
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of CeO2 assemblies through interaction with short-chain dicarboxylic acids under facile hydrothermal conditions†
Xinsong Huangab,
Liping Lic and
Guangshe Li *ac
*ac
aKey Laboratory of Design and Assembly of Functional Nanostructures, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, P. R. China. E-mail: guangshe@jlu.edu.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cState Key Laboratory of Inorganic Synthesis & Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, P. R. China
First published on 10th September 2019
Abstract
CeO2 assemblies with various morphologies were synthesized via a facile hydrothermal method using short-chain dicarboxylic acids as the only added agent. It is demonstrated that the morphology of CeO2 assemblies depends on the chain-length of the dicarboxylic acids. The reaction with propanedioic acid (PA) results in durian-like ceria assemblies. Comparatively, ethanedioic acid (EA) tends to precipitate with Ce3+ at the beginning, and then guides the formation of lamellar octahedral assemblies. The catalytic performance towards CO oxidation of the as-synthesized CeO2 with different morphologies was investigated. Compared with lamellar octahedral assemblies, durian-like CeO2 assemblies showed better catalytic performance, giving complete CO conversion at 350 °C, due to its properties of unique oxygen vacancies, loosely packed pore structure and larger specific surface area.
Introduction
Ceria has attracted great attention as a brilliant material with remarkable redox properties and oxygen storage and release capability (OSC) in numerous applications, including catalysis, fuel cells, and gas sensors.1,2 Besides the size, shape, and doping elements, the assembly of ceria is considered as another important factor determining its chemical and physical properties.3,4 Thus, the synthesis of hierarchical assemblies of ceria with controlled micro/nanostructures is an important theme for understanding and exploiting their unique properties.Some progress has been made in the fabrication of 3D hierarchical architectures of ceria, however, usually requiring special templates through a complex experimental course.5–11 To develop template-free strategies, organic molecules have been employed for the sequential crystallization and surface-modification processes to control the particle size and morphology of the metal oxides.4,12–16 Therefore, well understanding the interaction between the organic molecules and the surface of the nanocrystals is really helpful for the design of various functional hybrid nanomaterials, and could also discover the novel properties at the interface between organic molecules and inorganic compounds for the particular applications.
Carboxyl group is widely contained in organic molecules as surface modifiers to prepare various fine inorganic particles or assemblies, especially metal oxides. Carboxylic acids are regarded as suitable and stable surface modifiers for ceria assemblies. Long-chain dicarboxylic acids have been explored to control the morphology of cuboctahedral or cubic CeO2 nanocrystals in supercritical water.17 In the presence of hexanedioic acid, cubic ceria assemblies were also synthesized in a lab-scale plug-flow reactor by heating an aqueous solution of cerium nitrate.18 However, long-chain dicarboxylic acids have low solubility in water, thus harsh conditions, such as high temperature and pressure, typically near or superior to the critical point of water, are required to promote their dissolution and further the crystallization of nanomaterials although the reaction time can be controlled to be very short and the production can be continuous. In this viewpoint, dicarboxylic acids with short chain-length might be more suitable to prepare nanomaterials in facile synthetic conditions. For instance, lamellar octahedral ceria was hydrothermally synthesized utilizing ethanedioic acid and also a good support in Pd-based catalysts for methane combustion.19 The forming mechanism of ceria in presence of short-chain organics, however, is still unclear, which limits the preparation of nano-CeO2 with other assembly structures. More effort is required to sharpen the understanding of the behaviors and mechanisms in the reaction between short-chain dicarboxyl acids and Ce species.
Herein, dicarboxylic acids with 2–4 carbon atoms, i.e. ethanedioic acid (EA), propanedioic acid (PA), butanedioic acid (BA), were chosen to systemically investigate the effect of chain-length in the dicarboxylic acid on the morphology of CeO2 assemblies under mild hydrothermal conditions with cerium nitrate as raw material. The forming mechanism of different 3D morphologies affected by the varying number of carbon atoms and the effect of these assemblies on the CO oxidation are carefully explored and discussed.
Experimental
Sample preparation
Cerium nitrate (Ce(NO3)3·6H2O), ethanedioic acid (EA), propanedioic acid (PA), and butanedioic acid (BA) were analytical grade, purchased from Sinopharm Chemical Reagent Co., Ltd., China and used as received. All aqueous solution was prepared using ultrapure water (resistivity > 18 mΩ cm, 25 °C). CeO2 assemblies were prepared as following: 0.017 mol Ce(NO3)3·6H2O was dissolved in 60 mL of water and 0.073 mol of dicarboxylic acid was added. The mixed solution was stirred for 30 min, and then transferred into a 100 mL of Teflon-lined stainless steel auto-clave and maintained at 180 °C for 24 h. The obtained precipitates were collected by centrifugation and washed with water, vacuum dried at 90 °C for 12 h (denoted as Ce-EA-p and Ce-PA-p, no precipitates were gained in Ce + BA system) and then calcined in air at 400 °C for 2 h and the calcined samples were denoted as Ce-EA and Ce-PA, respectively.Characterization methods
Micro-morphology of the samples was investigated by a field-emission scanning electron microscopy (SEM) using a JEOL JSM 6700F instrument. Thermo gravimetric analysis (TGA) was carried out on a STA449F3 thermal analyzer at a heating rate of 5 °C min−1 in nitrogen over the temperature range of 30–600 °C. Crystalline phases of the synthesized materials were identified by an X-ray diffraction (XRD) on a Rigaku Miniflex apparatus equipped with a Cu Kα radiation source (λ = 1.5418 Å). Raman analysis was performed using a 532 nm excitation laser on a LabRAM-HR Raman spectrometer. Infrared spectroscopy and in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) over the samples were performed on a Nicolet 6700 spectrometer. TEM was performed on a Tecnai G2 F20 at an acceleration voltage of 200 kV. Temperature programmed reduction (TPR) tests were carried out on a FINESORB-3010C chemisorptions analyzer at a heating rate of 5 °C min−1 using 10 vol% H2 in Ar to examine the redox behavior of the samples. Nitrogen adsorption–desorption measurements were carried out on an ASAP2020 instrument. Prior to the measurement, all samples were degassed at 120 °C under vacuum for 2 h. Brunauer–Emmett–Teller (BET) specific surface areas of the samples were obtained from the adsorption data.Activity measurements
CO oxidation was carried out under atmospheric pressure in a fixed bed reactor containing 60 mg catalyst. The react stream was composed of CO (1%), O2 (21%) and He (78%) with the total flow rate of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL h−1 gcat−1. The reactants and products analysis were investigated by gas chromatography (GC-2014) with a thermal conductivity detector.
