
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced UHPLC-MS/MS determination of a therapeutic heptapeptide mimic for inflammatory-related diseases in rat plasma: application to a pharmacokinetic study
Liang Liabc,
Shumei Ma†
d,
Yan Qinabc and
Li Liu *abc
*abc
aCenter for Pharmacological Evaluation and Research, Shanghai Institute of Pharmaceutical Industry, China State Institute of Pharmaceutical Industry, Shanghai 200437, PR China. E-mail: liulisipi@foxmail.com; Tel: +8621-65043855
bState Key Laboratory of New Drug and Pharmaceutical Process, Shanghai 201203, PR China
cShanghai Professional and Technical Service Center for Biological Material Drug-Ability Evaluation, Shanghai 200437, PR China
dDepartment of Pharmacology, School of Pharmacy, Fudan University, Shanghai 201203, PR China
First published on 15th October 2019
Abstract
The seven amino acid peptide, GQTYTSG (named as SP), a peptide mimic derived from hypervariable region 1 (HVR1) of the hepatitis C virus (HCV) has presented remarkable anti-inflammatory activities in previous experiments, indicating that it could be a novel therapeutic peptide candidate for different inflammation-related diseases, such as HCV infection and asthma. A heptapeptide mimic discovery study highlighted the need for the development of quantitative bioanalytical assays for measuring the levels of SP. Herein, a reliable and sensitive ultrahigh-performance liquid chromatography (UHPLC) with tandem mass spectrometry (MS/MS) assay was established and validated for the determination of SP in rat plasma. C-11, with two amino acid substitutions compared to SP (Glycine 1 and Glycine 7) and a disulfide, acted as an internal standard (IS). SP and C-11 were isolated from acidified plasma using protein precipitation and the extracts were analyzed by reversed-phase UHPLC-MS/MS detection. We used an SHIM-PACK GISS C18 (2.1 × 100 mm, 1.9 μm) column with water containing 0.2% acetic acid as the aqueous mobile phase and methanol as the organic mobile phase with a 0.3 mL min−1 flow rate. We used an AB SCIEX TripleQuad™ 5500 mass spectrometer equipped with a TurboIon Spray interface and operated it in positive-ion mode. Multiple reaction monitoring (MRM) was used for the quantification of the precursor to the product ion at m/z 713.3 → 432.2 for SP and m/z 803.2 → 539.1 for IS. The method was fully validated according to the US Food and Drug Administration (FDA) guideline (2018), and provided satisfactory accuracy, precision, and reproducibility for the quantification of SP in rat plasma. Excellent linearity was achieved (r > 0.9977) over a linear dynamic range of 0.1–200 ng mL−1 with a lower limit of quantification (LLOQ) of 0.1 ng mL−1. The validated assay was applied to gain the pharmacokinetic (PK) parameters and the concentration–time profile for SP after subcutaneous administration in rats.
Introduction
Inflammation is a kind of defensive response of the body to exterior irritation, such as infection and damage, which might bring about negative changes to the involved tissues.1 Sometimes, it indicates a starting point of certain chronic diseases, such as tumours, skin disorders, arthritis, cardiovascular syndrome and asthma diseases.2 The inflammatory reaction includes the energizing of various biological mechanisms, such as the excretion and production of some pro-inflammatory mediators, such as growth factors, interleukins, cytokines and chemokines.3 However, this kind of inflammatory response is required to maintain homeostasis in injured biological compartments, and a change of regulation is involved with different inflammation-related diseases.4,5 The treatment of inflammation-related diseases usually depends heavily on conventional small molecule agents, which can potentially result in some unexpected side effects.6 Based on the low toxicity, high selectivity, high potency and broad range of targets, peptide-based therapies are being developed as an alternative treatment for different inflammatory disorders over the years.7A variety of bioactive peptides can be found in living organisms and nature is a key source of biologically active peptides.8 However, beyond the traditional focus on endogenous peptides, peptide drug discovery has turned into a broader range of entities developed from extra natural origins or from chemical libraries or from recombinant libraries.9,10 The bioinformatic approach, empirical approach and integrated approach are the three dominating means for seeking biologically active peptides according to present studies.11 Besides, there are other studies demonstrating the peptide display technology for exploring significant bioactive peptides.12 Peptides discovered by these approaches have become the foundation for the development of therapeutic molecule entities.
Hepatitis C virus (HCV) is a type of ribonucleic acid virus that is responsible for most chronic and acute viral hepatitis, cirrhosis and hepatic carcinomas.13 The high-frequency mutation of the HCV gene during replication allows it to smoothly escape from an immune attack.14 The high rate of mutation happens cumulatively in a certain restricted region known as the hyper-variable region 1 (HVR1).15 HVR1, approximately a 28 amino acid long segment of the envelope protein E2, plays a key role in eliciting immune evasion.16 Under our co-workers' research into HVR1,17 a battery of peptide mimics derived from HVR1 were discovered and verified for their anti-inflammatory properties in vitro and in vivo. One of these peptide mimics, the heptapeptide GQTYTSG (named SP, Fig. 1), was recognized for its stronger ability to produce a favourable cytokine profile, which could contribute to the anti-inflammatory and anti-virus immune response.18 The results showed that SP could decrease the serum levels of alkaline phosphatase, aminotransferases and bilirubin, and is beneficial to improve the histopathological evidence of a porcine serum-induced rat chronic liver injury.19 Furthermore, SP could notably suppress immunoglobulin E production from B cell, control T-helper 9 cells development in the lung and reduce the airway reactivity in a murine asthma model.20 The World Intellectual Property Organization has granted a patent for SP (No. PCT/CN2006/001176).
 | ||
| Fig. 1 Chemical structures of SP and C-11 (IS). | ||
The accurate determination of SP in a biological matrix is pivotal for evaluating its pharmacokinetics and toxicokinetics in a preclinical study. Hitherto that date, no bioanalytical assay or relevant pharmacokinetic (PK) research has been reported for SP. Allowing for the rapid decomposition of straight-chain SP in blood samples due to enzymatic degradation with a half-life of less than 10 min (25 °C, in vitro), the general sample preparation requires processing immediately after being drawn from rats.21 However, this method is not suitable as it is time-consuming and requires intensive manpower effort, which makes it especially challenging when a lab is faced with a large number of samples. Hence, it is crucial to figure out how to stabilize SP in blood samples during sampling while ensuring reliable and actual analytical data.
In this context, we established and fully validated a reliable and sensitive ultrahigh-performance liquid chromatography with the tandem mass spectrometry (UHPLC-MS/MS) assay to quantify SP in rat plasma. C-11, a stable cyclic peptide derived from SP, served as an internal standard (IS). The assay was then performed to evaluate the PK properties of SP. The results were intended to provide a key reference and an essential complement for an assay to support the clinical study of SP.
