
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHierarchical porous carbon derived from carboxylated coal-tar pitch for electrical double-layer capacitors
Haiyang Wang ab,
Hongzhe Zhub,
Yixuan Lib,
Debang Qia,
Shoukai Wang*b and
Kaihua Shen*a
ab,
Hongzhe Zhub,
Yixuan Lib,
Debang Qia,
Shoukai Wang*b and
Kaihua Shen*a
aState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian, 116012, PR China. E-mail: shen_kh@dlut.edu.cn; Tel: +86-411-84986102
bSinosteel Anshan Research Institute of Thermo-Energy Company Limited, Anshan, 114044, PR China. E-mail: wsk5840863@126.com
First published on 17th September 2019
Abstract
Hierarchical porous carbons have been synthesized using amphiphilic carboxylated coal-tar pitch as a precursor via a simple KOH activation process. Amphiphilic carboxylated coal-tar pitch has a high content of hydrophilic carboxyl groups that enable it to be easily wetted in KOH solution and that facilitate the activation process. In the present study, the effect of the activation agent to precursor ratio on the porosity and the specific surface area was studied by nitrogen adsorption–desorption. A maximum specific surface area of 2669.1 m2 g−1 was achieved with a KOH to carboxylated pitch ratio of three and this produced a structure with micropores/mesopores. Among the various hierarchical porous carbons, the sample prepared with an activation agent to precursor ratio of two exhibited the best electrochemical performance as an electrode for an electrical double-layer capacitor in a 6 M KOH electrolyte. The specific capacitance of the sample was 286 F g−1 at a current density of 2 A g−1 and it had a capacitance-retention ratio of 93.9%, even after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. Thus, hierarchical porous carbons derived from amphiphilic-carboxylated coal-tar pitch represent a promising electrode material for electrical double-layer capacitors.
000 cycles. Thus, hierarchical porous carbons derived from amphiphilic-carboxylated coal-tar pitch represent a promising electrode material for electrical double-layer capacitors.
1. Introduction
With the fast-growing demands for novel and high-efficiency energy-storage devices, supercapacitors (or electrochemical capacitors) have stimulated extensive interest due to their advantages of high-power capability, superior reversibility and long life cycle.1–3 On the basis of different energy storage mechanisms, supercapacitors can be divided into two types: electrical double layer capacitors (EDLCs) and pseudocapacitors.4 EDLCs utilize electrostatic adsorption of the electrolyte ions at the electrode–electrolyte interfaces, such as carbon materials,5,6 while pseudocapacitance electrode materials store the electrical energy by fast and reversible faradaic redox reactions occurring at the surface of electrodes, such as transition metal oxides7,8 and conducting polymers.9 Pseudocapacitance electrode materials possess high specific capacitance, but suffer from poor cyclability and rate capability which limit their further applications. On the contrary, porous carbon materials are the most promising candidates for commercial electrodes. An ideal electrode material is expected to have a large surface area, abundant accessible micropores for energy storage and mesopores for ion transport, interconnections between the pores for enhanced charge-storage sites and excellent charge/discharge rates.10,11Fabrication of hierarchical-porous carbons (HPCs) with high surface areas and large pore volumes as electrodes have long been pursued. Nowadays, HPCs are mainly synthesized via the pyrolysis and activation of carbon precursors including fossil fuels,12 biomass,13,14 synthetic polymers materials15–18 and some organic wastes.19 HPCs have been generally prepared by hard/soft templating approaches or templating/chemical activation combination methods.20–23 These strategies were successful in preparing HPCs with precise nanostructures. However, these techniques have limitations as they involve high production cost, energy consumption, longer carbonization/activation time, and more activation/template agents.24–26 Hence, it is necessary to find a rapid, efficient and economical route for the preparation of low-cost HPCs containing micropores/mesopores for supercapacitors for practical application purposes.
Coal-tar pitch (CP) contains carbonaceous polycyclic-aromatic hydrocarbons as its major component and relatively high in carbon content. Therefore, it is an inexpensive and promising precursor for preparing HPCs.27,28 However, CP is composed of lamellar macromolecules in parallel stacks and has a dense structure. CP upon direct pyrolysation and carbonization, became non-porous, irregular-shaped semi-coke or coke and cannot absorb a large number of ions and thereby ineffective for energy storage. Alternatively, HPCs were also obtained by a direct KOH-activated method of CP, and this method achieved an SBET value of 3200 m2 g−1.29 However this process had limitations (i) partial contact of CP particles upon KOH soaking, due to CP's irregular shapes and large sizes (ii) chemical activation from external to internal areas requires large amount of KOH, long activation time and high temperature (iii) obtained HPCs were microporous, and not effective for energy storage at a higher charge rate.
Another approach was introduction of hydrophilic polar groups such as OH and COOH in the CP architecture. This methodology was promising as the lamellar structure in CP was interrupted and aggregation from π–π stacking was avoided by the presence of hydrophilic groups. However, introduction of OH group required hazardous and strong oxidizing reagents (e.g., HNO3, KMnO4, and/or H2SO4) and a tedious multistep process.30–33 In addition, use of strong oxidizing agents caused extensive damage to the carbon-basal plane with a large number of chemical and topological defects on the prepared HPCs.34 The introduction of COOH groups in CPs were not explored yet. Hence, we report here a carboxylated coal-tar pitch (CCP) by simple Friedel–Crafts acylation followed by Baeyer–Villiger oxidation. HPCs were prepared from CCP by KOH activation and were investigated by nitrogen adsorption–desorption method, X-ray diffraction (XRD) and Raman spectroscopy. Electrochemical performances of these HPCs as an electrode for EDLCs were also evaluated.