000 mL h−1 gcat−1. The reactants and products analysis were investigated by gas chromatography (GC-2014) with a thermal conductivity detector.
Results and discussion
The interaction of EA, PA and BA with Ce species differs obviously at the very beginning of synthesis. It can be observed that under vigorous stirring at room temperature, white precipitates were produced immediately after EA addition into the aqueous solution of cerium nitrate, while the solution remains clear in Ce + PA and Ce + BA system. After hydrothermal reaction at 180 °C for 24 h, white precipitates of Ce-EA-p and Ce-PA-p were collected by centrifugation. No precipitate was found in Ce + BA system. Thus, it can be concluded that under current preparing conditions, only dicarboylic acids with 2 and 3 carbon atoms are suitable candidates of surface modifiers to prepare ceria assemblies.SEM images of Ce-PA are shown in Fig. 1. Durian-like micronized assemblies are observed. They inherited from the morphology of Ce-PA-p (Fig. S1a and b†), showing large amount of sharp pyramidate tips outwards on the surface. The inter-planar spacings in Fig. 1d are about 0.31 nm along the facets and 0.27 nm at tip direction. Both planes correspond to the (111) and (200) planes of CeO2, reflecting that the building units were well crystallized. In contrast, Ce-EA-p and Ce-EA exhibit lamellar octahedral assemblies and surface-stepped microstructures (Fig. S1c, d and S2†), which is consistent with the reported results.19
 | ||
| Fig. 1 Morphological characterization of Ce-PA sample: (a)–(c), SEM images; (d) HRTEM image (inset, FFT analysis of this particle showing that it is oriented along the [110] zone axis). | ||
As shown in Fig. 2a, Ce-PA and Ce-EA, after annealing Ce-EA-p and Ce-PA-p at 400 °C for 2 h, exhibit the same XRD patterns (CeO2, JCPDS file No. 34-0394), crystalizing in cubic fluorite structure. No other crystalline byproducts or obvious difference in characteristic diffraction peaks could be observed, confirming the high purity among various morphologies. The crystallite sizes calculated from the XRD patterns of Ce-PA and Ce-EA are 17.3 and 27.0 nm, slightly larger than that of their precursors, namely 14.7 nm for Ce-PA-p and 23.5 nm for Ce-EA-p, indicating that the bulk structure was little altered during the calcinations. In fact, the surface structure of Ce-PA and Ce-EA as well as their precursors changed a lot, revealed by the FTIR diffuse-reflection spectra, an extremely effective method for characterizing the functional groups at the surface. As depictured in Fig. 2b, strong peaks at 1446 cm−1 and at 1533 cm−1 for Ce-PA-p are assigned to the symmetric stretching vibration (νs) and asymmetric stretching vibration (νas) of the –COO− group, respectively.20–22 The binding states of –COO− can be identified using the wavenumber separation (Δ) between the asymmetric (νas) and symmetric (νs) IR bands.20–22 In Ce-PA-p, the Δ value is 87 (1533–1446) cm−1, suggesting that the –COO− groups coordinated to the CeO2 nanocrystals by chelating bidentate bonding mode. Comparatively, the intensities of these two bands for Ce-PA reduce obviously, but the shoulder peak at 1341 cm−1, assigned to the surface carbonates,23 remains strong. No peak was observed around 1700 cm−1, suggesting that there was no free –COOH on all the samples.19–21 Moreover, the weak peaks at 2925 and 2856 cm−1 (Fig. S3†), assigned to the asymmetric and symmetric stretching vibrations of the –CH2– group in the alkyl chain,24 demonstrates the presence of the aliphatic carboxylic groups as chelating agents on the surface of Ce-PA-p. In contrast, there is no residue of chelating carboxylic groups on the surface of Ce-EA-p or Ce-EA since no peaks related with –COO− group was detected. Instead, the peaks at 1466 and 1367 cm−1, attributed to the carbonate species,23 do not show obvious difference in position on both samples.
