
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of N-oxidation on the molecular and crystal structures and properties of isocinchomeronic acid, its metal complexes and their supramolecular architectures: experimental, CSD survey, solution and theoretical approaches†
Zahra Hosseini-Hashemia,
Masoud Mirzaei *a,
Ameneh Jafaria,
Peyman Hosseinpoura,
Mohammad Yousefi*b,
Antonio Fronterac,
Mahmoud Lari Dashtbayazd,
Mojtaba Shamsipure and
Mehdi Ardalanie
*a,
Ameneh Jafaria,
Peyman Hosseinpoura,
Mohammad Yousefi*b,
Antonio Fronterac,
Mahmoud Lari Dashtbayazd,
Mojtaba Shamsipure and
Mehdi Ardalanie
aDepartment of Chemistry, Faculty of Science, Ferdowsi University of Mashhad, Mashhad, Iran. E-mail: mirzaeesh@um.ac.ir
bDepartment of Chemistry, Yadegar-e-Imam Khomeini (RAH) Shahr-e-Rey Branch, Islamic Azad University, Tehran, Iran. E-mail: myousefi50@hotmail.com
cDepartment of Chemistry, Universitat de les Illes Balears, Crta de Valldemossa km 7.5, 07122 Palma de Mallorca (Baleares), Spain
dDepartment of Economics and Administrative Sciences, Ferdowsi University of Mashhad, Mashhad, Iran
eDepartment of Analytical Chemistry, Razi University, Kermanshah, Iran
First published on 14th August 2019
Abstract
Nine coordination complexes and polymer (M/L/X) based on Co, Ni, Zn, Cu (M), pyridine-N-oxide-2,5-dicarboxylic acid (H2pydco) (L) and either isonicotinamide (Ina), piperazine (pipz), 2,2′-bipyridine (bipy) and 1,10-phenanthroline (phen) (X) were synthesized and characterized by elemental analyses, infrared spectroscopy and single crystal X-ray diffraction. The resulting empirical formulae of the prepared complexes are [Co(H2O)6][Co(pydco)2(H2O)2]·2H2O (1), [M(pydco)(H2O)4]2 [M = Co (2), Ni (3), Zn (4)], [Co(pydco)(bipy)(H2O)2]·4H2O (5), [Co(pydco)(phen)(H2O)2]·5.135(H2O)·0.18(EtOH) (6), [Cu(Hpydco)(bipy)Cl]·2H2O (7), [Cu(Hpydco)(bipy)Cl]2·2H2O (8), and {[AgCu(H2O)2(phen)(pydco)]NO3}n (9). With the exception of 9, which forms an extended structure via multiple coordination modes, all the complexes contain (H)pydco as a bidentate ligand coordinated to the metal ion via the N-oxide and the adjacent carboxylate group oxygen atom, creating a chelate ring. The metal centers exhibit either distorted octahedral (1–6) or square pyramidal (7–9) geometry. Our results demonstrate that, when acting cooperatively, non-covalent interactions such as X–H⋯O hydrogen bonds (X = O, N, C), C–O⋯π and π⋯π stacking represent driving forces for the selection of different three-dimensional structures. Moreover, in compounds 2–4, 1D supramolecular chains are formed where O⋯π–hole interactions are established, which unexpectedly involve the non-coordinated carboxylate group. The non-covalent interaction (NCI) plot index analysis reveals the existence of the O⋯π–hole interactions that have been evaluated using DFT calculations. The Cremer and Pople ring puckering parameters are also investigated. The complexation reactions of these molecules with M were investigated by solution studies. The stoichiometry of the most abundant species in the solution was very close to the corresponding crystals. Finally, the effect of N-oxidation on the geometry of complexes has been also studied using the Cambridge Structural Database. It shows that complexes containing N-oxidized H2pydc are very rare.
Introduction
Over the last few decades, considerable attention has been devoted to crystal engineering, which is the design and synthesis of solid-state molecular structures with desired properties, based on the understanding and exploitation of intermolecular interactions. A variety of non-covalent interactions including hydrogen bonding, π⋯π stacking, C–H⋯π and other interactions produce many interesting structures with 1D (pillar, chain or band), 2D (layer) and 3D (network) topologies.1–3 Intermolecular forces are considerably weaker than covalent bonds, so that supramolecular species are thermodynamically less stable, kinetically more labile and dynamically more flexible than molecules. Supramolecular chemistry is therefore concerned with weak bonds. In the production of supramolecular scaffolds several factors such as the type of metal, solvent, organic ligand and auxiliary ligand play crucial roles.4 Auxiliary ligands such as 2,2′-bipyridine and 1,10-phenanthroline are bidentate with two aromatic units each carrying an N-donor atom. The crystal structures formed by these ligands can display weak π⋯π interactions that are very important for the construction of multidimensional arrays.5 In recent years, researchers have been attracted to the many special properties and fascinating applications of complexes containing dicarboxylic acid and pyridine rings.6 These applications exist in many areas, such as catalysis,7 antibacterial activity,8 anticancer properties,9 aqueous solution chemistry,10 surface chemistry,11 magnetism12,13 and fluorescence.14,15 Our research group therefore set out to synthesize coordination complexes based on derivatives of pyridinedicarboxylic acids with a view to their significant applications.16–20 Among the structural isomers of 2,n-pyridinedicarboxylic acids (n = 3–6), H2pydc (isocinchomeronic acid or pyridine-2,5-dicarboxylic acid) is an appropriate candidate for constructing metallotectons due to its interesting structural features: it is a multidentate ligand containing N- and O-donor atoms; it has two carboxylates on opposite sides of the pyridine ring. These features confer the potential to construct higher-dimensional extended structures.21,22 The carboxylate groups display various coordination modes including monodentate (terminal and bridging) and bidentate (chelating and bridging).23 Meanwhile, 5-carboxylate has a stronger bridging capability than 2-carboxylate due to its strong electron-donating and electrostatic power.24 N-oxidation of the pyridine ring adds a new coordination mode: whereas the nitrogen atom of the pyridine ring in H2pydc can donate one pair of electrons, the N-oxide group can donate two pairs and can therefore coordinate to a larger number of metal centers, resulting in a greater variety of bridging modes for H2pydco compared to H2pydc (see Scheme 1).25–27 We were inspired to synthesize the N-oxide form of the ligand by the work of Xiong et al. who demonstrated how N-oxide functionalization could greatly enhance the CO2 separation properties of isoreticular MOFs.28 Moreover, this family of pyridine N-oxides have been utilized as an anti-HIV agent, gas adsorbent, luminescent agent, etc.,28–30 which encouraged us to prepare new complexes with the pyridine-N-oxide-2,5-dicarboxylic acid. Our studies concur with previous investigations in showing that H2pydco tends to be an effective chelating and bridging ligand.27 Herein, we report the preparation of [Co(H2O)6][Co(pydco)2(H2O)2]·2H2O (1), [M(pydco)(H2O)4]2 [M = Co (2), Ni (3), and Zn (4)], [Co(pydco)(bipy)(H2O)2]·4H2O (5), [Co(pydco)(phen)(H2O)2]·5.135(H2O)·0.18(EtOH) (6), [Cu(Hpydco)(bipy)Cl]·2H2O (7), [Cu(Hpydco)(bipy)Cl]2·2H2O (8), and {[AgCu(H2O)2(phen)(pydco)]NO3}n (9), where H2pydco = pyridine-N-oxide-2,5-dicarboxylic acid, Ina = isonicotinamide, pipz = piperazine, bipy = 2,2′-bipyridine and phen = 1,10-phenanthroline (see Scheme S1†). By means of DFT calculations (M06-2X-D/def2-TZVP), we have studied the importance of π–hole interactions involving the carboxylate group (as π–hole) donor in compounds 2–4 and the influence of the metal center (M = Co, Ni, Zn) on the interaction strength. Moreover, we have also performed the non-covalent interaction (NCI) index analysis in complexes 2–4 to confirm the existence of π–hole O⋯CO interactions and their interplay with H-bonding interactions. The complexation reactions of these supramolecular systems in aqueous solution were investigated by potentiometric pH titration method (solution studies) in order to characterize the stoichiometry of new species. The solution behavior of the investigated species, providing additional evidence of the interactions between adduct and metal ions, supporting the results obtained from the solid-state studies. | ||
| Scheme 1 The structures of H2pydc and H2pydco. | ||
Experimental
General methods and materials
All chemicals and solvents were purchased from commercial sources and used without further purification. Melting points were determined using a Barnstead Electrothermal 9300 apparatus. IR spectra were recorded from KBr pellets in the 4000–600 cm−1 region using a Buck 500 IR spectrometer. Elemental analysis (CHN) was performed using a Thermo Finning Flash-1112 EA microanalyzer. The mass spectrum was scanned on a MS model CH7A Varian (EI, 70 eV). 1H NMR, and 13C{1H} NMR spectra were recorded in EtOD solution on a FT-NMR Bruker Avance III 300 MHz spectrometer, using TMS for 1H and 13C as the internal standard.X-ray structure determination and structure refinement
Single-crystal X-ray diffraction measurements were performed on a STOE IPDS-2T diffractometer with graphite monochromated Mo-Kα radiation. Single crystals were chosen using a polarizing microscope and were mounted on glass fibers for data collection. Cell constants and orientation matrices were obtained by least-squares refinement against setting angles for all reflections. Diffraction data were collected as a series of 1° ω scans and integrated using the Stoe X-AREA software package.31 Lorentz and polarization corrections were applied to the data and a numerical absorption correction was applied using X-RED32 and X-SHAPE.33 The structures were solved by direct methods34 and subsequent difference Fourier maps and then refined on F2 by full-matrix least-squares with anisotropic displacement parameters for all non-H atoms.35 The atomic scattering factors were taken from International Tables for X-ray Crystallography.36 All refinements were performed within the Stoe X-STEP32 structure evaluation package.37Synthesis of pyridine-N-oxide-2,5-dicarboxylic acid
H2pydc (1.003 g, 6 mmol) and a solution of Na2WO4·2H2O (0.065 g, 0.2 mmol) in hydrogen peroxide (30%, 9 mL) were heated at 90–100 °C with vigorous stirring for 50 min. Then an additional amount of hydrogen peroxide (30%, 22 mL) was added dropwise over a period of 2 h until all insoluble material had disappeared. After an additional 3 h of heating, the reaction mixture was allowed to stand for 24 h. The yellow crystalline solid was filtered off and dried in air. This product was obtained in 95% yield (based on H2pydc) (m.p. 198 °C). Anal. calcd for C7H5NO5: C 45.91; H 2.75; N 7.65%. Found: C 46.22; H 2.77; N 7.73%. IR bands (KBr pellet, cm−1): 3446–3081(br), 2634(m), 1725(s), 1643(m), 1612(m), 1513(m), 1402(s), 1231(s). MS (70 eV, EI): m/z (%) = 183 (45) [M]+, 182 (96) [M − 1]+, 138 (98) [M − COOH]+, 122 (100) [C6H4NO2]+, 94 (27) [C5H4NO]+, 78 (95) [C4H5N]+, 45 (85) [COOH]+. 1H NMR (300.81 MHz, EtOD, 296.6 K, TMS): δ 8.36 (dd, 3JH–H = 8.4 Hz, 4JH–H = 1.5 Hz, 1H, Ar-Hb), 8.52 (d, 3JH–H = 8.1 Hz, 1H, Ar-Hc), 8.96 (d, 4JH–H = 1.2 Hz, 1H, Ar-Ha). 13C{1H} NMR (75.64 MHz, EtOD, 297.9 K, TMS): δ 129.08 (s), 131.55 (s), 133.81 (s), 138.60 (s), 139.88 (s), 160.95 (s), 162.70 (s). The MS, 1H NMR and 13C NMR spectra of H2pydco are given in Fig. S1–S3.†Syntheses of [Co(H2O)6][Co(pydco)2(H2O)2]·2H2O (1) and [Co(pydco)(H2O)4]2 (2)
A solution of H2pydco (0.183 g, 1 mmol) in ethanol–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; 20 mL) was added dropwise with stirring to a solution of Co(CH3COO)2·4H2O (0.249 g, 1 mmol) in water (30 mL) at 50 °C. A suspension formed immediately and the solution was stirred for 2 h at room temperature. Ina (0.122 g, 1 mmol) in ethanol (30 mL) was added dropwise to this suspension and was stirred for 1 h at 60 °C and then cooled to room temperature.38 Two types of crystals [orange (1) and pink (2)] were obtained by slow evaporation after one week. The final yields were 0.25 g for 1 (38.4%) and 0.08 g (26.8%) for 2 (based on Co). Data for 1: (m.p. 157 °C), Anal. calcd for C14H26Co2N2O20: C 25.47; H 3.97; N 4.24%. Found: C 25.58; H 3.89; N 4.31%. IR bands (KBr pellet, cm−1): 3372(br), 1662(m), 1611(s), 1390(s), 1352(m), 1217(m). Data for 2: (m.p. 177 °C), Anal. calcd for C7H11CoNO9: C 26.94; H 3.55; N 4.49%. Found: C 26.78; H 3.41; N 4.63%. IR bands (KBr pellet, cm−1): 3258(br), 1639(s), 1591(s), 1552(m), 1390(s), 1352(m), 1211(w).
:1; 20 mL) was added dropwise with stirring to a solution of Co(CH3COO)2·4H2O (0.249 g, 1 mmol) in water (30 mL) at 50 °C. A suspension formed immediately and the solution was stirred for 2 h at room temperature. Ina (0.122 g, 1 mmol) in ethanol (30 mL) was added dropwise to this suspension and was stirred for 1 h at 60 °C and then cooled to room temperature.38 Two types of crystals [orange (1) and pink (2)] were obtained by slow evaporation after one week. The final yields were 0.25 g for 1 (38.4%) and 0.08 g (26.8%) for 2 (based on Co). Data for 1: (m.p. 157 °C), Anal. calcd for C14H26Co2N2O20: C 25.47; H 3.97; N 4.24%. Found: C 25.58; H 3.89; N 4.31%. IR bands (KBr pellet, cm−1): 3372(br), 1662(m), 1611(s), 1390(s), 1352(m), 1217(m). Data for 2: (m.p. 177 °C), Anal. calcd for C7H11CoNO9: C 26.94; H 3.55; N 4.49%. Found: C 26.78; H 3.41; N 4.63%. IR bands (KBr pellet, cm−1): 3258(br), 1639(s), 1591(s), 1552(m), 1390(s), 1352(m), 1211(w).
Synthesis of [Ni(pydco)(H2O)4]2 (3)
A solution of H2pydco (0.092 g, 0.5 mmol) in THF (10 mL) was added dropwise to a solution of pipz (0.043 g, 0.5 mmol) in THF (10 mL) and stirred at room temperature for 2 h. A solution of NiCl2·6H2O (0.119 g, 0.5 mmol) in water (15 mL) was added dropwise to the above solution and stirring was continued for 2 h at room temperature.39,40 After one-month, slow evaporation yielded green acicular crystals of 3 (m.p. > 300 °C) in ca. 43% yield based on Ni. Anal. calcd for C7H11NNiO9: C 26.96; H 3.56; N 4.49%. Found: C 26.64; H 3.63; N 4.39%. IR bands (KBr pellet, cm−1): 3362 (br), 1640(s), 1597(s), 1553(m), 1391(s), 1350(m), 1210(w).Synthesis of [Zn(pydco)(H2O)4]2 (4)
Preparation of 4 was similar to that of 3 except that Zn(NO3)2·6H2O (0.148 g, 0.5 mmol) was used instead of NiCl2·6H2O.39,41 Colourless platy crystals of compound 4 (m.p. > 300 °C) were obtained in ca. 54% yield based on Zn. Anal. calcd for C7H11NO9Zn: C 26.39; H 3.48; N 4.40%. Found: C 26.41; H 3.26; N 4.24%. IR bands (KBr pellet, cm−1): 3350(br), 1639(s), 1591(s), 1552(m), 1390(s), 1352(m), 1211(w).Synthesis of [Co(pydco)(bipy)(H2O)2]·4H2O (5)
A solution of bipy (0.031 g, 0.2 mmol) in ethanol–water (1:1; 3 mL) was added dropwise to a solution of H2pydco (0.037 g, 0.2 mmol) in ethanol–water (1:1; 10 mL) heated under reflux at 80–90 °C. After 1 h, a solution of Co(CH3COO)2·4H2O (0.049 g, 0.2 mmol) in ethanol–water (1:1; 3 mL) was added and refluxed for 6 h at 80–90 °C. After 10 days, orange blocky crystals of 5 (m.p. 177 °C) were obtained by slow evaporation in ca. 40% yield based on Co. Anal. calcd for C17H23CoN3O11: C, 40.49; H, 4.60; N, 8.33%. Found: C, 40.70; H, 4.35; N, 8.26%. IR (KBr pellet, cm−1): 3245(br), 1628(s), 1553(m), 1387(s), 1350(m), 1211(m).
Synthesis of [Co(pydco)(phen)(H2O)2]·5.135(H2O)·0.18(EtOH) (6)
Complex 6 was synthesized following a procedure similar to that employed for the synthesis of complex 5 but replacing bipy with phen (0.039 g, 0.2 mmol). Orange blocky crystals of compound 6 (m.p. 212 °C) were obtained in ca. 48% yield based on Co. Anal. calcd for C19.36H26.35CoN3O12.32: C, 41.73; H, 4.76; N, 7.54%. Found: C, 41.43; H, 4.99; N, 7.32%. IR (KBr pellet, cm−1): 3316(br), 1632(s), 1519(m), 1394(s), 1352(s), 1214(m).Syntheses of [Cu(Hpydco)(bipy)Cl]·2H2O (7) and [Cu(Hpydco)(bipy)Cl]2·2H2O (8)
A solution of H2pydco (0.092 g, 0.5 mmol), bipy (0.078 g, 0.5 mmol) and CuCl2·2H2O (0.085 g, 0.5 mmol) in ethanol–water (1:1; 25 mL) was prepared and stirred at room temperature. After 15 min a suspension formed and the mixture was stirred for 4 h at room temperature. Two differently-colored acicular crystals [green (7) and blue (8)] were obtained by slow evaporation in ca. 52% yield (based on Cu) after 2 weeks. Data for 7 (m.p. 230 °C). Anal. calcd for C17H16ClCuN3O7: C 43.14; H 3.41; N 8.88%. Found: C 43.93; H 3.30; N 9.05%. IR bands (KBr pellet, cm−1): 3379(br), 3066(m), 1695(m), 1643(s), 1603(s), 1565(m), 1398(s), 1345(m), 1206(m).
Data for 8 (m.p. 244 °C). Anal. calcd for C34H27ClCu2N6O12: C 46.72; H 3.11; N 9.61%. Found: C 47.27; H 3.00; N 9.89%. IR bands (KBr pellet, cm−1): 3505(br), 3071(m), 1741(m), 1652(s), 1567(w), 1392(m), 1341(m), 1205(m).
Synthesis of {[AgCu(H2O)2(phen)(pydco)]NO3}n (9)
H2pydco (0.092 g, 0.5 mmol) and NaOH (0.040 g, 1 mmol) were dissolved in deionized water (7.5 mL) and stirred for 30 min at room temperature. In a separate beaker CuCl2·2H2O (0.085 g, 0.5 mmol) was dissolved in deionized water (5 mL) and AgNO3 powder (0.255 g, 1.5 mmol) was added. The AgCl precipitate which formed was filtered off and phen (0.099 g, 0.5 mmol) was added to the clear filtrate. This mixture was added into the solution of H2pydco and NaOH and stirred for 1 h at room temperature. The mixture was sealed in a 25 mL Teflon-lined reactor. The reactor was heated at 130 °C for 8 h and then cooled to room temperature at a rate of 10 °C h−1. Blue acicular crystals of 9 (m.p. 245 °C) were obtained and the final yield of 9 was 0.25 g (40% based on Cu). Anal. calcd for C19H15AgCuN4O10: C 36.18; H 2.40; N 8.88%. Found: C 36.15; H 2.18; N 8.93%. IR bands (KBr pellet, cm−1): 3416(br), 1743(w), 1649(s), 1552(w), 1515(w), 1382(s), 1209(m).Theoretical methods
The calculations of the noncovalent interactions were computed using the Gaussian-09 program package.42 We have used the M06-2X DFT functional in combination with Grimme's dispersion correction43 because it is convenient for describing the weak noncovalent interactions properly. Moreover, it is recommended for system with transition metals.44 In order to describe correctly the π–hole interactions we have used the crystallographic coordinates and the def2-TZVP basis set for all atoms. This procedure and level of theory have been successfully used to evaluate similar interactions.45 The interaction energies were computed by calculating the difference between the energies of isolated monomers and their assembly. The interaction energies were calculated with correction for the basis set superposition error (BSSE) by using the Boys–Bernardi counterpoise technique.46 The NCI plot47 iso-surfaces have been used to characterize noncovalent interactions. They correspond to both favorable and unfavorable interactions, as differentiated by the sign of the second density Hessian eigenvalue and defined by the isosurface color. The color scheme is a red-yellow-green-blue scale with red for ρcut+ (repulsive) and blue for ρcut− (attractive).48 The Gaussian-09 M06-2X/def2TZVP level of theory wave function has been used to generate the NCI plot and the MEP surfaces.Potentiometric pH titrations
Potentiometric measurements were performed for solutions in a 50 mL double-walled glass vessel using a Model 686 Metrohm Tiroprocessor equipped with a combined glass-calomel electrode. The temperature was fixed at 25.0 ± 0.1 °C. The ionic strength was adjusted to 0.1 M by use of KNO3. A CO2-free atmosphere for the base (carbonate-free 0.095 M sodium hydroxide) was ensured throughout. The concentrations of H2pydco (L) and phen (Q) were 3.0 × 10−3 M and potentiometric pH titrations carried out in the absence and presence of 1.5 × 10−3 M of the metal ions. Metal complexes protonation and stability constants and ligands protonation constants were calculated using the program BEST introduced by Martell and Motekaitis.49 The value of KSH = [H+][OH−], for aqueous solution was 10−13.781.50–53Results and discussion
Synthesis
The H2pydco ligand was synthesized by treating H2pydc with 30% H2O2 as oxidizing agent and Na2WO4·2H2O as catalyst, thereby converting the pyridine nitrogen atom to an N-oxide. H2pydc is a N/O donor ligand whereas H2pydco is exclusively an O-donor, a difference which dramatically affects both the molecular and supramolecular structures of the complexes it forms. Our oxidation method affords a 95% yield of pyridine-N-oxide-2,5-dicarboxylic acid. The synthesis of this ligand (H2pydco) was achieved by a modified extent of oxidizing agent (H2O2) from H2pydc.54Infrared spectroscopy
The broad and strong bands at 3000–3500 cm−1 can be attributed to the stretching vibrations ν(OH) of lattice and coordinated water molecules and ν(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H) in the aromatic rings. They are also indicative of the presence of hydrogen bonding.13,55 The strong νas(COO−) and the νs(COO−) bands in free H2pydco 1726 and 1419 cm−1 are shifted in the complexes to the lower frequencies in the range 1674–1628 cm−1 and 1398–1372 cm−1, respectively. These results indicate that in these complexes the carboxylate group of pydco coordinates to the transition metal ions through deprotonation.56 Furthermore, the strong absorption bands in the range of 1550–1600 cm−1 can be attributed to the ν(CC) and ν(CN) vibration of aromatic pyridyl ring for all these complexes. The IR spectrum of H2pydco (see Fig. S4†) shows a strong band at 1230 cm−1 due to the presence of N–O group confirming the synthesis of the ligand.27 Bands in the 1228–1205 cm−1 region for the complexes (see Fig. S4†) were therefore assigned to the N–O stretching vibration this group. The bands at 900–700 cm−1 are characteristic of the bending vibration δ(C–H) in aromatic rings.
C–H) in the aromatic rings. They are also indicative of the presence of hydrogen bonding.13,55 The strong νas(COO−) and the νs(COO−) bands in free H2pydco 1726 and 1419 cm−1 are shifted in the complexes to the lower frequencies in the range 1674–1628 cm−1 and 1398–1372 cm−1, respectively. These results indicate that in these complexes the carboxylate group of pydco coordinates to the transition metal ions through deprotonation.56 Furthermore, the strong absorption bands in the range of 1550–1600 cm−1 can be attributed to the ν(CC) and ν(CN) vibration of aromatic pyridyl ring for all these complexes. The IR spectrum of H2pydco (see Fig. S4†) shows a strong band at 1230 cm−1 due to the presence of N–O group confirming the synthesis of the ligand.27 Bands in the 1228–1205 cm−1 region for the complexes (see Fig. S4†) were therefore assigned to the N–O stretching vibration this group. The bands at 900–700 cm−1 are characteristic of the bending vibration δ(C–H) in aromatic rings.
NMR study
In the 1H NMR spectrum of H2pydco, the aromatic protons of the pyridyl ring are revealed as doublet of doublets at 8.36 ppm for the Hb proton, due to the vicinal couplings with both Hc and Ha protons (3JH–H = 8.4 and 4JH–H = 1.5 Hz). The Hc proton shows a doublet signal at 8.52 ppm with coupling to the Hb proton (3JH–H= 8.1 Hz). The Ha proton appears as a doublet at 8.96 ppm, with coupling to the Hb proton (4JH–H = 1.2 Hz) (see Fig. S2†). In the 13C{1H} NMR spectrum of H2pydco, the signals at 129.08, 131.55, 133.81, 138.60, and 139.88 ppm are related to the carbon atoms of the pyridyl ring of H2pydco (C2, C3, C4, C5, and C1), respectively. Both carbon atoms of the two carboxylic acid groups (C6 and C7) are revealed at 160.95 and 162.70 ppm, respectively (see Fig. S3†).
