DOI:
10.1039/C9RA05097C
(Paper)
RSC Adv., 2019,
9, 32966-32976
Heterogeneous Fenton-like activity of novel metallosalophen magnetic nanocomposites: significant anchoring group effect†
Received
5th July 2019
, Accepted 8th October 2019
First published on 16th October 2019
Abstract
Two new pyridine and thiolate anchoring groups were prepared to functionalize γ-Fe2O3 nanoparticles for coordinative attachment of simple Fe(III)- and Mn(III)salophen complexes. Four new magnetically recoverable composites were characterized by several analytical techniques such as FT-IR, XRD, TGA, EDS compositional analysis and VSM to confirm superparamagnetic properties. TEM images revealed the nanostructure nature of composites with size ranging between 20 and 40 nm. A heterogeneous advanced oxidation process for degradation of some organic dyes as water pollution compounds using an aqueous solution of H2O2 were successfully exploited. Several key parameters including the metal center in the salophen complex, initial pH, catalyst dosage, H2O2 and dye concentration and temperature were investigated. A significant effect of the anchoring ligand on the degradation efficiency and catalyst stability was documented. The superior catalytic activity and particularly durability of the thiolate-based catalysts were demonstrated in comparison with their Py counterparts. Rate constants of 0.21, 0.17, 0.23 and 0.11 min−1 were obtained for degradation of rhodamine B (RhB), methylene blue (MB), methyl orange (MO), and crystal violet (CV). Finally, a photoluminescence probing technology and radical scavenging measurements were carried out to elucidate the active species involved in the process.
1. Introduction
Many efforts have been devoted to developing technologies that are able to minimize the environmental impact of industrial waste. Numerous chemicals such as dyes are extensively employed to impart color to various industrial products.1 These dyes are the most problematic pollutants of textile wastewaters. This fact occurs because after the reactive dyeing process is finished, more than 15% of the textile dyes are lost in the wastewater stream during the dyeing operation.2 During the last decade, a series of new methods for water and wastewater purification have been developed. Advanced oxidation processes (AOPs) which revolve around the generation of strongly oxidizing hydroxyl free radicals (OH˙) that quickly and non-selectively oxidize a broad range of organic pollutants have received considerable attention.3 H2O2 which produces water as the sole product has been extensively employed in environmentally benign oxidative degradation of organic pollutants via reactive oxygen species (ROS), such as OH˙ and HO2˙/O2˙− after activation by catalysts.4
An emerging field in AOPs is the application of magnetic nanoparticles (MNPs) as catalyst or support to offer easily and efficient isolation of catalyst after degradation of pollutants.5 The application of a magnetic field of low intensity induces the magnetization of the material and thus makes the use of a magnetic force possible, but when the magnetic field is cut off, the magnetization immediately decreases to zero.6 This last point is important for the release of particles after degradation of pollutants from aqueous media. Since, magnetite (Fe3O4) nanoparticles are highly susceptible to be oxidized to maghemite (γ-Fe2O3) nanoparticles in the presence of oxygen,7 the chemical applications of maghemite are significantly on the rising.8 However, because of the high surface energy, the naked magnetic nanoparticles are generally unstable and aggregate easily, which strongly affects their dispersion into aqueous medium. To overcome such limitations, various surface modification methods have been developed to modify the surface of nanoparticles via loading of different organic, inorganic and biological materials to improve the dispersibility, stability, biocompatibility and biodegradability for specific purposes.9 Moreover, the functionalization and surface modification of γ-Fe2O3 could enhance its capability as a support for immobilization of metal complexes, which decreases the catalyst leaching during reactions.10
Due to the crucial axial function of the thiolate groups of cysteine residues in cytochrome P-450, peroxidase, catalase and histidine imidazole in hemoglobin,11,12 more attentions have been paid to use of bio-relevant compounds for anchoring the large metal complexes such as porphyrin and salen complexes as model compounds of heme molecules.13 The mobility of the catalyst and the solidity of the attachment to the support are further benefits to encourage the use of anchoring ligands in immobilization process.14 Recently, as a part of our research projects we attached porphyrin, phthalocyanine and salen complexes to functionalized silica magnetic nanoparticles (SMNP) for catalytic applications in aqueous oxidation of organic compounds.15,16 In this work, we have designed two new anchoring ligands using condensation of a new silica carbonyl compound with 4-aminopyridine (SAPy) and 2-aminoethanethiol (SAET) to attach the Fe(III)- and Mn(III)salophenCl (FeSal, MnSal) complexes to SMNPs and provide an axial ligand for the metal center similar to the fifth iron-porphyrin ligand in heme-containing enzymes.11 The as-prepared magnetic nanocatalysts were employed successfully for dyes degradation using aqueous solution of H2O2 to imitate the peroxidase enzyme reaction. Our results revealed that Fe- and Mn-complexes and Fe2O3 nanoparticles synergistically catalyzed dyes degradation offering a new perspective into the design of new catalyst to control the reaction by mediation of axial ligands and magnetic support together. A systematic investigation on the different parameters such as pH, temperature, concentration of catalyst and oxidant and particularly nature of central metal and anchoring ligand on degradation of some dyes are presented. Bioinspired thiolate-based catalysts demonstrated higher catalytic activity and stability than those of Py counterparts. According to kinetic studies and scavenging experiments a possible mechanism is proposed.
2. Experimental
2.1. Preparation of catalysts
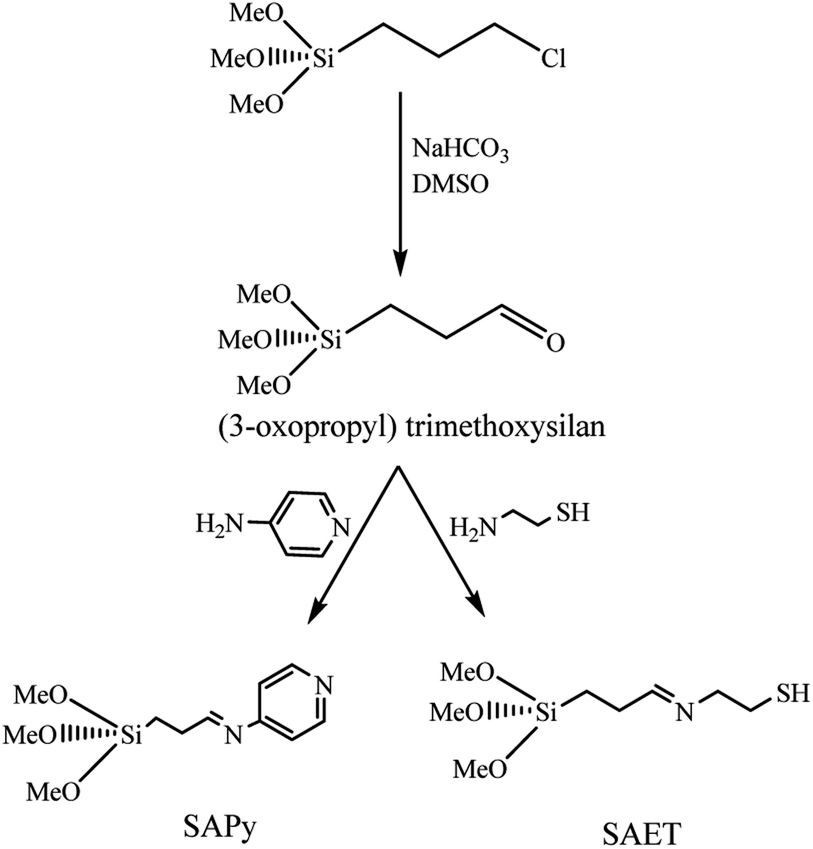
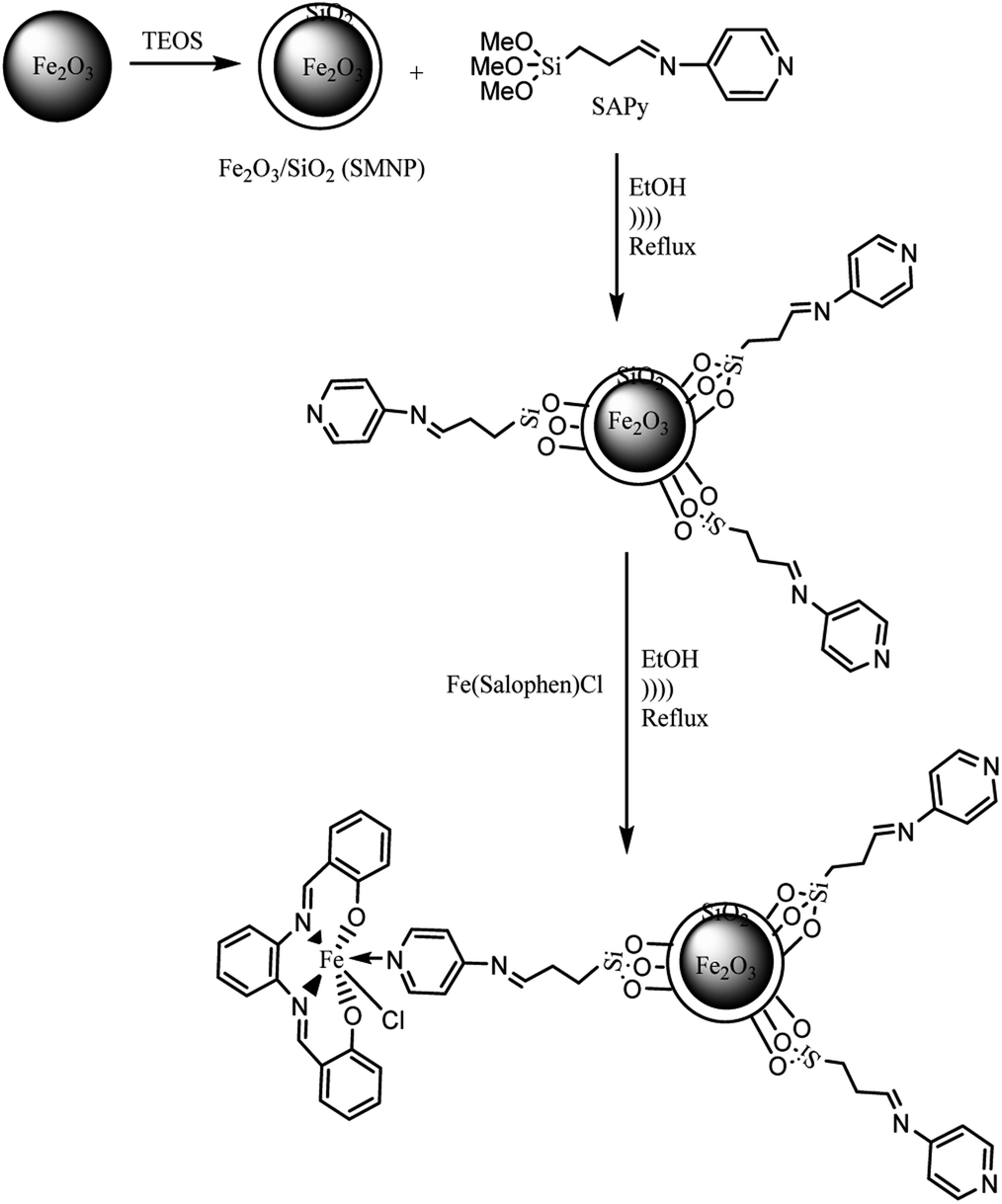
The procedures for the synthesis of γ-Fe2O3 (MNP), silica-coated maghemite nanoparticles (SMNP), Fe(III)salophenCl, and Mn(III)salophenCl complexes are given in ESI.† Herein, step by step procedures for preparation of catalysts are given as depicted in Schemes 1 and 2. Two new imine containing Si compounds have been prepared in this work as anchoring ligand for attachment of Fe(III)- and Mn(III)salophenCl to silica-coated maghemite nanoparticles (SMNP) (Scheme 1). For this purpose, (3-oxopropyl) trimethoxysilan prepared in our research group,17 was reacted with 4-aminopyridine and 2-aminoethanethiol producing two new imine compounds denoted here as SAPy and SAET, respectively.
