
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: MicroRNA-135a alleviates lipid accumulation and inflammation of atherosclerosis through targeting lipoprotein lipase†
Juan Li,
Peng Li,
Yanzhuo Zhao,
Xiang Ma,
Ruili He *,
Ketai Liang and
Erwei Zhang
*,
Ketai Liang and
Erwei Zhang
Department of Cardiology, Huaihe Hospital of Henan University, No. 8 Baobei Road, Gulou District, 475000, Kaifeng, Henan, China. E-mail: zcgksh@163.com; Tel: +86-0371-23906819
First published on 9th September 2019
Abstract
MicroRNAs (miRNAs) have recently attracted increasing attention for their involvement in atherosclerosis (AS). The purpose of this study was to further explore the function and underlying mechanism of miR-135a in AS progression. The expression levels of miR-135a and lipoprotein lipase (LPL) mRNA were detected by qRT-PCR, and LPL protein expression was measured by western blotting. The levels of blood lipids and inflammatory cytokines, and LPL activity were assessed using corresponding Assay Kits, and an HPLC assay was used to determine the levels of free cholesterol (FC), total cholesterol (TC) and cholesterol ester (CE). A Dil-oxLDL binding assay was performed to evaluate the ability of cholesterol uptake. The direct interaction between miR-135a and LPL was confirmed by a dual-luciferase reporter assay and RNA immunoprecipitation assay. Our data indicated that miR-135a was downregulated in serum samples of AS patients and mice. Upregulation of miR-135a alleviated lipid metabolic disorders and inflammation in AS mice. Moreover, miR-135a negatively regulated lipid accumulation and inflammation in ox-LDL-treated THP-1 macrophages. Mechanistically, miR-135a directly targeted LPL and repressed LPL expression. LPL mediated the regulatory effect of miR-135a on lipid accumulation and inflammation in ox-LDL-treated THP-1 macrophages. In conclusion, our study indicated that miR-135a upregulation ameliorated lipid accumulation and inflammation at least partly by targeting LPL in THP-1 macrophages, highlighting miR-135a as a potential antiatherogenic agent.
1. Introduction
Atherosclerosis (AS) is by far the most frequent underlying cause of cardiovascular disease and one of the leading causes of morbidity and death worldwide. AS is a multifocal, progressive inflammatory disease characterized by the accumulation of lipids and infiltration of inflammatory cells in the artery wall.1–3 The accumulation of oxidatively modified lipids in the arterial intima contributes to the initiation of AS, and extracellular lipids derived from dead foam macrophages accumulate to form a lipid core in the advanced lesion.2,4 Macrophage recruitment and activation that response to lipid accumulation is a hallmark of AS development, and macrophage-derived foam cell is a major contributor of AS progression.5,6 Therefore, in the present study, we aimed to explore the regulatory mechanisms involved in lipid accumulation and inflammation of AS.Lipoprotein lipase (LPL), a central enzyme in overall lipid metabolism, plays a key role in lipid homeostasis and energy balance. The physiological function of LPL is to catalyze the hydrolysis of plasma triglyceride (TG)-rich lipoproteins, giving rise to the formation of small and dense lipoproteins chylomicron remnants, low-density lipoproteins (LDLs) and intermediate-density lipoproteins (IDLs).7,8 LPL produced by macrophages has been manifested to be upregulated in the atherosclerotic lesions.9,10 It is widely accepted that macrophage-derived LPL in the arterial wall is pro-atherogenic during AS progression, possibly by promoting the formation of foam cells.11 Recent studies have also demonstrated that macrophage-derived LPL contributes to AS progression via increasing lipid accumulation and proinflammatory cytokine secretion.12–14
MicroRNAs (miRNAs) are a class of endogenous, noncoding small RNAs of ∼21–23 nucleotides in length, and negatively modulate gene expression at the posttranscriptional level by binding to 3′-untranslated region (3′-UTR) of target mRNAs, leading to translational repression and target mRNA degradation. To silence target mRNAs, they need to form a so-called RNA-induced silencing complex (RISC), which is composed of a miRNA and argonaute (Ago) protein.15,16 MiRNAs have recently attracted increasing attention for their involvement in a number of human diseases, including AS.17,18 Abnormal expression of miR-135a was found in human atherosclerotic plaques and increased miR-135a level suppressed oxidized low-density lipoprotein (ox-LDL)-induced foam cell formation, TG level and inflammation in RAW264.7 cells.19,20 Herein, we further explored the function and underlying mechanism of miR-135a in AS progression.
In this study, our data verified that miR-135a was downregulated in AS patients and AS mice, and miR-135a upregulation ameliorated lipid metabolic disorders and inflammation of AS mice. Moreover, our study suggested that miR-135a alleviated lipid accumulation and inflammation at least partly though targeting LPL in THP-1 macrophages.