2. Experimental procedures
2.1 Preparation of CCP
CCP was prepared by the Friedel–Crafts acylation followed by Baeyer–Villiger oxidation (Scheme 1). Under an argon atmosphere, 50 g of CP was dissolved in 500 mL of dichloroethane. Next, 50 g of AlCl3 and 30 g of oxalyl chloride were added dropwise. The reaction system was heated to 40 °C for 6 h. After the reaction, the mixture was quenched by 100 mL of ethanol. Subsequently, the reaction mixture was filtered and washed with ethanol and hydrochloric acid. The filter cake was washed with distilled water at least three times. The diketone functional coal-tar pitch (DKCP) was dried to a constant weight in vacuum under 80 °C. The weight yield of DKCP (based on CP) was 112% (wt%). | ||
| Scheme 1 Preparation of super capacitor from coal tar via carboxylated coal-tar pitch (CCP) followed by carbonization. | ||
The 15 g DKCP sample was dispersed into 75 mL of ethanol, and then 75 mL of 30% hydrogen peroxide solution was added dropwise. The mixture was then kept at 30 °C and stirred for 6 h in a water bath. After the reaction, the mixture was filtered and washed with distilled water at least three times. The final sediment, i.e., the carboxylated coal-tar pitch (CCP), was dried to a constant weight in vacuum under 80 °C. The weight yield of CCP (based on DKCP) was 96% (wt%).
2.2 Carbonization/activation
Aqueous 30% KOH was used for the activation of CCP. The mass ratio of KOH to CCP varied from 1 to 3. CCP was added to the aqueous 30% KOH solution and stirred for 1 h and then dried to a constant weight in vacuum under 80 °C for 12 h. The dried materials were carbonized and activated in a tube furnace at 800 °C for 2 h under a flow of nitrogen. After activation, the samples were washed three times with 1 M HCl solution and rinsed with deionized water 3 to 5 times until a pH of 7 was attained for the washing solution. Finally, the samples were dried at 120 °C for 12 h and were referred as HPC-x, where x = 1, 2, 3, x represents the weight ratio of KOH to CCP.2.3 Measurements and analyses
Fourier transform infrared spectroscopy (FTIR) was performed on a Thermo Nicolet-360 spectrometer (USA). Elemental analysis (EA) (carbon, hydrogen, oxygen and nitrogen) was done with a Vario Macro EL analyzer (Germany). Thermogravimetric analysis (TGA) was performed using a HCT-1 instrument (China). Surface morphology of HPCs were analyzed by a JSM-6700F field emission scanning-electron microscope (FESEM, Japan). Powder X-ray diffraction (XRD) spectra and Raman spectra were recorded on a D8 ADVANCE A25 X-ray powder diffractometer (Germany) using Cu-Kα radiation (λ = 1.541 Å) and a 769G05 laser Raman spectrometer (UK), respectively. Surface chemical composition was determined by X-ray photoelectron spectroscopy (XPS) on a Thermo ESCALAB250, USA. The surface area and porosity of HPCs were estimated from the isotherms of nitrogen adsorption–desorption at 77 K by Accelerated Surface Area & Porosimetry (ASAP2020). The specific surface area was calculated by Brunauer–Emmett–Teller (BET) equation. The pore-size (diameter) distributions (PSDs) were determined using the nonlocal-density functional theory, assuming slit-pores geometry.2.4 Electrochemical measurements
The carbon electrode was fabricated as follows. HPCs and polytetrafluoroethylene (PTFE) were mixed in a weight ratio of 9:1. The resulting mixture was rolled into a thin film and further cut into a circular shape (12 mm diameter). Each round film had a mass loading of 2.5 mg cm−2. The film was dried under vacuum at 120 °C for 2 h. This was then pressed onto nickel foams to fabricate supercapacitor electrodes. Finally, the electrodes were soaked in a 6 M KOH solution degassed using a vacuum for 2 h. Cyclic voltammetry (CV) and electrochemical-impedance spectroscopy (EIS) were conducted on a CHI760E electrochemical workstation (Chenhua, Shanghai, China). EIS was carried out over a frequency range of 100 KHz to 0.01 Hz with an amplitude of 5 mV. The galvanostatic charge–discharge (GCD) measurements and life-cycle tests were conducted on a supercapacitance test system (SCT) by Arbin Instruments, USA. The specific capacitance of the working electrodes was calculated from the galvanostatic-discharge process via the following equations:
 | (1) |
In a two-electrode cell, the two symmetrical electrodes and a porous polypropylene separator were sandwiched together in a poly(tetrafluoroethylene) cell. The specific capacitance of the single electrode was calculated as:35,36
 | (2) |
The energy and power density of the symmetric supercapacitor system were calculated by the following eqn (3) and (4):
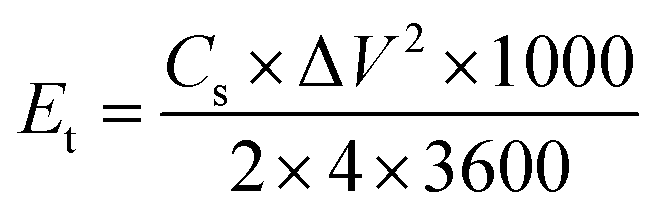 | (3) |
 | (4) |
3. Results and discussion
3.1 Material characterization
Table 1 represents the bulk and surface elemental compositions of CP and CCP. EA data for CCP showed an oxygen content of 18.4% with C/O ratio of 5.71, representing an average of one carboxylic-acid group for 11.42 carbons. Fig. 1a represents the FTIR spectra of CCP and HPCs. For CCP, the signals at 3040 and 2915 cm−1 result from aromatic C–H stretching vibrations and aliphatic C–H stretching vibrations, respectively.37 The signals at approximately 1720 cm−1 are assigned to carboxylic acid C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations and the signals at 1600 cm−1 are attributed to aromatic CC stretching vibrations.38 The peaks at 1440 cm−1 and 1380 cm−1 are attributed to the C–H bending vibration of methyl and methylene.39 For the HPCs, the significant decrease in the intensity of signals corresponding to the CO group confirmed the loss of COOH groups during the KOH-activation process. The introduction of carboxyl groups induces cross-linking of the CP structure, which prevents the melting and orderly rearrangement of the CP during the high-temperature carbonization process, and inhibits the graphitization process. The evolution of gas (CO2 and CO) during the high-temperature process changes the microstructure of the carbon materials and plays a dual regulation role.
O stretching vibrations and the signals at 1600 cm−1 are attributed to aromatic CC stretching vibrations.38 The peaks at 1440 cm−1 and 1380 cm−1 are attributed to the C–H bending vibration of methyl and methylene.39 For the HPCs, the significant decrease in the intensity of signals corresponding to the CO group confirmed the loss of COOH groups during the KOH-activation process. The introduction of carboxyl groups induces cross-linking of the CP structure, which prevents the melting and orderly rearrangement of the CP during the high-temperature carbonization process, and inhibits the graphitization process. The evolution of gas (CO2 and CO) during the high-temperature process changes the microstructure of the carbon materials and plays a dual regulation role.
| Sample | Element analysis (wt%) | XPS | ||||||
|---|---|---|---|---|---|---|---|---|
| C | H | O | N | C/H | C/O | C/at% | O/at% | |
| CP | 93.2 | 4.7 | 1.9 | 0.2 | 1.65 | 65.40 | 95.2 | 4.6 |
| CCP | 78.8 | 2.6 | 18.4 | 0.2 | 2.53 | 5.71 | 83.6 | 15.3 |
 | ||
| Fig. 1 (a) FTIR spectra; (b) XPS wide-scan spectra of CCP, HPC-1. HPC-2, HPC-3; (c) XPS spectra of CCP C 1s; (d) XPS spectra of CCP O 1s. | ||
XPS wide-scan spectra of CCP and HPC samples are represented in Fig. 1b. The intensity of oxygen O 1s peak located at 532 eV was significantly stronger in CCP indicating a high concentration of oxygen-containing groups in CCP than in HPC. In the C 1s spectrum of the CCP (Fig. 1c), there were two peaks representing different types of functional groups, namely C–C, CC, and C–H bonds (284.3 eV), and OC–O bonds (288.4 eV).40 The O 1s spectrum of the CCP (Fig. 1d) showed a peak at 533.8 eV and can be assigned to OC–O bonds. The minor peak at 535.0 eV can be attributed to the physically absorbed moisture on samples. These observations are consistent with FT-IR spectroscopy and elemental analysis indicating the presence of COOH groups in CCP and their absence in HPCs.
The thermal behavior CCP was obtained from their TGA (Fig. 2a) and TPD profiles (Fig. 2b). From room temperature to 200 °C, there was minimal weight loss (1.6%), which can be assigned to the removal of the adsorbed water. The mass loss between 200 °C and 600 °C was significant (29.9%) and can be assigned to the crosslinking and decomposition reaction of COOH groups and carboxylates.41 The mass loss between 600 °C and 800 °C was 3 wt% and can be attributed to the dehydroaromatization. The TPD profile of CO2 shows the highest peak at 262 °C with a strong shoulder at 402 °C and a large tail indicating the presence of different CO2 releasing groups such as anhydrides.
 | ||
| Fig. 2 TGA (a) and TPD profiles (b) of CCP. | ||
Based on the TGA and TPD results, the two possible pathways for the thermal decomposition of ArCOOH are (1) decarboxylation via a free-radical process to produce aryl radicals, which in turn being highly reactive extract hydrogen or combine themselves to form polyaromatic structures;42 (2) dehydration reaction leading to the formation of anhydride-crosslinked bridges between the adjacent aromatic rings and subsequent pyrolysis, forming polyaromatic structures.43 Formation of anhydrides can be reasoned to the dehydration of two neighboring carboxylic groups, and subsequent disintegration to generate CO and CO2,44 the decomposed route of –COOH, as observed in Fig. 3.
 | ||
| Fig. 3 Thermal decomposition pathways of CCP leading to HPC formation. | ||
3.2 Surface morphology characterization of HPCs
The XRD (a) and Raman spectra (b) of HPCs are presented in Fig. 4. The broad peaks at approximately 25° and 43° in XRD spectra are indicative of amorphous characteristic of HPCs.45 The amorphous property of HPCs was also analyzed by Raman spectra, as shown in Fig. 4b. The D band at approximately 1352 cm−1 represents the defective graphitic structures and disordered carbons, while the G band at approximately 1585 cm−1 refers to the bond stretching of sp2-hybridized carbons.46 The integration-area ratio for D and G bands (AD/AG) is 3.05, 3.25 and 3.64 for HPC-1, HPC-2 and HPC-3, respectively, indicating the low graphitization degree in all HPCs.47 Moreover, the AD/AG ratio of HPC-3 is the highest among the three samples because the highest activation degree of KOH results in the maximum lattice defects in HPC-3. Together with XRD results, these results indicate that the structure of the obtained carbon materials can be tuned by changing the dosage of KOH. | ||
| Fig. 4 XRD (a) and Raman spectra; (b) of HPC-1, HPC-2 and HPC-3. | ||
The morphology of HPCs was investigated by the FESEM technique and the results are represented in Fig. 5. The usage of the KOH was the main governing index that affected their porous nanostructure. The FESEM images indicate that HPC-2 possessed a homogeneous sheet-like structure and that the carbon sheets were interconnected (Fig. 5b and e). For HPC-1, the pore structure is undeveloped due to the small amount of activator (Fig. 5a and d). However, HPC-3, upon increasing the usage of KOH, has many coralloid micro-mesopores, and the carbon pores were interconnected (Fig. 5c and f). Therefore, the morphology and the pore sizes of the HPCs were tuned by the dosage of KOH usage.
 | ||
| Fig. 5 SEM images of HPC-1 (2 μm (a), 500 nm (d)), HPC-2 (2 μm (b), 500 nm (e)), and HPC-3 (2 μm (c), 500 nm (f)). | ||
The N2 adsorption–desorption isotherm (Fig. 6a) and pore-size distribution of the HPCs (Fig. 6b) were also determined. The isotherms exhibited typical characteristics of type I/IV, with a sharp adsorption capacity at low relative pressures, a knee at P/P0 in the range of 0.05–0.4 and a platform without obvious hysteresis loops in the high P/P0 pressure region, indicating abundant micropores and narrow mesopores in the HPCs.48 Fig. 6b is the pore diameter distributions of HPCs. The mesopore diameter of HPC-1 and HPC-2 mainly ranges from 2.0 to 4.0 nm, while that of HPC-3 ranges from 2.0 to 8.0 nm. For HPC-3, the enlarged mesopores are ascribed to extra activation of increased KOH dosage to some small mesopores (2.0–4.0 nm). Thus, the surface functional groups of the CP and the usage of KOH provide a meaningful contribution to pore structure.
 | ||
| Fig. 6 N2 adsorption/desorption isotherms (a) and pore-size distribution; (b) for HPC-1, HPC-2 and HPC-3. | ||
The pore-structure data of HPCs are listed in Table 2. The SBET and Vtotal of HPCs increased in the order of HPC-1 (1095.1 m2 g−1, 1.05 cm3 g−1) < HPC-2 (2098.2 m2 g−1, 1.63 cm3 g−1) < HPC-3 (2669 m2 g−1, 1.98 cm3 g−1). The high surface areas observed in HPCs can be due to the presence of COOH groups in CCP making CCP readily soluble in KOH solution favoring homogeneous activation.49 Based on the above analysis, we conclude that the pore structure of HPCs can be tuned by changing the dosage of KOH.
| Samples | SBET (m2 g−1) | Smeso (m2 g−1) | Vtotal (cm3 g−1) | Vmicro (cm3 g−1) | Vmeso (cm3 g−1) |
|---|---|---|---|---|---|
| HPC-1 | 1095.1 | 211.2 | 1.05 | 0.39 | 0.66 |
| HPC-2 | 2098.2 | 884.3 | 1.63 | 0.31 | 1.32 |
| HPC-3 | 2669.1 | 1582.1 | 1.98 | 0.29 | 1.69 |
In EDLCs, pore size plays an important role in addition to surface area as specific capacitance is governed by the easy accessibility of electrolyte to the pores of electrodes. Micropores can create bottlenecks and prevent solvated ions from entering into interior surfaces of the pores. According to Raymundo-Piñero et al.,50 the optimal pore size for an effective double-layer formation in an aqueous electrolyte media is approximately 0.7 nm. It is clear from Fig. 6b that all of the HPCs exhibited hierarchical porous structures. The ultrafine micropores had pore-width peaks of 0.8 and 0.9 nm and the micropores had a maximum pore width of 1.2 nm providing a highly ion-accessible surface, which is crucial for the formation of EDLCs.51 The mesopores were in the range of 2–8 nm, exhibiting fast ion transport and low resistance for charge transfer.
3.3 Electrochemical characterization
The electrochemical performances of HPCs were investigated by CV with a three electrode system in a 6.0 M KOH solution at a scan rate of 50 mV s−1 (Fig. 7a). The CV curves of all HPC electrodes exhibited a typical rectangular I–V curve without any redox peaks between −1.0 to 0 V, suggesting that all HPCs show a pure capacitive behavior.52 The rectangular curves at a high scanning rate indicated better electrochemical stability and rate capability due to the fast ion transport occurring in the pores of the electrode. The galvanostatic charge–discharge curve for the HPC electrodes at a current density of 1 A g−1 are shown in Fig. 7b. All curves were nearly triangular in shape, which is typical behavior for capacitive electrodes.53 HPC-1 had a smaller specific capacitance than HPC-2 and HPC-3. When the KOH to CCP ratio was increased from 1 to 2 in HPC-2 the gravimetric-specific capacitance increased rapidly from 230 to 320 F g−1. When the ratio was further increased from 2 to 3, there was no further increase in the capacitance. This can be ascribed to the low surface area and ultrafine micropores in HPC-1, which are not favorable for fast diffusion of the electrolyte into the pores. Micropores limited the motion of the ions thus, lending the inner parts of the electrode inaccessible at high charge/discharge rates. | ||
| Fig. 7 Electrochemical performance of HPC electrodes via a three-electrode system in a 6.0 M aqueous solution of KOH (a) CV curves at 50 mV s−1. (b) Charge–discharge profiles at 1 A g−1. (c) Capacitance at different current densities. (d) Nyquist plots of electrodes from HPCs with insets, in the frequency range 100 kHz to 0.01 Hz with an ac perturbation of 5 mV. | ||
Fig. 7c represent the relationship between specific capacitance and current density from which performance rate of all HPCs were determined. The general trend in all HPCs is that the specific capacitance decreases with the increasing current density.54 At a current density of 1 A g−1 HPC-2 exhibited a capacitance of 320 F g−1 which is higher than that of HPC-1 (230 F g−1) and HPC-3 (272 F g−1). BET surface area and Vtotal of HPC-2 (2098.2 m2 g−1,1.63 cm3 g−1) are both smaller than HPC-3 (2669 m2 g−1, 1.98 cm3 g−1), however, the EDLC of HPC-3 (272 F g−1 at 1 A g−1) is lower than HPC-2 (320 F g−1 at 1 A g−1). Again, the same trend was followed during the increase in current density. At a high current density of 10 A g−1, HPC-2 exhibited a specific capacitance of 268 F g−1, and retained about 83.8% of the specific capacitance. The capacitance retentions in the same current range were 60.0% and 71.3% for HPC-1 and HPC-3, respectively. This result is in good agreement with the results of charge–discharge curves of the HPC electrodes in Fig. 7b.
To further evaluate the electrochemical performances of HPC supercapacitors, the EIS was obtained at a frequency ranging from 100 kHz to 0.01 Hz. Fig. 7d presents the Nyquist plots of HPCs. It can be found that all the HPC supercapacitors present a straight line parallel to the Y axis at the low-frequency part, which is indicative of an ideal capacitive behavior.55
Size of semicircle in high to medium frequency regime which symbolizes charge transfer resistance (Rct). Nyquist plots revealed that electrodes from all HPCs have similar impedance behaviors. In the high-frequency region, the intercept of the semicircle with the real axis corresponds to the internal resistance. This is similar for all HPCs, suggesting that they have a steady conductivity. The HPC-2 electrode has a smaller semicircle in the high-frequency region compared with HPC-1 and HPC-3 (Fig. 7d, inset), indicating that it possesses excellent electrical conductivity and the fastest charge transfer speed among the three samples. This is because in HPC-2 there are abundant mesopores, facilitating the electrolytes to shuttle back and forth in comparison with HPC-1 and HPC-3, thus leading to a facile adsorption/desorption process at the electrolyte–carbon interface, and hence a smaller semicircle is observed.
Since the three-electrode configuration described above might produce errors leading to overestimating of capacitance, symmetric supercapacitor was assembled using HPC-2, both as the positive- and negative-electrodes. Fig. 8a shows the CV curves at scanning rates of 5–200 mV s−1 in a 6.0 M KOH solution. All the CV curves were in a perfect rectangular shape without any redox peaks or distortions suggesting faster ion/charge transport within electrodes and a near-ideal capacitive behavior with an excellent rate capability. Fig. 8b shows the galvanostatic charge–discharge curves of the HPC-2-based supercapacitor at current densities from 0.25–10 A g−1. The symmetric linear charge and discharge curves with negligible voltage drop demonstrated a high coulombic efficiency and a minimal internal resistance. The specific capacitances at different current densities are shown in Fig. 8c. From the discharge curve, the specific capacitance at a constant current density of 0.25 A g−1 was found to be 319 F g−1, which is much higher than that of the RGO-CMK-5 electrode (144.4 F g−1 at 0.2 A g−1),56 the curved graphene electrode (154.1 F g−1 at 1 A g−1),57 and the 3DG-MnO2-13% electrode (36 F g−1 at 0.5 A g−1)58 in a two-electrode system. Notably, the capacitance was 242 F g−1 at a high current density of 50 A g−1. Fig. 8c reveals that the capacitance decreases quickly from 319 to 295 F g−1 when the current density increases from 0.25 to 1 A g−1. And then, it drops slowly at higher current densities ranging from 1–50 A g−1. Fig. 8d represents the Ragone plot, which shows that the energy density exhibits only a minimal drop with the increase in power density. As is already known, the energy density of devices is decided by capacitance (C) and voltage (V). In aqueous electrolytes, the maximum voltage is 1 V due to the oxygen-evolution reaction. In the present study, the energy and power density were 8.4 W h kg−1 and 6.9 kW kg−1 at a current density of 50 A g−1, respectively, which confirmed an outstanding power performance. These values are higher than previously reported carbon materials59–61 studied under the same measurement conditions.
 | ||
| Fig. 8 Capacitive performances of symmetric electrodes for HPC-2 in 6.0 M aqueous solution of KOH: (a) CV curves at scanning rates of 5–200 mV s−1; (b) charge–discharge profiles at current densities of 0.25–10 A g−1; (c) plot of specific capacitance vs. current density; (d) Ragone plot of energy density vs. power density. | ||
Cycling stability is one of the most important parameters for supercapacitors. The cycling stability of the HPC-2-based supercapacitor was investigated by a consecutive charge–discharge measurement at a constant current density of 2 A g−1 for 10000 cycles. The specific capacitance of the HPC-2 electrode gradually decreased with increasing cycles (Fig. 9a) and a capacitance retention of 93.9% was obtained after 10000 cycles, indicating good electrochemical stability. The rectangular CV profile (Fig. 9a, inset) and Nyquist plots (Fig. 9b) with negligible changes after 10000 cycles also supported electrochemical cyclability. Again, the small semicircle in the high-frequency region and the almost vertical line in the low-frequency region indicated that HPC-2 based super capacitors had an excellent electrical conductivity.
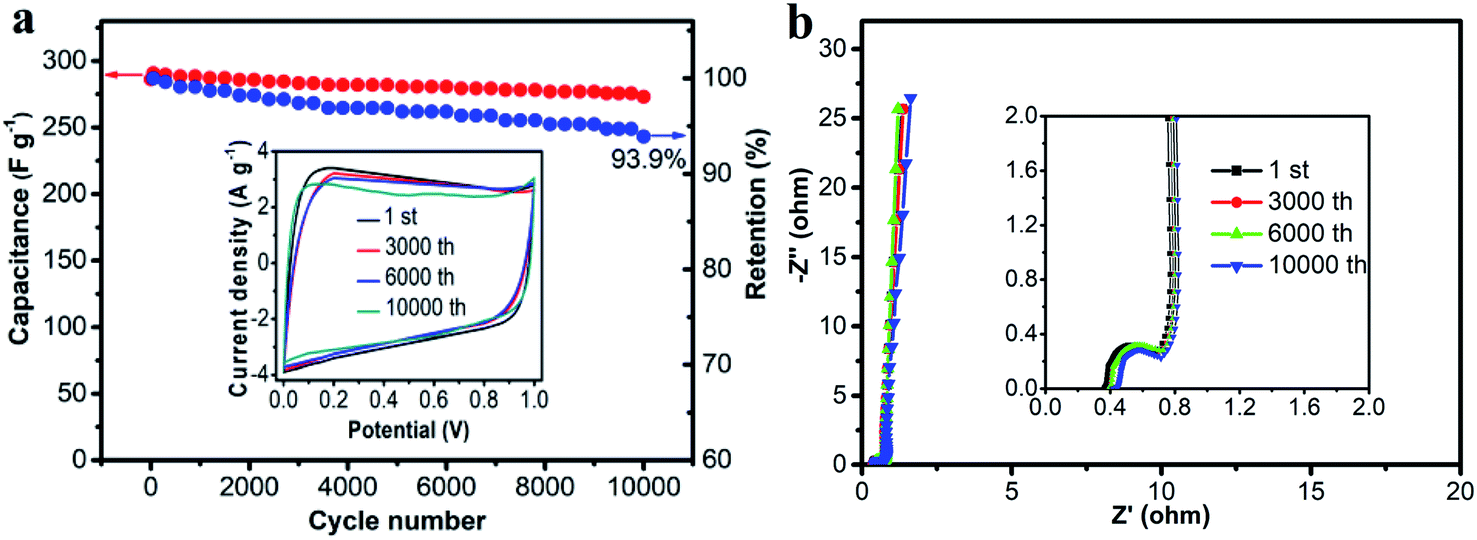 | ||
| Fig. 9 (a) Plot of specific capacitance (HPC-2 electrode) vs. the number of cycles Inset: CV curves at different cycles. (b) Nyquist plots of the different cycles with insets representing the high frequency region. | ||
4. Conclusions
CCPs were prepared by simple Friedel–Crafts acylation and subsequent Baeyer–Villiger oxidation of CPs. Presence of COOH groups in CCP was confirmed by elemental analysis, IR spectroscopy, TGA and TPD methods. HPCs were prepared by KOH activation of CCPs and subsequent carbonization. HPCs were thoroughly characterized by nitrogen adsorption–desorption method, X-ray diffraction (XRD) and Raman spectroscopy. The SBET and Vtotal of the HPCs increased with KOH to CCP ratio of 1–3. HPC-2 prepared with mass ratio of KOH to CCP two gave a specific surface area of 2098.2 m2 g−1 and a porosity volume of 1.28 cm3 g−1. A symmetrical EDLC using the HPC-2 as the electrode showed superior capacitive behavior when compared with HPC-1 and HPC-3. EDLC from HPC-2 possess specific capacitance of 286 F g−1 at a current density of 2 A g−1 and had capacitance-retention ratio of 93.9% after 10000 cycles. Thus, HPCs derived from CCPs exhibit great potential for applications in energy storage.
Conflicts of interest
There are no conflicts to declare.References
- Y. Wang, B. Chang, D. Guan and X. Dong, J. Solid State Electrochem., 2015, 19, 1783–1791 CrossRef CAS.
- J. Wong, Y. Chen, K. Cao, L. Yang, H. John and X. Tao, ACS Appl. Mater. Interfaces, 2018, 10(46), 39839–39850 CrossRef PubMed.
- Y. Zhang, X. Liu, S. Wang, S. Dou and L. Li, J. Mater. Chem., 2016, 4, 10869–10877 RSC.
- H. Hwang, C. H. Kima, J. Wee, J. H. Han and C. Yang, Appl. Surf. Sci., 2019, 489, 708–716 CrossRef CAS.
- X. Xiao, Y. Zeng, H. Feng, K. Xu, G. Zhong, S. Wu, C. Wang, W. Zhao, W. Su, Z. Wei and X. Lu, ChemNanoMat, 2019, 5, 152–157 CrossRef CAS.
- B. Fang, J. H. Kim, M.-S. Kim, A. Bonakdarpour, A. Lam, D. P. Wilkinson and J.-S. Yu, J. Mater. Chem., 2012, 22, 19031–19038 RSC.
- R. B. Rakhi, B. Ahmed, D. Anjum and H. N. Alshareef, ACS Appl. Mater. Interfaces, 2016, 8, 18806–18814 CrossRef CAS PubMed.
- W. Ma, S. Chen, S. Yang, W. Chen, W. Weng, Y. Cheng and M. Zhu, Carbon, 2017, 113, 151–158 CrossRef CAS.
- X. Chen, S. Gao, H. Mi, C. Jia, Z. Li, L. Sun and X. Zhang, Appl. Surf. Sci., 2019, 486, 490–498 CrossRef CAS.
- M. Li, Y. Zhang, L. Yang, Y. Liu and J. Yao, Electrochim. Acta, 2015, 166, 310–319 CrossRef CAS.
- H. Duan, T. Yan, Z. Li, G. Chen, J. Zhang, L. Shi and D. Zhang, Sustainable Energy Fuels, 2017, 1, 1557–1567 RSC.
- W. Geng, F. Ma, G. Wu, S. Song, J. Wan and D. Ma, Electrochim. Acta, 2016, 191, 854–863 CrossRef CAS.
- V. Sattayarut, T. Wanchaem, P. Ukkakimapan, V. Yordsri, P. Dulyaseree, M. Phonyiem, M. Obata, M. Fujishige, K. Takeuchi, W. Wongwiriyapan and M. Endo, RSC Adv., 2019, 9, 21724–21732 RSC.
- X. Song, X. Ma, Y. Li, L. Ding and R. Jiang, Appl. Surf. Sci., 2019, 487, 189–197 CrossRef CAS.
- X. Yang, X. Zhuang, Y. Huang, J. Jiang, H. Tian, D. Wu, F. Zhang, Y. Mai and X. Feng, Polym. Chem., 2015, 6, 1088–1095 RSC.
- Z. Lin, H. Tian, F. Xu, X. Yang, Y. Mai and X. Feng, Polym. Chem., 2016, 7, 2092–2098 RSC.
- H. Tian, Z. Lin, F. Xu, J. Zheng, X. Zhuang, Y. Mai and X. Feng, Small, 2016, 12, 3155–3163 CrossRef CAS PubMed.
- Z. Lin, H. Tian, F. Xu, X. Yang, Y. Mai and X. Feng, Polym. Chem., 2016, 7, 2092–2100 RSC.
- F. Ma, S. Ding, H. Ren and P. Peng, RSC Adv., 2019, 9, 18333–18343 RSC.
- W. Geng, F. Ma, G. Wu, S. Song, J. Wan and D. Ma, Electrochim. Acta, 2016, 191, 854–863 CrossRef CAS.
- T. Liu, L. Zhang, B. Cheng and J. Yu, Adv. Energy Mater., 2019, 9, 1803900 CrossRef.
- M. Oschatz, L. Borchardt, K. Pinkert, S. Thieme, M. R. Lohe, C. Hoffmann, M. Benusch, F. M. Wisser, C. Ziegler, L. Giebeler, M. H. Rümmeli, J. Eckert, A. Eychmüller and S. Kaskel, Adv. Energy Mater., 2014, 4, 130645–130654 Search PubMed.
- Y. He, X. Zhuang, C. Lei, L. Lei, Y. Hou, Y. Mai and X. Feng, Nano Today, 2019, 24, 103–119 CrossRef CAS.
- Y. Guo, Z. Shi, M. Chen and C. Wang, J. Power Sources, 2014, 252, 235–243 CrossRef CAS.
- S. Song, F. Ma, G. Wu, D. Ma, W. Geng and J. Wan, J. Mater. Chem. A, 2015, 3, 18154–18162 RSC.
- J. Deng, T. Xiong, F. Xu, M. Li, C. Han, Y. Gong, H. Wang and Y. Wang, Green Chem., 2015, 17, 4053–4060 RSC.
- Y. S. Wang, C. Y. Wang and M. M. Chen, New Carbon Mater., 2009, 24, 187–190 CrossRef CAS.
- J. Yang, K. Nakabayashi, J. Miyawaki and S. H. Yoon, Carbon, 2016, 106, 28–36 CrossRef CAS.
- B. Qin, Q. Wang, X. Zhang, X. Xie, L. Jin and Q. Cao, Electrochim. Acta, 2018, 283, 655–663 CrossRef CAS.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- S. Niyogi, E. Bekyarova, M. E. Itkis, J. L. McWilliams, M. A. Hamon and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 7720–7721 CrossRef CAS PubMed.
- G. Eda, G. Fanchini and M. Chhowallal, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed.
- B. William, S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1957, 80, 1339 Search PubMed.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- S. Song, F. Ma, G. Wu, D. Ma, W. Geng and J. Wan, J. Mater. Chem. A, 2015, 3, 18154–18162 RSC.
- W. Yang, W. Yang, F. Ding, L. Sang, Z. Ma and G. Shao, Carbon, 2017, 111, 419–427 CrossRef CAS.
- J. A. Monge, D. C. Amoros, A. Gungor and L. Toppare, Fuel, 2001, 80, 41–48 CrossRef.
- B. K. Pradhan and N. K. Sandle, Carbon, 1999, 37, 1323–1332 CrossRef CAS.
- H. M. Zhu, J. H. Yan, X. G. Jiang, Y. E. Lai and K. F. Cen, J. Hazard. Mater., 2008, 153, 670–676 CrossRef CAS PubMed.
- G. X. Zhao, L. Jiang, Y. D. He, J. X. Li, H. L. Dong, X. K. Wang and W. P. Hu, Adv. Mater., 2011, 23, 3959–3963 CrossRef CAS PubMed.
- T. Metzinger and K. J. Hüttinger, Carbon, 1997, 35, 885–892 CrossRef CAS.
- T. P. Eskay, P. F. Britt and A. C. Buchanan, Energy Fuels, 1996, 10, 1257–1261 CrossRef CAS.
- T. Metzinger, Untersuchungen zur Vernetzung der Bindepechmatrix van ohlenstoffkdrpern mit Molekularem Sauerstofl, PhD thesis, Universitgt Karlsruhe, Germany, 1996.
- C. Tangsathitkulchai, Y. Ngernyen and M. Tangsathitkulchai, Korean J. Chem. Eng., 2006, 23, 1046–1054 CrossRef.
- S. Yu, H. Wang, C. Hu, Q. Zhu, N. Qiao and B. Xu, J. Mater. Chem. A, 2016, 4, 16341–16348 RSC.
- T. Ouyang, K. Cheng, F. Yang, L. Zhou, K. Zhu, K. Ye, G. Wang and D. Cao, J. Mater. Chem. A, 2017, 5, 14551–14561 RSC.
- J. Luo, W. Zhong, Y. Zou, C. Xiong and W. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 317–326 CrossRef CAS PubMed.
- D. Shan, J. Yang, W. Liu, J. Yan and Z. Fan, J. Mater. Chem. A, 2016, 4, 13589–13602 RSC.
- H. Preiss, G. Oliew and K. Szulzewsky, Fuel, 1994, 73, 243–249 CrossRef CAS.
- E. Raymundo-Piñero, K. Kierzek, J. Machnikowski and F. Béguin, Carbon, 2006, 44, 2498–2507 CrossRef.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- N. Choudhary, C. Li, J. Moore, N. Nagaiah, L. Zhai, Y. Jung and J. Thomas, Adv. Mater., 2017, 29, 1–30 Search PubMed.
- X. Fan, C. Yu, J. Yang, Z. Ling, C. Hu, M. Zhang and J. Qiu, Adv. Energy Mater., 2015, 5, 1–7 CAS.
- M.-S. Park, S. Cho, E. Jeong and Y.-S. Lee, J. Ind. Eng. Chem., 2015, 23, 27–32 CrossRef CAS.
- Y. Guo, Z. Shi, M. Chen and C. Wang, J. Power Sources, 2014, 252, 235–243 CrossRef CAS.
- Z. Lei, Z. Liu, H. Wang, X. Sun, L. Lu and X. Zhao, J. Mater. Chem. A, 2013, 1, 2313–2321 RSC.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863–4868 CrossRef CAS PubMed.
- X. Sun, H. Wang, Z. Lei, Z. Liu and L. Wei, RSC Adv., 2014, 4, 20233–20240 Search PubMed.
- B. Lv, P. Li, Y. Liu, S. Lin, B. Gao and B. Lin, Appl. Surf. Sci., 2018, 437, 169–175 CrossRef CAS.
- X. Hao, J. Wang, B. Ding, Y. Wang, Z. Chang, H. Dou and X. Zhang, J. Power Sources, 2017, 352, 34–41 CrossRef CAS.
- Y. Chen, G. Zhang, J. Zhang, H. Guo, X. Feng and Y. Chen, J. Mater. Sci. Technol., 2018, 34, 2189–2196 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |