
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrecision synthesis of 3-substituted urushiol analogues and the realization of their urushiol-like performance†
Zengfeng Wei,
Xin Chen,
Jiang Duan*,
Caihong Mei,
Dan Xiao and
Aidong Zhang *
*
Key Laboratory of Pesticide & Chemical Biology of the Ministry of Education, College of Chemistry, Central China Normal University, Wuhan 430079, P. R. China. E-mail: adzhang@mail.ccnu.edu.cn; duanj@mail.ccnu.edu.cn
First published on 12th August 2019
Abstract
Urushiol is a resource-limited natural coating material with diverse applications; however, the synthesis of urushiol analogues and the realization of their urushiol-like performance remain challenging. Herein, four urushiol analogues, namely, 3-((4-alkenoylpiperazin-1-yl)methyl)catechols with the precise 3-substitution pattern on a catechol as that found in urushiol were synthesized by employing the Mannich reaction of catechol with formaldehyde and N-Boc-piperazine as the key step in a two-step route. By using optimization, the advantages of convenience in operation, cost-effectiveness, and scalability could be obtained. The electropolymerization of these analogues on copper was found to be practical due to their higher aerobic stability than urushiol, affording robust coatings with desirable hardness, adhesion strength, hydrophobicity, and thermal stability. Furthermore, the coatings exhibited effective corrosion protection on copper with initial anticorrosion efficiency up to 99.9% and comparatively higher efficiency (more than 97%) after 4 weeks of immersion in 3.5 wt% NaCl solution. The evidence from the electrochemical and infrared spectroscopic characterization data revealed that the electropolymerization process mechanically involved the free radical coupling of phenoxyl radicals to themselves and to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in the side chain, forming a robust crosslinking coating. This work paves a way for the synthesis of high-performance urushiol analogues with potential applications as metal protection materials.
C bonds in the side chain, forming a robust crosslinking coating. This work paves a way for the synthesis of high-performance urushiol analogues with potential applications as metal protection materials.
Introduction
Urushiol, a major component of naturally occurring lacquer1 having a catechol moiety similar to that observed in well-known dopamine,2 has found diverse applications as a protective,3 ornamental,4 and antibacterial5 coating material as well as the raw material of polybenzoxazine synthesis for electronic circuit packaging6 due to its high corrosion resistance, hardness, durability, and thermal stability.7 In fact, urushiol-containing lacquer has been used in China for thousands of years.8 Urushiol is a mixture of 3-alkenylcatechols mainly comprising C15 fatty chains with 1–3 CC bonds with both cis and trans configurations in different proportions, as shown in Scheme 1a.9 Although with desirable properties, urushiol is severely restricted in applications by its limited resources, low production, and high cost.10 Developing synthetic strategies for urushiol or urushiol analogues that have urushiol-like performance is imperative for academic study as well as engineering applications.
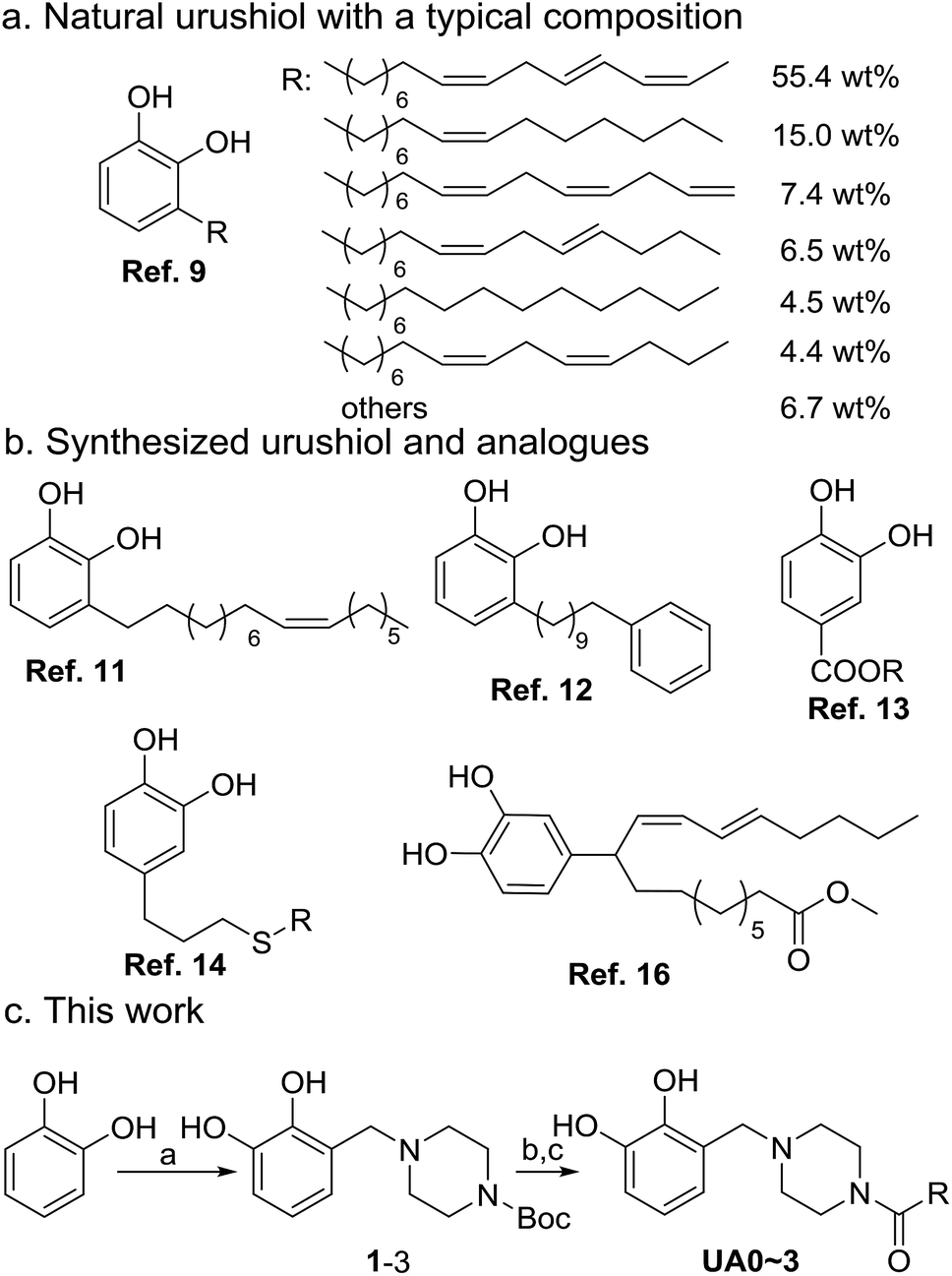 | ||
| Scheme 1 Composition of natural urushiol, structures of synthesized urushiol and urushiol analogues, and synthesis route for urushiol analogues used in this work. | ||
The synthesis of natural urushiol has proven to be challenging (Scheme 1b).11,12 With 3,4-dihydroxybenzaldehyde as the raw material, Dawson et al. developed a five-step procedure, involving hydroxyl protection, addition reaction, dehydration, catalytic hydrogenation, and subsequent deprotection, to yield urushiols containing one or no CC bond in the alkenyl side chain.11 To verify the structure of the extractants from the lacquer tree, Lu and co-workers also designed an elaborate five-step route for the synthesis of ω-phenylalkylcatechols as urushiols using 1,10-decanediol and 1,2-dimethoxybenzene as the raw materials.12 These syntheses employed lengthy routes, expensive reagents, and harsh reaction conditions, making their actual applications difficult. As an alternative strategy to the synthesis of urushiol, the design and synthesis of urushiol analogues have been extensively investigated. For example, Kobayashi and co-workers reported a one-step process to obtain urushiol analogues by the lipase-catalyzed esterification of 3,4-dihydroxybenzyl alcohol with unsaturated fatty acids.13 They also used eugenol (4-allyl-2-methoxyphenol) as the starting material after organosilane protection, thiol–ene click reaction, and deprotection to obtain a number of urushiol analogues with different alkyl chain lengths.14 In addition, there are many reports on the synthesis of the multi-component mixtures of urushiol analogues. Tsujimoto and colleagues used 6-hydroxy-1-naphthoic acid and linseed oil or fish oil and employed phenolic hydroxyl protection, acyl chlorination, and esterification to synthesize a mixture of urushiol analogues.15 Zhou et al. reported the synthesis of a multi-component mixture of urushiol analogues by the photoinitiated radical reaction of catechol with the alkenyl chains of tung oil.16 To the best of our knowledge, all these reported urushiol analogues are 4-substituted catechols, instead of the 3-substitution pattern inherent in urushiol. Nevertheless, these synthesis methods for urushiol analogues have effectively exploited the naturally occurring abundant triglycerides, providing the potential for large-scale synthesis and applications of urushiol analogues. Considering that the substitution pattern on catechol may have important influences on their properties, to synthesize urushiol analogues with the precise 3-substitution on catechol is highly desirable in realizing the natural urushiol-like performance.
Closely related to the application of urushiol analogues, their curing methods are also an important topic of interest, because the curing conditions of urushiol, where endogenous laccase is necessary, are usually ineffective toward analogues due to the specificity of laccase catalysis.17 To cure urushiol analogues, various oxidants, transition metal catalysts, and even catalase for generating the H2O2 oxidant, have been attempted. For example, the Fe(III)–salen complex or cobalt naphthenate was reported to cure 3,4-dihydroxybenzyl-alcohol-based urushiol analogues that have 2 or 3 CC bonds in the side chain.18 The Fe(III)–salen complex, as well as catalase, was also able to polymerize cardanol, a natural product of phenol with a 3-substituted unsaturated aliphatic chain as the urushiol analogue.19 On the other hand, to facilitate the curing of natural urushiol, electrochemical methods (e.g., potentiostatic electropolymerization) have been explored, but no performance of the coating has been reported;20 in particular, no such method has been applied to the curing of urushiol analogues. If the electropolymerization method could be used for the curing of urushiol analogues, it would have advantages such as operational convenience, easy control over the coating thickness, and applicability to different shapes of metal surfaces.21
In this work, four urushiol analogues, namely, 3-((4-alkenoylpiperazin-1-yl)methyl)catechols with a precise 3-substitution pattern on catechol as that of urushiol, were synthesized by exploiting the cheap and readily available catechol, formaldehyde, N-Boc-piperazine, and fatty acids as the raw materials, as well as the classical and high-yield Mannich reaction and amidation reaction (Scheme 1c). It should be noted that in order to address the main issue involving the substitution pattern of urushiol analogues, commercially available unsaturated fatty acids were used in the synthesis, all of which contain CC bonds in the cis configuration. Furthermore, an electropolymerization protocol was developed to cure these urushiol analogues on copper; in addition, their proportional mixture (UAm) with a composition ratio similar to natural urushiol, as well as urushiol itself, for effecting a comparison. The cured coating structure and morphologies and the anticorrosion performance for copper in 3.5 wt% NaCl were investigated and discussed.
Results and discussion
Optimization of the Mannich reaction and purification
A two-step procedure was developed for the synthesis of urushiol analogues, including the Mannich reaction of catechol with formaldehyde and N-Boc-piperazine and deprotection, in addition to amidation with various fatty acids bearing 0–3 CC bonds, such as stearic acid, oleic acid, linoleic acid, and α-linolenic acid (Scheme 1c). The Mannich reaction to yield the intermediate 3-((4-Boc-piperazin-1-yl)methyl)catechol (1–3) is the key to the success of this two-step procedure. Due to the directing role of the two ortho-hydroxyls, catechol has two active sites for the Mannich electrophilic substitution; hence, a mixture of 3- and 4-substituted catechols would be obtained. The 4-substituted catechol is an unwanted byproduct for the purpose of this study since urushiol mainly consists of 3-alkenylcatechols (Scheme 1a). To this end, the reaction conditions of the Mannich reaction were optimized with the expectation to minimize the yield of 4-substituted catechols.
In our previous work, water was used as the reaction medium for the Mannich reaction of catechol with formaldehyde and diethanolamine to yield 4-diethanolaminomethyl catechol.22 To obtain the 3-substituted intermediate 1–3, N-Boc-piperazine instead of diethanolamine was used as the partner in the Mannich reaction. Due to the poor solubility of N-Boc-piperazine in water, a number of organic solvents and CH3OH/H2O mixture were explored, and the results are listed in Table 1. The reaction was first conducted using CH3OH as the medium at 40 °C under N2 and continued until there were no obvious changes in the product composition as determined by thin-layer chromatography (ca. 4 h), resulting in a conversion rate of 85% for catechol and the generation of 1–3 and 4-substituted byproduct 1–4 in yields of 61 and 10%, respectively (Table 1, entry 1) (conversion rate determination and product isolation are discussed below). Then, the other organic solvents, namely, 1,4-dioxane, CH3CN, THF, and isopropanol, were examined. The reaction in 1,4-dioxane for 6 h afforded a catechol conversion rate of 86%, as well as 1–3 and 1–4 in yields of 54 and 10%, respectively (entry 2). In CH3CN, THF, or isopropanol, the reaction yielded similar results to those in CH3OH and 1,4-dioxane (entries 3–5). When the mixture of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) CH3OH/H2O was used as the medium, the reaction at 25 °C for 4 h afforded a low conversion rate of 65% for catechol, as well as 1–3 and 1–4 in yields of 23 and 36%, respectively (entry 6). Fortunately, when the temperature was raised to 40 °C for 4 h, a conversion rate of up to 88% and the highest yield of 76% for 1–3 were achieved, where only a trace of 1–4 was generated (entry 7). Although the highest conversion of catechol (91%) was reached at 60 °C for 4 h, the yield of 1–3 inversely decreased to 68% (entry 8). In addition, conducting the reaction without N2 protection always resulted in decreased yields.
:1 (v/v) CH3OH/H2O was used as the medium, the reaction at 25 °C for 4 h afforded a low conversion rate of 65% for catechol, as well as 1–3 and 1–4 in yields of 23 and 36%, respectively (entry 6). Fortunately, when the temperature was raised to 40 °C for 4 h, a conversion rate of up to 88% and the highest yield of 76% for 1–3 were achieved, where only a trace of 1–4 was generated (entry 7). Although the highest conversion of catechol (91%) was reached at 60 °C for 4 h, the yield of 1–3 inversely decreased to 68% (entry 8). In addition, conducting the reaction without N2 protection always resulted in decreased yields.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Temperature (°C) | Time (h) | Conversionb (%) | Yield of 1–3/–4c (%) |
| a Reaction conditions: catechol (0.02 mol), N-Boc-piperazine (0.02 mol), formaldehyde (0.02 mol, 37 wt% aqueous), solvent (30 mL), N2 atmosphere.b The conversion rate of catechol was calculated based on the recovered amount of catechol.c Yield of isolated product. | |||||
| 1 | Methanol | 40 | 4 | 85 | 61/10 |
| 2 | 1,4-Dioxane | 40 | 6 | 86 | 54/10 |
| 3 | Acetonitrile | 40 | 6 | 88 | 59/20 |
| 4 | THF | 40 | 6 | 86 | 49/29 |
| 5 | Isopropanol | 40 | 6 | 70 | 31/15 |
| 6 | Methanol/H2O | 25 | 4 | 65 | 23/36 |
| 7 | Methanol/H2O | 40 | 4 | 88 | 76/trace |
| 8 | Methanol/H2O | 60 | 4 | 91 | 68/trace |
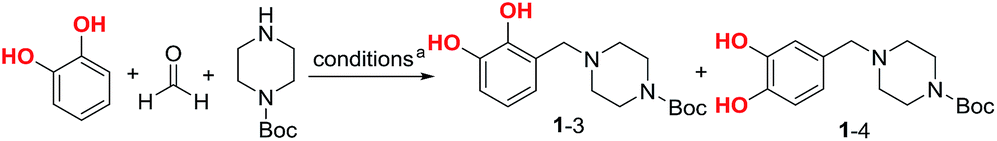
Hence, the optimal reaction conditions were determined as a mixture of CH3OH/H2O in a volumetric ratio of 1:1 as the medium at 40 °C for 4 h. The obtained results suggest that the presence of H2O in the medium is important and it remarkably improved the 3-position selectivity. In the mechanism of this three-component Mannich reaction, an iminium cation is first generated23 from the reaction between the aldehyde and amine; further, due to its highly positive charge, high dielectric water as the medium is more favorable to its formation than common solvents such as CH3OH, THF, etc., among which CH3OH is relatively better. To balance the requirements of simultaneously high dielectricity and solubility for the reaction, the mixture of CH3OH/H2O in a volumetric ratio of 1:1 as the medium was considered suitable. On the other hand, the reaction in the CH3OH/H2O medium performed better at 40 °C (entry 7) than that at 25 °C (entry 6), because the former afforded 1–3 in 76% yield and 1–4 in trace amounts, while the latter afforded 1–3 and 1–4 in yields of 23 and 36%, respectively. This regioselectivity of the Mannich reaction favored at a higher reaction temperature suggests the kinetic control process in the formation of 1–3.
During the optimization of the reaction conditions, separating the desired product 1–3 from the byproduct 1–4 was firstly attempted using column chromatography. However, due to the strong adsorption of 1–3 and 1–4 on silica gel, low yields were obtained. To improve the yields, a pH-controlled extraction–recrystallization method, which was developed in our previous work,22 was found to be fairly suitable. Briefly, the reaction solution mixture was first stripped of CH3OH and adjusted to pH 2, followed by catechol recovery by EtOAc extraction for calculating the reaction conversion rate; however, the mixture of 1–3 and 1–4 as ammonium salts remained in the aqueous phase. Next, the aqueous phase was adjusted to pH 8, converting the products to their zwitterionic forms, which made them less soluble in water and extractable with EtOAc. This solubility reduction in H2O for the zwitterionic forms is analogous to the purification process of amino acids.24 We speculated that the extracted product mixture in EtOAc might get converted into the non-dissociated neutral states, thereby resulting in high solubility. After the removal of the solvent, the mixture was recrystallized in CH3CN to afford the byproduct 1–4 as a crystal, while the filtrate yielded 1–3 as a liquid after the removal of CH3CN.
Following the synthesis of 1–3, a hydrolysis reaction25 was conducted using 37% HCl in EtOAc at room temperature, giving the intermediate 3-(piperazin-1-ylmethyl)catechol (2). The hydrolysis was complete in 4 h; thereafter, a suitable amount of H2O was added, and the pH-controlled extraction–recrystallization was repeated. In this circumstance, after the aqueous phase was adjusted to pH 8, extraction with EtOAc and rotary drying afforded a residue, which was recrystallized in CH3CN to afford 2 as a yellow crystal in high yield (96%).
Owing to the high yield of 1–3 and a trace amount of 1–4 produced in the optimized Mannich reaction, as well as the fact that 2 is a solid, the two reactions could be combined without using column chromatography to purify 1–3, not only avoiding the loss resulting from the adsorption on silica gel, but also simplifying the post-synthesis purification process. Hence, after the Mannich reaction, CH3OH was removed, and 37% HCl and EtOAc were added. Further, the reaction mixture was stirred for 4 h at room temperature and then subjected to the pH-controlled extraction–recrystallization procedure, directly affording the desired intermediate 2 with sufficient purity, as confirmed by NMR, required for the subsequent synthesis of urushiol analogues. This simplification made the synthesis of 2 expedient and suitable to be scaled up. In this case, a 4-fold synthesis of 2 was carried out with the simplified procedure using 17.6 g catechol (160 mmol) and other reactants, furnishing 24.3 g of 2 at a total yield of 73% for the two combined steps.
Synthesis and characterization of urushiol analogues
The second step in the synthesis procedure was the amidation reaction of intermediate 2 with equimolar fatty acids bearing 0–3 CC bonds, including stearic acid, oleic acid, linoleic acid, and α-linolenic acid, affording the respective four urushiol analogues (UA0 ∼ 3). The amidation reaction was carried out using the coupling agents of EDC·HCl and NHS in CH2Cl2 and the product was purified by flash column chromatography on silica with EtOAc/petroleum ether as the eluent initially at the ratio of 1:4 (v/v) and then of 1:2, affording UA0 ∼ 3 in satisfactory yields (59–71%). The adsorption of these analogues on silica gel was largely reduced due to the presence of long fatty chains in the molecular structure. The structures of UA0 ∼ 3 and their yields and physical appearances are shown in Table 2. UA0, which has no CC bond in the chain, is a light yellow solid, while UA1 ∼ 3 appear as yellow liquids. Urushiol analogues are soluble in common organic solvents such as CH3OH, CHCl3, and EtOAc, but insoluble in H2O.
|
|||
|---|---|---|---|
| Compound | R structure | Yield (%) | Physical appearance |
| UA0 | 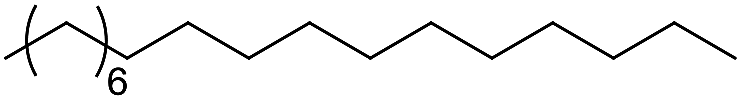 |
71 | Light yellow solid |
| UA1 |  |
67 | Yellow liquid |
| UA2 | 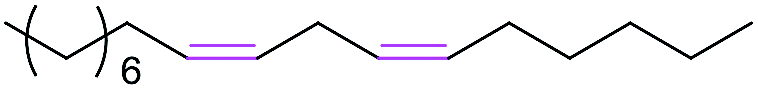 |
61 | Yellow liquid |
| UA3 |  |
59 | Yellow liquid |
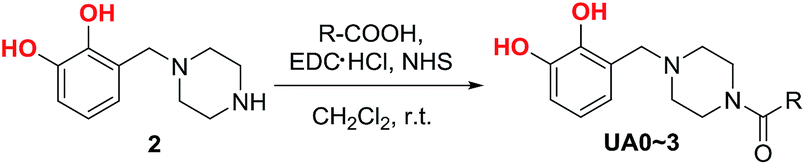
The structures of UA0 ∼ 3 and their dissimilarities can be effectively determined using their stacked 1H NMR spectra, as shown in Fig. 1. The identical aromatic resonances appear as a set of three signals of neighboring three Ar-Hs with 2:3:2 splitting pattern in δ 6.5–7.0 ppm, representing the 3-substituted catechol unit of UA0 ∼ 3.22 The difference in the UA0 ∼ 3 spectra lies in the signals of the fatty chains; most evidently, UA0 has no signal but UA1 ∼ 3 exhibit the vinylic hydrogen signal at δ 5.36 ppm, which increases in intensity with an increase in the number of CC bonds. Another distinction is that UA2 ∼ 3 have the resonance of the methylene group flanked by 2 CC bonds at δ 2.80 ppm, while UA1 has none, which only exhibits CC-bond-neighboring methylene absorption at 2.06 ppm. Other characteristic resonances for UA0 ∼ 3 are the peaks of the linking methylene 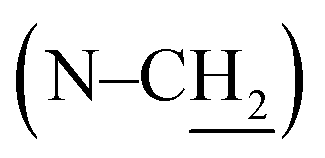 at 3.73 ppm, piperazine ring methylenes at 3.53 and 2.56 ppm, acyl methylene at 2.32 ppm, and the subsequent one at 1.62 ppm; the other methylenes in the chain are at ∼1.27 ppm. The terminal –CH3 of UA0 ∼ 2 appears at δ 0.88 ppm, while the absorption occurs at δ 0.98 ppm for UA3. It is worth mentioning that the 1H NMR signal of the terminal –CH3 and its intensity can be used to determine the purity of these commercial fatty acids, such that linoleic acid containing 25% α-linolenic acid and α-linolenic acid having 10% linoleic acid can be directly calculated from their 1H NMR spectra. The FTIR spectra of UA0 ∼ 3 (Fig. S1†) provide no significant disparity but their common characteristic absorptions. For example, catecholic hydroxyls absorb at 3250 cm−1 as a broad peak, the amide group intensively appears at 1648 cm−1, and the vinylic C–H bonds stretch at 3016 cm−1 with their intensity increasing with the number of CC bonds in the side chain, which is an important indicator to evidence their participation in the crosslinking reaction.
at 3.73 ppm, piperazine ring methylenes at 3.53 and 2.56 ppm, acyl methylene at 2.32 ppm, and the subsequent one at 1.62 ppm; the other methylenes in the chain are at ∼1.27 ppm. The terminal –CH3 of UA0 ∼ 2 appears at δ 0.88 ppm, while the absorption occurs at δ 0.98 ppm for UA3. It is worth mentioning that the 1H NMR signal of the terminal –CH3 and its intensity can be used to determine the purity of these commercial fatty acids, such that linoleic acid containing 25% α-linolenic acid and α-linolenic acid having 10% linoleic acid can be directly calculated from their 1H NMR spectra. The FTIR spectra of UA0 ∼ 3 (Fig. S1†) provide no significant disparity but their common characteristic absorptions. For example, catecholic hydroxyls absorb at 3250 cm−1 as a broad peak, the amide group intensively appears at 1648 cm−1, and the vinylic C–H bonds stretch at 3016 cm−1 with their intensity increasing with the number of CC bonds in the side chain, which is an important indicator to evidence their participation in the crosslinking reaction.
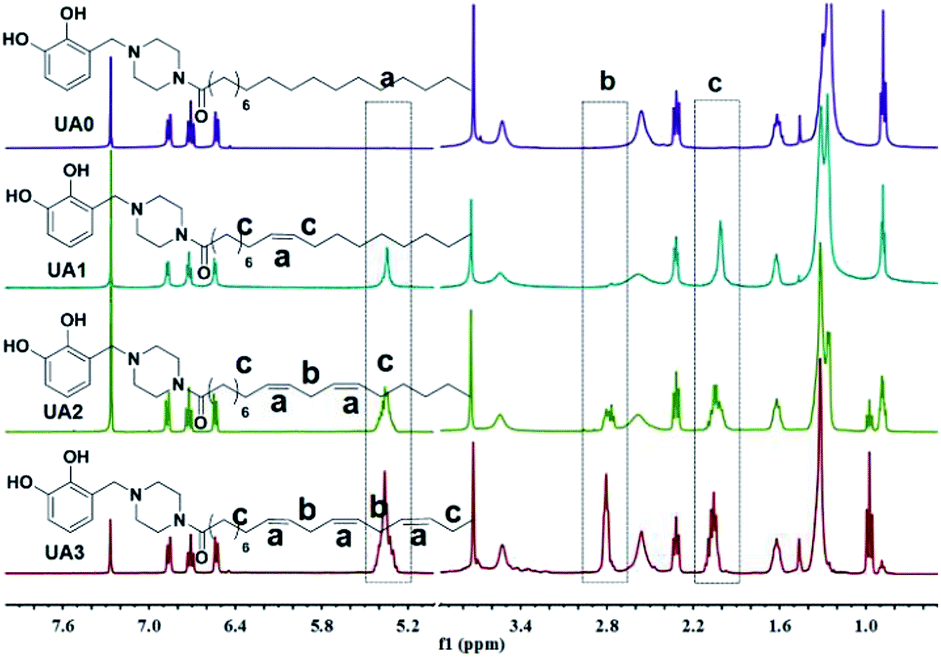 | ||
| Fig. 1 Stacked 1H NMR spectra of UA0 ∼ 3 for structural comparative analyses. | ||
Electropolymerization and its mechanism
As mentioned in the Introduction section, laccase usually is ineffective in the polymerization of urushiol analogues due to its substrate specificity, which was also true in our case; hence, developing a curing approach was imperative. Here, UA0 ∼ 3 were investigated for their electropolymerization on copper surfaces, as well as UAm and urushiol for effecting a comparison. They were first separately dissolved in ethanol containing 10% volumetric parts of 10 mM Tris–HCl buffer (pH 8.5; concentration: 30 mg mL−1) and then cyclic voltammetry (CV) was applied with potential scanning from 0 to 3.0 V vs. SCE (Fig. 2a and S2†). An irreversible and broad oxidation peak within 1.0–1.9 V was observed for UA0 ∼ 3 and UAm for the first scan cycle, as well as for urushiol, which can be attributed to the oxidation of the catechol unit.26 When compared with the analogues, urushiol exhibited a smaller oxidation peak at around 1.0 V, reflecting that it is easier to be oxidized at the electrode and the available quantity is small. This is possibly due to the inferior solution stability of urushiol as compared to that of UA0 ∼ 3 and UAm, which was observed by the color change in the solution during the solution preparation and electropolymerization process. The ethanol/Tris buffer solution of urushiol tended to quickly darken in about 10 s, while the solutions of the analogues remained clear yellow, even when exposed to air (Fig. 2a: inset, UAm). During electropolymerization for about 1 h, the urushiol solution became opaque and evolved to precipitate in the form of black particles (Fig. 2a; inset: urushiol), while the analogue solutions were still transparent and only turned claret red at the end. This can be attributed to the fact that most of the urushiol underwent a self-polymerization reaction prior to reaching the electrode surface under the slightly basic condition of pH 8.5, similar to that observed with dopamine that polymerizes under this pH condition in various applications.27 For the analogues in the solution, the analogous self-polymerization was considerably reduced, and their electropolymerization on the electrode became dominant, yielding a stronger oxidation peak. It is speculated that the analogues can form intramolecular hydrogen bonds between the phenolic hydroxyl and piperazine nitrogen (Fig. S3†), stabilizing the catechol ring and suppressing self-polymerization. With an increase in the number of scans, all the oxidation currents rapidly dropped; when the number of scan cycles was seven, the oxidation currents decreased to the minimum values and remained constant, suggesting that the formation of an insulating coating was a fast process.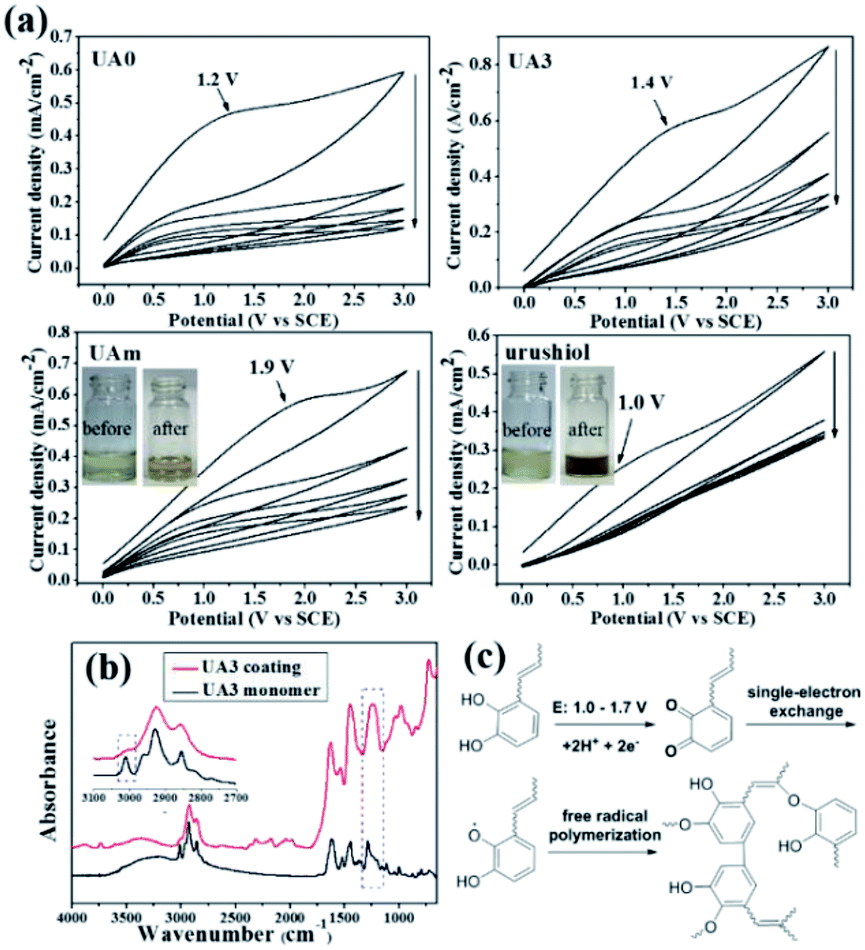 | ||
| Fig. 2 CV plots of UA0, UA3, UAm, and urushiol, with potential scan cycles of 1, 3, 5, 7, and 9, in the range of 0–3.0 V vs. SCE at a scan rate of 30 mV s−1 (a); ATR-FTIR spectra of UA3 and its coating (b); proposed mechanism for the electropolymerization of urushiol analogues (c). | ||
The polymerization of these urushiol analogues and urushiol was also investigated by comparing their ATR-FTIR spectra before and after polymerization (Fig. S4†). As an example, the spectra of UA3 monomer and its coating are shown in Fig. 2b for understanding. Before polymerization, UA3 exhibits the characteristic broad phenolic O–H at around 3250 cm−1, Ar-H and alkenyl C–H at 3016 cm−1, strong saturated C–H absorption at about 2800–2980 cm−1, carbonyl at 1650 cm−1, phenyl ring band at about 1445 cm−1, and the phenylic O–H bending at ∼1280 cm−1.28 After polymerization, several variations in the spectrum are noticeable; for example, the significant decrease in C–H stretching at 3016 cm−1, combined enhancement of the phenyl ring band at about 1370–1520 cm−1, and the newly appeared strong bands at about 1236 and 976 cm−1. The decrease in the C–H stretching absorption can be attributed to the participation of CC bonds in the polymerization; the enhancement of the phenyl ring band may result from the coupling of phenyl rings, and the appearance of adsorptions at about 1236 and 976 cm−1 can be ascribed to the formation of aromatic ether bonds and trans CC bonds, respectively, in the polymerization.29
Taken together, the data of CV and FTIR, in combination with the mechanisms of urushiol evidenced by other methods,30,31 suggest that the analogue undergoes an electrochemically initiated radical polymerization process on the copper surface, which is shown in Fig. 2c. First, one catechol unit is anodically oxidized to its quinone form, which immediately reacts with another catechol unit to form a semiquinone radical species via a single-electron exchange;32 next, a variety of radical couplings occur, including those among the phenyl rings, phenoxyl radical to phenyl ring and to the CC bond in the side chain, etc., leading to the formation of a complex crosslinking network. Overall, this electrochemical process involving urushiol analogues, at the stage of semiquinone radical formation, is similar to the electropolymerization of catechol-containing dopamine,26 but slightly different to the curing processes of urushiol by laccase and UV irradiation, which are considered to directly generate the semiquinone radical.29 At the stage of free radical polymerization, the analogues react in a similar way as that of urushiol in the polymerization via laccase catalysis or UV irradiation.30,31
Characterization of electropolymerization coatings
The surface morphology of the electropolymerization coatings of UA0 ∼ 3, UAm, and urushiol were characterized by AFM, as shown in Fig. 3, along with their values of average roughness (Ra). The surface of UA0 coating appears to have rugged and irregularly convex topology, where Ra = 503 nm. The UA1 surface is relatively flattened, where Ra = 285 nm. For the UA2 coating, its surface is flatter, exhibiting only a few large protrusions, where Ra = 38.9 nm. Interestingly, the topology of the UA3 coating is fairly smooth, without the existence of protrusions, and its Ra is very small (17.1 nm). UAm exhibits a relatively flat coating with small protrusions, where Ra = 47.0 nm. In contrast, urushiol affords a coating surface with large and broad convex aggregates and Ra value as large as 383 nm.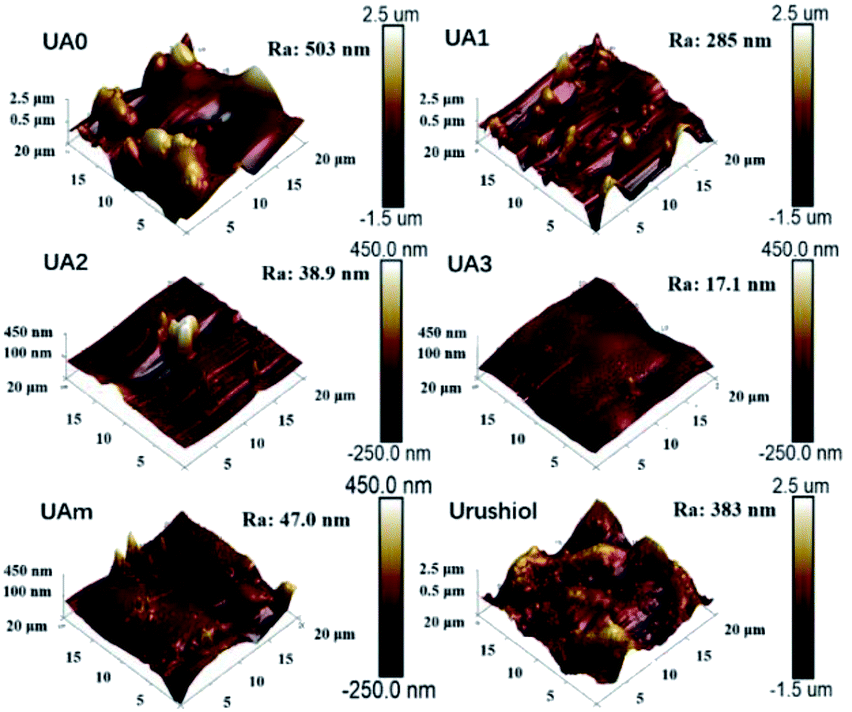 | ||
| Fig. 3 AFM height images of UA0 ∼ 3, UAm, and urushiol coatings on copper. | ||
The differences in the morphologies of these coatings may arise from two aspects. The first is the influence of the molecular structure, which is clearly evident because the morphology of the coating has the flatness order of UA0 < UA1 < UA2 < UA3, which is proportional to the number of CC bonds in the side chain; probably, the more the number of CC bonds in the side chain, the denser is the crosslinking network as well as increased suppression of the formation and aggregation of polymer particles. However, for UAm, the nature of the mixture favors the suppression of the crystalline aggregation, and therefore, afforded relatively flat topology. On the other hand, due to the instability of urushiol in the electrolyte solution, self-crosslinking and subsequent aggregation accompanied the electropolymerization process of urushiol, leading to a highly rough surface of the coating, as shown in Fig. 3.
The physical properties of the coatings, such as the coating thickness, hardness, adhesion strength, water contact angle, and thermal stability, were also investigated, and the results are summarized in Table 3. The coating thicknesses of UA0 ∼ 3, as well as that of UAm, were between 12 and 15 μm, indicating that they underwent a crosslinking reaction at a similar rate in the electropolymerization process. In this aspect, urushiol behaved differently, yielding a coating with a thickness of only 7.5 μm. On the other hand, their coating hardness values varied dramatically, ranging from 1H to 6H. For the urushiol coating, the highest hardness of 6H was determined, whereas the UA0, UA3, and UAm coatings exhibited a moderate hardness value of 4H, and UA1 and UA2 coatings had a hardness value as low as 1H. Apparently, no positive correlation exists between the hardness and the number of CC bonds in the side chain. We ascribe this abnormal phenomenon to the complex process required to form coatings of urushiol and its analogues, as mentioned in the mechanism discussion. For example, UA0 has no CC bonds in the side chain and crosslinks only by means of the catechol itself, but it still yielded a moderately hard surface, probably arising from the strong interactions of the saturated alkyl side chains due to their regular orientation.33 It is noteworthy that the hardness of 4H (UA0, UA3, and UAm coatings) can be considered to be a good result, which is comparable to the hardness recently reported for organosilicone-cured urushiol coatings.34
| Coating | Thickness (μm) | Pencil hardness | Adhesion strength | Contact angle (°) | T10% (°C) | T50% (°C) | Tmax (°C) | Wchar (%) |
|---|---|---|---|---|---|---|---|---|
| UA0 | 11.7 ± 1.8 | 4H | 5B | 114.1 ± 1.5 | 289.6 | 399.3 | 379.9 | 32.91 |
| UA1 | 14.0 ± 4.0 | 1H | 4B | 100.9 ± 1.4 | 284.5 | 445.0 | 423.7 | 39.84 |
| UA2 | 15.6 ± 2.8 | 1H | 4B | 94.7 ± 0.5 | 291.8 | 445.4 | 409.2 | 40.83 |
| UA3 | 12.5 ± 2.3 | 4H | 4B | 95.4 ± 1.4 | 284.1 | 457.6 | 401.6 | 44.29 |
| UAm | 15.3 ± 3.6 | 4H | 4B | 98.9 ± 1.5 | 286.1 | 456.7 | 379.9 | 44.66 |
| Urushiol | 7.5 ± 1.3 | 6H | 5B | 98.5 ± 1.1 | 321.8 | 446.5 | 437.2 | 34.62 |
All the coatings exhibited excellent adhesion strengths and high hydrophobicities. For example, the coatings of UA0 and urushiol have strength of 5B and that of UA1 ∼ 3 is 4B, which were measured with the standard ASTM D3359 test method. All the coatings were hydrophobic, with water contact angles ranging from 94.7° to 114.1°. The thermal stability parameters of the coatings were obtained by TGA (Fig. S5†), including the weight loss temperatures (T10% and T50%), temperature of the maximum weight loss rate (Tmax), and char yield at 600 °C (Wchar), and the results are listed in Table 3. All the individual coatings of the analogues and their mixture have similar T10% values >280 °C, which are slightly inferior to that of urushiol (321.8 °C). UA0 has a T50% value of 399.3 °C, while UA1 ∼ 3 have values >440 °C, similar to that of urushiol. All the Tmax values for the analogues are around 400 °C, somewhat lower than that of urushiol (437.2 °C). Except for the UA0 coating, one obvious advantage of the analogue coatings is their high Wchar values, much higher than urushiol, and the coatings of UA3 and UAm have Wchar values larger than 44%, probably due to the higher number of CC bonds in the side chain. The abovementioned results reveal that these analogues and their mixture have high thermal stability.
Anticorrosion performance of coatings
The most fascinating properties of urushiol coatings are their high corrosion resistance and durability, which provides desirable protection for various materials, such as wood, ceramics, and metals.35 Copper is widely used in the industry and its corrosion protection is a longstanding interest. Therefore, the coatings of urushiol analogues were investigated for their protection toward copper, and the performance was compared with that of urushiol. The corrosion inhibition behavior was examined by potentiodynamic polarization, and the resulting Tafel curves of the Cu substrates with or without coatings in 3.5 wt% NaCl solution are shown in Fig. 4a. The corrosion current density (icorr) and potential (Ecorr) were obtained from the intersection of the cathodic and anodic Tafel curves using the Tafel extrapolation method, and the results are summarized in Table 4, along with the estimated corrosion rate (CR) and corrosion inhibition efficiency (IE).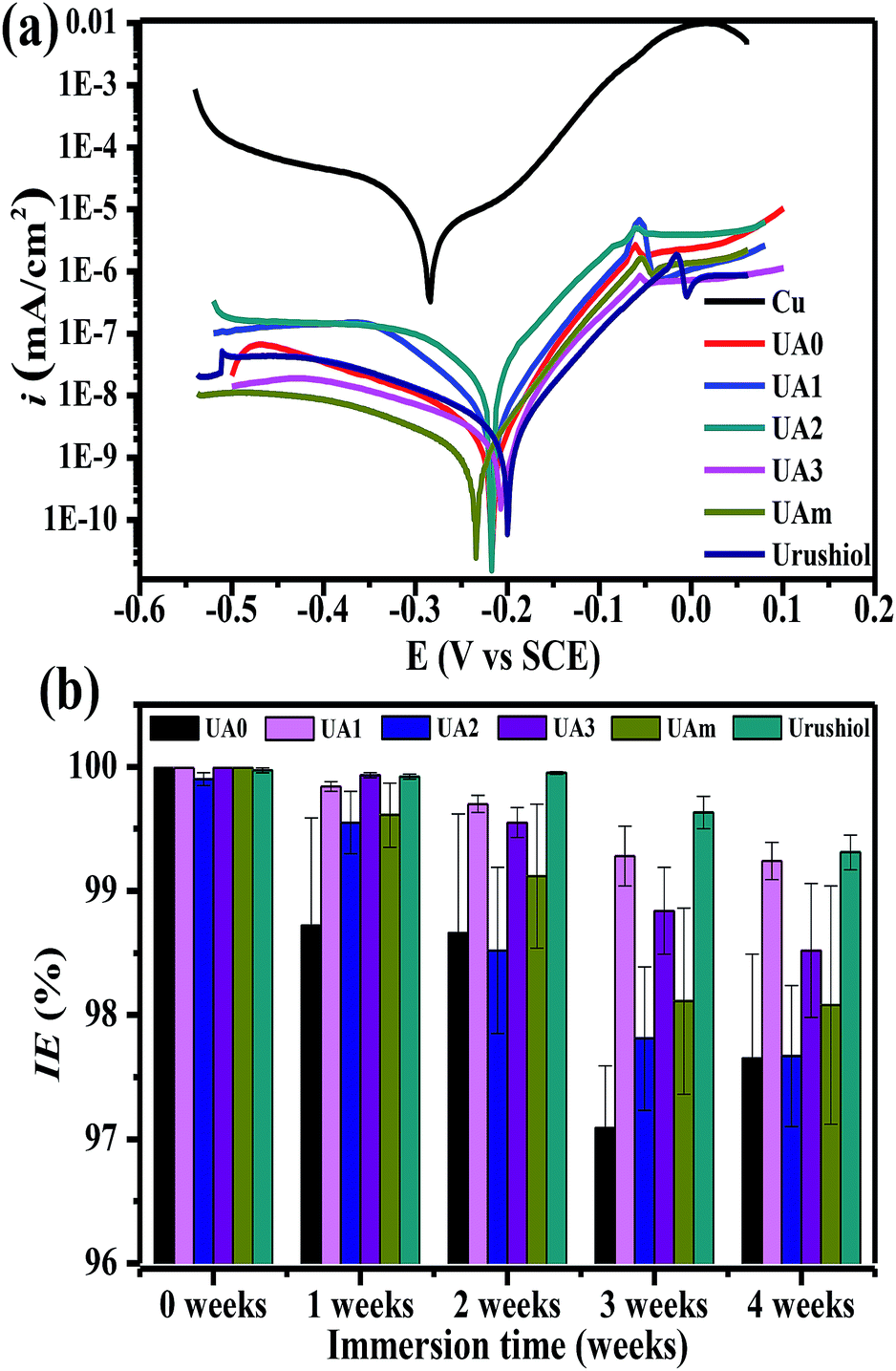 | ||
| Fig. 4 Polarization curves of the Cu substrate without and with a coating in 3.5 wt% NaCl solution at 25 °C (a). Long-term corrosion inhibition performance of the UA0 ∼ 3, UAm, and urushiol coatings on copper in 3.5 wt% NaCl (b). | ||
| Sample | icorr (A cm−2) | Ecorr (V vs. SCE) | CR (mm per year) | IE (%) |
|---|---|---|---|---|
| Copper | 2.23 × 10−5 | −0.283 | 2.59 × 10−1 | — |
| UA0 | 2.86 × 10−9 | −0.216 | 3.32 × 10−5 | 99.99 |
| UA1 | 4.42 × 10−9 | −0.216 | 5.13 × 10−5 | 99.99 |
| UA2 | 2.51 × 10−8 | −0.213 | 2.91 × 10−4 | 99.89 |
| UA3 | 2.01 × 10−9 | −0.220 | 2.33 × 10−5 | 99.99 |
| UAm | 9.44 × 10−10 | −0.234 | 1.10 × 10−5 | 99.99 |
| Urushiol | 2.50 × 10−9 | −0.208 | 2.90 × 10−5 | 99.99 |
Without any coating, the Cu substrate displayed an icorr value of 2.23 × 10−5 A cm−2 and Ecorr at −0.283 V; for the newly prepared coatings, regardless of UA0 ∼ 3, UAm, or urushiol, icorr significantly decreased by 3–4 orders of magnitude and Ecorr shifted to a higher positive potential by about 0.06 V. Relatively, except the UA2 coating, which showed a slightly larger icorr value (2.51 × 10−8), the other analogues, mixture, and urushiol, exhibited similar icorr values in the order of 10−9. The Cu substrates with these coatings were estimated to have CR values in the range of 10−4 to 10−5 mm per year, which are much less than those for bare copper. Based on the values of icorr and CR, excellent IE values up to 99.9% could be derived for the UA0 ∼ 3 and UAm coatings, similar to the urushiol coating.
The long-term corrosion inhibitions of UA0 ∼ 3 and UAm coatings on copper were also tested and compared with that of the urushiol coating (Fig. 4b). The test was performed on each coating on copper in the immersing solution of 3.5 wt% NaCl with an interval of 1 week in a period of 4 weeks. The results at week 0 have been discussed above with respect to the freshly prepared coatings. At week 1, the effects of the UA0, UA2, and UAm coatings began to decrease, but still possessed IE values higher than 98%; while for UA1, UA3, and urushiol, their coatings retained the similar excellent performance. With the passage of immersion time, all the coatings became slightly inferior to the original ones in inhibiting corrosion, but their IE values were still higher than 97%; in particular, for UA1 and urushiol, the IE values of their coatings remained as high as 99%. It should be noted that the long-term corrosion inhibition effects of these coatings are comparable to that of recently reported graphene/lacquer composites.36 The desirable anticorrosion performance of urushiol analogues can be attributed to their ability to form a multiple crosslinking network, which prevents the corrosive species in the solution from coming into contact with the Cu surface.37
Conclusions
In this work, urushiol analogues with side chains of different number of CC bonds were designed and synthesized; the Mannich reaction of catechol with formaldehyde and N-Boc-piperazine was exploited, which is a method that affords convenience, cost-effectiveness, and scalability in order to obtain urushiol analogues as an alternative to the resource-limited and expensive natural urushiol. The electropolymerization of these analogues on copper was demonstrated to be practical in forming coatings on copper due to their higher aerobic stability than that of urushiol itself; this merit could be ascribed to the role of intramolecular hydrogen bonding. Importantly, the analogue coatings exhibited excellent corrosion inhibition performance on copper, as well as their long-term effect, which is crucial for applications involving copper in diverse fields. In addition, by analyzing the results using electrochemistry and ATR-FTIR characterization, a plausible mechanism for the electropolymerization of analogues on copper was proposed, which involves various free radical couplings occurring not just among the catecholic phenyl rings, but also the coupling of phenoxyl radical to phenyl ring and to CC bonds in the side chain. The developed synthesis strategy of urushiol analogues and the relevant electropolymerization method may have promising potential in the field of metal corrosion protection.
Experimental
Reagents and compound analysis
Catechol, N-Boc-piperazine, oleic acid (98%), linoleic acid (98%), and α-linolenic acid (85%) were purchased from Aladdin Reagent Co., Ltd. (China). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl), N-hydroxysuccinimide (NHS), formaldehyde (37 wt% aqueous), and stearic acid were obtained from Sinopharm Chemical Reagents Co., Ltd. (China). Other reagents and solvents were of the analytical grade and used without any further purification. Raw lacquer was obtained from the Institute of Lacquer (Xi'an, China). Urushiol was extracted from raw lacquer with ethanol.38 Ultrapure water (18.2 MΩ cm) was prepared by a LabPure KL-UP-UV-20 system (Kangning, China). The NMR spectra were recorded on a Varian NMR spectrometer (Mercury plus-400 or plus-600) using CDCl3, CD3OD, or DMSO-d6 as the solvent. Chemical shifts were reported in ppm relative to tetramethylsilane (TMS). The high-resolution ESI mass spectra were obtained on an Agilent 6530 accurate-mass Q-TOF spectrometer.Synthesis and isolation of 1–3 and 1–4
A solvent (60 mL), formaldehyde (3.24 g, 40 mmol, 37 wt% aqueous), and N-Boc-piperazine (7.4 g, 40 mmol) were put in a round-bottom flask; after stirring at room temperature for 30 min under N2, catechol (4.4 g, 40 mmol) was added. The reaction continued at a certain temperature for 4 h. After removing CH3OH, the resultant solution was adjusted to pH 2 using dilute HCl and then extracted with EtOAc (3 × 100 mL) to recover catechol. Subsequently, the aqueous phase was adjusted to pH 8 with a dilute NaOH solution and then extracted with 5 × 100 mL EtOAc. The combined organic phase was dried over anhydrous Na2SO4 and evaporated to afford a mixture. The mixture was dissolved in CH3CN and recrystallized to afford 1–4; while the filtrate was evaporated to yield a residue as 1–3. The obtained catechol, 1–3, and 1–4 were used to calculate the conversion rate of catechol and the yields of 1–3 and 1–4, as shown in Table 1.For the purpose of structural characterization, the product mixtures of 1–3 and 1–4 obtained under nonoptimized conditions were subjected to purification on silica gel using petroleum ether/EtOAc (4/1, v/v) as the eluent, affording isolated 1–3 and 1–4.
When the synthesis was conducted under optimized conditions (CH3OH/H2O in 1:1 volumetric ratio as the medium at 40 °C for 4 h), 1–3 was obtained in a yield of 76% (10.4 g) and 1–4 in a trace amount.
Synthesis of 3-(piperazin-1-ylmethyl)catechol (2)
A flask was charged with EtOAc (100 mL), 1–3 (6.16 g, 20 mmol), and concentrated HCl (37 wt%, 25 mL), and the mixture was magnetically stirred for 4 h at room temperature. Thereafter, 50 mL water was added into the flask and the mixture was washed with EtOAc (3 × 100 mL). Next, the aqueous phase was adjusted to pH 8 with a dilute NaOH solution and extracted by EtOAc (5 × 100 mL). The organic phases were combined, dried with anhydrous sodium sulfate, and concentrated by rotary evaporation to give a residue, which was recrystallized in CH3CN to give product 2 (3.99 g) as a yellow solid in a yield of 96%.Simplified procedure for the scale-up synthesis of 2
A 4-fold synthesis of 2 was carried out with a simplified procedure that combined the synthesis of 1–3 with the following hydrolysis. CH3OH/water (240 mL, 1:1 in volume), formaldehyde (12.96 g, 160 mmol, 37 wt% aqueous), and N-Boc-piperazine (29.6 g, 160 mmol) were charged to a round-bottom flask; after stirring at room temperature for 30 min under N2, catechol (17.6 g, 160 mmol) was added. The reaction continued at 40 °C for 4 h. After removing CH3OH, 50 mL of 37% HCl and 200 mL EtOAc were added, and the reaction mixture was stirred for 4 h at room temperature. The mixture was washed with EtOAc (3 × 200 mL) and the aqueous phase was adjusted to pH 8 with a dilute NaOH solution and extracted by EtOAc (5 × 200 mL). The organic phases were combined, dried with anhydrous sodium sulfate, and concentrated by rotary evaporation to afford a residue, which was recrystallized in CH3CN, yielding 24.3 g of 2 in a yield of 73%.
Synthesis of urushiol analogues (UA0 ∼ 3)
Coating preparation
Cu substrates (>99.7%) were polished with emery papers, cut into square substrates in dimensions of 15.0 × 10.0 × 1.0 mm3, followed by washing ultrasonically with ultrapure water and ethanol for 5 min. Next, the Cu substrates were ultrasonicated in diluted HCl for 10 s, rinsed with water, and blow-dried with N2. An Autolab PGSTAT30 electrochemical workstation (Echo Chemie B.V., Netherlands) with a three-electrode system was used for the electropolymerization of UA0 ∼ 3, where the Cu substrate was used as the working electrode, a saturated calomel electrode (SCE) as the reference electrode, and a platinum wire as the counter electrode. A solution of 10 mM Tris–HCl (pH 8.5) diluted with ethanol (1:10 in volume) containing one of the UA0 ∼ 3 products in 30 mg mL−1 was used as the electrolyte. The analogue coating was formed by cyclic voltammetry with potential sweeping from 0 to 3.0 V vs. SCE at the rate of 30 mV s−1 for 15 cycles. The coatings were rinsed with water and blow-dried for performance characterization.
A proportional mixture of UA0 ∼ 3, denoted as UAm, consisting of the analogues UA0 (4.5 wt%), UA1 (15.0 wt%), UA2 (10.8 wt%), and UA3 (62.8 wt%), was made according to the typical content of each 3-alkenyl catechol derivative in urushiol (Scheme 1a). UAm and natural urushiol were also electrodeposited on a Cu substrate for comparative analyses.
Characterization of coatings
The thickness of the coating was measured by a coating thickness gauge (EC-770, YUWESE, China); the pencil hardness was determined by the standard ASTM D3363 test method, and the adhesion strength of coating to copper was determined according to ASTM D3359. The static contact angle of a liquid droplet of water (3 μL) on the coating was measured using the OCA 20 contact angle system (Dataphysics, Germany) and reported with the average value of 5 measurements on different sites. The ATR-FTIR was measured by a Nicolet iS10 spectrometer (Thermo Scientific, USA). The thermal stability of the coating was studied by thermogravimetric analysis (TGA 4000, PerkinElmer, USA) at a heating rate of 10 °C min−1 under constant N2 flow. The morphological images of the coatings on Cu were acquired using a MultiMode-8 SPM atomic force microscope system (AFM, Bruker) in the tapping mode and processed by the associated NanoScope Analysis software.Electrochemical corrosion measurements
The anticorrosion performance of the coatings was characterized by potentiodynamic polarization39 with the electropolymerization system, where the coated Cu was used as the working electrode and a corrosive solution containing 3.5 wt% NaCl as the electrolyte. The potentiodynamic polarization curves were obtained by sweeping the potential from −300 mV to +300 mV relative to the open circuit potential (vs. SCE) at a scan rate of 1 mV s−1 at 25 °C. For each measurement, three individual working electrodes without or with the same coating were measured to ensure reproducibility. The anticorrosion efficiency of the coating on copper was represented using its inhibition efficiency (IE), which was calculated according to the following equation:| IE (%) = (icorr(0) − icorr(c))/icorr(0) × 100 | (1) |
| CR = K(icorr/ρ)Ew | (2) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial supports from the National Natural Science Foundation of China (21772060), the Wuhan Municipal Front Program for Application Basis Research (2018010401011283), and the Central China Normal University Basic Research Fund of Education Ministry of China (CCNU18GF001).Notes and references
- S. Sato, R. Ido, T. Ose, Y. Takahashi, S. Kanehashi, T. Ishimura, T. Honda, T. Miyakoshi and K. Nagai, Prog. Org. Coat., 2017, 104, 43–49 CrossRef CAS.
- J. Saiz-Poseu, J. Mancebo-Aracil, F. Nador, F. Busqué and D. Ruiz-Molina, Angew. Chem., Int. Ed., 2019, 58, 696–714 CrossRef CAS PubMed.
- X. Zheng, J. Weng, S. Li, H. Liu, B. Hu, Y. Li, X. Meng and H. Ruan, Chem. Eng. J., 2014, 245, 265–275 CrossRef CAS.
- H. Watanabe, M. Takahashi, H. Kihara and M. Yoshida, Langmuir, 2019, 35, 4534–4539 CrossRef CAS PubMed.
- H. Jeong, J. Heo, B. Son, D. Choi, T. H. Park, M. Chang and J. Hong, ACS Appl. Mater. Interfaces, 2015, 7, 26117–26123 CrossRef CAS PubMed.
- G. Lligadas, A. Tüzün, J. C. Ronda, M. Galià and V. Cádiz, Polym. Chem., 2014, 5, 6636–6644 RSC.
- X. Zheng, X. Xiong, J. Yang, D. Chen, R. Jian and L. Lin, Chem. Eng. J., 2018, 333, 153–161 CrossRef CAS.
- H. Langhals and D. Bathelt, Angew. Chem., Int. Ed., 2003, 42, 5676–5681 CrossRef CAS PubMed.
- H. Watanabe, A. Fujimoto, R. Yamamoto, J. Nishida, M. Kobayashi and A. Takahara, ACS Appl. Mater. Interfaces, 2014, 6, 3648–3653 CrossRef CAS PubMed.
- S. Kobayashi, Struct. Chem., 2017, 28, 461–474 CrossRef CAS.
- B. Loev and C. R. Dawson, J. Org. Chem., 1959, 24, 980–985 CrossRef CAS.
- L. Rong, K. Yukio and M. Tetsuo, Talanta, 2007, 71, 1536–1540 CrossRef CAS PubMed.
- S. Kobayashi, R. Ikeda, H. Oyabu, H. Tanaka and H. Uyama, Chem. Lett., 2000, 1–2 CAS.
- H. Watanabe, M. Takahashi, H. Kihara and M. Yoshida, ACS Appl. Bio Mater., 2018, 1, 808–813 CrossRef CAS.
- T. Tsujimoto, H. Uyama and S. Kobayashi, Macromolecules, 2004, 37, 1777–1782 CrossRef CAS.
- C. Zhou, Y. Hu, Z. Yang, T. Yuan, J. Huang, P. Li and Y. Liu, Prog. Org. Coat., 2018, 120, 240–251 CrossRef CAS.
- S. I. Shoda, H. Uyama, J. I. Kadokawa, S. Kimura and S. Kobayashi, Chem. Rev., 2016, 116, 2307–2413 CrossRef CAS PubMed.
- R. Ikeda, T. Tsujimoto, H. Tanaka, H. Oyabu, H. Uyama and S. Kobayashi, Proc. Jpn. Acad., 2000, 76, 155–160 CrossRef.
- R. Ikeda, H. Tanaka, H. Uyama and S. Kobayashi, Polym. J., 2000, 32, 589–593 CrossRef CAS.
- F. Gao, W. G. Zhang, G. C. Shen and J. Y. Tang, Journal of Fujian Teachers University (Natural Science), 2003, 1, 017 Search PubMed.
- R. Yue, H. Huang, G. Ke, H. Zhang, J. Pei, G. Xue and G. Yuan, Mater. Charact., 2017, 134, 114–122 CrossRef CAS.
- J. Duan, W. Wu, Z. Wei, D. Zhu, H. Tu and A. Zhang, Green Chem., 2018, 20, 912–920 RSC.
- T. Yurino, Y. Aota, D. Asakawa, T. Kano and K. Maruoka, Tetrahedron, 2016, 72, 3687–3700 CrossRef CAS.
- J. B. Grosse Daldrup, C. Held, G. Sadowski and G. Schembecker, Ind. Eng. Chem. Res., 2011, 50, 3503–3509 CrossRef CAS.
- O. Fraczak, A. Lasota, D. Tymecka, P. Kosson, A. Muchowska, A. Misicka and A. Olma, J. Pept. Sci., 2016, 22, 222–227 CrossRef CAS PubMed.
- J. Yang, M. A. Cohen Stuart and M. Kamperman, Chem. Soc. Rev., 2014, 43, 8271–8298 RSC.
- X. Du, L. Li, F. Behboodi-Sadabad, A. Welle, J. Li, S. Heissler, H. Zhang, N. Plumeré and P. A. Levkin, Polym. Chem., 2017, 8, 2145–2151 RSC.
- C. Narita, K. Yamada and T. Tsujii, Mater. Chem. Phys., 2017, 199, 387–392 CrossRef CAS.
- J. Xia, J. Lin, Y. Xu and Q. Chen, ACS Appl. Mater. Interfaces, 2011, 3, 482–489 CrossRef CAS PubMed.
- J. Yang, F. Shen, J. Deng, J. Cai, Q. Zhang and W. Liu, J. Appl. Polym. Sci., 2018, 135, 1–8 Search PubMed.
- X. L. Zheng, J. B. Weng, Q. M. Huang, B. H. Hu, T. Qiao and P. Deng, Colloids Surf., A, 2009, 337, 15–20 CrossRef CAS.
- C. Zhang, M. Q. Ma, T. T. Chen, H. Zhang, D. F. Hu, B. H. Wu, J. Ji and Z. K. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 34356–34366 CrossRef CAS PubMed.
- H. Watanabe, A. Fujimoto, J. Nishida, T. Ohishi and A. Takahara, Langmuir, 2016, 32, 4619–4623 CrossRef CAS PubMed.
- Y. Deng, W. Bai, X. Zhang, J. Chen, S. Wang, J. Lin and Y. Xu, ACS Omega, 2018, 3, 4129–4140 CrossRef CAS.
- H. P. Wang, Q. Wu, C. M. Li and N. Gu, Mater. Corros., 2013, 64, 347–352 CrossRef CAS.
- Y. Deng, W. Bai, J. Chen, X. Zhang, S. Wang, J. Lin and Y. Xu, RSC Adv., 2017, 7, 45034–45044 RSC.
- N. C. Rosero-Navarro, S. A. Pellice, A. Durán, S. Ceré and M. Aparicio, J. Sol-Gel Sci. Technol., 2009, 52, 31–40 CrossRef CAS.
- A. Yaǧan, N. Ö. Pekmez and A. Yildiz, Corros. Sci., 2007, 49, 2905–2919 CrossRef.
- J. Chen, W. Cheng, S. Chen, W. Xu, J. Lin, H. Liu and Q. Chen, Nanoscale, 2018, 10, 22818–22829 RSC.
- Z. Wei, X. Chen, J. Duan, G. Zhan, Y. Wei and A. Zhang, J. Mol. Liq., 2019, 280, 327–333 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04981a |
| This journal is © The Royal Society of Chemistry 2019 |