
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnorthodox synthesis, biological activity and DFT studies of novel and multifunctionalized naphthoxocine derivatives†
Mohamed Ahmed Abozeid a,
Aya Atef El-Sawia,
Mohamed Ramadan Elmorsya,
Mohamed Abdelmotelebbc,
Abdel-Rahman Hassan Abdel-Rahmana and
El-Sayed Ibrahim El-Desoky*a
a,
Aya Atef El-Sawia,
Mohamed Ramadan Elmorsya,
Mohamed Abdelmotelebbc,
Abdel-Rahman Hassan Abdel-Rahmana and
El-Sayed Ibrahim El-Desoky*a
aDepartment of Chemistry, Faculty of Science, Mansoura University, Mansoura-35516, Egypt. E-mail: prof.desoky.orgchem@gmail.com; Fax: +20502246254; Tel: +201060614054
bFood Allergy Research & Resource Program (FARRP), Department of Food Science & Technology, University of Nebraska, Lincoln, NE, USA
cDepartment of Botany, Faculty of Science, Mansoura University, Mansoura-35516, Egypt
First published on 6th September 2019
Abstract
A new promising protocol has been developed for the synthesis of scarce oxocine derivatives 3a–e and 6 through addition of amine-based nucleophiles such as hydroxylamine hydrochloride, primary amine and hydrazide to chromonylidene benzothiazol-2-ylacetonitrile 2 in refluxing dioxane under metal free reaction conditions in moderate to good yields. Other nitrogen nucleophiles such as piperidine, hydrazine and thiosemicarbazide failed to afford the corresponding oxocinols, and instead pyridine derivatives 7, 8 and 10 were obtained exclusively. Predictive study for the biological activities using PASS (prediction of activity spectra for biologically active substances) online software showed optimistic activities for oxocinols 3a–e in the treatment of cancer, influenza A and microbial infections. Additionally, DFT studies of oxocine derivatives 3a–e and 6 indicated the presence of required thermodynamics parameters for the application in dye-sensitized solar cells (DSSCs).
Introduction
The medium sized (7 to 9 membered) cyclic ethers are biologically interesting as well as challenging organic skeletons due their occurrence in numerous bioactive natural products in addition to their complex and macrocyclic structure which hinder their synthesis.1 In this class of macro- and heterocycles, oxocine constitutes a common scaffold in different biologically active natural products such as heliannoul H, acremine G and protosapannin B in addition to alkaloids such as murraya and arcyroxocin (Fig. 1).2–8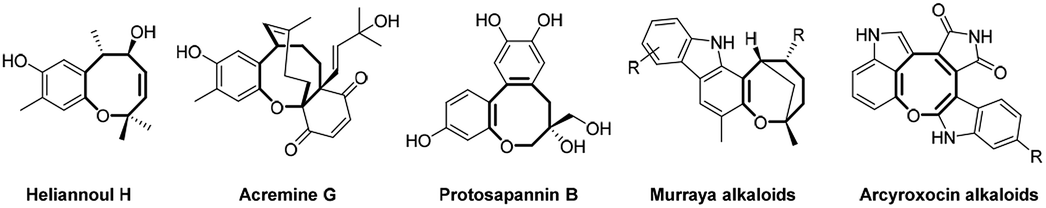 | ||
| Fig. 1 Representative examples of biologically active oxocine natural products. | ||
Consequently, many chemists have paid increased attention to develop new methodologies in order to build up this promising scaffold over the last ten years such as (1) retro-Claisen rearrangement,9 (2) ring closing olefin metathesis10 (3) Ni-catalyzed reductive Heck reaction,6 (4) DABCO-mediated [4+4] domino annulation of ynone and α-cyano-α,β-unsaturated ketone,7 (5) Lewis acid promoted acetal–alkene cyclization,11 and others.12–23 However, even in the presence of these various protocols, many observable drawbacks have been encountered such as applying drastic conditions, using precious heavy metal catalysts, needing to have finely designed starting materials and tedious purification. Therefore, it would be worthy to develop new cost effective and functional group tolerant synthetic methodology toward oxocine framework.
In the view of the aforementioned natural occurrence and biological importance of oxocine heterocyclic compounds, we herein report our recent findings in the synthesis of oxocine derivatives via simple and novel domino nucleophilic addition of amine-based nucleophiles to chromonylidene benzothiazol-2-ylacetonitrile 2 affording the corresponding oxocine derivatives 3a–e and 6 in moderate to good yields.
Results and discussion
Chemistry
The acid mediated condensation of formylbenzochromone 1 (ref. 24) with 2-cyanomethyl-1,3-benzothiazole afforded the corresponding E,Z-mixture of 2-cyano-2-(1,3-benzothoazol-2-yl)-3-vinylnaphthopyran-4-one (2) in 90% yield (Scheme 1). Under acidic reaction conditions, equimolar E,Z-mixture of chromonylidene benzothiazol-2-ylacetonitrile 2 was obtained. The formation of both isomers in almost equal amounts might be rationalized to the absence of energy barrier between both isomers due to the presence of bulky 1,3-benzothiazole heterocyclic moiety. This assumption was further supported by DFT studies of the optimized geometrical structures of E-2 and Z-2 as shown in Scheme 1. The calculations were computed by Gaussian 09 software using B3LYP as functional energy and 6-31G (d,p) as a basis set,25–28 revealed the equal energies of both geometrical isomers (−1532.9 au for E-2 and −1533 au for Z-2).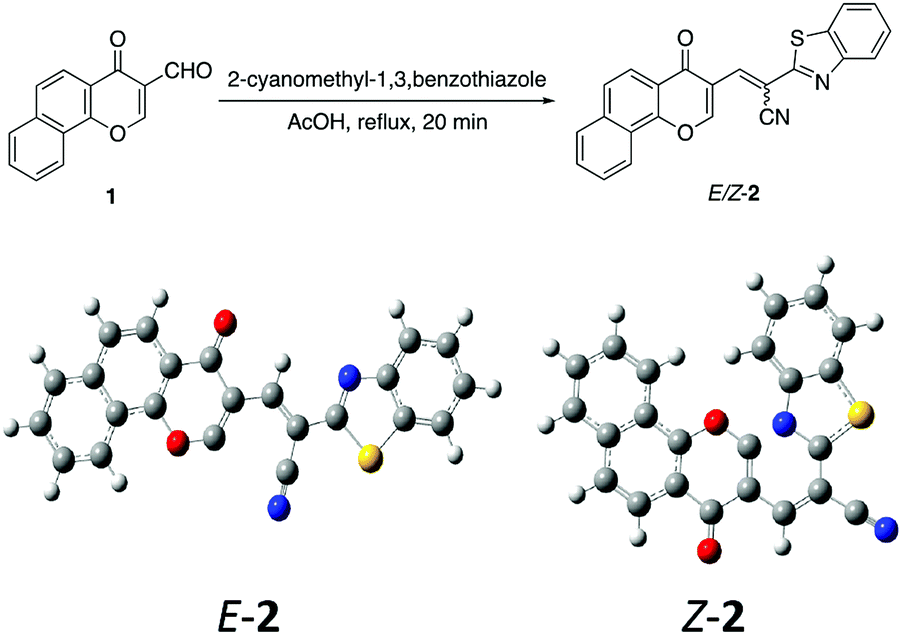 | ||
| Scheme 1 Synthesis and energy-optimized geometrical structures of E- and Z-chromonylidene 2. | ||
Based on the skeletal features of the key compound 2, we hypothesized the pathway shown in Scheme 2 towards oxocine skeleton. In order to build up this 8-membered heterocycle, our hypothesis was based upon creating a new connectivity between the oxygen pro-nucleophile and cyano electrophilic position. In order to unveil the oxygen nucleophile, the γ-pyrone ring has to be opened using some nucleophile to generate free hydroxyl group as shown in intermediate B (Pathway a). After that, the in situ generated hydroxyl group will attack cyano group to give the desired oxocine skeleton. Actually, this process most probably will be accompanied by various side reactions (for example: Pathways b and c) due to the presence of different nucleophiles which can easily attack the cyano group as well.