2. Materials and methods
2.1. Serum samples collection
A total of 24 AS patients diagnosed by the Division of Cardiology at Huaihe Hospital of Henan University (Kaifeng, China) and 10 healthy volunteers were enrolled in the study. Peripheral blood samples (5 ml) were collected from all participators before treatment and serum samples were obtained by centrifugation at 1500g for 10 min. Fresh serum samples were immediately frozen in liquid nitrogen and stored at −80 °C until use. Informed consent was signed by all participators, and the study was approved by the Ethics Committee of Huaihe Hospital of Henan University in accordance with the Declaration of Helsinki Principles.2.2. Animals
Female C57BL/6J ApoE−/− mice (6–8 weeks, 20–25 g) were purchased from Biocytogen (Biocytogen Co., Ltd., Beijing, China) and maintained in laminar flow cabinets under specific pathogen-free conditions. All mice were randomly divided into the following four groups: (1) control group (n = 6), mice fed on a normal diet; (2) AS model group (n = 6), mice fed on a high fat diet (HFD, 21% pork lard and 1.5% cholesterol); (3) LV-miR-NC group (n = 6); and (4) LV-miR-135a group (n = 6). When the HFD started, the mice were injected with miR-135a-overexpression lentivirus vector (LV-miR-135a, Fulengen, Guangzhou, China) or negative control vector (LV-miR-NC, Fulengen) via the tail vein once every 4 weeks. 16 weeks later, all mice were euthanized, and blood samples and aortas tissues were collected for further experiments. For en face analysis, the whole aorta was excised from the aortic arch and stained with oil-red O as described previously.13 All animal experiments were performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal study was approved by the Animal Care and Use Committee of Huaihe Hospital of Henan University.2.3. RNA extraction, reverse transcription and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from serum samples and cells using RNeasy Mini Kit (Qiagen, Hilden, Germany) following the protocols of manufacturers. The quality and quantity of RNA extracts were measured using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). For miR-135a detection, cDNA was synthesized from 0.5 μg of RNA extracts using miScript Reverse Transcription Kit (Qiagen), and subjected to qRT-PCR on an iCycler iQ Multicolor Real-Time Detection System (Bio-Rad Laboratories, Hercules, CA, USA) using miScript SYBR Green PCR Kit (Qiagen) with specific primer for miR-135a (Qiagen). U6 snRNA expression was used as an internal control. For LPL mRNA measurement, the High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA) and SYBR Green PCR Master Mix (Applied Biosystems) were used with GAPDH as the housekeeping gene control. Gene expression fold changes were calculated by using the 2−ΔΔCt method.2.4. Determination of blood lipids and inflammatory cytokines levels
Blood lipids profile including high-density lipoprotein-cholesterol (HDL-C), low-density lipoprotein-cholesterol (LDL-C) and TG were determined using HDL-C Assay Kit (Huili Biotechnology, Changchun, China), LDL-C Assay Kit (Huili Biotechnology) and TG Assay Kit (Huili Biotechnology), respectively, following the manufacturers' guidance.The levels of pro-inflammatory cytokines including tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β) and IL-6 were measured using corresponding commercial Mouse or Human ELISA Assay Kits (R&D Systems, Minneapolis, MN, USA) in accordance with the instructions of manufacturers.
2.5. Cell culture and treatment
Human THP-1 cell line purchased from ATCC (Manassas, VA, USA) was maintained in RPMI-1640 medium (Gibco, BRL Life Sciences Technologies, Breda, The Netherlands) supplemented with 10% fetal calf serum (FCS, Wisent, St-Bruno, QC, Canada), 1% L-glutamine (Gibco), 1% sodium pyruvate (Gibco) and 1% (v/v) antibiotics (penicillin/streptomycin, Gibco) in a humidified 5% CO2 controlled incubator at 37 °C.For macrophage differentiation, THP-1 cell line was induced using 160 nM of phorbol-12-myristate acetate (PMA, Sigma-Aldrich, St. Louis, MO, USA) for 24 h. For foam cell formation, THP-1 macrophages were incubated with 40 μg ml−1 of ox-LDL for 24 h.
2.6. Cell transfection
For miR-135a overexpression, THP-1 macrophages were transfected with 50 nM of human matured miR-135a mimic (Ambion, Austin, TX, USA) or a scrambled sequence as negative control (miR-NC mimic, Ambion). MiR-135a silencing were carried on using 50 nM of miR-135a inhibitor (anti-miR-135a, Ambion), and a scrambled sequence (anti-miR-NC, Ambion) was used as negative control. For LPL investigation, THP-1 macrophages were transfected with 50 ng of LPL overexpression vector (pcDNA-LPL, Ambion), 50 nM of pre-designed siRNA targeting LPL (si-LPL, Ambion) or respective negative control (pcDNA-NC or si-NC, Ambion). All transfections were performed using the Lipofectamine RNAimax transfection reagent (Invitrogen, Grand Island, NY, USA) following the instructions of manufacturers. Oligonucleotide sequences (5′–3′) were as follows: miR-135a mimic: GUGCCGAGGAUUAGGGAUAUACU; miR-NC mimic: UUCUCCGAACGUGUCACGUTT; anti-miR-135a: UCACAUAGGAAUAAAAAGCCAUA; anti-miR-NC: CAGUACUUUUGUGUAGUACAA; si-LPL: CCACGAACGUUCCGUUCAU and si-NC: GGAGUUGCCGUCGUAAGAU.2.7. High-performance liquid chromatography (HPLC) assay
The levels of free cholesterol (FC), total cholesterol (TC) and cholesterol ester (CE) were measured using HPLC analysis as described previously.21 To be brief, THP-1 macrophages were transfected with miR-NC mimic, miR-135a mimic, anti-miR-NC or anti-miR-135a prior to ox-LDL treatment. Treated cells were harvested, washed three times with ice-cold PBS, and then resuspended in 0.5% (w/v) NaCl solution. Cells were sonicated with a Sonic Dismembrator (Thermo Fisher Scientific, Waltham, MA, USA) for 5 min and protein extracts were obtained by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 10 min. The concentrations of protein extracts were determined by a BCA Protein Assay Kit (Thermo Fisher Scientific) following the instructions of manufacturers. A total of 0.5 ml of cell lysates (containing 20–50 μg protein) was used for the detection of FC, TC and CE. The sterol analyses were performed using an Agilent 1200 HPLC system (Agilent Technologies, Waldbronn, Germany) with a photodiode array detector. The column was eluted with isopropanol:n-heptane:acetonitrile (35:13:52) at a flow rate of 1 ml min−1 for 8 min, followed by the determination of the absorbance at 216 nm. TotalChrom software (PerkinElmer Inc., Shelton, CT, USA) was used to analyze the data and each test was repeated three times.