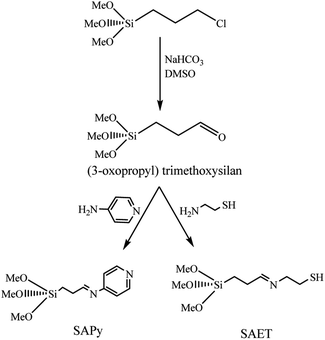 |
| | Scheme 1 Preparation of SAPy and SAET. | |
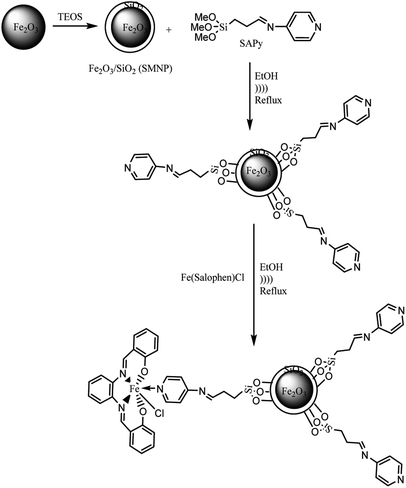 |
| | Scheme 2 Preparation of SMNP@SAPy/FeSal. | |
2.2. Preparation of SAPy
The organosilicon aldehyde, (3-oxopropyl)trimethoxysilan, was synthesized according to method described in our previous report.17 For the synthesis of SAPy, to a solution of (3-oxopropyl)trimethoxysilan (0.8 mL, 5 mmol) in ethanol (10 mL) was gradually added a solution of 4-aminopyridine (0.470 g, 5 mmol) in ethanol (10 mL) at 60 °C under ultrasonic agitation and remained under the same conditions for 60 min (Scheme 1). The product precipitated after 24 hours at room temperature, which was filtered and washed with ethanol and dried in a vacuum desiccator (yield 85%). The structure of product was characterized by FT-IR (Fig. S1†) and the composition and its purity were confirmed by elemental analysis. Cald: C, 51.94; H, 7.13; N, 11.01; found: C, 52.82; H, 6.93; N, 10.87.
2.3. Preparation of SAET
The SAET was prepared according to the same procedure used for preparation of SAPy, except for 4-aminopyridine which was replaced by 2-aminoethanethiol (0.385 g, 5 mmol) (Scheme 1). The structure of product was characterized by FT-IR (Fig. S8†) and the composition and its purity were confirmed by elemental analysis. Cald: C, 40.48; H, 8.07; N, 5.90, found: 41.27; H, 7.89; N, 5.54.
2.4. Preparation of SMNP@SAPy and SMNP@SAET
4.0 g of dry SMNP powder (see ESI†) was mixed with 100 mL ethanol to produce a homogeneous suspension. Then 3.0 g of Schiff base ligands (SAPy or SAET) suspended in 50 mL ethanol was added dropwise to SMNP suspension, followed by sonication for 2 h at 60 °C. Then the mixture was heated to 70 °C for 12 h while stirring (Scheme 2). The desired magnetic products were separated by an external magnetic field, sonicated in ethanol to remove unreacted Schiff base ligands and dried under vacuum.
2.5. The attachment of Fe(III)salophenCl to SMNP@SAPy and SMNP@SAET
To 1.0 g of SMNP@SAPy in 30 mL ethanol, was gradually added 0.5 g Fe(III)salophenCl (2.0 mmol) (in a period of 10 min) under ultrasonic agitation at 60 °C followed by refluxing for 12 h (Scheme 2). The final sample was separated by an external magnetic field, washed with ethanol and dried under vacuum (Scheme 2). The same procedure was used for Mn(III)salophenCl complex.
2.6. Procedure for catalytic dyes degradation
In a typical process, 10 mg SMNP@SAET/FeSal was added into 50 mL aqueous solution containing required concentration of RhB with pH = 3.0 under stirring, followed by addition of 62 μL H2O2 solution (30%). The pH was adjusted by the addition of appropriate amounts of NaOH or HCl solution. At given intervals, an appropriate amount of the suspension was taken out and filtered to remove the solid particles before analysis. The concentration of RhB was measured using spectrophotometer set at an absorbance maximum of 554 nm. The decoloration ratio (DC) of RhB was calculated by eqn (1):| | |
DC = (C0 − Ct)/C0 × 100
| (1) |
Where C0 (mg L−1) and Ct (mg L−1) are the initial and final RhB concentrations, respectively. This was used to evaluate the activity of catalyst.
3. Results and discussion
3.1. Characterization of the SMNP@SAPy/FeSal
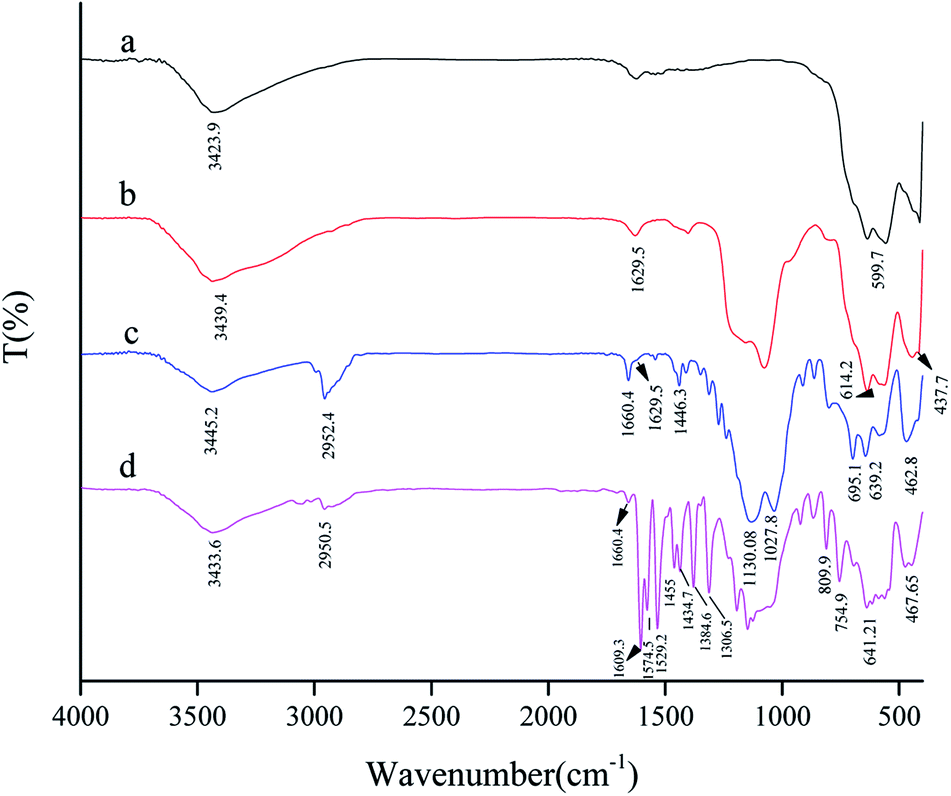
The FT-IR spectrum of the SAPy Schiff base ligand is given in Fig. S1.† Peaks at 1027.8 and 1125.2 cm−1 are assigned to the stretching vibrations of the Si–O bond at the SAPy structure.17 The band at 1659.4 cm−1 is characteristic of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond featuring a strong evidence for the formation of Schiff base ligand.17 The peak at the 2950 cm−1 is attributed to the stretching vibrations of the aliphatic C–H bonds of propyl. The broad band at the 3436.5 cm−1, is related to the O–H stretching of the water in the structure. The comparative FT-IR spectra of MNP, SMNP, SMNP@SAPy and the SMNP@SAPy/FeSal is given in Fig. 1. The bands at 554.4 and 625.7 cm−1 substantiate the presence of the maghemite core (Fig. 1a).18 The broad peaks at about 900–1200 cm−1, assigned to the Si–O–Si and Si–OH stretching vibrations, show the silica coating of maghemite nanoparticles (Fig. 1b).19,20 The bands at 3439 and 1629 cm−1 attributed to O–H stretching and bending of adsorbed water molecules.20 The FT-IR spectra of SMNP@SAPy (Fig. 1c) show the C–H stretching vibration bands of the SAPy group at 2952 cm−1.21 The frequency of CN bond of SAPy Schiff base compound shifted slightly to 1660.4 cm−1 after attachment to maghemite nanoparticles. The sharp peaks at 1130 and 1027 cm−1 are related to Si–O in the structure (Fig. 1c). Immobilization of Fe(III)salophenCl on functionalized magnetic nanoparticles was demonstrated by the appearance of signals at about 800–750 and 1610–1300 cm−1 (Fig. 1d) corresponding to the vibration modes of neat Fe(III)salophenCl (Fig. S2†).22 The CN stretch of Fe(III)salophenCl (1603 cm−1, Fig. S2†) shifted slightly to 1609 cm−1 after coordination to pyridine axial ligand of SAPy (Fig. 1d). The similar results for successful immobilization of Mn(III)salophen complex on magnetic nanoparticles (SMNP@SAPy/MnSal) can be extracted using FT-IR spectra given in SI (Fig. S3,† the peak at 1652.69 cm−1 is related to the CN frequency of imine bonds of Mn(III)salophenCl).
N bond featuring a strong evidence for the formation of Schiff base ligand.17 The peak at the 2950 cm−1 is attributed to the stretching vibrations of the aliphatic C–H bonds of propyl. The broad band at the 3436.5 cm−1, is related to the O–H stretching of the water in the structure. The comparative FT-IR spectra of MNP, SMNP, SMNP@SAPy and the SMNP@SAPy/FeSal is given in Fig. 1. The bands at 554.4 and 625.7 cm−1 substantiate the presence of the maghemite core (Fig. 1a).18 The broad peaks at about 900–1200 cm−1, assigned to the Si–O–Si and Si–OH stretching vibrations, show the silica coating of maghemite nanoparticles (Fig. 1b).19,20 The bands at 3439 and 1629 cm−1 attributed to O–H stretching and bending of adsorbed water molecules.20 The FT-IR spectra of SMNP@SAPy (Fig. 1c) show the C–H stretching vibration bands of the SAPy group at 2952 cm−1.21 The frequency of CN bond of SAPy Schiff base compound shifted slightly to 1660.4 cm−1 after attachment to maghemite nanoparticles. The sharp peaks at 1130 and 1027 cm−1 are related to Si–O in the structure (Fig. 1c). Immobilization of Fe(III)salophenCl on functionalized magnetic nanoparticles was demonstrated by the appearance of signals at about 800–750 and 1610–1300 cm−1 (Fig. 1d) corresponding to the vibration modes of neat Fe(III)salophenCl (Fig. S2†).22 The CN stretch of Fe(III)salophenCl (1603 cm−1, Fig. S2†) shifted slightly to 1609 cm−1 after coordination to pyridine axial ligand of SAPy (Fig. 1d). The similar results for successful immobilization of Mn(III)salophen complex on magnetic nanoparticles (SMNP@SAPy/MnSal) can be extracted using FT-IR spectra given in SI (Fig. S3,† the peak at 1652.69 cm−1 is related to the CN frequency of imine bonds of Mn(III)salophenCl).