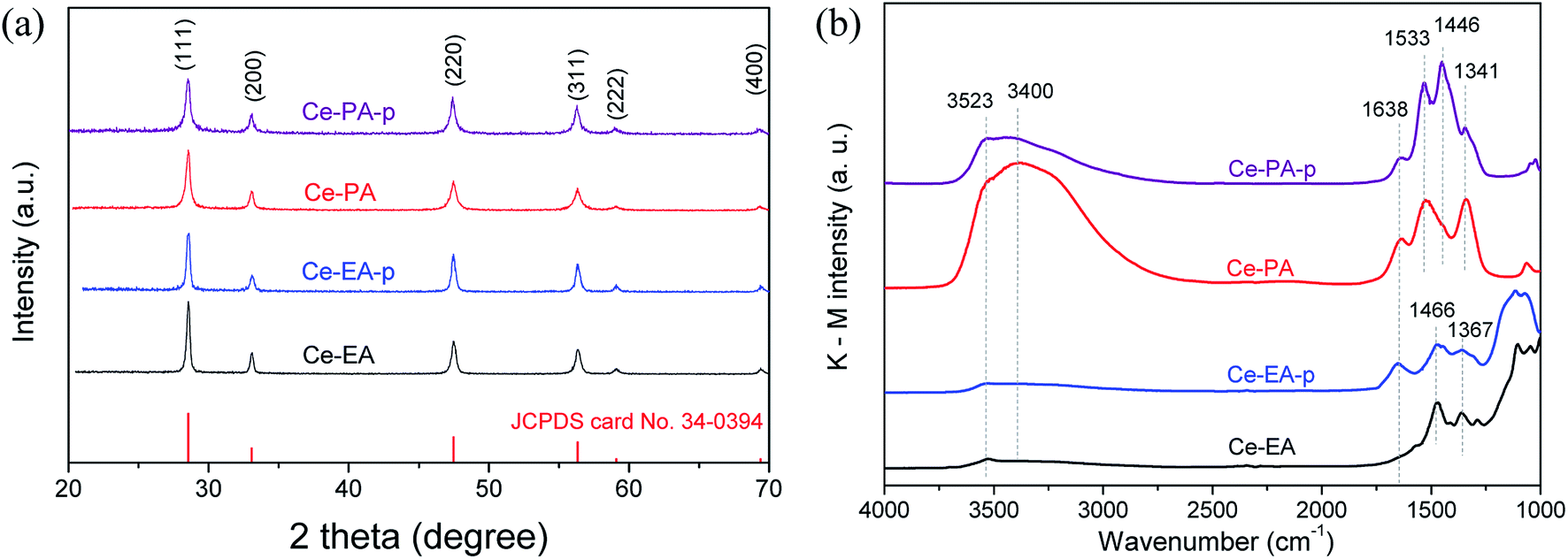 | ||
| Fig. 2 XRD patterns (a) and DRIFTS spectra (b) for Ce-PA and Ce-EA as well as their precursors. | ||
Additionally, the peaks at the band of 3400 cm−1 with a shoulder peak at 3523 cm−1, together with the peak at 1638 cm−1, are attributed to the O–H vibration and scissor bending mode of adsorbed water and –OH species. It is interesting to note that after calcination, the intensity of O–H bands for Ce-PA increased greatly, completely different from Ce-EA. For Ce-EA, it is reasonable that it contains less adsorbed water and –OH on the surface compared with the uncalcined Ce-EA-p. For Ce-PA, the desorption of organic molecules during the annealing process could generate newly exposed surfaces, which are able to adsorb water in air, resulting in large amount of the surface-adsorbed water or –OH species.
TG-DSC measurements were performed to examine the heating behaviours on the precursors Ce-PA-p and Ce-EA-p (Fig. 3). The negligible weight loss observed for Ce-EA-p indicates very limited existence of adsorbed molecules on the surface of Ce-EA-p. Comparatively, Ce-PA-p exhibits an obvious weight loss (about 2%) accompanied by an endothermic process in the range of 200–300 °C, which should be attributed to the desorption or decomposition of residual organic molecules or water on the surface of sample. To further understand the different adsorption features, nitrogen adsorption–desorption isotherms of calcined samples Ce-PA and Ce-EA were measured. Both samples give the type IV curves with H3 hysteresis loop (Fig. S4†). Ce-PA owns the feature of slit mesopores of 2–5 nm, while the pores in Ce-EA distribute widely with a much smaller pore volume, originating from the different stacking behaviors of the building blocks. Moreover, the specific area of Ce-PA was 53.7 m2 g−1, much larger than that of Ce-EA (1.6 m2 g−1).
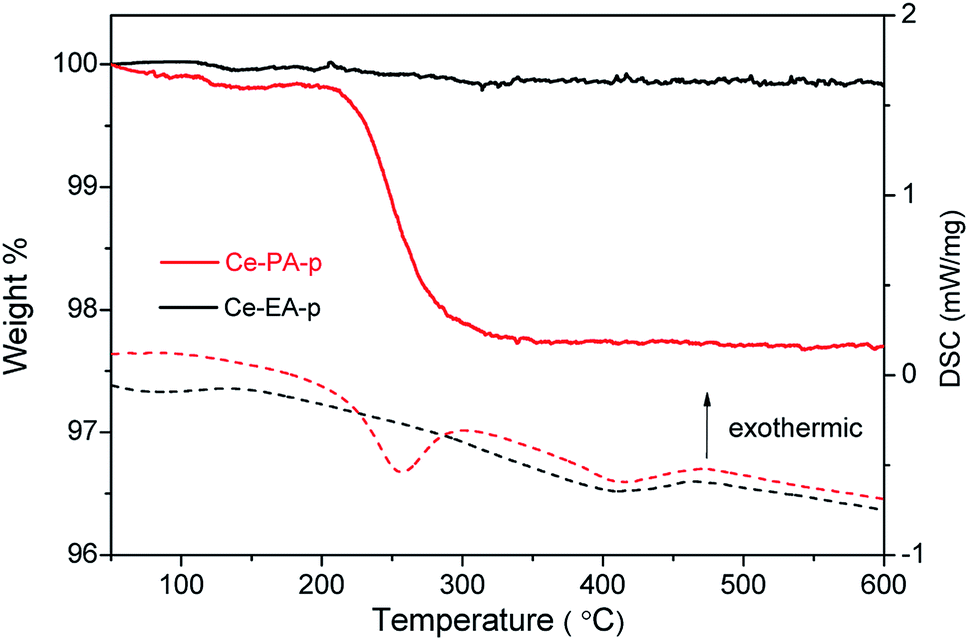 | ||
| Fig. 3 TGA (solid line) and DSC (dash line) curves of Ce-PA-p and Ce-EA-p samples. | ||
CeO2 is one of the oxygen-vacancy-rich substances. The distribution of oxygen vacancies in the lattices of samples was examined by Raman spectra. In addition to the strongest peak at 460 cm−1 that is related to the F2g vibration of the fluorite-type structure,25 both of Ce-PA and Ce-EA samples give a weak and less prominent band near 600 cm−1, as shown in Fig. 4. This band, denoted as RD, is assigned to the contribution of oxygen vacancy.26 The Raman blue shift for RD should be related with the O atom relocation from the interior of tetrahedral cationic sub-lattice sites to the interior of ideally empty octahedral cationic sites (Frenkel interstitial sites).27 The integral area ratios of RD to F2g is commensurate to the abundance of structural defects.28 The ratio of Ce-PA is 0.11, higher than that of Ce-EA (0.08), indicating that there are more oxygen vacancies in Ce-PA lattice.
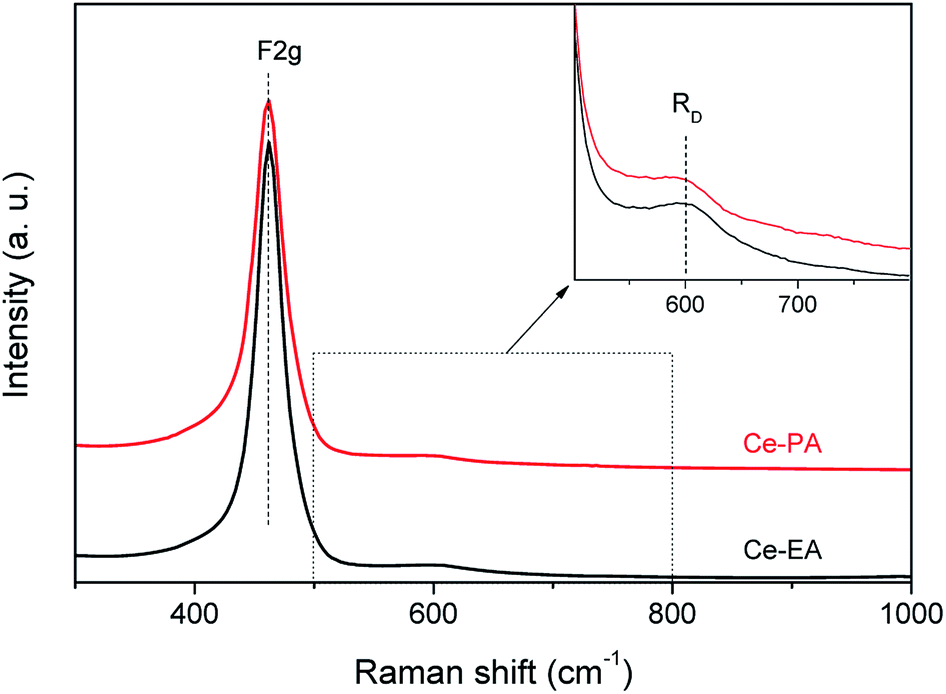 | ||
| Fig. 4 Raman spectra of Ce-PA and Ce-EA, inset: focus on the spectral region of the RD band. | ||
The concentration of Ce3+ could be estimated by analyzing the core level XPS spectra of Ce3d, as shown in Fig. 5a. Evaluated according to the method of Hilaire et al.,29 the ratios of Ce3+/Ce4+ of Ce-PA and Ce-EA were 0.308 and 0.361, respectively. It seems that even though Ce-EA has less oxygen vacancy in the bulk, as revealed by Raman spectra above, but gives more Ce3+ ions. These Ce3+ could locate at surface sites. On the other hand, high vacuum conditions and X-ray irradiation might make it difficult to determine the precise concentration of Ce3+ by XPS.30 Fig. 5b shows O1s electron core level XPS spectra for Ce-PA and Ce-EA, which also provide some information about the surface oxygen vacancies. The presence of main peaks at lower bounding energy at ca. 529 eV elucidates the existence of lattice oxygen (denoted as Olatt), and the shoulder peaks at higher bounding energy can be assigned to the adsorbed oxygen or oxygen in hydroxyl groups on the surface of ceria (denoted as Oad).31 It is clearly that Ce-EA has more Oad, which might be generated along with the higher surface Ce3+ ratio.