000g for 10 min. The concentrations of protein extracts were determined by a BCA Protein Assay Kit (Thermo Fisher Scientific) following the instructions of manufacturers. A total of 0.5 ml of cell lysates (containing 20–50 μg protein) was used for the detection of FC, TC and CE. The sterol analyses were performed using an Agilent 1200 HPLC system (Agilent Technologies, Waldbronn, Germany) with a photodiode array detector. The column was eluted with isopropanol:n-heptane:acetonitrile (35:13:52) at a flow rate of 1 ml min−1 for 8 min, followed by the determination of the absorbance at 216 nm. TotalChrom software (PerkinElmer Inc., Shelton, CT, USA) was used to analyze the data and each test was repeated three times.
2.8. Dil-oxLDL uptake analysis
Cholesterol uptake analysis was performed using the Dil-oxLDL uptake assay as described previously.22 Briefly, THP-1 macrophages were transfected with the indicated oligonucleotide or plasmid for 24 h. Then, transfected cells were placed in glass chamber slides at 100 cells per mm2 and incubated with 40 μg ml−1 of Dil-oxLDL (Yiyuan Biotech, Guangzhou, China) at 4 °C for 0.5 h. Some cells were fixed with 3% paraformaldehyde and 2% sucrose at room temperature for 10 min, and then fluorescent photomicrographs were taken with an Olympus BX43 microscope (Olympus, Tokyo, Japan) with an excitation filter for rhodamine. Other cells were lysed and Dil-oxLDL uptake ability was determined with 540 nm excitation laser line and 590 nm emission filters of fluorometry.2.9. LPL activity assay
The activity of LPL secreted by macrophages in the culture media was detected using a commercial LPL Activity Assay Kit (Roar Biomedical Inc., New York, NY, USA) following the protocols of manufacturers. In brief, transfected THP-1 macrophages were incubated with 10 U ml−1 of heparin for 1 h. Then, cells were harvested and lysed, and the supernatant was collected for the determination of LPL activity. Enzyme activity is presented as relative activity normalized to protein concentration.2.10. Dual-luciferase reporter assay
Luciferase constructs were made by Fulengen via ligating oligonucleotides of LPL 3′-UTR containing the wild-type or mutant target site for miR-135a into the pMIR-REPORT vector (LPL-WT and LPL-MUT). THP-1 macrophages were cotransfected with 50 ng of LPL-WT or LPL-MUT and 50 nM of miR-135a mimic or miR-NC mimic using Lipofectamine RNAimax transfection reagent. After 48 h transfection, luciferase assays were carried out using the Dual Luciferase Reporter Assay System (Dual-Light System, Applied Biosystems) according to the guidance of manufacturers.2.11. RNA immunoprecipitation (RIP) assay
THP-1 macrophages were transfected with 50 nM of miR-NC mimic or miR-135a mimic, and then cells were lysed using ice-cold RIPA buffer (50 mM Tris-HCl, pH = 7.4, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 0.5% sodium deoxycholate). Cell lysates were incubated with anti-Ago2 (Cell Signaling Technology, Danvers, MA, USA) or IgG (Cell Signaling Technology) antibody at 4 °C for 4 h before adding protein A/G agarose for 2 h. Beads were washed three times with the same buffer and total RNA was extracted for the determination of LPL mRNA enrichment by qRT-PCR.2.12. Western blot
Cell extracts were prepared using ice-cold RIPA buffer supplemented with complete protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany), and total protein was quantified by the BCA Protein Assay Kit following the instructions of manufacturers. Protein was separated by a 10% SDS–polyacrylamide gel electrophoresis and then transferred to PVDF membranes (Millipore, Billerica, MA, USA). The membranes were probed with anti-LPL (1:5000, Abcam, Cambridge, UK) or anti-β-actin (1:2000, Abcam) antibody. Horseradish peroxidase-labeled IgG (1:5000, Abcam) antibody was used as a secondary antibody. Protein bands were determined by ECL reagent (Amersham Bioscience, Munich, Germany) and the intensity of the bands was analyzed by ImageJ software (National Institutes of Health, Bethesda, MD, USA).2.13. Statistical analysis
