imp(250) fiber for uranium increased from 0.36 mg g−1 to 1.00 mg g−1 in simulated seawater and from 5.02 mg g−1 to 12.03 mg g−1 in simulated acid effluent, while its adsorption capacities for other co-existing metal ions were particularly low. This study provides an approach to prepare ion-imprinted adsorbents without introducing crosslinking reagents, which may be a promising method for uranium extraction.
1. Introduction
Uranium is an indispensable raw material in nuclear plants.1 However, the development of the nuclear industry may result in problems related to the discharge of radioactive nuclear wastewater.2–4 Uranium is considered to be one of the most important pollutants in wastewater from the nuclear industry; it is harmful to the environment and human health due to its radioactivity and chemical toxicity.5 Uranium concentrations in the discharge from uranium ore mining and from abandoned mines reach hundreds of μg L−1 to several mg L−1, while the values in low-level radioactive wastewater are even higher than tens of mg L−1. Hence, a strict effluent discharge limit on the concentration of uranium of 30 μg L−1 has been established by the United States Environmental Protection Agency (EPA).6,7 On the other hand, uranium recovery from nuclear wastewater and other uranium-containing systems such as seawater is a supplemental source of nuclear raw material.8–10 Especially, the unconventional uranium resource of seawater has attracted worldwide attention for several decades. Although the concentration of uranium in seawater is relatively low (approximately 3.3 μg L−1), the amount of uranium in the ocean is almost 1000 times that on land.11,12 Therefore, research on uranium recovery could provide a steady supplement of nuclear raw materials for the sustainable development of the nuclear industry.Among numerous uranium recovery methods,13,14 adsorption by organic adsorbents has been considered to play an important role for a long time due to the strong chelating properties of modified functional groups.15–20 Extensive investigations have found that the amidoxime (AO) group has extraordinary affinity and high selectivity toward uranyl ions.10,21,22 Meanwhile, AO-functionalized polymers have been prepared and applied in research on uranium extraction.23–30 However, uranium-containing aqueous systems such as nuclear wastewater and seawater are always complicated with numerous co-existing ions, such as vanadium, iron, copper, cobalt, nickel, and zinc.27,31 Meanwhile, AO can only show a relatively high selectivity of uranium over alkali metals and alkaline earth metals, such as sodium, magnesium, and calcium.32 Therefore, the improvement of uranium selectivity over co-existing ions such as vanadium, iron, copper, cobalt, nickel, and zinc in the process of uranium extraction from seawater as well as radioactive wastewater is extremely significant.
Ion imprinting technology has emerged as a solution to the problem of low selectivity in uranium extraction.33,34 The typical procedure of this method is mainly based on the introduction of template particles in the preparation of crosslinked materials by crosslinking reagents followed by elution of the templates, thus endowing the adsorbent material with the ability of selectively identifying and coordinating particles that are similar in spatial size and structure to the template.35 In recent years, much attention has been paid to the synthesis of uranium ion-imprinted adsorbents, such as ion-imprinted mesoporous silica,36 surface ion-imprinted polymers,37 imprinted porous aromatic frameworks,34 and ion-imprinted magnetic microspheres.38 However, almost all ion imprinting techniques involve a crosslinking reagent, leading to complicated preparation processes.
In this paper, a method of ion imprinting based on radiation-induced crosslinking was investigated to improve adsorption selectivity without the use of a crosslinking reagent. Ultra-high molecular weight polyethylene (UHMWPE) fiber was used as a substrate due to its high mechanical strength, excellent chemical corrosion resistance and impact resistance.39,40 UHMWPE has been widely used for uranium extraction deployment for a long time.40,41 In this work, firstly, UHMWPE fibers were irradiated in air atmosphere at room temperature with an absorbed dose of 20 kGy. Then, graft polymerization of 4-hydroxybutyl acrylate glycidyl ether (4HB) was carried out in an emulsion solution, with subsequent modification of the amidoxime group. Radiation-induced crosslinking of the amidoxime-functionalized UHMWPE fiber (AO fiber) loaded with a template of uranyl ion was performed in water at three different absorbed doses of 50, 150 and 250 kGy, respectively, to prepare ion-imprinted AO fibers (AO-Imp(50) fiber, AO-Imp(150) fiber and AO-Imp(250) fiber). Investigations of the properties of the materials were carried out, and their adsorption performance for uranium in the presence of co-existing metal ions was evaluated. The results showed that the ion imprinting technique in this work can promote the adsorption selectivity of AO fiber for uranium, which may provide new ideas for the preparation of adsorbents with high selectivity in uranium extraction.
2. Experimental
2.1 Materials and reagents
UHMWPE fiber (TYZ Safetex FT-103, size of 3.6 Denier), supplied by Beijing Tongyizhong Advanced Material Company, was used as a base material for pre-irradiation graft polymerization. 4-Hydroxybutyl acrylate glycidyl ether (4HB) was purchased from Suzhou University and was used without further purification. Ethylene diamine (EDA, AR), acrylonitrile (AN, CP), acrylic acid (AAc, AR), dimethylformamide (DMF, AR), dimethyl sulfoxide (DMSO), 1,4-dioxane, toluene, hydroxylamine hydrochloride (NH2OH HCl), sodium bicarbonate (NaHCO3), sodium carbonate (Na2CO3), sodium chloride (NaCl), potassium hydroxide (KOH) and concentrated nitric acid were purchased from Sinopharm Chemical Reagent Company and were used without further purification. All standard metal ion solutions with 1000 ppm concentrations were purchased from SPEX CertiPrep Company. Deionized water was used for all experiments except where otherwise noted.2.2 Preparation of amidoxime adsorbents
As illustrated in Scheme 1, the preparation of the radiation-induced crosslinked ion-imprinted adsorbent involved two steps, including: (I) synthesis of AO-functionalized UHMWPE fiber (AO fiber) using 4HB, EDA and AN, according to a method reported in a previous study40 (detailed in the ESI†). (II) Use of ion imprinting technology based on radiation-induced crosslinking to obtain ion-imprinted AO-functionalized UHMWPE fiber, noted as AO-Imp fiber. | ||
| Scheme 1 Illustrative synthesis of AO-Imp fiber based on radiation-induced crosslinking. | ||
Specifically, firstly, AO fiber (about 0.4 g) was soaked in a PET plastic bottle containing 100 mL uranyl ion solution (200 mg L−1). The pH value of the solution was 5.0, adjusted with 0.1 mol L−1 KOH solution. After shaking for 12 h to reach the template (uranyl ions) loading equilibrium, the sample was rinsed with distilled water three times and then placed in a vacuum oven at 60 °C for 24 h. Secondly, the uranyl-loaded fiber was transferred to a PE bag filled with 10 mL distilled water and irradiated by an electron beam with absorbed doses of 50 kGy, 150 kGy and 250 kGy, respectively. Thirdly, the template ions were removed from the irradiated samples using 0.5 mmol L−1 hydrochloric acid solution. After shaking for 2 h, the samples were rinsed with distilled water three times and dried in a vacuum oven at 60 °C for 24 h. The resulting fibers were referred to as AO-Imp(50), AO-Imp(150) and AO-Imp(250), respectively.
2.3 Characterization
The Fourier transform infrared (FT-IR) spectra of the fibers were collected on a Nicolet Avatar 370 FTIR spectrometer (Thermo Nicolet Company, USA), with a resolution of 4 cm−1 and 32 scans.Thermogravimetric analysis (TGA) was carried out by a NETZSCH TG 209 F3 instrument in the temperature range from 25 °C to 800 °C with a heating rate of 10 °C min−1 under a nitrogen flow.
The morphologies of the fibers were determined on a JSM-6700F scanning electron microscope (JEOL, Japan) at an acceleration voltage of 5 kV. All samples were sputtered with gold to enhance the electron conductivity before observation.
The nanometer-scaled free-volume holes of the fibers were characterized by positron annihilation lifetime (PAL) spectroscopy (DPLSAIO3000, Anhui Nuclear and Technology Ltd.) with a time resolution of about 179 ps according to a previous study.29,42 The LTv9 program was used to analyze all the PAL spectra and obtain three lifetime components of τ1, τ2 and τ3 with corresponding intensities of I1, I2 and I3, respectively. The longest component τ3 corresponds to the o-Ps annihilation in free-volume holes determined by the size of the free-volume holes, and its intensity I3 is closely correlated with the fraction of the free-volume holes. Using the Tao-Eldrup model, the average radius (R) of free-volume holes and the o-Ps lifetime (τ3) have the relationship described in eqn (1):
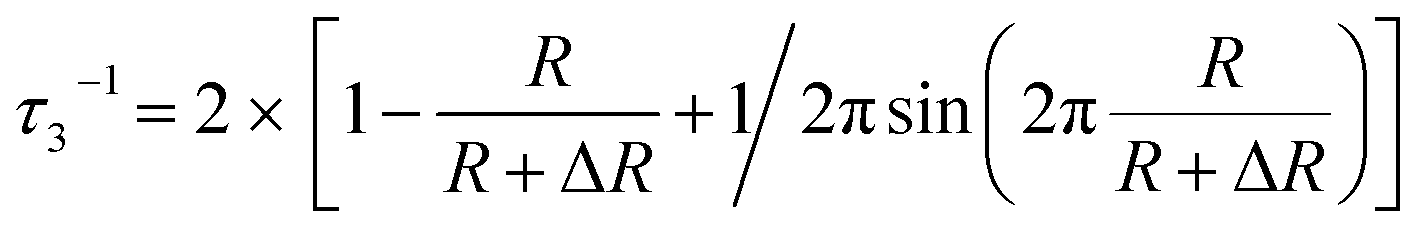 | (1) |
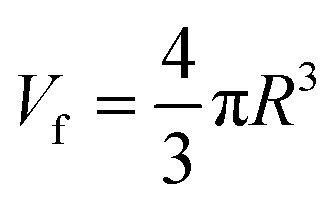 | (2) |
The gel fractions of the fibers before and after ion imprinting were estimated by the swelling ratio through the equilibrium swelling method.43 The fibers were extracted with toluene in a Soxhlet apparatus for 12 h. The gel fraction was defined by eqn (3):
| Gel fraction = (Wa − Wb)/Wb × 100% | (3) |
2.4 Adsorption experiments
Experiments to determine the effects of pH on uranium adsorption were conducted in 25 mL solution containing uranium (10.0 mg L−1) with 0.01 g of the adsorbent. The pH of the solution was adjusted using sodium hydroxide and hydrochloric acid solutions to 2.5, 3.5, 4.5, 5.5, 6.5, and 7.0, respectively. The trials were carried out for 24 h on a rotary shaker at a rate of 100 rpm and at 25 °C. The metal adsorption capacities (Q, mg g−1) were obtained using the following equation (eqn (4)):| QM = (C0 − Ct)ΔV/m | (4) |
The experiments to determine the adsorption performance of AO fiber and the AO-Imp fibers with three different absorbed doses (50 kGy, 150 kGy, 250 kGy) were conducted in 25 mL solutions containing uranium (10.0 mg L−1), vanadium (5.1 mg L−1), nickel (5.1 mg L−1), copper (2.3 mg L−1) and zinc (10.0 mg L−1) with an adsorbent dose of 2.0 g L−1. The pH of the solution was 3.5, adjusted with HNO3 solution. The trial was carried out for 48 h on a rotary shaker at a rate of 100 rpm and 25 °C. The metal adsorption capacities (QM, mg g−1) were obtained using eqn (4).
The adsorption kinetics experiments for simulated acidic effluent were carried out in 25 mL solutions containing uranium, vanadium, nickel, copper and zinc with an adsorbent dose of 0.4 g L−1. The pH of the solution was adjusted to 3.5 with HNO3 solution. Then, the metal ion concentrations were determined using ICP-AES; they are shown in Table 1. The adsorption capacities for metal ions (Qt, mg g−1) at different times were obtained using eqn (4). The distribution ratios (Kd(M)) and selectivity coefficients (β) were calculated using eqn (5) and (6), respectively:
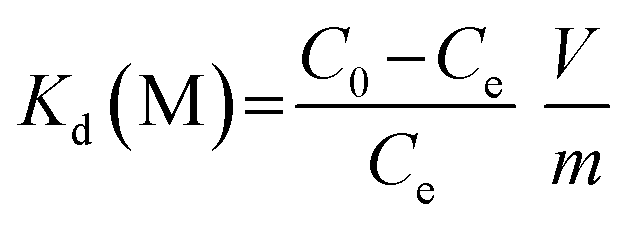 | (5) |
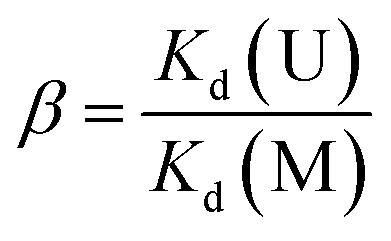 | (6) |
| Concentrations of ions in simulated acidic effluent | ||||||||
| Element | U | V | Ni | Cu | Zn | — | — | — |
| Concentration (mg L−1) | 10.5 | 5.1 | 5.0 | 2.3 | 7.2 | — | — | — |
| Concentrations of ions in simulated seawater | ||||||||
| Element | U | V | Fe | Co | Ni | Zn | Ca | Mg |
| Concentration (μg L−1) | 330 | 150 | 140 | 5.3 | 101 | 408 | 0.6 × 105 | 1.2 × 105 |
| Concentrations of ions in seawater | ||||||||
| Element | U | V | Fe | Co | Ni | Zn | Ca | Mg |
| Concentration (μg L−1) | 3.3 | 1.8 | 3.4 | 0.05 | 5.0 | 4.0 | 0.4 × 106 | 1.3 × 106 |
The adsorption kinetics and adsorption rates were fitted and calculated using the pseudo-first-order model, which is expressed as eqn (7) in its linear form:
ln(Qe − Qt) = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Qe − kt Qe − kt
| (7) |
Selective adsorption experiments in simulated seawater were performed in plastic tanks which contained 5 L of deionized water containing a certain amount of sea salt with uranium and competing ions added at initial concentrations 100 times higher than those in real seawater, as listed in Table 1. The pH value of the simulated seawater was adjusted to about 8.1 by the addition of anhydrous Na2CO3, ensuring that [UO2(CO3)3]4− was the dominant species of uranium, prior to addition of the adsorbent (0.1 g). After shaking by a rotary shaker at a rate of 100 rpm for 48 h at 25 °C, the adsorbents were retrieved from the plastic tanks, washed with deionized water, and then digested with concentrated nitric acid by a MARS 6 Microwave Digestion System (CEM, USA). The concentrations of metal ions in the digestion solution were measured by ICP-AES after diluting to the desired concentration range with deionized water for the analysis. The adsorption capacities (QM, mg g−1) of different ions for the adsorbents were calculated using the following equation (eqn (8)):
| QM = CMV/m | (8) |
Reusability experiments of the adsorbents were conducted. In each cycle, the adsorption of uranium was determined in 25 mL solution containing uranium (10.0 mg L−1) with 0.01 g of adsorbent. The pH of the solution was adjusted to 3.5 using sodium hydroxide and hydrochloric acid solutions. The trials were carried out for 24 h on a rotary shaker at a rate of 100 rpm and 25 °C. Desorption of uranium was carried out by immersing the uranium-loaded adsorbents in 50 mL hydrochloric acid solution (0.5 mol L−1) and a large amount of water with continuous shaking at a rate of 100 rpm and 25 °C for 30 min, respectively.
Marine adsorption tests were conducted near the coast of Hainan, China, where the average temperature of seawater is 23 ± 5 °C. A pump was used to draw water from a seawater reservoir into a flume (1.53 × 0.28 × 0.15 m) without further filtration and eventually back into the ocean. AO fiber and AO-Imp(250) fiber were packed in the flume by nylon ropes and freely dispersed in flowing sea water (Fig. S1†). The flowing water was controlled by a pump at a constant inlet flow rate of 2.0 L min−1, with a corresponding flow velocity of 0.0028 m s−1. After extraction for 7 days, the adsorbents were washed with water and dried in the vacuum oven. After that, a portion of the adsorbent (0.04 g) was cut and digested by concentrated nitric acid. ICP-AES was used to identify the metal ion concentrations of the digestion solution. The adsorption capacities for metal ions were calculated by eqn (8). The concentrations of uranium and co-existing ions are listed in Table 1.
3. Results and discussion
3.1 Characterization
Fig. 1(A) shows the obvious distinctions between the FT-IR spectra of the pristine and modified UHMWPE fibers. The pristine UHMWPE fiber shows absorption peaks at 2910, 2845, 1468 and 717 cm−1 because of the vibrations of –CH2– in the UHMWPE chains.45 New peaks of UHMWPE-g-P(AAc-co-4HB) at 908 and 845 cm−1 correspond to the stretching vibrations of the epoxy groups,46 suggesting the grafting of 4HB onto the base materials. In addition, strong absorption peaks at 1726 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) and 1248 cm−1 (–C–O– stretching) were observed.47 The disappearance of the characteristic vibration of epoxy groups in the spectrum of UHMWPE-g-P(AAc-co-4HB-EDA) indicated the ring opening reaction between the N–H of EDA and the epoxy groups.48 The C
O stretching) and 1248 cm−1 (–C–O– stretching) were observed.47 The disappearance of the characteristic vibration of epoxy groups in the spectrum of UHMWPE-g-P(AAc-co-4HB-EDA) indicated the ring opening reaction between the N–H of EDA and the epoxy groups.48 The C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching on UHMWPE-g-P(AAc-co-4HB-EDA) was observed at 2245 cm−1 after the Michael addition reaction between the amino groups and the nitrile group.27,40 In comparison with the spectrum of UHMWPE-g-P(AAc-co-4HB-EDA-AN), the spectrum of AO fiber shows the disappearance of the band at 2245 cm−1 and new characteristic bands at 3000 to 3500 cm−1 and 1645 cm−1, suggesting the successful amidoximation of the nitrile groups.41 When loading with template uranyl ions, with subsequent radiation-induced crosslinking, OUO bands appeared at 912 cm−1, providing evidence of the successful coordination of the AO ligands with uranyl ions.33 After leaching of the uranyl ions, it was obvious that the AO fiber and AO-Imp fiber presented very similar FTIR spectra, indicating that the identical chemical structure was restored. Furthermore, the absence of the OUO bands at 914 cm−1 in the spectra of the AO-Imp fibers suggests the complete removal of the template uranyl ions from the adsorbents.
N stretching on UHMWPE-g-P(AAc-co-4HB-EDA) was observed at 2245 cm−1 after the Michael addition reaction between the amino groups and the nitrile group.27,40 In comparison with the spectrum of UHMWPE-g-P(AAc-co-4HB-EDA-AN), the spectrum of AO fiber shows the disappearance of the band at 2245 cm−1 and new characteristic bands at 3000 to 3500 cm−1 and 1645 cm−1, suggesting the successful amidoximation of the nitrile groups.41 When loading with template uranyl ions, with subsequent radiation-induced crosslinking, OUO bands appeared at 912 cm−1, providing evidence of the successful coordination of the AO ligands with uranyl ions.33 After leaching of the uranyl ions, it was obvious that the AO fiber and AO-Imp fiber presented very similar FTIR spectra, indicating that the identical chemical structure was restored. Furthermore, the absence of the OUO bands at 914 cm−1 in the spectra of the AO-Imp fibers suggests the complete removal of the template uranyl ions from the adsorbents.
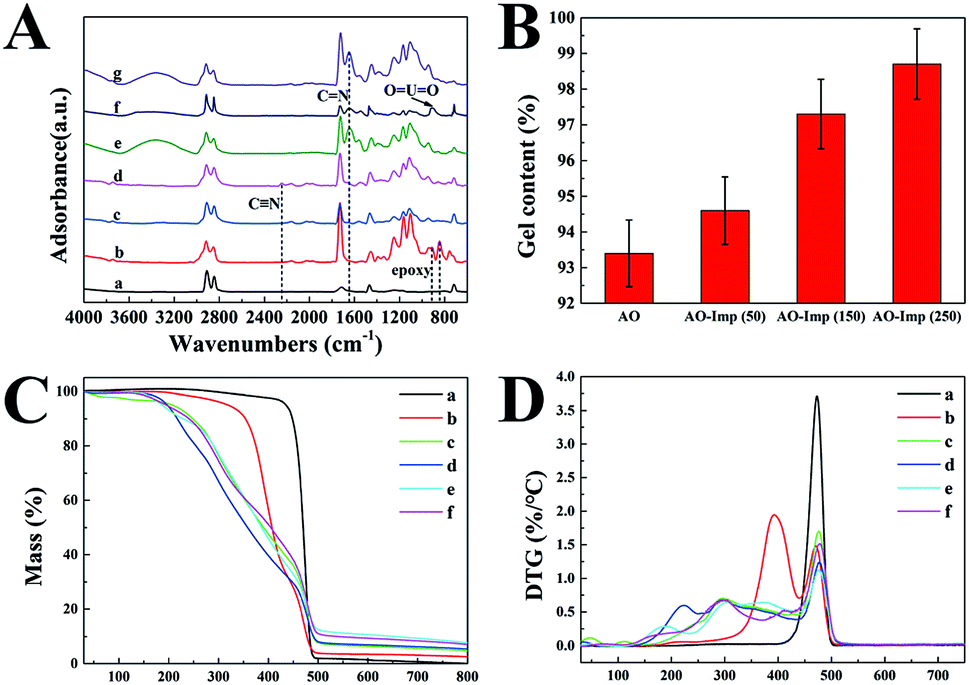 | ||
| Fig. 1 (A) FT-IR curves of the synthesized adsorbents: (a) UHMWPE, (b) UHMWPE-g-P(AAc-co-4HB), (c) UHMWPE-g-P(AAc-co-4HB-EDA), (d) UHMWPE-g-P(AAc-co-4HB-EDA-AN), (e) AO fiber, (f) AO fiber loaded with U(VI) and (g) AO-Imp fiber. (B) Gel contents of the AO fiber and the AO-Imp fibers synthesized with absorbed doses of 50 kGy, 150 kGy and 250 kGy. (C) TGA and (D) DTG curves of the synthesized adsorbents: (a) UHMWPE, (b) UHMWPE-g-P(AAc-co-4HB), (c) UHMWPE-g-P(AAc-co-4HB-EDA), (d) UHMWPE-g-P(AAc-co-4HB-EDA-AN), (e) AO fiber, (f) AO-Imp(250) fiber. | ||
The TGA and DTG curves and data of the pristine UHMWPE fiber and modified UHMWPE fibers are shown in Fig. 1(C and D) and Table S1,† respectively. The thermogram of the pristine UHMWPE fiber shows a clean, single-step degradation with an initial decomposition temperature (Tdi) of 438 °C and a 100% weight degradation temperature of 500 °C. After modification with functional groups, the TGA and DTG curves of each fiber showed different initial decomposition temperatures and different temperatures of maximum decomposition (Tmax). All the modified fibers had three or four Tmax values, which was attributed to the loss of each grafted fragment, except for the last Tmax, which is due to the degradation of the UHMWPE matrix. For the UHMWPE-g-P(AAc-co-4HB) fiber, the weight loss at 393 °C is due to the decomposition of the grafted P(AAc-co-4HB) fragments. In the TGA and DTG curves of the UHMWPE-g-P(AAc-co-4HB-EDA) fiber, the weight loss at 241 °C is due to the decomposition of the P(AAc) fragments. Also, the second weight loss at 294 °C can be ascribed to the decomposition of P(4HB-EDA). The TGA and DTG curves of the UHMWPE-g-P(AAc-co-4HB-EDA) fiber and the nitrile-modified UHMWPE-g-P(AAc-co-4HB-EDA-AN) fiber are similar, except that the Tdi and Tmax of the UHMWPE-g-P(AAc-co-4HB-EDA-AN) fiber showed slight decrements. After amidoximation, the fiber became less thermally stable and degraded at about 131 °C. For the AO-Imp fibers, the TGA and DTG curves were similar to those of the AO fiber, indicating that the ion imprinting procedure did not affect the chemical stability of the fiber.