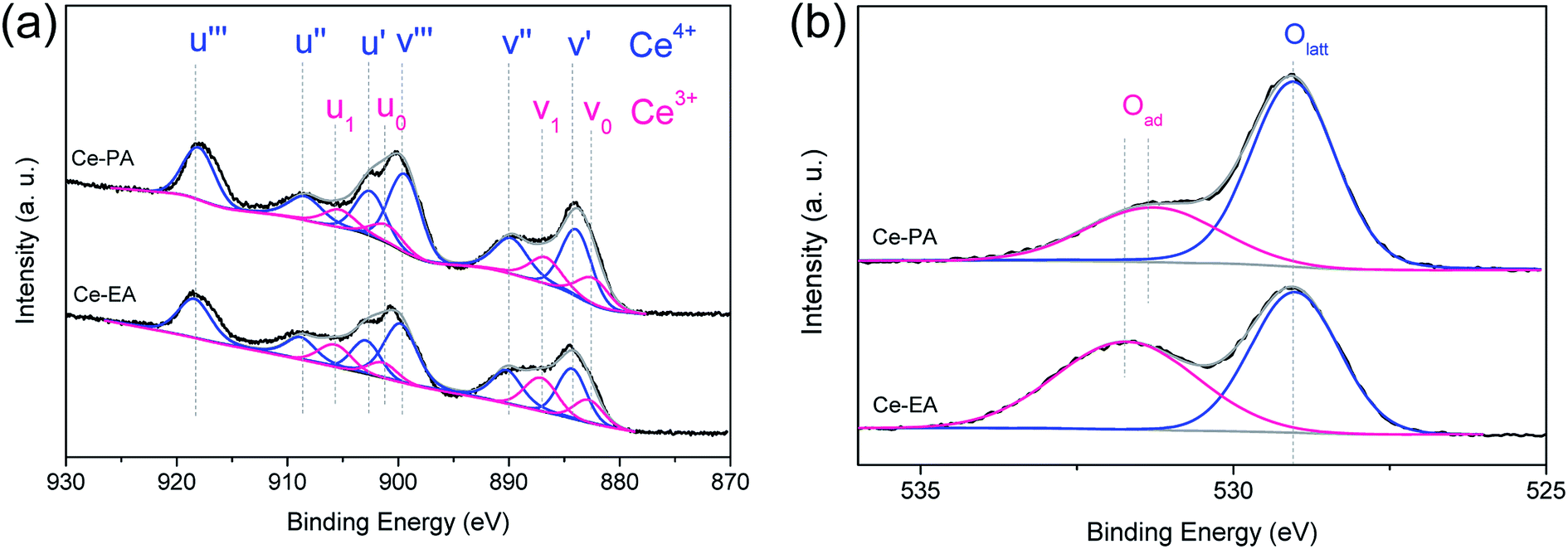 | ||
| Fig. 5 Ce3d (a) and O1s (b) core level XPS spectra of Ce-PA and Ce-EA samples. | ||
Temperature-programmed reduction (TPR) in a flow of dilute hydrogen up to 900 °C was used to characterize ceria assemblies and probe their redox properties. As shown in Fig. 6, Ce-PA and Ce-EA show a similar peak of bulk reduction at ca. 870 °C, but quite different TPR traces in the low temperature range from 300 to 700 °C. One sharp peak at 620 °C was observed for Ce-EA. This peak is usually attributed to the reduction of surface oxygen species. Comparatively, the peak for Ce-PA shifted to lower temperatures, demonstrating that it possessed more active surface reductivity, although the total amount of the reactive oxygen species is less than that of Ce-EA. Moreover, the reduction temperature of the active oxygen species was lower than ceria with other morphologies reported like spindles, spheres, cubes nanorods or octahedrons,32 suggesting that Ce-PA may have an outstanding performance in catalytic applications, especially in oxidation. The comparison for the TPR traces of Ce-PA and Ce-EA is well consistent with the results obtained in XPS and Raman analysis, i.e. Ce-PA contains less amount of Ce3+, but, more loosely-confined oxygen species.
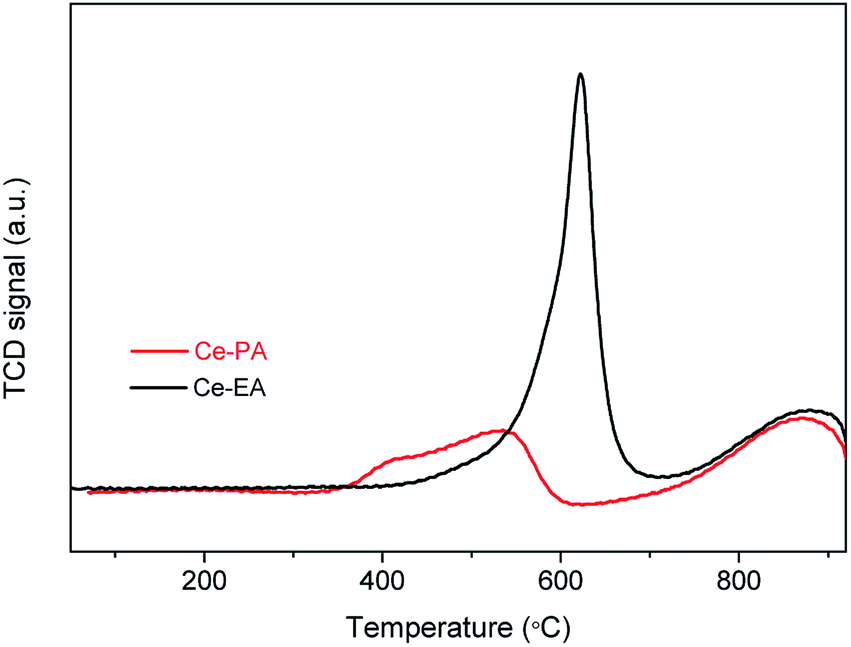 | ||
| Fig. 6 H2-TPR profiles of Ce-PA and Ce-EA samples. | ||
CO oxidation was carried out to investigate the catalytic properties of ceria assemblies from short-chain dicarboxylic acids, as shown in Fig. 7. The catalytic performance of Ce-PA-p and Ce-EA-p are generally similar with their annealed samples. A slight enhancement can be found for Ce-PA-p at temperatures below 250 °C, which might be attributed to the participation of the unstable surface adsorbed species. The enhancement could not be repeated on the used Ce-PA-p catalysts (not shown). The temperature of complete CO conversion for durian-like Ce-PA was 350 °C, much lower than that of Ce-EA (550 °C). In addition, the performance of Ce-PA is also superior to a lot of reported ceria particles or assemblies with various morphologies.4,15,16,26,33,34 To better evaluate the catalytic property, the specific rate at 300 °C together with the surface area for Ce-PA in comparison with the data in literature are listed in Table 1. It is obviously that Ce-PA possesses the highest specific rate at 300 °C per surface area among all the listed samples with or without assembling structures.