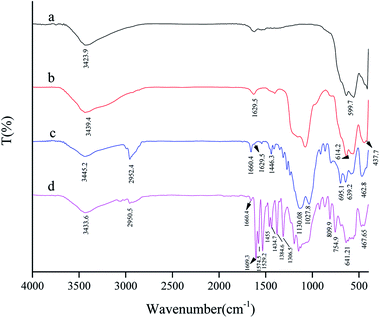 |
| | Fig. 1 FT-IR spectra of MNP (a), SMNP (b), SMNP@SAPy (c) and SMNP@SAPy/FeSal (d). | |
The incorporation of the Msalophen (M = Fe, Mn) complexes into the magnetic catalyst was confirmed by elemental composition determined by EDX (Fig. S4†). The distinct peaks of iron and Mn were appeared clearly alongside carbon, nitrogen, oxygen and silicon in the EDX spectrum. Chlorine is a counter ion for Fe(III) and Mn(III)salophen complexes as evidenced by EDX spectra.
The XRD patterns of SAPy@SMNP and SMNP@SAPy/MSal (M = Fe, Mn) are given in ESI (Fig. S5).† It can be seen clearly that the cubic crystalline structure of iron oxide remains intact after modification with silica, organic compounds as well as transition metal complexes.23,24
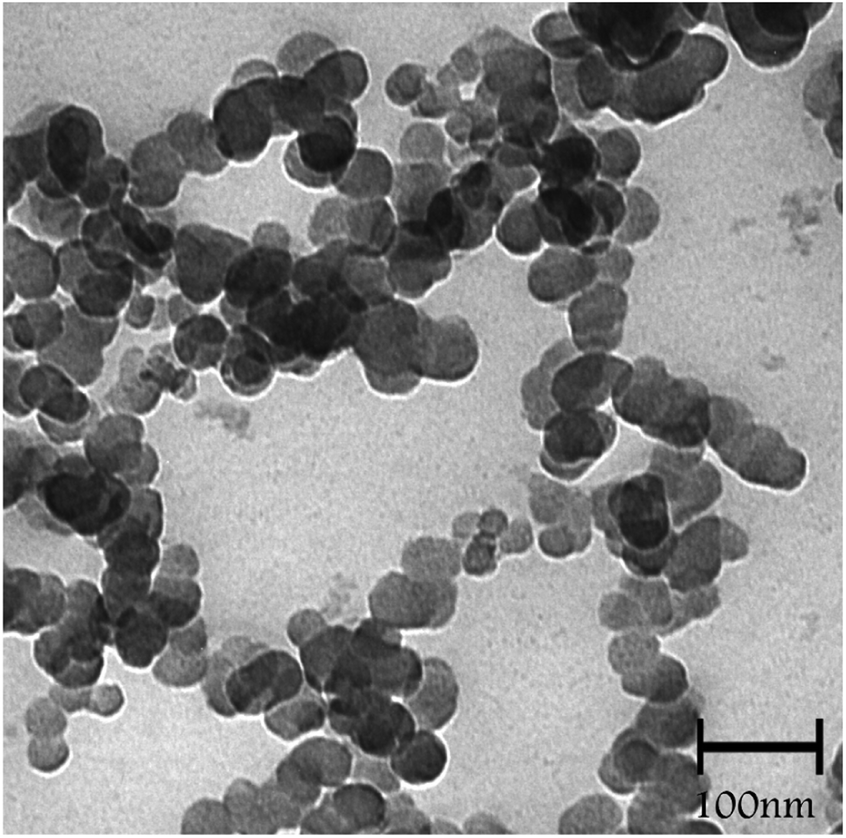
Transmission electron microscopy (TEM) analysis of SMNP@SAPy/FeSal (Fig. 2) revealed the spherical particles ranging in size between 20–40 nm.
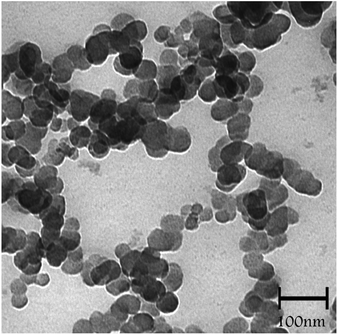 |
| | Fig. 2 TEM image of SMNP@SAPy/FeSal nanocatalyst. | |
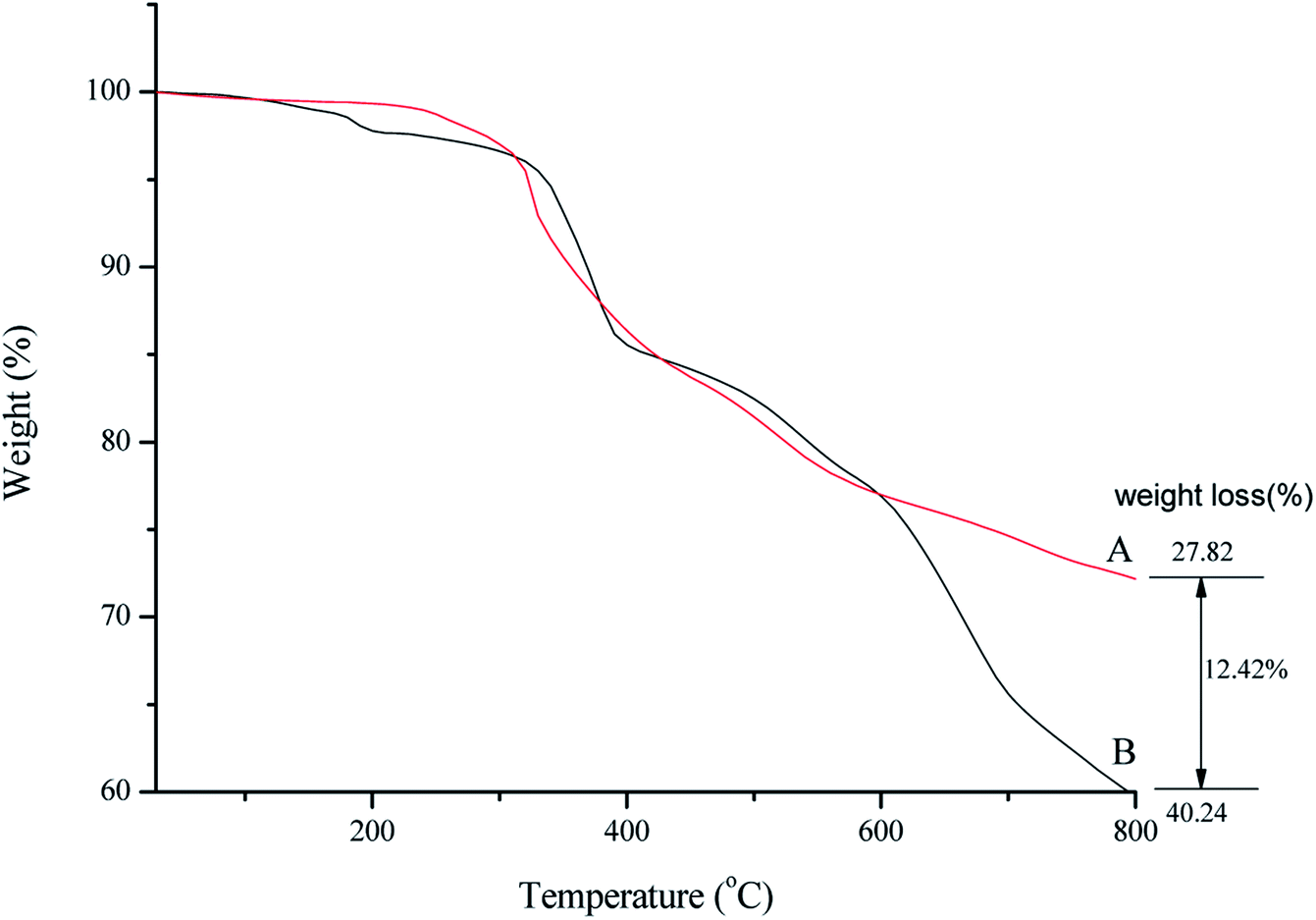
Thermal behavior of SMNP@SAPy/FeSal (Fig. 3B) demonstrated the stability of catalyst up to around 400 °C and after that the degradation of organic compounds including Fe(III)salophenCl occurred.16 A decrease in the weight percentage at about 150–250 °C related to desorption of water molecules from the catalyst surface. Decomposition of organic parts continued until 800 °C. Comparative TGA thermograms of SMNP@SAPy (A) and SMNP@SAPy/FeSal (B) (Fig. 3), shows the same thermal behavior until 600 °C. After that a weight loss of 12.4% occurred for SMNP@SAPy/FeSal (Fig. 3B) which can be rationalized to incorporated Fe(III)salophenCl. According to this result, the amount of Fe supported on the surface of functionalized γ-Fe2O3 is 0.30 mmol g−1 close to result obtained by ICP-AES analysis (0.33 mmol g−1) obtained by difference in iron-containing SMNP@SAPy/FeSal and SMNP@SAPy. The TGA thermograms of SMNP@SAPy/MnSal is also given in ESI (Fig. S6).† The amount of Mn is estimated to be 0.30 mmol g−1 (0.28 mmol g−1 according to ICP-AES analysis).
