
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, characterization, and photoluminescent studies of three-coordinate Cu(I)–NHC complexes bearing unsymmetrically-substituted dipyridylamine ligands†
Kwame Glinton,
Reza Latifi ,
David S. Cockrell,
Matthew Bardeaux,
Bachkhoa Nguyen and
Laleh Tahsini*
,
David S. Cockrell,
Matthew Bardeaux,
Bachkhoa Nguyen and
Laleh Tahsini*
Department of Chemistry, Oklahoma State University, Stillwater, Oklahoma 74078, USA. E-mail: tahsini@okstate.edu
First published on 19th July 2019
Abstract
A series of heteroleptic three-coordinate Cu(I) complexes bearing monodentate N-heterocyclic carbene (NHC) ligands of the type 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr) and 1,3-bis(2,6-diisopropylphenyl)imidazolidin-2-ylidene (SIPr), and bidentate N-donor ligands of the type unsymmetrically-substituted dimethyl dipyridylamine (Me2Hdpa) and bis(mesityl)biazanaphthenequinone (mesBIAN) have been synthesized. The complexes [Cu(IPr)(3,4′-Me2Hdpa)]PF6, 1; [Cu(IPr)(3,5′-Me2Hdpa)]PF6, 2; [Cu(IPr)(3,6′-Me2Hdpa)]PF6, 3; [Cu(IPr)(mesBIAN)]PF6, 6; [Cu(SIPr)(3,4′-Me2Hdpa)]PF6, 7; [Cu(SIPr)(3,5′-Me2Hdpa)]PF6, 8; and [Cu(SIPr)(3,3′-Me2Hdpa)]PF6, 11 have been characterized by 1H and 13C NMR spectroscopies, elemental analysis, cyclic voltammetry, and photophysical studies in solid and solution phase. Single crystal X-ray structures were obtained for all complexes except 11. The crystallographic data reveal a mononuclear structure for all complexes with the copper atom ligated by one C and two N atoms. The UV-Vis absorption spectra of all dipyridylamine complexes in CH2Cl2 show a strong ligand-centered absorption band around 250 nm and a strong metal-to-ligand charge transfer (MLCT) band around 300 nm. When irradiated with UV light, the complexes exhibit strong emission maxima at 453–482 nm with photoluminescence quantum yields (PLQY) ranging from 0.21 to 0.87 in solid state. While the PLQY values are comparable to those of the symmetrical [Cu(IPr)(Me2Hdpa)]PF6 complexes, a stabilizing CH–π interaction has been reduced in the current systems. In particular, complex 3 lacks any strong CH–π interaction, but emits more efficiently than 1 and 2 wherein the interactions exist. Structural data analysis was performed to clarify the role of ligands' plane angle and the NH/CH⋯F interactions to the observed light interaction of unsymmetrical [Cu(NHC)(Me2Hdpa)]PF6 complexes. DFT calculations were performed to assist in the assignment of the electronic structure and excited state behavior of the complexes.
Introduction
Photoluminescent (PL) complexes containing precious transition metals (Ru(II), Ir(III), Pt(II), etc.) have been extensively studied due to their potential application in electroluminescent devices, photochemical catalysis, OLED light displays, and solar energy conversion.1–9 However, owing to their limited availability, toxicity, and relative cost, the substitution of these metals by more abundant alternatives is desired. In this context, d10 metals such as Cu(I) are of considerable interest. Over the last several decades, a great variety of Cu(I) complexes have been developed aiming to improve the emissive properties of Cu luminophores towards strong emission and long-lived excited states. Majority of these efforts have focused on the four-coordinate tetrahedral Cu(I) complexes with bisimine and phosphine ligands to block their non-radiative decay pathways. The flattening of the photoexcited Cu(II) complexes via a Jahn–Teller distortion promotes the formation of a five-coordinate exciplex with solvent molecules or other nucleophiles that often leads to a radiationless deactivation of the excited state.10–16 A useful strategy to prevent the excited state configurational changes, first examined by McMillin,17 is by increasing the steric bulk of ligands at the coordination site. There has been an extensive work on the effect of bulky substituents around the tetrahedral Cu(I) coordination sphere, leading to highly emissive luminophores with long-lived excited state.18–24 Another useful approach towards blocking the four-coordinate geometry is via bridging halides or other potential linkers in bi- and polynuclear complexes that will not be discussed in this text.25–27Recently, emissive three-coordinate heteroleptic Cu(I) complexes supported by bulky phosphine ligands have been reported by groups of Peters and others.28,29 While the possibility of Jahn–Teller distortion in the excited state is eliminated in the three-coordinate geometry, the exciplex formation can still occur especially in coordinating solvents. Alternatively, Thompson,30–33 Gaillard34–36 and their co-workers have investigated three-coordinate mononuclear Cu(I) complexes bound to sterically challenged NHC and bidentate N-donor ligands. The choice of the ligand, guided by the high stability of Cu(I)–NHC complexes and easy modulation of the nitrogen ligand, has led to [Cu(NHC)(N^N)]X complexes (N^N = dipyridylamine and its geometrically symmetrical derivatives) with very promising luminescent properties. In their work, Gaillard and co-workers had synthesized a series of Cu(I) complexes using symmetrically-substituted Hdpa ligands bearing methyl groups on two pyridyl rings. Interestingly, the position of the methyl groups plays an important role in the luminescent properties of the complexes, with the highest and lowest quantum yields obtained using 3,3′-Me2Hdpa and 4,4′-Me2Hdpa ligands, respectively.
The strong emissive properties of these complexes have been assigned partly to their stable excited-state geometry that resulted from the stabilizing CH–π system interactions between the NHC and the Hdpa ligands. It could be envisioned that disrupting such stabilizing interactions in the [Cu(NHC)(Hdpa)]X complexes would affect their emissive properties. However, knowledge about the extent of their contribution to the observed photophysical activity has yet to be developed. We report herein the synthesis of a new series of cationic Cu(I) complexes bearing bulky NHC ligands (IPr, SIPr) and Me2Hdpa ligands with methyl groups positioned unsymmetrically on two pyridyl rings (Chart 1, L1–L3). Also, we have prepared Cu(I)–NHC complexes containing Hdpa (L4) and 3,3′-Me2Hdpa (L5) for direct comparison of chemical and photophysical properties to the unsymmetrically-substituted analogues under the same conditions. To illustrate the effect of steric challenge and rigidity on the coordination chemistry and photophysical properties of Cu(I) complexes, the bulky mesBIAN (L6) ligand has also been synthesized. The extent of stabilizing CH–π and other long-range interactions are examined by analyzing the structural parameters from single-crystal X-ray diffraction. The electrochemical and photophysical properties of these complexes are examined and compared to the Cu(I) complexes bearing unsubstituted Hdpa and symmetrically substituted Me2Hdpa ligands. In addition, density functional theory (DFT) calculations are performed to rationalize the relationship between the electronic structure and photophysical property in these complexes.
 | ||
| Chart 1 Symmetrical and unsymmetrical Me2Hdpa and NHC ligands used in this study. | ||
Results and discussion
Synthesis of heteroleptic Cu(I) complexes
The [Cu(NHC)(N^N)]PF6 complexes (1–12) were synthesized in a single-step reaction using [Cu(NHC)Cl], the N-donor ligand, and KPF6 under an inert atmosphere at room temperature (Scheme 1). Both [Cu(IPr)Cl] and [Cu(SIPr)Cl] precursors were prepared according to a slightly modified Cazin–Nolan procedure using CuCl, the imidazolium/imidazolinium salt, and K2CO3 as a base.37 Dipyridylamine ligands were obtained using a Buchwald–Hartwig coupling reaction between 2-bromopyridine and 2-aminopyridine derivatives. The optimal amount of Pd2(dba)3 catalyst for these syntheses was found to be 1.0 mol%, half of the amount used previously for the symmetrical Me2Hdpa ligands.34 Interestingly, some ligands such as L2 and L3 were obtained only at 20% or lower yields using 2.0 mol% catalyst loading, whereas the reduced amount of the catalyst provided great to excellent yields of all ligands.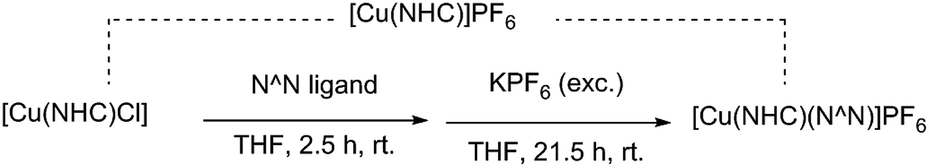 | ||
| Scheme 1 The formation of heteroleptic Cu(I) complexes via a mono-coordinate Cu–NHC intermediate. | ||
Regarding [Cu(NHC)(N^N)]PF6 complexes, the yield is dependent on the steric hindrance of the N-donor ligands as depicted in Table 1. However, the effect appears to be less pronounced than what is found for the symmetrical Me2Hdpa analogues as shown by a close yield of [Cu(IPr)(L1)]PF6 (1) and [Cu(IPr)(L2)]PF6 (2) complexes. A clear deviation from the expected trend based on steric hindrance was found for [Cu(NHC)(L2)]PF6 complexes that were isolated at a higher yield than those with L4 and L5 ligands. Also, the formation of [Cu(NHC)(N^N)]PF6 complexes with bulky N-donor ligands appears to be dependent on the nature of the carbene's backbone, i.e. saturated vs. unsaturated. While both [Cu(IPr)(L3)]PF6 (3) and [Cu(IPr)(L6)]PF6 (6) complexes were obtained at adequate yields, no [Cu(SIPr)(L3)]PF6 (9) and only 10% [Cu(SIPr)(L6)]PF6 (12) formed after a two-day reaction as confirmed by 1HNMR analysis. The synthesis of the heteroleptic Cu(I) complexes is expected to follow a two-step mechanism, wherein a mono-coordinate [Cu(NHC)]PF6 forms initially from the removal of Cl− with tenfold excess KPF6 (Scheme 1). This was confirmed by a white solid isolated from the [Cu(IPr)Cl] and KPF6 reaction that shows a different composition from the starting material by elemental analysis (ESI†).
| Entrya | Complex | Yieldb |
|---|---|---|
| a Reaction conditions: [Cu(NHC)Cl] (1 equiv.), N^N ligand (1 equiv.), KPF6 (10 equiv.), THF, stir at r.t.b Isolated yield.c Calculated from the 1H NMR spectrum of the reaction mixture containing the product and [Cu(IPr)Cl]. | ||
| 1 | [Cu(IPr)(L1)]PF6 (1) | 77 |
| 2 | [Cu(IPr)(L2)]PF6 (2) | 83 |
| 3 | [Cu(IPr)(L3)]PF6 (3) | 62 |
| 4 | [Cu(IPr)(L4)]PF6 (4) | 70 |
| 5 | [Cu(IPr)(L5)]PF6 (5) | 53 |
| 6 | [Cu(IPr)(L6)]PF6 (6) | 68 |
| 7 | [Cu(SIPr)(L1)]PF6 (7) | 69 |
| 8 | [Cu(SIPr)(L2)]PF6 (8) | 85 |
| 9 | [Cu(SIPr)(L3)]PF6 (9) | 0 |
| 10 | [Cu(SIPr)(L4)]PF6 (10) | 63 |
| 11 | [Cu(SIPr)(L5)]PF6 (11) | 78 |
| 12 | [Cu(SIPr)(L6)]PF6 (12) | ∼10c |
X-ray crystallography of Cu(I) complexes
Single crystals of complexes 1–3, 6–8, and 12 were obtained by slow diffusion of hexane into a solution of copper complexes in dichloromethane at room temperature. The red crystals of 12 were isolated carefully from a mixture of the complex and starting precursor, [Cu(SIPr)Cl], to further verify its formation and structure by X-ray crystallography. Also, we obtained single crystals of [Cu(SIPr)Cl] precursor from a mixture of tetrahydrofuran and hexane solution and report its crystal structure here (Fig. S1†). ORTEP diagrams of IPr- and SIPr-based complexes are shown in Fig. 1. A collection of distances and angles are presented in Table 2 with crystallographic data collection parameters summarized in Tables S1 and S2.†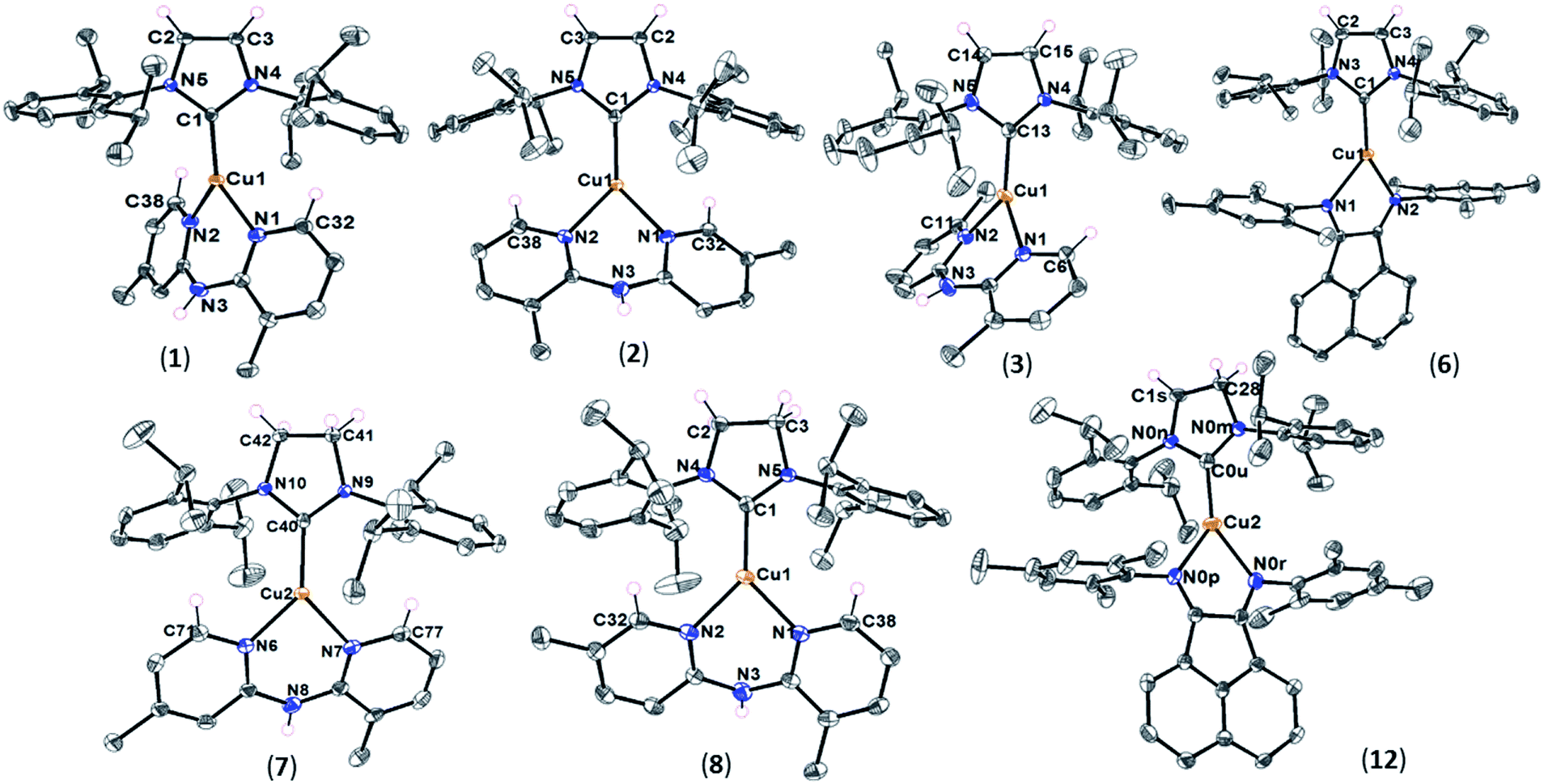 | ||
| Fig. 1 ORTEP diagrams of [Cu(IPr)(L1)]PF6 (1), [Cu(IPr)(L2)]PF6 (2), [Cu(IPr)(L3)]PF6 (3), [Cu(IPr)(L6)]PF6 (6), [Cu(SIPr)(L1)]PF6 (7), [Cu(SIPr)(L2)]PF6 (8), and [Cu(SIPr)(L6)]PF6 (12). Only one of the cationic complexes in the unit cell are shown for 1, 7, and 12. Anions and some hydrogens are omitted for clarity. Ellipsoids are shown at the 55% probability level. | ||
| Complex | Cu–CNHC (Å) | Cu–NN^N (Å) | CH⋯π (Å) | NH/CH⋯Fa (Å) | CNHC–Cu–NN^N (°) | Plane angle θ (deg) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a The distances have been determined only for the asymmetric unit.b The CH⋯π distances are shown only for one of the cationic complexes (Cu1) in the asymmetric unit. | ||||||||||||
| [Cu(IPr)(L1)]PF6 (1) | Cu1 | 1.891(2) | Cu1 | 2.019(2) | Cu1 | 2.78 | Cu1 | 2.25 (NH) | Cu1 | 138.23(7) | Cu1 | 35.52 |
| Cu2 | 1.906(2) | 2.038(2) | Cu2 | 2.45 | 2.48, 2.54 (CH) | 131.94(7) | Cu2 | 59.76 | ||||
| Cu2 | 2.015(2) | 2.70 | Cu2 | 2.25 (NH) | Cu2 | 134.08(7) | ||||||
| 2.044(2) | 2.31(CH) | 135.24(7) | ||||||||||
| [Cu(IPr)(L2)]PF6 (2) | 1.900(2) | 2.032(2) | 2.58 | 2.43 (NH) | 136.22(7) | 47.13 | ||||||