All data were expressed as mean ± standard deviation (SD) of at least three independent experiments. Statistical differences were compared by a Student's t-test or one-way ANOVA using GraphPad Prism 5.0 software (GraphPad, San Diego, CA, USA). Correlations between miR-135a expression and HDL-C, LDL-C, TC or TG level were analyzed using Spearman test. Statistical significance was defined as *P < 0.05, **P < 0.01 or ***P < 0.001.3. Results
3.1. Downregulation of miR-135a in serum of AS patients and ApoE−/− mice fed a HFD
For a preliminary investigation for the involvement of miR-135a in AS, we firstly determined the expression of miR-135a in serum of AS patients by qRT-PCR. In contrast to normal controls, AS patients had lower serum expression of miR-135a (Fig. 1A). Then, we examined the expression of miR-135a in serum samples of ApoE−/− mice fed a HFD. Likewise, miR-135a levels were lower in mice fed a HFD than those in normal controls (Fig. 1B). Moreover, our data revealed that AS patients had lower HDL-C expression and higher levels of LDL-C, TC and TG (ESI Fig. 1A–D†). Additionally, miR-135a expression was positively correlated with HDL-C level and inversely correlated with the expression levels of LDL-C, TC and TG (ESI Fig. 1E–H†). These data suggested that miR-135a might be involved in AS progression.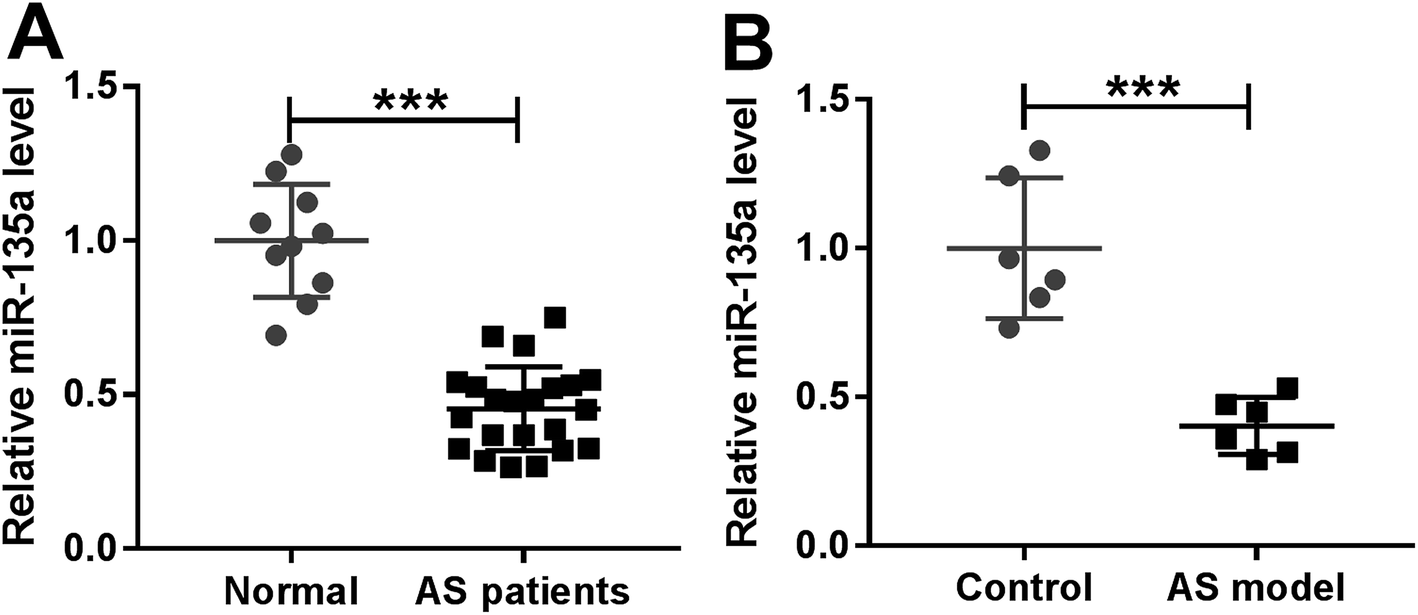 | ||
| Fig. 1 The expression of miR-135a in serum of AS patients and ApoE−/− mice fed a HFD. MiR-135a expression by qRT-PCR in serum samples of AS patients and healthy volunteers (A), and ApoE−/− mice fed a HFD and normal diet (B). Each experiment was performed with 3 biological replicates × 3 technical replicates. ***P < 0.001. | ||
3.2. Upregulation of miR-135a alleviated lipid metabolic disorders and inflammation in AS mice
Given our data that miR-135a level was reduced in mice fed a HFD (Fig. 2A), we then assessed several blood parameters (HDL-C, LDL-C, TG, IL-1β, IL-6 and TNF-α) in mice fed a HFD. In comparison to normal controls, mice fed a HFD had blood lower HDL-C level (Fig. 2B) and higher levels of LDL-C, TG, IL-1β, IL-6 and TNF-α (Fig. 2C–G). The atherosclerotic lesions of the whole aorta in en face were increased in mice fed a HFD compared with negative controls (ESI Fig. 2†), supporting a successful construct for AS mice model. | ||
| Fig. 2 The functional role of miR-135a in lipid metabolic disorders and inflammation of AS mice. ApoE−/− mice were fed a HFD or normal diet for 16 weeks, or injected with LV-miR-NC or LV-miR-135a via the tail vein every 4 weeks when the HFD started. At the end, all mice were euthanized and blood samples were harvested. (A) MiR-135a expression by qRT-PCR in serum samples of AS mice. (B–D) The levels of HDL-C, LDL-C and TG using corresponding commercial Assay Kits in blood samples. (E–G) The levels of IL-1β, IL-6 and TNF-α using corresponding Mouse ELISA Assay Kits. Each experiment was performed with 3 biological replicates × 3 technical replicates. *P < 0.05, **P < 0.01 or ***P < 0.001. | ||
To further observe the role of miR-135a in AS progression, ApoE−/− mice were injected with LV-miR-NC or LV-miR-135a via the tail vein every 4 weeks when the HFD started. In contrast to negative control, infection of LV-miR-135a triggered about a 2-fold increase of miR-135a expression in serum of AS mice (Fig. 2A). Moreover, miR-135a upregulation resulted in increased HDL-C level (Fig. 2B) and decreased LDL-C and TG levels (Fig. 2C and D), indicating the ameliorative effect of miR-135a upregulation on lipid metabolic disorders of AS mice. Additionally, miR-135a upregulation led to the significant reduction of IL-1β, IL-6 and TNF-α levels (Fig. 2E–G), illuminating its anti-inflammatory property in AS mice. These results together established that miR-135a upregulation ameliorated lipid metabolic disorders and inflammation in AS mice.