 | ||
| Fig. 7 CO conversion as a function of temperature for Ce-PA-p, Ce-EA-p (empty dots) and Ce-PA, Ce-EA (solid dots) samples. | ||
| Samples | Conversion rate at 300 °C [μmol (g−1 s−1)] | SBET [m2 g−1] | Specific rate at 300 °C per surface area [μmol (m2 s−1)] | Ref. |
|---|---|---|---|---|
| a Gas velocity & CO content: 500 cm3 min−1 & 2000 ppm.b Gas velocity & CO content: 40 cm3 min−1 & 1%.c Gas velocity & CO content: 20 cm3 min−1 & 1%.d Gas velocity & CO content: 33 cm3 min−1 & 1%.e Gas velocity & CO content: 50 cm3 min−1 & 1%.f Gas velocity & CO content: 50 cm3 min−1 & 0.5%. | ||||
| NCa (nanocubes) | 0.186 | 39.5 | 0.0047 | 26 |
| NPa (nanopolyhedra) | 0.372 | 109.4 | 0.0034 | 26 |
| NRa (nanorods) | 0.669 | 91.8 | 0.0073 | 26 |
| Nanotubesb | 1.81 | 98.2 | 0.0184 | 34 |
| Nanoparticlesb | 0.933 | 105.1 | 0.0089 | 34 |
| Nanorodsb | 1.79 | 115.9 | 0.0154 | 34 |
| Ce-PAc | 1.488 | 53.7 | 0.0277 | This work |
| Bowknot assembliesd | 0.793 | 85.0 | 0.0093 | 15 |
| Nanosheets assembliese | 0.892 | 36.7 | 0.0243 | 33 |
| Flowerlike assembliesf | 0.6324 | 34.1 | 0.0185 | 4 |
To understand the outstanding performance of Ce-PA, two features should be mentioned based on the above characterizations. The first one is the morphology effect of the durian-like assemblies. In Table 1, it can be found that ceria samples with assembling structures generally hold higher specific rates, indicating the positive role of the assembly morphology, which could intensify the mass transfer of the reactants. The mass transfer is an important factor affecting the specific rates of the catalytic reaction. For instance, for samples synthesized without organic modifiers (Row 1–6, ref. 26 and 34), the morphology was adjusted by the concentration of alkali. The limited specific rate of NC, NP and NR can be ascribed to the ultra high gas velocity (500 cm3 min−1) and low content of CO (2000 ppm),26 which is negative for the efficient mass transfer through the samples' surface. The morphology of durian-like assemblies for Ce-PA could provide moderate specific surface area and loosely packed porous structure, so the mass transfer of CO can be greatly improved. The second feature is the intrinsic defects which are able to generate more active oxygen species, as indicated by H2-TPR results. Both features for Ce-PA with the unique durian-like morphology are formed under the guidance of PA in the facile hydrothermal conditions.
To reveal the function of EA and PA, the formation process of Ce-EA and Ce-PA was tracked by investigating the precipitates collected in the varying hydrothermal times. XRD patterns of Ce-EA-x h and Ce-PA-x h (x = 1, 1.5, 3, or 6, which represents the times in hour at the fixed temperature of 180 °C) without annealing process are shown in Fig. 8. For the 1 hour reaction (Ce-PA-1 h), there was no precipitates. Prolonging the reaction time, all obtained Ce-PA-x h samples (x = 1.5, 3, or 6) are ceria (Fig. 8b). In contrast, Ce-EA-1 h is a mixed phase containing cerium oxalate (JCPDS file No. 40-0570) and cerium oxalate hydrate (JCPDS file No. 20-0268). Meanwhile, a little amount of cerium carbonate hydroxide (JCPDS file No. 40-0570) was also found. These oxalates or carbonates were not stable and could transform to ceria, as confirmed by the increased ceria content when prolonging the hydrothermal time from 3 h to 6 h for Ce-EA-3 h and Ce-EA-6 h. SEM results of Ce-EA-x h and Ce-PA-x h are also examined to explore the evolution of the morphology (Fig. S5 and S6†). For Ce-EA-x h samples, micropillars of cerium oxalate were rapidly formed after 1 h reaction, and then the product decomposed to form the attached small lamellar octahedral assemblies, which would grow larger in the continuing hydrothermal process. In contrast, all the Ce-PA-x h samples exhibit sphere assemblies or aggregates of ceria. The crystallinity of these assemblies became better with the hydrothermal time (Fig. S6†), being in accordance with the XRD observation (Fig. 8b).
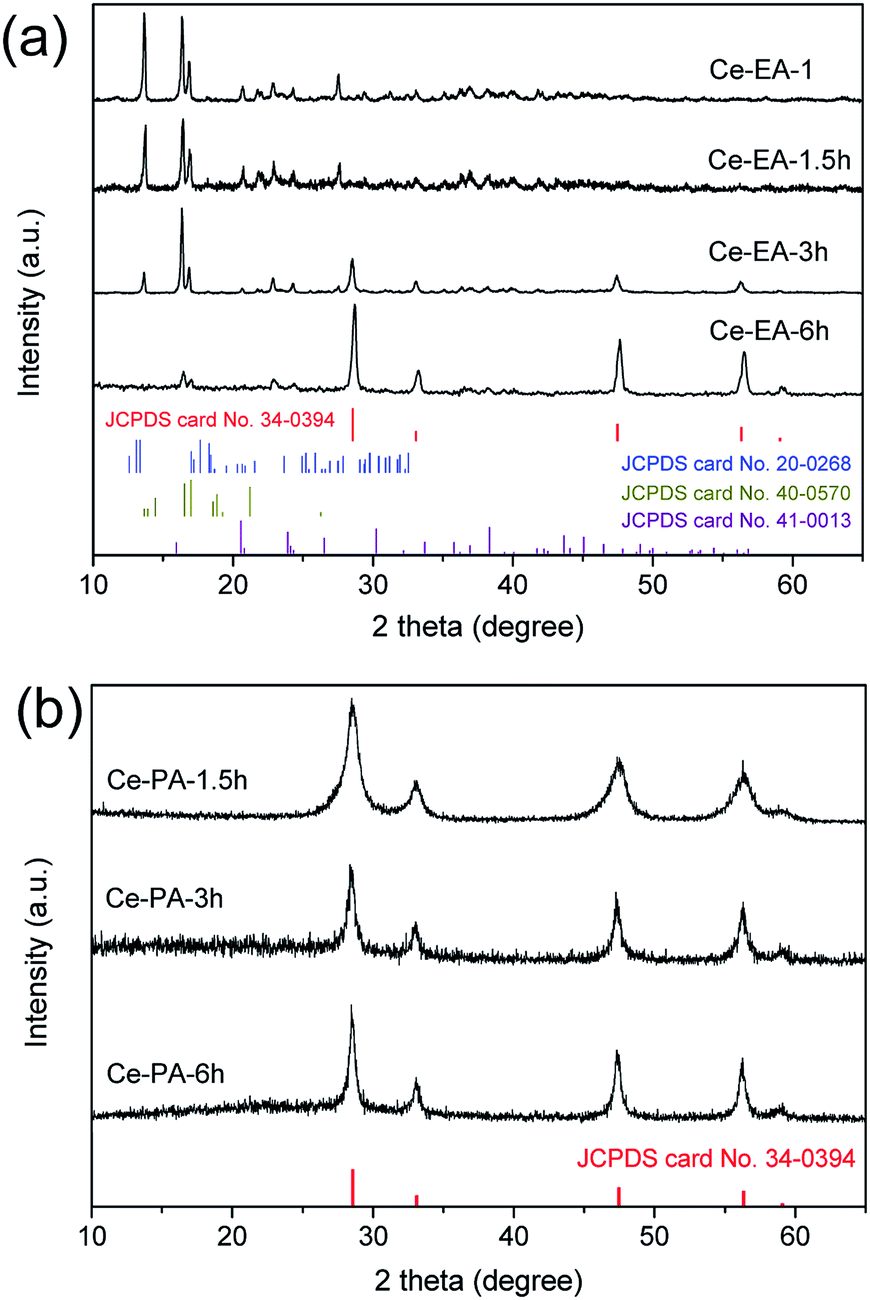 | ||
| Fig. 8 XRD patterns for Ce-EA-x h (a) and Ce-PA-x h (b) samples, x = 1, 1.5, 3, or 6, which represents the times in hour at the fixed temperature of 180 °C. | ||
The forming processes of ceria here are completely different from the systems utilizing alkali for morphology control, in which all products synthesized were 1-D structures with no assembling behavior.34 In this work, cerium nitrate, water and dicarboxylic acids are the only added agents. Therefore, the chemical properties of the short-chain dicarboxylic acids under hydrothermal condition govern the nucleation and crystal growth of cerium species. Specifically, in Ce + EA system, cerium oxalate or its hydrate precipitates firstly, due to the strong chelating effect of EA. Then the precipitates decompose to convert into ceria, while no residual carboxylic acids are produced. In contrast, the chelating ability of PA is weaker compared to EA, due to the space effect and the flexibility of the longer carbon chain. No precipitates can be generated in the beginning of the PA-added hydrothermal process. As well known, PA is easy to decompose when heated slightly above its melting point (137 °C), producing CO2 and acetic acid. Thus, in Ce + PA system, where the hydrothermal temperature is 180 °C, PA is surely decomposed into CO2 and acetic acid to produce a micro-emulsion like environment. In such an environment, acetic acid could acts as the real surface modifier, and the produced CO2–acetic acid–water species could react with Ce ions to promote the crystallization of ceria and aggregation into spherical durian-like assemblies. The proposed mechanism may illuminate that why Ce-PA-p has larger amount of residual organic materials on the surface compared with Ce-EA-p. Finally, in the case of Ce + BA, the chelating ability of BA is even weaker than that of PA. More importantly, according to the rule of Blanc, the formation of the succinic anhydride is the dominating process when BA is given enough heat to decompose.35 Neither reactive agent nor suitable environment can be provided for the nucleation or crystallization of Ce species in Ce + BA system.
Conclusions
In summary, ceria assemblies with different morphologies, namely durian-like for Ce-PA and lamellar octahedral for Ce-EA, have been successfully synthesized by adding short-chain dicarboxylic acids without any other reagents under a facile hydrothermal process. Owing to the outstanding characteristics like unique oxygen vacancies, loosely packed pore structure and larger specific surface area originated from the PA guidance, which is obviously different from the interaction of EA with Ce3+, Ce-PA possesses the excellent catalytic activity with the complete CO conversion temperature of 350 °C. The study herein may provide a good reference for the numerous organic-molecular aided synthesis of functional assemblies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grants 21871106, 21671077 and 21771075).Notes and references
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- W. Yang, X. Wang, S. Song and H. Zhang, Chem, 2019, 5, 1–32 Search PubMed.
- H. Liu and H. Liu, J. Alloys Compd., 2016, 681, 342–349 CrossRef CAS.
- L.-S. Zhong, J.-S. Hu, A.-M. Cao, Q. Liu, W.-G. Song and L.-J. Wan, Chem. Mater., 2007, 19, 1648–1655 CrossRef CAS.
- B. Xu, Q. Zhang, S. Yuan, M. Zhang and T. Ohno, Chem. Eng. J., 2015, 260, 126–132 CrossRef CAS.
- Y. Ma, W. Gao, Z. Zhang, S. Zhang, Z. Tian, Y. Liu, J. C. Ho and Y. Qu, Surf. Sci. Rep., 2018, 73, 1–36 CrossRef.
- F. Meng, J. Gong, Z. Fan, H. Li and J. Yuan, Ceram. Int., 2016, 42, 4700–4708 CrossRef CAS.
- C. Cheng, F. Chen, H. Yi and G. Lai, J. Alloys Compd., 2017, 694, 276–281 CrossRef CAS.
- M.-M. Titirici, M. Antonietti and A. Thomas, Chem. Mater., 2006, 18, 3808–3812 CrossRef CAS.
- L. González-Rovira, J. M. Sánchez-Amaya, M. López-Haro, E. Del Rio, A. B. Hungría, P. Midgley, J. J. Calvino, S. Bernal and F. J. Botana, Nano Lett., 2009, 9, 1395–1400 CrossRef PubMed.
- J. Qian, Z. Chen, C. Liu, X. Lu, F. Wang and M. Wang, Mater. Sci. Semicond. Process., 2014, 25, 27–33 CrossRef CAS.
- M. Taguchi, N. Yamamoto, D. Hojo, S. Takami, T. Adschiri, T. Funazukuri and T. Naka, RSC Adv., 2014, 4, 49605–49613 RSC.
- B. Giroire, C. Slostowski, S. Marre, C. Aymonier, T. Aida, D. Hojo, N. Aoki, S. Takami and T. Adschiri, Phys. Chem. Chem. Phys., 2016, 18, 1727–1734 RSC.
- Y. Guo, S. Lin, X. Li and Y. Liu, Appl. Surf. Sci., 2016, 384, 83–91 CrossRef CAS.
- X. Jiang, X. Huang, W. Zeng, J. Huang, Y. Zheng, D. Sun and Q. Li, RSC Adv., 2018, 8, 21658–21663 RSC.
- X. Lu, X. Li, J. Qian and Z. Chen, Powder Technol., 2013, 239, 415–421 CrossRef CAS.
- M. Taguchi, S. Takami, T. Naka and T. Adschiri, Cryst. Growth Des., 2009, 9, 5297–5303 CrossRef CAS.
- S. Takami, S. Ohara, T. Adschiri, Y. Wakayama and T. Chikyow, Dalton Trans., 2008, 5442–5446 RSC.
- T. Guo, J. Du, J. Wu, S. Wang and J. Li, Chem. Eng. J., 2016, 306, 745–753 CrossRef CAS.
- K. Nakamoto, Infrared and Raman spectra of inorganic and coordination compounds, Wiley, Hoboken, N. J., 6th edn, 2009 Search PubMed.
- L. Zhang, R. He and H.-C. Gu, Appl. Surf. Sci., 2006, 253, 2611–2617 CrossRef CAS.
- Y. Ren, K.-i. Iimura and T. Kato, Langmuir, 2001, 17, 2688–2693 CrossRef CAS.
- F. Bozonverduraz and A. Bensalem, J. Chem. Soc., Faraday Trans., 1994, 90, 653–657 RSC.
- R. Lakshmi, S. Aruna and S. Sampath, Appl. Surf. Sci., 2017, 393, 397–404 CrossRef.
- H. Li, A. Petz, H. Yan, J. C. Nie and S. Kunsági-Máté, J. Phys. Chem. C, 2011, 115, 1480–1483 CrossRef CAS.
- M. Lykaki, E. Pachatouridou, S. A. C. Carabineiro, E. Iliopoulou, C. Andriopoulou, N. Kallithrakas-Kontos, S. Boghosian and M. Konsolakis, Appl. Catal., B, 2018, 230, 18–28 CrossRef CAS.
- E. Mamontov, T. Egami, R. Brezny, M. Koranne and S. Tyagi, J. Phys. Chem. B, 2000, 104, 11110–11116 CrossRef CAS.
- C. Andriopoulou, A. Trimpalis, K. C. Petallidou, A. Sgoura, A. M. Efstathiou and S. Boghosian, J. Phys. Chem. C, 2017, 121, 7931–7943 CrossRef.
- S. Hilaire, X. Wang, T. Luo, R. Gorte and J. Wagner, Appl. Catal., A, 2001, 215, 271–278 CrossRef CAS.
- F. Zhang, P. Wang, J. Koberstein, S. Khalid and S.-W. Chan, Surf. Sci., 2004, 563, 74–82 CrossRef CAS.
- E. Beche, G. Peraudeau, V. Flaud and D. Perarnau, Surf. Interface Anal., 2012, 44, 1045–1050 CrossRef CAS.
- Y. Xie, J. Wu, G. Jing, H. Zhang, S. Zeng, X. Tian, X. Zou, J. Wen, H. Su, C.-J. Zhong and P. Cui, Appl. Catal., B, 2018, 239, 665–676 CrossRef CAS.
- X. Zhou, J. Ling, W. Sun and Z. Shen, J. Mater. Chem. A, 2017, 5, 9717–9722 RSC.
- C. Pan, D. Zhang, L. Shi and J. Fang, Eur. J. Inorg. Chem., 2008, 2429–2436 CrossRef CAS.
- S. Gál, T. Meisel and L. Erdey, J. Therm. Anal. Calorim., 1969, 1, 159–170 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04245h |
| This journal is © The Royal Society of Chemistry 2019 |