Scanning electron microscopy (SEM) images of the pristine UHMWPE fiber and the modified fibers are shown in Fig. 2, revealing significant differences in the microstructures of the fibers. It can be clearly seen that the pristine fiber has a very smooth surface with microgrooves.40 Fig. 2(B) shows SEM images of UHMWPE-g-P(AAc-co-4HB) with a DG of 252%. For the grafted UHMWPE fibers, the microgrooves were covered by a thick layer of grafted chains, resulting in an increment of the diameter of the fibers from 16.5 μm to 42.3 μm; also, the surface became rough, indicating that the P(AAc-co-4HB) chains were uniformly grafted on the UHMWPE fibers. After the ring opening reaction of EDA and the Michael addition reaction of AN, numerous smooth particles appeared on the surface of the UHMWPE-g-P(AAc-co-4HB-EDA-AN) fibers, which can be ascribed to the increment in the grafting chains of the functional groups (Fig. 2(C and D)). In comparison with the UHMWPE-g-P(AAc-co-4HB-EDA-AN) fiber, the particles on the AO fiber (Fig. 2(E and G)) became more distinct due to amidoximation. After ion imprinting based on radiation-induced crosslinking, the SEM image of the AO-Imp fiber in Fig. 2(F and H) exhibits numerous micro/nanopores with diameters of 207 ± 44 nm; also, the shapes of the particles became roughly spherical, which may be due to traces of the eluted template ions. The four methylene groups of 4HB in the grafted chains provide sufficient sites for crosslinking.43,49 After crosslinking, the AO groups in the polymer were firmly fixed, and the motion of the grafted chains was restrained.43 In aqueous solution, the chains swell and the firmly fixed AO groups can adsorb the template ions of uranyl efficiently. In the dry state of the fiber, shrinkage of the chains endows them with numerous pores with diameters of about 200 nm. Elemental distribution maps and energy-dispersive X-ray spectroscopy (Fig. 3(A–D)) showed that the elements C, O, N and U are distributed homogeneously, and no uranium residue was found on the surface of the AO-Imp fiber (Fig. 3(C)); this indicates that the template uranyl ions have been completely removed.
 | ||
| Fig. 2 Scanning electron microscopy images of (A) UHMWPE, (B) UHMWPE-g-P(AAc-co-4HB), (C) UHMWPE-g-P(AAc-co-4HB-EDA), (D) UHMWPE-g-P(AAc-co-4HB-EDA-AN), (E) AO fiber, and (F) AO-Imp(250) fiber; (G) AO fiber at high magnification and (H) AO-Imp(250) fiber at high magnification. | ||
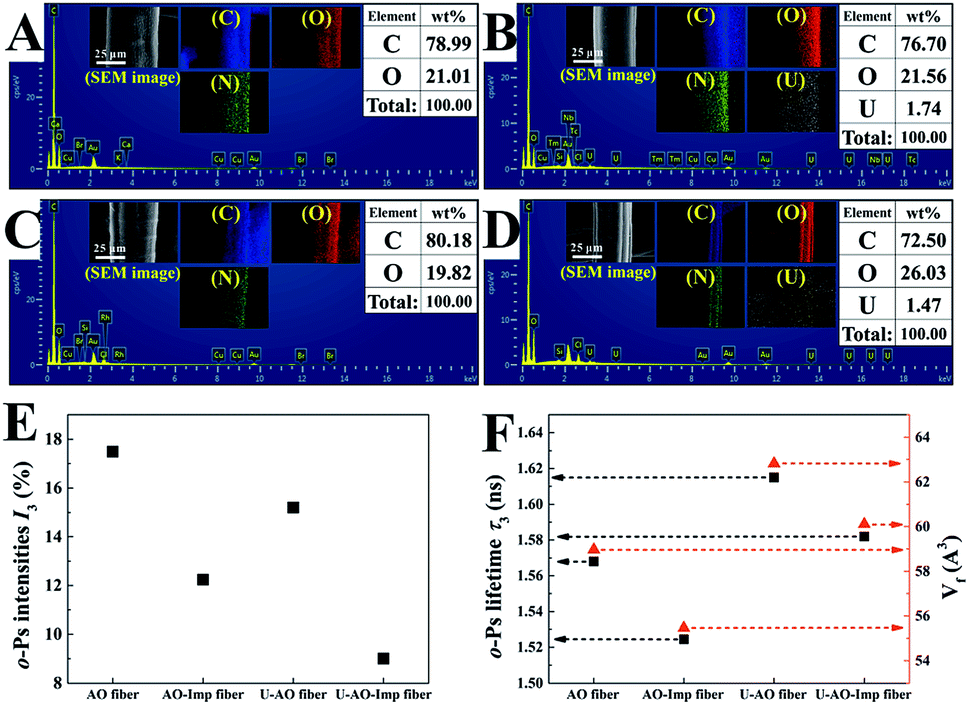 | ||
| Fig. 3 EDS of (A) AO fiber, (B) AO fiber loaded with U(VI), (C) AO-Imp(250) fiber and (D) AO-Imp(250) fiber loaded with U(VI) and their elemental distribution maps (inset) of C, O and U, respectively; (E) o-Ps intensities and (F) lifetimes with the corresponding average volumes of free-volume holes in AO fiber, the AO-Imp fibers and their uranium-loaded fibers (U-AO fiber and U-AO-Imp(250) fiber), respectively. | ||
The degree of crosslinking can be reflected in the gel fraction, which was measured by the equilibrium swelling method. As shown in Fig. 1(B), the gel fraction increased with increasing absorbed dose of irradiation. The AO-Imp(250) fiber achieved a high gel fraction of up to 98.7%, indicating that a much more crosslinked structure was formed compared with AO fiber.50 Notably, the gel fraction of AO fiber was also extremely high, which may be due to the inevitable crosslinking in the pre-irradiation grafting of 4HB and AAc under an electron beam.
PAL spectroscopy was performed to characterize the nanometer-scaled free-volume holes of the fibers. As shown in Fig. 3(E), after the ion imprinting process, the o-Ps intensity of the fiber decreased from 17.5% to 12.2%; this indicates that the number of pores in the fibers decreased, including the micropores, nanopores and sub-nanopores. This can be attributed to the densification changes of the fibers, which were compacted via radiation-induced crosslinking. Also, the average volume (Vf) of free-volume holes in the AO-Imp fiber decreased slightly from 59.0 A3 to 55.5 A3 (Fig. 3(F)); this suggests that sub-nano structures matching the sizes of the uranyl ions were retained due to the introduction of the template during the preparation process.
In previous studies, the inhibition of o-Ps formation and chemical quenching of o-Ps annihilation could occur once metals and metal oxides were introduced. After adsorption of uranium, the I3 values of both the AO fiber and the AO-Imp fibers decreased significantly; this indicates that the porous-structured holes in the fibers may be occupied by uranium. Meanwhile, the Vf values in the fibers loaded with uranium were both higher than that of the fibers without uranium-loading. These results are probably due to the diminution of the sub-nanopores, which were mostly occupied by the sub-nano-meter sized uranium, leading to an increment in the average volume of free-volume holes after adsorption. Based on the PAL spectroscopy results, although the overall content of porous structures decreased, the AO-Imp fiber may preserve sub-nano sized holes that are nearly the same size as uranium from this ion imprinting technology; this can increase the adsorption selectivity for uranium.
3.2 Effects of solution pH
The effects of pH on uranium adsorption by AO-Imp(250) fiber were investigated in uranium solutions with pH values ranging from 2.5 to 7.0. In Fig. 4, it can be clearly seen that the uranium adsorption capacity of AO-Imp(250) fiber was strongly affected by the pH of the solution. Also, the adsorption capacity of uranium increased dramatically from pH 2.5 to 3.5, with a subsequent decrease from pH 3.5 to 7.0. Therefore, the optimum pH is 3.5, and the subsequent adsorption selectivity and kinetics experiments were conducted at a pH of 3.5.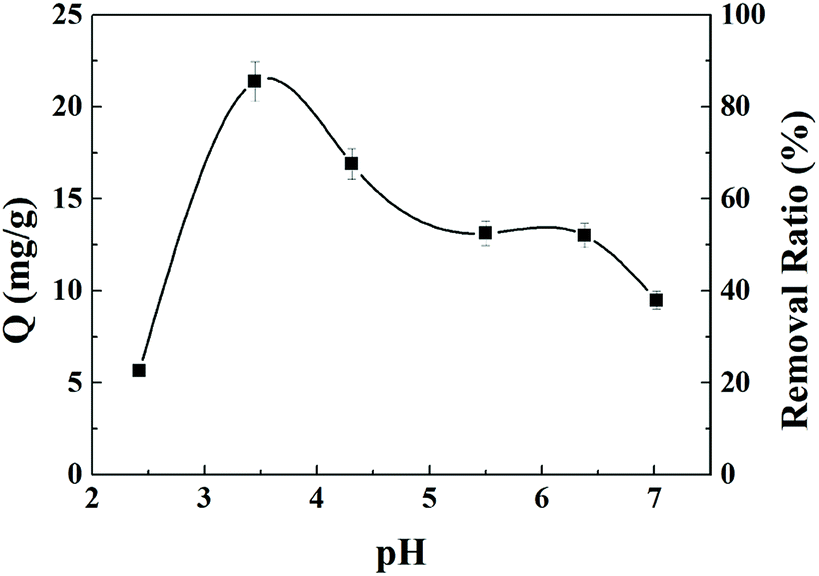 | ||
| Fig. 4 pH effects on the adsorption capacity and removal ratio of uranium by AO-Imp(250) fiber. | ||
3.3 Adsorption selectivity in acidic liquid
The adsorption performance of the AO fiber and AO-Imp fibers was evaluated in a solution containing uranium, vanadium, nickel, copper and zinc at pH 3.5. After 35 h of adsorption, the uranium adsorption capacities of the AO-Imp(50), AO-Imp(150) and AO-Imp(250) fibers were 11.49 mg g−1, 10.52 mg g−1 and 12.03 mg g−1, respectively. Compared with AO fiber (5.02 mg g−1), these AO-Imp fibers exhibited 129%, 110% and 140% improvements in adsorption capacity, respectively (Fig. 5). In contrast, the adsorption capacities of the AO-Imp fibers for nickel, copper and zinc decreased sharply. Notably, there was a slight increase in adsorption capacity for vanadium. However, the adsorption capacity ratios of uranium to vanadium (U/V) increased to 1.33, 1.61 and 1.35 for AO-Imp(50) fiber, AO-Imp(150) fiber and AO-Imp(250) fiber, respectively, compared to the U/V adsorption ratio of 1.00 for AO fiber. Because the adsorption capacity of AO-Imp(250) fiber was higher than those of AO-Imp(50) and AO-Imp(150), in the following evaluation and discussion, AO-Imp(250) fiber was chosen for detailed selectivity and adsorption kinetics experiments.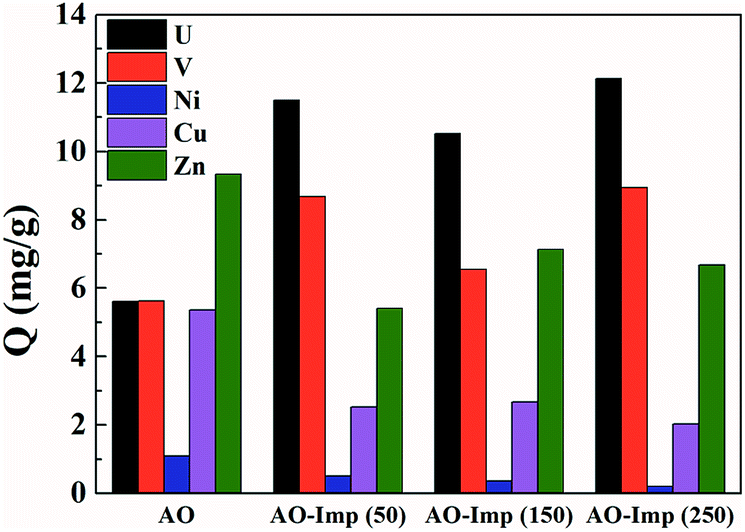 | ||
| Fig. 5 The adsorption capacities of AO fiber and the AO-Imp (50, 150 and 250) fibers. | ||
The time-dependent curves of adsorption capacity for each metal ion using AO fiber and AO-Imp(250) fiber at different times are shown in Fig. 6 and S2†. The pseudo-first-order kinetic rate constants k and the correlation coefficients R2 are listed in Table 2. When extracting uranium from solutions containing interfering ions, AO-Imp(250) fiber showed better adsorption performance than AO fiber. The adsorption capacity (12.03 mg g−1) and k (k = 0.08 h−1) of AO-Imp(250) fiber were both higher than those of AO fiber (Q = 5.05 mg g−1 and k = 0.05 h−1). Meanwhile, AO-Imp(250) fiber also had much higher adsorption rates for other coexisting metal ions than AO fiber (Fig. S2 and Table S2†). The faster adsorption performance for metal ions can be attributed to the fixed and specific coordination structure toward uranium rather than other ions due to the ion-imprinting technology based on radiation-induced crosslinking.
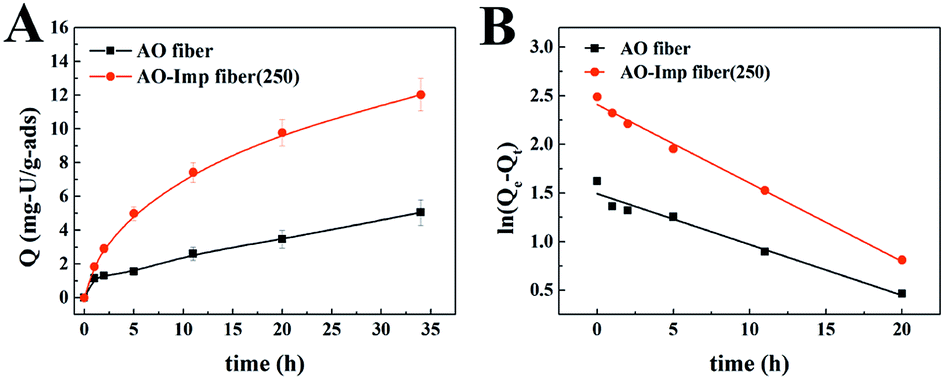 | ||
| Fig. 6 The time-dependent curves of adsorption capacity for uranium. | ||
| Sample | Qe, exp (mg g−1) | Qe, cal (mg g−1) | k (h−1) | R2 |
|---|---|---|---|---|
| AO fiber | 5.02 | 4.45 | 0.05 | 0.9581 |
| AO-Imp(250) fiber | 12.03 | 11.12 | 0.08 | 0.9931 |
Kd(M) is an indicator of affinity for metal (M) ions and can be used to derive β, as shown in Table 3. After ion imprinting, the distribution ratio for uranium increased more than three times, from 0.628 L g−1 to 2.297 L g−1, although the distribution ratio for vanadium also increased from 1.849 L g−1 to 3.188 L g−1. However, the selectivity coefficient for uranium over vanadium increased more than two times, from 0.340 to 0.721, as a result of the greater increment in adsorption capacity for uranium than for vanadium. Meanwhile, the distribution ratios for the other metal ions decreased dramatically, and the selectivity coefficients for uranium over all the other metal ions increased; this suggests extraordinary adsorption selectivity for the template ions UO22−. The high adsorption selectivity can be ascribed to the fact that the functional groups in AO-Imp(250) fiber formed complementary spatial structures and fixed coordination structures for UO22− by the ion imprinting technology based on radiation-induced crosslinking. Accordingly, the coordination ability to other ions was weakened.
| AO fiber | AO-Imp(250) fiber | |||
|---|---|---|---|---|
| Kd (L g−1) | β | Kd (L g−1) | β | |
| U | 0.628 | — | 2.297 | — |
| V | 1.849 | 0.340 | 3.188 | 0.721 |
| Ni | 0.199 | 3.156 | 0.057 | 40.298 |
| Cu | 28.861 | 0.022 | 1.201 | 1.913 |
| Zn | 0.223 | 2.816 | 0.021 | 109.381 |
3.4 Adsorption selectivity in simulated seawater
Adsorption experiments for [UO2(CO3)3]4− and other metal ions were conducted with AO fiber as well as AO-Imp(250) fiber in simulated seawater (100 times the actual concentrations in natural seawater, Table 1). The adsorption capacities for different ions are shown in Fig. 7. The Kd values for metal ions and the β values for uranium over other metal ions are shown in Table 4. The results obviously show that the adsorption capacity of AO-Imp(250) fiber for [UO2(CO3)3]4− increased remarkably to 1.00 mg g−1, nearly three times that of AO fiber (0.36 mg g−1). The adsorption capacities for all the other co-existing ions decreased except for the adsorption capacity for vanadium, which remained almost stable. However, the adsorption capacity ratio of uranium to vanadium (U/V) increased from 1.05 to 2.81, and the β for uranium over vanadium increased nearly two times, from 0.467 to 1.296. Moreover, the β for uranium over other metal ions also showed a dramatic increment. It is noteworthy that although the template ion used in the preparation of AO-Imp(250) fiber was UO22− instead of ([UO2(CO3)3]4−), the selectivity was also enhanced in simulated seawater. This can be attributed to the dissociation of CO32− from [UO2(CO3)3]4− by the AO ligands, which have stronger coordination ability to uranyl than CO32−.51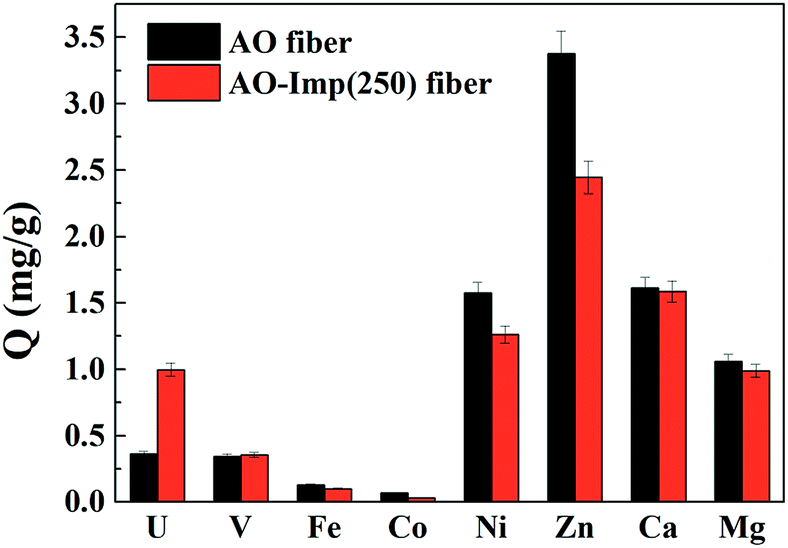 | ||
| Fig. 7 The selective adsorption of metal ions in simulated seawater. | ||
| AO fiber | AO-Imp(250) fiber | |||
|---|---|---|---|---|
| Kd (L g−1) | β | Kd (L g−1) | β | |
| U | 1.128 | — | 3.206 | — |
| V | 2.413 | 0.467 | 2.475 | 1.296 |
| Fe | 0.943 | 1.196 | 0.723 | 4.437 |
| Co | 17.586 | 0.064 | 7.039 | 0.456 |
| Ni | 22.5842 | 0.050 | 16.421 | 0.195 |
| Zn | 9.910 | 0.114 | 6.765 | 0.474 |
| Ca | 0.027 | 41.906 | 0.026 | 121.234 |
| Mg | 0.009 | 127.861 | 0.008 | 389.229 |
3.5 Reusability experiment
Adsorption and desorption can provide sustainable and efficient utility and decreased cost of fibers for uranium adsorption. Hydrochloric acid solution (0.5 mol L−1) was used as the eluting agent for uranium desorption. As shown in Fig. 8, a relatively modest decrease occurred in the adsorption capacity and removal ratio after each adsorption–desorption cycle. Eventually, the adsorption capacity decreased by 16.8% to 17.2 mg g−1 after five cycles, which suggests potential reusability of AO-Imp fiber for uranium extraction. | ||
| Fig. 8 Reusability experiments of the AO-Imp(250) fiber. | ||
3.6 Adsorption selectivity in natural seawater
Selectivity in natural seawater is significant to screen adsorption selectivity in field tests. Marine adsorption experiments were performed near the coast of Hainan, China. After adsorption for 7 days, the adsorbents became dark in color and were covered by large amounts of marine fouling (Fig. S1†) because no filtration was used on the flowing seawater. The color of the adsorbents was still dark after removing the fouling by water, suggesting that ocean metal ions had been extracted by the adsorbents. The adsorption capacities for metal ions are shown in Fig. 9; AO fiber achieved an adsorption capacity of 0.076 mg g−1 for uranium in the marine fouling environment. Meanwhile, the adsorption capacity of AO-Imp(250) fiber increased by 43%, to 0.109 mg g−1. In marine environments, there are numerous microorganisms as well as natural organic matter, which can affect the adsorption performance of adsorbents.52–54 Both AO-Imp(250) fiber and AO fiber exhibited lower adsorption capacity of uranium in the marine environment than in the laboratory. However, the adsorption capacity ratio of uranium to vanadium (U/V) increased from 0.70 to 0.94. For other metal ions, the adsorption capacities for cobalt, nickel and zinc decreased; however, the adsorption capacities for iron, calcium and magnesium increased. This may be caused by the complex and fouling environment in the ocean. The inevitable microorganisms and natural organic matter remaining on the surface of the adsorbents, including some marine algae and bacteria containing uncertain amounts of iron, calcium and magnesium, caused deviations in the measurements of their adsorption capabilities.55,56 Apart from this, the adsorption selectivity for uranium was extremely enhanced by ion imprinting technology. These results can be ascribed to the fixed coordination structure produced through ion imprinting under radiation-induced crosslinking. The enhanced adsorption selectivity performance in natural seawater indicates that AO-Imp fiber is a promising candidate for potential uranium extraction from seawater.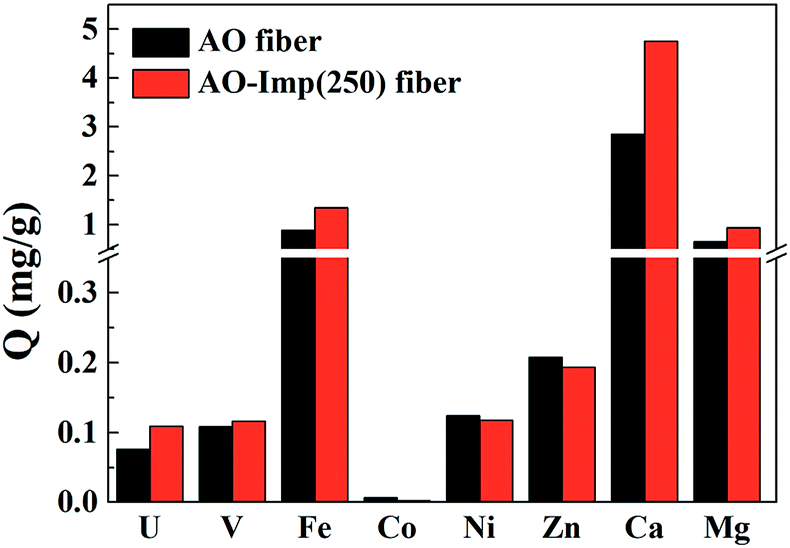 | ||
| Fig. 9 Marine adsorption of metal ions in natural seawater. | ||
4. Conclusion
In summary, a novel uranium-imprinted adsorbent, AO-Imp fiber, was developed for highly selective adsorption of uranium by radiation-induced grafting and crosslinking. Micro-nanopores, sub-nanometer-scaled free-volume holes and higher degrees of crosslinking were characterized, suggesting that the ion imprinting was successfully conducted by the AO-functionalized fibers themselves based on radiation-induced crosslinking without adding crosslinking reagents. High selectivity, adsorption capacity and adsorption rates for uranium could be achieved by the AO-Imp(250) fiber in co-existing ion solutions, regardless of whether uranium was present in the form of UO22− or [UO2(CO3)3]4−. During adsorption tests in simulated seawater, the adsorption capacity of the AO-Imp(250) fiber for uranium (1.00 mg g−1) was almost three times that of the AO fiber (0.36 mg g−1). Meanwhile, the AO-Imp fiber had a particularly low adsorption capacity for other metal ions compared with AO fiber, which can be attributed to the specific complementary spatial structure and fixed coordination structure of the AO ligands for UO22− achieved through ion imprinting technology. Moreover, the selectivity coefficient for uranium over vanadium was increased. In the marine adsorption experiment, the AO-Imp(250) fiber showed enhanced adsorption selectivity performance, where the adsorption capacity for uranium increased by 43%; this suggests potential applications in uranium extraction from seawater. To the best of our knowledge, this is the first report on an ion-imprinted AO fiber synthesized by radiation-induced crosslinking. This work may provide a new approach for increasing the adsorption selectivity of many other ions and extracted particles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We greatly appreciate support from the National Natural Science Foundation of China (U1732151, U1832124) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA02030000).Notes and references
- S.-S. Chen, Y. Ren, H.-Y. Ding and Y.-T. Su, Atomic Energy Sci. Technol., 2015, 49, 415–423 Search PubMed.
- X. L. Han, M. Y. Xu, S. Yang, J. Qian and D. B. Hua, J. Mater. Chem. A, 2017, 5, 5123–5128 RSC.
- S. Z. Su, Q. Liu, J. Y. Liu, H. S. Zhang, R. M. Li, X. Y. Jing and J. Wang, Sci. Rep., 2018, 8, 10 CrossRef PubMed.
- N. Reynier, R. Lastra, C. Laviolette, J. F. Fiset, N. Bouzoubaa and M. Chapman, Minerals, 2015, 5, 744–757 CrossRef CAS.
- Z. S. Chen, J. Wang, Z. X. Pu, Y. S. Zhao, D. S. Jia, H. X. Chen, T. Wen, B. W. Hu, A. Alsaedi, T. Hayat and X. K. Wang, Chem. Eng. J., 2017, 320, 448–457 CrossRef CAS.
- S. A. Arabi, I. I. Funtua, B. B. M. Dewu, S. A. Alagbe, M. Y. Kwaya, M. L. Garba and A. D. Baloga, J. Radioanal. Nucl. Chem., 2013, 295, 135–142 CrossRef CAS.
- S. H. Frisbie, E. J. Mitchell and B. Sarkar, Environ. Sci.: Processes Impacts, 2013, 15, 1817–1823 RSC.
- X. F. Yi, Z. Q. Xu, Y. Liu, X. Y. Guo, M. R. Ou and X. P. Xu, RSC Adv., 2017, 7, 6278–6287 RSC.
- C. W. Abney, R. T. Mayes, T. Saito and S. Dai, Chem. Rev., 2017, 117, 13935–14013 CrossRef CAS PubMed.
- J. Kim, C. Tsouris, R. T. Mayes, Y. Oyola, T. Saito, C. J. Janke, S. Dai, E. Schneider and D. Sachde, Sep. Sci. Technol., 2013, 48, 367–387 CrossRef CAS.
- M. F. Byers and E. Schneider, Ind. Eng. Chem. Res., 2016, 55, 4351–4361 CrossRef.
- F. Endrizzi, C. J. Leggett and L. Rao, Ind. Eng. Chem. Res., 2016, 55, 4249–4256 CrossRef CAS.
- F. Best and S. Maroldo, Trans. Am. Nucl. Soc., 1981, 39, 199–201 Search PubMed.
- W. J. Williams and A. H. Gillam, Analyst, 1978, 103, 1239–1243 RSC.
- Y. Wang, Z. S. Wang, R. Ang, J. J. Yang, N. Liu, J. L. Liao, Y. Y. Yang and J. Tang, RSC Adv., 2015, 5, 89309–89318 RSC.
- M. Y. Xu, X. L. Han and D. B. Hua, J. Mater. Chem. A, 2017, 5, 12278–12284 RSC.
- E. Cal, J. Qi, O. Preedy, S. Chen, D. Boldrin, W. R. Branford, L. Vandeperre and M. P. Ryan, J. Mater. Chem. A, 2018, 6, 3063–3073 RSC.
- B. W. Zhang, X. J. Guo, S. Y. Xie, X. Y. Liu, C. J. Ling, H. J. Ma, M. Yu and J. Y. Li, RSC Adv., 2016, 6, 81995–82005 RSC.
- Z. Q. Bai, L. Y. Yuan, L. Zhu, Z. R. Liu, S. Q. Chu, L. R. Zheng, J. Zhang, Z. F. Chai and W. Q. Shi, J. Mater. Chem. A, 2014, 3, 525–534 RSC.
- W. Zhang, G. Ye and J. Chen, J. Mater. Chem. A, 2013, 1, 12706–12709 RSC.
- H. J. Schenk, L. Astheimer, E. G. Witte and K. Schwochau, Sep. Sci. Technol., 1982, 17, 1293–1308 CrossRef CAS.
- L. Shen, X. L. Han, J. Qian and D. B. Hua, RSC Adv., 2017, 7, 10791–10797 RSC.
- S. Das, Y. Oyola, R. T. Mayes, C. J. Janke, L. J. Kuo, G. Gill, J. R. Wood and S. Dai, Ind. Eng. Chem. Res., 2016, 55, 4103–4109 CrossRef CAS.
- S. Das, Y. Oyola, R. T. Mayes, C. J. Janke, L. J. Kuo, G. Gill, J. R. Wood and S. Dai, Ind. Eng. Chem. Res., 2016, 55, 4110–4117 CrossRef CAS.
- S. Brown, Y. F. Yue, L. J. Kuo, N. Mehio, M. J. Li, G. Gill, C. Tsouris, R. T. Mayes, T. Saito and S. Dai, Ind. Eng. Chem. Res., 2016, 55, 4139–4148 CrossRef CAS.
- D. Wang, J. Song, J. Wen, Y. Yuan, Z. Liu, S. Lin, H. Wang, H. Wang, S. Zhao, X. Zhao, M. Fang, M. Lei, B. Li, N. Wang, X. Wang and H. Wu, Adv. Energy Mater., 2018, 1802607, DOI:10.1002/aenm.201802607.
- J. Hu, H. Ma, Z. Xing, X. Liu, L. Xu, R. Li, C. Lin, M. Wang, J. Li and G. Wu, Ind. Eng. Chem. Res., 2015, 55, 4118–4124 CrossRef.
- X. Xu, X. J. Ding, J. X. Ao, R. Li, Z. Xing, X. Y. Liu, X. J. Guo, G. Z. Wu, H. J. Ma and X. Y. Zhao, Nucl. Sci. Tech., 2019, 30, 40–52 CrossRef.
- X. Xu, H. Zhang, J. Ao, L. Xu, X. Liu, X. Guo, J. Li, L. Zhang, Q. Li, X. Zhao, B. Ye, D. Wang, F. Shen and H. Ma, Energy Environ. Sci., 2019 10.1039/C9EE00626E.
- C. Gunathilake, J. Gorka, S. Dai and M. Jaroniec, J. Mater. Chem. A, 2015, 3, 11650–11659 RSC.
- G. Tian, J. X. Geng, Y. D. Jin, C. L. Wang, S. Q. Li, Z. Chen, H. Wang, Y. S. Zhao and S. J. Li, J. Hazard. Mater., 2011, 190, 442–450 CrossRef CAS PubMed.
- S. Y. Xie, X. Y. Liu, B. W. Zhang, H. J. Ma, C. J. Ling, M. Yu, L. F. Li and J. Y. Li, J. Mater. Chem. A, 2015, 3, 2552–2558 RSC.
- H. Meng, Z. Li, F. Y. Ma, X. N. Wang, W. Zhou and L. Zhang, RSC Adv., 2015, 5, 67662–67668 RSC.
- Y. Yuan, Y. Yang, X. Ma, Q. Meng, L. Wang, S. Zhao and G. Zhu, Adv. Mater., 2018, 30, 1706507 CrossRef PubMed.
- H. L. Liang, Q. D. Chen, J. Y. Ma, Y. Y. Huang and X. H. Shen, RSC Adv., 2017, 7, 35394–35402 RSC.
- S. Yang, J. Qian, L. J. Kuang and D. B. Hua, ACS Appl. Mater. Interfaces, 2017, 9, 29337–29344 CrossRef CAS PubMed.
- L. X. Zhang, S. Yang, J. Qian and D. B. Hua, Ind. Eng. Chem. Res., 2017, 56, 1860–1867 CrossRef CAS.
- J. Qian, S. Zhang, Y. Zhou, P. Dong and D. B. Hua, RSC Adv., 2015, 5, 4153–4161 RSC.
- R. Li, Y. Li, M. Zhang, Z. Xing, H. Ma and G. Wu, Molecules, 2018, 23, 1245 CrossRef PubMed.
- Q. H. Gao, J. T. Hu, R. Li, Z. Xing, L. Xu, M. H. Wang, X. J. Guo and G. Z. Wu, Radiat. Phys. Chem., 2016, 122, 1–8 CrossRef.
- Z. Xing, J. T. Hu, M. H. Wang, W. L. Zhang, S. N. Li, Q. H. Gao and G. Z. Wu, Sci. China: Chem., 2013, 56, 1504–1509 CrossRef CAS.
- Y. C. Jean, J. D. Van Horn, W. S. Hung and K. R. Lee, Macromolecules, 2013, 46, 7133–7145 CrossRef CAS.
- H. J. Ma, H. Hoshina and N. Seko, J. Appl. Polym. Sci., 2013, 128, 4253–4260 CrossRef CAS.
- C. J. Ling, X. Y. Liu, X. J. Yang, J. T. Hu, R. Li, L. J. Pang, H. J. Ma, J. Y. Li, G. Z. Wu, S. M. Lu and D. L. Wang, Ind. Eng. Chem. Res., 2017, 56, 1103–1111 CrossRef CAS.
- R. Li, H. J. Ma, Z. Xing and G. Z. Wu, J. Radioanal. Nucl. Chem., 2018, 315, 111–117 CrossRef CAS.
- H. J. Ma, H. Y. Chi, J. X. Wu, M. Wang, J. Y. Li, H. Hoshina, S. Saiki and N. Seko, ACS Appl. Mater. Interfaces, 2013, 5, 8761–8765 CrossRef CAS PubMed.
- M. X. Zhang, Q. H. Gao, C. G. Yang, L. J. Pang, H. L. Wang, H. Li, R. Li, L. Xu, Z. Xing, J. T. Hu and G. Z. Wu, Ind. Eng. Chem. Res., 2016, 55, 10523–10532 CrossRef CAS.
- H. J. Ma, S. D. Yao, J. Y. Li, C. Q. Cao and M. Wang, Radiat. Phys. Chem., 2012, 81, 1393–1397 CrossRef CAS.
- H. Y. Chi, X. Y. Liu, H. J. Ma, X. J. Yang, M. Yu, J. Y. Zhang, M. Wang, J. Y. Li, H. Hoshina and N. Seko, Nucl. Sci. Tech., 2014, 25, 7 Search PubMed.
- J. H. Chen, D. R. Li, H. Koshikawa, M. Asano and Y. Maekawa, J. Membr. Sci., 2010, 362, 488–494 CrossRef CAS.
- L. J. Zhang, J. Su, S. T. Yang, X. J. Guo, Y. P. Jia, N. Chen, J. Zhou, S. Zhang, S. Wang, J. Li, J. Y. Li, G. Z. Wu and J. Q. Wang, Ind. Eng. Chem. Res., 2016, 55, 4224–4230 CrossRef CAS.
- H. B. Pan, C. M. Wai, L. J. Kuo, G. Gill, G. X. Tian, L. F. Rao, S. Das, R. T. Mayes and C. J. Janke, ChemistrySelect, 2017, 2, 3769–3774 CrossRef CAS.
- J. Park, G. A. Gill, J. E. Strivens, L. J. Kuo, R. T. Jeters, A. Avila, J. R. Wood, N. J. Schlafer, C. J. Janke, E. A. Miller, M. Thomas, R. S. Addleman and G. T. Bonheyo, Ind. Eng. Chem. Res., 2016, 55, 4328–4338 CrossRef CAS.
- J.-X. Ao, Y.-H. Yuan, X. Xu, L. Xu, Z. Xing, R. Li, G.-Z. Wu, X.-J. Guo, H.-J. Ma and Q.-N. Li, Ind. Eng. Chem. Res., 2019, 58, 8026–8034 CrossRef CAS.
- S. J. Xie, S. L. Xie and B. M. Xie, Spectrosc. Spectr. Anal., 2003, 23, 615–616 CAS.
- S. R. Erga, S. B. Haugen, G. Bratbak, J. K. Egge, M. Heldal, K. A. Mork and S. Norland, Mar. Chem., 2017, 196, 1–12 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed description of modification with amidoxime group; the schematic diagrams of the marine adsorption flume; the data of TGA and DTG of the pristine UHMWPE fiber and the modified fibers; the time-dependent curves and the kinetic parameters fitted by pseudo-first-order of adsorption capacity for vanadium, copper, zinc and nickel ions. See DOI: 10.1039/c9ra05440e |
| This journal is © The Royal Society of Chemistry 2019 |