CSD search
A search of the Cambridge Structural Database (CSD) for complexes of pyridine-2,5-dicarboxylic acid ligands (Fig. 1) returned 419 entries of complexes with 2,5-pydc, but only four complexes containing 2,5-pydco, the latter being lanthanoid complexes.27 This dearth of structurally-characterized complexes of all isomers of pydco spurred our efforts to prepare and characterize new supramolecular coordination complexes of 2,5-pydco with various transition metals. | ||
| Fig. 1 Frequency of complexes containing all isomers of the pydc and pydco ligands. | ||
Description of the crystal structures
The crystallographic data for compounds 1–9 is shown in Table 1. In addition, selected bond lengths, valence angles and hydrogen bond geometries are given in Tables S1 and S2 in the ESI.†| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| Empirical formula | C14H26Co2N2O20 | C7H11CoNO9 | C7H11NNiO9 | C7H11NO9Zn | C17H23CoN3O11 |
| Formula weight | 660.23 | 312.10 | 311.86 | 318.56 | 504.31 |
| T (K) | 293(2) | 298(2) | 298(2) | 298(2) | 298(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group | C2/c | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P |
| a (Å) | 12.9792(19) | 9.902(2) | 9.858(2) | 9.889(2) | 9.3305(19) |
| b (Å) | 9.746(3) | 10.698(2) | 10.656(2) | 10.653(2) | 9.882(2) |
| c (Å) | 19.835(4) | 11.742(2) | 11.692(2) | 11.751(2) | 11.820(2) |
| α (°) | 90 | 68.84(3) | 68.85(3) | 68.87(3) | 83.25(3) |
| β (°) | 105.290(14) | 84.28(3) | 84.18(3) | 84.25(3) | 78.22(3) |
| γ (°) | 90 | 66.30(3) | 66.00(3) | 66.44(3) | 89.16(3) |
| V (Å3) | 2420.2(10) | 1060.7(5) | 1045.0(3) | 1057.1(5) | 1059.5(4) |
| Z | 4 | 4 | 4 | 4 | 2 |
| Dcalc (g cm−3) | 1.812 | 1.954 | 1.982 | 2.001 | 1.581 |
| μ (mm−1) | 1.469 | 1.663 | 1.902 | 2.369 | 0.875 |
| F(000) | 1352 | 636 | 640 | 648 | 522 |
| Crystal size (mm3) | 0.36 × 0.36 × 0.15 | 0.50 × 0.20 × 0.20 | 0.50 × 0.25 × 0.20 | 0.50 × 0.30 × 0.20 | 0.50 × 0.50 × 0.50 |
| θ Range for data collection (°) | 2.129 to 24.963 | 2.22 to 29.19 | 2.23 to 29.20 | 2.47 to 29.24 | 2.55 to 29.17 |
| Index ranges | −15 ≤ h ≤ 14 | −13 ≤ h ≤ 13 | −13 ≤ h ≤ 13 | −11 ≤ h ≤ 13 | −10 ≤ h ≤ 12 |
| 0 ≤ k ≤ 11 | −14 ≤ k ≤ 14 | −14 ≤ k ≤ 13 | −14 ≤ k ≤ 14 | −13 ≤ k ≤ 13 | |
| 0 ≤ l ≤ 11 | −16 ≤ l ≤ 12 | −16 ≤ l ≤ 14 | −16 ≤ l ≤ 15 | −16 ≤ l ≤ 16 | |
| Reflections collected | 1471 | 11589 |
11560 |
11619 |
11927 |
| Independent reflections | 1403 [Rint = 0.0331] | 5672 [Rint = 0.0505] | 5590 [Rint = 0.0409] | 5656 [Rint = 0.1040] | 5672 [Rint = 0.0545] |
| Data/restraints/parameters | 1403/9/226 | 5672/2/371 | 5590/0/325 | 5656/6/373 | 5672/10/325 |
| GOF on F2 | 1.037 | 0.892 | 1.000 | 1.028 | 1.083 |
| Final R indices [I > 2σ(I)] | R1 = 0.0423 wR2 = 0.1067 | R1 = 0.0324 wR2 = 0.0668 | R1 = 0.0283 wR2 = 0.0710 | R1 = 0.0528 wR2 = 0.1217 | R1 = 0.0428 wR2 = 0.1107 |
| R Indices (all data) | R1 = 0.0591 wR2 = 0.1154 | R1 = 0.0574 wR2 = 0.0716 | R1 = 0.0412 wR2 = 0.0742 | R1 = 0.0763 wR2 = 0.1339 | R1 = 0.0537 wR2 = 0.1162 |
| Largest diff. peak and hole (e Å−3) | 0.597 and −0.677 | 0.432 and −0.376 | 0.415 and −0.549 | 1.030 and −1.315 | 0.462 and −0.424 |
| 6 | 7 | 8 | 9 | |
|---|---|---|---|---|
| Empirical formula | C19.36H26.35CoN3O12.32 | C17H16ClCuN3O7 | C34H27ClCu2N6O12 | C19H15AgCuN4O10 |
| Formula weight | 556.86 | 473.33 | 874.17 | 630.76 |
| T (K) | 120(2) | 298(2) | 298(2) | 298(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group | P |
P |
P |
P |
| a (Å) | 7.9216(16) | 8.5223(17) | 8.7022(17) | 9.0136(18) |
| b (Å) | 9.862(2) | 9.4697(19) | 9.913(2) | 10.366(2) |
| c (Å) | 16.736(3) | 12.214(2) | 10.553(2) | 12.609(3) |
| α (°) | 91.96(3) | 75.25(3) | 98.76(3) | 113.63(3) |
| β (°) | 102.07(3) | 84.25(3) | 90.30(3) | 91.47(3) |
| γ (°) | 108.51(3) | 80.60(3) | 108.64(3) | 106.35(3) |
| V (Å3) | 1205.3(5) | 938.7(3) | 851.1(3) | 1022.8(5) |
| Z | 2 | 2 | 1 | 2 |
| Dcalc (g cm−3) | 1.535 | 1.675 | 1.706 | 2.048 |
| μ (mm−1) | 0.781 | 1.353 | 1.404 | 2.067 |
| F(000) | 578 | 482 | 444 | 626 |
| Crystal size (mm3) | 0.50 × 0.50 × 0.45 | 0.30 × 0.10 × 0.05 | 0.45 × 0.25 × 0.20 | 0.5 × 0.25 × 0.25 |
| θ Range for data collection (°) | 2.504 to 29.177 | 2.43 to 29.16 | 2.20 to 29.14 | 2.21 to 29.19 |
| Index ranges | −10 ≤ h ≤ 10 | −11 ≤ h ≤ 11 | −11 ≤ h ≤ 11 | −12 ≤ h ≤ 12 |
| −11 ≤ k ≤ 13 | −12 ≤ k ≤ 12 | −13 ≤ k ≤ 12 | −14 ≤ k ≤ 14 | |
| −22 ≤ l ≤ 22 | −12 ≤ l ≤ 16 | −14 ≤ l ≤ 14 | −17 ≤ l ≤ 16 | |
| Reflections collected | 13150 |
10450 |
9424 | 11268 |
| Independent reflections | 6451 [Rint = 0.0606] | 5021 [Rint = 0.1092] | 4556 [Rint = 0.0554] | 5486 [Rint = 0.0504] |
| Data/restraints/parameters | 6451/29/396 | 5021/2/277 | 4556/2/256 | 5486/6/328 |
| GOF on F2 | 1.041 | 0.966 | 0.977 | 0.988 |
| Final R indices [I > 2σ (I)] | R1 = 0.0478 wR2 = 0.1120 | R1 = 0.0676 wR2 = 0.1196 | R1 = 0.0411 wR2 = 0.0911 | R1 = 0.0499 wR2 = 0.1291 |
| R Indices (all data) | R1 = 0.0619 wR2 = 0.1200 | R1 = 0.1329 wR2 = 0.1388 | R1 = 0.0622 wR2 = 0.0975 | R1 = 0.0763 wR2 = 0.1406 |
| Largest diff. peak and hole (e Å−3) | 0.66 and −0.86 | 0.791 and −0.517 | 0.545 and −0.405 | 1.377 and −0.931 |
Crystal structure of 1
Single crystal X-ray diffraction analysis reveals that 1 crystallizes in the monoclinic space group C2/c. The asymmetric unit of 1 contains two Co(II) ion, one pydco2− anion, four coordinated water molecules (one and three for Co1 and Co2, respectively) and one uncoordinated water molecule (see Fig. 2). Co1 is coordinated by two mutually trans carboxylate O donors and two mutually trans N-oxide O donors from two pydco2− ligands to form the equatorial plane, and by two apical oxygen atoms from two water molecules to complete the distorted octahedral geometry. Co2 is coordinated by six water molecules in a regular octahedral geometry. The O–H⋯O hydrogen bonds create motifs of graph-set notation R21(4), R22(11), R22(13) and C22(8) which extend complex 1 along the crystallographic b direction. Interestingly, the structure exhibits bifurcated O–H⋯O interactions with the oxygen atom of the 5-carboxylate group acting as a hydrogen bond acceptor from two different O–H donors, while two of the water molecules coordinated to Co2 act as bifurcated donors (see Fig. 3). Each unit comprising one cationic and one anionic complex is linked together through O–H⋯O hydrogen bonds between uncoordinated water molecules and 2-carboxylate oxygen atoms or water molecules coordinated to Co2, resulting in a zigzag chain structure along the c direction, as shown in Fig S5.† In addition, there are strong O–H⋯O hydrogen bonds in another chain which form motifs with the graph-set notations R22(18), C11(9) and C22(18) (see Fig. 4). Consequently, the three-dimensional network in 1 made by all above intermolecular O–H⋯O and C–H⋯O interactions. | ||
| Fig. 2 Molecular structures for complexes 1–9, with selected atoms labeled and displacement ellipsoids at 50% probability. For the sake of clarity, NO3− counterion, solvent molecules and H atoms are omitted. | ||
 | ||
| Fig. 3 Representation of the linear chain along the b axis in 1. | ||
 | ||
| Fig. 4 Extended one-dimensional structure of 1 along the side view formed by hydrogen bonding described by graph-set notations R22(18), C11(9) and C22(18). | ||
Crystal structures of 2, 3, and 4
Complexes 2, 3 and 4 are isomorphous and isostructural, crystallizing in the triclinic space group P. The asymmetric unit of each complex contains two independent [M(pydco)(H2O)4] units (M = Co, Ni, and Zn), each consisting of one metal ion on a general position and in a distorted octahedral environment, with one bidentate pydco2− ligand and two water molecules occupying the equatorial plane, while the axial positions are occupied by two water molecules (see Fig. 2). Taking 2 as representative of the three isostructural complexes, we find that it forms dimers via O–H⋯O hydrogen bonds producing motifs with graph-set notations R22(8) and R22(18). In addition, we identified weak C–O⋯π interactions (with a centroid⋯O12 separation of 3.732 Å).16,57 These dimers form chains along the c direction via O15–H15A⋯O9 hydrogen bonds (H15A⋯O9 = 2.235 Å) between water molecules coordinated to CoII (see Fig. S6†). O–H⋯O and N–H⋯O hydrogen bonds generate chains along the b axis, forming motifs with graph-set notation R22(8), C22(13) and C22(15) (see Fig. 5 and S7†).
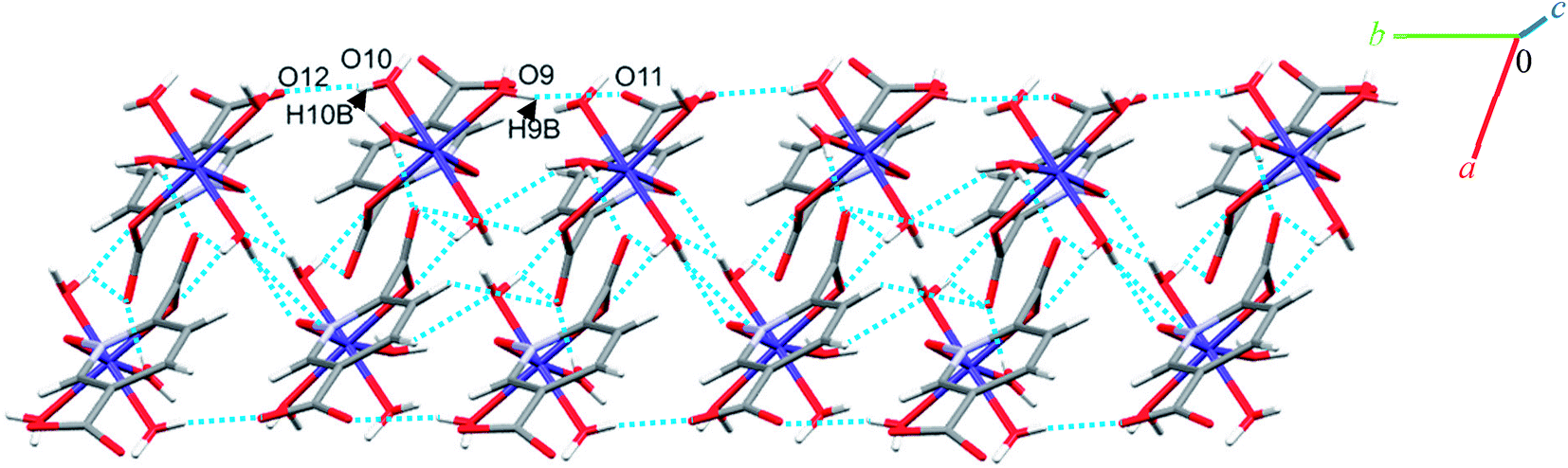 | ||
| Fig. 5 Representation of the one-dimensional linear chain along the b axis in 2. | ||
Additionally, weak C–H⋯π interactions (with a centroid⋯H12C12 separation of 3.334 Å)58 link dimers into chains along the a axis, thereby completing a three-dimensional network (see Fig. S8†). Additional interactions are analyzed below in the theoretical study.
Crystal structure of 5
Complex 5 crystallizes in the triclinic space group P. The Co(II) ion (see Fig. 2) occupies a general position and exhibits distorted octahedral coordination from one bidentate pydco2− ligand, one bipy ligand and two water molecules. The equatorial plane is occupied by one water molecule, two bipy nitrogen atoms and O1 from an N-oxide, while axial positions are occupied by one oxygen atom (O4) from position 2 of carboxyl group and another water molecule.
O–H⋯O hydrogen bonding interactions between water molecules, two carboxylate groups oxygen atoms and an N-oxide from a pydco ligand result in an extended chain along the b direction via motifs with graph-set notations R22(8), R22(18) and R44(16). Weak C–O⋯π interactions (with a centroid⋯O2C3 separation of 3.716 Å) reinforce this chain structure (see Fig. S9†). A C22(11) motif is generated along the c axis by hydrogen bonding interactions (H7B⋯O9: 1.93(2) Å and H9A⋯O3: 1.94(4) Å) between water molecules and oxygen acceptors of the 5-carboxylate group (Fig. 6).
 | ||
| Fig. 6 Representation of the one-dimensional chain along the c axis in 5. | ||
In addition, hydrogen bonding (C11H11⋯O4; 2.440 Å) and two types of π⋯π stacking between pyridine rings (with two different types of centroid⋯centroid separations of 3.585, and 3.606 Å) leads to a third chain along the a axis (see Fig. S10†).59 In general, the chains are packed via O–H⋯O, C–O⋯π, π⋯π stacking interactions in three directions to cooperate three-dimensional supramolecular structure.
Crystal structure of 6
A view of complex 6 with labeling of selected atoms is shown in Fig. 2. The asymmetric unit contains a Co(II) ion occupying a general position and exhibiting distorted octahedral coordination geometry, in which the apical positions are occupied by O6 from a water molecule and N2 from a phen ligand; the equatorial plane is defined by O7 from water molecule, N3 from a phen ligand, O1 from an N-oxide group and O4 from the 2-carboxylate group of a pydco2− ligand.Due to the presence of disorder, it was a challenge to identify the solvent molecules, but we have been able to conclude that the asymmetric unit contains five molecules of water and 0.2 molecules of ethanol. Molecules of 6 are linked into dimers by O–H⋯O hydrogen bonds between two water molecules from one complex and two oxygen atoms of the 5-carboxylate group from an adjacent complex, resulting in homosynthons with graph-set notation R22(8). Dimers are linked into chains along the b axis by two lattice water molecules which form O–H⋯O hydrogen bonds between oxygen atoms from an N-oxide group and from an O atom from the 2-carboxylate group (see Fig. 7). The dimers repeat along a axis via O–H⋯O hydrogen bonds between a coordinated water molecule and oxygen atoms of 2-carboxylate group from pydco2− ligand forming one-dimensional ladders (see Fig. 8). These ladders are attached to each other via hydrogen bonding (C17H17⋯O4; 2.510 Å), π⋯π (with two different types of centroid⋯centroid separations of 3.627 and 3.671 Å), CH⋯π (with a centroid⋯H18C18 separation of 3.5325 Å), and C–O⋯π (with a centroid⋯O5C7 separation of 3.317 Å) interactions between pyridine and phenyl rings of phen and thereby generate two-dimensional sheets (Fig. S11 and S12†).
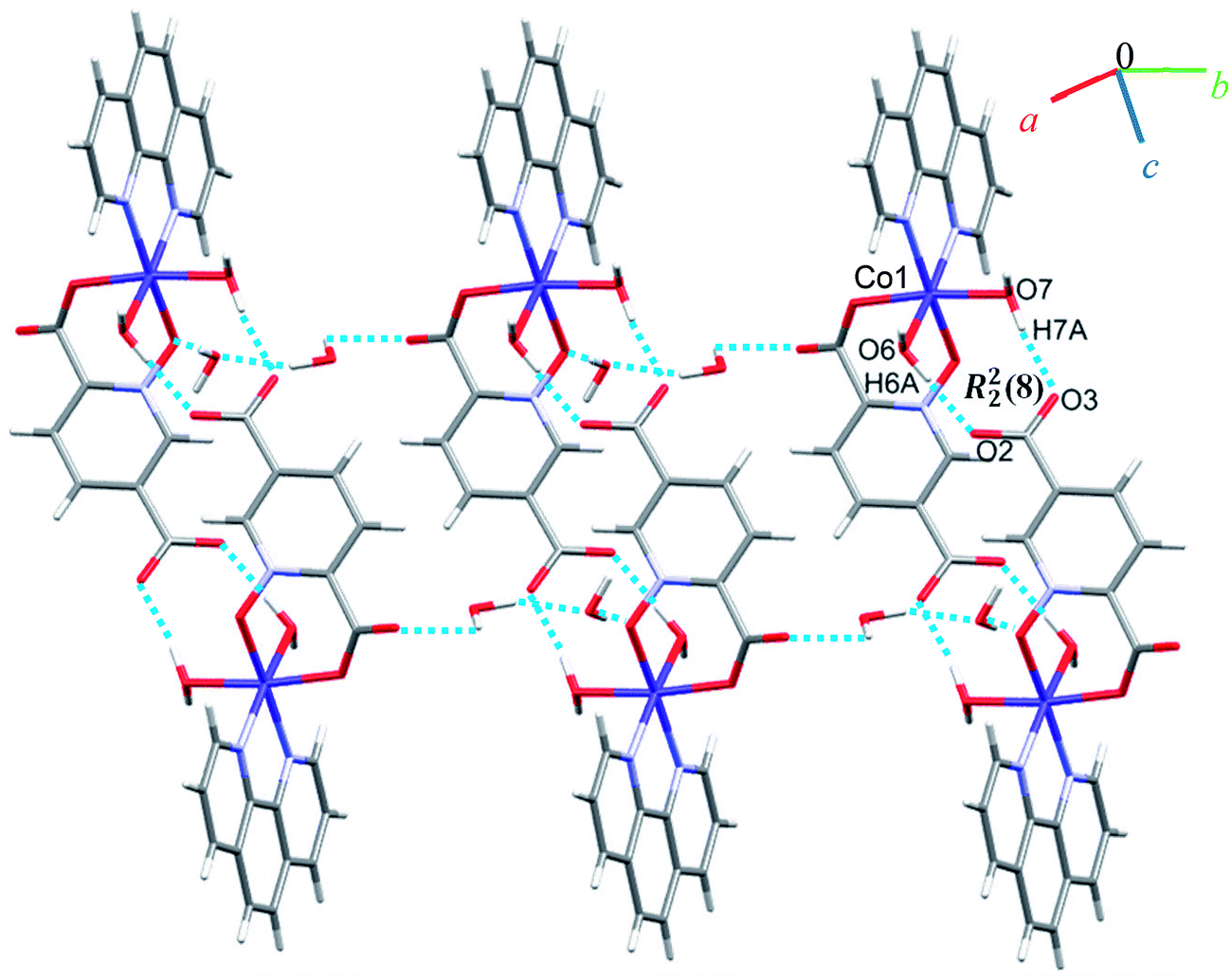 | ||
| Fig. 7 Representation of the chain along the b axis in 6. | ||
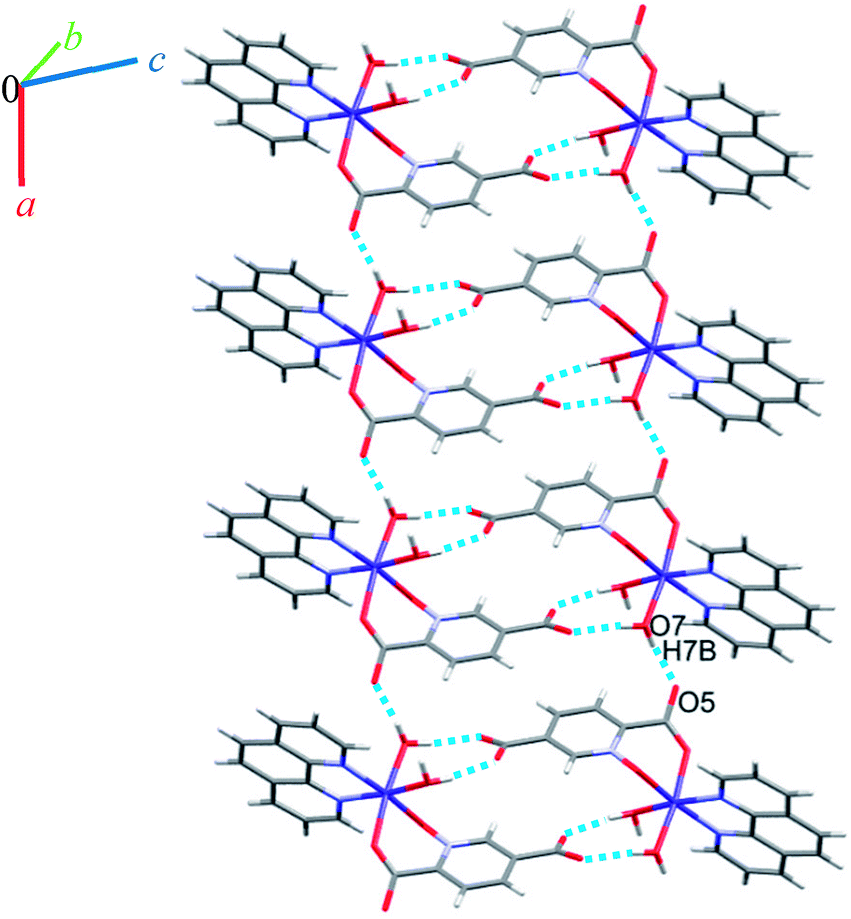 | ||
| Fig. 8 View of the ladder running parallel to the a axis in 6. | ||
Crystal structure of 7
Complex 7 crystallizes in the triclinic space group P. The asymmetric unit contains a five-coordinated complex of Cu(II) which adopts a distorted square pyramidal coordination geometry‡ in which the basal plane is occupied by two bipy nitrogen atoms and two oxygen atoms from one Hpydco− ligand, while the axial position is occupied by a chloride ion. The asymmetric unit is completed by two uncoordinated water molecules. A displacement ellipsoid plot of complex 7 with selected atoms labeling scheme is shown in Fig. 2. The chloride anions and uncoordinated water molecules participate in O–H⋯O and O–H⋯Cl hydrogen bonding and thereby form chains along the b axis with motifs described by the graph-set notation C22(8) (see Fig. 9).
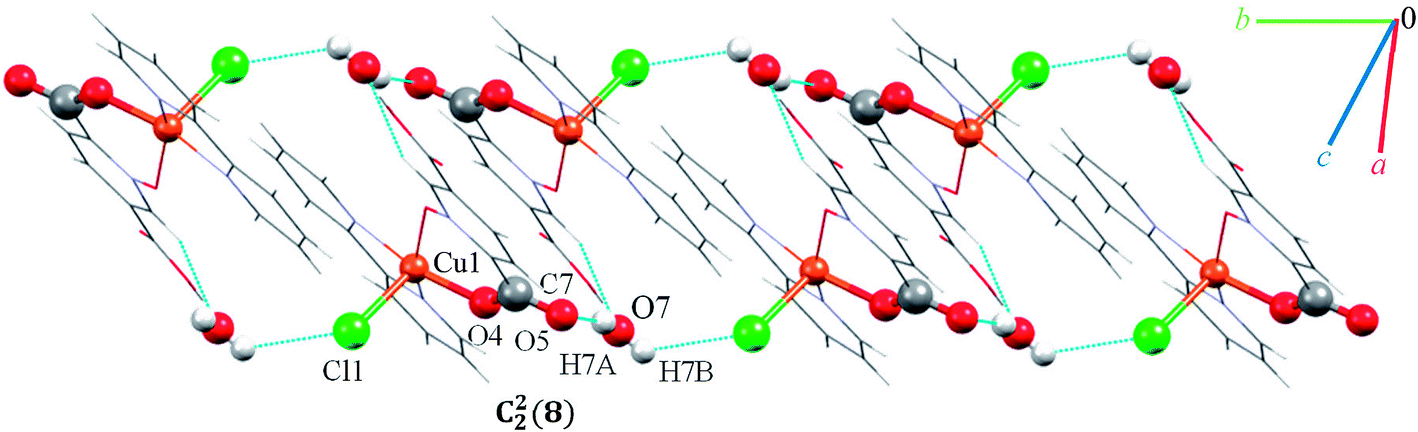 | ||
| Fig. 9 Representation of the chain running parallel to the b axis in 7. | ||
Another chain is generated through weak C–H⋯O, C–H⋯Cl, C–H⋯π (with a centroid⋯H5C5 separation of 3.535 Å), and π⋯π (with a centroid⋯centroid separations of 3.631 Å) interactions (see Fig. S13 in the ESI†). Moreover, the third dimension is generated via strong O–H⋯O hydrogen bonds between water molecules and 5-carboxylate groups of pydco2− which lead to the formation of a motif described by the graph-set notation R44(12) (see Fig. S14 in the ESI†). According to Fig. S15 in the ESI,† the three-dimensional structure of compound 7 has expanded via different interactions by connection of all above chains.
Crystal structure of 8
Single crystal X-ray diffraction analysis shows that compound 8 crystallizes in the triclinic space group P. A displacement ellipsoid drawing of 8 with selected atoms labelled is shown in Fig. 2. The asymmetric unit of 8 contains one Cu(II) ion, one bipy ligand, one Hpydco− anion, one chloride anion and one uncoordinated water molecule wherein hydroxyl proton H2 is shared between two O2 atoms with 50% occupancy.61 A distorted square pyramidal geometry, is defined by two nitrogen atoms from a bipy ligand and two oxygen atoms from a Hpydco− ligand forming the equatorial plane with a chloride anion in the axial position.
Molecules form zigzag chains via alternating chloride bridges between Cu(II) ions and O2H2⋯O2 hydrogen bonds (see Fig. 10).
 | ||
| Fig. 10 Representation of the zigzag chain along the side view in 8. | ||
(H6B⋯O6; 2.59(3) Å) and (C1H1⋯O6; 2.554 Å) hydrogen bonds between water molecules and C1 of pyridine ring from pydco2− ligand cross-ink the zigzag chains into two-dimensional sheets. These O–H⋯O hydrogen bonds lead to the formation of a motif with the graph-set notation R22(4). Weak π⋯π interactions with a centroid⋯centroid separations of 3.775 Å occur between the pyridine rings of two bipy ligands in adjacent chains (see Fig. S16†). The structure is extended parallel to a axis via weak C–H⋯O interactions between uncoordinated water molecules and the pyridine rings of pydco2− and bipy ligands (see Fig. S17†). Finally, covalent bonds and non-covalent interactions such as hydrogen bonds and π⋯π stacking in these three chains expand the structure to give a three-dimensional network (Fig. S18†).
Crystal structure of 9
Complex 9 is a coordination polymer that crystallizes in the triclinic space group P. The structure consists of a cationic complex [AgCu(H2O)2(phen)(pydco)]n+ with a nitrate as counterion (see Fig. 2). The asymmetric unit contains one Cu(II) and one Ag(I) cation, one pydco2− and one phen ligand and the aforementioned nitrate anion. Cu(II) (τ5 = 0.105) is five-coordinate with distorted square pyramidal geometry. For the Cu(II) center, the basal plane is defined by two oxygen atoms (O1 and O2) from one pydco2− ligand and two nitrogen atoms (N2 and N3) from a phen ligand, while the axial position is occupied by one oxygen atom (O6) of a coordinated water molecule. The Ag(I) complex forms an acetate-bridged dimer with an Ag⋯Ag separation of 2.886(1) Å. The coordination around the Ag(I) metal ion is four-coordinate (by O3, O7, O4 and O50) and its geometry is distorted tetrahedral. In 9, the pydco2− ligand is completely deprotonated and coordinated to metal ions as a pentadentate ligand. It is worth noting that pydco2− chelates to the Cu(II) ion through the oxygen atoms of the N-oxide (O1) and carboxylate (O2) while another oxygen of this carboxylate (O3) is coordinated to Ag. The oxygen atoms of the 5-carboxyl group coordinates in a bidentate fashion to Ag1. In this structure the free nitrate anion has a degree of disorder, perhaps because the data were acquired at room temperature, and no suitable modelling of this disorder could be achieved.
Monomer units form ladder parallel to the b axis via coordinate bonds (Ag1–O3) and two types of interactions increase the stability of these monomers: the first is O–H⋯O hydrogen bonding interactions between coordinated water molecules and nitrate ions and the second type is π⋯π stacking interactions between two pyridine rings of pydco2− ligands (with a centroid⋯centroid separation of 3.639 Å).
We identified cyclic synthons with graph-set notations R21(4), R11(10), R22(14) and R22(4) (see Fig. 11). Another one-dimensional chain was formed via weak interactions C13H13⋯O8 and C4H4⋯O9 between phenyl ring of phen, pyridine ring of pydco2− ligands and nitrate anion as well as weak π⋯π interactions between phenyl rings of two phen ligands (with a centroid⋯centroid separations of 3.535 Å) and C–H⋯π interactions (with a centroid⋯H13C13 separations of 3.617 Å) (see Fig. S19†). Molecules are linked into chains running parallel to the a axis via different types of interactions including O7–H7A⋯O8, C19H19⋯O10, C–H⋯π interactions (with centroid⋯H4C4 and centroid⋯H5C5 separations of 3.108 and 30124 Å, respectively) (see Fig. 12).
 | ||
| Fig. 11 Structural representations and graph-set notation for the ladder parallel to the b axis assembled via hydrogen bonding and π⋯π interactions in 9. The hydrogen bonds and π⋯π interactions are shown as blue dotted lines and gray dashed lines, respectively. | ||
 | ||
| Fig. 12 Representation of the chain in compound 9, assembled via O–H⋯O and C–H⋯O hydrogen bonding and C–H⋯π interactions, running parallel to the a axis. Distances are given in Å. The hydrogen bonds and C–H⋯π interactions are shown as blue and green dotted lines, respectively. | ||
Thus, a one-dimensional coordination polymer is linked by further interactions into a three-dimensional network (see Fig. S20†).
Comparisons
A search of the CSD (version 5.38, with updates until February 2017) for the pydco ligand returned no published examples of transition metal complexes coordinated to this ligand. Previously in our group we have synthesized a series of complexes with N/O donor ligand H2pydc62–65 and here we report the corresponding transition metal complexes with exclusively O-donor H2pydco ligand. The aim was to explore how altering the coordination mode of the ligand changes the molecular structures, non-covalent interactions and therefore the supramolecular structures of these complexes. The first-row transition metals are borderline acids and they have a greater affinity for nitrogen than for oxygen atoms. However, it could be anticipated that the presence of two donor oxygen atoms in the H2pydco ligand would lead to their chelating the metal ion and the formation of a six-membered ring. Indeed, if the ligand can assume a suitable conformation, the formation of six-membered ring causes little or no strain on the bond angles of the ligand or at the metal.66 We observed two coordination modes for the pydco ligand in our complexes (see Fig. 13).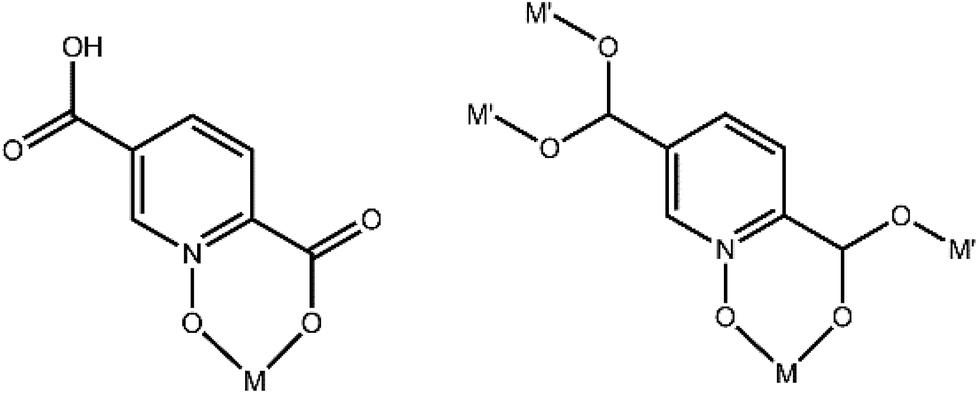 | ||
| Fig. 13 Hpydco− and pydco2− coordination modes observed in 1–9. | ||
Based on the CSD search, there are three complexes with the same structures as complexes 1, 3, and 6 in this work. In Table 2, M–ON-oxide, M–Ocoo and M–Npyridine distances are shown. Since complexes 2, 3, and 4 are isomorphous, a comparison was performed between corresponding M–ON-oxide and M–OCOO distances against the relevant ionic radii for Co(II), Ni(II), and Zn(II) (Table 2). Average bond distances for these three complexes show that M–OmeanN-oxide is longer than M–OmeanCOO.
| Complexes | M–NN-oxide | M–OCOO | M–Npyridin | Ref. |
|---|---|---|---|---|
| 1 | 2.018(3) | 2.032(4) | — | This work |
| 2 | 2.0991(17) | 2.0901(17) | — | This work |
| 3 | 2.0679(14) | 2.0507(14) | — | This work |
| 4 | 2.105(3) | 2.141(3) | — | This work |
| 5 | 2.101(2) | 2.083(2) | — | This work |
| 6 | 2.095(2) | 2.059(2) | — | This work |
| 7 | 1.957(4) | 1.922(3) | — | This work |
| 8 | 1.941(2) | 1.927(2) | — | This work |
| 9 | 1.932(3) | 1.907(4) | — | This work |
| [Co(H2O)6][Co(pydc)2(H2O)2]·4H2O | — | 2.075 | 2.136 | 67 |
| [Co(H2O)2(phen)(pydc)]·H2O | — | 2.064(2) | 2.159(2) | 68 |
| [Ni(pydc)(H2O)4]·2H2O | — | 2.048(1) | 2.070(1) | 69 |
The Cremer and Pople ring puckering parameters70,71 shown in Table 3 indicate that the six-membered chelating rings created by coordination of ligand to the metal ions adopt the following conformations:
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|
| q2 (Å) | 0.529(4), 0.529(4) | 0.7070(19), 0.7239(19) | 0.6996(17), 0.7033(17) | 0.733(3), 0.735(3) | 0.7629(17) | 0.7126(19) | 0.397(4) | 0.571(2) | 0.268(4) |
| q3 (Å) | −0.187(4), 0.187(4) | −0.1569(19), 0.1171(19) | −0.1582(17), 0.1288(17) | −0.122(3), 0.152(3) | 0.2048(17) | −0.2127(19) | −0.137(4) | −0.248(2) | 0.102(4) |
| Φ2 (°) | 143.7(5), 323.7(5) | 221.42(15), 46.12(15) | 220.86(14), 44.57(14) | 225.9(3), 42.2(3) | 37.01(13) | 215.20(15) | 214.3(6) | 204.8(2) | 40.4(9) |
| Θ2 (°) | 109.5(4), 70.5(4) | 102.52(15), 80.81(15) | 107.75(14), 79.63(14) | 99.4(2), 78.4(2) | 74.97(12) | 106.62(15) | 109.0(5) | 113.51(18) | 69.1(8) |
| Q (Å) | 0.561(4), 0.561(4) | 0.7239(17), 0.7330(18) | 0.7170(16), 0.7154(16) | 0.748(3), 0.751(3) | 0.7896(16) | 0.7437(17) | 0.420(3) | 0.6219(18) | 0.287(4) |
(i) 1 has two rings with half-chair and skew–boat conformations;
(ii) Each of 2, 3 and 4 complexes have two rings in which they have a skew–boat conformation in one ring. In another ring, they have state between skew–boat and boat conformations.
(iii) 5 and 6 each have one ring with a skew–boat conformation;
(iv) 7, 8, and 9 each have one ring with a half-chair conformation;
In Fig. 14 we compare the structures of compounds 1, 3, and 6 with previously synthesized complexes of H2pydc.67–69 The formation of 5-membered chelate rings by the H2pydc ligand frequently resulted in complexes with planar conformations, while the H2pydco ligand formed complexes with 6-membered chelate rings displaying twisted conformations. N-oxide functionalization of the pyridine ring of the ligand led to the formation of complex and interesting supramolecular frameworks.
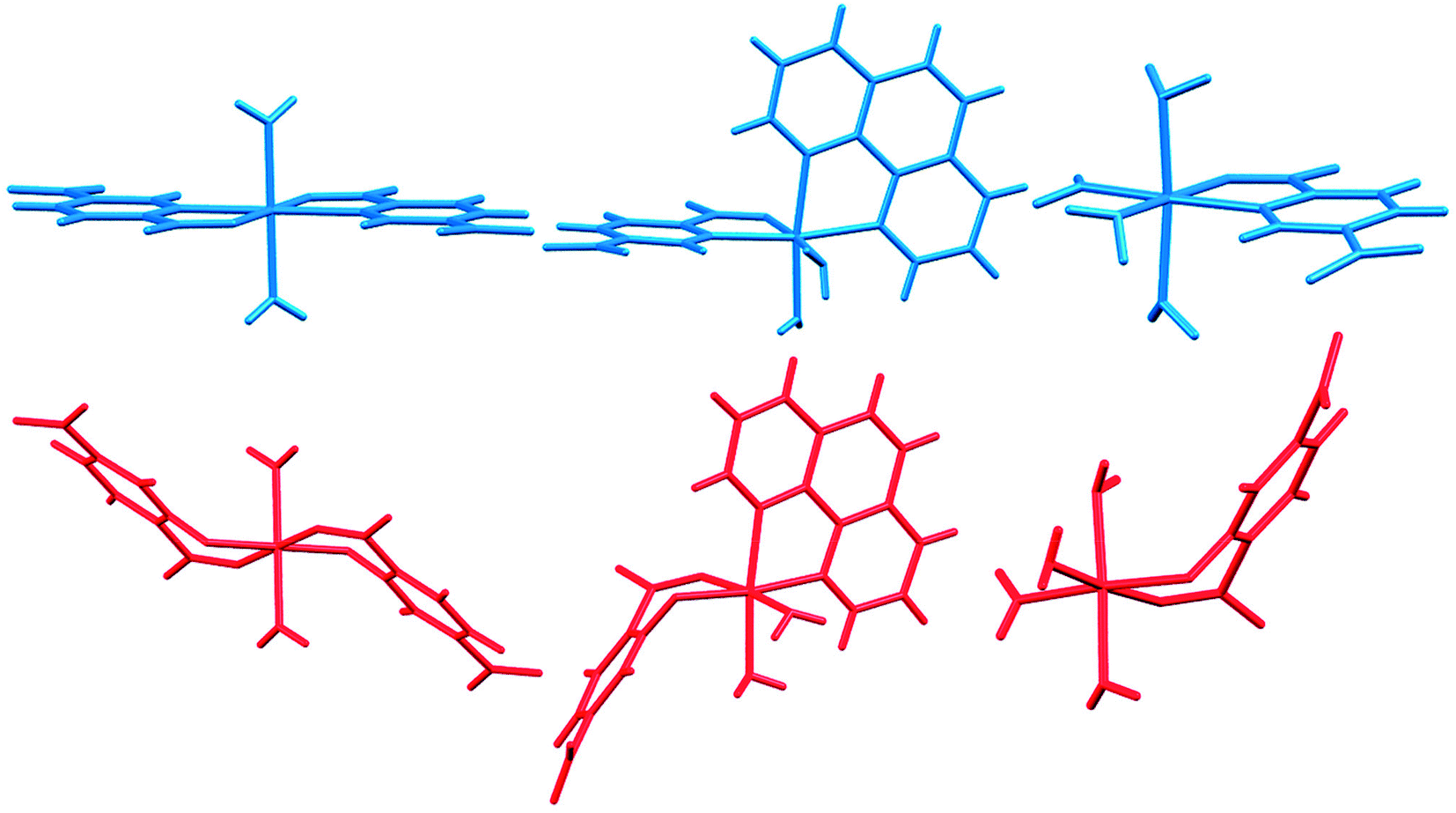 | ||
| Fig. 14 Comparison of the structures of 1, 3, and 6 (red) with complexes of the H2pydc ligand (blue). | ||
Theoretical study
The theoretical study is devoted to analyze some unconventional non-covalent interactions observed in the solid-state structures of compounds 2–4. In particular, we have estimated the energy associated to π–hole interactions that are established between the coordinated carboxylate group and the uncoordinated one, which is the π–hole donor. It should be mentioned that π–hole interactions in X-ray structures have been studied by Bürgi and Dunitz in 1975,72 thus revealing the trajectory along which a nucleophile attacks the π–hole of carbonyl group. More recently, the importance of n → π* interactions in proteins from a lone pair of electrons (n) to the antibonding orbital (π*) of carbonyl group has been demonstrated.73 Moreover, operative π-holes have been described nitroderivatives,74 group 13 molecules75 and acyl carbon containing molecules.76First of all, we have computed the molecular electrostatic potential (MEP) plotted onto the approximate van der Waals surface (isosurface 0.001 au) in order to investigate the electron rich and electron poor region of the complex. Since compounds 2–4 are isostructural, we have used compound 4 as a model because the closed-shell electronic configuration of this complex (d10 metal center) facilitates the computational analysis. The MEP surface of compound 4 is given in Fig. 14a and it can be observed the most positive region is located at the H-atoms of the coordinated water molecules (+99 kcal mol−1). This is due to the coordination of the water molecules to the metal center that increases the acidity of the H atoms. The most negative region is located at the O-atoms of the uncoordinated carboxylate group (−95 kcal mol−1), as expected. Therefore, the most favored interaction from an electrostatic point of view is an H-bond between the M − OH2 and the carboxylate group. As a matter of fact, this molecule forms self-assembled dimer in the solid state due to the formation of four strong H-bonds, as shown in Fig. 15b. The formation energy of this dimer is very large ΔE1 = −112.8 kcal mol−1, in good agreement with the MEP analysis and solid-state structure. In order to further characterize this assembly, we have used the NCI plot index computational tool. Non-covalent interactions are efficiently visualized and identified by using the NCI plot tool. It allows an easy assessment of host–guest complementarity and the extent to which weak interactions stabilize a complex. For the theoretical model of the assembly used in Fig. 15b we have computed the NCI plot that is represented in Fig. 15c. It can be observed several small and dark blue isosurfaces that characterize the intramolecular H-bonds that confirm the strong nature of these bonds.
 | ||
| Fig. 15 (a) MEP surface of compound 4. (b) H-bonded dimer of 4. Distances in Å. (c) NCI plot of the H-bonded dimer of compound 4. The gradient cut-off is s = 0.35 au, and the color scale is −0.04 < ρ < 0.04 au. | ||
It is important to highlight that the MEP surface of the monomer of compound 4 does not exhibits a positive π–hole over the carboxylate group, which is expected taking into consideration that the negative charge is located in this group. However, if the MEP surface is computed for the H-bonded dimer shown Fig. 15b, a small π–hole appears over the C atoms (see Fig. 16). The MEP surface shown in Fig. 16a also reveals a negative MEP at the O-atom of the coordinated carboxylate group (−55 kcal mol−1). The MEP over the C-atom of the carboxylate that establishes the double H-bond with the coordinated water molecules is very small +2.5 kcal mol−1 (see Fig. 16b). Nevertheless, it explains the formation of the infinite supramolecular chain in the solid state (see Fig. 16b) where the H-bonded self-assembled dimer interacts with the neighbor molecules in the X-ray structure by means of double π–hole interactions. Both O⋯C distances are shorter than the sum of their van der Waals radii (3.22 Å) and quite directional.
 | ||
| Fig. 16 (a) MEP surface of the dimer of compound 4. The MEP values at selected points of the surface are indicated in kcal mol−1 (b) partial view of the X-ray structure of compound 4. Distances in Å. | ||
We have evaluated energetically the π–hole complexes in complexes 2–4, see Fig. 17. It can be observed that the geometric features of the three complexes are almost identical and also the interaction energies, thus suggesting that the type of metal center has a little influence on the interaction energy. The interaction energies are weak (around 2.5 kcal mol−1 each O⋯π–hole) in good agreement with the small MEP value at the π–hole.
 | ||
| Fig. 17 Formation energies of the π–hole dimers in compounds 2–4. Distances in Å. | ||
We have also computed the NCI plots of the π–hole dimers represented in Fig. 18. The existence and weak attractive nature of the O⋯π–hole interactions is confirmed by the presence of green isosurfaces located between the O and C-atoms of carboxylate. Moreover, the NCI plot also reveals the existence of π–π stacking interactions since a more extended isosurface located between the aromatic ligands also appears upon complexation.
 | ||
| Fig. 18 NCI plots of the π–hole dimers of compounds 2 (a), 3 (b) and 4 (c). The gradient cut-off is s = 0.35 au, and the color scale is −0.04 < ρ < 0.04 au. | ||
Solution studies
In these experiments, the completely protonated forms of 2,5-pydco (L), 2,2′-bipyridine (Q) and 1,10-phenanthroline (Q′) were titrated with a standard NaOH solution in order to investigate their stoichiometry and protonation constants. The resulting values for the overall stability and stepwise protonation constants of L, Q and Q′ as well as the recognition constants for the L–Q and L–Q′ proton transfer systems are listed in Table 4. The resulting protonation constants are in satisfactory agreement with those reported in the literature; the observed difference is due to different conditions. The corresponding experimental pH titration profiles are shown in Fig. S21(a–c).† As can be seen, in all cases the potentiometric titration curves are depressed in the presence of the metal ions, indicating their strong interactions with metal ions. As it is obvious from Table 4, the most abundant proton transfer species for 2,5-pydco/2,2′-bipy system present at pH 2.3–2.6 (46.2%), and 3.7 (33.2%) are 2,5-pydcoH3—2,2′-bipy (logK = 7.72) and 2,5-pydcoH2—2,2′-bipy (logK = 3.93). In the 2,5-pydco/1,10-phen system the most abundant proton transfer species are 2,5-pydcoH2—1,10-phen (52.3%, logK = 10.47) and 2,5-pydcoH3—1,10-phen (34.0%, logK = 13.23).
| Stoichiometry | logβ |
Equilibrium quotient K | logK |
Max% | At pH | |||
|---|---|---|---|---|---|---|---|---|
| 2,5-pydco | 1,10-phen | 2,2′-bipy | h | |||||
| 1 | 0 | 0 | 1 | 4.47 | — | 4.47 | 96.2 | 3.7 |
| 1 | 0 | 0 | 2 | 7.38 | — | 2.91 | 40.3 | 2.0 |
| 1 | 0 | 0 | 3 | 8.13 | — | 0.75 | 22.1 | 2.0 |
| 0 | 1 | 0 | 1 | 4.54 | — | 4.54 | 87.5 | 2.0 |
| 0 | 1 | 0 | 2 | 8.76 | — | 4.22 | 17.9 | 2.0 |
| 0 | 0 | 1 | 1 | 2.01 | — | 2.01 | 44.0 | 2.0 |
| 0 | 0 | 1 | 2 | 3.49 | — | 1.48 | 13.1 | 2.0 |
| 1 | 1 | 0 | 1 | 13.52 | [2,5-pydcoH(1,10-phen)]/[2,5-pydcoH][1,10-phen] | 9.05 | 32.6 | 4.6 |
| 1 | 1 | 0 | 2 | 17.85 | [2,5-pydcoH2(1,10-phen)]/[2,5-pydcoH2][1,10-phen] | 10.47 | 52.3 | 4.0 |
| 1 | 1 | 0 | 3 | 21.36 | [2,5-pydcoH3(1,10-phen)]/[2,5-pydcoH3][1,10-phen] | 13.23 | 34.0 | 2.9–3.1 |
| 1 | 1 | 0 | 4 | 27.92 | [2,5-pydcoH3(1,10-phenH)]/[2,5-pydcoH3][1,10-phenH] | 15.25 | 25.7 | 2.5 |
| 2 | 1 | 0 | 3 | 32.14 | [2,5-pydcoH2(2,5-pydcoH)(1,10-phen)]/[2,5-pydcoH2][2,5-pydcoH][1,10-phen] | 20.29 | 16.6 | 4.3 |
| 1 | 2 | 0 | 4 | 34.37 | [2,5-pydcoH2(1,10-phenH)2]/[2,5-pydcoH2][1,10-phenH]2 | 17.91 | 8.9 | 4.8 |
| 1 | 0 | 1 | 0 | 9.24 | [2,5-pydco(2,2-bipy)]/[2,5-pydco][2,2-bipy] | — | 9.5 | 6.2–12.0 |
| 1 | 0 | 1 | 2 | 11.31 | [2,5-pydcoH2(2,2-bipy)]/[2,5-pydcoH2][2,2-bipy] | 3.93 | 33.2 | 3.7 |
| 1 | 0 | 1 | 3 | 15.85 | [2,5-pydcoH3(2,2-bipy)]/[2,5-pydcoH3][2,2-bipy] | 7.72 | 46.2 | 2.3–2.6 |
| 2 | 0 | 1 | 2 | 24.16 | [(2,5-pydcoH)2(2,2-bipy)]/[2,5-pydcoH]2[2,2-bipy] | 15.22 | 24.9 | 4.8 |
| 2 | 0 | 1 | 4 | 26.74 | [(2,5-pydcoH2)2(2,2-bipy)]/[2,5-pydcoH2]2[2,2-bipy] | 11.98 | 16.6 | 3.6 |
| 2 | 0 | 1 | 6 | 29.12 | [(2,5-pydcoH3)2(2,2-bipy)]/[2,5-pydcoH3]2[2,2-bipy] | 12.86 | 9.3 | 2.2 |
| 1 | 0 | 2 | 3 | 32.63 | [2,5-pydcoH3(2,2-bipy)2]/[2,5-pydcoH3][2,2-bipy]2 | 24.50 | 8.0 | 3.7 |
| 1 | 0 | 2 | 4 | 34.87 | [2,5-pydcoH2(2,2-bipyH)2]/[2,5-pydcoH2][2,2-bipyH]2 | 23.47 | 3.6 | 2.1 |
The pH titration data in the absence of metal ions were used to get the protonation constants for L and Q (KnH [HmL]/[H(m−n)L][H]n, the charges are omitted for simplicity) via the program BEST.49 The corresponding distribution diagrams for 2,5-pydco (a), 2,2′-bipyridine (b), 1,10-phenanthroline (c), 2,5-pydco/2,2′-bipyridine (d), 2,5-pydco/1,10-phenanthroline (e), 2,5-pydco/Co2+ [1:2] (f), 2,5-pydco/Co2+ [1:1] (g), 2,5-pydco/Ni2+ (h), 2,5-pydco/Zn2+ (i), 2,2′-bipyridine/Co2+ (j), 2,5-pydco/2,2′-bipyridine/Co2+ (k), 1,10-phenanthroline/Co2+ (l), 2,5-pydco/1,10-phenanthroline/Co2+ (m), 2,5-pydco/Cu2+ (n), 2,2′-bipyridine/Cu2+ (o), 2,5-pydco/2,2′-bipyridine/Cu2+ (p), 1,10-phenanthroline/Cu2+ (q), 2,5-pydco/Ag+ (r), 1,10-phenanthroline/Ag+ (s), 2,5-pydco/1,10-phenanthroline/Ag+/Cu2+ (t) are shown in Fig. 19 (see also Table 5).
 | ||
| Fig. 19 (contd.) | ||
| System | m | n | l | q | h | cl | logβ |
Max% | At pH |
|---|---|---|---|---|---|---|---|---|---|
| Co2+–2,5-pydco [1:2] |
1 | 0 | 2 | 0 | 0 | 17.23 | 49.9 | 6.5–6.9 | |
| 1 | 0 | 2 | 0 | 2 | 19.74 | 39.4 | 2.5 | ||
| 1 | 0 | 2 | 0 | −2 | −2.16 | 65.3 | 11.1–12.0 | ||
| 2 | 0 | 2 | 0 | 0 | 23.84 | 21.0 | 6.7 | ||
| 2 | 0 | 2 | 0 | 1 | 25.28 | 29.0 | 3.9 | ||
| 2 | 0 | 4 | 0 | 0 | 29.97 | 8.9 | 6.8 | ||
| 2 | 0 | 4 | 0 | 2 | 31.10 | 24.9 | 4.3 | ||
| 2 | 0 | 4 | 0 | −4 | 7.02 | 23.4 | 10.3 | ||
| 3 | 0 | 2 | 0 | 0 | 33.76 | 13.9 | 7.0–7.3 | ||
| 3 | 0 | 6 | 0 | −6 | 3.12 | 12.4 | 11.4–12.0 | ||
| Co2+–2,5-pydco [1:1] |
1 | 0 | 1 | 0 | 0 | 10.49 | 15.7 | 5.3 | |
| 1 | 0 | 1 | 0 | 1 | 13.21 | 13.9 | 2.8 | ||
| 1 | 0 | 1 | 0 | −4 | −21.05 | 44.7 | 10.3 | ||
| 2 | 0 | 2 | 0 | 0 | 23.79 | 24.4 | 6.6 | ||
| 2 | 0 | 2 | 0 | 1 | 25.30 | 29 | 3.9 | ||
| 2 | 0 | 2 | 0 | −6 | 1.94 | 6.2 | 11.1–12.0 | ||
| 2 | 0 | 2 | 0 | −8 | −9.62 | 33.1 | 10.8–11.2 | ||
| 3 | 0 | 3 | 0 | 0 | 34.98 | 20.7 | 6.9 | ||
| 3 | 0 | 3 | 0 | −12 | −28.96 | 18.3 | 11.5–12.0 | ||
| Ni2+–2,5-pydco | 1 | 0 | 1 | 0 | 0 | 11.01 | 15.1 | 5.0 | |
| 1 | 0 | 1 | 0 | 1 | 13.85 | 13.9 | 3.1 | ||
| 1 | 0 | 1 | 0 | −4 | −19.96 | 48.1 | 9.9–12.0 | ||
| 2 | 0 | 2 | 0 | 0 | 23.04 | 33.2 | 6.4 | ||
| 2 | 0 | 2 | 0 | 1 | 25.00 | 23.3 | 4.3 | ||
| 2 | 0 | 2 | 0 | −4 | 3.11 | 19.4 | 10.4–12.0 | ||
| 2 | 0 | 2 | 0 | −6 | −2.02 | 5.9 | 11.2–12.0 | ||
| 2 | 0 | 2 | 0 | −8 | −10.17 | 29.8 | 117–12.0 | ||
| 3 | 0 | 3 | 0 | 0 | 34.82 | 15.9 | 6.3–6.5 | ||
| 3 | 0 | 3 | 0 | −12 | −27.84 | 9.5 | 10.8–12.0 | ||
| Zn2+–2,5-pydco | 1 | 0 | 1 | 0 | 0 | 11.92 | 33.2 | 6.1 | |
| 1 | 0 | 1 | 0 | 1 | 13.97 | 36.0 | 2.8 | ||
| 1 | 0 | 1 | 0 | −4 | −18.46 | 58.4 | 10.2–12.0 | ||
| 2 | 0 | 2 | 0 | 0 | 23.99 | 40.5 | 5.4 | ||
| 2 | 0 | 2 | 0 | −4 | 2.89 | 10.6 | 9.8–12.0 | ||
| 2 | 0 | 2 | 0 | −6 | −3.14 | 4.2 | 8.0–12.0 | ||
| 2 | 0 | 2 | 0 | −8 | −10.98 | 27.0 | 12.0 | ||
| 3 | 0 | 3 | 0 | 0 | 34.25 | 15.8 | 6.2 | ||
| Co2+–2,2-bipyridine | 1 | 0 | 0 | 1 | 0 | 12.91 | 20.0 | 5.4 | |
| 1 | 0 | 0 | 1 | −4 | −16.59 | 30.9 | 10.8–11.2 | ||
| 1 | 0 | 0 | 2 | 0 | 20.88 | 32.6 | 4.5–4.9 | ||
| 1 | 0 | 0 | 2 | −2 | −0.63 | 61.4 | 9.6 | ||
| 2 | 0 | 0 | 3 | −6 | 2.39 | 9.5 | 11.2 | ||
| 2 | 0 | 0 | 4 | −4 | 8.56 | 22.4 | 12.0 | ||
| Co2+–2,5-pydco–2,2-bipyridine | 1 | 0 | 1 | 1 | 0 | 22.53 | 76.8 | 6.3 | |
| 1 | 0 | 1 | 1 | 1 | 25.18 | 57.9 | 2.7 | ||
| 1 | 0 | 1 | 1 | −2 | 6.01 | 55.7 | 10.8–12.0 | ||
| 2 | 0 | 2 | 2 | −4 | 8.47 | 32.5 | 11.4–12.0 | ||
| 3 | 0 | 3 | 3 | −6 | 10.35 | 14.5 | 11.7–12.0 | ||
| Co2+–1,10-phenanthroline | 1 | 0 | 0 | 1 | 0 | 13.89 | 31.1 | 4.1–4.3 | |
| 1 | 0 | 0 | 1 | −4 | −18.03 | 41.6 | 12.0 | ||
| 1 | 0 | 0 | 2 | 0 | 25.46 | 74.7 | 6.0 | ||
| 1 | 0 | 0 | 2 | −2 | −2.38 | 63.3 | 10.0–12.0 | ||
| Co2+–2,5-pydco–1,10-phenanthroline | 1 | 0 | 1 | 1 | 0 | 23.15 | 64.9 | 6.2 | |
| 1 | 0 | 1 | 1 | 1 | 25.07 | 42.5 | 3.2 | ||
| 1 | 0 | 1 | 1 | −2 | 4.52 | 65.4 | 10.9–12.0 | ||
| 2 | 0 | 2 | 2 | −4 | 6.31 | 20.6 | 11.4–12.0 | ||
| 3 | 0 | 3 | 3 | −6 | 7.95 | 13.9 | 11.8–12.0 | ||
| Cu2+–2,5-pydco | 1 | 0 | 1 | 0 | −2 | 1 | 2.46 | 39.0 | 10.4–10.7 |
| 1 | 0 | 2 | 0 | 0 | 1 | 28.83 | 48.4 | 6.5 | |
| 1 | 0 | 2 | 0 | −1 | 0 | 13.41 | 46.4 | 8.4 | |
| 1 | 0 | 2 | 0 | 2 | 1 | 31.60 | 40.9 | 3.9 | |
| 2 | 0 | 2 | 0 | −4 | 2 | 11.43 | 27.5 | 12.0 | |
| 2 | 0 | 4 | 0 | 0 | 2 | 34.27 | 29.5 | 6.4–6.6 | |
| 2 | 0 | 4 | 0 | 4 | 2 | 39.76 | 31.3 | 2.7 | |
| 3 | 0 | 6 | 0 | 0 | 3 | 46.01 | 19.7 | 6.7 | |
| 3 | 0 | 6 | 0 | 6 | 3 | 48.89 | 20.9 | 2.0 | |
| 4 | 0 | 8 | 0 | 0 | 4 | 52.93 | 12.0 | 6.7 | |
| 4 | 0 | 8 | 0 | 8 | 4 | 55.02 | 13.1 | 2.0 | |
| Cu2+–2,2-bipyridine | 1 | 0 | 0 | 1 | −3 | 0 | −10.71 | 30.9 | 10.8–11.2 |
| 1 | 0 | 0 | 1 | −2 | 1 | −4.63 | 61.4 | 9.0 | |
| 1 | 0 | 0 | 2 | 0 | 1 | 29.58 | 50.2 | 5.1 | |
| 2 | 0 | 0 | 2 | −4 | 2 | 13.03 | 20.6 | 9.0–9.3 | |
| 2 | 0 | 0 | 4 | 0 | 2 | 36.82 | 35.1 | 3.6 | |
| 3 | 0 | 0 | 6 | 0 | 3 | 47.12 | 29.2 | 4.8 | |
| Cu2+–2,5-pydco–2,2-bipyridine | 1 | 0 | 1 | 1 | 0 | 1 | 32.69 | 52.3 | 7.2 |
| 1 | 0 | 1 | 1 | 1 | 1 | 33.88 | 54.2 | 2.7 | |
| 2 | 0 | 2 | 2 | 0 | 2 | 41.86 | 35.0 | 9.7–12.0 | |
| 2 | 0 | 2 | 2 | 2 | 2 | 43.94 | 27.4 | 2.4–2.6 | |
| 3 | 0 | 3 | 3 | 0 | 3 | 49.97 | 22.9 | 10.3–12.0 | |
| 3 | 0 | 3 | 3 | 3 | 3 | 52.35 | 19.9 | 2.0 | |
| 4 | 0 | 4 | 4 | 0 | 4 | 54.71 | 14.3 | 8.9 | |
| 4 | 0 | 4 | 4 | 4 | 4 | 57.99 | 11.4 | 2.0 | |
| Cu2+–1,10-phenanthroline | 1 | 0 | 0 | 1 | −3 | 0 | −10.83 | 30.9 | 10.8–11.2 |
| 1 | 0 | 0 | 1 | −2 | 1 | −3.29 | 26.3 | 10.1 | |
| 1 | 0 | 0 | 2 | −1 | 0 | 12.74 | 47.6 | 9.7–12.0 | |
| 1 | 0 | 0 | 2 | 0 | 1 | 29.96 | 57.1 | 5.1 | |
| 2 | 0 | 0 | 4 | 0 | 2 | 37.21 | 34.9 | 4.8 | |
| 3 | 0 | 0 | 6 | 0 | 3 | 47.48 | 11.6 | 5.3 | |
| Ag+–2,5-pydco | 0 | 1 | 1 | 0 | −3 | −11.26 | 30.4 | 8.3 | |
| 0 | 1 | 2 | 0 | −1 | 12.50 | 41.1 | 7.9–8.1 | ||
| 0 | 2 | 2 | 0 | 0 | 25.11 | 32.5 | 5.0–5.2 | ||
| 0 | 2 | 2 | 0 | 2 | 27.19 | 24.6 | 3.0 | ||
| 0 | 2 | 2 | 0 | −4 | −1.04 | 27.7 | 12.0 | ||
| 0 | 2 | 4 | 0 | 0 | 32.81 | 55.2 | 5.6 | ||
| 0 | 2 | 4 | 0 | 4 | 36.09 | 65.3 | 2.0 | ||
| 0 | 3 | 3 | 0 | 0 | 39.98 | 12.0 | 6.0 | ||
| 0 | 3 | 3 | 0 | 3 | 42.19 | 10.1 | 2.3 | ||
| 0 | 3 | 3 | 0 | −5 | 9.67 | 21.2 | 9.3 | ||
| 0 | 4 | 1 | 0 | −15 | −18.34 | 5.1 | 12.0 | ||
| 0 | 4 | 2 | 0 | 0 | 41.13 | 15.3 | 6.2 | ||
| 0 | 4 | 2 | 0 | −12 | −12.69 | 11.0 | 12.0 | ||
| 0 | 4 | 4 | 0 | −7 | 8.36 | 6.2 | 9.8 | ||
| Ag+–1,10-phenanthroline | 0 | 1 | 0 | 1 | −3 | −14.08 | 45.6 | 9.4 | |
| 0 | 1 | 0 | 2 | −1 | 13.77 | 67.8 | 7.3 | ||
| 0 | 2 | 0 | 2 | 0 | 27.06 | 51.9 | 4.0 | ||
| 0 | 2 | 0 | 2 | −4 | −0.79 | 45.0 | 12.0 | ||
| 0 | 2 | 0 | 4 | 0 | 33.23 | 42.5 | 4.5 | ||
| 0 | 3 | 0 | 4 | −3 | 17.18 | 35.6 | 11.6–12.0 | ||
| 0 | 3 | 0 | 5 | −1 | 29.19 | 13.9 | 7.8 | ||
| Ag+–Cu2+–2,5-pydco–1,10-phenanthroline | 1 | 1 | 1 | 1 | 0 | 39.83 | 45.3 | 3.7 | |
| 1 | 1 | 1 | 1 | 0 | 1 | 44.98 | 6.6 | 3.7 | |
| 1 | 1 | 1 | 1 | −2 | 18.06 | 42.3 | 9.6 | ||
| 1 | 1 | 1 | 1 | −1 | 1 | 20.17 | 33.2 | 8.3 | |
| 2 | 2 | 2 | 2 | 0 | 52.14 | 30.4 | 3.5–3.9 | ||
| 2 | 2 | 2 | 2 | −4 | 19.92 | 26.1 | 9.9 | ||
| 3 | 3 | 3 | 3 | 0 | 59.79 | 17.4 | 3.9 | ||
| 3 | 3 | 3 | 3 | −6 | 21.06 | 20.6 | 11.1–12.0 | ||
| 4 | 4 | 4 | 4 | 0 | 63.18 | 10.2 | 3.9 | ||
| 4 | 4 | 4 | 4 | −8 | 25.89 | 11.4 | 11.3–12.0 | ||
| 1 | 3 | 1 | 1 | 0 | 48.72 | 20.3 | 6.7 | ||
| 1 | 3 | 1 | 1 | −10 | 14.8 | 5.4 | 11.5–12.0 | ||
| 2 | 5 | 2 | 2 | 0 | 59.99 | 11.1 | 7.0 | ||
| 2 | 5 | 2 | 2 | −15 | 9.87 | 3.0 | 11.4 | ||
| 3 | 7 | 3 | 3 | 0 | 65.24 | 6.2 | 6.9 | ||
| 3 | 7 | 3 | 3 | −19 | 5.82 | Neg. | 12.0 | ||
As it is obvious from Table 5, the main species in the Co2+ (M) with 2,5-pydco (L) binary system (Fig. 19f and g) are ML2(OH)2 (at pH 11.1–12.0; 65.3%), ML2 (at pH 6.5–6.9; 49.9%), ML(OH)4 (at pH 10.3; 44.7%), ML2H2 39.4% at pH 2.5 and M2L2(OH)8 at pH 10.8–11.2 (33.1%). In the case of Ni2+ (M) with 2,5-pydco (L) in the binary system, the most abundant species are ML(OH)4 existed at pH 9.9–12.0 by an extent of 48.1% and M2L2 (33.2% at pH 6.4). For the Zn2+—2,5-pydco binary system (Fig. 19i), ML(OH)4 (58.4% at pH 10.2–12.0), M2L2 (40.5% at pH 5.4) and MLH (36.0% at pH 2.8) are the most abundant species. For the Co2+ (M) with 2,2′-bipy (Q) in the binary system (Fig. 19j), the main species are MQ2(OH)2 at pH 9.6 by an extent of 61.4% and MQ2 with an extent of 32.6% existed at pH 4.5–4.9. In the Co2+ (M) with 1,10-phen (Q′) for the binary system, the most abundant species are MQ2 existed at pH 6.0 by an extent of 74.7% and MQ2(OH)2 (63.3% at pH 10.0–12.0). In the case of Co2+ with L and Q in the ternary complexes system, the most abundant species are MLQ existed at pH 6.3 by an extent of 76.8%, MLQH (57.9% at pH 2.7) and MLQ(OH)2 at pH 10.8–12.0 with 55.7%. For the Co2+ with L and Q′ in the ternary complexes system, the major species are MLQ(OH)2 existed at pH 10.9–12.0 by an extent of 65.4%, MLQ at pH 6.2 by an extent of 64.9% and MLQH (42.5% at pH 3.2).
In the case of Cu2+ (M) with 2,5-pydco (L) in the binary system, the most abundant species are ML2Cl existed at pH 6.5 by an extent of 48.4%, ML2OH (46.4% at pH 8.4), ML2H2Cl (40.9% at pH 3.9) and ML(OH)2Cl (39.0% at pH 10.4–10.7). For the Cu2+ (M) with 2,2′-bipy (Q) in the binary system, the main species are MQ (OH)2Cl at pH 9.0 by an extent of 61.4%, MQ2Cl with an extent of 50.2% existed at pH 5.1 and M2Q4Cl2 (35.1% at pH 3.6). In the Cu2+ (M) with 1,10-phen (Q′) in the binary system, the major species are MQ2Cl (57.1% at pH 5.1), MQ2OH (47.6% at pH 9.7–12.0) and M2Q4Cl2 (34.9% at pH 4.8). In the case of Cu2+ with L and Q in the ternary complexes system, the most abundant species are MLQHCl at pH 2.7 by an extent of 54.2%, MLQCl (52.3% at pH 7.2) and M2L2Q2Cl2 at pH 9.7–12.0 with 35.0%. In the case of Ag+–2,5-pydco binary system, the main species are N2L4H4 at pH 2.0 by an extent of 65.3% N2L4 (55.2% at pH 5.6) and NL2OH with an extent of 41.1% existed at pH 7.9–8.1. In the Ag+ (M) with 1,10-phen (Q′) in the binary system, the major species are NQ2OH (67.8% at pH 7.3), N2Q2 (51.9% at pH 4.0), NQ(OH)3 (45.6% at pH 9.4), N2Q2(OH)4 (45.0% at pH 12.0) and N2Q4 (42.5% at pH 4.5). In the case of Ag+–Cu2+–2,5-pydco–1,10-phenanthroline quaternary system (M/N/L/Q), the most abundant species are MNLQ (45.3% at pH 3.7), MNLQ(OH)2 (42.3% at pH 9.6), MNLQ(OH)Cl (33.2% at pH 8.3), M2N2L2Q2 (30.4% at pH 3.5–3.9) and M2N2L2Q2(OH)4 (26.1% at pH 9.9). A comparison between the stoichiometry of the crystalline complexes and that of the most abundant species detected in solution revealed that they are very similar to those reported for the corresponding isolated complexes in the solid state.
Conclusions
In summary, we have successfully synthesized and modified the H2pydco ligand from the N/O-donor ligand H2pydc in high yield by a new procedure to create a new O-only donor set than subsequent synthesis of the chlorinated derivative of this ligand. The results indicate that altering the donor set of the ligand profoundly affects the molecular and supramolecular structures of the corresponding transition metal complexes. The H2pydco ligand exhibits exceptional abilities to link metallic centers via its versatile chelating and bridging coordination modes. In this regard, we synthesized nine complexes with different architectures. In these complexes, H2pydco acts as a bidentate ligand through one oxygen atom of the carboxylate group and one oxygen atom of the N-oxide group. The formation of 5-membered chelate rings by the H2pydc ligand frequently resulted in complexes with planar conformations, while the H2pydco ligand formed complexes with 6-membered chelate rings displaying twisted conformations. N-oxide functionalization of the pyridine ring of the ligand led to the formation of complex and interesting supramolecular frameworks. The present study demonstrates that in these complexes cooperativity between various strong hydrogen bonds and a range of relatively weak π⋯π, C–H⋯π and C–O⋯π interactions is necessary for the construction of the supramolecular frameworks. DFT studies combined with MEP and NCI plot computational tools have been used to characterize and rationalize the O⋯π–hole interaction that is energetically very weak due to the small π–hole over the carboxylate group. Such π–hole appears due to the participation of the COO− group in strong H-bonding interactions. The formation of binary, ternary and quaternary complexes in solution with stoichiometries very close to those of the solid state is strongly supported by the results of the potentiometric pH titration studies in aqueous solutions and the stoichiometry of the most abundant species in the solution was very similar to the corresponding crystalline complexes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MM gratefully acknowledges the financial support by the Ferdowsi University of Mashhad, Mashhad, Iran (Grant Nos. 31817/3, 31843/3, and 31848/3). MM also thanks the Cambridge Crystallographic Data Centre (CCDC) for access to the Cambridge Structural Database. AF thanks the MINECO/AEI from Spain for financial support (project CTQ2017-85821-R FEDER funds). We thank Professor Alexander J. Blake, University of Nottingham, UK for helpful discussions.References
- M. Mirzaei, H. Eshtiagh-Hosseini, A. Bauza, S. Zarghami, P. Ballester, J. T. Mague and A. Frontera, CrystEngComm, 2014, 16, 6149–6158 RSC
.
- M. Mirzaei, H. Eshtiagh-Hosseini, M. Chahkandi, N. Alfi, A. Shokrollahi, N. Shokrollahi and A. Janiak, CrystEngComm, 2012, 14, 8468–8484 RSC
- D. Dogan, A. T. Colak, O. Sahin, T. Tunc and O. Celik, Polyhedron, 2015, 93, 37–45 CrossRef CAS
- J. M. Lehn, Supramolecular chemistry: Concepts and perspectives, VCH, Weinheim, 1995 Search PubMed
- D. Wang, Z. F. Tian, F. Wang, L. L. Wen and D. F. Li, J. Inorg. Organomet. Polym., 2009, 19, 196–201 CrossRef CAS
- M. Mirzaei, H. Aghabozorg and H. Eshtiagh-Hosseini, J. Iran. Chem. Soc., 2011, 8, 580–607 CrossRef CAS
- G. S. Fink, L. G. Cuervo, B. Therrien, H. S. Evans and G. B. Shulpin, Inorg. Chim. Acta, 2004, 357, 475–484 CrossRef
- A. T. Colak, F. Colak, D. Akduman, O. Z. Yesilel and O. Buyukgungor, Solid State Sci., 2009, 11, 1908–1918 CrossRef CAS
- T. S. Kamatchi, N. Chitrapriya, H. Lee, C. F. Fronczek, F. R. Fronczekc and K. Natarajan, Dalton Trans., 2012, 41, 2066–2077 RSC
- E. Kita, H. Marai and K. Zajac, Transition Met. Chem., 2008, 33, 211–217 CrossRef CAS
- L. P. Sun, S. Y. Niu, J. Jin, G. D. Yang and L. Ye, Eur. J. Inorg. Chem., 2006, 2006, 5130–5137 CrossRef
- D. J. Zhang, T. Y. Song, L. Wang, J. Shi, J. N. Xu, Y. Wang, K. R. Ma, W. R. Yin, L. R. Zhang and Y. Fan, Inorg. Chim. Acta, 2009, 362, 299–302 CrossRef CAS
- F. A. Mautner, J. H. Albering, R. Vicente, C. Andrepont, J. G. Gautreaux, A. A. Gallo and S. S. Massoud, Polyhedron, 2013, 54, 158–163 CrossRef CAS
- A. C. Wibowo, S. A. Vaughn, M. D. Smith and H. C. zur Loye, Inorg. Chem., 2010, 49, 11001–11008 CrossRef CAS PubMed
- P. Mahata and S. Natarajan, Eur. J. Inorg. Chem., 2005, 2005, 2156–2163 CrossRef
- M. Mirzaei, H. Eshtiagh-Hosseini, Z. Karrabi, K. Molcanov, E. Eydizadeh, J. T. Mague, A. Bauza and A. Frontera, CrystEngComm, 2014, 16, 5352–5363 RSC
- M. Mirzaei, H. Eshtiagh-Hosseini, M. Shamsipur, M. Saeedi, M. Ardalani, A. Bauza, J. T. Mague, A. Frontera and M. Habibi, RSC Adv., 2015, 5, 72923–72936 RSC
- M. Shahbazi, F. Mehrzad, M. Mirzaei, H. Eshtiagh-Hosseini, J. T. Mague, M. Ardalani and M. Shamsipur, Inorg. Chim. Acta, 2017, 458, 84–96 CrossRef CAS
- M. Mirzaei, H. Eshtiagh-Hosseini, M. Bazargan, F. Mehrzad, M. Shahbazi, J. T. Mague, A. Bauza and A. Frontera, Inorg. Chim. Acta, 2015, 438, 135–145 CrossRef CAS
- M. Bazargan, M. Mirzaei, H. Eshtiagh-Hosseini, J. T. Mague, A. Bauza and A. Frontera, Inorg. Chim. Acta, 2016, 449, 44–51 CrossRef CAS
- L. Xue, F. Luo, Y. X. Che and J. M. Zheng, J. Mol. Struct., 2007, 832, 132–137 CrossRef CAS
- K. Shankar, B. Das and J. B. Baruah, RSC Adv., 2013, 3, 26220–26228 RSC
- Y. Wei, H. Hou, L. Li, Y. Fan and Y. Zhu, Cryst. Growth Des., 2005, 5, 1405–1413 CrossRef CAS
- S. L. Huang, L. Zhang, Y. J. Lin and G. X. Jin, CrystEngComm, 2013, 15, 78–85 RSC
- L. L. Wen, Z. D. Lu, X. M. Ren, C. Y. Duan, Q. J. Meng and S. Gao, Cryst. Growth Des., 2009, 9, 227–238 CrossRef CAS
- L. L. Wen, D. B. Dang, C. Y. Duan, Y. Z. Li, Z. F. Tian and Q. J. Meng, Inorg. Chem., 2005, 44, 7161–7170 CrossRef CAS PubMed
- H. L. Sun, X. L. Wang, L. Jia, W. Cao, K. Z. Wang and M. Du, CrystEngComm, 2012, 14, 512–518 RSC
- Y. Xiong, Y. Z. Fan, R. Yang, S. Chen, M. Pan, J. J. Jiang and C. Y. Su, Chem. Commun., 2014, 50, 14631–14634 RSC
- J. Balzarini, M. Stevens, E. De Clercq, D. Schols and C. Pannecouque, J. Antimicrob. Chemother., 2005, 55, 135–138 CrossRef CAS PubMed
- S. Lis, Z. Hnatejko, P. Barczynski and M. Elbanowski, J. Alloys Compd., 2002, 344, 70–74 CrossRef CAS
- X–AREA: Program for the Acquisition and Analysis of Data, Version 1.30, Stoe & Cie GmbH, Darmstadt, Germany, 2005 Search PubMed
- X–RED: Program for Data Reduction and Absorption Correction, Version 1.28b, Stoe & Cie GmbH, Darmstadt, Germany, 2005 Search PubMed
- X–SHAPE: Program for Crystal Optimization for Numerical Absorption Correction, Version 2.05, Stoe & Cie GmbH, Darmstadt, Germany, 2004 Search PubMed
- G. M. Sheldrick, SHELXS97. Program for Crystal Structure Solution, University of Göttingen, Germany, 1997 Search PubMed
- G. M. Sheldrick, SHELXL97. Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed
- E. Prince and A. J. C. Wilson, International Tables for X-ray Crystallography, vol. C, 1995 Search PubMed
- X-STEP32: Crystallographic Package, Version 1.07b, Stoe & Cie GmbH, Darmstadt, Germany, 2000 Search PubMed
- A. T. Çolak, F. Çolak, O. Z. Yeşilel, D. Akduman, F. Yılmaz and M. Tümer, Inorg. Chim. Acta, 2010, 363, 2149–2162 CrossRef
- S. Sheshmani, H. Aghabozorg and M. Ghadermazi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o2869 CrossRef CAS
- H. Aghabozorg, Z. Derikvand, A. Nemati and M. Ghadermazi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2919–m2920 CrossRef CAS
- H. Aghabozorg, Z. Derikvand, A. Nemati, Z. Bahrami and J. Attar Gharamaleki, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m111 CrossRef CAS PubMed
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision C.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed
-
(a) D. Sadhukhan, M. Maiti, G. Pilet, A. Bauzá, A. Frontera and S. Mitra, Eur. J. Inorg. Chem., 2015, 11, 1958–1972 CrossRef
- S. B. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed
- E. Martell and R. J. Motekaitis, Determination and Use of Stability Constants, VCH, New York, 2nd edn, 1992 Search PubMed
- M. Devereuxa, M. McCann, V. Leon, M. Geraghty, V. McKee and J. Wikaira, Polyhedron, 2000, 19, 1205–1211 CrossRef
- F. H. Sweeton, R. E. Mesmer and C. F. Baes Jr, J. Solution Chem., 1974, 3, 191–214 CrossRef CAS
- C. W. Miller and L. V. Benson, Water Resour. Res., 1983, 19, 381–391 CrossRef CAS
- M. Shahbazi, F. Mehrzad, M. Mirzaei, H. Eshtiagh-Hosseini, J. T. Mague, M. Ardalani and M. Shamsipur, Inorg. Chim. Acta, 2017, 458, 84–96 CrossRef CAS
- J. B. Hendrickson and J. Wang, Org. Lett., 2004, 6, 3–5 CrossRef CAS PubMed
- Z. Q. Xia, Q. Wei, S. P. Chen, X. M. Feng, G. Xie, C. F. Qiao, G. C. Zhang and S. L. Gao, J. Solid State Chem., 2013, 197, 489–498 CrossRef CAS
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, Wiley, New York, 6th edn, 2009 Search PubMed
- M. Arefian, M. Mirzaei and H. Eshtiagh-Hosseini, J. Mol. Struct., 2018, 1156, 550–558 CrossRef CAS
- M. Nishio, CrystEngComm, 2004, 6, 130–158 RSC
- C. A. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC
- A. W. Addison and T. N. Rao, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC
- R. Clarke, K. Latham, C. Rix, M. Hobday and J. White, CrystEngComm, 2005, 7, 28–36 RSC
- H. Eshtiagh-Hosseini, M. Mirzaei, S. Zarghami, A. Bauza, A. Frontera, J. T. Mague, M. Habibi and M. Shamsipur, CrystEngComm, 2014, 16, 1359–1377 RSC
- M. Mahjoobizadeh, M. Mirzaei, A. Bauza, V. Lippolis, M. C. Aragoni, M. Shamsipur, M. Ghanbari and A. Frontera, ChemistrySelect, 2016, 1, 1556–1566 CrossRef CAS
- M. Mirzaei, H. Eshtiagh-Hosseini, Z. Karrabi, B. Notash, A. Bauza and A. Frontera, J. Mol. Struct., 2015, 1080, 30–36 CrossRef CAS
- M. Eftekhar, M. Mirzaei, A. Hassanpoor, I. Khosravi, A. Bauza, J. T. Mague and A. Frontera, J. Coord. Chem., 2015, 68, 3599–3610 CrossRef CAS
- L. Cattalini, A. Cassol, G. Marangoni, G. Rizzardi and E. Rotondo, Inorg. Chim. Acta, 1969, 3, 681–684 CrossRef CAS
- W. M. Zhang, Z. M. Lu, W. Li, M. Chen and D. M. Cao, Z. Kristallogr.–New Cryst. Struct., 2012, 227, 311–312 CrossRef CAS
- F. N. Shi, Y. F. Han and C. B. Liu, J. Chem. Crystallogr., 2012, 42, 438–442 CrossRef CAS
- K. B. Shiu, Z. W. Chen, F. L. Liao and S. L. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, m1072–m1074 CrossRef CAS
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS
- B. M. Sattelle and A. Almond, Phys. Chem. Chem. Phys., 2012, 14, 5843–5848 RSC
- H. B. Burgi, Angew. Chem., Int. Ed., 1975, 14, 460–473 CrossRef
- G. J. Bartlett, A. Choudhary, R. T. Raines and D. N. Woolfson, Nat. Chem. Biol., 2010, 6, 615–620 CrossRef CAS PubMed
-
(a) A. Bauza, A. Frontera and T. J. Mooibroek, Cryst. Growth Des., 2016, 16, 5520–5524 CrossRef CAS
-
(a) S. J. Grabowski, Molecules, 2015, 20, 11297–11316 CrossRef CAS PubMed
- D. Dutta, H. Nath, A. Frontera and M. K. Bhattacharyya, Inorg. Chim. Acta, 2019, 487, 354–361 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1554217, 1554221–1554227, and 1568348 contain the supplementary crystallographic data for 1–9. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra05143k |
| ‡ The distorted square pyramidal coordination geometry is indicated by the value of τ5 (0.230). The parameter τ5 is defined as (β − α)/60, where β and α are the largest angles subtended at the metal center). For ideal square pyramidal τ5 is 0 and for ideal trigonal bipyramidal geometry is 1.60 |
| This journal is © The Royal Society of Chemistry 2019 |