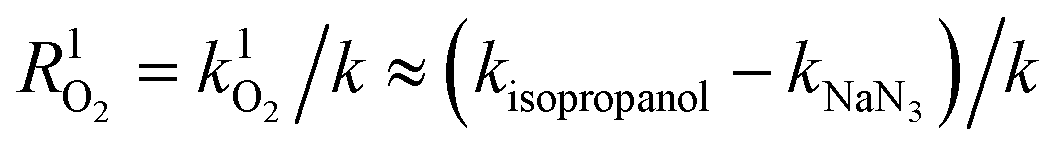DOI:
10.1039/C9RA05169D
(Paper)
RSC Adv., 2019,
9, 31325-31332
Mechanism of methylphosphonic acid photo-degradation based on phosphate oxygen isotopes and density functional theory†
Received
8th July 2019
, Accepted 6th September 2019
First published on 2nd October 2019
Abstract
Methylphosphonic acid (MPn) is an intermediate in the synthesis of the phosphorus-containing nerve agents, such as sarin and VX, and a biosynthesis product of marine microbes with ramifications to global climate change and eutrophication. Here, we applied the multi-labeled water isotope probing (MLWIP) approach to investigate the C–P bond cleavage mechanism of MPn under UV irradiation and density functional theory (DFT) to simulate the photo-oxidation reaction process involving reactive oxygen species (ROS). The results contrasted with those of the addition of the ROS-quenching compounds, 2-propanol and NaN3. The degradation kinetics results indicated that the extent of MPn degradation was more under alkaline conditions and that the degradation process was more rapid at the initial stage of the reaction. The phosphate oxygen isotope data confirmed that one exogenous oxygen atom was incorporated into the product orthophosphate (PO4) following the C–P bond cleavage, and the oxygen isotopic composition of this free PO4 was found to vary with pH. The combined results of the ROS-quenching experiments and DFT indicate that the C–P bond was cleaved by OH−/˙OH and not by other reactive oxygen species. Based on these results, we have established a mechanistic model for the photolysis of MPn, which provides new insights into the fate of MPn and other phosphonate/organophosphate compounds in the environment.
1. Introduction
Methylphosphonic acid (MPn) is one of the most structurally simple C–P bonded compounds. It is a precursor to more complex phosphonate molecules, such as glyphosate.1–3 MPn is an intermediate in the synthesis of the chemical warfare agents (CWA), such as sarin and VX,4–7 and a biosynthesis product of most marine microorganisms, such as Nitrosopumilus maritimus.8 MPn is very stable,9 and recent research shows that the microbial metabolism of MPn has a significant link to the methane production in the open ocean and the large-scale biogeochemical cycles of key elements—Fe, N and P,10,11 and thus indirectly affects the environment.12,13
The net flux of methane is generally supersaturated at the ocean surface. Photolysis of MPn maybe one of the pathways of methane production under aerobic environments, however, the detailed mechanism for the cleavage of the C–P bond still remains elusive.14 The contribution of UV radiation to the MPn degradation in the uppermost euphotic zone of aquatic systems could be highly significant. In aquatic systems, UV photons usually interact with the reactant molecules to form radical species that degrade the dissolved phosphonate compounds by stimulating an attack on the C–P bond.15,16 Photo-degradation of the dissolved organic phosphorus (DOP) is an important pathway for the generation of the biologically-available dissolved inorganic phosphate (DIP).17,18 Therefore, the degradation of MPn is accompanied by the release of the dissolved inorganic phosphate (PO4 or Pi). Under most environmental conditions and in the absence of microbial/enzymatic activity, the oxygen in the P–O of the dissolved inorganic orthophosphate (DIP or PO4) is difficult to exchange with oxygen in the other substances, such as water.19 The ratio of the oxygen isotopes, 18O/16O, in orthophosphate (δ18OP) has been widely applied to study the reaction pathways and P-cycling processes over the past decade.20,21
The multi-labeled water isotope probing (MLWIP) approach, developed by Blake22 and widely employed by others,23,24 was first used to probe the C–P bond cleavage mechanism of glyphosate and phosphonoacetic acid by Sandy et al.25 Previous results from our group indicate that O from an external source, possibly 2 sources (ambient water and O2), is incorporated into the orthophosphate and released along with the glyphosate C–P bond cleavage. However, different models have been put forth to account for the source(s) of the external O incorporated into the released orthophosphate.26,27 In order to verify which form of oxygen is involved in the C–P bond cleavage process, we studied the role of the ROS in the C–P bond cleavage mechanism.
In surface waters, some organic contaminants undergo self-sensitization in sunlight and form an excited triplet state species that can perform direct photolysis.28 To investigate the process of the organic contaminant degradation and test whether ROS are produced during the degradation process, NaN3 (an 1O2 and ˙OH scavenger) and 2-propanol (an ˙OH scavenger) are widely used as ROS quenchers to constrain photolysis.29,30 As for the environmental chemical effect on degradation, pH is one of the most important factors influencing photo-oxidation due to the different proportion of ROS produced at different pH.31,32 Therefore, different pH conditions can help predict the reaction path of MPn in models of its transport and degradation in natural environment, and also aid in the understanding of the C–P bond cleavage mechanisms.
In this study, we present the results of the experiments performed on the degradation of MPn using the 18O-labeled water to probe the mechanism and kinetics of the C–P bond cleavage and track the oxygen isotope composition of the inorganic product PO4 or Pi (δ18OP). We also studied the role of the reactive oxygen species (ROS) and pH in the MPn degradation in aquatic systems. DFT was used to verify the role of ROS in the MPn photo-degradation as it may not be possible to isolate and explicitly determine the O isotopic composition of ROS the way it is typically done to assess the contribution of the potential O sources, involved in the C–P cleavage.26,27 Also, since the MPn structure is very simple, the degradation products are detected by nuclear magnetic resonance (NMR). Hence, based on the combined results of the abovementioned studies, we posit plausible MPn photo-degradation pathways.
2. Materials and methods
2.1 Reagents
MPn was purchased from Sigma-Aldrich, and NaN3 and 2-propanol were purchased from Sinopharm Chemical Reagent Co., China. Deuterium oxide for the NMR analysis was obtained from Sahn Chemical Technology. All other materials were ≥99% pure and obtained from commercial sources.