| 2.041(2) | 2.95 | 2.43 (CH) | 134.21(7) | |||||||||
| [Cu(IPr)(L3)]PF6 (3) | 1.887(2) | 2.020(2) | 2.68 (CH) | 134.44(7) | 76.06 | |||||||
| 2.029(2) | 134.02(7) | |||||||||||
| [Cu(IPr)(L6)]PF6 (6) | 1.922(3) | 2.231(3) | 2.53 (CHiPr) | 2.32 (CHNHC) | 133.86(6) | 45.89 | ||||||
| 2.062(2) | 2.74 (CHMe) | 2.47 (CHNHC) | 147.70(6) | |||||||||
| 2.77 (CHiPr) | ||||||||||||
| 2.94 (CHace) | ||||||||||||
| 2.98 (CHace) | ||||||||||||
| [Cu(SIPr)(L1)]PF6 (7) | Cu1 | 1.893(2) | Cu1 | 2.026(2) | Cu1 | 2.74 | 2.26 (NH) | Cu1 | 130.70(8) | Cu1 | 38.77 | |
| Cu2 | 1.917(2) | 2.020(2) | Cu2 | 2.42 | 2.50 (CH) | 139.25(8) | Cu2 | 59.59 | ||||
| Cu2 | 2.021(2) | 2.74 | 2.28 (CH) | Cu2 | 134.27(9) | |||||||
| 2.052(2) | Cu2 | 135.30(9) | ||||||||||
| [Cu(SIPr)(L2)]PF6 (8) | 1.918(2) | 2.045(2) | 2.46 | 2.24 (NH) | 134.13(7) | 45.04 | ||||||
| 2.046(2) | 2.94 | 2.61 (CH) | 135.57(7) | |||||||||
| [Cu(SIPr)(L6)]PF6 (12) | Cu1 | 1.914(4) | Cu1 | 2.126(3) | 2.65 (CHiPr)b | 2.26 (NH) | Cu1 | 138.4(1) | Cu1 | 48.46 | ||
| Cu2 | 1.914(4) | 2.137(3) | 2.71 (CHMe) | 2.65 (CHNHC) | 142.5(1) | Cu2 | 49.74 | |||||
| Cu2 | 2.087(3) | 2.76 (CHiPr) | Cu2 | 138.4(1) | ||||||||
| 2.155(3) | 2.87 (CHace) | 142.7(1) | ||||||||||
| 2.92 (CHace) | ||||||||||||
As expected for the heteroleptic Cu(I) complexes, each Cu center is bound to the NHC and the bidentate N-donor ligand in a three-coordinate geometry. The dipyridylamine ligation in complexes 1–3, 7, and 8 results in a 6-membered chelate unlike mesBIAN that forms a 5-membered ring with Cu in 6 and 12. A common feature among the heteroleptic Cu(I) complexes in this study is the presence of long-distance interactions in their solid state. This includes, but is not limited to, the non-bonding CH⋯π interactions between the hydrogen atom(s) at the α position of the pyridyl rings and the π system of the diisopropylphenyl groups on the NHC ligands. The structures in which the calculated distance between the α hydrogen atom and the center of the NHC aromatic ring is shorter than 3.05 Å are identified to possess this interaction.38 As shown in Table 2, all the dipyridylamine complexes present this interaction, except 3 that has a much longer CH⋯π distance than the cut-off. The sp2-CH⋯π interactions were considered crucial for the formation of complexes with symmetrical Me2Hdpa ligands to an extent that [Cu(IPr)(6,6′-Me2Hdpa)]PF6 could not form due to the lack of α hydrogen atoms on the Hdpa ligand.34 Our findings support the significance of CH⋯π interactions to the stability of the Cu(I) complexes bearing unsymmetrical Me2Hdpa ligands as well. Nonetheless, the synthesis of [Cu(IPr)(3,6′-Me2Hdpa)]PF6 (3) with no sp2-CH⋯π interaction at a good yield (Table 1) indicates the contribution of other parameters to its stability.
Not surprisingly, due to the lack of α hydrogen atom with respect to nitrogen in the mesBIAN ligand, the interligand sp2-CH⋯π interactions do not exist in complexes 6 and 12. However, the presence of several aromatic rings may facilitate the sp3-CH⋯π interactions between the aliphatic hydrogens and the π systems of the NHC and diimine ligands in these complexes. Furthermore, 12 benefits from an intermolecular sp2-CH⋯π interaction between the CH groups at position 6 of the BIAN ring and the π system of the phenyl ring in SIPr (Fig. S2†). The sp3-CH⋯π interactions in 6 occur between the isopropyl groups of IPr and the aromatic mesityl substituents of mesBIAN as well as the methyl groups of mesBIAN and aromatic phenyl ring of IPr (Fig. S3†). The complexes 6 and 12, also, benefit from an intraligand interaction between the sp2-CH at positions 5 and 10 of the BIAN ring and the aromatic ring of the mesityl substituents (Fig. S2 and S3†). Additionally, the effect of NH⋯F and CH⋯F hydrogen-bonding interactions on the stability of the current heteroleptic Cu(I) systems should not be disregarded. For all the complexes in Table 2, the H⋯F distance between the F atoms of PF6− anion and the NH or sp2-CH groups of Hdpa falls within the range 2.24–2.72 Å. Since the distance is shorter than the sum of van der Waals radii of H and F (2.76 Å), the interaction can be classified as a legitimate hydrogen bond.39 It should also be noted that 3 is the only Hdpa-based complex in the table in which no NH⋯F interaction was found. The significance of hydrogen-bonding to the shaping, stability, and self-assembly of organic molecules has been extensively investigated,39–41 however, their effect on the transition metal-containing structures is far less understood. In this work, the interplay between the long-range interactions, the plan angle θ, and the light-emitting properties of the copper complexes will be elucidated via structural analysis and photophysical studies (vide infra). The θ parameter in the table describes the angle between the NHC ring and the plane formed by three nitrogen atoms in the Hdpa ligand or the plane of the mesBIAN ligand (Fig. S4†). There seem to be no direct correlation between θ and the ligand type, e.g. saturated NHC vs. unsaturated NHC or substituted Hdpa vs. unsubstituted derivatives.