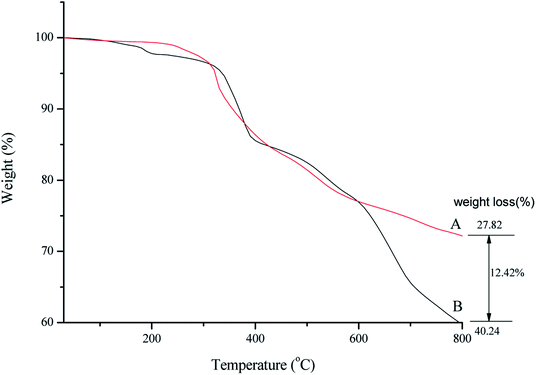 |
| | Fig. 3 TGA thermograms of SMNP@SAPy (A) and SMNP@SAPy/FeSal (B). | |
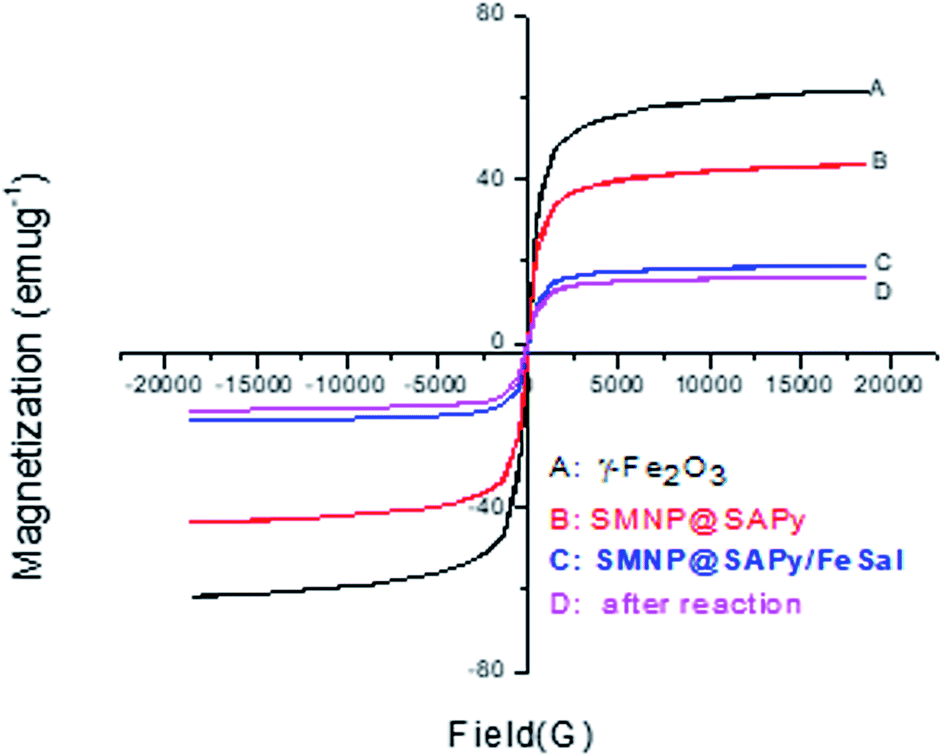
The magnetic properties of the as-synthesized γ-Fe2O3, SMNP@SAPy, SMNP@SAPy/FeSal and recycled nanocatalyst were measured using a magnetometer at 298 K (Fig. 4). The four materials exhibited a superparamagnetic behavior and no obvious remanence was recorded when the applied magnetic field was removed. The magnetization saturation (Ms) values of the as-synthesized γ-Fe2O3, SMNP@SAPy, SMNP@SAPy/FeSal and recycling nanocatalyst were 61.42, 43.35, 18.72, and 16.53 emu g−1, respectively. All synthesized materials shows exactly the same behavior of the γ-Fe2O3 magnetic nanoparticles albeit with less intensity owing to the organic coating on the surface of the magnetic nanoparticles that is the reason for the successful synthesis of the catalyst. Slight change in the magnetization saturation (Ms) value of the reused catalyst (Fig. 4D, 16.53 emu g−1) compared with fresh one (Fig. 4C, 18.72 emu g−1) affirms well that the catalyst preserved its integrity during the oxidation reaction. The magnetic properties of Mn(III)salophenCl attached to SMNP@SAPy is given in ESI (Fig. S7).†
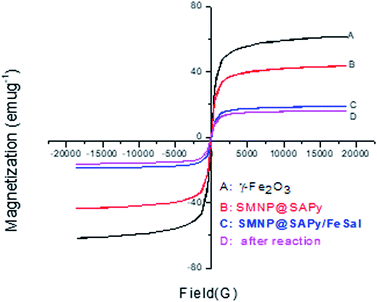 |
| | Fig. 4 Magnetic hysteresis curves of the pristine γ-Fe2O3 (A), SMNP@SAPy (B), SMNP@SAPy/FeSal (C) and recycled nanocatalyst (D) at 298 K. | |
3.2. Synthesis and characterization of bio-inspired SMNP@SAET/Metallosalophen
To investigate the effect of anchoring group, 4-aminopyridine was replaced by 2-aminoethanthiol to react with (3-oxopropyl)trimethoxysilan,17 producing a new imine compound containing a thiolate group denoted as SAET (Scheme 1). Thiolate ligands are of extreme interest in coordination chemistry,25 because of key function of thiolate groups of cysteine residues in iron and zinc enzymes.26 They have also found important applications in the fields of nanoscience.27 By imitating of the peroxidase enzyme structure, coordinative attachment of Fe(III)salophenCl to SAET-functionalized SMNP through thiolate group gave SMNP@SAET/FeSal as a bio-inspired magnetically recoverable composite. The results for characterization of SAET, SMNP@SAET and SMNP@SAET/MSal (M = Fe, Mn) are given in ESI (FT-IR, EDX, XRD, TGA, Fig. S8–S13).† According to FT-IR spectra, the CN frequency of the anchored ligand SAET at 1595.8 cm−1 shifted to 1601.59 cm−1 after coating the SMNP (Fig. S9a and b†). The incorporation of MsalophenCl (M = Fe, Mn) into functionalized magnetic nanoparticles were evidenced by the appearance of new signals in FT-IR spectra depicted in Fig. S9c and d.†22 More evidence was obtained by EDX compositional analysis. Fig. S11† illustrates the distinct peaks of carbon, chlorine, nitrogen, oxygen, silica, sulfur, Fe and Mn in the EDX spectrum that confirmed the successful incorporation of the both complexes on the surface of the magnetic nanoparticles. The amounts of Fe and Mn loaded on SAET-functionalized SMNP were found to be 0.13 and 0.06 mmol g−1, respectively.
3.3. Degradation performance of SMNP@SAET/FeSal
3.3.1 Preliminary tests for RhB degradation. Knowing that H2O2 solution is not able to decompose RhB (Fig. 5),24,28 the reaction mixture subjected to catalytic amount of the as-synthesized SMNP@SAET/FeSal composite compared with parent materials at the same conditions (Fig. 6). The results presented in Fig. 6, clearly confirmed the superior catalytic activity of title magnetically recoverable salophen catalyst.
 |
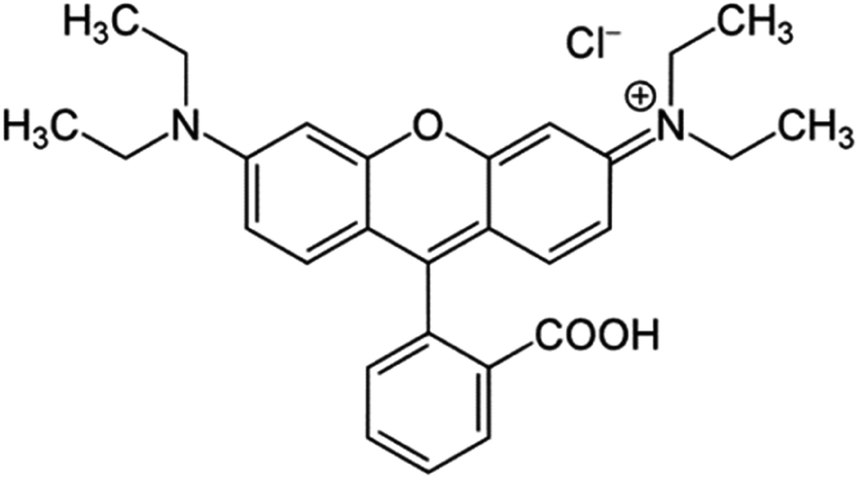
| | Fig. 5 Molecular Structure of the RhB. | |
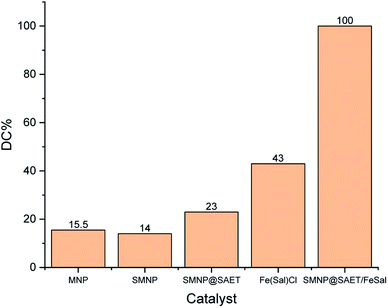 |
| | Fig. 6 Comparing of catalytic activity of SMNP@SAET/FeSal with parent materials. The aqueous solution with a pH of 3.0 containing 2 ppm RhB and 62 μL H2O2 and 2 mmol catalyst were run under air for 70 min. | |
The influences of pH, catalyst dosage and H2O2 concentration on RhB degradation in the presence of SMNP@SAET/FeSal were confirmed. As shown in Table 1, an acidic solution with a pH of 3.0 seems more favorable for degradation activity of catalyst to remove RhB completely after 70 min at 25 °C. The faster formation of Fe(II) at acidic media may be a good reason for such an exhibition considering this fact that, the classic Fenton reaction occurs in the presence of Fe(II) to produce hydroxyl radical from H2O2 (eqn. 2). Thus, the catalytic efficiency of Fe(III)-containing complexes such as peroxidase and the related bioinspired compounds like those synthesized in this work, largely depends upon the rate of FeIII to FeII transformation (eqn. 3).29,30 However, the stability of the Fe(III)salophen complex in the acidic pH,29should be taken into account.
| | |
Fe2+ + H2O2 + H+ → Fe3+ + ·OH + H2O
| (2) |
| | |
Fe3+ + H2O2 → Fe2+ + ·OOH + H+
| (3) |
Table 1 The effect of pH on RhB removal efficiency of SMNP@SAPy/FeSal.a
| pH |
DC (%) |
| The reaction were run under air for 70 min at 25 °C using 10 mg catalyst and 62 μL H2O2. |
| 2 |
95 |
| 3 |
100 |
| 4 |
90 |
| 5 |
89 |
| 6 |
45 |
| 7 |
38 |
| 8 |
29 |
| 9 |
15 |
The effect of the initial pollutant concentration was also investigated. According to the results presented in Table 2, the method is efficiently applicable for a solution of 2 to 20 ppm RhB within 70–280 min using 62 μL H2O2 and 10 mg of solid catalyst (0.026 mmol Fesalophen complex). Degradation rate was dependent to catalyst loading and reduced when the lower catalyst was used. For example, removal of 2 ppm RhB took 70 and 210 min using 10 and 5 mg catalyst. Moreover, for 50 ppm RhB, using 1.5 mL H2O2 and 10 and 20 mg catalyst, 23.5 and 40% removal efficiency were observed within 280 min respectively, indicating that the method is amenable for scale up procedure.
Table 2 The screening of RhB, H2O2 and catalyst concentrationa
| RhB (ppm) |
Time (min) |
| 70 |
140 |
210 |
280 |
| The reaction were run in a solution of pH 3.0 under air at 25 °C using 62 μL H2O2 and 10 mg SMNP@SAET/FeSal catalyst. 6, 32, 81 and 93% at 30, 40, 50, and 60 min respectively. 5 mg catalyst. 1.5 mL H2O2. 1.5 mL H2O2 and 20 mg catalyst. |
| 2 |
100b |
— |
— |
— |
| 2c |
19 |
88 |
100 |
|
| 5 |
17 |
93 |
95 |
— |
| 10 |
9 |
46 |
79 |
87 |
| 20 |
0 |
8 |
44 |
74.5 |
| 50d |
5 |
16 |
18 |
23.5 |
| 50e |
8 |
22 |
32 |
40 |
3.4. Effect of reaction temperature
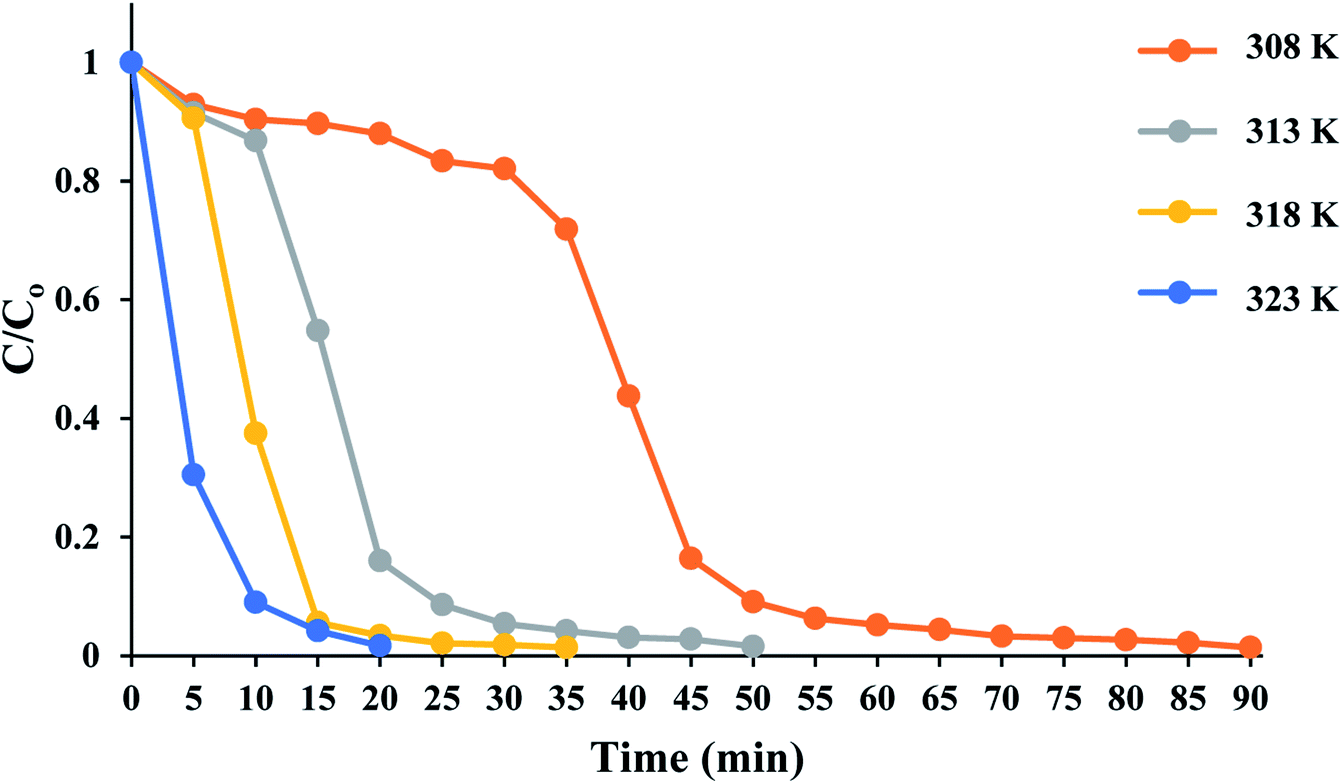
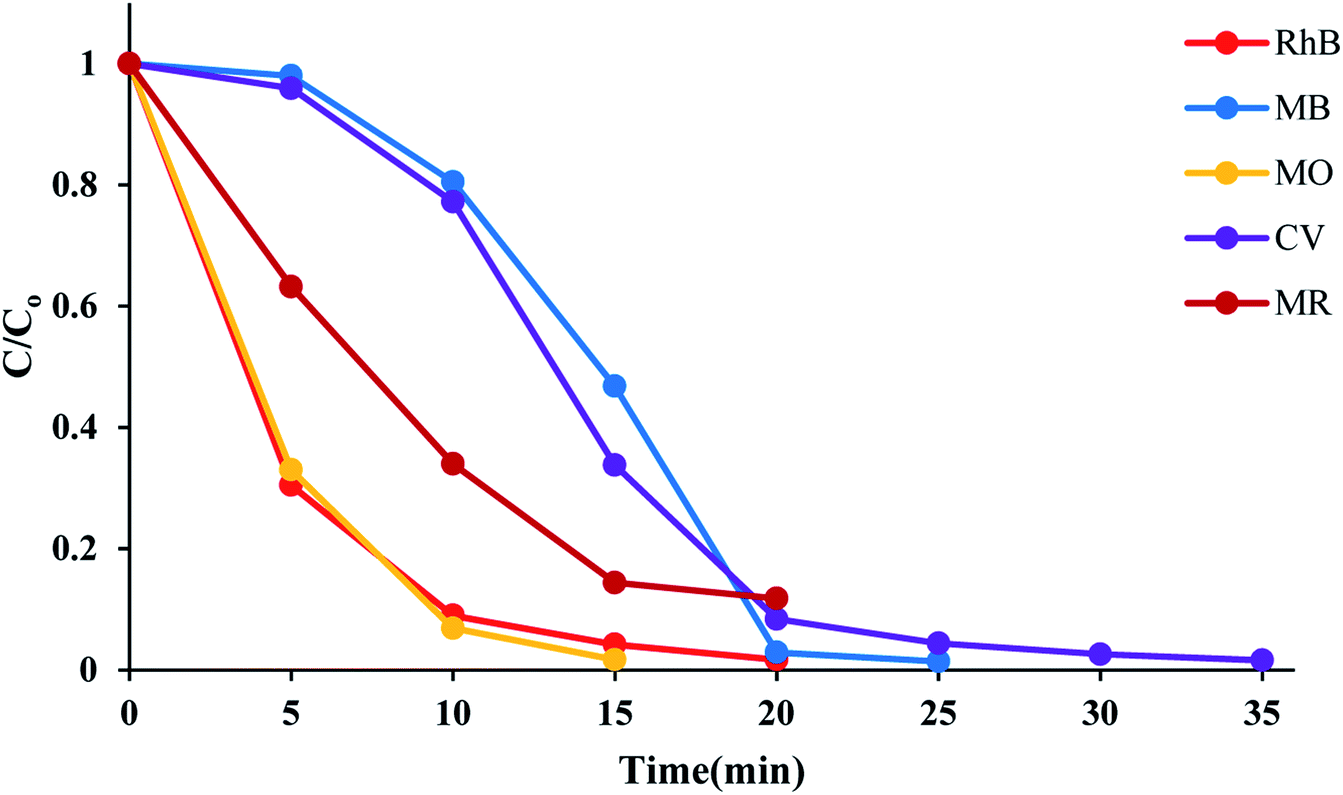
The time course of RhB degradation in SMNP@SAET/FeSal/H2O2 system at different temperature (35–50 °C) is depicted in Fig. 7. The degradation of RhB significantly accelerated by rising the temperature and a lag time observed in the degradation reaction at lower temperatures could be rationalized to retarded transformation of FeIII to FeII (eqn 2),30 and consequently slow formation of active species. When the solution was heated from 25 to 50 °C, the reaction accelerated 100-fold and the reaction time decreased from 210 to 20 min for a solution of 5 ppm RhB (pseudo-first-order rate constant increased from 0.0016 to 0.2 min−1, Fig S14†). It could be caused by facile formation of active oxidants such as radical OH˙,31 or high valent FeO species,32 under thermal activation. Nevertheless, further heating dropped the removal efficiency to 81 and 74% at 60 and 70 °C resulting from H2O2 destruction at high temperatures.33 We also evaluated title catalytic systems for oxidative degradation of other organic dyes such as methylene blue (MB), methyl orange (MO), and crystal violet (CV). According to results presented in Fig. 8 and Table 3, all pollutants can also be decomposed thoroughly and effectively within 25 min using Fe containing catalyst at the same condition used for RhB. The pseudo-first-order rate constant of dyes degradation (Table 3 and Fig. S15†) revealed that the complete degradations of dyes in this work occurred in a rapid manner. For example, the k/min for degradation of RhB in title system are 0.087 and 0.21 min−1 at 40 and 50 °C, respectively, which are much larger than those reported for magnetic nanoparticle containing systems at different temperatures (6–200 fold).24,34 They are also consistent with those reported for photodegradation of organic dyes in the presence of Fe(III)-complex/H2O2 under visible light irradiation (k = 0.17 and 0.084 min−1 for RhB and CV, respectively).29
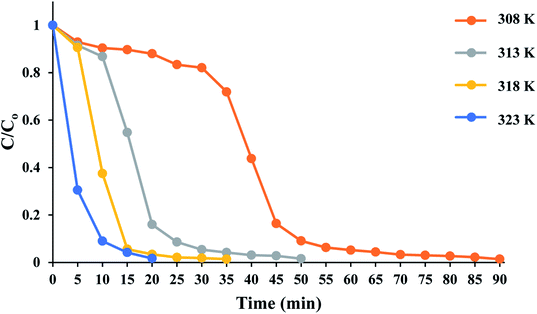 |
| | Fig. 7 Time course of RhB degradation at different temperatures. The reactions containing RhB (50 mL 5 ppm), 10 mg SMNP@SAET/FeSal catalyst, and 62 μL H2O2 were run under air. | |
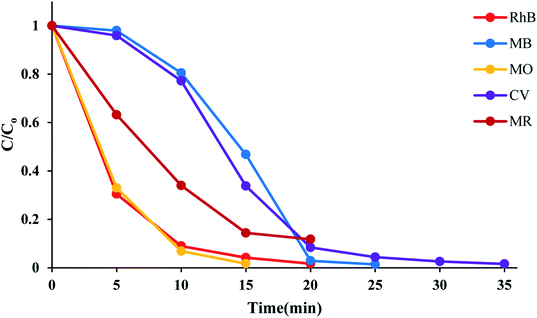 |
| | Fig. 8 Time course of dyes degradation at 50 °C. The reactions containing 50 mL, 5 ppm dye solution, 10 mg SMNP@SAET/FeSal catalyst, 62 μL H2O2 were run under air. | |
Table 3 The pseudo-first-order rate constants of dyes degradation in SMNP@SAET/FeSal/H2O2 systema
| Dye |
T (°C) |
Time (min) |
k min−1 |
| 50 mL solution with pH of 3.0 containing 5 ppm dye, 10 mg SMNP@SAET/FeSal catalyst, 62 μL H2O2 were run under air. |
| RhB |
25 |
210 |
0.0016 |
| RhB |
35 |
90 |
0.014 |
| RhB |
40 |
50 |
0.087 |
| RhB |
50 |
20 |
0.21 |
| MB |
50 |
20 |
0.17 |
| MO |
50 |
15 |
0.23 |
| CV |
50 |
25 |
0.11 |
3.5. The effect of anchoring ligand and salophen metal center
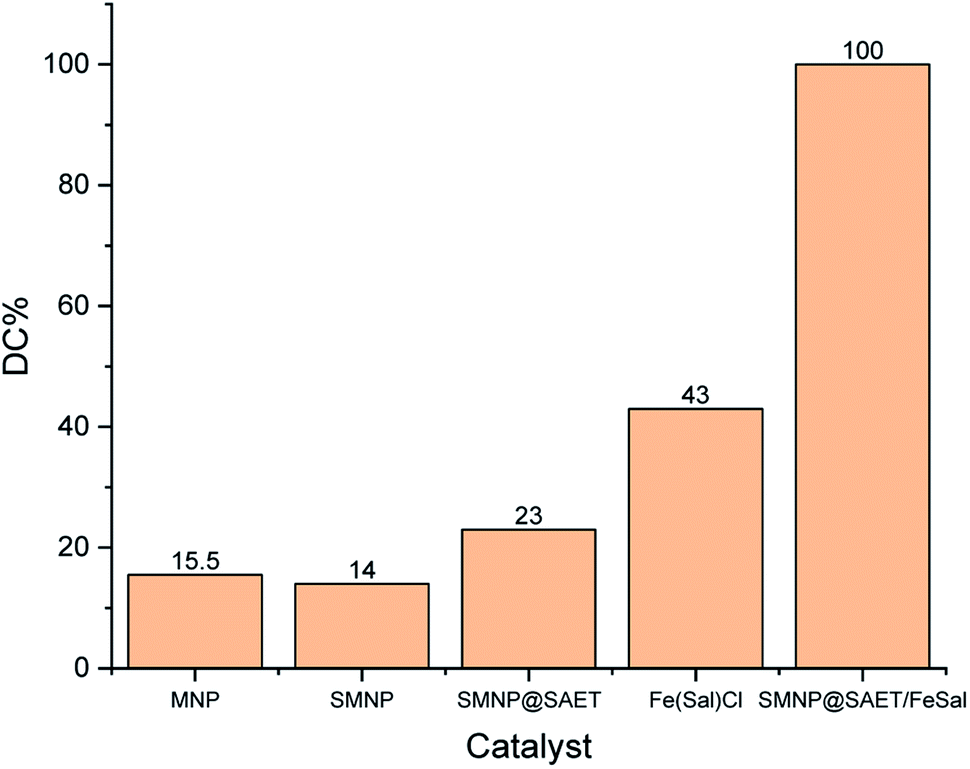
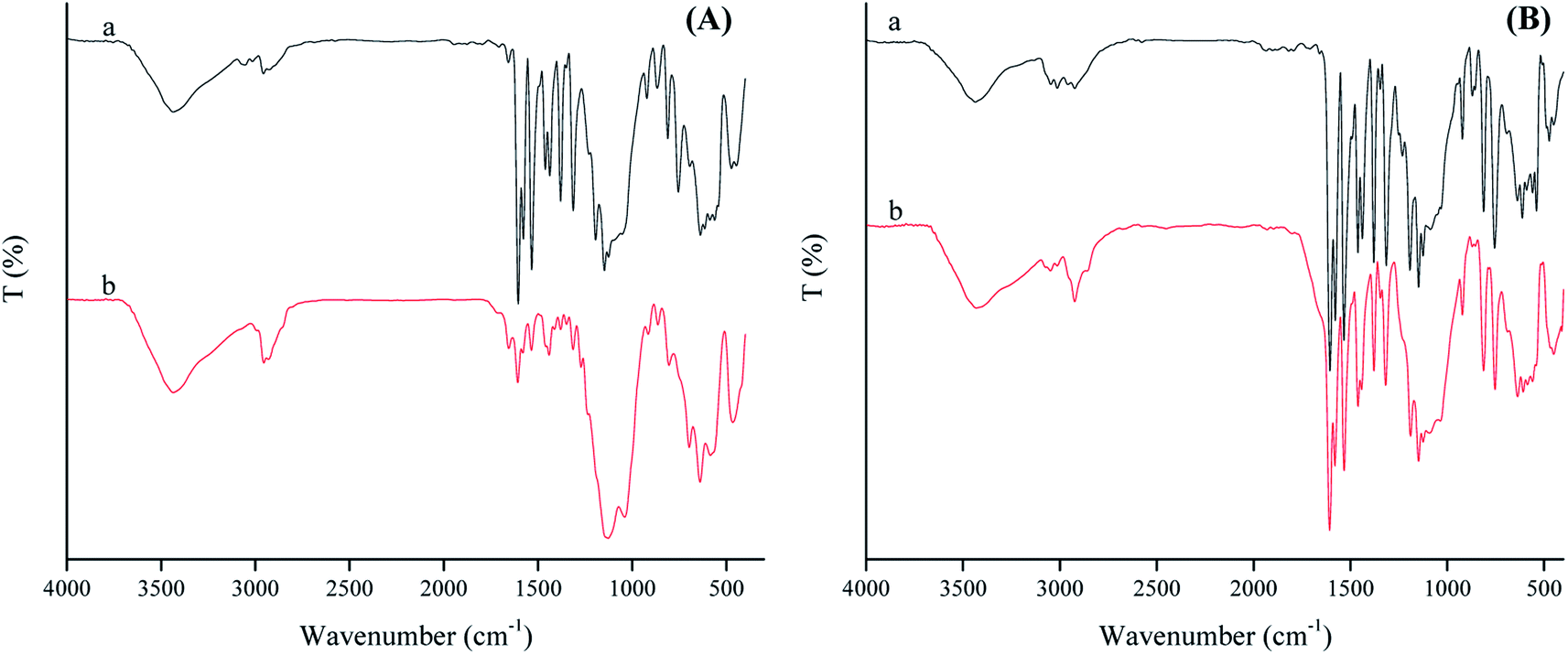
When SAET containing strong π-donor –SH group (Scheme 1) was replaced by SAPy having weak π-donor Py (Scheme 1), the catalytic degradation performance of magnetically recoverable catalyst decreased to less than half (46.5%) under the same conditions (Table 4, entry 1 and 2). This result clearly shows that how a ferric site surrounded by thiolate ligand,35 surpass the corresponding catalysts containing Py.36 However, the activity of Mn-complex attached to thiolate ligand was 4 times lower than iron counterpart (26, entry 3) while, the corresponding Py axial ligand (SAPy) promoted partially its efficiency compared to iron catalyst (56 vs. 46 entries 2 and 4) (see ESI† for characterization of SMNP@SAPy/MnSal, SMNP@SAET/MnSal). The higher catalytic activity of iron containing catalyst than that of Mn counterpart may be rationalized to Fenton mechanism mediated by iron center (eqn. 2). However, when H2O2 was replaced by more active Na2S2O8 (Table 4, entries 5–8), both Fe and Mn sites surrounded by thiolate ligand SAET showed superior activity (100 and 82% respectively) than those containing SAPy (46 and 48% respectively). Further and more important advantage of using SAET observed in this work is excellent durability of catalyst. The inspection of FT-IR spectra of used iron catalysts revealed some changes in vibration bands of SMNP@SAPy/FeSal after reaction, while the spectra of thiolate containing counterpart, remained quite unchanged (Fig. 9). These results clearly demonstrate that anchoring ligand structure and coordinated atom help to change the ligand environment of central metal and then enhance catalytic efficiency and stability.
Table 4 The effect of anchoring group and metal center of M(III)salophenCla