2.2 Methylphosphonic acid degradation experiments
A series of MPn degradation experiments were carried out to investigate the effect of pH and water O isotopic composition on the degradation kinetics. Aqueous solutions of MPn were prepared in Milli-Q deionized water spiked with 18O-labeled water with oxygen isotopic composition (δ18OW) of −6, 10.8 and 32.4‰ (VSMOW). The pH values of the MPn solutions were adjusted to 3 and 8, respectively. The concentrations of MPn were selected to release between 50 and 100 μmoles of Pi after complete photolysis. About five milliliters of each MPn solution was treated with a 1200 W high-pressure mercury lamp in an Ace glass 7900 UV photo-oxidation apparatus (in the Blake laboratory at Yale University) kept at ≤40 °C. Each experiment was conducted in triplicate, and the degradation rates of MPn were determined based on the concentration of the inorganic phosphate released into the solution every 6 hours. The phospho-molybdate blue assay was used to perform the colorimetric phosphate analysis.33
The orthophosphate samples were processed using the following method described by Jaisi and Blake.34 The released phosphate was first precipitated as ammonium phosphomolybdate (APM) at an acidic pH, then dissolved and precipitated as magnesium ammonium phosphate (MAP) at an alkaline pH, and treated with a cation exchange resin to remove Mg2+ and other contaminants before the final precipitation as silver phosphate (Ag3PO4), the compound used for the phosphate oxygen isotope analysis. The Ag3PO4 was roasted at ∼550 °C for 3 min under vacuum for further purification and the removal of water and any possible remaining organic matter. Silver capsules, containing 250–350 μg of the roasted Ag3PO4, were prepared in triplicate for the oxygen isotope analysis and examined on a Thermo-Chemolysis/Elemental Analyzer (TC/EA) coupled to a Delta V continuous flow isotope ratio mass spectrometer (IRMS, Finnigan-MAT, Bremen, Germany). The δ18OP values of the silver phosphate samples were calibrated against two laboratory silver phosphate standards, YR1a and YR-32, with the δ18OPO4 values of −5.49‰ and 33.63‰, respectively. The analytical precision of the silver phosphate δ18OP values based on replicate analyses was better than ±0.3‰ (1SD).35 Isotope analysis was performed at the Yale Institute for Biospheric Studies (YIBS)–Yale Analytical and Stable Isotope Center (YASIC).
2.3 Methylphosphonic acid degradation experiments w/ROS
A series of experiments were carried out to investigate the influence of ROS under UV irradiation. Twenty mL aliquots of 0.1 mM MPn were prepared and transferred to separate 35 mL sterilized quartz tubes to which 20 mM 2-propanol (an ˙OH scavenger) and 20 mM NaN3 (an 1O2 and ˙OH scavenger) were added to quantify the contribution of ˙OH and 1O2 to the photolysis, respectively. The samples were treated with a photo-chemical reaction instrument (BL-GHX-V; Shanghai, China) under a 1000 W mercury lamp with a temperature of ≤25 °C controlled by a cooling water circulation system at the University of Science and Technology Beijing.
2.4 DFT calculations
The DFT calculations were used to verify the contribution of ROS and the theoretical reaction mechanism of MPn solution under UV conditions. All calculations were performed using the Gaussian 09 package. The 3D-structures of the optimized species were generated using GaussView 5.0. Geometry optimizations and the vibrational frequencies of all the stationary points were performed at the M06-2X/6-311G+(d,p) level. Simulation of the solvent effect was performed using the SMD model with water as the solvent. With geometry optimization at each point, the relaxed potential energy surface (PES) scan was performed to ascertain the existence of the transition state in each step. A lower level, M06-2X/6-31G(d), was adopted for the PES scan. Intrinsic reaction coordinate (IRC) calculations were performed to assure that the transition states connected the reactants and the products for each elementary reaction. The activation free energy barrier was defined as the free energy difference between the transition state and the lowest-energy stationary point in the reaction pathway.
2.5 NMR analysis of complexation in the solution
Spectroscopic studies were carried out in order to investigate the structure of the product of MPn photo-oxidation. This included the analysis of the proton NMR spectrum obtained. MPn (1.87 mM) was directly dissolved in 10 mL D2O and exposed to UV irradiation. The photo-degradation conditions were consistent with those of the ROS-quenching reactions. Carbon (13C) NMR and proton (1H) NMR spectra were recorded in D2O at room temperature on a Bruker-500 MHz nuclear magnetic resonance (NMR) spectrometer (Bruker).
3. Results and discussion
3.1 MPn degradation kinetics
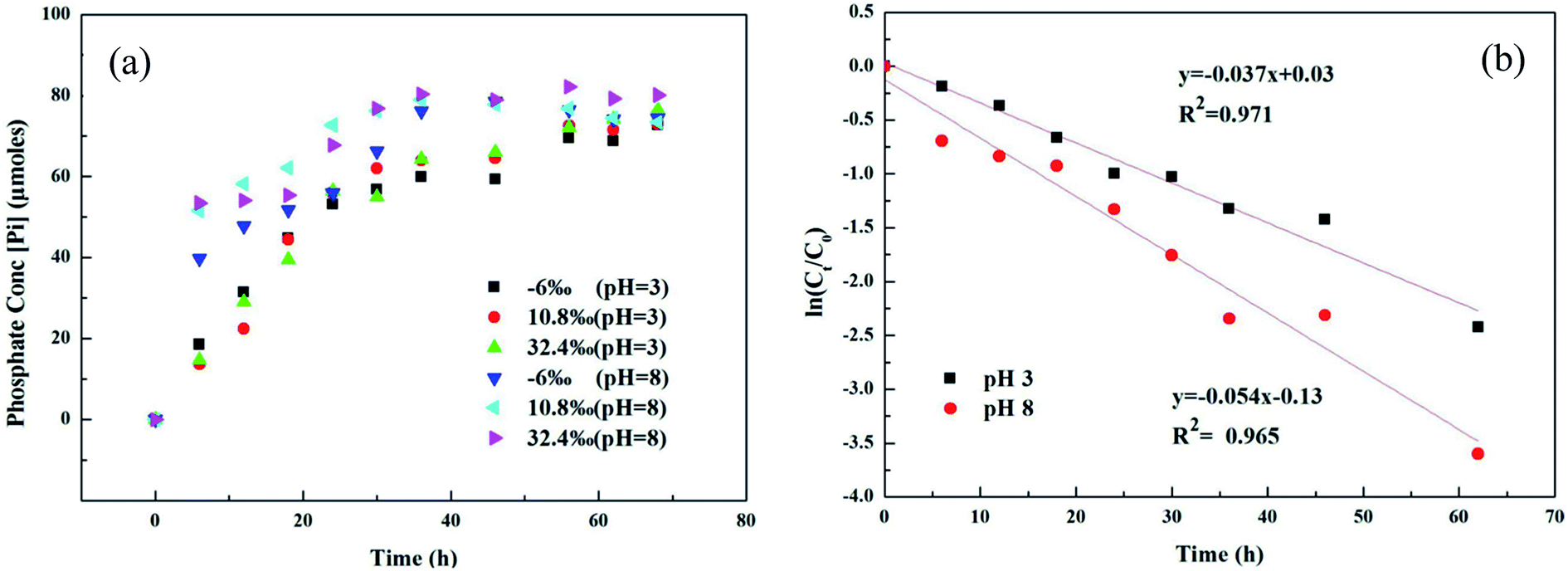
Microbial metabolism was reported to be an important pathway for MPn degradation in the environment, while chemical degradation is uncommon.36 Phosphate (Pi) release in different 18O-labeled water samples based on pH during UVR photo-oxidation of MPn is shown in Fig. 1a. Degradation kinetics of MPn (Fig. 1a) followed pseudo-first order kinetics with a rapid initial rate followed by a gradual slower rate before approaching maximum degradation. There was no significant difference in the degradation rate of the different 18O-labeled solutions. Fig. 1b shows the photolysis of MPn at pH 3 and pH 8; the corresponding photolysis rate constants were 0.037 h−1 and 0.054 h−1, respectively. Pajares et al. found that under the alkaline conditions, phenol exists in a more active anion state, and its degradation rate is faster.37 Previous studies indicated that 2-chloroethylphosphonic acid hydrolyzes at 35–55 °C, appreciably only at alkaline pH (6–9) based on the different ionic species present at different pH.38 Nevertheless, the control experiments revealed that without UV exposure, MPn hydrolysis is almost nonexistent in solution at about 40 °C. On the other hand, the cleavage of the C–P bond in alkylphosphonates is assumed to be a redox-dependent free-radical process, which is probably dependent on the aeration and the pH of the medium.39,40 Therefore, we posit that MPn is more likely to undergo redox reactions under alkaline conditions.
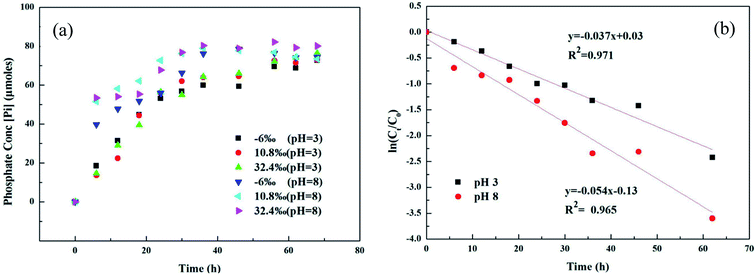 |
| | Fig. 1 (a) UVR degradation of MPn in different labeled water samples based on the released inorganic phosphate (Pi) concentration; (b) photolysis kinetics of MPn irradiation by a UV lamp at pH 3 and pH 8. | |
3.2 Stable oxygen isotope analysis based on the amount of PO4 released from MPn as a result of UV photo-oxidation
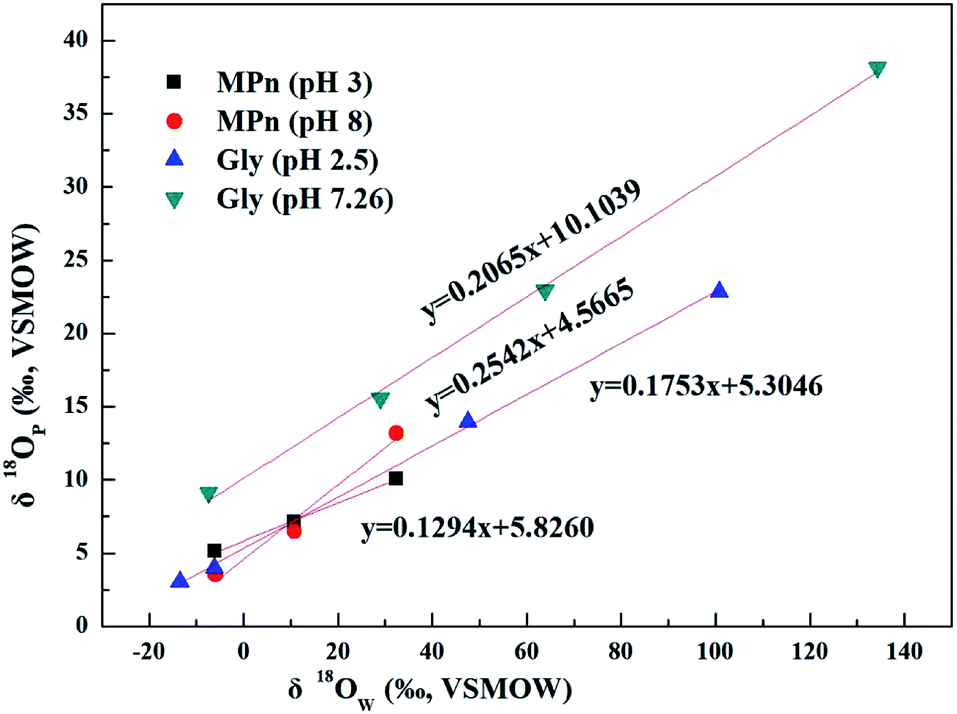
The UVR degradation data of MPn in different 18O-labeled water solutions based on the phosphate concentration measurements, expressed as the percentage of the total available P, are shown in Table 1. As shown in the table, at least 90% of the MPn hydrolysis was achieved in 68 h of the UVR treatment. The phosphorus atom in MPn is bonded to three oxygen atoms. Thus, the cleavage of the C–P bond results in the incorporation of an external oxygen atom from an ambient source to generate phosphate. In water, O from water have the greatest chance of binding to released PO4 when the C–P bond breaks. So the releasing phosphate oxygen isotope values (δ18OP) have a positive correlation with 18O-labeled water (δ18OW). The slope of δ18OPvs. δ18OW was used to express the amount of O from water incorporated into the released PO4. If one oxygen atom (1 out of 4) from water is incorporated into the released PO4, the slope of the linear correlation curve of δ18OPvs. δ18OW should be ca. 0.25.25 The slopes obtained for MPn indicated a 13% (acidic) and 25% (alkaline) ambient water oxygen incorporation into the released phosphate (Fig. 2). Yu41 found that only 7% of the ambient water oxygen was incorporated into the released PO4 for MPn under the 400 W and 1200 W mercury lamp UV photo-oxidation after 2162 h. This observation indicates that different reaction conditions affect the contribution rate of each substance involved in the reaction. Both Sandy25 and Jaisi26 studied the mechanism of the glyphosate degradation under ultraviolet radiation; the slope of δ18OPvs. δ18OW was 0.175 at pH 2.5 and 0.21 at pH 7.26, respectively. These results are similar to those of the present experiments, and it was concluded that more O from ambient water is incorporated into the product PO4 at higher pH. The lower the slope, the fewer O atoms come from water, and we assume that the other source of O is from O2.42 The dissolved oxygen is usually converted to the radical O species and decomposes organic matter in the aqueous solution.43 Thus, at low pH, there are more radical O species from the exogenous oxygen present in the solution. Based on the acid–base equilibrium relationship, there are few hydroxyl groups present at pH 3, and the dissolved oxygen is one of the main sources of hydroxyl groups under the photolysis conditions. Therefore, the slope of δ18OPvs. δ18OW is lower due to the higher amount of exogenous oxygen incorporated into the released product PO4 at pH 3.
Table 1 Concentration of released Pi at different pH values after the UVR treatment and measured Pi–δ18OP in different labeled water solutions
| Phosphonate |
Water δ18OW (‰, VSMOW) |
Released Pi (μmoles) |
Percent released based on [Pi] (%) |
Pi-δ18OP, ‰ (VSMOW) |
SD |
n
|
| MPn |
−6 (pH 3) |
73.75 |
91.28 |
5.123 |
0.302 |
3 |
| 10.8 (pH 3) |
76.03 |
91.54 |
7.093 |
0.097 |
3 |
| 32.4 (pH 3) |
75.59 |
90.63 |
10.076 |
0.149 |
4 |
| −6 (pH 8) |
79.91 |
95.30 |
3.521 |
0.113 |
4 |
| 10.8 (pH 8) |
79.66 |
96.27 |
6.459 |
0.126 |
4 |
| 32.4 (pH 8) |
88.65 |
98.83 |
13.179 |
0.068 |
4 |
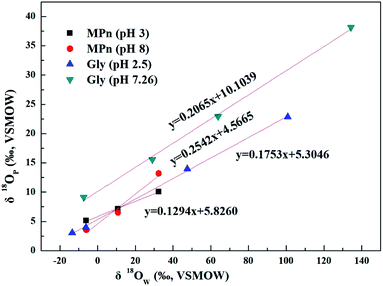 |
| | Fig. 2 Plots of δ18OPvs. δ18OW of phosphates obtained from UVR photo-oxidative hydrolysis of MPn at different pH values and the comparison to the glyphosate data from Sandy' paper. | |
3.3 Participation of ROS
Ultraviolet radiation is known to stimulate the formation of free radicals. Under the UVR conditions, atmospheric oxygen combines with the excited organic compounds to form peroxy radicals, which are subsequently hydrolyzed.25 To qualitatively investigate the role of ROS in the MPn photodegradation, solutions containing 0.1 mM MPn were irradiated in the presence of 2-propanol and NaN3, respectively. The contribution of various radical species was calculated using eqn (1) and (2).44 While k represents the rate constant of the original reaction without the addition of the radical quenchers, kisopropanol and kNaN3 represent the rate constants in the presence of the scavengers.| | | R˙OH = k˙OH/k ≈ (k − kisopropanol)/k | (1) |
| | 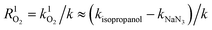 | (2) |
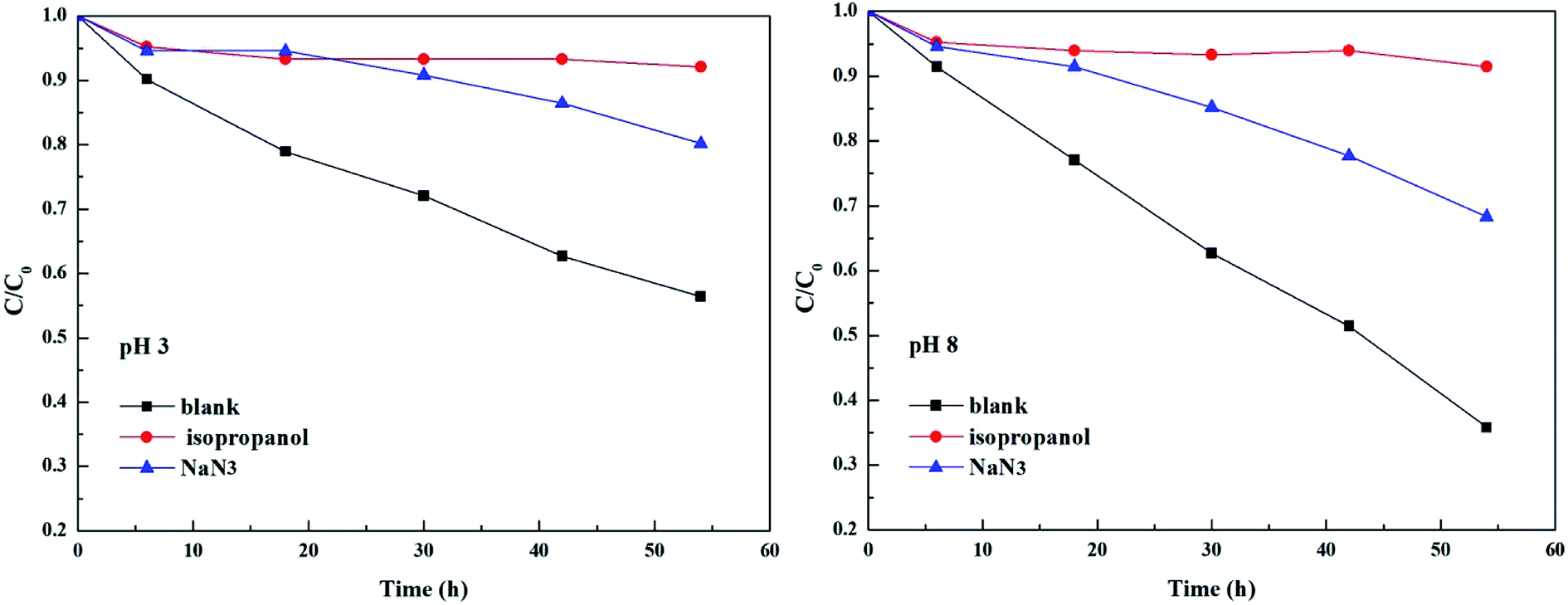
As shown in Fig. 3 and Table 2, the photolysis reaction conforms to the first order kinetics under acidic and alkaline conditions. The photolysis rate constant without additives and MPn alone was 0.98 × 10−2 h−1 at pH 3 and 1.77 × 10−2 h−1 at pH 8. All samples containing radical scavengers showed a decrease in the photolysis rate. The value for kisopropanol and kNaN3 was 0.07 × 10−2 h−1 and 0.31 × 10−2 h−1 at pH 3, and 0.06 × 10−2 h−1 and 0.6 × 10−2 h−1 at pH 8, suggesting that the reactive oxygen species play an important role in the reaction. It was calculated that the contribution of the ˙OH radical is 92.86% at pH 3 and 96.61% at pH 8. Because kisopropanol < kNaN3, we hypothesized that, on the one hand, the singlet oxygen consumes sodium azide and alleviates the quenching effect of sodium azide on the hydroxyl group, and on the other hand, methylphosphonic acid is unreactive under these reaction conditions. Also, the degradation of MPn is faster under alkaline conditions than under acidic conditions. This may be because hydroxyl radicals are more abundant under alkaline conditions relative to acidic conditions. In summary, hydroxyl groups play a major role in the C–P bond cleavage process. As Jaisi observed, the hydroxyl ion and the ˙OH radical have similar barriers opposing the C–P bond cleavage, indicating that a strong competitive relationship affects their respective rates.26 It is known that molecular oxygen may act as an electron acceptor in the photosensitization process and produce superoxide anions or hydroxyl radicals.45 It was not known whether the superoxide anions are involved in the MPn photolysis reactions. On the other hand, there could be a reverse phenomenon: kisopropanol < kNaN3. So, we used the DFT method to calculate the reaction barrier and verify the results of the ROS-quenching experiments.
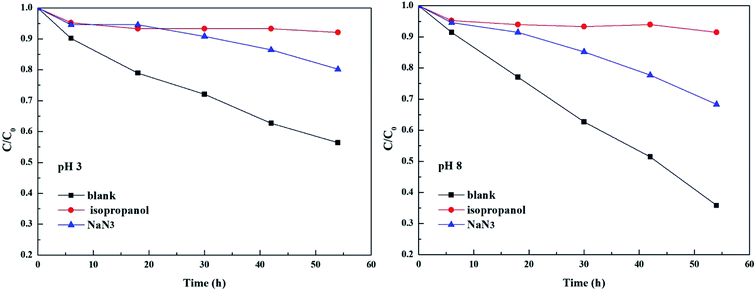 |
| | Fig. 3 Comparison of photolysis of MPn in the absence and the presence of 20 mM isopropanol and 20 mM NaN3 at pH 3 and pH 8. | |
Table 2 The contribution of various radical species to the degradation of MPn and the rate constants of the degradation after adding various scavengers
| Scavengers |
MPn |
| pH 3 |
pH 8 |
| Inhibition rate (%) |
Rate constant (h−1) |
Inhibition rate (%) |
Rate constant (h−1) |
| Blank |
— |
0.98 × 10−2 |
— |
1.77 × 10−2 |
| Isopropanol |
92.86 |
0.07 × 10−2 |
96.61 |
0.06 × 10−2 |
| NaN3 |
68.37 |
0.31 × 10−2 |
66.1 |
0.6 × 10−2 |
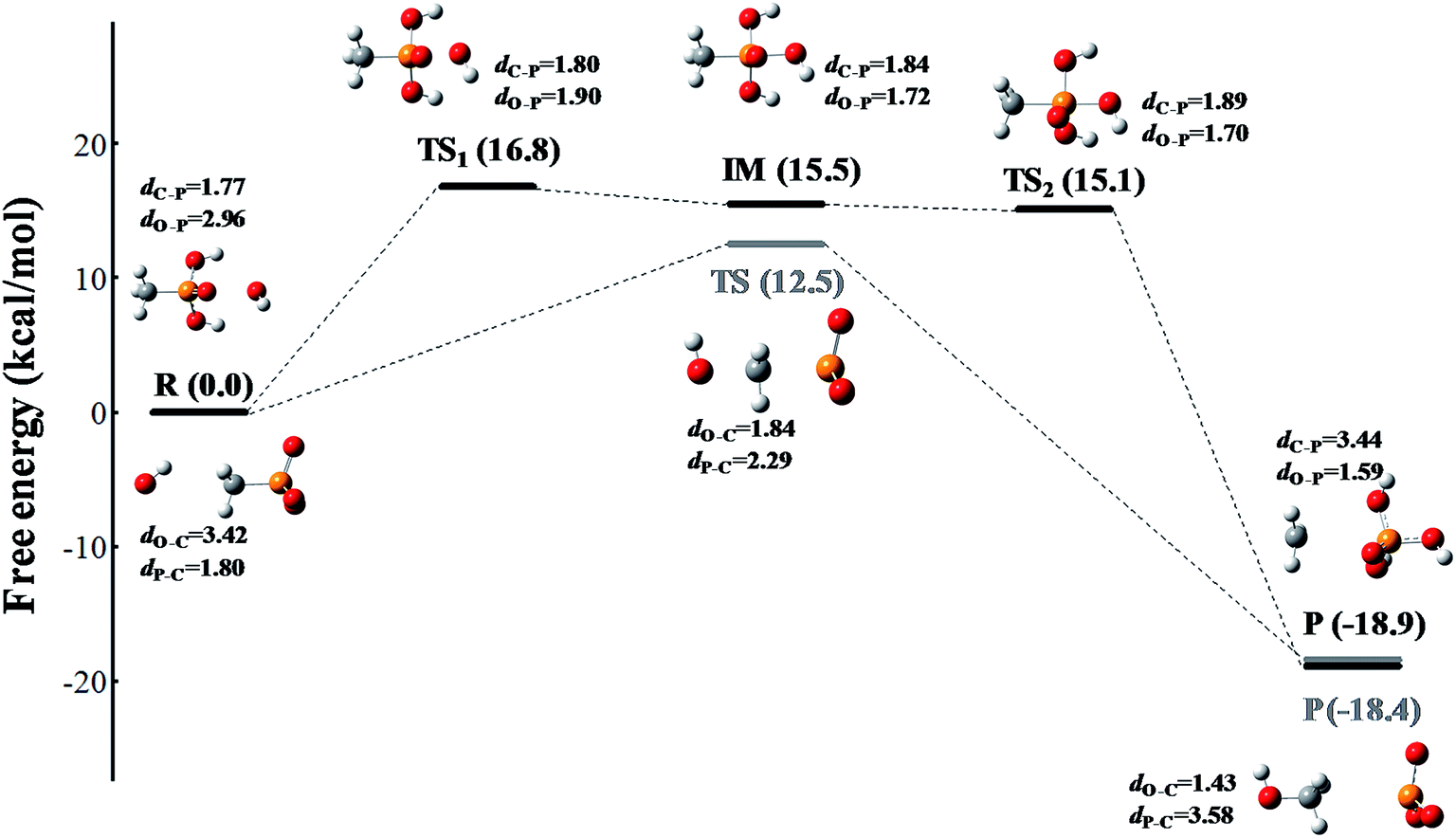
The reaction occurs according to the nucleophilic substitution mechanism, and the nucleophile ROS can attack either the methyl group or the phosphoryl group of the methylphosphonic acid molecule. However, the calculated free energy barriers involved 1O2 and O2˙− radical triggered cleavage of the C–P bond in MPn are extremely high, which can be considered in the ordinary conditions is difficult to happen as listed in Tables S1 and S2.† In comparison, the ˙OH radical is more likely to trigger the cleavage of the C–P bond of methylphosphonic acid. As shown in Fig. 4 and Table S3,† the C–P bond can be broken by the ˙OH radical either from the phosphoryl side under acidic conditions or the methyl side under alkaline conditions. The former refers to an asynchronous process with a free energy barrier of 16.8 kcal mol−1 for the first half and a barrier-free step for the second half; the process is exothermic with a free energy change of −18.9 kcal mol−1. The latter is a synchronous process with a free energy barrier of 12.5 kcal mol; it is exothermic with a free energy change of −18.4 kcal mol−1. The lower the potential energy barrier, the more spontaneous will be the reaction. Our calculations suggest that the alkaline conditions are more favorable for the cleavage of the C–P bond in methylphosphonic acid than the acidic conditions, which is consistent with the results of the ROS-quenching experiments.
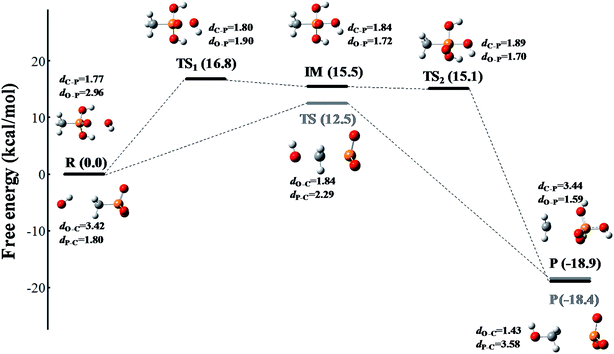 |
| | Fig. 4 The free energy profile of the ˙OH radical triggered cleavage of the C–P bond in MPn. The pathway colored in black represents the nucleophilic attack on the phosphoryl side of MPn under acidic conditions, and the pathway colored in gray represents the nucleophilic attack on the methyl side of MPn under alkaline conditions. The free energy is measured in kcal mol−1 and the distance is measured in Å. The structures are presented in a ball-and-stick model with carbon in gray, oxygen in red, phosphorus in orange and hydrogen in white. | |
3.4 Mechanism of C–P bond cleavage
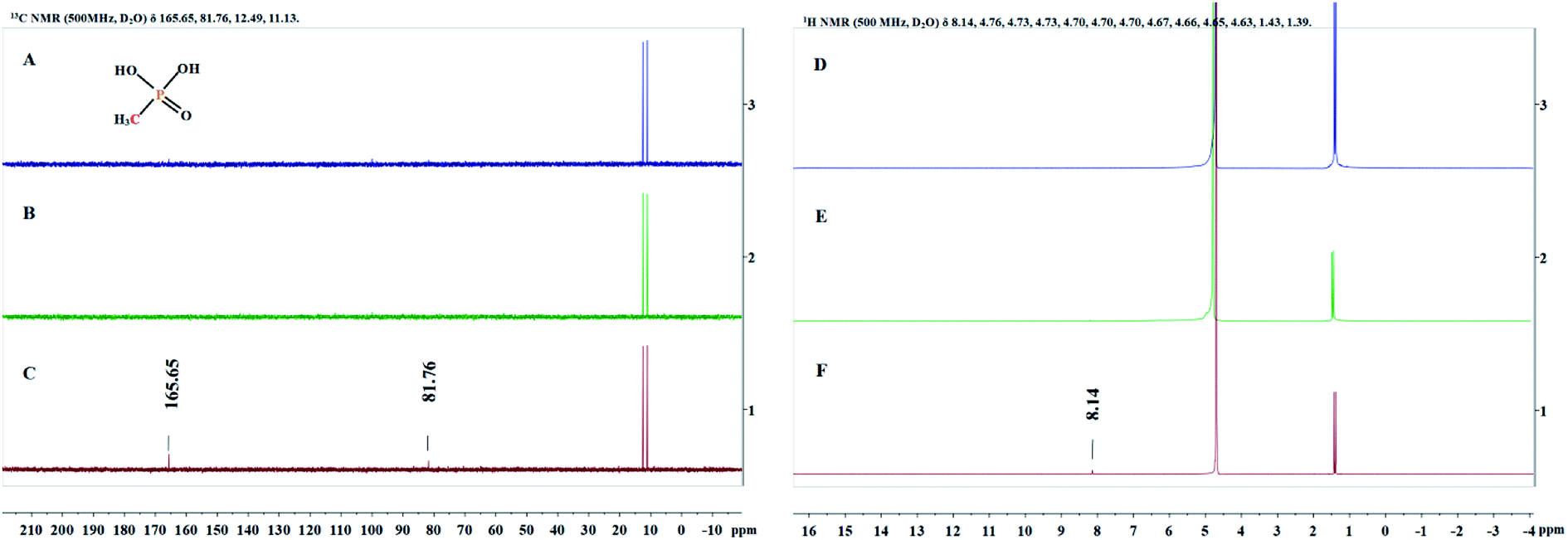
Since the structure of MPn is relatively simple, we detected the change of groups in the reaction by 1H NMR and 13C NMR spectroscopy, and then deduced the degradation products. Sample solutions (0.5 mL aliquots) were used for the NMR analysis at different times during the reaction and the results are shown in Fig. 5. The 1H NMR results showed a new single peak at 8.14 ppm on the fourth day of reaction, and two new peaks at 81.76 ppm and 165.65 ppm were found in the 13C NMR spectrum. As mentioned above, we speculate that the organic degradation products of MPn are methanol and formic acid, which are different from the microbial degradation pathway product, methane.10,36 The generation of ROS and degradation of MPn are represented by formulas (3)–(9).46–48
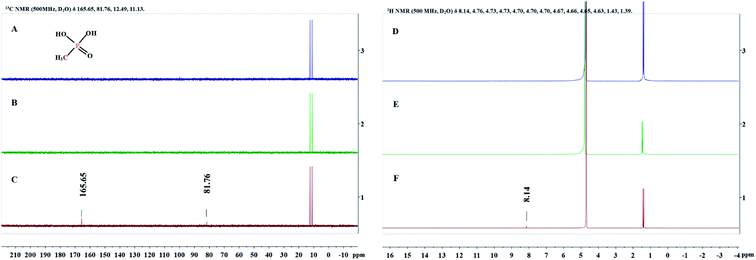 |
| | Fig. 5
13C NMR spectra (A–C) and 1H NMR spectra (D–F) obtained at the start of the reaction (A and D), the first day (B and E) and the fourth day (C and F). | |
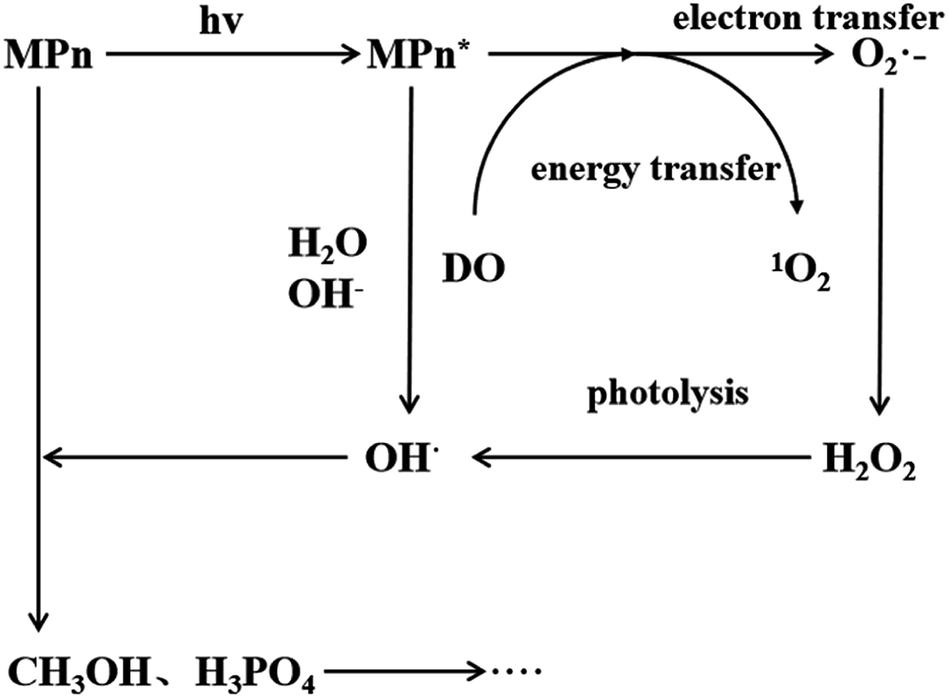
Under ultraviolet radiation, MPn absorbs energy and transitions into a single-line excited state. Then, the energy, transferred to the dissolved oxygen in solution, generates singlet oxygen, or electrons are transferred to produce superoxide anion radicals. The superoxide anion radicals combine with the hydrogen ion to form hydrogen peroxide, and finally hydroxyl, which then reacts with MPn, producing methanol and phosphoric acid. Methanol is further oxidized to formic acid and ultimately generates stable substances.
The proposed mechanism for the UVR photolysis of the C–P bond in MPn is presented in Fig. 6. It can be proposed that a series of reactions between phosphonate and other substances in the solution were induced by ultraviolet photons, and a nucleophilic substitution occurred with the generated radical O species and phosphonate.49 The nucleophilic attack could be influenced by the nature of the group adjacent to the C–P bond in phosphonate. The hydrolysis mechanism and the water incorporation pattern/slope for phosphate released from the phosphonate studied here differ markedly from those reported for the phosphomonoesters hydrolyzed by UVR in similar labeled water in the previous experiments.27 We posit that C–P bond cleavage during UV photo-oxidation results from the excitation of the phosphonate molecules into more reactive species, which transfer electrons to acceptors present in the reaction medium to form free radicals. The free radicals subsequently recombine or hydrolyze to form radicals that react with oxygen to produce peroxy radicals. Upon further photolysis, the peroxy radicals are converted to hydrogen peroxide, and finally decomposed to hydroxyl radicals that attack the methyl group.43,50 The reaction between water and methyl phosphonate cationic states also produces hydroxyl radicals, and we hypothesized that as the pH increases, the proportion of O from water incorporated into the released PO4 increases.
| |  | (3) |
| | | MPn* + O2 → MPn˙+ + O2˙− | (5) |
| | | HO2˙ + O2˙− + H+ → H2O2 + O2 | (7) |
| | | H2O2 + O2˙− → OH˙ + OH− + O2 | (8) |
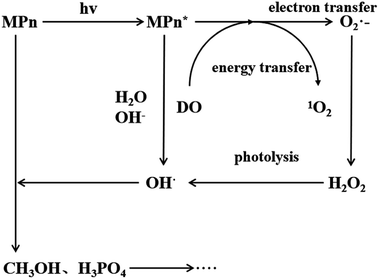 |
| | Fig. 6 Proposed mechanism for MPn photo-degradation and ROS formation. | |
4. Conclusions
The kinetics of MPn photo-degradation under UV irradiation was investigated in this study. MPn was approximately 90% degraded within 68 h at both pH 3 and 8 and slightly impacted by the 18O-labeled water, which suggests the involvement of ROS in the C–P bond cleavage. The results of the stable isotope analysis indicate that oxygen from a source other than ambient water is incorporated into the produced inorganic phosphate during the C–P bond cleavage in MPn. The results of the ROS-quenching experiments and DFT simulations of the photo-oxidation reaction suggest that the dissolved oxygen reacts with the radical species produced by the UV excitation of MPn and eventually forms hydroxyl groups that attack the C–P bond, leading to the incorporation of the hydroxyl O into the released phosphate. The NMR results showed that the degradation products of MPn are methanol, formic acid and inorganic phosphoric acid. This study indicates that the phosphate oxygen isotope technique can be used to trace the source of O, which is incorporated into PO4, when the compound containing P is degraded, releasing PO4. DFT theoretical simulation can qualitatively obtain the degree of difficulty of reactive oxygen radicals and reactive substances. Combining with the ROS-quenching experiment and phosphate oxygen isotope experiment, the source and reaction amount of oxygen that incorporated into the orthophosphate can be derived.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The labelled-water experiments and the stable oxygen isotope analysis were carried out at Yale University and supported in part by the grants, awarded to R. E. Blake from the US National Science Foundation (NSF-EAR-0746241, NSF-OCE-0928247 and NSF-OCE-1129499) and NASA (Award No. NNX13AJ36G). This study was also supported in part by the grants from the National Natural Science Foundation of China (41822706, 41473096, 41703109 and 21903089) and Beijing Natural Science Foundation (8182034).
References
- S. V. Kononova and M. A. Nesmeyanova, Biochemistry, 2002, 67, 184–195 CAS.
- R. Wagner, S. J. Wetzel, J. Kern and H. M. Kingston, J. Mass Spectrom., 2012, 47, 147–154 CrossRef CAS.
- G. Zhang, B. Xu, H. Chong, W. Wei, C. Wang and G. Wang, RSC Adv., 2019, 9(25), 14016–14023 RSC.
- O. Ruth, K. Doron, N. Ido, R. Hadar, E. Shmuel and C. Ishay, Environ. Sci. Technol., 2014, 48, 10912 CrossRef.
- Y. Seto, M. Tachikawa, M. Kanamori-Kataoka, K. Sasamoto and N. Ochiai, J. Chromatogr. A, 2017, 1501, 99–106 CrossRef CAS.
- Q. Zhao, G. Liu, H. Zhang, F. Zhou, Y. Li and W. Cai, J. Hazard. Mater., 2016, 324, 194–202 CrossRef.
- D. F. S. Gallis, J. A. Harvey, C. J. Pearce, M. G. Hall, J. B. Decoste, M. K. Kinnan and J. A. Greathouse, J. Mater. Chem. A, 2018, 6, 3038–3045 RSC.
- W. W. Metcalf, B. M. Griffin, R. M. Cicchillo, G. Jiangtao, J. Sarath Chandra, H. A. Cooke, B. T. Circello, B. S. Evans, M. H. Willm and D. A. Stahl, Science, 2012, 337, 1104–1107 CrossRef CAS.
- I. Rodin, T. Baygildiev, A. Stavrianidi, A. Braun, I. Rybalchenko and O. Shpigun, Chromatographia, 2015, 78, 1–7 CrossRef.
- D. A. A. K. Del Valle, Aquat. Microb. Ecol., 2014, 73, 93–105 CrossRef.
- D. M. Karl, L. Beversdorf, K. M. Björkman, M. J. Church, A. Martinez and E. F. Delong, Nat. Geosci., 2008, 1, 473–478 CrossRef CAS.
- E. B. Burwicz, K. Kretschmer, L. Rüpke and A. Biastoch, Global Biogeochem. Cycles, 2015, 29, 610–625 CrossRef.
- W. Gian-Reto, P. Eric, C. Peter, M. Annette, P. Camille, T. J. C. Beebee, F. Jean-Marc, H. G. Ove and B. Franz, Nature, 2002, 416, 389–395 CrossRef.
- X. Xu, F. Ji, Z. Fan and L. He, Int. J. Environ. Res. Public Health, 2011, 8, 1258–1270 CrossRef CAS.
- A. Paytan and K. Mclaughlin, Chem. Rev., 2007, 107, 563–576 CrossRef CAS.
- C. N. Rusu and J. T. Yates Jr, J. Phys. Chem. B, 2000, 104, 12292–12298 CrossRef CAS.
- G. Liu, Q. Tang, Y. Zhou, X. Cao, J. Zhao and D. Zhu, Photochem. Photobiol. Sci., 2017, 16, 467–475 RSC.
- E. Rott, R. Minke, U. Bali and H. Steinmetz, Water Res., 2017, 122, 345 CrossRef CAS.
- R. J. O'neil, W. T. Vennemann and F. W. Mckenzie, Geochim. Cosmochim. Acta, 2003, 67, 3135–3144 CrossRef.
-
D. P. Jaisi and R. E. Blake, Chapter One – Advances in Using Oxygen Isotope Ratios of Phosphate to Understand Phosphorus Cycling in the Environment, Advances in Agronomy, Academic Press, 2014, pp. 1–53 Search PubMed.
- S. R. Joshi, R. K. Kukkadapu, D. J. Burdige, M. E. Bowden, D. L. Sparks and D. P. Jaisi, Environ. Sci. Technol., 2015, 49, 5887 CrossRef CAS.
- R. E. Blake, J. R. O'Neil and G. A. Garcia, Am. Mineral., 1998, 83, 1516–1531 CrossRef CAS.
- D. P. Jaisi and R. E. Blake, Geochim. Cosmochim. Acta, 2010, 74, 3212 Search PubMed.
- Y. Liang and R. E. Blake, Geochim. Cosmochim. Acta, 2009, 73, 3782–3794 CrossRef CAS.
- E. H. Sandy, R. E. Blake, S. J. Chang, J. Yao and C. Yu, J. Hazard. Mater., 2013, 260, 947–954 CrossRef CAS.
- D. P. Jaisi, H. Li, A. F. Wallace, P. Paudel, M. Sun, A. Balakrishna and B. N. Lerch, J. Agric. Food Chem., 2016, 64, 8474 CrossRef CAS.
- H. Li, S. R. Joshi and D. P. Jaisi, J. Agric. Food Chem., 2015, 64, 529–538 CrossRef.
- M. Salimi, M. Behbahani, H. R. Sobhi, M. Gholami and A. Esrafily, New J. Chem., 2018, 43 10.1039/C8NJ05020A.
- J. Heo, Y. Yoon, D. H. Kim, H. Lee, D. Lee and N. Her, Desalin. Water Treat., 2015, 1–13 Search PubMed.
- Z. Lei, J. Yuefei, Z. Chao, Z. Ya, W. Zunyao and Y. Xi, Water Res., 2013, 47, 153–162 CrossRef.
- Y. Chen, C. Hu, J. Qu and M. Yang, J. Photochem. Photobiol., A, 2008, 197, 81–87 CrossRef CAS.
- X. Han, Y. Li, D. Li and C. Liu, Environ. Sci. Technol., 2017, 51, 3784–3793 CrossRef CAS.
- J. Murphy and J. P. Riley, Anal. Chim. Acta, 1962, 27, 91–95 CrossRef.
- D. P. Jaisi, R. K. Kukkadapu, L. M. Stout, V. Tamas and R. E. Blake, Environ. Sci. Technol., 2011, 45, 6254–6261 CrossRef CAS.
- Y. Liang and R. E. Blake, Geochim. Cosmochim. Acta, 2006, 70, 3957–3969 CrossRef CAS.
- A. Martínez, L. A. Ventouras, S. T. Wilson, D. M. Karl and E. F. Delong, Front. Microbiol., 2013, 4, 340 Search PubMed.
- A. Pajares, M. Bregliani, J. Natera, S. Criado, S. Miskoski, J. P. Escalada and N. A. García, J. Photochem. Photobiol., A, 2011, 219, 84–89 CrossRef CAS.
- E. Biddle, D. G. Kerfoot, Y. H. Kho and K. E. Russell, Plant Physiol., 1976, 58, 700–702 CrossRef CAS.
- M. Gmurek, M. Olak-Kucharczyk and S. Ledakowicz, Chem. Eng. J., 2017, 310, 437–456 CrossRef CAS.
- S. V. Matys, N. M. Kuzmina, K. S. Laurinavichius and M. A. Nesmeyanova, Process Biochem., 2004, 39, 1063–1071 CrossRef CAS.
- Y. Chan, W. Fei, C. S. Jung, Y. Jun and B. R. Elaine, Sci. Total Environ., 2018, 644, 747–753 CrossRef.
- A. M. Braun, F. H. Frimmel and J. Hoigné, Int. J. Environ. Anal. Chem., 1986, 27, 137–149 CrossRef CAS.
- P. Attri, Y. H. Kim, D. H. Park, J. H. Park, Y. J. Hong, H. S. Uhm, K. N. Kim, A. Fridman and E. H. Choi, Sci.
Rep., 2015, 5, 9332 CrossRef CAS.
- L. F. Hua, Y. Kun, L. W. Ying, L. G. Guang, C. Ping, H. H. Ping and K. Ya Pu, Bull. Environ. Contam. Toxicol., 2015, 94, 479–483 CrossRef.
- A. P. S. Batista, F. C. C. Pires and A. C. S. C. Teixeira, J. Photochem. Photobiol., A, 2014, 290, 77–85 CrossRef CAS.
- Y. Du, H. Chen, Y. Zhang and Y. Chang, Chemosphere, 2014, 99, 254–260 CrossRef CAS.
- Y. Guo, M. Xiao, G. Li and C. Xi, J. Chem., 2017, 2017, 1–8 Search PubMed.
- L. Z. X. M. Song, J. Cent. South Univ., 2013, 20, 495–500 CrossRef CAS.
- C. Li, J. Chen, H. B. Xie, Y. H. Zhao, D. Xia, T. Xu, X. Li and X. Qiao, Environ. Sci. Technol., 2017, 51, 7b–347b Search PubMed.
- M. C. Derosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233, 351–371 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05169d |
| ‡ These authors contributed to the work equally. |
| § Present address: Division of Earth and Environmental Sciences, Korea Basic Science Institute, 162, Yeongudanji-ro, Ochang-eup, Cheongwon-gu Cheongju-si, Chungbuk 28119, Republic of Korea. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Xiaowen
Tang
*c,
R. E.
Blake
bd,
Hui
Li
d,
Sae Jung
Chang§
d and
Chan
Yu
b
*b,
Xiaowen
Tang
*c,
R. E.
Blake
bd,
Hui
Li
d,
Sae Jung
Chang§
d and
Chan
Yu
b