Electrochemical study of Cu(I) complexes
The cyclic voltammetry (CV) data were measured to investigate the electrochemical features and to estimate the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of the complexes 1–11. The electrochemical data are summarized in Table 3. As shown in Fig. 2a, all Cu–IPr complexes bearing Hdpa and its derivatives display an electrochemically quasi-reversible/reversible oxidation at 0.61–0.65 V (vs. Fc+/Fc), assigned to the Cu(II)/Cu(I) couple. The reversibility of this event is more prevalent for 4 and its unsymmetrical analogs shown by a linear plot of current versus the square root of scan rate (Fig. S5†). The partial reversibility was confirmed by a shift of the reduction or oxidation potentials at varying scan rates for all the Cu–IPr complexes, but is more distinct for complex 5 (Fig. S6†). On the other hand, all the Cu–SIPr complexes containing Hdpa ligand and its derivatives reveal a predominantly quasi-reversible Cu(II)/Cu(I) couple (Fig. 2b). The shape of the first oxidation peak of 4, 5, and 10 appears different in our work from that reported earlier.34 While this event was irreversible and attributed to the oxidation of NH group of Hdpa ligands, we have determined a reversible/quasi-reversible couple and assigned it to a metal-centered oxidation feature. In addition to the presented data in Fig. S5 and S6,† the comparative cyclic voltammograms of the solvent and the Hdpa ligand support the assignment (Fig. S7†). A metal-based oxidation is also more consistent with a similar feature observed in the CV studies of [Ru(Hdpa)n(bpy)3−n]2+ (n = 3–0) and [Ni(Hdpa)2(salo)]Cl complexes and with the HOMO composition in the DFT calculations (vide infra).42,43 Nonetheless, our assessment of the irreversible reduction features at around −2.3 V and −2.5 V vs. Fc+/Fc in the CV of the Hdpa/Me2Hdpa complexes is in line with the previous assignments to the Cu(I)/Cu(0) and Hdpa reduction.34,44| Complex | λabsmaxa (nm) | λemmaxb (nm) | Eox1/2, ΔEpc (V) | EPc, Epad (V) | HOMO/LUMOd,g (eV) | PLQY/ΦPLe | TPLe (μs) |
|---|---|---|---|---|---|---|---|
| a Data obtained in CH2Cl2 at room temperature.b Data obtained in solid state at room temperature.c Recorded in CH2Cl2 containing 2 mM Cu(I) complexes and 0.1 M TBAPF6 at 50 mV s−1 scan rate and 22 °C.d Recorded at 100 mV s−1 scan rate and 22 °C.e Estimated errors ±5%.f The half-wave potential of 0 and ΔEp = Epc − Epa = 0.174 V was determined for ferrocene under the same conditions.g The values in () were calculated by the difference between the HOMO levels and the HOMO–LUMO gap obtained from the absorption edge of the UV/Vis spectra. | |||||||
| [Cu(IPr)(L1)]PF6 (1) | 253, 302 | 468 | 0.62, 0.276f | −2.29, 0.524 | −5.32/−2.51 (−2.04) | 0.48 | 18 |
| [Cu(IPr)(L2)]PF6 (2) | 254, 306 | 475 | 0.63, 0.278 | −2.20, 0.530 | −5.33/−2.60 (−2.04) | 0.49 | 19 |
| [Cu(IPr)(L3)]PF6 (3) | 254, 307 | 482 | 0.63, 0.266 | −2.22, 0.502 | −5.30/−2.58 (2.03) | 0.64 | 23 |
| [Cu(IPr)(L4)]PF6 (4) | 252, 305 | 453 | 0.65, 0.244 | −2.35, 0.546 | −5.35/−2.45 (−2.03) | 0.21 | 15 |
| [Cu(IPr)(L5)]PF6 (5) | 251, 303 | 458 | 0.61, 0.267 | −2.35, 0.514 | −5.31/−2.45 (−1.99) | 0.87 | 35 |
| [Cu(IPr)(L6)]PF6 (6) | 241, 321, 336 | ∼490 | 1.07, 0.364 | −1.27, 1.03 | −5.83/−3.53 (−3.3) | 0 | |
| [Cu(SIPr)(L1)]PF6 (7) | 254, 307 | 472 | 0.61, 0.425 | −2.15, 0.603 | −5.40/−2.65 (−2.20) | 0.55 | 24 |
| [Cu(SIPr)(L2)]PF6 (8) | 254, 309 | 471 | 0.65, 0.294 | −2.16, 0.562 | −5.36/−2.64 (−2.19) | 0.33 | 19 |
| [Cu(SIPr)(L4)]PF6 (10) | 254, 307 | 466 | 0.67, 0.298 | −2.29, 0.629 | −5.43/−2.51 (−2.13) | 0.85 | 15 |
| [Cu(SIPr)(L5)]PF6 (11) | 254, 304 | 473 | 0.68, 0.376 | −2.25, 0.601 | −5.40/−2.55 (−2.25) | 0.83 | 25 |
 | ||
| Fig. 2 Cyclic voltammogram of (a) [Cu(IPr)(Me2Hdpa)]PF6 and (b) [Cu(SIPr)(Me2Hdpa)]PF6 complexes (2.0 mM) in deaerated CH2Cl2/nBu4NPF6 (0.1 M) at a scan rate of 100 mV s−1 indicating Cu(II)/Cu(I) redox couple. | ||
As expected the electron-transfer behavior of complex 6 is different from the Hdpa-containing analogs. The CV of this complex (Fig. S8†) presents a quasi-reversible Cu(II)/Cu(I) couple at 1.30 V (vs. Fc+/Fc) resembling the same oxidation event in the CV of Cu(I)–(aryl-BIAN) complexes bearing nitrile and phosphine co-ligands.45 The quite large difference (0.4 V) between the oxidation and reduction waves of this couple and the substantial decrease in the cathodic peak current is likely due to some rearrangements or chemical processes during the oxidation. When continuing the cathodic scan of complex 6, two quasi-reversible reduction waves appear at −1.41 and −2.03 V (vs. Fc+/Fc) assigned to the Cu(I)/Cu(0) and mesBIAN/mesBIAN−˙ couples. The reduction potential of the ligand to its radical anion form is reasonably close to that of the free ligand reported under similar conditions.45 While the authors did not observe this reduction couple in the cathodic scan of the bis-chelate Cu(I)–aryl-BIAN complexes, this could be due to a shorter potential window utilized than that for 6 in this study.
The onset oxidation and reduction potentials of complexes 1–11 were used to calculate the HOMO and LUMO levels according to the equation EHOMO/LUMO = −(E[onset, ox/red vs. Fc+/Fc] + 4.8) eV.46,47 Since the reduction potential of the dipyridylamine complexes is close to the onset of solvent reduction, erroneous reading of the onset potential might occur. Thus, the LUMO levels were also calculated by considering the difference between their HOMO levels and the corresponding HOMO–LUMO gap determined from the absorption edge of their UV-Vis spectra. The calculated HOMO and LUMO values for complexes 1–11 in solution are listed in Table 3.
Photophysical study of Cu(I) complexes
The UV-Vis absorption spectra, recorded in CH2Cl2, and the solid-state emission spectra of complexes 1–11 are shown in Fig. 3, S9,† and Table 3. The intense absorption bands around 251–254 nm (ε = (2.4–4.3) × 104 L mol−1 cm−1) in the Hdpa-based complexes are attributed to the π–π* ligand-centered (LC) transitions. The relatively weaker bands around 302–309 nm (ε = (1.4–1.9) × 104 L mol−1 cm−1) in these complexes are ascribed to the metal-to-ligand charge transfer (MLCT) transitions.34 In complex 6, the π–π* LC transitions result in an intense shoulder below 250 nm beside the two equal-intensity bands at 321 nm and 336 nm (ε = 1.8 × 104 L mol−1 cm−1) due to the spin-allowed MLCT transitions (Fig. S9a†).48 Additionally, the broad band between 350 and 480 nm could be assigned to the intraligand π–π* charge transfer (ILCT) from the aryl groups to the naphthyl backbone and/or a low-energy MLCT band.48,49 | ||
| Fig. 3 (a and b) Absorption spectra of complexes 1–11 (except 6) in CH2Cl2 solution at room temperature. (c and d) Photoluminescent (PL) spectra of the complexes in the solid state. | ||
The PL spectra of the complexes were recorded in solid state at room temperature using an excitation of 340 nm. As shown in Fig. 3, the Hdpa-bound complexes display broad emission spectra with PL maxima in the range of 453–482 nm. In contrast, the PL spectrum of complex 6 shows a very weak emission around 490 nm (Fig. S9†). Furthermore, the methyl substitution of the Hdpa ligands leads to the red-shifted emission maxima in Cu–IPr complexes in the order L4 < L5 < L1 < L2 < L3. In contrast, the emission pattern and PL maxima of the substituted Cu–SIPr complexes are very similar indicating that the position of methyl groups has a minimal effect on the emissive energy of these complexes. Exploring the emission and absorption profiles of 1–11, the PL quantum yields (PLQY) were estimated between 21% and 87% with emission lifetimes in the range 15–35 μs (Table 3). In this series, complex 4 exhibits the lowest yield and complexes 5, 10, and 11 show the highest quantum yields with 87% (T = 35 μs), 85% (T = 15 μs), and 83% (T = 25 μs), respectively. The calculated QY values for 4, 5, and 10 are consistent with the previous report,34 however, the emission lifetime of 10 is substantially different.
Structure and light interaction relationship
Regarding the structure effect on the emission profile, different trends were found for the Cu–IPr and Cu–SIPr complexes bearing the same Hdpa ligands. While 4 is the weakest emitter among the [Cu(IPr)(N^N)]+ complexes, 10 has the highest QY in the [Cu(SIPr)(N^N)]+ series. The difference cannot be simply attributed to the greater electron-donating ability of SIPr than IPr given the reverse order of the quantum yields for 2 vs. 8 and 5 vs. 11 (Table 3). Given the PL profile of these complexes in solid state, close attention should be paid to the long-range interactions within the solid matrix. Three structural parameters relevant to the emissive properties of the Hdpa-based copper complexes are listed in Table S3.† Of these parameters, the NH/CH⋯F interactions are detrimental to PL, most likely due to inhibiting the planarization of the central nitrogen atom and change of its hybridization from sp3 to sp2 during the photoexcitation. This change is necessary to access a stable triplet state and an effective emissive system. On the other hand, the CH⋯π interactions are in favor of the PL property, possibly due to enhancing the π–π* character of electronic transitions, e.g. T1 → S0 via the interligand CT (vide infra). Finally, the large plane angle θ has a positive impact on the emission yield by breaking the symmetry in the solid matrix and allowing the symmetry-forbidden transitions to intensify. This parameter is mostly prominent in 3, with no favorable CH⋯π interaction and with relatively long CH⋯F distances. Complex 3 displays PL with a QY of 0.64 (T = 23 s), making it the third highly emissive [Cu(IPr)(N^N)]PF6 complex after [Cu(IPr)(3,3′-Me2Hdp)]PF6 and [Cu(MeIPrOMe)(Hdpa)]PF6 reported by Gaillard and coworkers.34,36 It is also worth noting that 3 possesses the largest plane angle θ among the reported [Cu(NHC)(N^N)]PF6 complexes and those presented here (Table S3†).Computational study of Cu(I) complexes
Density functional theory (DFT) and time-dependent DFT (TD-DFT) were used to rationalize the fundamental properties of these copper complexes and provide a theoretical foundation to explain the experimentally detected luminesce behavior. The details of the theoretical calculations are described in the ESI.† Despite the diverse range of functionals and basis sets used for copper complexes in literature that often provide good results,50–53 the functional sensitivity of TD-DFT, especially for CT systems,54 requires their careful screening in the luminescence studies.For the HOMO of complex 6, a major contribution (68–78%) from the π* orbitals of mesBIAN and a much smaller distribution (20–25%) over copper orbitals were calculated at three levels. Fig. 4 depicts the electron density distributions of the HOMO and LUMO of complexes 1 and 6 at wB97XD/6-31+G(d,p) level.
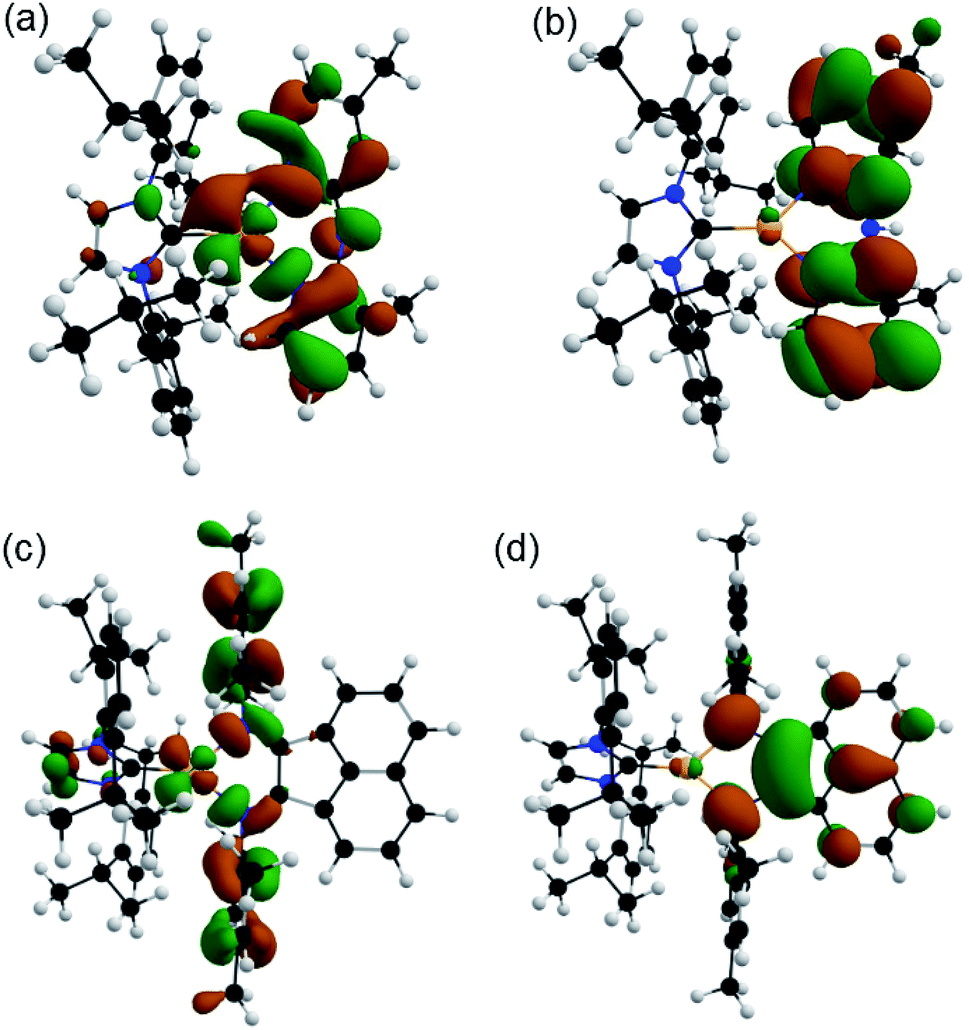 | ||
| Fig. 4 (a) HOMO and (b) LUMO of [Cu(IPr)(3,4′-Me2Hdpa)]+ (1), and (c) HOMO and (d) LUMO of [Cu(IPr)(mesBIAN)]+ (6) calculated at wB97XD/6-31+G (d,p) level. | ||
Regarding the effect of methyl groups on the HOMO–LUMO gap, there are marginal differences between unsubstituted and substituted Hdpa complexes at three theory levels. Furthermore, all saturated SIPr complexes have a slightly larger HOMO–LUMO gap than their unsaturated IPr analog due to the more electron-donating carbene and a more stabilized HOMO level. A comparative HOMO–LUMO level energy diagram of all the complexes is shown in Fig. S12.†
Regardless of the different excitation energy and the main MO contributor to the S0 → S1 transition, both wB97XD and M06 exhibit comparable emission energy and transition weight for the S0 → T1 transition (Table 4). Furthermore, both methods describe distinct characters for these transitions in the Hdpa-based complexes from those in complex 6. For 1–5 and 7–10, the S0 → S1 transition is primarily MLCT in nature, whereas the S0 → T1 transition is a complex mixture of MLCT and ligand-based charge transfer (LCCT). In contrast, both S0 → S1 and S0 → T1 in 6 are spatially identical and have predominantly LCCT character due to the H → L transition being the main MO contributor (Tables S9 and S10†).
| Complex | S0 → S1 | S0 → T1a | Method | |||||
|---|---|---|---|---|---|---|---|---|
| Transition typeb (weight, %) | E (eV) | λ (nm) | f | Transition type (weight, %) | E (eV) | λ (nm) | ||
| a The oscillator strength for the singlet to triplet transitions is 0 due to them being forbidden by the spin selection rule.b For each excitation energy, the corresponding most relevant molecular orbital transitions are shown with the relative weight. | ||||||||
| [Cu(IPr)(L1)]+ (1) | H → L + 1 (45) | 4.25 | 291.8 | 0.003 | H − 1 → L (40) | 3.52 | 351.8 | wB97XD |
| H → L (88) | 3.80 | 326.5 | 0.003 | H − 1 → L (46) | 3.56 | 369.5 | M06 | |
| [Cu(IPr)(L2)]+ (2) | H − 1 → L (29) | 4.23 | 293.0 | 0.002 | H → L (43) | 3.49 | 355.7 | wB97XD |
| H → L (78) | 3.76 | 329.8 | 0.001 | H − 1 → L (38) | 3.31 | 375.2 | M06 | |
| [Cu(IPr)(L3)]+ (3) | H − 1 → L (32) | 4.29 | 289.2 | 0.012 | H → L (38) | 3.51 | 353.7 | wB97XD |
| H → L (86) | 3.83 | 323.9 | 0.004 | H − 1 → L (46) | 3.36 | 369.2 | M06 | |
| [Cu(IPr)(L4)]+ (4) | H → L + 1 (58) | 4.24 | 292.4 | 0.001 | H − 1 → L (37) | 3.55 | 349.7 | wB97XD |
| H → L (97) | 3.77 | 328.9 | 0.000 | H − 1 → L (54) | 3.39 | 365.8 | M06 | |
| [Cu(IPr)(L5)]+ (5) | H − 1 → L + 1 (41) | 4.23 | 292.8 | 0.000 | H → L (39) | 3.49 | 355.5 | wB97XD |
| H → L (97) | 3.77 | 328.7 | 0.002 | H − 1 → L (54) | 3.31 | 374.8 | M06 | |
| [Cu(IPr)(L6)]+ (6) | H → L (87) | 2.42 | 511.4 | 0.001 | H → L (85) | 2.23 | 556.5 | wB97XD |
| H → L (95) | 2.04 | 608.8 | 0.001 | H → L (93) | 1.90 | 654.0 | M06 | |
| [Cu(SIPr)(L1)]+ (7) | H − 1 → L + 1 (36) | 4.14 | 299.3 | 0.001 | H → L (34) | 3.53 | 351.8 | wB97XD |
| H → L (60) | 3.83 | 324.1 | 0.002 | H − 1 → L (44) | 3.35 | 370.1 | M06 | |
| [Cu(SIPr)(L2)]+ (8) | H − 1 → L + 1 (40) | 4.15 | 299.1 | 0.002 | H → L (50) | 3.49 | 354.8 | wB97XD |
| H − 1 → L + 1 (56) | 3.86 | 320.9 | 0.004 | H → L (52) | 3.30 | 375.8 | M06 | |
| [Cu(SIPr)(L4)]+ (10) | H → L + 1 (49) | 4.15 | 298.7 | 0.001 | H − 1 → L (36) | 3.56 | 348.5 | wB97XD |
| H → L (98) | 3.80 | 326.6 | 0.001 | H − 1 → L (53) | 3.40 | 364.9 | M06 | |
 | ||
| Fig. 5 Experimental and TD-DFT simulated UV-Vis absorption spectra of [Cu(IPr)(L4)]+ in gas-phase and in CH2Cl2 at (a) M06 level and (b) wB97XD level. | ||
Experimental methods
General procedure
The Cu(I) complex syntheses were performed using standard Schlenk techniques or in an MBraun glove box under Ar atmosphere. Imidazolium salts, IPr·HCl and SIPr·HCl were purchased from TCI America. [Cu(IPr)Cl] and [Cu(SIPr)Cl] precursor complexes were prepared according to our modified version of Cazin–Nolan's procedure.37 The ligands, Me2Hdpa and mesBAIN, were synthesized according to the modified literature procedure (ESI†). The solvents used for the syntheses and analytical studies of copper complexes were purified and distilled under N2 atmosphere before being stored over activated molecular sieves (4 Å) in the glovebox. Tetrahydrofuran and diethylether were purified by distillation over Na/benzophenone under N2 atmosphere. Acetonitrile, dichloromethane, and hexane were distilled from CaH2 under N2. Potassium tert-butoxide (97%) was obtained from Sigma-Aldrich and crystallized from THF. Tetrabutylammonium hexafluorophosphate for electrochemical studies was doubly recrystallized from dichloromethane and diethylether. NMR samples were prepared under Ar in CDCl3, containing 0.05% TMS as internal standard. NMR spectra were measured using a 400 MHz Bruker Avance III spectrometer. Chemical shifts (δ) for 1H and 13C NMR spectra were referenced to the resonance of TMS or the residual protio solvent. Elemental analyses were performed by Atlantic Microlabs, Inc. (Norcross, Georgia). UV-visible spectra were measured at room temperature in CH2Cl2 and CH3CN on a CARY 100 Bio UV-Vis spectrometer. Steady state emission spectra and emission lifetimes were recorded in solid state on a CARY ECLIPSE fluorescence spectrophotometer. For measuring absolute solid-state emission quantum yield, a solid sample holder accessory was equipped to the CARY ECLIPSE spectrofluorometer.Synthesis of [Cu(NHC)(N^N)]PF6 complexes
Under Ar atmosphere, [Cu(IPr)Cl] (1 equiv.) and N^N ligand (1 equiv.) were combined using 4 ml THF in a 20 ml reaction vial. The solution was stirred for 1.5 hours and then charged with KPF6 (10 equiv.) providing a white suspension. The reaction mixture was kept stirring for 22.5 hours at room temperature, except for complex 3, after which time it was filtered through a plug of Celite and washed with THF and CH2Cl2. The filtrate was placed under vacuum to remove the solvent and the resultant solid was recrystallized from a mixture of dichloromethane and hexane.Solid state quantum yield measurements
The quantum yield measurements were performed after calibrating the incident light to strike at the center of a custom-designed sample holder. The holder was made from a pair of quartz cover glasses, glued together in a squared area (0.5 × 0.5 cm) after sample placement. A 5 mg portion of a finely-ground powder was placed inside the area and distributed evenly using a flat-head spatula to cover the entire surface. For quantum yield measurements, sodium salicylate was used as the reference with quantum yield (Φ) of 0.53. The excitation wavelength of 340 nm was used to obtain phosphorescence spectra for each compound ranging from 370 nm to 650 nm. Quantum yields were calculated using the formula:55where I, A, and n refer to the integrated emission, the absorbance at excitation wavelength, and the refractive index, respectively. The integrated emissions were obtained from the emission spectra. The absorbance values of the reference and the sample solutions were used at the same wavelength to calculate the second term in the formula. For refractive indices, the n values of CH2Cl2 (n = 1.424) and CH3OH (n = 1.331) were used as the solvents that dissolve the complexes and the reference, respectively. The emission measurements were repeated at least three times and an average of three close peak areas were used to calculate the integrated emission values. The compatibility of this method and solid samples was assessed by comparing the quantum yields of complexes 4, 5, and 11 to those obtained using integrating sphere equipment. The difference in the QY values was determined to be ±3%.

Electrochemistry
Cyclic voltammetry studies of complexes 1–11 and ferrocene at 2 mM concentration were performed using 0.1 M nBu4PF6/CH2Cl2 as the supporting electrolyte under nitrogen. The electrochemistry experiment set-up consisted of a three-electrode cell connected to an external CHI 730C potentiostat run by a personal computer with CHI software. A glassy carbon electrode (0.5 mm diameter) was employed as the working electrode, with Ag/AgNO3 (0.01 M) as the reference and Pt wire as the counter electrode. The working electrode was cleaned between experiments using a polishing pad and carefully dried. All potentials are reported versus [Fe(C5H5)2]+/[Fe(C5H5)2] by subtracting 0.252 V from the measured value versus Ag/AgNO3 electrode.Conclusions
A new series of heteroleptic three-coordinate Cu–IPr and Cu–SIPr complexes bearing unsymmetrically-substituted Me2Hdpa and mesBIAN ligands were prepared and characterized by solid- and solution-phase analytical techniques. The solid-state analyses indicate the CH⋯π interactions between the center of the diisopropylphenyl groups of NHC and the α hydrogen atoms of Hdpa in the 3,4′-Me2Hdpa and 3,5′-Me2Hdpa complexes, like their symmetrically substituted analogs. In contrast, no such interaction was found for the 3,6′-Me2Hdpa version within a reasonable distance. While the lack of α-hydrogen atoms in 6,6′-Me2Hdpa and thus, the absence of CH–π interaction was reasoned for the complexation failure previously, a decent yield was obtained for [Cu(IPr)(3,6′-Me2Hdpa)]PF6 in our work. The stability of this complex was attributed to the large plane angle between the IPr ring and the 3,6-Me2Hdpa ligand reducing the repulsive interactions and allowing its formation. The large plane angle was also identified as an important parameter towards improving the emissive properties of these copper complexes in solid state. This finding and the solid state luminescent properties of the complexes are consistent with their analogs bearing modified IPr and Hdpa ligands.Furthermore, the character of the electronic states involved in light absorption and emission were elucidated via DFT and TD-DFT calculations at three theory levels. The data obtained using M06, a hybrid functional, and wB97XD, a non-hybrid functional, support the dominance of MLCT character in S0 → S1 transitions and a combination of MLCT and LLCT in S0 → T1 transitions for the Hdpa-based complexes. The simulated UV-Vis absorption spectra indicate a close match with the experimental data in solution at M06 level. While the wB97XD level calculation was adopted previously to rationalize the photophysical properties of the [Cu(NHC)(N^N)]+ complexes, our work supports the competency of M06 functional in these types of calculations as well. The combined experimental and theoretical findings offer a deeper understanding of the structure and light-interaction relationship in [Cu(IPr)(N^N)]+ and [Cu(SIPr)(N^N)]+ complexes and a more rational-based approach towards efficient copper-based luminophores.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Oklahoma State University start-up funding (LT), the National Science Foundation REU program through the grant CHE-1559874 (BN), the OSU fund (Single Crystal X-ray Diffraction in the OSU core facility center), and the computing at the OSU High Performance Computing Center (HPCC) supported in part through the NSF grant OAC–1531128.References
- M. H. Keefe, K. D. Benkstein and J. T. Hupp, Coord. Chem. Rev., 2000, 205, 201–228 CrossRef CAS.
- J. A. Gareth Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef CAS.
- K. M.-C. Wong and V. W.-W. Yam, Coord. Chem. Rev., 2007, 251, 2477–2488 CrossRef CAS.
- M. S. Lowry and S. Bernhard, Chem.–Eur. J., 2006, 12, 7970–7977 CrossRef CAS.
- Z. Liu, Z. Bian and C. Huang, in Molecular Organometallic Materials for Optics, ed. H. Bozec and V. Guerchais, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, pp. 113–142, DOI:10.1007/978-3-642-01866-4_4.
- Y. You and W. Nam, Chem. Soc. Rev., 2012, 41, 7061–7084 RSC.
- K. E. Erkkila, D. T. Odom and J. K. Barton, Chem. Rev., 1999, 99, 2777–2796 CrossRef CAS.
- G. M. Brown, B. S. Brunschwig, C. Creutz, J. F. Endicott and N. Sutin, J. Am. Chem. Soc., 1979, 101, 1298–1300 CrossRef CAS.
- N. Robertson, ChemSusChem, 2008, 1, 977–979 CrossRef CAS PubMed.
- M. Iwamura, S. Takeuchi and T. Tahara, Acc. Chem. Res., 2015, 48, 782–791 CrossRef CAS.
- G. Capano, U. Rothlisberger, I. Tavernelli and T. J. Penfold, J. Phys. Chem. A, 2015, 119, 7026–7037 CrossRef CAS.
- G. B. Shaw, C. D. Grant, H. Shirota, E. W. Castner, G. J. Meyer and L. X. Chen, J. Am. Chem. Soc., 2007, 129, 2147–2160 CrossRef CAS.
- M. Iwamura, H. Watanabe, K. Ishii, S. Takeuchi and T. Tahara, J. Am. Chem. Soc., 2011, 133, 7728–7736 CrossRef CAS.
- L. X. Chen, G. B. Shaw, I. Novozhilova, T. Liu, G. Jennings, K. Attenkofer, G. J. Meyer and P. Coppens, J. Am. Chem. Soc., 2003, 125, 7022–7034 CrossRef CAS.
- Z. A. Siddique, Y. Yamamoto, T. Ohno and K. Nozaki, Inorg. Chem., 2003, 42, 6366–6378 CrossRef CAS.
- E. M. Stacy and D. R. McMillin, Inorg. Chem., 1990, 29, 393–396 CrossRef CAS.
- M. W. Blaskie and D. R. McMillin, Inorg. Chem., 1980, 19, 3519–3522 CrossRef CAS.
- M. Nishikawa, Y. Wakita, T. Nishi, T. Miura and T. Tsubomura, Dalton Trans., 2015, 44, 9170–9181 RSC.
- N. Armaroli, Chem. Soc. Rev., 2001, 30, 113–124 RSC.
- A. J. M. Miller, J. L. Dempsey and J. C. Peters, Inorg. Chem., 2007, 46, 7244–7246 CrossRef CAS.
- M. K. Eggleston, D. R. McMillin, K. S. Koenig and A. J. Pallenberg, Inorg. Chem., 1997, 36, 172–176 CrossRef CAS.
- A. Lavie-Cambot, M. Cantuel, Y. Leydet, G. Jonusauskas, D. M. Bassani and N. D. McClenaghan, Coord. Chem. Rev., 2008, 252, 2572–2584 CrossRef CAS.
- D. V. Scaltrito, D. W. Thompson, J. A. O'Callaghan and G. J. Meyer, Coord. Chem. Rev., 2000, 208, 243–266 CrossRef CAS.
- J. Huang, M. W. Mara, A. B. Stickrath, O. Kokhan, M. R. Harpham, K. Haldrup, M. L. Shelby, X. Zhang, R. Ruppert, J. P. Sauvage and L. X. Chen, Dalton Trans., 2014, 43, 17615–17623 RSC.
- C. Bizzarri, C. Strabler, J. Prock, B. Trettenbrein, M. Ruggenthaler, C.-H. Yang, F. Polo, A. Iordache, P. Brüggeller and L. D. Cola, Inorg. Chem., 2014, 53, 10944–10951 CrossRef CAS.
- T. Hofbeck, U. Monkowius and H. Yersin, J. Am. Chem. Soc., 2015, 137, 399–404 CrossRef CAS.
- S. B. Harkins and J. C. Peters, J. Am. Chem. Soc., 2005, 127, 2030–2031 CrossRef CAS.
- K. J. Lotito and J. C. Peters, Chem. Commun., 2010, 46, 3690–3692 RSC.
- M. Hashimoto, S. Igawa, M. Yashima, I. Kawata, M. Hoshino and M. Osawa, J. Am. Chem. Soc., 2011, 133, 10348–10351 CrossRef CAS.
- V. A. Krylova, P. I. Djurovich, M. T. Whited and M. E. Thompson, Chem. Commun., 2010, 46, 6696–6698 RSC.
- V. A. Krylova, P. I. Djurovich, J. W. Aronson, R. Haiges, M. T. Whited and M. E. Thompson, Organometallics, 2012, 31, 7983–7993 CrossRef CAS.
- V. A. Krylova, P. I. Djurovich, B. L. Conley, R. Haiges, M. T. Whited, T. J. Williams and M. E. Thompson, Chem. Commun., 2014, 50, 7176–7179 RSC.
- M. J. Leitl, V. A. Krylova, P. I. Djurovich, M. E. Thompson and H. Yersin, J. Am. Chem. Soc., 2014, 136, 16032–16038 CrossRef CAS.
- R. Marion, F. Sguerra, F. Di Meo, E. Sauvageot, J.-F. Lohier, R. Daniellou, J.-L. Renaud, M. Linares, M. Hamel and S. Gaillard, Inorg. Chem., 2014, 53, 9181–9191 CrossRef CAS.
- R. Marion, F. Sguerra, F. Di Meo, E. Sauvageot, J.-F. Lohier, R. Daniellou, J.-L. Renaud, M. Linares, M. Hamel and S. Gaillard, Inorg. Chem., 2016, 55, 4068 CrossRef CAS.
- M. Elie, F. Sguerra, F. Di Meo, M. D. Weber, R. Marion, A. Grimault, J.-F. Lohier, A. Stallivieri, A. Brosseau, R. B. Pansu, J.-L. Renaud, M. Linares, M. Hamel, R. D. Costa and S. Gaillard, ACS Appl. Mater. Interfaces, 2016, 8, 14678–14691 CrossRef CAS.
- J. L. Minnick, D. Domyati, R. Ammons and L. Tahsini, Front. Chem., 2019, 17, 12 CrossRef.
- H. Suezawa, T. Yoshida, Y. Umezawa, S. Tsuboyama and M. Nishio, Eur. J. Inorg. Chem., 2002, 2002, 3148–3155 CrossRef.
- E. Kryachko and S. Scheiner, J. Phys. Chem. A, 2004, 108, 2527–2535 CrossRef CAS.
- T. Steiner, J. Chem. Crystallogr., 1997, 27, 673–675 CrossRef CAS.
- B. Tolnai, J. T. Kiss, K. Felföldi and I. Pálinkó, J. Mol. Struct., 2009, 924–926, 27–31 CrossRef CAS.
- D. E. Morris, Y. Ohsawa, D. P. Segers, M. K. DeArmond and K. W. Hanck, Inorg. Chem., 1984, 23, 3010–3017 CrossRef CAS.
- A. Zianna, G. Psomas, A. Hatzidimitriou and M. Lalia-Kantouri, J. Inorg. Biochem., 2016, 163, 131–142 CrossRef CAS.
- F. Franco, C. Cometto, F. Sordello, C. Minero, L. Nencini, J. Fiedler, R. Gobetto and C. Nervi, ChemElectroChem, 2015, 2, 1372–1379 CrossRef CAS.
- L. Li, P. S. Lopes, V. Rosa, C. A. Figueira, M. A. N. D. A. Lemos, M. T. Duarte, T. Avilés and P. T. Gomes, Dalton Trans., 2012, 41, 5144–5154 RSC.
- S. Wang, Z. Cheng, X. Song, X. Yan, K. Ye, Y. Liu, G. Yang and Y. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 9892–9901 CrossRef CAS.
- D. M. de Leeuw, M. M. J. Simenon, A. R. Brown and R. E. F. Einerhand, Synth. Met., 1997, 87, 53–59 CrossRef CAS.
- J. W. Kee, Y. Y. Ng, S. A. Kulkarni, S. K. Muduli, K. Xu, R. Ganguly, Y. Lu, H. Hirao and H. S. Soo, Inorg. Chem. Front., 2016, 3, 651–662 RSC.
- P. Papanikolaou, P. D. Akrivos, A. Czapik, B. Wicher, M. Gdaniec and N. Tkachenko, Eur. J. Inorg. Chem., 2013, 2013, 2418–2431 CrossRef CAS.
- T. Lu, J.-Y. Wang, D. Tu, Z.-N. Chen, X.-T. Chen and Z.-L. Xue, Inorg. Chem., 2018, 57, 13618–13630 CrossRef CAS.
- T. Gneuß, M. J. Leitl, L. H. Finger, N. Rau, H. Yersin and J. Sundermeyer, Dalton Trans., 2015, 44, 8506–8520 RSC.
- D. M. Zink, T. Grab, T. Baumann, M. Nieger, E. C. Barnes, W. Klopper and S. Bräse, Organometallics, 2011, 30, 3275–3283 CrossRef CAS.
- P. R. Varadwaj and H. M. Marques, Theor. Chem. Acc., 2010, 127, 711–725 Search PubMed.
- F. Neese, JBIC, J. Biol. Inorg. Chem., 2006, 11, 702–711 CrossRef CAS.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic information files (CIF) for 1–3, 6–8, and 12, crystallographic data collection table, 1H and 13C{1H} NMR spectra for ligands, Cu precursors, and complexes 1–12 (Fig. S16–S51), ORTEP diagrams of [Cu(SIPr)Cl] and 6 and 12 with long-distance interactions, CV of complexes 4 and 5 at varying scan rates and CV of the free Hdpa (Fig. S5–S7), and further details of ligand and Cu complexes synthesis. CCDC 1861502, 1865893, 1865901, 1866337, 1866344, 1866360, 1556652 and 1918545. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra04886c |
| This journal is © The Royal Society of Chemistry 2019 |