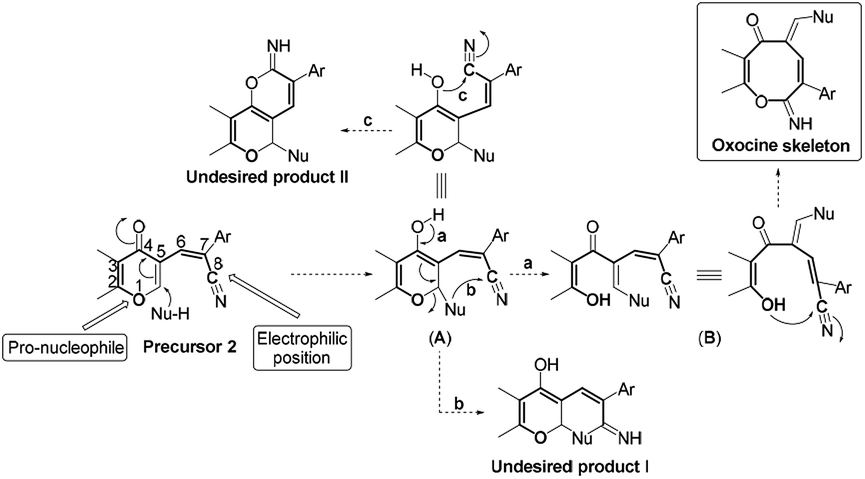 | ||
| Scheme 2 Working hypothesis. | ||
In order to test the possibility of our hypothesis, we selected the reaction of chromonylidene benzothiazol-2-ylacetonitrile 2 with hydroxylamine hydrochloride to build up the oxocinol heterocycle 3a (Table 1). However, no reaction was observed by boiling 2 with hydroxylamine hydrochloride in ethanol (Entry 1), trace amount of the desired product 3a was detected by performing the reaction in 1,4-dioxane at 80 °C (Entry 2). By increasing the temperature to 101 °C, the desired product 3a was isolated in 55% yield (Entry 3). Additionally, the use of other solvents with increasing boiling points failed to improve the yield of 3a (Entries 4–8). The attempts by using different organic and inorganic basic additives failed to increase the chemical yield as well (Entries 9–13). Moreover, the use of basic additives promoted different reaction pathways and leaded to tedious chromatographic purification of 3a (Entries 9–13). Increasing the amount of hydroxylamine hydrochloride (1.5 equivalent) didn't impart any benefits (Entry 14).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Temp. °C | Base | Time | Yield (%) |
| a Reaction conditions: 1 (1 mmol) and hydroxylamine·HCl (1 mmol) in 15 ml solvent.b NR: no reaction.c TCE: tetrachloroethylene.d DMF: N,N-dimethylformamide.e ND: not detected.f Using hydroxylamine·HCl (1.5 eq.). | |||||
| 1 | Ethanol | 78 | None | 12 h | NRb |
| 2 | 1,4-Dioxane | 80 | None | 12 h | Trace |
| 3 | 1,4-Dioxane | 101 | None | 30 min | 55 |
| 4 | Isobutanol | 108 | None | 3 h | 30 |
| 5 | Toluene | 111 | None | 6 h | 12 |
| 6 | Pyridine | 115 | None | 1 h | 45 |
| 7 | TCEc | 121 | None | 12 h | NRb |
| 8 | DMFd | 153 | None | 12 h | NDe |
| 9 | 1,4-Dioxane | 101 | Et3N | 30 min | 41 |
| 10 | 1,4-Dioxane | 101 | DBU | 5 min | 22 |
| 11 | 1,4-Dioxane | 101 | NaOAc | 30 min | 41 |
| 12 | 1,4-Dioxane | 101 | K2CO3 | 1.5 h | 40 |
| 13 | 1,4-Dioxane | 101 | NaOH | 10 min | 47 |
| 14f | 1,4-Dioxane | 101 | None | 30 min | 55 |
 |
|||||
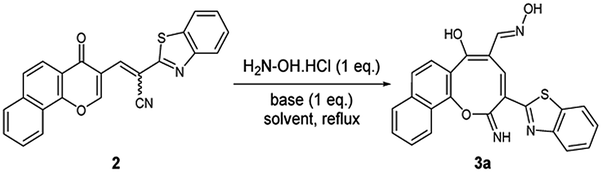
The skeleton of oxocinol 3a was secured based upon different analytical analyses. Firstly, IR spectrum showed absorption peaks at regions 3430–3570, 3319 and 3198 cm−1 assigned for two OH's and NH groups. In addition, 1H NMR spectrum of 3a showed two characteristic singlets at 8.78 and 8.83 ppm which were assigned to protons CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and H-4 (oxocinol), respectively. The 13C NMR spectrum of 3a exhibited all carbons of the proposed structure. EI-MS supported the proposed structure of compound 3a by the presence of molecular ion peak at m/z 413, which was compatible with its molecular weight. The formation of side products 4 (ref. 29) and 5 (ref. 30) was excluded due to the absence of aliphatic CH in 1H- and 13C-NMR.
N and H-4 (oxocinol), respectively. The 13C NMR spectrum of 3a exhibited all carbons of the proposed structure. EI-MS supported the proposed structure of compound 3a by the presence of molecular ion peak at m/z 413, which was compatible with its molecular weight. The formation of side products 4 (ref. 29) and 5 (ref. 30) was excluded due to the absence of aliphatic CH in 1H- and 13C-NMR.
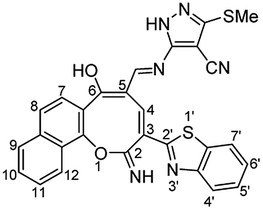
Similarly, the newly developed methodology could be applied with different primary homo and heteroaryl amines such as p-ansidine, 5-amino-3-methylthio-1H-pyrazole-4-carbonitrile, 2-amino-1,3,4-triazole and 2-amino benzimidazole affording the corresponding oxocinol products 3b–e in moderate to good yields (Scheme 3).
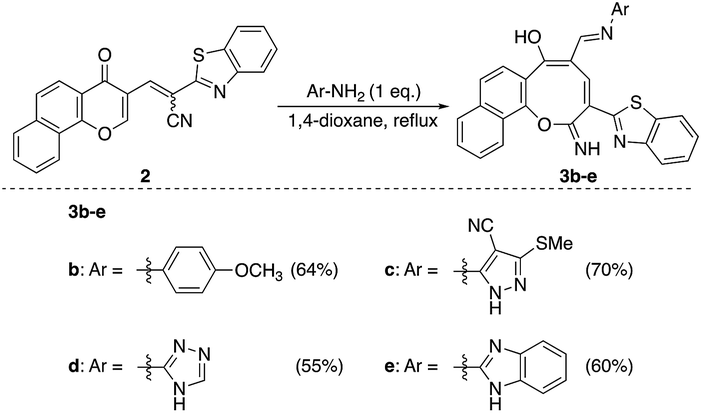 | ||
| Scheme 3 Reaction of chromonylidene 2 with different 1° amines. | ||
Additionally, the reaction of 2 with cyanoacetic acid hydrazide in the presence of a catalytic amount of triethylamine afforded oxocinone 6 in 50% yield (Scheme 4). This reaction involved initially the aforementioned cascade process to build up the corresponding oxocinol intermediate I which underwent enol to keto tautomerization giving intermediate II, form which the final product 6 was formed by further cyclization between hydrazonyl NH and cyano group (Scheme 4). IR spectrum of oxocinone 6 confirmed the absence of cyano group, and instead hydroxyl group was observed at 3550–3420 cm−1 as broad peak in addition to aliphatic CH functionality peaks at 2961, 2898 cm−1. The 1H NMR analysis showed a singlet at δ 4.25 ppm assignable to methylene protons of pyrazole ring.
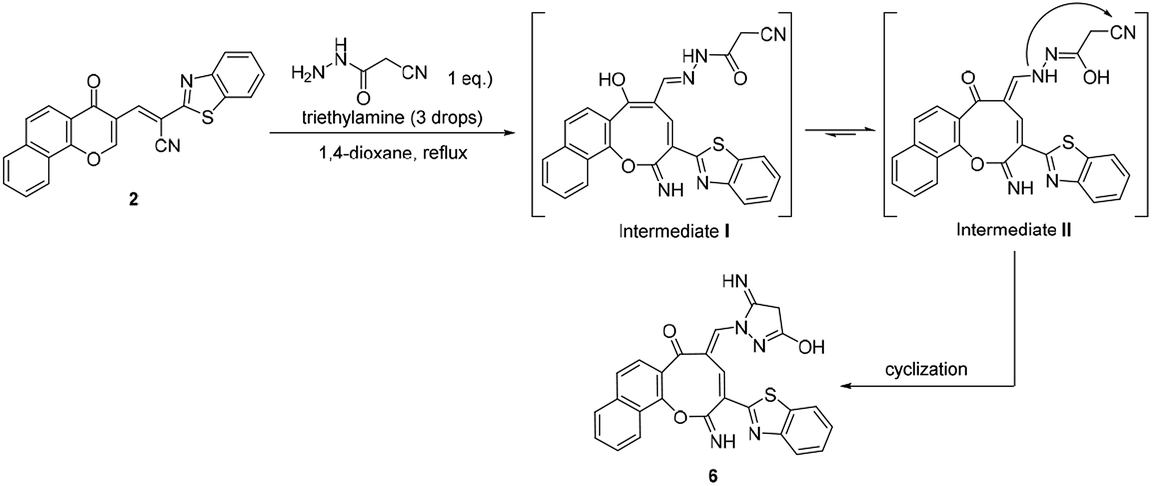 | ||
| Scheme 4 Reaction of chromonylidene 2 with cyanoacetic acid hydrazide. | ||
On the other hand, the reaction of chromonylidene 2 with piperidine in refluxing 1,4-dioxane didn't afford the corresponding oxocinol product, and instead, 2-(benzo[d]thiazol-2-yl)-12-(piperidin-1-yl)-4,12-dihydro-3H-benzo[7,8]chromeno[4,3-b]pyridine-3-one (7) was obtained in 66% yield (Scheme 5). The formation of pyridinone 7 was proposed to proceed via nucleophilic attack at C-2 of γ-pyrone accompanied by the generation hydroxyl group at C-4 (intermediate I), which attack the nearby cyano group giving α-iminopyrone intermediate II. By Dimroth rearrangement, α-iminopyrone intermediate II was transformed into the corresponding product 7. The IR spectrum of pyridinone 7 showed a broad peak at 3350–3460 cm−1 which was attributable to NH group in addition to the amidic carbonyl peak at 1643 cm−1. Additionally, the newly inserted aliphatic carbons of piperidinyl moiety were expressed in IR by the peaks at the range of 2931–2816 cm−1. The 1H NMR spectrum of compounds 7 showed a characteristic singlet δ 6.97 ppm assigned to the methine proton (H-5). In a different scenario, no reaction was observed in the case of the reaction of chromonylidene 2 with triethylamine (3° amine) even after prolonged reaction time (12 h).