Experimental
Chemicals and reagents
SP (99.4% purity) was provided by Rui Yunhai Bio-Technology Co., Ltd (Beijing, China). C-11 (purity ≥ 99%) was synthesized by Naval Medical University (Shanghai, China). The chemical structures are presented in Fig. 1. The molecular weight of SP was 712.30 Da, and that of C-11 was 802.26 Da.Methanol (HPLC grade) was purchased from Merck Company (Darmstadt, Germany). LC/MS-grade acetic acid was purchased from Fisher Scientific (Loughborough, UK). cOmplete™ protease inhibitor cocktail tablets were purchased from Roche (Mannheim, Germany). Pepstatin A (purity ≥ 98.0%) and aprotinin (3–7 TIU per mg protein) were purchased from Sigma-Aldrich (Missouri, USA). Biotech grade 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF, purity ≥ 97.0%) was purchased from Fisher Scientific (Loughborough, UK). Analytical reagent sulphuric acid (H2SO4, 95–98%) was purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China), and guaranteed reagent perchloric acid (HClO4, 70.0–72.0%) was purchased from Macklin Biochemical Co., Ltd (Shanghai, China). HPLC-grade water was produced in-house by a Milli-Q purifying system (Bedford, USA). Blank rat blood (EDTA-K2 or heparin sodium as the anticoagulant) containing 0.3% perchloric acid (v/v), obtained from the Centre for Pharmacological Evaluation and Research of Shanghai Institute of Pharmaceutical Industry (SIPI), was centrifuged at 1000×g for 15 min to gain the acidified rat plasma, and was then stored below −70 °C until analysis.
UHPLC-MS/MS analysis
The UHPLC-MS/MS system was composed of an AB SCIEX ExionLC™ AD system coupled to an AB SCIEX TripleQuad™ 5500 (Applied Biosystems, Foster City, CA, USA) equipped with an electrospray ionization (ESI) source. Data processing was performed using the MultiQuant™ software (Version 3.0.3, AB SCIEX).The separation was performed on a SHIM-PACK GISS C18, 1.9 μm, 2.1 × 100 mm column (Shimadzu Corporation, Kyoto, Japan) coupled with a SHIM-PACK GISS-HP (G), 1.9 μm, 2.1 × 10 mm pre-column (Shimadzu Corporation, Kyoto, Japan) maintained at 50 °C. The mobile phase consisted of methanol (mobile phase B) and water containing 0.2% acetic acid (v/v) (mobile phase A). The chromatographic analysis was performed with gradient elution, and the gradation of the elution is shown in Table 1. The temperature of the auto-sampler was set at 10 °C. The injection volume was 10 μL, and the total run time was 6.0 min.
| Time (min) | Flow rate (mL min−1) | A% | B% |
|---|---|---|---|
| Initial | 0.300 | 98 | 2 |
| 0.50 | 0.300 | 98 | 2 |
| 1.50 | 0.300 | 81 | 19 |
| 3.00 | 0.300 | 81 | 19 |
| 3.50 | 0.300 | 20 | 80 |
| 4.50 | 0.300 | 20 | 80 |
| 4.60 | 0.300 | 98 | 2 |
| 6.00 | 0.300 | 98 | 2 |
The mass spectrometry was performed in the positive ionization, multiple reaction monitoring (MRM) mode. The ionization parameters were optimized as follows: curtain gas (CUR), 35 psi; collision gas (CAD), 8 psi; gas1 (GS1), 60 psi; gas2 (GS2), 50 psi; ionization voltage, 5500 V; source temperature, 550 °C. Nitrogen was used as the CUR, CAD and nebulizing gas. The precursor ion (Q1) and product ion (Q3) quadrupoles were set at unit resolution. The dwell time was 150 ms for SP and C-11. The MRM transitions for SP and C-11 were at m/z 713.3 → 432.2 and 803.2 → 539.1, respectively. Also, the fragmentation parameters, including the de-clustering potential, collision energy, collision cell exit potential and entrance potential, were 120 V, 36.0 V, 16.7 V and 10 V for SP, and 150 V, 44.7 V, 9.7 V and 10 V for IS.
Preparation of calibration samples and quality control samples
The reference standard of SP was accurately prepared in water to gain separate stock solutions (1 mg mL−1) for the calibration standards and for quality control (QC) of the samples. Standard material as the IS was accurately weighed and dissolved in water–methanol (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, v/v) at 1 mg mL−1 as the stock solution. The working solutions of SP (1, 5, 25, 100, 500, 1000 and 2000 ng mL−1) were gained by diluting the stock solution with water containing 0.2% acetic acid (v/v). The working solutions of QC samples (2.5, 50 and 1500 ng mL−1) were prepared as above. The working solution for IS (500 ng mL−1) was freshly prepared when necessary by diluting the stock solution with water containing 0.2% acetic acid (v/v). All the working solutions were kept at 4 °C, and the aliquots of stock solutions after preparation were stored below −70 °C prior to further use.
:50, v/v) at 1 mg mL−1 as the stock solution. The working solutions of SP (1, 5, 25, 100, 500, 1000 and 2000 ng mL−1) were gained by diluting the stock solution with water containing 0.2% acetic acid (v/v). The working solutions of QC samples (2.5, 50 and 1500 ng mL−1) were prepared as above. The working solution for IS (500 ng mL−1) was freshly prepared when necessary by diluting the stock solution with water containing 0.2% acetic acid (v/v). All the working solutions were kept at 4 °C, and the aliquots of stock solutions after preparation were stored below −70 °C prior to further use.
Seven non-zero calibration standards were freshly prepared on each day of analysis at the following concentrations: 0.1, 0.5, 2.5, 10, 50, 100 and 200 ng mL−1 by adding 5 μL of the working solution of SP to a 45 μL aliquot of blank acidified plasma. QC samples were obtained in a similar way at low, medium and high concentrations (0.25, 5 and 150 ng mL−1).
Estimation of adsorption
To estimate the loss of SP and IS during the solution preparation, the extent of adsorption was measured. Adsorption was investigated at low and high concentrations (1 and 2000 ng mL−1) of SP and at 500 ng mL−1 of IS. Six consecutive steps, transferring a portion of the working solutions from a ordinary polypropylene tube or Eppendorf Protein LoBind tube or glass tube to another, followed by UHPLC-MS/MS analysis, were followed to perform the adsorption test. Adsorption was evaluated by relative recovery, defined by comparing the peak area before and after transfer.Working solutions and stock solutions stability
The stability of SP and IS in the working solutions under different conditions was inspected. The test concentrations were 2000 ng mL−1 for SP and 500 ng mL−1 for IS. Aliquots (500 μL) of SP or IS working solution were set at ambient temperature (25 °C) and in an ice-water bath (0 °C) for 6 h, respectively. The loss of SP and IS was determined by comparing the peak area of the samples before and after incubation. Other aliquots (500 μL) of SP or IS working solution were stored in an ice chamber (4 °C) for 30 days. The long-term stability of the working solutions was evaluated by comparing the peak areas of the freshly prepared solutions and original solutions.The stability of SP and IS in the stock solutions was also inspected. Aliquots (1 mL) of SP or IS stock solution were stored in a freezer (−70 °C) for 90 days. The loss of SP and IS was checked by comparing the peak areas of the freshly prepared solutions and original solutions. The stock solutions stability was assessed by HPLC with a diode array detector (λ = 224 nm).
Precipitant selection
One of the most common and convenient sample processing methods is precipitation with organic solvents to remove plasma proteins, and for this, a selection of organic solvents was investigated herein. Medium concentration acidified plasma samples (5 ng mL−1) were prepared and precipitated by different volumes of acetonitrile or methanol with 0.2% acetic acid (v/v). Also, the samples were centrifuged for 10 min at 1000×g at ambient temperature. The supernatant was then transferred into a vial containing low-volume inserts for the UHPLC-MS/MS analysis. The extraction efficiency was examined by comparing the recovery of SP in samples precipitated with different volumes of different organic solvents.Sample processing
Here, a 5 μL working solution of IS (except for double blanks) was added into a volume of 50 μL of each sample before being extracted by triple volumes (150 μL) of cooled methanol containing 0.2% acetic acid (v/v). After shaking for 10 min on a horizontal shaker at 160 strokes per min, the samples were centrifuged for 10 min at 1000×g at ambient temperature. Then, 150 μL of the supernatant was transferred into a tube and evaporated to dry under a nitrogen stream at 40 °C. The residue was re-dissolved into 80 μL of methanol:water:acetic acid (2:98:0.2, v:v:v) by thoroughly mixing for 30 s. The mixture was then centrifuged for 10 min at 1000×g at room temperature. The supernatant was directly collected into a vial containing low-volume inserts for UHPLC-MS/MS analysis.
Stabilization tactics for biological samples
During method development, a couple of factors affecting the stabilization of SP in biological samples were probed, including anticoagulant, enzyme inhibitors and acidified samples. High concentration blood samples (150 ng mL−1) were prepared and employed to evaluate the different factors. The sample-processing procedure was the same as in the previous section.System suitability tests
System suitability tests were utilized to confirm that the performance of the UHPLC-MS/MS system was applicable for batches to be done. A separate medium concentration sample (5 ng mL−1) was analysed as a reference standard before each run. The resolution and reproducibility of the UHPLC-MS/MS system were regarded as acceptable if the relative standard deviations (RSD%) of the retention time and peak area of analytes from six replicate injections was no more than 15%.Validation of the UHPLC-MS/MS assay
The UHPLC-MS/MS method validation was performed on the basis of the bioanalytical guideline documented by the US Food and Drug Administration (2018).22 The following parameters were evaluated: selectivity and specificity, linearity, sensitivity (lower limit of quantification, LLOQ), carryover, precision, accuracy, stability, dilution integrity, matrix effect and extraction recovery.Acceptable runs
The calibration curve for each analytical batch should meet the acceptance criteria for linearity described in the previous section. Also, samples with at least three QC concentration levels (n ≥ 2) were tested in each analytical batch. In addition, the data from batches were qualified when at least two-thirds of the QC samples and at least half of the QC samples per level were within 15% deviation from the theoretical concentrations. Also, the acceptance criterion for the analytical batch containing the diluted samples was the same as above.Pharmacokinetic study
Twelve Sprague-Dawley rats (female and male in half, 200 ± 20 g for each) were purchased from Shanghai Slac Laboratory Animal Co., Ltd. (Shanghai, China). The rats were maintained under specific pathogen-free conditions with 12 h light–dark cycles, kept at 22 ± 2 °C and a relative humidity of 55 ± 15%. The rats took commercial food and autoclaved water freely and adjusted themselves to their new situation for a week before experiment. The experimental workings were authorized by the Experimental Animal Care and Use Committee of SIPI, and appropriate animal welfare was seriously applied according to the related guidelines.The rats were forced to stop food ingestion for about 12 h before receiving SP via subcutaneous administration (10 mg kg−1). The blood samples were collected via orbital bleeding into pre-acidified EDTA-K2 tubes at 0 (before administration), 0.5, 1, 2, 3, 5, 10, 20, 30, 45 and 60 min, following administration and placed in an ice-water bath (0 °C) immediately. Then, the plasma samples were obtained by centrifugation at 1000×g for 15 min and stored at −70 °C until analysis.