| Entry |
SMNP catalyst |
Oxidant |
DC (%) |
| The 50 mL solution of 2 ppm RhB at pH = 3.0 were run under air for 70 min using 2 mmol catalyst (based on metal center content of MsalophenCl) attached to nanocomposites, 62 μL H2O2. The reaction were run using 0.62 mmol (147 mg) Na2S2O8 at the same conditions used for H2O2. |
| 1 |
@SAET/FeSal |
H2O2 |
100 |
| 2 |
@SAPy/FeSal |
H2O2 |
46.5 |
| 3 |
@SAET/MnSal |
H2O2 |
26 |
| 4 |
@SAPy/MnSal |
H2O2 |
56 |
| 5b |
@SAET/FeSal |
Na2S2O8 |
100 |
| 6b |
@SAPy/FeSal |
Na2S2O8 |
46 |
| 7b |
@SAET/MnSal |
Na2S2O8 |
82 |
| 8b |
@SAPy/MnSal |
Na2S2O8 |
48 |
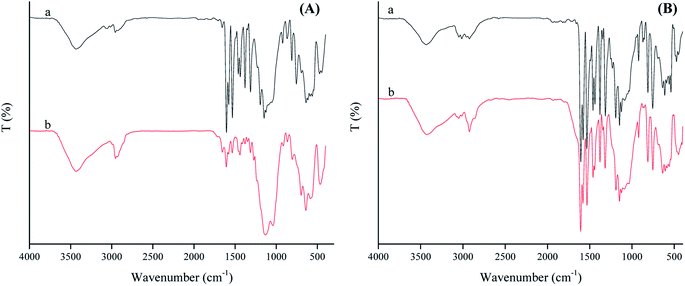 |
| | Fig. 9 FT-IR spectrum represents fresh (a) and reused (b) spectrum of SMNP@SAPy/FeSal (A) and SMNP@SAET/FeSal (B) from RhB degradation reaction using H2O2. | |
The electron π-donating ability and “push” capability of the S atom in thiolate group is stronger than the N atom in Pys and amines,37 making a metal center to be more appropriate for oxidation reactions results in the easier production of active oxidant such as OH˙ radicals. These advantages make the catalyst more active and persistent against the oxidative degradation. Moreover, the lone pair of S atom is prone to form hydrogen bonding with H2O2 adjacent to catalytic site which facilitates bond cleavage of peroxide.38 Thus, Fe(III)salophenCl anchored on thiolate-functionalized SMNP is an effective catalyst for removal of different structurally dyes pollutants, with further advantage of high catalyst durability.
3.6. Radical quenching studies and possible degradation mechanism
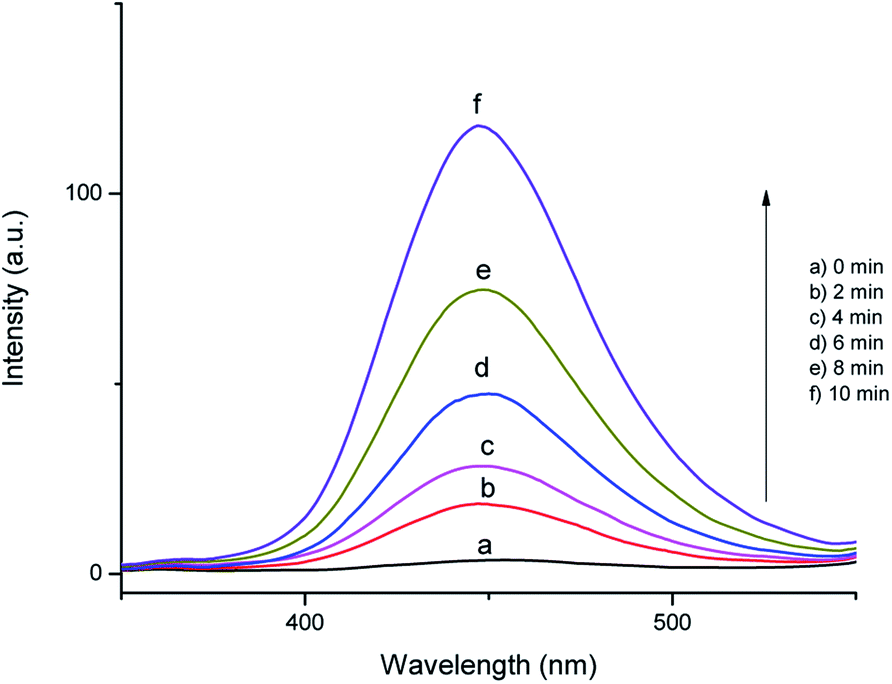
In order to identify the reactive oxygen species formed in the SMNP@SAET/FeSal catalyst/H2O2-system, the photoluminescence probing technology and radical scavenging measurements were conducted. Disodium salt of the terephthalic acid (NaTA) could react with OH• to furnish 2-hydroxy terephthalic acid (HTA), which relies on the PL signal at 425 nm. This reaction is unaffected by the presence of other reactive species such as H2O2, HO2˙ and O2˙−, so it could be used as a sensitive probe in detecting OH˙ radicals.39 Fig. 10 shows the fluorescence spectra of the solution containing the SMNP@SAET/FeSal/H2O2 system and NaTA. The control experiments confirmed that there is no NaTA fluorescence peak without the H2O2 (Fig. 10a). It can be seen that the fluorescence intensity increases sharply within 10 min, implying that OH radicals were indeed generated in the system. Also, when we have used isopropanol as an effective hydroxyl radical scavenger, the degradation rate of dye gets reduced to 45% in the presence of the catalyst and H2O2 providing a strong evidence for involvement of hydroxyl radicals in degradation process.40 According to these experiments, Fe(III) is reduced to Fe(II) in the presence of H2O2 followed by reaction with H2O2 to produce Fe(III) and hydroxyl radicals i.e. a classic Fenton reaction. Nevertheless, an excess amount of the isopropanol (124 mM) didn't stop the degradation process completely and DC percentage reached down ultimately to 36% indicating the involvement of secondary oxidant in this reaction. The contribution of O2˙− to the degradation of RhB was confirmed by addition of p-benzoquinone leading to a 40% decrease in decoloration rate.40 The involvement of oxo species Fe(V) or Fe(IV) cation radical as well-known active species in peroxidase-like catalysis mediated by salen and porphyrin metal complexes in the presence of H2O2 may be a suggestion.29,32 Delayed reactions at lower temperatures in this work (Fig. 7) is related to formation of less reactive FeIII–OOH species. At higher temperature, the formation of the active high-valent iron oxo species is favored32 and accordingly degradation of RhB was triggered from the beginning at 50 °C. The trace of GC-MS analysis of intermediates formed during degradation of RhB are shown in Fig. S16.† The lack of any peak with molecular mass of 433 confirmed quite removal of RhB. Moreover, no evidences for N-de-ethylated intermediates from RhB degradation were observed (molecular mass of 415, 387, 359, and 331 for one, di, tri and fully N-de-ethylated RhB products, respectively). More evidence was obtained by UV-vis spectral change of RhB during degradation (Fig. S17†). The characteristic absorbance peak of RhB at 554 nm gradually decreased without any peak shift, ruling out any N-deethylation of RhB (λ = 498 nm).41 Thus, SMNP@SAET/FeSal is efficient enough to degrade the chromophore of the dye (RhB) successfully.42
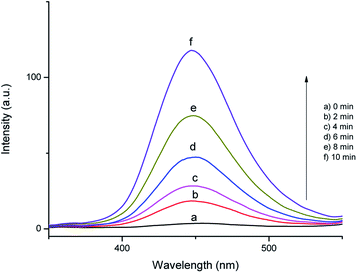 |
| | Fig. 10 The time course of photoluminescence spectra of the RhB solution at pH 3.0 containing 10 mg SMNP@SAET/FeSal catalyst, 10 mM NaTA, and 12.0 mM H2O2 at 50 °C. | |
3.7. Recycling of the catalyst
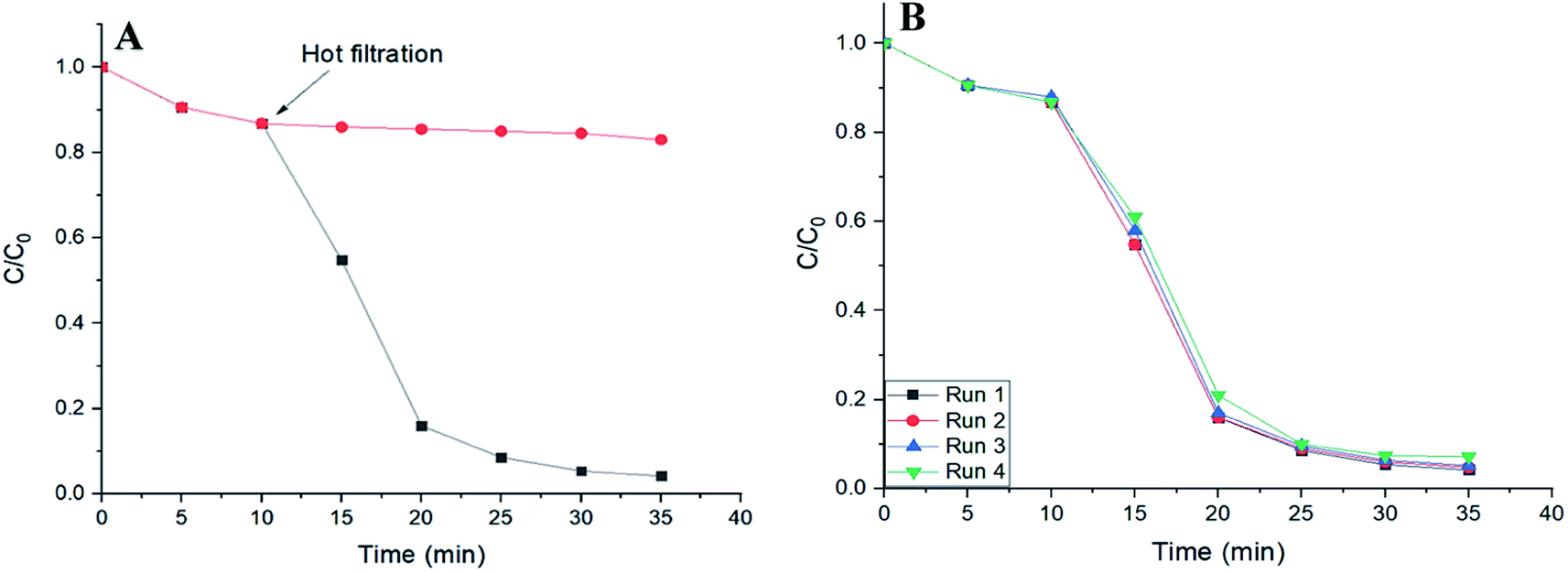
To verify the heterogeneous nature of catalysis in this work, hot filtration tests were performed. A control reaction was carried out in the presence of SMNP@SAET/FeSal solid catalyst. When the degradation rate of RhB reached to 15% at 10 min, the solid was removed at the reaction temperature and the filtrate allowed to stir under the reaction temperature. As depicted in Fig. 11A, no further noticeable degradation was occurred. This indicates that no active species was present in the supernatant. ICP analysis of the filtrate showed trace iron leaching (1.8%) which may be resulted from both Fe2O3 core and Fe(III)salophenCl. The second point was the deactivation and reusability of the catalyst. After each reaction, the catalyst could be easily separated and recovered from the reaction mixture by an external magnet and reused in subsequent reactions with just 3% loss of activity after 4 runs (Fig. 11B). These results confirmed the high reusability performance of title magnetically separable nanocatalyst in dye decolorization (Fig. 11B) as well as the structural integrity and durability of SMNP@SAET/FeSal catalyst as evidenced by FT-IR spectra (Fig. 9B). These advantages highlight the merit of as-synthesized nanocomposites in comparison with previously reported catalysts, in terms of catalyst loading (0.026 mM), reusability and stability of catalyst, efficiency and degradation rate of catalytic system and conditions used in the degradation of organic dyes.24,29,34,43
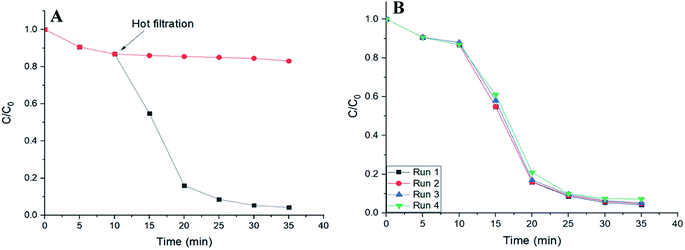 |
| | Fig. 11 (A) Time plot for degradation of 50 mL RhB solution (2 ppm) using 62 μL H2O2 in the presence of SMNP@SAET/FeSal at 40 °C. (B) Four runs recovery of SMNP@SAET/FeSal catalyst for the degradation of RhB under the same conditions. | |
4. Conclusion
In conclusion, new thiolate- and Py-functionalized γ-Fe2O3 nanoparticles were prepared and used for coordinative anchoring of Fe(III) and Mn(III)salophen complexes. Four novel magnetically recoverable nanocomposites were characterized completely. Their catalytic efficiency in activation of H2O2 for degradation of organic dyes as water pollution through a heterogeneous advanced oxidation process (AOP) was evaluated. The decolorization efficiency was influenced by several parameters, such as temperature, the initial pH, catalyst dosage, H2O2 and dyes concentration. Moreover, we found that the catalytic efficiency of Fe- and Mn–salophen complexes were much more strongly affected by anchoring ligand environments. Particularly, π-donor thiolate anchoring ligand promoted markedly the efficiency and stability of Fe(III)salophen catalysts evidenced by experimental results and spectral data. A Fenton-like mechanism involving hydroxyl radicals was postulated according to fluorescence spectra of terephthalic acid as well as scavenging experiments. However, involvement of any secondary oxidant such as superoxide radical as well as high valent oxo iron should be taken into account. Rate constants for degradation of different dyes were larger than those reported for the related systems. The magnetically separable iron-based nanocatalysts proved to act heterogeneously with high reusability and excellent durability evidenced by FT-IR spectra. Thus, the present work offers efficient, economical, environment-friendly and easy separable catalysts to treat environmental contaminants which is an enduring attention in recent years due to their great potential for environmental protection and remediation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Support of this work by research council of University of Birjand and the “Iran National Science Foundation” (grant no. 96005005) is highly appreciated. We would like to thank Prof. Dr Philipp Kurz and Sabine Zuelsdorf from Albert-Ludwigs University of Freiburg for friendly collaboration in some analysis.
References
- F. H. AlHamedi, M. A. Rauf and S. S. Ashraf, Desalination, 2009, 239, 159–166 CrossRef CAS.
- Z. Qiu, Y. He, X. Liu and S. Yu, Chem. Eng. Process., 2005, 44, 1013–1017 CrossRef CAS.
- N. Daneshvar, D. Salari and A. R. Khataee, J. Photochem. Photobiol., A, 2003, 157, 111–116 CrossRef CAS.
-
(a) J. An, L. Zhu, Y. Zhang and H. Tang, J. Environ. Sci., 2013, 25, 1213–1225 CrossRef CAS;
(b) N. M. Mahmoodi, Mater. Res. Bull., 2013, 48, 4255–4260 CrossRef CAS;
(c) G. Y. Mao, F. X. Bu, W. Wang, D. M. Jiang, Z. J. Zhao, Q. H. Zhang and J. S. Jiang, Desalin. Water Treat., 2016, 57, 9226–9236 CrossRef CAS.
-
(a) E. Casbeer, V. K. Sharma and X. Z. Li, Sep. Purif. Technol., 2012, 87, 1–14 CrossRef CAS;
(b) S. Xu, D. Feng and W. Shangguan, J. Phys. Chem. C, 2009, 113, 2463–2467 CrossRef CAS;
(c) C. H. Chen, Y. H. Liang and W. D. Zhang, J. Alloys Compd., 2010, 501, 168–172 CrossRef CAS;
(d) J. Qiu, C. Wang and M. Gu, Mater. Sci. Eng., B, 2004, 112, 1–4 CrossRef.
-
(a) A. F. Ngomsik, A. Bee, M. Draye, G. Cote and V. Cabuil, C. R. Chim., 2005, 8, 963–970 CrossRef CAS;
(b) S. Thurm and S. Odenbach, J. Magn. Magn. Mater., 2002, 252, 247–249 CrossRef CAS.
-
(a) C. Yang, J. Wu and Y. Hou, Chem. Commun., 2011, 47, 5130–5514 RSC;
(b) S. Chen, Z. Xu, H. Dai and S. Zhang, J. Alloys Compd., 2010, 497, 221–227 CrossRef.
-
(a) Q. Zhang, X. Yang and J. Guan, ACS Appl. Nano Mater., 2019, 2, 4681–4697 CrossRef CAS;
(b) N. Panda, H. Sahoo and S. Mohapatra, J. Hazard. Mater., 2011, 1, 359–365 CrossRef;
(c) A. Asfaram, M. Ghaedi, S. Hajati and A. Goudarzi, Ultrason. Sonochem., 2016, 32, 418–431 CrossRef CAS;
(d) L. Yue, K. Wang, J. Guo, J. Yang, X. Luo, J. Lian and L. Wang, J. Ind. Eng. Chem., 2014, 20, 725–731 CrossRef CAS;
(e) S. Sobhani and M. Honarmand, Applied Catalysis, A: General, 2013, 467, 456–462 CrossRef CAS;
(f) E. Rafiee and S. Eavani, Green Chem., 2011, 13, 2116–2122 RSC.