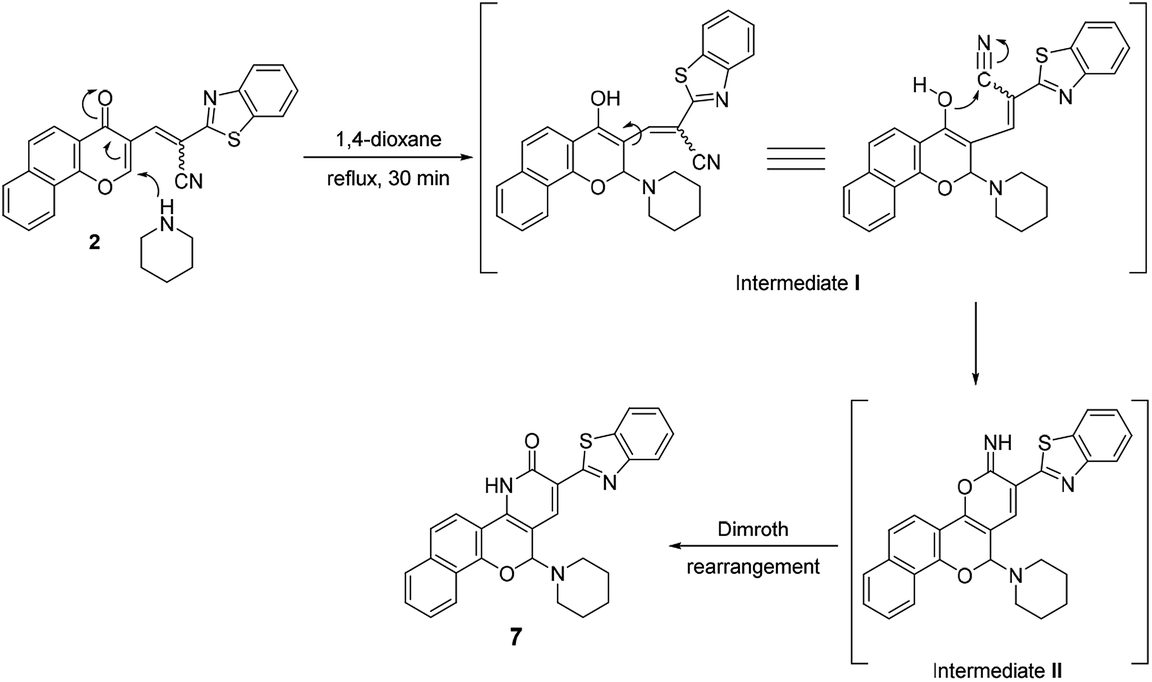 | ||
| Scheme 5 Reaction of chromonylidene 2 with piperidine. | ||
Moreover, the reaction of chromonylidene 2 with hydrazine hydrate in refluxing 1,4-dioxane afforded product 8 (73% yield) which was generated in a completely different scenario to the aforementioned reactions with primary and secondary amines (Scheme 6). Treatment of precursor 2 with thiosemicarbazide produced the ring junction nitrogen product 10 in 53% yield as shown in the reaction pathway (Scheme 7). The reaction proceeded in a similar style to the reaction with hydrazine hydrate (Scheme 6).
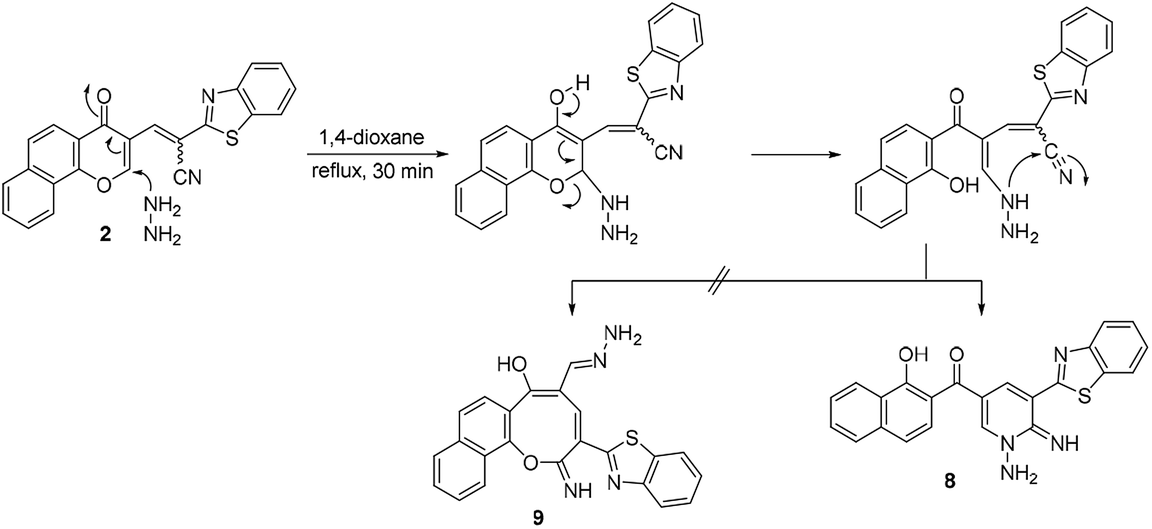 | ||
| Scheme 6 Reaction of chromonylidene 2 with hydrazine hydrate. | ||
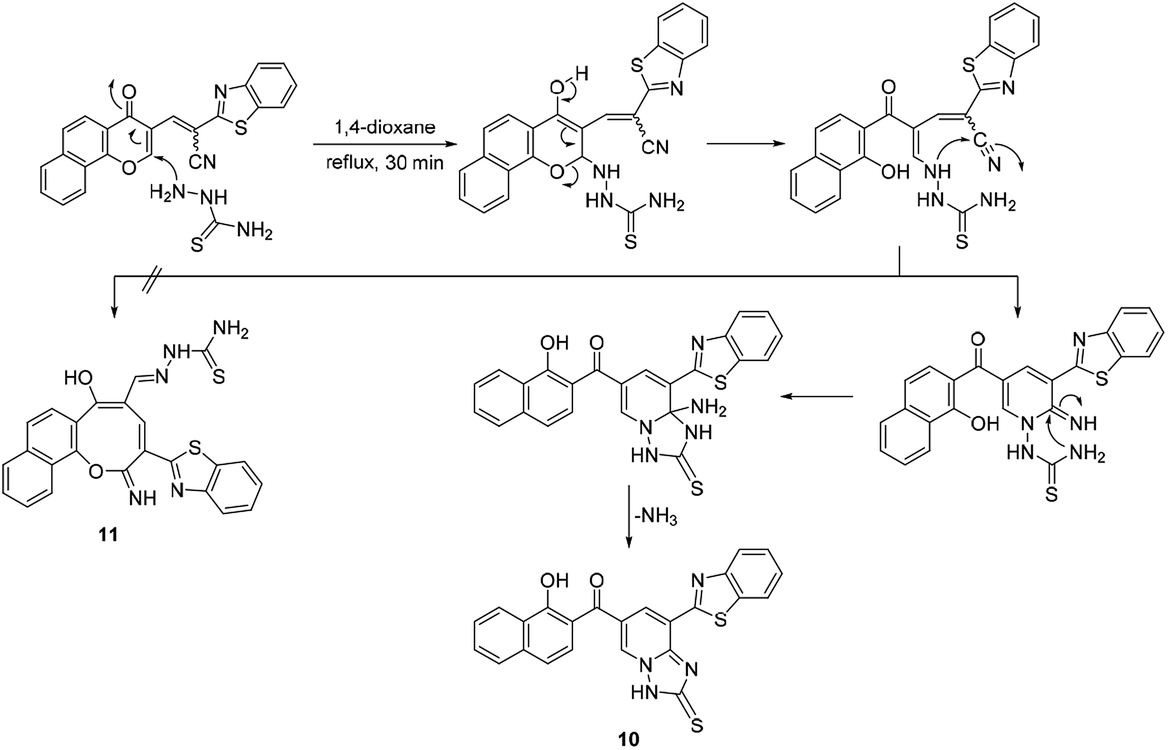 | ||
| Scheme 7 Reaction of chromonylidene 2 with thiosemicarbazide. | ||
The different reaction profiles of chromonylidene 2 with hydroxyl amine and its similar nucleophiles (products 3a–e and 6) compared to hydrazine nucleophile (product 8) were explained on the basis of the electron density on the nucleophilic centers in the corresponding intermediates B (Fig. 2). These electron densities were measured on the optimized structures using Gaussian 09 software at DFT level via B3LYP as energy function and 6-31G (d,p) as a basis set.25–28 In the case of key intermediate for oxocinol 3a, the electron density on phenolic OH group was found to be (−0.61 e) which is higher than the corresponding density on NH group (−0.47 e). This significant difference in the electron density value enhanced the nucleophilicity of phenolic OH compared to that of NH group leading to the desired oxocine derivatives 3a–e and 6. In the case of key intermediate for pyridine 8, the electron density on NH center was increased due to the presence of adjacent NH2 group to value of −0.58 e which became very close to the value of phenolic OH group. As a result, the 6-membered cyclization (affording product 8) became more energetically favored than 8-membered cyclization (affording product 9). In the case of the reaction with thiosemicarbazide (Scheme 7), the calculated electron densities on the nucleophilic centers could not rationalize solely the formation of product 10 where its formation might be attributed to some other thermodynamic parameters.
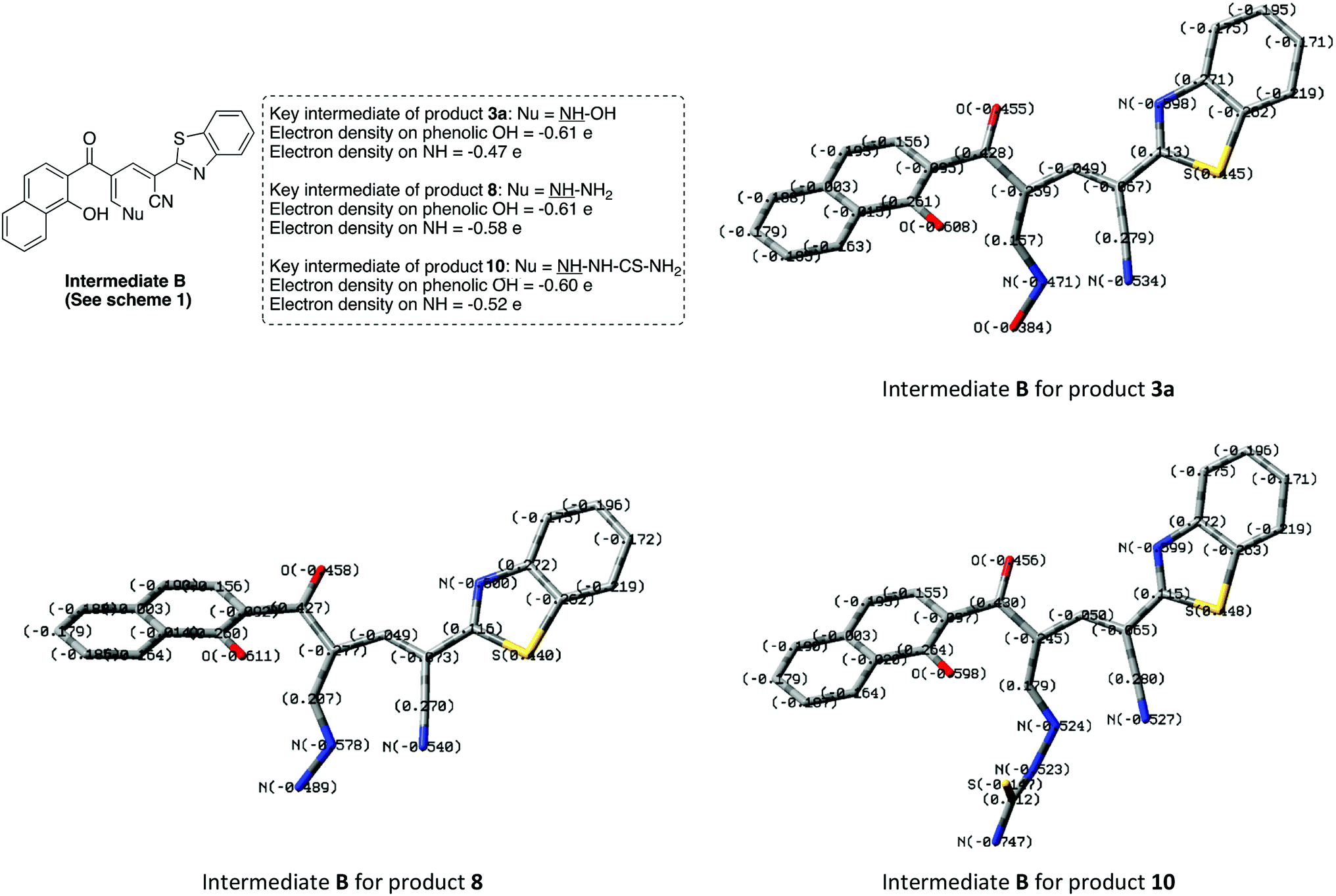 | ||
| Fig. 2 Electron densities on OH and NH functionalities of the key intermediate B in case of products 3a, 8 and 10. | ||
Computational prediction of biological activities of oxocine derivatives 3a–e and 6
PASS online software was used to predict the putative biological activity spectrum of the oxocine derivatives 3a–e and 6 which was represented in Table 2. From the results of biological activity prediction, the synthesized oxocine derivatives 3a–e and 6 showed various anticancer (antimetastatic, antiischemic (cerebral), antineoplastic, and protection from prostate cancer), antiviral, and antimicrobial promising activities. Oxocinols 3a and 3b showed the best probable activity in the treatment of cancer. In addition, oxocinol 3b revealed the best predicted antimicrobial activity. Ultimately, compounds 3d and 3e may possess antiviral (influenza A) activity.| Biological activity | 3a | 3b | 3c | 3d | 3e | 6 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | ||
| Anticancer | Antimetastatic | NA | NA | 0.310 | 0.077 | 0.238 | 0.119 | 0.201 | 0.158 | 0.220 | 0.135 | 0.212 | 0.143 |
| Antiischemic (cerebral) | 0.472 | 0.125 | 0.378 | 0.204 | NA | NA | 0.414 | 0.174 | NA | NA | 0.503 | 0.104 | |
| Antineoplastic | 0.604 | 0.044 | 0.640 | 0.037 | 0.492 | 0.074 | 0507 | 0.069 | 0.468 | 0.081 | 0.565 | 0.053 | |
| Antineoplastic (brain cancer) | NA | NA | 0.226 | 0.084 | 0.198 | 0.133 | 0212 | 0.108 | 0.202 | 0.126 | NA | NA | |
| Prostate cancer treatment | 0.515 | 0.008 | 0.511 | 0.009 | 0.343 | 0.031 | 0.459 | 0.014 | 0.480 | 0.012 | 0.459 | 0.014 | |
| Antiviral | Influenza A | 0.245 | 0.106 | 0.214 | 0.181 | 0.242 | 0.112 | 0.536 | 0.004 | 0.549 | 0.004 | 0.417 | 0.007 |
| Antimicrobial | Antibiotic | 0.183 | 0.033 | 0.254 | 0.019 | 0.147 | 0.051 | 0.181 | 0.034 | 0.176 | 0.036 | 0.169 | 0.039 |
| Antibacterial | 0.358 | 0.041 | 0.487 | 0.018 | 0.258 | 0.079 | 0.359 | 0.041 | 0.306 | 0.058 | 0.275 | 0.070 | |
Applications of oxocine derivatives 3a–e and 6 in dye-sensitized solar cells (DSSCs)
DFT calculations followed by TD-DFT studies were performed for oxocine derivatives 3a–e and 6 using B3LYP as energy function and 6-31G (d,p)25–28 as a basis set to investigate their theoretical GSOP/ESOP energy levels (Fig. 3) and their optical absorption spectra (Fig. 4).31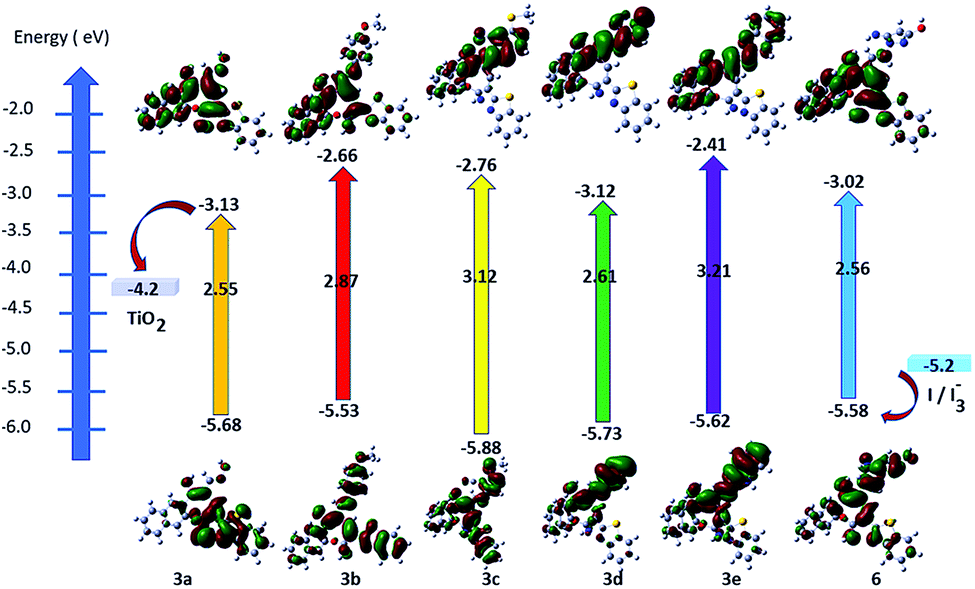 | ||
| Fig. 3 Energy level diagram of oxocine derivatives 3a–e and 6. | ||
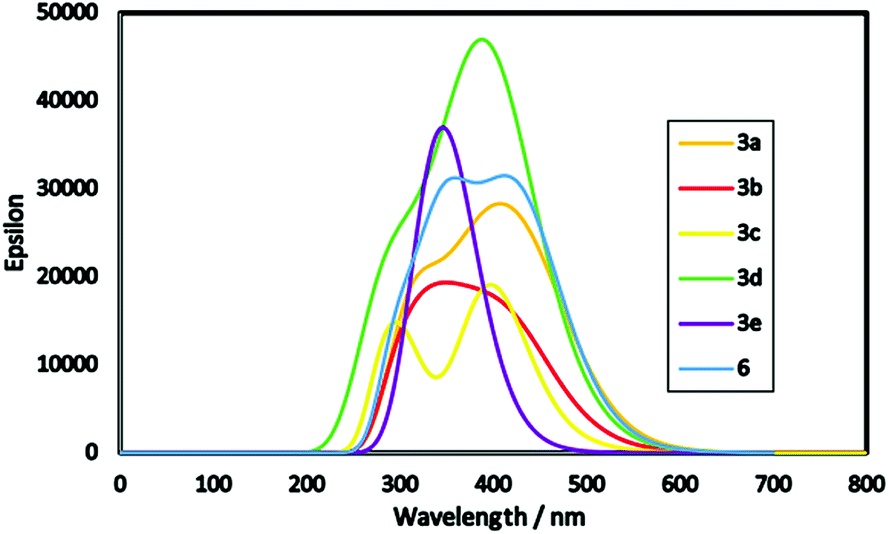 | ||
| Fig. 4 Simulated absorption spectra of oxocine derivatives 3a–e and 6. | ||
In addition, from the results displayed in Table 3, it is quite interesting to note that, all products 3a–e and 6 possess the thermodynamic requirements as an application for DSSCs.32,33 The estimated ESOP levels of 3a–e and 6 were found to be 3a (−3.13 eV), 3b (−2.66 eV), 3c (−2.76 eV), 3d (−3.12 eV), 3e (−2.41 eV) and 6 (−3.02 eV). These values are more positive than the conduction band (CB) potential of TiO2 (−4.2 eV); showing that their ability for the electron injection. Furthermore, the calculated GSOP/HOMO levels of 3a (−5.68 eV), 3b (−5.53 eV), 3c (−5.88 eV), 3d (−5.73 eV), 3e (−5.62 eV) and 6 (−5.68 eV) were found to be more negative than the oxidation potential of the electrolyte (−5.2 eV) (I3−/I−), exhibited the regeneration of the electrons.
| Product | Basis set | GSOP (eV) | E0–0 | ESOP (eV) |
|---|---|---|---|---|
| 3a | B3LYP/6-31G (d,p) | −5.68 | 2.55 | −3.13 |
| 3b | −5.53 | 2.87 | −2.66 | |
| 3c | −5.88 | 3.12 | −2.76 | |
| 3d | −5.73 | 2.61 | −3.12 | |
| 3e | −5.62 | 3.21 | −2.41 | |
| 6 | −5.58 | 2.56 | −3.02 |
Furthermore, energy band gap (E0–0) can be estimated from the onset of the lowest energy peak.34 Small band gap increases the optical properties of the products allowing for absorption most of the incident light. From Fig. 4, product 3a is found to be red shifted than other products which attributed to its lowest optical energy gap of 2.55 eV. Moreover, molar extinction coefficient (epsilon) plays a vital role for enhancing the light harvesting ability of the dye where its higher value indicates maximum light harvesting capability. For example, product 3d showed the highest molar extinction co-efficient with broadening in its absorption peak depending on the nature of its donor and acceptor structures, and as a result it is expected to show improved photovoltaic performance in DSSCs.35
Conclusion
A promising protocol for the synthesis of oxocine derivatives 3a–e and 6 has been developed via reaction of chromonylidene 2 with different NH2 bearing nucleophiles under metal free conditions refluxing in 1,4-dioxane. Other nucleophiles such as piperidine, hydrazine and thiosemicarbazide failed to give the desired oxocine derivatives and instead, they afforded different pyridine derivatives 7, 8 and 10. Some oxocine products showed some promising probability of biological activity in the treatment of cancer, influenza A, and as antimicrobial agents. Furthermore, TD-DFT simulated spectra and calculated GSOP/ESOP energy levels indicated the ability of using products 3a–e and 6 in the application of DSSCs. The present study is just preliminary where detailed studies for the synthesis of different oxocine derivatives in addition to biological activity and DSSCs applications are currently underway, with collaboration with different specialized research groups, and will be reported in the due time.Experimental
General
The melting points of the newly synthesized compounds were measured on a Gallenkamp electrical melting point apparatus using open glass capillaries and are reported in degree Celsius (°C) (uncorrected). The infrared spectra (IR) were determined using the pressed KBr disc method on a Mattson 5000 FT-IR spectrophotometer (Faculty of Pharmacy, Mansoura University) or Nicolet iS10 FT-IR Spectrometer (Faculty of Science, Mansoura University). The 1H-NMR spectra were measured on Bruker AC 300 MHz (Faculty of Science, Cairo University), Bruker Avance III 400 MHz (Faculty of Pharmacy, Beni Suef University) and/or JEOL ECA II 500 MHz (Faculty of Science, Mansoura University) using tetramethylsilane (TMS) as an internal reference, and using DMSO-d6 or trifluoroacetic acid (TFA) as solvents. The signals' multiplicities are reported as follows: s = singlet, d = doublet, dd = doublet of doublets and m = multiplet. Exchangeable protons were detected through D2O test. 13C-NMR spectra were measured on JEOL ECA II 125 MHz (Faculty of Science, Mansoura University). The electron impact mass spectra (EI-MS) were determined on Thermo Fisher scientific DSQ II GC/MS with focus GC (70 eV) (Faculty of Science, Mansoura University) or Kratos MS (70 eV) equipment (Faculty of Pharmacy, Al-Azhar University). Elemental analysis was executed at the microanalytical lab at Cairo University. All reactions were monitored using thin layer chromatography (TLC) on aluminum sheets pre-coated with Al2O3 with fluorescent indicator F254, Merck (Darmstadt, Germany). DFT-followed by TD-DFT were performed using Gaussian 09 package software. The simulated absorption spectra of products 3a–e and 6 obtained at B3LYP/6-31G (d,p) level.Synthesis
A mixture of 3-formylbenzochromone (673 mg, 3 mmol), 2-benzothiazoleacetonitrile (523 mg, 3 mmol) in glacial acetic acid (25 ml) was refluxed for 20 min. The formed precipitate was filtered off and recrystallized from a mixture of DMF/EtOH giving product 2 (E
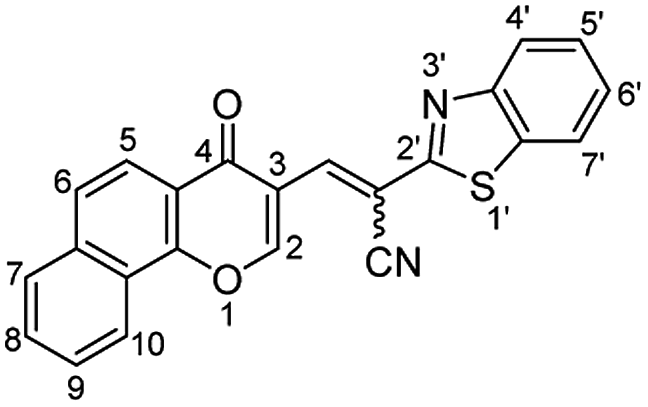
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z = 1:1). Yield (1027 mg, 90%); reddish orange crystals; mp = 286–288 °C; IR (KBr, ν/cm−1): 3061 (aromatic CH), 2206 (CN), 1650 (CO), 1603 (CN); 1H NMR (500 MHz, DMSO-d6): δ (ppm) 7.40 (s, 1H, H-2a),7.52 (dd, 1H,
:Z = 1:1). Yield (1027 mg, 90%); reddish orange crystals; mp = 286–288 °C; IR (KBr, ν/cm−1): 3061 (aromatic CH), 2206 (CN), 1650 (CO), 1603 (CN); 1H NMR (500 MHz, DMSO-d6): δ (ppm) 7.40 (s, 1H, H-2a),7.52 (dd, 1H, 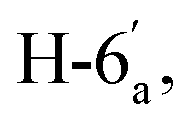 J = 7.00, 8.00 Hz), 7.55 (d, 1H,
J = 7.00, 8.00 Hz), 7.55 (d, 1H, 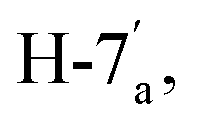 J = 8.20 Hz), 7.60–7.67 (m, 4H, Ar–H), 7.73 (dd, 1H,
J = 8.20 Hz), 7.60–7.67 (m, 4H, Ar–H), 7.73 (dd, 1H,  J = 7.50, 8.00 Hz),7.85–7.91 (m, 3H, Ar–H), 7.93 (d, 1H,
J = 7.50, 8.00 Hz),7.85–7.91 (m, 3H, Ar–H), 7.93 (d, 1H,  J = 8.20 Hz), 8.00 (d, 2H, Ar–H, J = 8.90 Hz), 8.04 (s, 1H, H-2b),8.05 (d, 1H, Ar–H, J = 8.90 Hz), 8.10–8.18 (m, 4H, Ar–H), 8.22 (d, 1H, H-10a, J = 7.60 Hz), 8.35 (s, 1H, CHaC–CN), 8.57 (d, 1H, H-10b, J = 7.60 Hz), 9.46 (s, 1H, CHbC–CN); EI-MS m/z (%): 382.14 [M+ + 2] (11.91), 381.19 [M+ + 1] (29.07), 380.20 [M+] (100.00), 352.22 (30.16), 351.19 (94.43), 350.30 (11.86), 114.12 (26.95); anal. calcd for C23H12N2O2S (380.06): C, 72.62; H, 3.18; N, 7.36; S, 8.43%. Found: C, 72.65; H, 3.20; N, 7.32; S, 8.50%.
J = 8.20 Hz), 8.00 (d, 2H, Ar–H, J = 8.90 Hz), 8.04 (s, 1H, H-2b),8.05 (d, 1H, Ar–H, J = 8.90 Hz), 8.10–8.18 (m, 4H, Ar–H), 8.22 (d, 1H, H-10a, J = 7.60 Hz), 8.35 (s, 1H, CHaC–CN), 8.57 (d, 1H, H-10b, J = 7.60 Hz), 9.46 (s, 1H, CHbC–CN); EI-MS m/z (%): 382.14 [M+ + 2] (11.91), 381.19 [M+ + 1] (29.07), 380.20 [M+] (100.00), 352.22 (30.16), 351.19 (94.43), 350.30 (11.86), 114.12 (26.95); anal. calcd for C23H12N2O2S (380.06): C, 72.62; H, 3.18; N, 7.36; S, 8.43%. Found: C, 72.65; H, 3.20; N, 7.32; S, 8.50%.Yellow needles; (227 mg, 55%); mp > 330 °C; IR (KBr, ν/cm−1): 3430–3570 (OH, broad), 3319 (NH), 3065 (aromatic CH),1627 (C
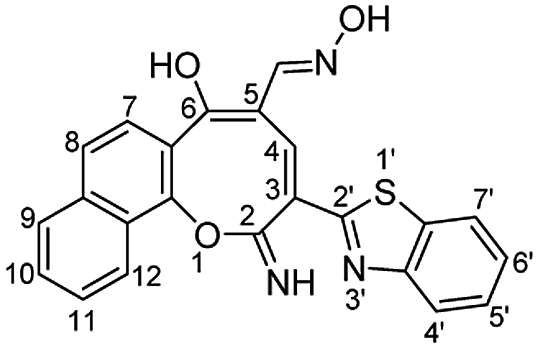 N), 1583 (CC); 1H NMR (500 MHz, TFA): δ (ppm) 7.45 (d, 1H, H-8, J = 8.70 Hz), 7.51 (d, 1H, H-7, J = 8.90 Hz), 7.66–7.72 (m, 2H, Ar–H), 7.75–7.82 (m, 2H, Ar–H), 7.87 (d, 1H, H-7′, J = 8.00 Hz), 8.09 (d, 1H, H-4′, J = 8.00 Hz), 8.22 (d, 1H, H-9, J = 8.00 Hz), 8.56 (d, 1H, H-12, J = 8.50 Hz), 8.78 (s, 1H, CHN), 8.83 (s, 1H, H-4); 13C NMR (126 MHz, TFA): δ (ppm) 119.10, 122.29, 123.51, 124.72, 125.06, 125.95, 126.11, 126.74, 128.99, 129.68, 130.01, 130.06, 133.99, 135.35, 140.41, 140.85, 141.76, 152.60, 153.17, 153.21, 163.29, 166.97, 194.84; EI-MS m/z (%): 415.18 [M+ + 2] (5.69), 414.26 [M+ + 1] (23.88), 413.22 [M+] (85.42), 397.24 (100.00), 396.25 (41.78), 381.21 (25.85), 368.18 (7.77); anal. calcd for C23H15N3O3S (413.45): C, 66.82; H, 3.66; N, 10.16; S, 7.75%. Found: C, 66.80; H, 3.69; N, 10.12; S, 7.79%.
N), 1583 (CC); 1H NMR (500 MHz, TFA): δ (ppm) 7.45 (d, 1H, H-8, J = 8.70 Hz), 7.51 (d, 1H, H-7, J = 8.90 Hz), 7.66–7.72 (m, 2H, Ar–H), 7.75–7.82 (m, 2H, Ar–H), 7.87 (d, 1H, H-7′, J = 8.00 Hz), 8.09 (d, 1H, H-4′, J = 8.00 Hz), 8.22 (d, 1H, H-9, J = 8.00 Hz), 8.56 (d, 1H, H-12, J = 8.50 Hz), 8.78 (s, 1H, CHN), 8.83 (s, 1H, H-4); 13C NMR (126 MHz, TFA): δ (ppm) 119.10, 122.29, 123.51, 124.72, 125.06, 125.95, 126.11, 126.74, 128.99, 129.68, 130.01, 130.06, 133.99, 135.35, 140.41, 140.85, 141.76, 152.60, 153.17, 153.21, 163.29, 166.97, 194.84; EI-MS m/z (%): 415.18 [M+ + 2] (5.69), 414.26 [M+ + 1] (23.88), 413.22 [M+] (85.42), 397.24 (100.00), 396.25 (41.78), 381.21 (25.85), 368.18 (7.77); anal. calcd for C23H15N3O3S (413.45): C, 66.82; H, 3.66; N, 10.16; S, 7.75%. Found: C, 66.80; H, 3.69; N, 10.12; S, 7.79%.Yellowish orange crystals; (374 mg, 70%); mp 326–327 °C; IR (KBr, ν/cm−1): 3380–3550 (OH, broad), 3450 (NH), 3226 (NH), 2215 (CN), 1630 (C
 N), 1600 (CC); 1H NMR (500 MHz, DMSO-d6): δ (ppm) 2.60 (s, 3H, SCH3), 7.49–7.78 (m, 6H, Ar–H), 7.96 (d, 1H, Ar–H, J = 8.00 Hz), 8.15 (d, 1H, Ar–H, J = 8.50 Hz), 8.22 (d, 1H, Ar–H, J = 8.00 Hz), 8.41 (d, 1H, Ar–H, J = 8.50 Hz), 8.63 (d, 1H, Ar–H, J = 2.50 Hz), 8.76 (d, 1H, Ar–H, J = 2.50 Hz), 9.49 (s, 1H, NH or OH), 10.00 (s, 1H, NH or OH), 12.78 (s, 1H, NH or OH); EI-MS m/z (%): 534.82 [M+] (48.94), 507.08 (40.21), 502.40 (32.33), 473.18 (16.44), 271.20 (54.74), 243.06 (68.660), 183.42 (77.08), 180.05 (100.00), 129.09 (58.57), 81.14 (58.28); anal. calcd for C28H18N6O2S2 (534.09): C, 62.91; H, 3.39; N, 15.72; S, 11.99%. Found: C, 62.95; H, 3.35; N, 15.73; S, 11.96%.
N), 1600 (CC); 1H NMR (500 MHz, DMSO-d6): δ (ppm) 2.60 (s, 3H, SCH3), 7.49–7.78 (m, 6H, Ar–H), 7.96 (d, 1H, Ar–H, J = 8.00 Hz), 8.15 (d, 1H, Ar–H, J = 8.50 Hz), 8.22 (d, 1H, Ar–H, J = 8.00 Hz), 8.41 (d, 1H, Ar–H, J = 8.50 Hz), 8.63 (d, 1H, Ar–H, J = 2.50 Hz), 8.76 (d, 1H, Ar–H, J = 2.50 Hz), 9.49 (s, 1H, NH or OH), 10.00 (s, 1H, NH or OH), 12.78 (s, 1H, NH or OH); EI-MS m/z (%): 534.82 [M+] (48.94), 507.08 (40.21), 502.40 (32.33), 473.18 (16.44), 271.20 (54.74), 243.06 (68.660), 183.42 (77.08), 180.05 (100.00), 129.09 (58.57), 81.14 (58.28); anal. calcd for C28H18N6O2S2 (534.09): C, 62.91; H, 3.39; N, 15.72; S, 11.99%. Found: C, 62.95; H, 3.35; N, 15.73; S, 11.96%.Canary yellow crystals; (255 mg, 55%); mp > 330 °C; IR (KBr, ν/cm−1): 3200–3450 (OH, broad), 3424 (NH), 3209 (NH), 3059 (aromatic CH), 1601 (C
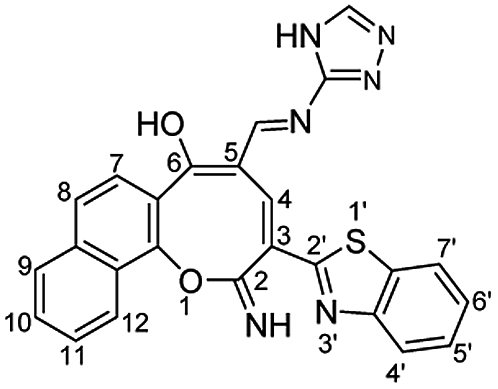 N), 1550 (CC); 1H NMR (500 MHz, DMSO-d6): δ (ppm) 7.48 (d, 1H, H-8, J = 8.90 Hz), 7.54 (dd, 1H, Ar–H, J = 7.49, 7.51 Hz), 7.62 (dd, 2H, Ar–H, J = 6.50, 8.50 Hz), 7.66 (d, 1H, H-7, J = 8.90 Hz), 7.72 (dd, 1H, Ar–H, J = 7.00, 8.00 Hz), 7.82 (s, 1H, triazole), 7.95 (d, 1H, H-9, J = 8.20 Hz), 8.16 (d, 1H, H-7′, J = 7.60 Hz), 8.21 (d, 1H, H-4′, J = 7.60 Hz), 8.38 (d, 1H, H-12, J = 8.20 Hz), 8.61 (s, 1H, CHN), 8.75 (s, 1H, H-4), 12.51 (s, 1H, NH or OH), 12.77 (s, 1H, NH or OH), 13.76 (s, 1H, NH or OH); EI-MS m/z (%): 464.29 [M+] (100.00), 462.82 (17.73), 435.33 (13.86), 380.18 (13.32), 379.22 (9.70), 306.23 (18.92), 115.18 (17.33); anal. calcd for C25H16N6O2S (464.11): C, 64.64; H, 3.47; N, 18.09; S, 6.90%. Found: C, 64.66; H, 3.42; N, 18.03; S, 7.01%.
N), 1550 (CC); 1H NMR (500 MHz, DMSO-d6): δ (ppm) 7.48 (d, 1H, H-8, J = 8.90 Hz), 7.54 (dd, 1H, Ar–H, J = 7.49, 7.51 Hz), 7.62 (dd, 2H, Ar–H, J = 6.50, 8.50 Hz), 7.66 (d, 1H, H-7, J = 8.90 Hz), 7.72 (dd, 1H, Ar–H, J = 7.00, 8.00 Hz), 7.82 (s, 1H, triazole), 7.95 (d, 1H, H-9, J = 8.20 Hz), 8.16 (d, 1H, H-7′, J = 7.60 Hz), 8.21 (d, 1H, H-4′, J = 7.60 Hz), 8.38 (d, 1H, H-12, J = 8.20 Hz), 8.61 (s, 1H, CHN), 8.75 (s, 1H, H-4), 12.51 (s, 1H, NH or OH), 12.77 (s, 1H, NH or OH), 13.76 (s, 1H, NH or OH); EI-MS m/z (%): 464.29 [M+] (100.00), 462.82 (17.73), 435.33 (13.86), 380.18 (13.32), 379.22 (9.70), 306.23 (18.92), 115.18 (17.33); anal. calcd for C25H16N6O2S (464.11): C, 64.64; H, 3.47; N, 18.09; S, 6.90%. Found: C, 64.66; H, 3.42; N, 18.03; S, 7.01%.Yellow needles; (308 mg, 60%); mp 318 °C; IR (KBr, ν/cm−1): 3270–3420 (OH, NH, broad), 3347 (NH), 3058 (aromatic CH), 1631 (C
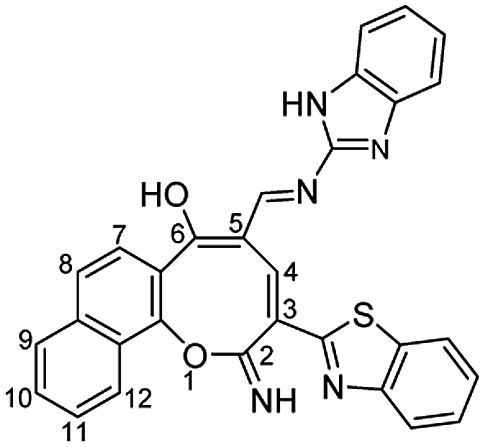 N), 1590 (CC); 1H NMR (400 MHz, DMSO-d6): δ (ppm) 7.22–7.56 (m, 7H, Ar–H), 7.67 (dd, 1H, Ar–H, J = 8.00, 8.50 Hz), 7.77 (d, 2H, Ar–H, J = 8.00 Hz), 8.01 (d, 1H, H-9, J = 8.00 Hz), 8.08 (dd, 2H, Ar–H, J = 8.00, 8.50 Hz), 8.43 (d, 1H, H-12, J = 8.00 Hz), 8.85 (s, 1H, CHN), 9.18 (s, 1H, H-4), 12.01 (s, 1H, NH or OH), 12.53 (s, 1H, NH or OH), 13.00 (s, 1H, NH or OH); EI-MS m/z (%): 515.92 [M+ + 2] (0.95), 514.32 [M+ + 1] (1.03), 513.61 [M+] (2.44), 398.31 (14.90), 397.25 (20.37), 355.34 (13.15), 354.30 (16.10), 337.26 (21.39), 339.26 (48.12), 337.26 (16.73), 40.20 (100.00); anal. calcd for C30H19N5O2S (513.58): C, 70.16; H, 3.73; N, 13.64; S, 6.24%. Found: C, 70.18; H, 3.70; N, 13.62; S, 6.21%.
N), 1590 (CC); 1H NMR (400 MHz, DMSO-d6): δ (ppm) 7.22–7.56 (m, 7H, Ar–H), 7.67 (dd, 1H, Ar–H, J = 8.00, 8.50 Hz), 7.77 (d, 2H, Ar–H, J = 8.00 Hz), 8.01 (d, 1H, H-9, J = 8.00 Hz), 8.08 (dd, 2H, Ar–H, J = 8.00, 8.50 Hz), 8.43 (d, 1H, H-12, J = 8.00 Hz), 8.85 (s, 1H, CHN), 9.18 (s, 1H, H-4), 12.01 (s, 1H, NH or OH), 12.53 (s, 1H, NH or OH), 13.00 (s, 1H, NH or OH); EI-MS m/z (%): 515.92 [M+ + 2] (0.95), 514.32 [M+ + 1] (1.03), 513.61 [M+] (2.44), 398.31 (14.90), 397.25 (20.37), 355.34 (13.15), 354.30 (16.10), 337.26 (21.39), 339.26 (48.12), 337.26 (16.73), 40.20 (100.00); anal. calcd for C30H19N5O2S (513.58): C, 70.16; H, 3.73; N, 13.64; S, 6.24%. Found: C, 70.18; H, 3.70; N, 13.62; S, 6.21%.Yellow crystals; (240 mg, 50%); mp 310–311 °C; IR (KBr, ν/cm−1): 3550–3420 (OH, broad), 3378 (NH), 3209 (NH), 3051 (aromatic CH), 2961, 2898 (aliphatic CH), 1636 (C
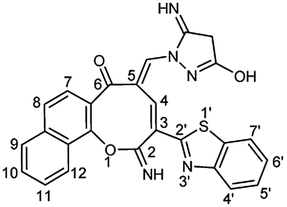 O), 1600 (CN), 1596 (CC); 1H NMR (500 MHz, TFA): δ (ppm) 4.25 (s, 2H, CH2), 7.43 (d, 1H, H-8, J = 8.90 Hz), 7.50 (d, 1H, H-7, J = 8.90 Hz), 7.65 (dd, 2H, Ar–H, J = 7.50, 8.00 Hz), 7.72 (dd, 1H, Ar–H, J = 7.00, 7.50 Hz), 7.79 (dd, 1H, Ar–H, J = 7.00, 7.50 Hz), 7.85 (d, 1H, H-7′, J = 8.20 Hz), 8.04 (d, 1H, H-4′, J = 7.50 Hz), 8.17 (d, 1H, H-9, J = 8.20 Hz), 8.54 (d, 1H, H-12, J = 8.20 Hz), 8.58 (d, 1H, CHN, J = 1.40 Hz), 8.92 (d, 1H, H-4, J = 1.40 Hz); anal. calcd for C26H17N5O3S (479.51): C, 65.13; H, 3.57; N, 14.61; S, 6.69%. Found: C, 65.16; H, 3.56; N, 14.65; S, 6.66%.
O), 1600 (CN), 1596 (CC); 1H NMR (500 MHz, TFA): δ (ppm) 4.25 (s, 2H, CH2), 7.43 (d, 1H, H-8, J = 8.90 Hz), 7.50 (d, 1H, H-7, J = 8.90 Hz), 7.65 (dd, 2H, Ar–H, J = 7.50, 8.00 Hz), 7.72 (dd, 1H, Ar–H, J = 7.00, 7.50 Hz), 7.79 (dd, 1H, Ar–H, J = 7.00, 7.50 Hz), 7.85 (d, 1H, H-7′, J = 8.20 Hz), 8.04 (d, 1H, H-4′, J = 7.50 Hz), 8.17 (d, 1H, H-9, J = 8.20 Hz), 8.54 (d, 1H, H-12, J = 8.20 Hz), 8.58 (d, 1H, CHN, J = 1.40 Hz), 8.92 (d, 1H, H-4, J = 1.40 Hz); anal. calcd for C26H17N5O3S (479.51): C, 65.13; H, 3.57; N, 14.61; S, 6.69%. Found: C, 65.16; H, 3.56; N, 14.65; S, 6.66%.Note: both of keto and enol forms were only observed in NMR analyses due to the use of trifluoroacetic acid as solvent which might induce this tautomerization process.
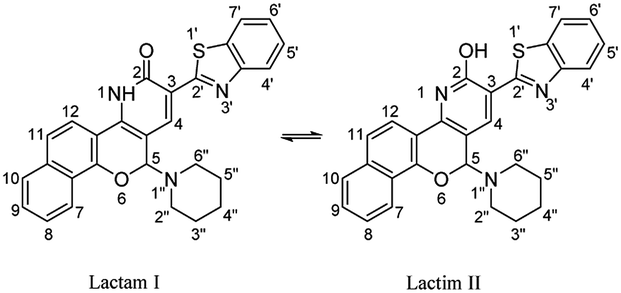
Yellow crystals; (307 mg, 66%); mp > 330 °C; IR (KBr, ν/cm−1): 3350–3460 (NH, broad), 3064 (aromatic CH), 2931–2816 (aliphatic CH), 1643 (CO), 1604 (CN), 1560 (CC); 1H NMR (500 MHz, TFA): δ (ppm) 1.49–2.16 (m, 12H, H-3′′,4′′,5′′, I & II), 3.09–4.04 (m, 8H, H-2′′, H-6′′, I & II), 6.97 (s, 2H, H-4, I & II), 7.85 (dd, 2H, Ar–H, J = 7.49, 7.51 Hz, I & II), 7.92 (m, 4H, Ar–H, I & II), 7.97 (d, 4H, Ar–H, I & II) 8.05 (m, 2H, Ar–H, I & II), 8.12 (m, 2H, Ar–H, I & II), 8.22 (m, 2H, Ar–H, I & II), 8.29 (m, 2H, Ar–H, I & II), 8.46 (m, 2H, H-7, I & II), 9.03 (s, 2H, H-4, I & II); EI-MS m/z (%): 465.57 [M+] (45.76), 462.54 (64.03), 331.08 (59.94), 315.14 (45.02), 99.57 (69.91), 83.35 (100.00), 59.43 (82.74), 55.20 (76.75); 13C NMR (126 MHz, TFA): δ (ppm) 22.91, 23.34, 24.39, 24.68, 24.82, 48.52, 51.84, 52.98, 68.60, 91.64, 105.22, 109.30, 114.16, 114.32, 119.54, 120.53, 122.93, 123.98, 124.69, 124.90, 125.15, 125.38, 125.62, 128.92, 130.94, 131.22, 131.39, 131.75, 132.75, 132.93, 133.06, 133.21, 133.92, 134.22, 135.79, 137.48, 140.42, 140.66, 141.64, 142.14, 144.41, 146.13, 148.36, 153.73, 168.88; anal. calcd for C28H23N3O2S (465.57): C, 72.24; H, 4.98; N, 9.03; S, 6.89%. Found: C, 72.22; H, 4.99; N, 9.01, S, 6.87%.
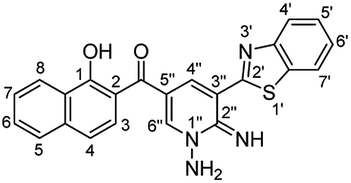 O), 1628 (CN), 1599 (CC); 1H NMR (500 MHz, DMSO-d6): δ (ppm) 7.29 (s, 2H, NH2), 7.51 (d, 1H, H-3, J = 8.20 Hz), 7.57 (dd, 1H, H-6′, J = 7.49, 7.51 Hz), 7.61–7.65 (dd, 2H, Ar–H), 7.66 (d, 1H, H-4′, J = 8.90 Hz), 7.73 (dd, 1H, H-5′, J = 7.49, 7.51 Hz), 7.96 (d, 1H, H-4, J = 8.20 Hz), 8.22 (d, 1H, H-8, J = 8.20 Hz), 8.39 (d, 1H, H-5, J = 8.20 Hz), 8.68 (d, 1H, H-4′′, J = 1.40 Hz), 8.78 (s, 1H, H-6′′), 10.18 (brs, 1H, OH), 12.30 (s, 1H, NH); EI-MS m/z (%): 412.34 [M+] (8.22), 350.34 (42.07), 305.04 (73.80), 240.95 (100.00), 221.44 (77.21), 107.52 (50.84), 83.05 (76.40); 13C NMR (126 MHz, DMSO-d6): δ (ppm) 114.79, 114.91, 119.21, 121.40, 122.24, 123.21, 123.56, 124.73, 126.08, 126.17, 126.83, 127.16, 127.57, 129.86, 133.17, 136.64, 139.87, 144.94, 151.63, 151.84, 158.63, 163.06, 191.91; anal. calcd for C23H16N4O2S (412.47): C, 66.98; H, 3.91; N, 13.58; S, 7.77%. Found: C, 66.95; H, 3.93; N, 13.59; S, 7.79%.
O), 1628 (CN), 1599 (CC); 1H NMR (500 MHz, DMSO-d6): δ (ppm) 7.29 (s, 2H, NH2), 7.51 (d, 1H, H-3, J = 8.20 Hz), 7.57 (dd, 1H, H-6′, J = 7.49, 7.51 Hz), 7.61–7.65 (dd, 2H, Ar–H), 7.66 (d, 1H, H-4′, J = 8.90 Hz), 7.73 (dd, 1H, H-5′, J = 7.49, 7.51 Hz), 7.96 (d, 1H, H-4, J = 8.20 Hz), 8.22 (d, 1H, H-8, J = 8.20 Hz), 8.39 (d, 1H, H-5, J = 8.20 Hz), 8.68 (d, 1H, H-4′′, J = 1.40 Hz), 8.78 (s, 1H, H-6′′), 10.18 (brs, 1H, OH), 12.30 (s, 1H, NH); EI-MS m/z (%): 412.34 [M+] (8.22), 350.34 (42.07), 305.04 (73.80), 240.95 (100.00), 221.44 (77.21), 107.52 (50.84), 83.05 (76.40); 13C NMR (126 MHz, DMSO-d6): δ (ppm) 114.79, 114.91, 119.21, 121.40, 122.24, 123.21, 123.56, 124.73, 126.08, 126.17, 126.83, 127.16, 127.57, 129.86, 133.17, 136.64, 139.87, 144.94, 151.63, 151.84, 158.63, 163.06, 191.91; anal. calcd for C23H16N4O2S (412.47): C, 66.98; H, 3.91; N, 13.58; S, 7.77%. Found: C, 66.95; H, 3.93; N, 13.59; S, 7.79%.
Orange crystals; (241 mg, 53%); mp 264 °C; IR (KBr, ν/cm−1): 3450–3300 (OH, broad), 3293 (NH), 3054 (aromatic CH), 1637 (C
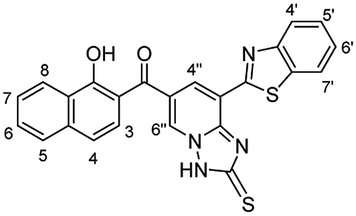 N), 1593 (CC); 1H NMR (500 MHz, DMSO-d6): δ (ppm) 7.48–7.50 (m, 2H, Ar–H), 7.57 (dd, 1H, H-7, J = 7.00, 8.00 Hz), 7.63 (dd, 1H, Ar–H, J = 7.00, 8.50 Hz), 7.67 (dd, 1H, J = 1.50, 7.60 Hz), 7.73 (dd, 1H, H-6, J = 7.00, 8.00 Hz), 7.96 (d, 1H, H-5, J = 8.20 Hz), 8.14 (dd, 2H, Ar–H, J = 8.00, 9.00 Hz), 8.38 (d, 1H, Ar–H, J = 8.20 Hz), 8.40 (d, 1H, H-4′′, J = 2.00 Hz), 8.59 (d, 1H, H-6′′, J = 2.00 Hz), 8.76 (brs, 2H, NH2), 12.85 (s, 1H, OH); EI-MS m/z (%): 471.08 [M+] (14.29), 420.45 (18.15), 263.38 (19.92), 147.26 (37.62), 102.65 (21.94), 97.34 (38.44), 67.14 (100.00), 60.16 (52.60), 53.36 (84.10); anal. calcd for C24H17N5O2S2 (471.55): C, 61.13; H, 3.63; N, 14.85; S, 13.60%. Found: C, 61.15; H, 3.64; N, 14.84 S, 13.56%.
N), 1593 (CC); 1H NMR (500 MHz, DMSO-d6): δ (ppm) 7.48–7.50 (m, 2H, Ar–H), 7.57 (dd, 1H, H-7, J = 7.00, 8.00 Hz), 7.63 (dd, 1H, Ar–H, J = 7.00, 8.50 Hz), 7.67 (dd, 1H, J = 1.50, 7.60 Hz), 7.73 (dd, 1H, H-6, J = 7.00, 8.00 Hz), 7.96 (d, 1H, H-5, J = 8.20 Hz), 8.14 (dd, 2H, Ar–H, J = 8.00, 9.00 Hz), 8.38 (d, 1H, Ar–H, J = 8.20 Hz), 8.40 (d, 1H, H-4′′, J = 2.00 Hz), 8.59 (d, 1H, H-6′′, J = 2.00 Hz), 8.76 (brs, 2H, NH2), 12.85 (s, 1H, OH); EI-MS m/z (%): 471.08 [M+] (14.29), 420.45 (18.15), 263.38 (19.92), 147.26 (37.62), 102.65 (21.94), 97.34 (38.44), 67.14 (100.00), 60.16 (52.60), 53.36 (84.10); anal. calcd for C24H17N5O2S2 (471.55): C, 61.13; H, 3.63; N, 14.85; S, 13.60%. Found: C, 61.15; H, 3.64; N, 14.84 S, 13.56%.Computational prediction of biological activities of oxocine derivatives 3a–e and 6
The 2D chemical structures of the oxocine derivatives 3a–e and 6 were drawn using both ChemDoodle36 and Marvin Sketch (ChemAxon, Version 18.30). After that SwissADME predictor software (http://www.swissadme.ch/) was used to convert the 2D structures into SMILEY mode.37 The biological activities of the synthesized products were predicted using PASS online software. The main target of PASS (prediction of activity spectra for biologically active substances) is prediction of the most probable kind of biological activity from the chemical structure. It reflects all biological activities that arise from interactions with biological entities among the compounds from the viable database. In this study, PASS develops prediction of biological activities with the probabilities of Pa (to be active) and Pi (to be inactive) values.38Conflicts of interest
The authors have no conflict of interest to mention.References
- J. Cossy, M. Brimble and M. Cordes, Synthesis of Saturated Oxygenated Heterocycles, Springer, 2014 Search PubMed.
- B. Harrison and P. Crews, J. Org. Chem., 1997, 62, 2646–2648 CrossRef CAS.
- A. Arnone, G. Nasini, W. Panzeri, O. V. d. Pava and L. Malpezzi, J. Nat. Prod., 2007, 71, 146–149 CrossRef.
- W. Steglich, Pure Appl. Chem., 1989, 61, 281–288 CAS.
- L.-c. Fu, X.-a. Huang, Z.-y. Lai, Y.-j. Hu, H.-j. Liu and X.-l. Cai, Molecules, 2008, 13, 1923–1930 CrossRef CAS.
- T. Ghosh, Synth. Commun., 2018, 48, 1338–1345 CrossRef CAS.
- L. Liang, E. Li, X. Dong and Y. Huang, Org. Lett., 2015, 17, 4914–4917 CrossRef CAS.
- T. Ghosh, New J. Chem., 2017, 41, 2927–2933 RSC.
- R. K. Boeckman, J. Zhang and M. R. Reeder, Org. Lett., 2002, 4, 3891–3894 CrossRef CAS.
- S. K. Chattopadhyay, S. Karmakar, T. Biswas, K. Majumdar, H. Rahaman and B. Roy, Tetrahedron, 2007, 63, 3919–3952 CrossRef CAS.
- M. Bratz, W. H. Bullock, L. E. Overman and T. Takemoto, J. Am. Chem. Soc., 1995, 117, 5958–5966 CrossRef CAS.
- K. Nicolaou, D. McGarry, P. Somers, B. Kim, W. Ogilvie, G. Yiannikouros, C. Prasad, C. Veale and R. Hark, J. Am. Chem. Soc., 1990, 112, 6263–6276 CrossRef CAS.
- S. Ma and E.-i. Negishi, J. Org. Chem., 1994, 59, 4730–4732 CrossRef CAS.
- N. Ortega, T. Martín and V. S. Martín, Org. Lett., 2006, 8, 871–873 CrossRef CAS.
- S. K. Mandal and S. C. Roy, Tetrahedron, 2007, 63, 11341–11348 CrossRef CAS.
- M. Sasaki, A. Hashimoto, K. Tanaka, M. Kawahata, K. Yamaguchi and K. Takeda, Org. Lett., 2008, 10, 1803–1806 CrossRef CAS.
- M. A. Ibrahim, ARKIVOC, 2008,(xvii), 192–204 Search PubMed.
- J. Li, J. M. Suh and E. Chin, Org. Lett., 2010, 12, 4712–4715 CrossRef CAS.
- H.-H. Liao and R.-S. Liu, Chem. Commun., 2011, 47, 1339–1341 RSC.
- A. Thakur and J. Louie, Acc. Chem. Res., 2015, 48, 2354–2365 CrossRef CAS.
- G. Kim, T. i. Sohn, D. Kim and R. S. Paton, Angew. Chem., Int. Ed., 2014, 53, 272–276 CrossRef CAS.
- M. Ibrahim, N. El-Gohary, S. Ibrahim and S. Said, Chem. Heterocycl. Compd., 2015, 50, 1624–1633 CrossRef CAS.
- M. A. Ibrahim and T. E.-S. Ali, Turk. J. Chem., 2015, 39, 412–425 CrossRef CAS.
- S. P. Alexander, E. Kelly, N. Marrion, J. A. Peters, H. E. Benson, E. Faccenda, A. J. Pawson, J. L. Sharman, C. Southan and O. P. Buneman, Br. J. Pharmacol., 2015, 172, 5729–5743 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT USA, 2009 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- N. Godbout, D. R. Salahub, J. Andzelm and E. Wimmer, Can. J. Chem., 1992, 70, 560–571 CrossRef CAS.
- S. Gupta and J. M. Khurana, Green Chem., 2017, 19, 4153–4156 RSC.
- M. A. Ibrahim and A.-S. Badran, ARKIVOC, 2018,(vii), 214–224, DOI:10.24820/ark.5550190.p010.745.
- A good agreement was found between the experimental and predicted IR spectra of compound 3a which support the reliability of the calculated results. Predicted IR absorptions: 3450 cm−1 (OH), 3280 cm−1 (NH), 33105 cm−1 (aromatic CH), 1608 cm−1 (CN), 1530 cm−1 (CC) (see ESI†).
- S. E. Koops, B. C. O'Regan, P. R. Barnes and J. R. Durrant, J. Am. Chem. Soc., 2009, 131, 4808–4818 CrossRef CAS.
- Q.-H. Yao, L. Shan, F.-Y. Li, D.-D. Yin and C.-H. Huang, New J. Chem., 2003, 27, 1277–1283 RSC.
- M. Al-Eid, S. Lim, K. Park, B. Fitzpatrick, C. Han, K. Kwak, J. Hong and G. Cooke, Dyes Pigm., 2014, 104, 197–203 CrossRef CAS.
- X. Liu, Z. Cao, H. Huang, X. Liu, Y. Tan, H. Chen, Y. Pei and S. Tan, J. Power Sources, 2014, 248, 400–406 CrossRef CAS.
- M. C. Burger, J. Cheminf., 2015, 7, 35 Search PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef.
- S. Parasuraman, J. Pharmacol. Pharmacother., 2011, 2, 52–53 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05154f |
| This journal is © The Royal Society of Chemistry 2019 |