Data treatment
The data was acquired using the Analyst® software (Version 1.7, AB SCIEX), and data processing was performed using the MultiQuant™ software (Version 3.0.3, AB SCIEX). During data analysis, the concentrations before administration below the LLOQ were defined as zero, and the concentrations after administration below LLOQ were defined as below LLOQ. The T test results were evaluated with Microsoft® Excel for Mac 2011 (Microsoft Corp., USA). PK parameters were calculated using the Phoenix WinNonlin software (Version 6.3, Pharsight).Results and discussion
Method development
In the present study context, a reliable and sensitive UHPLC-MS/MS assay was developed for the determination of SP in rat plasma, and a stable cyclic peptide (C-11) acted as IS to improve the variability during bio-analysis.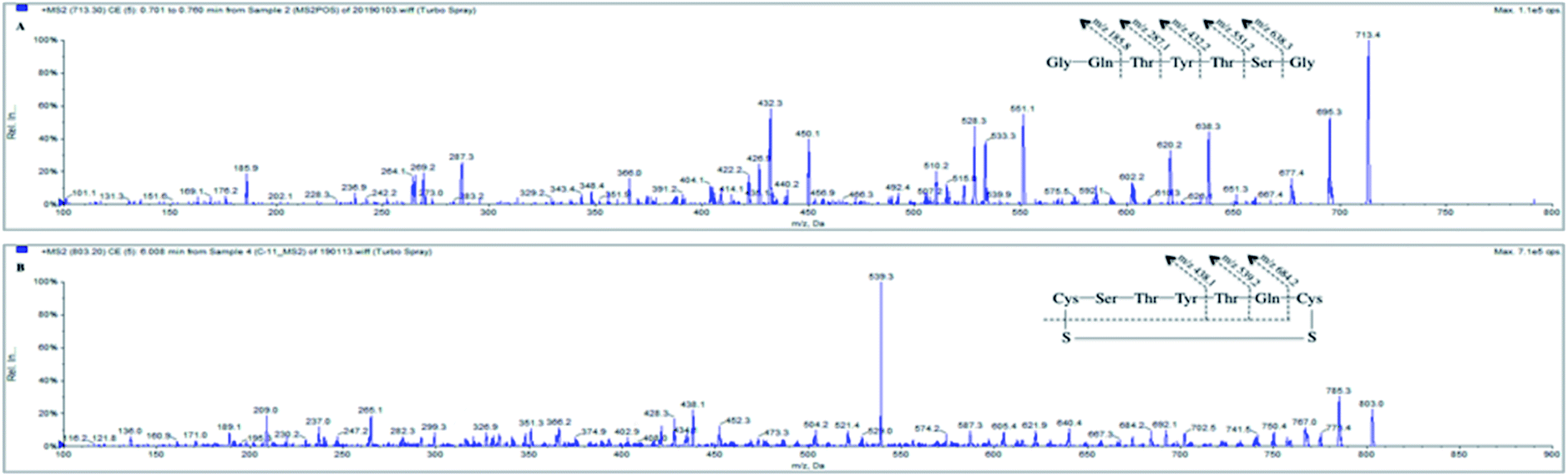 | ||
| Fig. 2 Product ion mass spectrum of SP (a) and IS (b). Illustration: the proposed fragmentation pathway. | ||
Estimation of the adsorption
One of the most common traps run into during the inchoate development of analysis methods for peptides is the adsorption to the frequently used materials in instrumental analysis. A large number of factors could affect the detected concentrations of target peptides and the robustness of analysis methods, including, but not limited to, extraction recovery and matrix effects. The first thought concerns the nonspecific binding of target peptides to the solid surfaces of the containers.27 In addition, because the capacity of binding to the wall is limited, the relative loss caused by adsorption in low concentration solutions is more widespread than for high concentration samples.28 In the present study, a fast method composed of six consecutive steps for transferring the SP solutions (1 and 2000 ng mL−1) and IS solution (500 ng mL−1) from a container to another, followed by UHPLC-MS/MS analysis was developed. Also, a process for the experimental capacity measurement of the extent of adsorption was developed. The adsorption results of SP and IS are depicted in Fig. 3. No remarkable adsorption for SP and IS was observed for the test concentrations and three kinds of containers. Due to the hard-to-predict nature of the adsorption behaviour caused by the huge variability of amino acid sequences in peptides and their structural modifications, the adsorption aspect must be considered during quantitative analysis.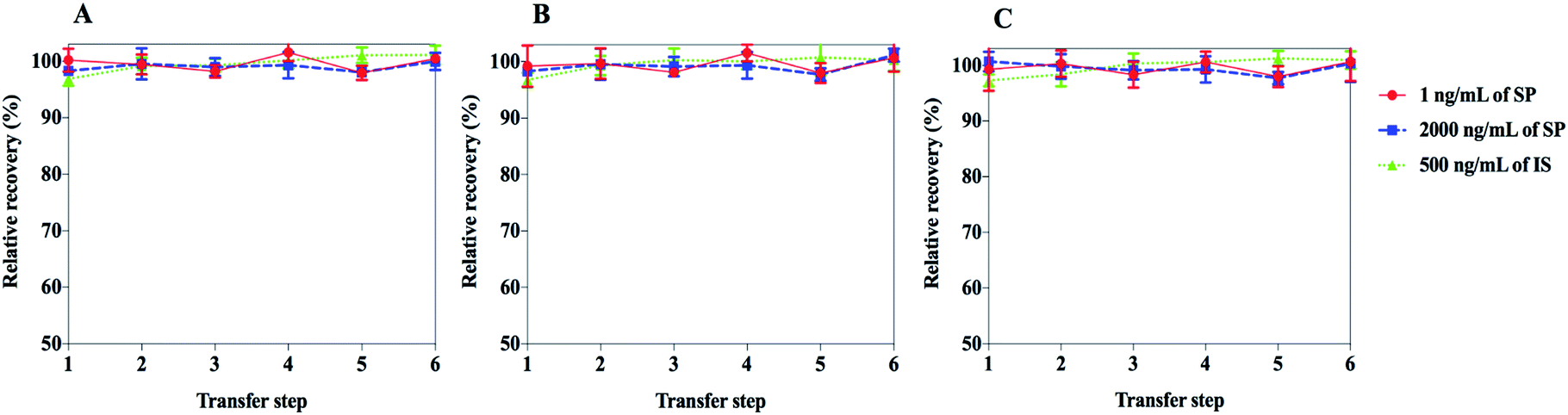 | ||
| Fig. 3 Relative recovery of SP and IS after six transfer steps in different vessels (n = 3). SP was dissolved in the vessels indicated: (a) ordinary polypropylene tubes; (b) Eppendorf protein LoBind tubes; (c) glass tubes. After every 30 min storage at 25 °C, 300 μL was transferred from one vessel to the next for UHPLC-MS/MS analysis. | ||
Stock solutions and working solutions stability
Considering the cysteine residue in C-11 is potentially sensitive to oxidation and the disulphide bond could be cleaved, leading to the amide bonds breaking down and releasing smaller fragments, stability tests for IS were necessary. The stabilities of the SP and IS in stock solutions and working solutions are shown in Table 2. The results demonstrated that there was no obvious degradation in the working solutions within the test concentrations after 6 h incubation at 25 °C and 0 °C, respectively. As shown in Table 2, the working solutions of SP and IS were stable after being stored in an ice chamber (4 °C) for 30 days, and the stock solutions of SP and IS were also stable after being stored in a freezer (−70 °C) for 90 days. Consequently, the two peptides were confirmed to have sufficient chemical stability during the process required for the UHPLC-MS/MS analysis.| Solution-type | Compound (concentration) | Storage condition | Peak area of the fresh solutiona (mean ± SD, n = 5) | Peak area of the original solutiona (mean ± SD, n = 5) | Percentage of initial level (%) |