-
(a) L. Wang, H. Qiu, C. Liang, P. Song, Y. Han, Y. Han, J. Gu, J. Kong, D. Pan and Z. Guo, Carbon, 2019, 141, 506–514 CrossRef CAS;
(b) J. Gu, L. Wang, C. Liang, Q. Zhuang and J. Kong, J. Alloys Compd., 2018, 745, 430–435 CrossRef CAS;
(c) Y. Huangfu, C. Liang, Y. Han, H. Qiu, P. Song, L. Wang, J. Kong and J. Gu, Compos. Sci. Technol., 2019, 169, 70–75 CrossRef CAS;
(d) C. Liang, P. Song, A. Ma, X. Shi, H. Gu, L. Wang, H. Qiu, J. Kong and J. Gu, Compos. Sci. Technol., 2019, 181, 107683 CrossRef;
(e) L. M. Rossi, N. J. Costa, F. P. Silva and R. Wojcieszak, Green Chem., 2014, 16, 2906–2933 RSC;
(f) S. Palanisamy and Y. M. Wang, Dalton Trans., 2019, 48, 9490–9515 RSC;
(g) J. K. Xu, F. F. Zhang, J. J. Sun, J. Sheng, F. Wang and M. Sun, Molecules, 2014, 19, 21506–21528 CrossRef.
-
(a) P. Shi and N. Ye, Talanta, 2015, 143, 219–225 CrossRef CAS;
(b) M. Araghi and F. Bokaei, Polyhedron, 2013, 53, 15–19 CrossRef CAS;
(c) Y. Wang, P. Jiang, W. Zhang and J. Zheng, Appl. Surf. Sci., 2013, 270, 531–538 CrossRef CAS;
(d) F. Jia, L. Wu, J. Meng, M. Yang, H. Kong, T. Liu and H. Xu, J. Mater. Chem., 2009, 19, 8950–8957 RSC;
(e) Z. B. Liu, J. G. Tian, Z. Guo, D. M. Ren, F. Du, J. Y. Zheng and Y. S. Chen, Adv. Mater., 2008, 20, 511–515 CrossRef CAS.
- T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS.
- S. Yoshioka, S. Takahashi, H. Hori, K. Ishimori and I. Morishima, Eur. J. Biochem., 2001, 268, 252–259 CrossRef CAS.
- K. U. Ingold, P. A. MacFaul and B. Meunier, ed. Meunier B, Biomimetic Oxidations Catalyzed by Transition Metal Complexes, World Scientific Publishing and Imperial College Press, London, 2000 Search PubMed.
- E. Brulé and Y. R. De Miguel, Org. Biomol. Chem., 2006, 4, 599–609 RSC.
-
(a) A. Rezaeifard, M. Jafarpour, P. Farshid and A. Naeimi, Eur. J. Inorg. Chem., 2012, 2012, 5515–5524 CrossRef CAS;
(b) A. Rezaeifard, M. Jafarpour, A. Naeimi and R. Haddad, Green Chem., 2012, 14, 3386–3394 RSC;
(c) A. Rezaeifard, P. Farshid, M. Jafarpour and G. K. Moghaddam, RSC Adv., 2014, 4, 9189–9196 RSC.
- A. Rezaeifard, M. Jafarpour, A. Farrokhi, S. Parvin and F. Feizpour, RSC Adv., 2016, 6, 64640–64650 RSC.
- M. Jafarpour, A. Rezaeifard, V. Yasinzadeh and H. Kargar, RSC Adv., 2015, 5, 38460–38469 RSC.
- K. Do Kim, S. S. Kim, Y. H. Choa and H. T. Kim, J. Ind. Eng. Chem., 2007, 13, 1137–1141 Search PubMed.
- J. H. Cai, J. W. Huang, P. Zhao, Y. J. Ye, H. C. Yu and L. N. Ji, J. Sol-Gel Sci. Technol., 2009, 50, 430–436 CrossRef CAS.
- C. Yuan, Z. Huang and J. Chen, Catal. Lett., 2011, 141, 1484–1490 CrossRef CAS.
-
(a) L. Saikia, D. Srinivas and P. Ratnasamy, Microporous Mesoporous Mater., 2007, 104, 225–235 CrossRef CAS;
(b) P. Das, A. R. Silva, A. P. Carvalho, J. Pires and C. Freire, J. Mater. Sci., 2009, 44, 2865–2875 CrossRef CAS.
- E. Kadwa, M. D. Bala and H. B. Friedrich, Appl. Clay Sci., 2014, 95, 340–347 CrossRef CAS.
-
(a) S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204–8205 CrossRef CAS;
(b) B. Z. Tang, Y. Geng, J. W. Y. Lam, B. Li, X. Jing, X. Wang, F. Wang, A. B. Pakhomov and X. X Zhang, Chem. Mater., 1999, 11, 1581–1589 CrossRef CAS.
- N. Wang, L. Zhu, M. Wang, D. Wang and H. Tang, Ultrason. Sonochem., 2010, 17, 78–83 CrossRef CAS PubMed.
- A. Kreider-Mueller, P. J. Quinlivan, J. S. Owen and G. Parkin, Inorg. Chem., 2017, 56, 4643–4653 CrossRef.
-
(a) R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef CAS;
(b) W. N. Lipscomb and N. Sträter, Chem. Rev., 1996, 96, 2375–2434 CrossRef CAS.
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC.
-
(a) N. Jaafarzadeh, A. Takdastan, S. Jorfi, F. Ghanbari, M. Ahmadi and G. Barzegar, J. Mol. Liq., 2018, 256, 462–470 CrossRef CAS;
(b) X. Du, J. Wan, J. Jia, C. Pan, X. Hu, J. Fan and E. Liu, Mater. Des., 2017, 119, 113–123 CrossRef CAS.
- S. Gazi, A. Rajakumar and N. P. Singh, J. Hazard. Mater., 2010, 183, 894–901 CrossRef CAS.
-
(a) C. P. Huang, Y. F. Huang, H. P. Cheng and Y. H. Huang, Catal. Commun., 2009, 10, 561–566 CrossRef CAS;
(b) J. H. Ramirez, F. M. Duarte, F. G. Martins, C. A. Costa and L. M. Madeira, Chem. Eng. J., 2009, 148, 394–404 CrossRef CAS;
(c) P. K. Malik, J. Phys. Chem. A, 2004, 108, 2675–2681 CrossRef CAS;
(d) D. Ni, J. Zhang, X. Wang, D. Qin, N. Li, W. Lu and W. Chen, Ind. Eng. Chem. Res., 2017, 56, 2899–2907 CrossRef CAS.
-
(a) H. Li, J. Liao and T. Zeng, Catal. Commun., 2014, 46, 169–173 CrossRef;
(b) B. H. Hameed and T. W. Lee, J. Hazard. Mater., 2009, 164, 468–472 CrossRef CAS PubMed;
(c) S. P. Sun, C. J. Li, J. H. Sun, S. H. Shi, M. H. Fan and Q. Zhou, J. Hazard. Mater., 2009, 161, 1052–1057 CrossRef CAS PubMed;
(d) L. Narayanasamy and T. Murugesan, Environ. Prog. Sustainable Energy, 2014, 33, 482–489 CrossRef CAS.
- J. Bernadou and B. Meunier, Chem. Commun., 1998, 20, 2167–2173 RSC.
- M. Arshadi, M. K. Abdolmaleki, F. Mousavinia, A. Khalafi-Nezhad, H. Firouzabadi and A. Gil, Chem. Eng. Res. Des., 2016, 112, 113–121 CrossRef CAS.
-
(a) M. Wang, N. Wang, H. Tang, M. Cao, Y. She and L. Zhu, Catal. Sci. Technol., 2012, 2, 187–194 RSC;
(b) F. Chen, S. Xie, X. Huang and X. Qiu, J.
Hazard. Mater., 2017, 322, 152–162 CrossRef CAS;
(c) P. P. Gan and S. F. Y. Li, Chem. Eng. J., 2013, 229, 351–363 CrossRef CAS.
- P. Kennepohl, F. Neese, D. Schweitzer, H. L. Jackson, J. A. Kovacs and E. I. Solomon, Inorg. Chem., 2005, 44, 1826–1836 CrossRef CAS.
- A. Rezaeifard, M. Jafarpour, H. Raissi, E. Ghiamati and A. Tootoonchi, Polyhedron, 2011, 30, 592–598 CrossRef CAS.
-
(a) L. Saikia, D. Srinivas and P. Ratnasamy, Microporous Mesoporous Mater., 2007, 104, 225–235 CrossRef CAS;
(b) Z. Wang, W. D. Schenkeveld, S. M. Kraemer and D. E. Giammar, Environ. Sci. Technol., 2015, 49, 7236–7244 CrossRef CAS;
(c) K. N. T. Tseng, J. W. Kampf and N. K. Szymczak, ACS Catal., 2014, 5, 411–415 CrossRef.
- I. A. Lobo, P. A. Robertson, L. Villani, D. J. Wilson and E. G. Robertson, J. Phys. Chem. A, 2018, 122, 7171–7180 CrossRef CAS.
- D. H. Gonzalez, X. M. Kuang, J. A. Scott, G. O. Rocha and S. E. Paulson, Anal. Lett., 2018, 51, 2488–2497 CrossRef CAS.
- D. Wang, J. Zou, H. Cai, Y. Huang, F. Li and Q. Cheng, Environ. Sci. Pollut. Res., 2019, 26, 1445–1454 CrossRef CAS.
- J. Zhuang, W. Dai, Q. Tian, Z. Li, L. Xie, J. Wang, P. Liu, X. Shi and D. Wang, Langmuir, 2010, 26, 9686–9694 CrossRef CAS.
-
(a) J. Y. Li, P. X. Lei and J. C. Zhao, J. Environ. Sci., 2007, 19, 892–896 CrossRef CAS;
(b) Z. Chang and J. Zeng, RSC Adv., 2014, 4, 38974–38977 RSC.
-
(a) Z. Han, X. Han, X. Zhao, J. Yu and H. Xu, J. Hazard. Mater., 2016, 320, 27–35 CrossRef CAS PubMed;
(b) Q. Song, M. Jia, W. Ma, Y. Fang and Y. Huang, Sci. China: Chem., 2013, 56, 1775–1782 CrossRef CAS;
(c) Q. Song, W. H. Ma, M. K. Jia, D. Johnson and Y. P. Huang, Applied Catalysis, A: General, 2015, 505, 70–76 CrossRef CAS;
(d) S. Yang, Y. Xu, Y. Sun, G. Zhang and D. Gao, CrystEngComm, 2012, 14, 7915–7921 RSC;
(e) A. Umar, M. S. Akhtar, G. N. Dar and S. Baskoutas, Talanta, 2013, 116, 1060–1066 CrossRef CAS;
(f) N. Wang, L. Zhu, D. Wang, M. Wang, Z. Lin and H. Tang, Ultrason. Sonochem., 2010, 17, 526–533 CrossRef CAS;
(g) X. Tao, W. Ma, T. Zhang and J. Zhao, Chem. - Eur. J., 2002, 8, 1321–1326 CrossRef CAS;
(h) D. Lu, Y. Zhang, S. Lin, L. Wang and C. Wang, J. Alloys Compd., 2013, 579, 336–342 CrossRef CAS;
(i) Y. S. Lu, Z. Wang, Y. F. Xu, Q. Liu and G. R. Qian, Desalin. Water Treat., 2016, 57, 7898–7909 CrossRef CAS;
(j) W. J. Sun, J. Li, G. Mele, Z. Q. Zhang and F. X. Zhang, J. Mol. Catal. A: Chem., 2013, 366, 84–91 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05097c |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Maasoumeh Jafarpour
* and
Maasoumeh Jafarpour