3.3. MiR-135a negatively regulated lipid accumulation and inflammation in ox-LDL-treated THP-1 macrophages
Next, we examined the effect of miR-135a on lipid accumulation in vitro by transfection of miR-135a mimic or anti-miR-135a prior to ox-LDL treatment. In contrast to their counterparts, transient introduction of miR-135a mimic induced about a 4-fold increase of miR-135a expression, and anti-miR-135a transfection triggered a 70% decrease of miR-135a level in ox-LDL-treated THP-1 macrophages (Fig. 3A). As demonstrated by HPLC, the levels of FC, TC and CE were significantly repressed by miR-135a upregulation, while they were highly enhanced when miR-135a knockdown (Fig. 3B–D). Subsequently, we determined whether miR-135a influenced the lipid uptake by Dil-oxLDL uptake assay. In comparison to their counterparts, miR-135a upregulation significantly repressed the Dil-oxLDL entering into the macrophages, while miR-135a knockdown promoted the ability to uptake the Dil-oxLDL (Fig. 3E), indicating the impact of miR-135a in lipid uptake. Additionally, LPL activity was weakened by miR-135a upregulation, but elevated following miR-135a knockdown (Fig. 3F). All these data suggested that miR-135a negatively regulated lipid accumulation in ox-LDL-treated THP-1 macrophages. | ||
| Fig. 3 Effects of miR-135a on lipid accumulation and inflammation in ox-LDL-treated THP-1 macrophages. THP-1 macrophages were transfected with miR-NC mimic, miR-135a mimic, anti-miR-NC or anti-miR-135a, and then treated with or without 40 μg ml−1 of ox-LDL for 24 h. (A) The expression of miR-135a by qRT-PCR in treated cells. (B–D) The levels of FC, TC and CE by HPLC in treated cells. (E) Representative images (intact cells) and the ability of lipid uptake by Dil-oxLDL uptake assay in transfected cells. (F) The activity of LPL secreted by transfected cells using the LPL Assay Kit. (G–I) The levels of IL-1β, IL-6 and TNF-α using corresponding Human ELISA Assay Kits in treated cells. Each experiment was performed with 3 biological replicates × 3 technical replicates. *P < 0.05, **P < 0.01 or ***P < 0.001. | ||
Then, we assessed the effect of miR-135a on inflammation in ox-LDL-treated THP-1 macrophages. As expected, miR-135a upregulation resulted in decreased levels of IL-1β, IL-6 and TNF-α, while miR-135a knockdown exerted opposite effects (Fig. 3G–I), suggesting the negative regulation of miR-135a on inflammation.
3.4. LPL was a direct target of miR-135a
To further explore the underlying mechanism by miR-135a regulated lipid accumulation and inflammation in THP-1 macrophages, we carried out a detailed analysis for its molecular targets. Using microT-CDS software, the predicted data revealed a putative miR-135a binding site in the 3′-UTR of LPL mRNA (Fig. 4A). When we performed a dual-luciferase reporter assay in THP-1 macrophages, cotransfection of LPL 3′-UTR luciferase reporter and miR-135a mimic into cells produced lower luciferase activity than in cells cotransfected with a scrambled control sequence (Fig. 4B). Whereas, mutation of miR-135a binding sequence significantly abrogated the effect of miR-135a on luciferase activity under the same conditions (Fig. 4B). Moreover, RIP assays showed that the enrichment of LPL mRNA were highly abundant in the RISC of THP-1 macrophages when them were transfected with miR-135a mimic (Fig. 4C), indicating the endogenous interaction between miR-135a and LPL. After that, we observed whether miR-135a modulated LPL expression in THP-1 macrophages. As presented by western blot, LPL expression was significantly decreased by miR-135a upregulation, while it was dramatically increased following miR-135a knockdown compared with their counterparts (Fig. 4D and E). These results together strongly pointed that miR-135a directly targeted LPL and inhibited LPL expression.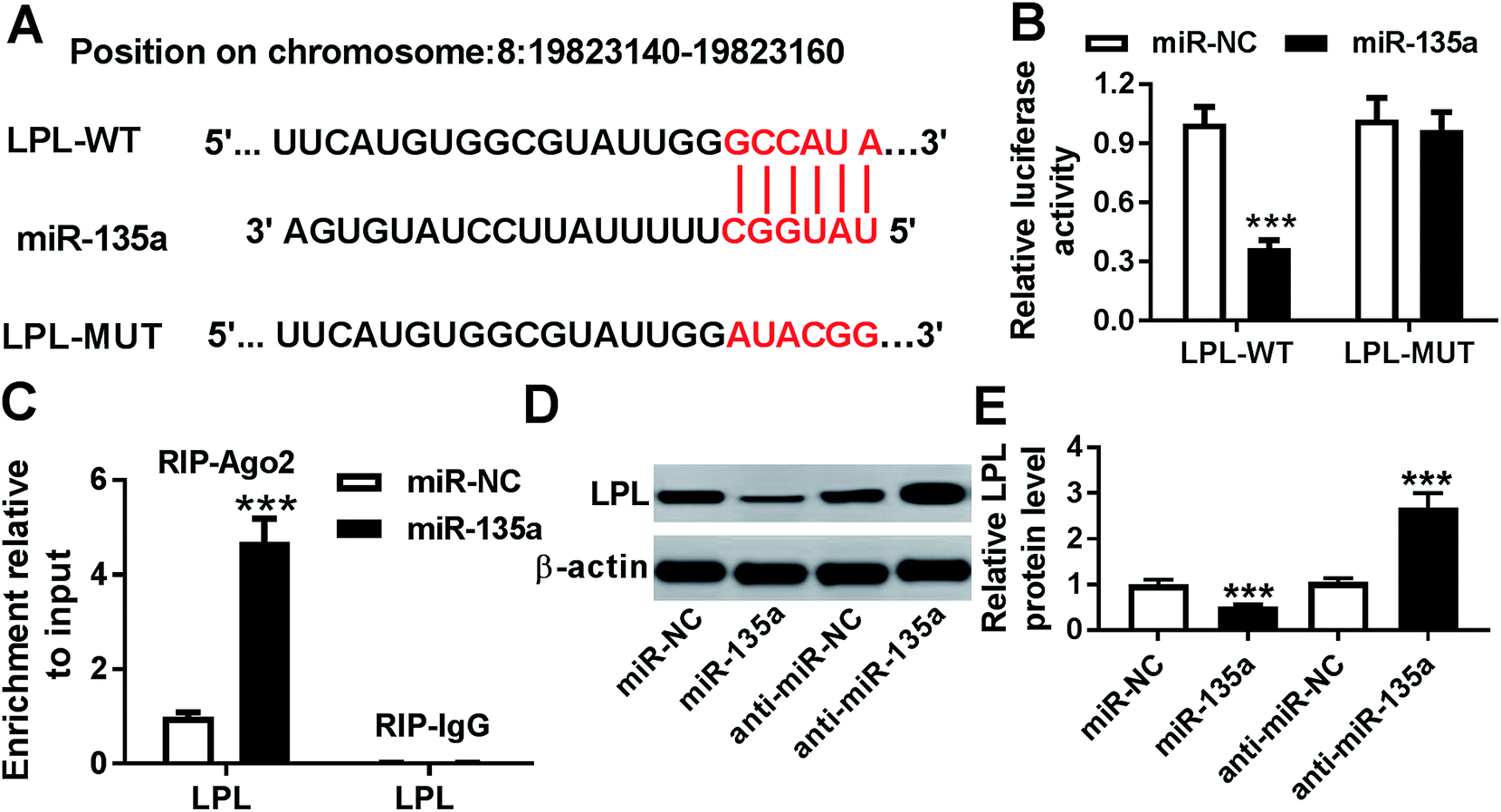 | ||
| Fig. 4 LPL was a direct target of miR-135a. (A) Schematic of the putative miR-135a binding site in the 3′-UTR of LPL mRNA and the mutation of miR-135a binding sequence. (B) LPL 3′-UTR luciferase reporter vector (LPL-WT) or its mutation in seeded region (LPL-MUT) were transfected into THP-1 macrophages together with miR-NC mimic or miR-135a mimic, followed by the measurement of luciferase activity. (C) The enrichment of LPL mRNA by qRT-PCR in THP-1 macrophages transfected with miR-NC mimic or miR-135a mimic using anti-Ago2 or IgG antibody. (D and E) LPL expression by western blot in THP-1 macrophages transfected with miR-NC mimic, miR-135a mimic, anti-miR-NC or anti-miR-135a. Each experiment was performed with 3 biological replicates × 3 technical replicates. ***P < 0.001. | ||
3.5. LPL mediated the regulatory effect of miR-135a on lipid accumulation and inflammation in ox-LDL-treated THP-1 macrophages
To provide further mechanistic insight into the link between miR-135a and LPL on lipid accumulation and inflammation of AS in vitro, THP-1 macrophages were cotransfected with miR-135a mimic and pcDNA-LPL or anti-miR-135a and si-LPL prior to ox-LDL treatment. In comparison to their counterparts, cotransfection of pcDNA-LPL significantly reversed the decreased effect of miR-135a mimic on LPL expression, while anti-miR-135a-mediated increased effect on LPL level was markedly abolished by cotransfection of si-LPL in ox-LDL-treated THP-1 macrophages (Fig. 5A). Subsequent experiments revealed that LPL expression restoration significantly antagonized the changes of cell lipid uptake and LPL activity, which were induced by miR-135a level alteration (Fig. 5B and C). Besides, miR-135a-mediated anti-inflammation effect was significantly abrogated by LPL expression restoration (Fig. 5D–F). All these data suggested that LPL mediated the regulatory effect of miR-135a on lipid accumulation and inflammation in ox-LDL-treated THP-1 macrophages. | ||
| Fig. 5 The regulatory effect of miR-135a on lipid accumulation and inflammation was mediated by LPL. THP-1 macrophages were transfected with miR-NC mimic, miR-135a mimic, miR-135a mimic + pcDNA-NC, miR-135a mimic + pcDNA-LPL, anti-miR-NC, anti-miR-135a, anti-miR-135a + si-NC or anti-miR-135a + si-LPL and then treated with or without 40 μg ml−1 of ox-LDL for 24 h. (A) Western blot for LPL expression in treated cells. (B) The ability of lipid uptake in transfected cells using Dil-oxLDL uptake assay. (C) LPL activity in transfected cells using the LPL Assay Kit. (D–F) The levels of IL-1β, IL-6 and TNF-α in treated cells using Human ELISA Assay Kits. Each experiment was performed with 3 biological replicates × 3 technical replicates. *P < 0.05, **P < 0.01 or ***P < 0.001. | ||
4. Discussion
AS is a chronic inflammatory disease characterized by the accumulation of lipids and inflammatory response in the artery wall, resulting in a series of cardiovascular diseases. Accumulating evidence has recently showed that miRNAs are closely associated with lipid metabolism, cholesterol lipoprotein transport and chronic inflammatory processes involved in AS.17,23 Our previous study had manifested that the knockdown of long noncoding RNA GAS5 repressed lipid metabolic disorders and inflammatory response of AS through acting as a molecular sponge of miR-135a (awaiting publication). In this study, we further explored how miR-135a influenced lipid accumulation and inflammatory response of AS in vivo and in vitro. Our data demonstrated that miR-135a upregulation ameliorated lipid accumulation and inflammation of AS possibly through targeting LPL.MiR-135a has been identified as a crucial regulator in the progression of multiple human cancers.24–26 A recent document reported that dysregulation of miR-135a enhanced the development of rat pulmonary arterial hypertension.27 Moreover, the alteration of HDL-carried miR-135a after consumption of dietary trans fat was correlated with changes in blood lipid and inflammation in healthy men.28 MiR-135a was also manifested as a potential suppressor involved in vascular calcification which was a common feature of atherosclerotic lesions.29 Besides, a previous report demonstrated that miR-135a upregulation resulted in a repression of foam cell formation and cell apoptosis, as well as a reduction of TG level and pro-inflammatory cytokines secretion in ox-LDL-treated RAW264.7 cells, highlighting its role as an antiatherogenic agent.20 Therefore, the present research started from the hypothesis that miR-135a might alleviate lipid accumulation and inflammatory response of AS. To verify this, we firstly determined miR-135a expression in serum samples of AS patients and mice model, and our data demonstrated a significant downregulation of miR-135a in AS. Moreover, our results indicated that miR-135a upregulation alleviated lipid metabolic disorders and inflammation in AS mice, suggesting the inhibitory effect of miR-135a on malignant progression of AS in vivo. More importantly, we firstly elucidated that miR-135a upregulation repressed lipid accumulation and inflammatory cytokines secretion in ox-LDL-treated THP-1 macrophages, while miR-135a knockdown exerted opposite effect, supporting the antiatherogenic role of miR-135a.
It is widely acknowledged that miRNAs exert biological function through regulation of target genes expression. Hence, online software microT-CDS was performed to predict the molecular targets of miR-135a. More interestingly, the predicted data revealed that LPL 3′-UTR harbored a putative target sequence for miR-135a. Subsequently, we confirmed that miR-135a directly targeted LPL by dual-luciferase reporter assay and RIP assay. Additionally, western blot analysis indicated that miR-135a inhibited LPL expression in THP-1 macrophages. LPL, a major TG hydrolysis enzyme, is mainly secreted by parenchymal cells of adipose tissues, cardiac and skeletal muscle and macrophages.14 It was reported that macrophage-derived LPL accelerated lipid accumulation, proinflammatory cytokine secretion and foam cell formation, thereby contributing to AS progression.12,30 In the present study, we firstly demonstrated that LPL mediated the regulatory effect of miR-135a on lipid accumulation and inflammation in ox-LDL-treated THP-1 macrophages. Similar with our findings, He et al. reported that miR-590 repressed lipid accumulation and pro-inflammatory cytokine production through targeting LPL.31 Chen et al. manifested that miR-29a modulated the secretion of pro-inflammatory cytokine and the expression of scavenger receptor in ox-LDL-induced dendritic cells via targeting LPL.32 Moreover, Tian et al. demonstrated that miR-467b weakened several inflammatory cytokine expression and lipid accumulation by targeting LPL in ox-LDL-induced RAW macrophages.33 Additionally, Xie et al. described that miR-27 played an antiatherogenic role through regulating LPL expression.13 Lan et al. found that miR-134 could activate LPL-mediated lipid accumulation, illuminating miR-134 as a potentially therapeutic target for AS.34 These researches suggested a wide variety of miRNAs were involved in LPL-mediated lipid accumulation and inflammation in AS.
5. Conclusion
In conclusion, our study suggested that miR-135a upregulation alleviated lipid metabolic disorders and inflammation in AS mice. Furthermore, miR-135a upregulation ameliorated lipid accumulation and inflammation at least partly by targeting LPL in THP-1 macrophages. The clinical significance of miR-135a and its potential value as an antiatherogenic agent should be further explored.Conflicts of interest
The authors have no conflicts of interest to declare.References
- E. Falk, J. Am. Coll. Cardiol., 2006, 47, C7–C12 CrossRef CAS PubMed.
- K. E. Paulson, S. N. Zhu, M. Chen, S. Nurmohamed, J. Jongstra-Bilen and M. I. Cybulsky, Circ. Res., 2010, 106, 383–390 CrossRef CAS PubMed.
- G. K. Hansson, N. Engl. J. Med., 2005, 352, 1685–1695 CrossRef CAS PubMed.
- Y. Nakashima, H. Fujii, S. Sumiyoshi, T. N. Wight and K. Sueishi, Arterioscler., Thromb., Vasc. Biol., 2007, 27, 1159–1165 CrossRef CAS PubMed.
- E. M. Maguire, S. W. A. Pearce and Q. Xiao, Vasc. Pharmacol., 2019, 112, 54–71 CrossRef CAS.
- D. A. Chistiakov, A. A. Melnichenko, V. A. Myasoedova, A. V. Grechko and A. N. Orekhov, J. Mol. Med., 2017, 95, 1153–1165 CrossRef CAS PubMed.
- I. J. Goldberg and M. Merkel, Front. Biosci., 2001, 6, D388–405 CAS.
- P. P. He, T. Jiang, X. P. OuYang, Y. Q. Liang, J. Q. Zou, Y. Wang, Q. Q. Shen, L. Liao and X. L. Zheng, Clin. Chim. Acta, 2018, 480, 126–137 CrossRef CAS.
- M. Takahashi, H. Yagyu, F. Tazoe, S. Nagashima, T. Ohshiro, K. Okada, J. Osuga, I. J. Goldberg and S. Ishibashi, J. Lipid Res., 2013, 54, 1124–1134 CrossRef CAS PubMed.
- K. D. O'Brien, D. Gordon, S. Deeb, M. Ferguson and A. Chait, J. Clin. Invest., 1992, 89, 1544–1550 CrossRef PubMed.
- T. Ichikawa, J. Liang, S. Kitajima, T. Koike, X. Wang, H. Sun, M. Morimoto, H. Shikama, T. Watanabe, N. Yamada and J. Fan, Atherosclerosis, 2005, 179, 87–95 CrossRef CAS PubMed.
- G. Qiu, A. C. Ho, W. Yu and J. S. Hill, J. Lipid Res., 2007, 48, 385–394 CrossRef CAS PubMed.
- W. Xie, L. Li, M. Zhang, H. P. Cheng, D. Gong, Y. C. Lv, F. Yao, P. P. He, X. P. Ouyang, G. Lan, D. Liu, Z. W. Zhao, Y. L. Tan, X. L. Zheng and W. D. Yin, et al., PLoS One, 2016, 11, e0157085, DOI:10.1371/journal.pone.0157085.
- Y. Li, P.-P. He, D.-W. Zhang, X.-L. Zheng, F. S. Cayabyab, W.-D. Yin and C.-K. Tang, Atherosclerosis, 2014, 237, 597–608 CrossRef CAS PubMed.
- H. O. Iwakawa and Y. Tomari, Trends Cell Biol., 2015, 25, 651–665 CrossRef CAS PubMed.
- Z. Li and T. M. Rana, Nat. Rev. Drug Discovery, 2014, 13, 622–638 CrossRef CAS PubMed.
- H. Giral, A. Kratzer and U. Landmesser, Best Pract. Res., Clin. Endocrinol. Metab., 2016, 30, 665–676 CrossRef CAS PubMed.
- N. Rotllan, N. Price, P. Pati, L. Goedeke and C. Fernandez-Hernando, Atherosclerosis, 2016, 246, 352–360 CrossRef CAS PubMed.
- E. Raitoharju, L. P. Lyytikainen, M. Levula, N. Oksala, A. Mennander, M. Tarkka, N. Klopp, T. Illig, M. Kahonen, P. J. Karhunen, R. Laaksonen and T. Lehtimaki, Atherosclerosis, 2011, 219, 211–217 CrossRef CAS PubMed.
- X. J. Du and J. M. Lu, J. Cell. Biochem., 2018, 119, 6154–6161 CrossRef CAS PubMed.
- Y. C. Lv, Y. Y. Tang, J. Peng, G. J. Zhao, J. Yang, F. Yao, X. P. Ouyang, P. P. He, W. Xie, Y. L. Tan, M. Zhang, D. Liu, D. P. Tang, F. S. Cayabyab and X. L. Zheng, et al., Atherosclerosis, 2014, 236, 215–226 CrossRef CAS PubMed.
- S. W. Sun, X. Y. Zu, Q. H. Tuo, L. X. Chen, X. Y. Lei, K. Li, C. K. Tang and D. F. Liao, Acta Pharmacol. Sin., 2010, 31, 1336–1342 CrossRef CAS PubMed.
- J. Novak, J. Bienertova-Vasku, T. Kara and M. Novak, Mediators Inflammation, 2014, 2014, 275867, DOI:10.1155/2014/275867.
- S. Wu, Y. Lin, D. Xu, J. Chen, M. Shu, Y. Zhou, W. Zhu, X. Su, Y. Zhou, P. Qiu and G. Yan, Oncogene, 2012, 31, 3866–3874 CrossRef CAS PubMed.
- A. Holleman, I. Chung, R. R. Olsen, B. Kwak, A. Mizokami, N. Saijo, A. Parissenti, Z. Duan, E. E. Voest and B. R. Zetter, Oncogene, 2011, 30, 4386–4398 CrossRef CAS PubMed.
- W. Tang, Y. Jiang, X. Mu, L. Xu, W. Cheng and X. Wang, Cell. Signalling, 2014, 26, 1420–1426 CrossRef CAS PubMed.
- H. M. Liu, Y. Jia, Y. X. Zhang, J. Yan, N. Liao, X. H. Li and Y. Tang, Acta Pharmacol. Sin., 2019, 40, 477–485 CrossRef CAS PubMed.
- V. Desgagne, S. P. Guay, R. Guerin, F. Corbin, P. Couture, B. Lamarche and L. Bouchard, Epigenetics, 2016, 11, 438–448 CrossRef PubMed.
- L. Lin, Y. He, B. L. Xi, H. C. Zheng, Q. Chen, J. Li, Y. Hu, M. H. Ye, P. Chen and Y. Qu, Curr. Vasc. Pharmacol., 2016, 14, 211–218 CrossRef CAS PubMed.
- V. R. Babaev, M. B. Patel, C. F. Semenkovich, S. Fazio and M. F. Linton, J. Biol. Chem., 2000, 275, 26293–26299 CrossRef CAS PubMed.
- P. P. He, X. P. Ouyang, Y. Y. Tang, L. Liao, Z. B. Wang, Y. C. Lv, G. P. Tian, G. J. Zhao, L. Huang, F. Yao, W. Xie, Y. L. Tang, W. J. Chen, M. Zhang and Y. Li, et al., Biochimie, 2014, 106, 81–90 CrossRef CAS PubMed.
- T. Chen, Z. Li, J. Tu, W. Zhu, J. Ge, X. Zheng, L. Yang, X. Pan, H. Yan and J. Zhu, FEBS Lett., 2011, 585, 657–663 CrossRef CAS PubMed.
- G. P. Tian, W. J. Chen, P. P. He, S. L. Tang, G. J. Zhao, Y. C. Lv, X. P. Ouyang, K. Yin, P. P. Wang, H. Cheng, Y. Chen, S. L. Huang, Y. Fu, D. W. Zhang and W. D. Yin, et al., Biochimie, 2012, 94, 2749–2755 CrossRef CAS PubMed.
- G. Lan, W. Xie, L. Li, M. Zhang, D. Liu, Y. L. Tan, H. P. Cheng, D. Gong, C. Huang, X. L. Zheng, W. D. Yin and C. K. Tang, Biochem. Biophys. Res. Commun., 2016, 472, 410–417 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05176g |
| This journal is © The Royal Society of Chemistry 2019 |