|---|---|---|---|---|---|
| a The 6 h stability at 25 °C and 0 °C was determined by comparing the peak area of the solutions before and after incubation. | |||||
| Working solution | SP (2000 ng mL−1) | 25 °C for 6 hours | 29855000 ± 964421 |
29942000 ± 101825 |
100.3 |
| 0 °C for 6 hours | 30184000 ± 227618 |
29916000 ± 382537 |
99.1 | ||
| 4 °C for 30 days | 31668000 ± 853542 |
31174000 ± 799235 |
98.4 | ||
| IS (500 ng mL−1) | 25 °C for 6 hours | 2958500 ± 92984 |
2975100 ± 128621 |
100.6 | |
| 0 °C for 6 hours | 3077500 ± 103641 |
2944500 ± 96614 |
95.7 | ||
| 4 °C for 30 days | 2850300 ± 86554 |
2911700 ± 100528 |
102.1 | ||
| Stock solution | SP (1 mg mL−1) | −70 °C for 90 days | 143 ± 1.12 | 145 ± 0.976 | 101.4 |
| IS (1 mg mL−1) | −70 °C for 90 days | 129 ± 0.991 | 127 ± 0.852 | 98.4 | |
Precipitant selection
The precipitation of the plasma samples with organic solvents could result in the removal of large abundant proteins, such as albumin, leaving only the low molecular weight proteins or peptides in solution. Moreover, the addition of ion-pairing agents, such as certain organic acids, could also separate the smaller proteins and peptides from the high molecular weight proteins, which is beneficial for the extraction of peptides.29 Also, the organic solvents should be kept below 4 °C to reduce thermally stressing the peptide.30 The precipitation of plasma samples was performed by spiking the samples with increasing the volumes of cooled acetonitrile or methanol with 0.2% acetic acid (v/v) to affect the deproteinization (Fig. 4). The results indicated that methanol was the best choice as the precipitant due to its more satisfactory recovery compared to acetonitrile. Although many papers have reported that the generic acetonitrile precipitation strategy is favourable for the extraction of small peptides in a biological matrix for mass spectrometry analysis,31–34 methanol was considered a better precipitating agent for SP in this study. Moreover, Fig. 4 also illustrates that the increase in the volumes of acetonitrile or methanol caused a tendency to reduce the recovery of SP. This suggests that increase in the volumes of organic solvents might cause a loss of the peptide to some extent on account of the co-precipitation or an insufficient solubility in a high concentration of precipitants. Solid-phase extraction (SPE) was frequently applied during the sample preparation process to avoid possible interferences that could impact the result of the UHPLC-MS/MS analysis, but it finally fell out of favour owing to its unsatisfactory accuracy and poor precision of the assay. Hence, the handy protein precipitation by applying triple volumes of cooled methanol containing 0.2% acetic acid (v/v) was applied to extract the peptide from the plasma samples.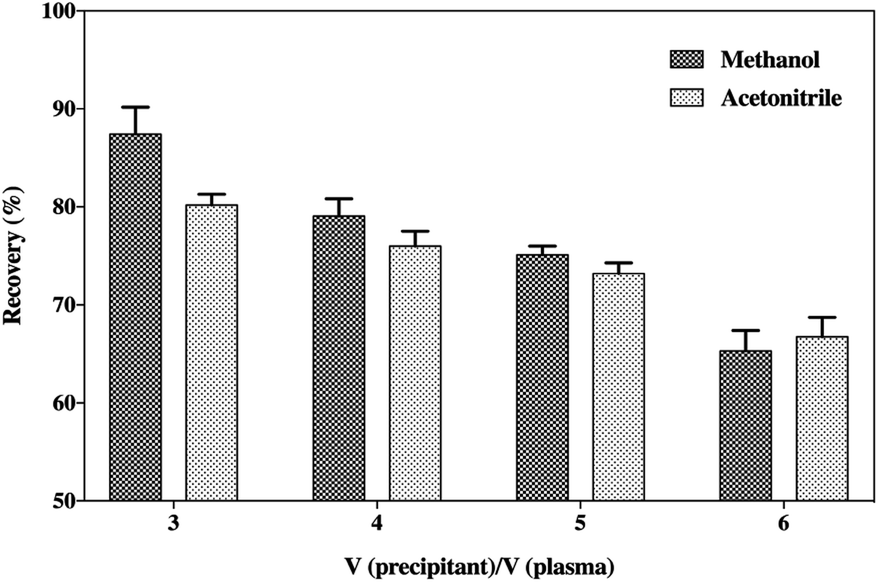 | ||
| Fig. 4 Recovery of SP after plasma precipitation (n = 3). Blank acid rat plasma was spiked with SP (5 ng mL−1) and precipitated with methanol or acetonitrile containing 0.2% acetic acid (v/v) at the ratios indicated. SP in the supernatant was determined by UHPLC-MS/MS analysis and the recovery was calculated by comparing the peak areas of SP. | ||
Stabilization of the SP in the samples
SP was detected to have insufficient chemical stability in the samples as a result of enzymatic degradation. As it is well-known, the preclinical samples collected from rats are in the form of whole blood, and stabilization should be considered in the sampling step. Therefore, different stabilization tactics for the blood samples were investigated so as to identify the degradation process and to determine the optimal sample processing method.The effects of different anticoagulants on the stability of SP in the samples are shown in Fig. 5a. EDTA-K2 was used as an anticoagulant and the samples were kept at 0 °C, which simultaneously slightly improved the degradation of SP, which prolonged the half-life to 11.48 ± 0.22 min. This suggested that EDTA-K2 could be a better choice for its potential inhibition of metal-catalysed oxidation, but it could not meet all our needs. The addition of protease inhibitors, such as pepstatin A, aprotinin, cOmplete™ protease inhibitor cocktail tablets and AEBSF, did not slow down the degradation process compared to the control group (Fig. 5b). The results (Fig. 6) showed that there was no significant change in the half-life of SP. As illustrated in Fig. 5c and d and Fig. 5e and f, the samples pre-treated with 0.3% H2SO4 (v/v) or 0.3% HClO4 (v/v) combined with an ice-water bath were stable within 1 h. Also, the half-life of SP in both the blood samples acidified with 0.3% H2SO4 (v/v) or 0.3% HClO4 (v/v) was far longer than 350 min (Fig. 6). This suggested that the pre-treatment with acidification agents could significantly improve the degradation and afford a fairly stable environment for SP. However, we found that the samples acidified with 0.3% H2SO4 (v/v) produced extra signal suppression and impacted the sensitivity during UHPLC-MS/MS analysis. We also speculated that the samples were moderately haemolysed, which might have resulted in severe matrix effects. Accordingly, the addition of 0.3% HClO4 (v/v) into the samples was finally applied to stabilize SP.
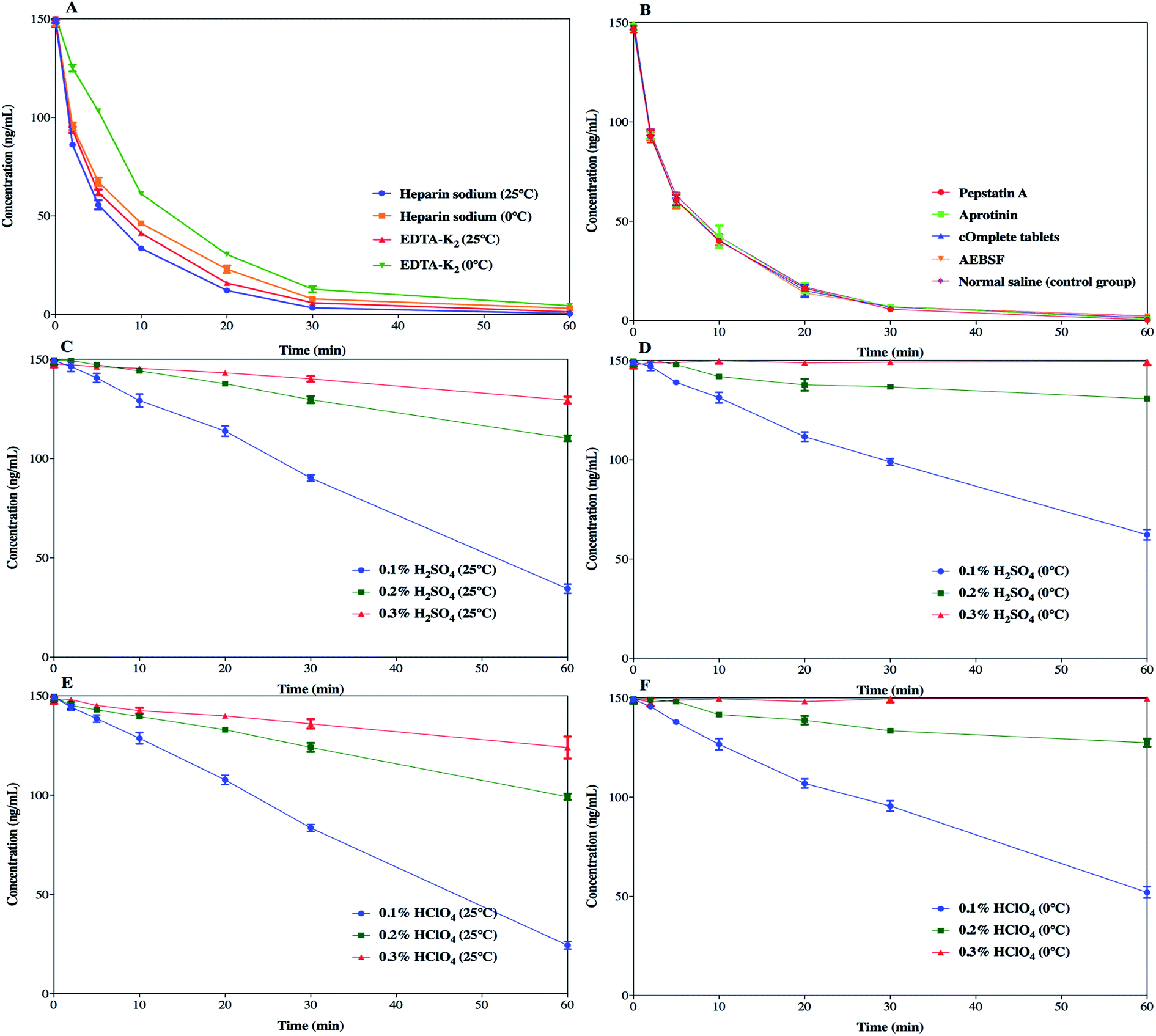 | ||
| Fig. 5 Stability results of SP in blood samples under different test conditions (n = 3). (a) Blood samples using different anticoagulants at different temperatures; (b) blood samples inhibited by different enzyme inhibitors, including pepstatin A, aprotinin, cOmplete™ protease inhibitor cocktail tablets (cOmplete™ tablets) and AEBSF; (c) blood samples pre-treated with H2SO4 (0.1–0.3%, v:v) at ambient temperature; (d) blood samples pre-treated with H2SO4 (0.1–0.3%, v:v) in an ice-water bath; (e) blood samples pre-treated with HClO4 (0.1–0.3%, v:v) at ambient temperature; (f) blood samples pre-treated with HClO4 (0.1–0.3%, v:v) in an ice-water bath. | ||
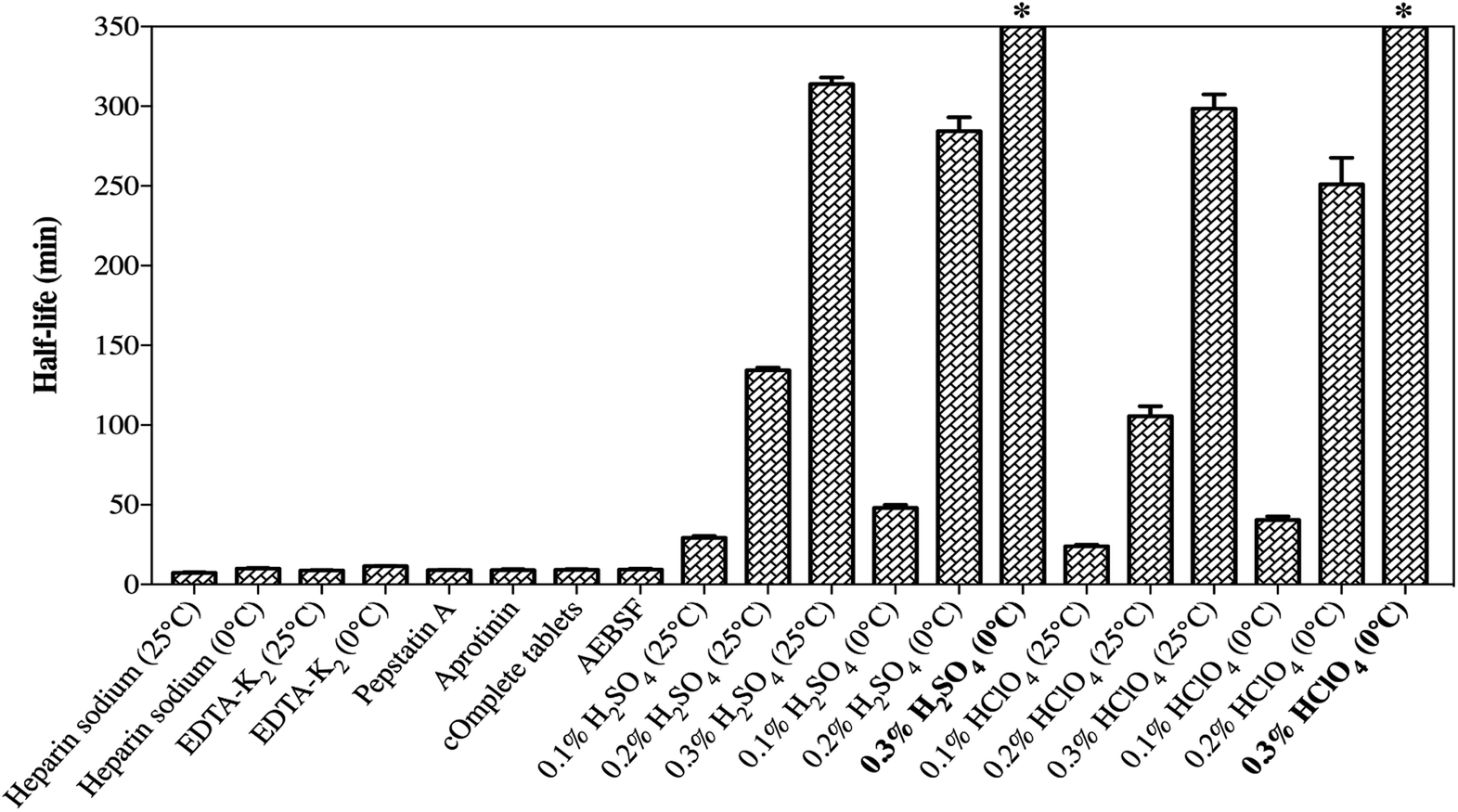 | ||
| Fig. 6 Half-lives of SP in blood samples under different test conditions (n = 3). * SP in the blood samples acidified with 0.3% H2SO4 (v:v) (0 °C) and 0.3% HClO4 (v:v) (0 °C) was highly stable, and the half-lives could not be calculated (that is, much more than 350 min). | ||
The enzymatic degradation of SP in blood is a sophisticated process and the mechanism has not been fully probed fully before. The relevant protease inhibitors did not block the degradation of SP, manifesting that the corresponding protease hydrolysis might not be the critical mechanism behind the degradation. In addition, other kinds of enzymes may be conducive to the degradation. Hence, an extensive investigation of different stabilization factors was accomplished to elucidate the SP process. An effective, low-cost and time-saving stabilization tactic is presented in this study despite the mechanism of degradation still not being explicitly set out. Moreover, hydrochloric acid and formic acid were not adopted due to their volatility, which goes against the long-term stability (unpublished data). Moreover, phosphoric acid could bring about acute haemolysis and nearly curdle the blood samples, and was thus discarded. Finally, it was demonstrated that simply pre-treating the samples with 0.3% HClO4 (v/v) combined with EDTA-K2 as an anticoagulant and sitting in an ice-water bath was suitable for inhibiting the activity of the enzymes and stabilizing SP.
Method validation
 | ||
| Fig. 7 Typical MRM chromatograms of SP and IS from (a) a double blank sample (no SP and IS); (b) blank sample (IS only); (c) LLOQ sample (0.1 ng mL−1); (d) sample taken at 10 min after administration (20.3 ng mL−1). | ||
| Run | Item | LLOQ (0.1 ng mL−1) | Low QC (0.25 ng mL−1) | Medium QC (5 ng mL−1) | High QC (150 ng mL−1) | Dilution (400 ng mL−1) |
|---|---|---|---|---|---|---|
| Within-run (n = 5) | Mean ± SD | 0.109 ± 0.003 | 0.263 ± 0.006 | 4.95 ± 0.352 | 146 ± 2.98 | 387 ± 11.4 |
| Precision (RSD%) | 2.8 | 2.3 | 7.1 | 2.0 | 2.9 | |
| Accuracy (%) | 109.0 | 105.3 | 99.1 | 97.0 | 96.8 | |
| Between-run (n = 15) | Mean ± SD | 0.112 ± 0.005 | 0.269 ± 0.016 | 5.27 ± 0.487 | 142 ± 3.73 | — |
| Precision (RSD%) | 4.5 | 5.9 | 9.2 | 2.6 | — | |
| Accuracy (%) | 112.0 | 107.6 | 105.4 | 94.7 | — |
| Matrix | Test conditions | Theoretical concentration (ng mL−1) | Calculated concentration (ng mL−1) (Mean ± SD, n = 5) | Precision (RSD%) | Accuracy (%) |
|---|---|---|---|---|---|
| Plasma | Short-term stability (0 °C for 1 h) | 0.25 | 0.241 ± 0.011 | 4.6 | 96.4 |
| 150 | 144 ± 3.17 | 2.2 | 96.0 | ||
| Post-preparation stability (25 °C for 1 h) | 0.25 | 0.227 ± 0.018 | 7.9 | 90.8 | |
| 150 | 158 ± 6.12 | 3.9 | 105.3 | ||
| Auto-sampler stability (4 °C for 24 h) | 0.25 | 0.275 ± 0.009 | 3.3 | 110.0 | |
| 150 | 160 ± 7.17 | 4.5 | 106.7 | ||
| Freeze–thaw stability (three cycles) | 0.25 | 0.266 ± 0.005 | 1.9 | 106.4 | |
| 150 | 142 ± 4.21 | 3.0 | 94.7 | ||
| Long-term stability (−70 °C for 60 days) | 0.25 | 0.240 ± 0.005 | 2.1 | 96.1 | |
| 150 | 141 ± 3.85 | 2.7 | 94.0 | ||
| Blood | Blood stability (0 °C for 1 h) | 0.5 | 0.572 ± 0.078 | 13.6 | 114.4 |
| 300 | 312 ± 10.0 | 3.2 | 104.1 |
Application to pharmacokinetic study
The fully validated UHPLC–MS/MS method was applied to a PK study of SP. Single-dose data in 12 Sprague-Dawley rats (equal number females and males) weighing 200 ± 20 g was obtained after a subcutaneous dose of 10 mg kg−1 was administered. The typical MRM chromatogram from a rat plasma sample taken at 10 min after administration is shown in Fig. 7d, and the concentration of SP was 20.3 ng mL−1. The mean plasma concentration–time profiles of the female and male Sprague-Dawley rats after a single subcutaneous administration are illustrated in Fig. 8. It was demonstrated that there was no significant statistical difference between the plasma concentrations of female rats and male rats (p > 0.5). Also, it was indicated that the PK behaviour in rats was independent of gender differences. Furthermore, an examination of Fig. 8 shows that the developed UHPLC–MS/MS method possessed the desired sensitivity to display the absorption, distribution and elimination process of SP following subcutaneous infusion. | ||
| Fig. 8 Mean plasma concentration–time profiles of female and male Sprague-Dawley rats after a subcutaneous administration of 10 mg kg−1 (n = 6). | ||
The PK parameters were evaluated by using a non-compartmental model and the results are summarized in Table 5. After subcutaneous administration, the mean maximum concentration (Cmax) of SP in plasma was 210 ± 21.6 ng mL−1 and the area under the plasma concentration–time profile (AUC0-t) was 712 ± 43.0 ng min mL−1. The plasma concentration of SP declined by 50% within 10 min (T1/2 = 8.65 ± 1.79 min), and the LLOQ level of the assay was reached at around 60 min (Fig. 8). This indicates SP could be a therapeutic peptide candidate for inflammatory-related diseases, because the continuous existence of compounds in the body might cause adverse events related to immunomodulatory therapies. Fast absorption was found following the subcutaneous administration of 10 mg kg−1 SP with the Tmax at 1.75 ± 0.957 min.
| Parameters | Unit | Values (mean ± SD, n = 12) |
|---|---|---|
| a AUC0−t, area under the plasma concentration–time profile from 0 to 60 min; AUC0−∞, area under the plasma concentration–time profile from 0 to infinity; Cmax, maximum concentration; Tmax, time to Cmax; T1/2, elimination half-life; CL, apparent total plasma clearance; Vz, total volume of distribution. | ||
| AUC0-t | ng min mL−1 | 712 ± 43.0 |
| AUC0-∞ | ng min mL−1 | 714 ± 43.3 |
| Cmax | ng mL−1 | 210 ± 21.6 |
| Tmax | min | 1.75 ± 0.957 |
| T1/2 | min | 8.65 ± 1.79 |
| CL | L min−1 | 17.9 ± 9.96 |
| Vz | L | 230 ± 118 |
Conclusion
In conclusion, a reliable, simple and sensitive UHPLC-MS/MS method with a stable cyclic peptide acting as an IS was successfully developed for the determination of SP in rat plasma and fully validated according to the latest bioanalytical guideline documented by the US Food and Drug Administration. Also, it fulfilled all the acceptance criteria for working as a bioanalytical method in pharmaceutical analysis. The instability of SP in blood samples, a primary challenge for the analysis, was cracked by acidifying the blood samples with perchloric acid (0.3%, v/v). Besides, a simple deproteinization was employed to extract the peptide from the plasma samples, which did not require a complicated pre-treatment, thus shortening the sample processing and weakening the variability of the assay. The expected sensitivity of SP was accomplished with an LLOQ of 0.1 ng mL−1 using 50 μL of plasma. Also, clearly, the proposed UHPLC-MS/MS assay for the quantitation of SP could provide reference for the determination of other unstable small peptides in biological matrices. The application of the fully verified method to a PK study of SP in rats during preclinical stage enabled calculation of the main PK parameters. It was found that SP was eliminated quickly from body with a half-life of less than 10 min, which was a promising result for an immunomodulatory candidate in the field of safety for the reason that some inflammatory-related diseases, such as asthma, would require repeated doses. The work could provide the basis for further in-depth studies into a clinical assay of SP.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Science and Technology Commission of Shanghai Municipality (No. 18DZ2290900). We greatly appreciate the sacrificial rats for their contribution to the study of SP in this work. And we also thank Jinqiu Mo for their help on the publication. Additionally, Liang Li wants to thank, in particular, the support and company from Yueying Zhi over the years.Notes and references
- Y. Wu, S. Antony, J. L. Meitzler and J. H. Doroshow, Cancer Lett., 2014, 345, 164–173 CrossRef CAS.
- S. La Manna, C. Di Natale, D. Florio and D. Marasco, Int. J. Mol. Sci., 2018, 19, 2714 CrossRef.
- P. C. Calder, R. Albers, J. M. Antoine, S. Blum, R. Bourdet-Sicard, G. A. Ferns, G. Folkerts, P. S. Friedmann, G. S. Frost, F. Guarner, M. Løvik, S. MacFarlane, P. D. Meyer, L. M'Rabet, M. Serafini, W. Van Eden, J. Van Loo, W. Vas Dias, S. Vidry, B. M. Winklhofer-Roob and J. Zhao, Br. J. Nutr., 2009, 101, S1–S45 CrossRef PubMed.
- Q. Alam, M. Zubair Alam, G. Mushtaq, G. A. Damanhouri, M. Rasool, M. Amjad Kamal and A. Haque, Curr. Pharm. Des., 2016, 22, 541–548 CrossRef CAS.
- D. Okin and R. Medzhitov, Curr. Biol., 2012, 22, 733–740 CrossRef.
- S. La Manna, P. L. Scognamiglio, C. Di Natale, M. Leone, F. A. Mercurio, A. M. Malfitano, F. Cianfarani, S. Madonna, S. Caravella, C. Albanesi, E. Novellino and D. Marasco, Biochimie, 2017, 138, 106–115 CrossRef CAS.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS.
- E. B. M. Daliri, B. H. Lee and D. H. Oh, Crit. Rev. Food Sci. Nutr., 2018, 58, 2273–2284 CrossRef CAS.
- Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K.-H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. Cai, P. Berry, E. Feyfant, X. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3445–E3454 CrossRef CAS.
- T. Uhlig, T. Kyprianou, F. G. Martinelli, C. A. Oppici, D. Heiligers, D. Hills, X. R. Calvo and P. Verhaert, EuPa Open Proteomics, 2014, 4, 58–69 CrossRef CAS.
- H. R. Hoogenboom, Nat. Biotechnol., 2005, 23, 1105–1116 CrossRef CAS.
- C. H. Wu, Y. H. Kuo, R. L. Hong and H. C. Wu, Sci. Transl. Med., 2005, 7, 290–291 Search PubMed.
- X. Forns, R. H. Purcell and J. Bukh, Trends Microbiol., 1999, 7, 402–410 CrossRef CAS.
- L. Frasca, C. Scottà, P. Del Porto, A. Nicosia, C. Pasquazzi, I. Versace, A. M. Masci, L. Racioppi and E. Piccolella, Hepatology, 2003, 38, 653–663 CrossRef CAS.
- Y. K. Shimizu, H. Yoshikura, M. Shapiro, H. Igarashi, D. C. Wong, T. Kiyohara and R. H. Purcell, J. Gen. Virol., 2015, 79, 1383–1386 CrossRef PubMed.
- R. Thimme, V. Lohmann and F. Weber, Antiviral Res., 2006, 69, 129–141 CrossRef CAS.
- J. L. Haibin Sun and S. Chi, J. Chin. PLA Postgrad. Med. Sch., 2006, 27, 254–256 Search PubMed.
- C. Song, L. Chen, B. Yang, S. Chi and Y. Cheng, J. Med. Coll. PLA, 2010, 25, 257–266 CrossRef CAS.
- W. Zhaob, L. Wanga, P. Chenc, J. Zhaoa, Y. Qia, S. Chia, J. Suna, L. Zhua and Y. Chenga, Inflammation Allergy: Drug Targets, 2010, 9, 135–145 CrossRef.
- Y. C. Chuihui Song and B. Yang, Med. J. Chin. People's Liberation Army, 2009, 34, 1333–1334 Search PubMed.
- H. Jiang, J. Li, X. Zhao, Y. Zhou, Y. Li, F. Sun, X. Hu, J. Huang, Y. Rui and G. Fan, J. Pharm. Biomed. Anal., 2013, 83, 305–313 CrossRef CAS PubMed.
- U.S. Department of Health and Human Services, Food and Drug Administration, Guidance for Industry: Bioanalytical Method Validation, http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf Search PubMed.
- D. S. W. Palagama, D. Baliu-Rodriguez, A. Lad, B. S. Levison, D. J. Kennedy, S. T. Haller, J. Westrick, K. Hensley and D. Isailovic, J. Chromatogr. A, 2018, 1573, 66–77 CrossRef CAS.
- L. A. Dahabiyeh, D. Tooth, R. W. Carrell, R. J. Read, Y. Yan, F. B. Pipkin and D. A. Barrett, Anal. Bioanal. Chem., 2019, 411, 427–437 CrossRef CAS.
- H. Fouda, M. Nocerini, R. Schneider and C. Gedutis, J. Am. Soc. Mass Spectrom., 1991, 2, 164–167 CrossRef CAS.
- J. H. Gross, Mass Spectrometry, Springer-Verlag, Berlin, 1st edn, 2004 Search PubMed.
- A. Garcia-Ac, S. V. Duy, S. Sauvé, F. Moldovan, V. G. Roullin and X. Banquy, Talanta, 2018, 186, 124–132 CrossRef CAS PubMed.
- P. Hyenstrand, J. S. Metcalf, K. A. Beattie and G. A. Codd, Toxicon, 2001, 39, 589–594 CrossRef CAS.
- O. Chertov, A. Biragyn, L. W. Kwak, J. T. Simpson, T. Boronina, V. M. Hoang, D. R. A. Prieto, T. P. Conrads, T. D. Veenstra and R. J. Fisher, Proteomics, 2010, 4, 1195–1203 CrossRef.
- M. Shimizu, P. Swanson, H. Fukada, A. Hara and W. W. Dickhoff, Gen. Comp. Endocrinol., 2000, 119, 26–36 CrossRef CAS.
- C. Polson, P. Sarkar, B. Incledon, V. Raguvaran and R. Grant, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 785, 263–275 CrossRef CAS.
- J. Ma, J. Shi, H. Le, R. Cho, J. C. jou Huang, S. Miao and B. K. Wong, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 862, 219–226 CrossRef CAS.
- R. S. Tirumalai, K. C. Chan, D. A. Prieto, H. J. Issaq, T. P. Conrads and T. D. Veenstra, Mol. Cell. Proteomics, 2003, 2, 1096–1103 CrossRef CAS PubMed.
- R. Kay, C. Barton, L. Ratcliffe, B. Matharoo-Ball, P. Brown, J. Roberts, P. Teale and C. Creaser, Rapid Commun. Mass Spectrom., 2008, 22, 3255–3260 CrossRef CAS.
Footnote |
| † Shumei Ma contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |