
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNucleophilic cyclopropanation of [60]fullerene by the addition–elimination mechanism
Yulya N. Biglova *ab and
Akhat G. Mustafina
*ab and
Akhat G. Mustafina
aUfa Institute of Chemistry, Russian Academy of Sciences, Ufa, Russian Federation. E-mail: bn.yulya@mail.ru
bDepartment of Chemistry, Bashkir State University, Ufa, Russian Federation
First published on 19th July 2019
Abstract
Information on the synthesis of monofunctionalized methanofullerenes C60 obtained by the addition–elimination mechanism is generalized. The main reagents for cyclopropanation, mechanisms and optimal conditions for the processes, and the prospects for practical application of the products are considered.
Introduction
The discovery of fullerenes was recognized as one of the most amazing breakthrough scientific events of the 20th century that was awarded the Nobel Prize in Chemistry in 1996.Fullerenes are attributed both to inorganic materials as new allotropic modifications of carbon and to organic compounds, since they manifest many properties of unsaturated hydrocarbons in chemical reactions. The chemistry of derivatives of this compound has become one of the most productive and significant areas of organic chemistry (both in theoretical and practical aspects). It is sufficient to say that several thousand publications on the chemistry of fullerene appeared over the past five years, and these studies are becoming more and more practical. For example, the use of fullerene derivatives in medicine seems to be a very promising approach. Some compounds based on C60 are used in the photodynamic therapy of malignant tumors, others proved to be quite effective bacteriostatic and fungicidal agents (effective against the influenza virus, inhibit HIV-1 protease), and still others are antioxidants preventing neurodegenerative processes that lead, in particular, to Parkinson's disease.
The use of fullerene as an acceptor unit with polyconjugated polymers in solar batteries is also an important approach. This field of organic photovoltaics is actively developing in research institutes and commercial structures in Russia and abroad. One should hope that in the coming years, fullerene compounds will become classical materials for organic electronics.
A primary role is currently given to the development of new effective approaches to organic modifications of fullerenes that would provide high yields of target products and facile incorporation of required functional groups. As an electron-deficient polyene, C60 is prone to radical addition, nucleophilic addition and cycloaddition reactions, while a decrease in the fullerene skeleton strain is the driving force of fullerene reactivity. It appears that various [2 + n]-cycloaddition processes are particularly promising in the functionalization of the fullerene sphere. As a rule, processes with n = 1, 2, 3 or 4 occur. The [2 + 1] variant, which is most common for synthetic organic chemists, is attractive from all points of view. Reactions of [2 + 1] cycloaddition result in compounds in which the fullerene cage is annulated with three-membered carbo- and heterocyclic moieties, i.e., they give methanofullerenes, fullerenoaziridines or fullerenooxiranes. The reactions indicated above can occur by various mechanisms including, for example, the addition of carbenes, nitrenes, stabilized carbanions, etc.
The vast majority of researchers who develop various methods for the functionalization of the fullerene spheroid prefer [2 + 1]-cycloaddition processes and, above all, the Bingel reaction. It is reasonable, since this process is the most efficient way to synthesize methanofullerenes.
Ample information on fullerene chemistry has been accumulated to date. It is partially covered in reviews1–14 and monographs.15–18 Now that the useful properties of C60 derivatives have been clearly determined, it is time for their real practical use and creation of effective optical and electrophysical instruments as well as essential pharmaceuticals.
This review, in which we touch upon the primary functionalization of the C60 core for purposes of selective synthesis of monofunctionalized carbon-chain products, includes the main achievements in the field of cycloaddition to [60]fullerene for the past 20 years, including principally important studies performed before that.
Results and discussion
The molecule of fullerene C60 is a closed hollow sphere composed of sp2-hybridized carbon atoms interconnected into three-dimensional frames. Twelve five-membered and 20 six-membered rings isolated from each other can be distinguished in it. All the carbon atoms are equivalent, and the molecule geometry implies that two types of bonds exist: [6,6] bonds located on the borders of two hexagons and [5,6] bonds located on the borders of hexagons and pentagons (Fig. 1). According to experimental and calculated data, the [6,6] bonds (1.38 Å long) in the C60 frame are shorter than the [5,6] bonds (1.45 Å). Therefore, the former are considered as double bonds, while the latter, as single ones.19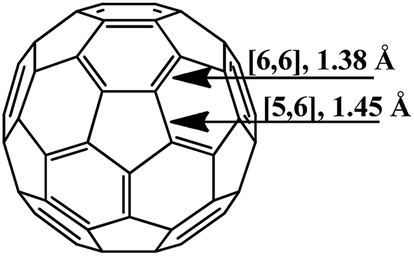 | ||
| Fig. 1 The C60 fullerene molecule. | ||
The spherical molecule of fullerene is highly strained, since the usually planar aromatic six-membered (benzene) rings must be twisted to build a sphere. As a result, the thermodynamic stability of C60 is lower than that of graphite.
The C60 molecule is highly symmetric (a truncated icosahedron) and contains a considerable number of double bonds. However, these bonds are not similar to those in benzene: the rings in the latter are planar, whereas the cells that compose the fullerene cage are three-dimensional and their double bonds are localized to a considerable extent. Strictly speaking, there is no π-electron system in the polyhedral molecule since this molecule is non-planar and its surface is close to a sphere. Therefore, the assignment of fullerenes to aromatic compounds is clearly incorrect. If one has to emphasize the formation of a single p-electronic delocalized system leading to a certain energy gain, it is better to use the term “pseudo-aromaticity”. Comparison of the lengths of the bonds (double and single ones) in C60 indicates that some conjugation between single and double bonds in fullerene still exists. However, it is more similar to the conjugation of bonds in buta-1,3-diene than in totally aromatic benzene. It is this non-aromaticity that determines the chemical properties of fullerenes, which mainly involve opening of double bonds in the frame on treatment with various reagents.
The driving force behind the reactivity of fullerene involves the reduction of strain in its frame.16 Four isomers can theoretically exist in monofunctionalized fullerenes: [5,6]-open, [5,6]-closed, [6,6]-open and [6,6]-closed ones; only [5,6]-open and [6,6]-closed isomers are experimentally observed (Fig. 2). As interpreted in ref. 14 and 20, this fact is not due to the nature of substituents, but is explained by the conservation of energetically favorable energy levels in the isomers indicated above. Calculations showed that [6,6]-isomers of methanofullerenes have a closed transannular bond (π-homoaromatic structure) and [5,6]-isomers have an open transannular bond (σ-homoaromatic structure), and the former are thermodynamically much more stable. In most cases, the [5,6]-open adducts formed initially can be converted into thermodynamically more stable [6,6]-closed isomers.
 | ||
| Fig. 2 The four theoretically possible isomeric methanofullerenes C60. | ||
Of the numerous methods for functionalization of the fullerene core developed over the past 20 years, we will focus on the [2 + 1]-monocycloaddition processes as the most significant ones.
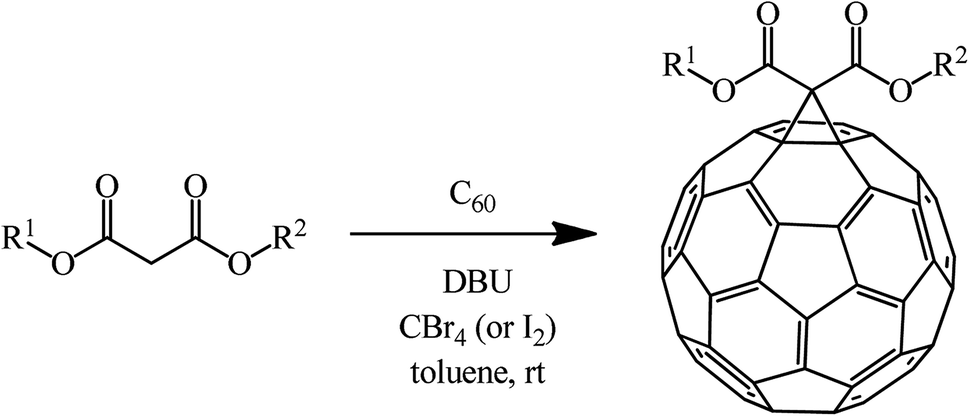
The methods for synthesizing methanofullerenes can be divided into two categories (Scheme 1): (I) nucleophilic cyclopropanation with stabilized carbanions that occur by the addition–elimination mechanism (Bingel reaction); (II) thermal addition of diazo compounds followed by thermolysis or photolysis of the pyrazoline intermediates and N2 extrusion.
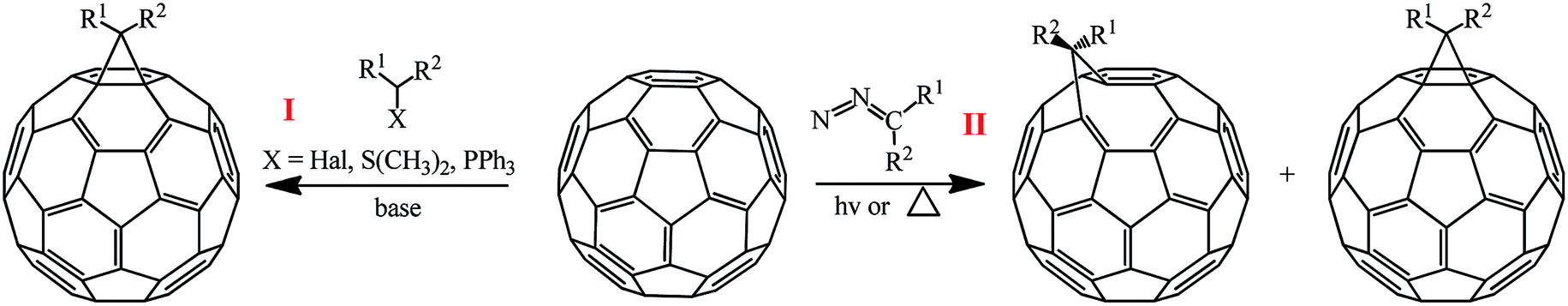 | ||
| Scheme 1 | ||
1. Synthesis of methanofullerenes based on halide-activated substrates according to Bingel
The most efficient way to synthesize methanofullerenes is by C60 cyclopropanation with stabilized halo-carbanions. In the classical version, the process occurs upon treatment of C60 with 2-bromomalonic ester in the presence of a base.The nucleophilic cyclopropylation of fullerene was first reported in 1993 by Bingel in an original work describing the reaction of C60 with 2-diethyl bromomalonate, methyl 2-chloroacetoacetate, ω-bromoacetophenone and desyl chloride in the presence of various bases, namely, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), NaH, and potassium tert-butylate at room temperature under the conditions and with the yields shown in Scheme 2.21
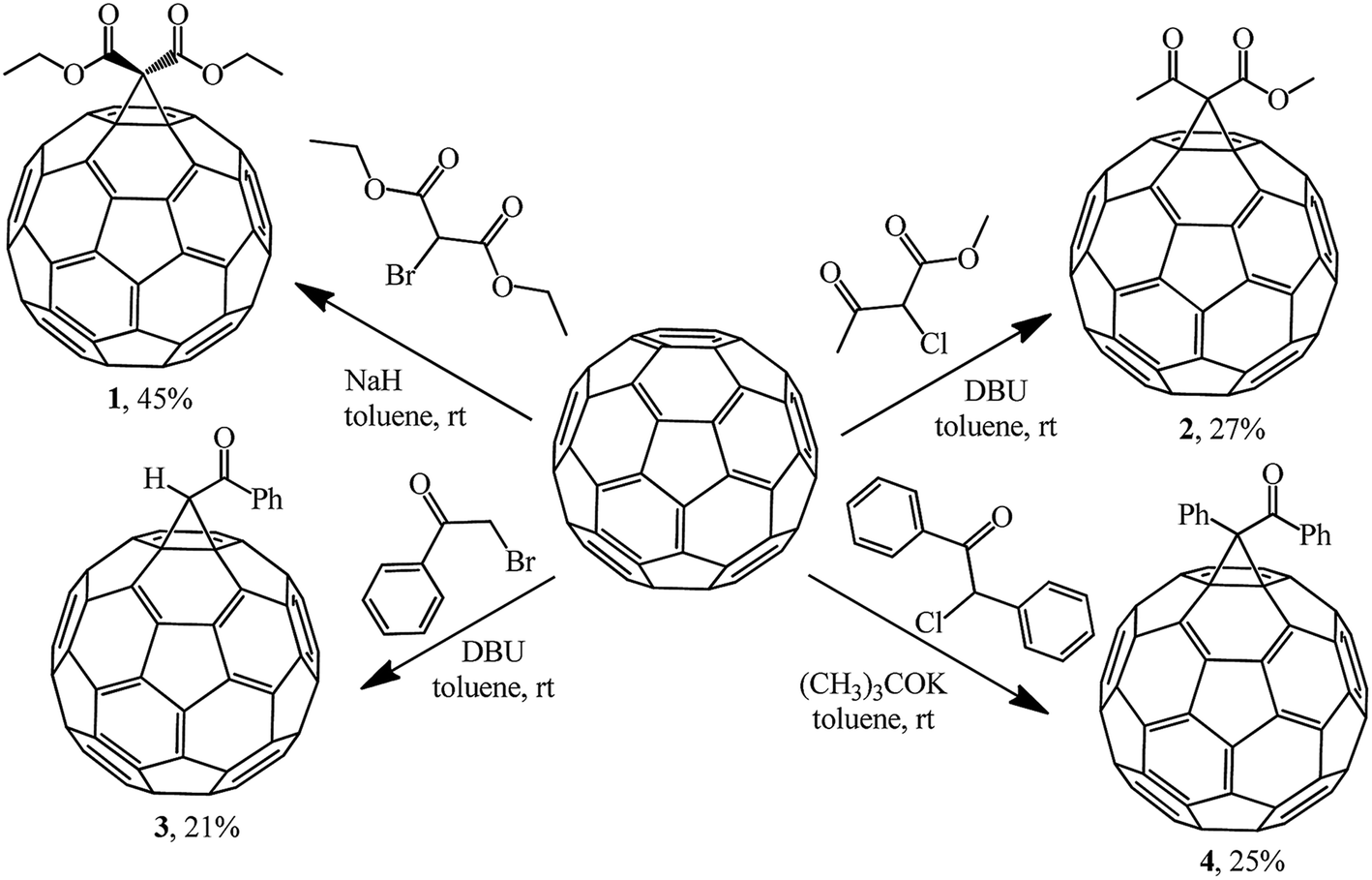 | ||
| Scheme 2 | ||
In all the cases, the reaction products were isolated by column chromatography on silica gel from the residues obtained after acidification of the reaction mixture (2 N H2SO4), drying and evaporation of the organic phases. Compounds 1–4 were obtained as crystalline dark brown powders, which is characteristic of the majority of monofunctionalized methanofullerenes C60.
The suggested reaction mechanism involves the initial deprotonation of halo derivative A by a base with generation of reactive nucleophile B, which attacks an electron-deficient double bond in fullerene C60. The resulting carbanion C undergoes intramolecular SN2 substitution of the halogen atom to give methano-derivative D (Scheme 3).16
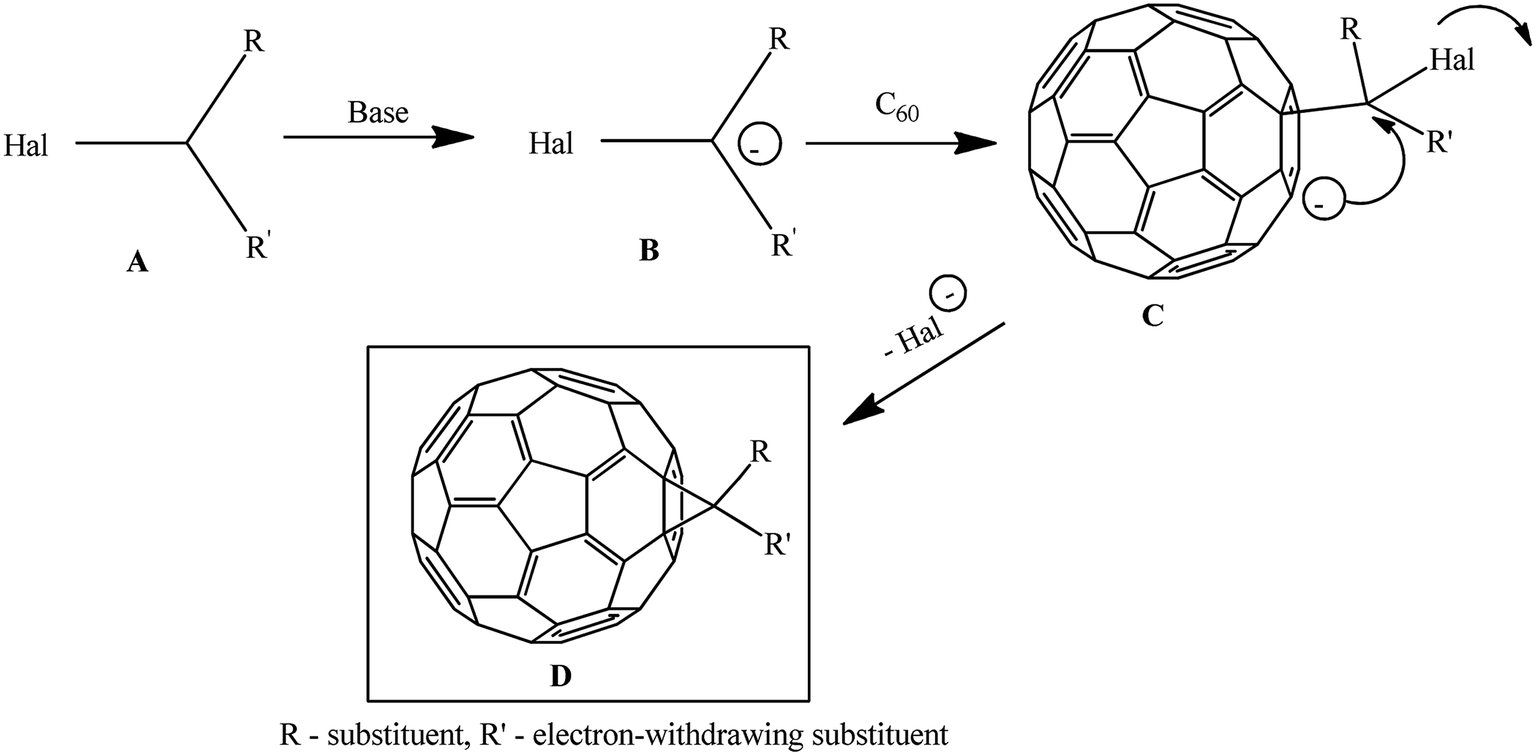 | ||
| Scheme 3 | ||
The key agents for C60 cyclopropanation under the “classical” Bingel reaction conditions include substrates with an activated halomethine function capable of forming stabilized carbanions, while the readily leaving halide group (mainly Cl−) ensures the intramolecular cyclization.
The Bingel's approach was successfully expanded by F. Diederich who used 3-bromo-1,5-bis (trimethylsilyl)penta-1,4-diyne22 to give adduct 5 in the presence of DBU. Proto-desilylation of 5 with potassium carbonate in tetrahydrofuran (THF)/MeOH results in the related hydrocarbon 6. Controlled desilylation of 5 makes it possible to obtain monosubstituted diynofullerene 7 in 35% yield. The latter compound is interesting due to the prospects of its application in syntheses of hybrid structures (Scheme 4).23–25
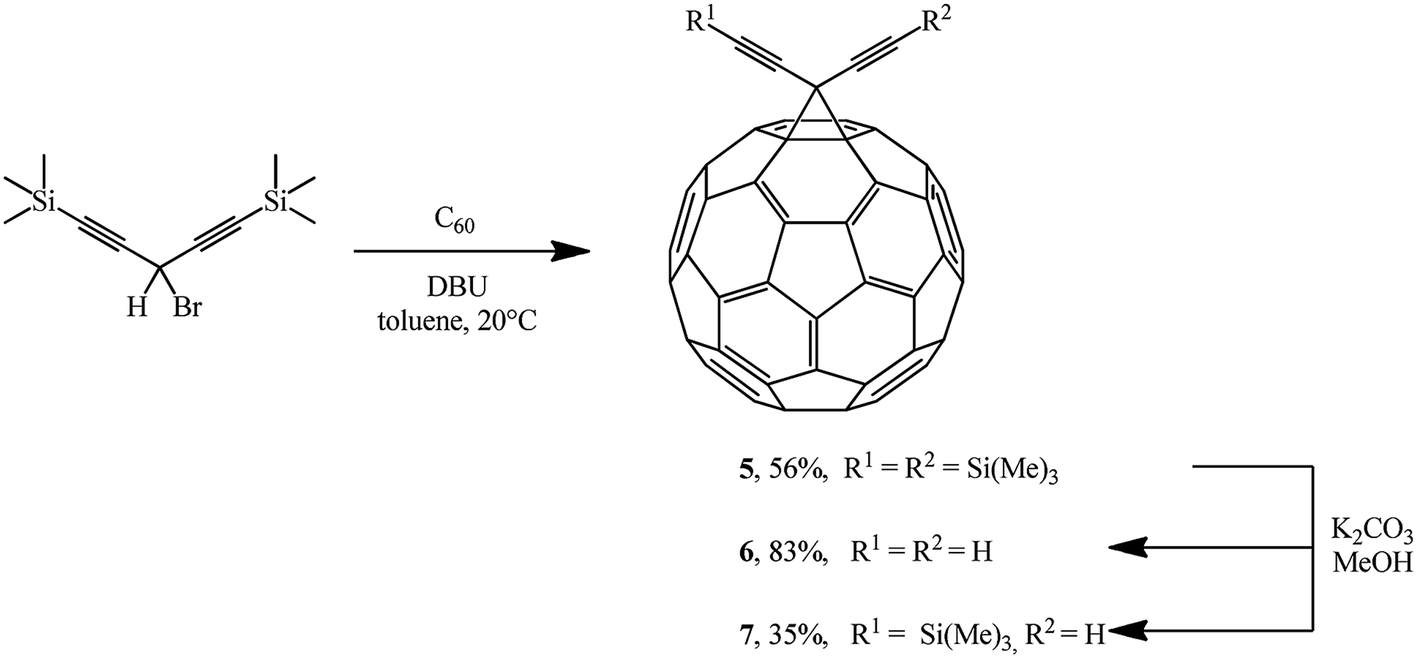 | ||
| Scheme 4 | ||
Later, the same scientists26 used dipyridylchloromethane that was obtained in two stages from 4-pyridinecarboxaldehyde (Scheme 5) and was capable of generating a stabilized carbanion under the Bingel reaction conditions in order to synthesize an adduct of monoaddition to fullerene 8.
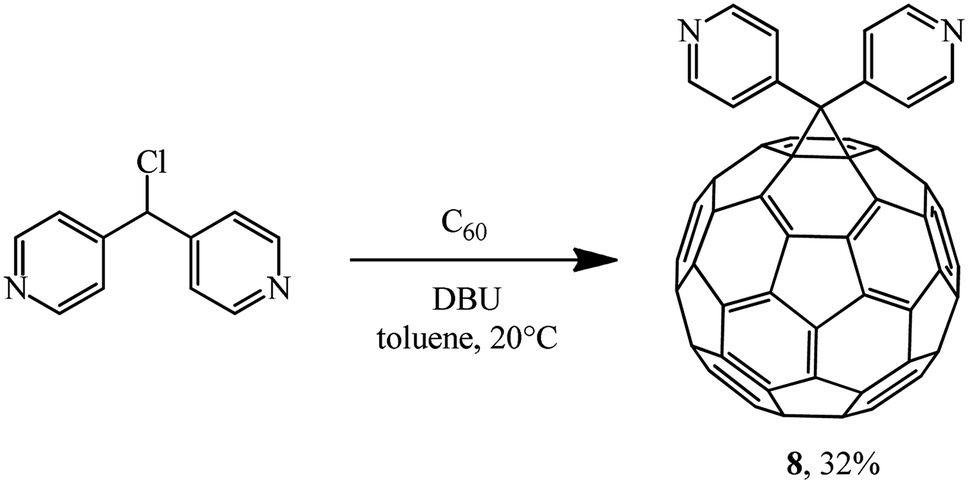 | ||
| Scheme 5 | ||
The versatility of the method was demonstrated by synthesizing a new series of ligands incorporating conjugated 2,2′;6′,2′′-terpyridine (tpy) moieties.27–29 The latter were converted to 9 and 10 using the Bingel reaction and further converted to diads and triads with ruthenium 11 and 12, where the pendant fullerene acted as an electron acceptor (Scheme 6).
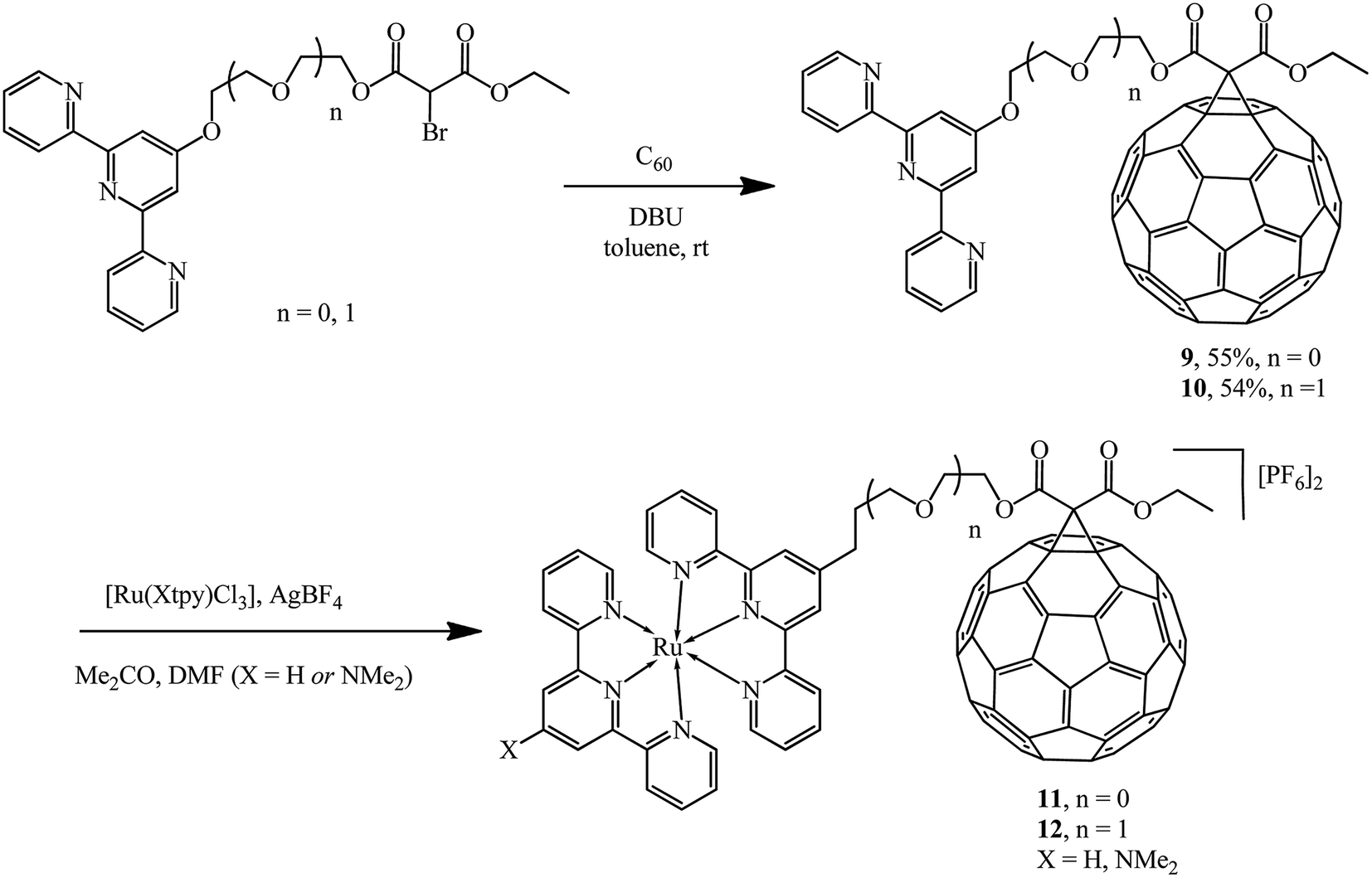 | ||
| Scheme 6 | ||
In continuation of studies on fullerene functionalization, the reaction of bis(2-bromomalonate) with two equivalents of C60 was performed to give bis-adducts 13, 14 and the corresponding minor 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adducts (Scheme 7).30
:1 adducts (Scheme 7).30
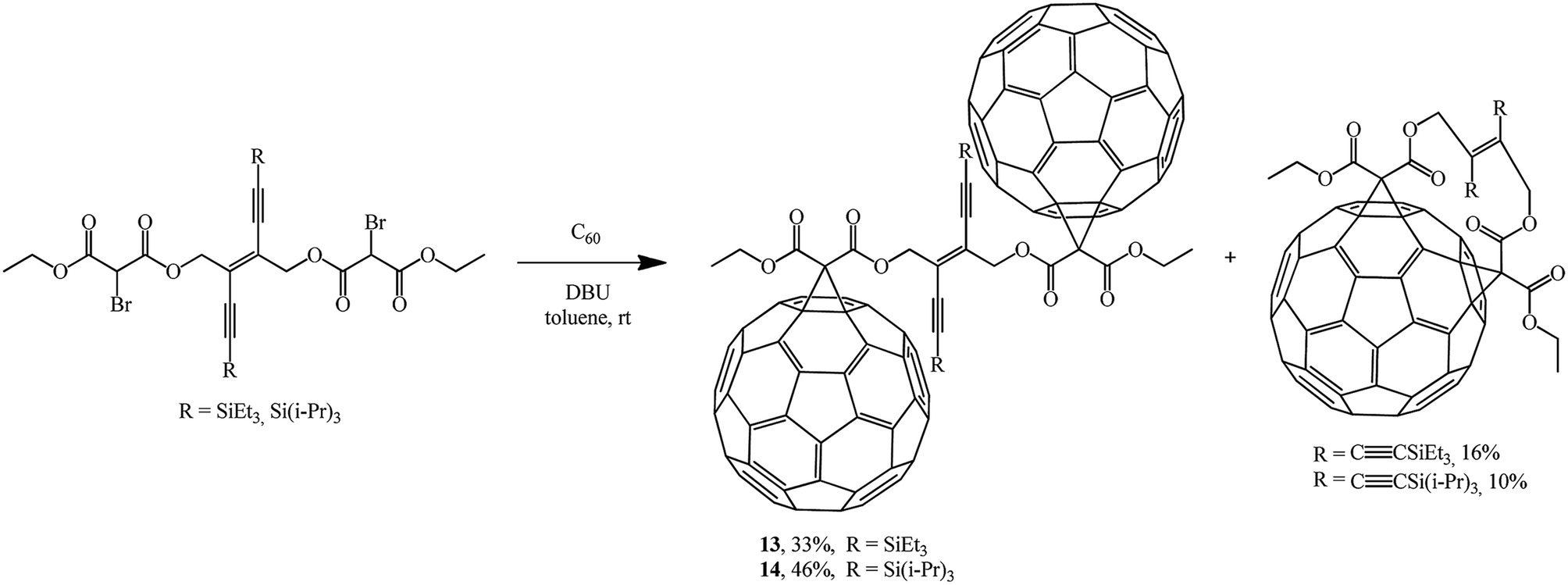 | ||
| Scheme 7 | ||
Rather an interesting publication31 reported a synthesis of methanofullerene 15 whose frame includes two cholesterol residues. Its liquid-crystal and thermal properties were studied (Scheme 8).
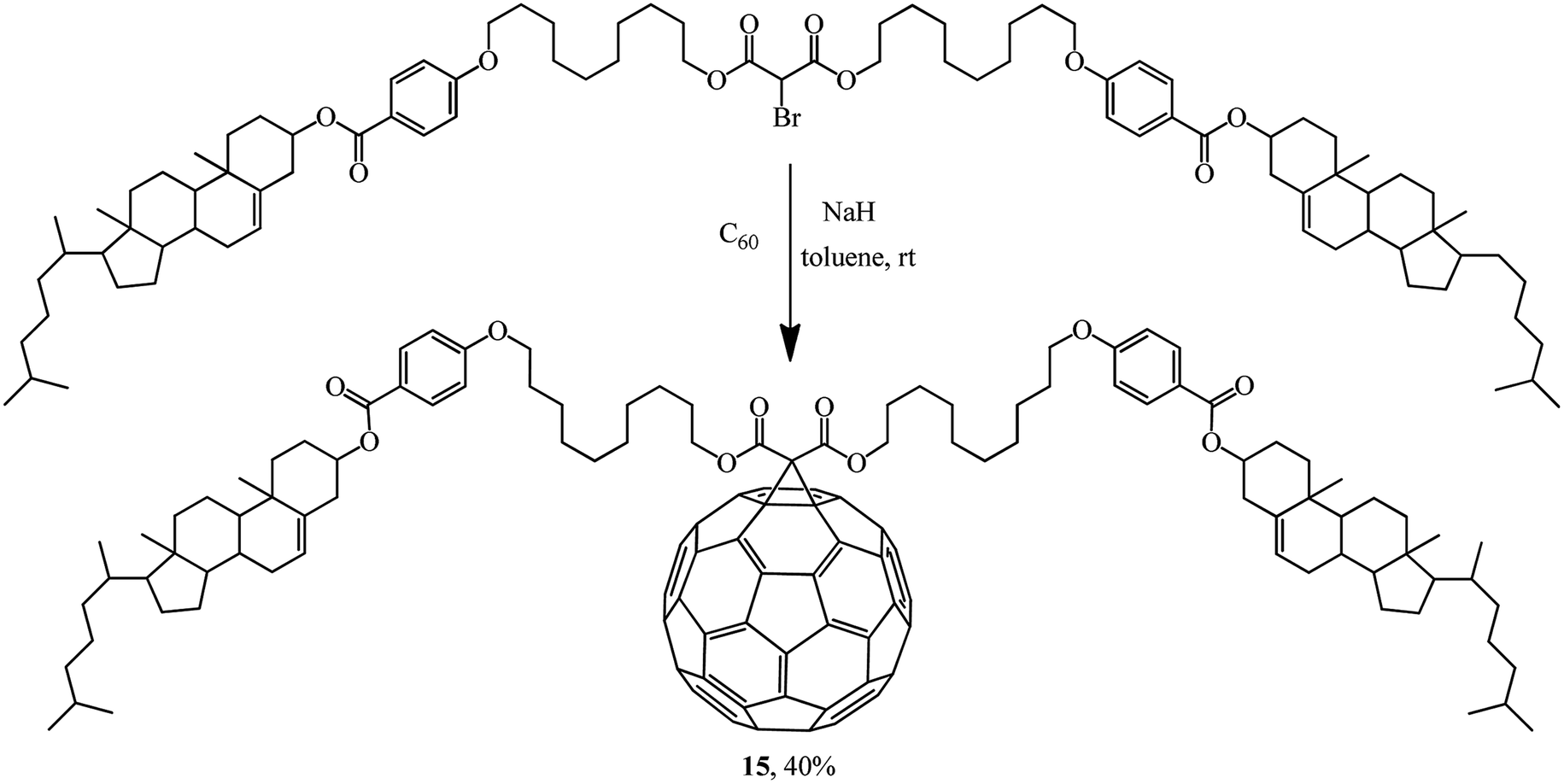 | ||
| Scheme 8 | ||
Addition of the resulting dendrons based on 3,5-substituted benzyl esters to C60 gave fullerene-containing dendrimers as monoadducts 16–18.32 The cyclopropanating agents were synthesized by bromination of the corresponding bis(3,5-dibenzyloxybenzyl)propanedionate with the CBr4-DBU system (Scheme 9).
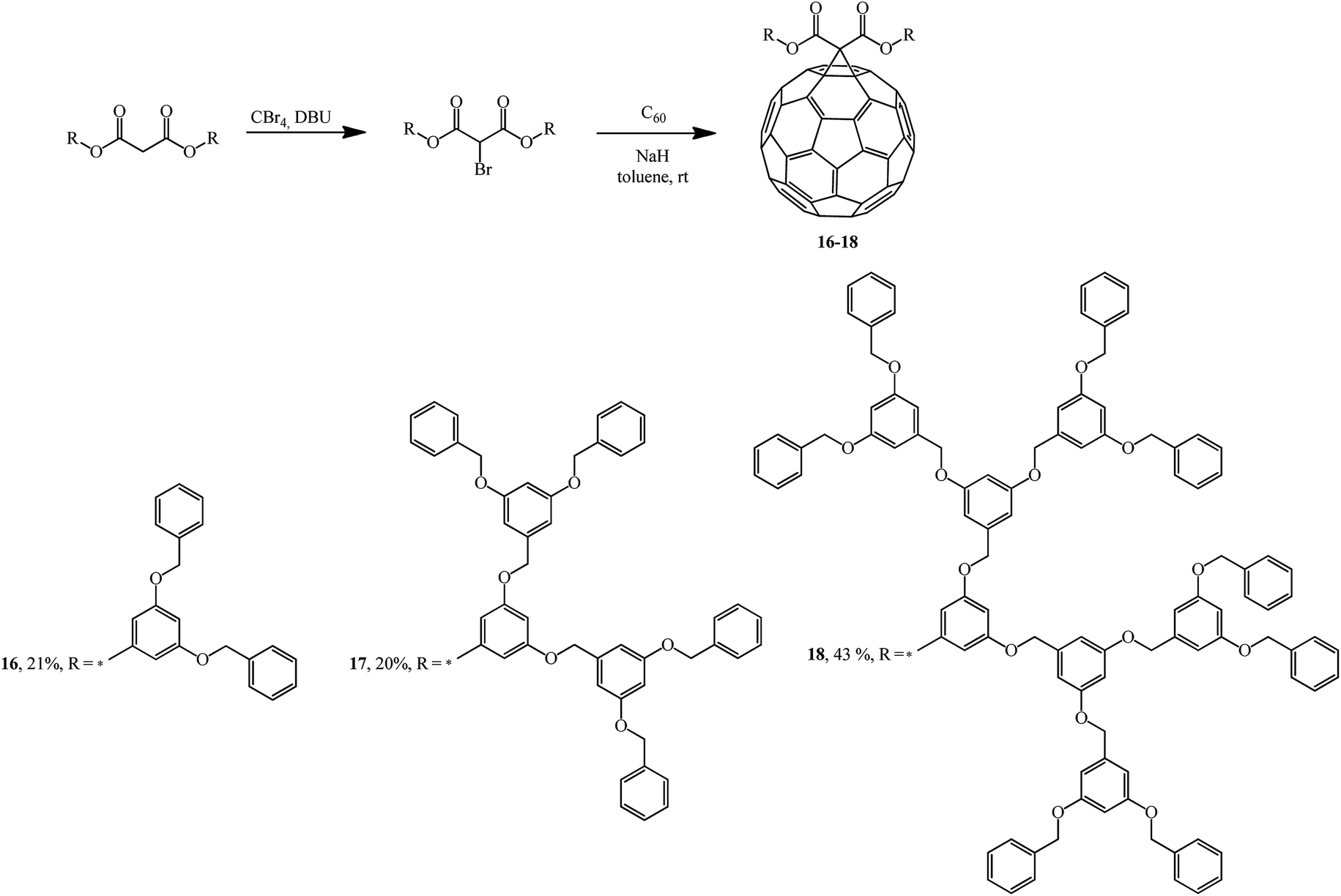 | ||
| Scheme 9 | ||
Article33 describes a synthesis of a number of monofunctionalized dialkyl 1,2-[6,6]-methano-[60]-fullerene dicarboxylate derivatives whose structures contain substituents with various chain lengths, namely, saturated and unsaturated hydrocarbon groups (compounds 19–32). Reactions of fullerene C60 with dialkyl bromomalonates in the presence of sodium hydride give the target products in 35–69% yields (Fig. 3).
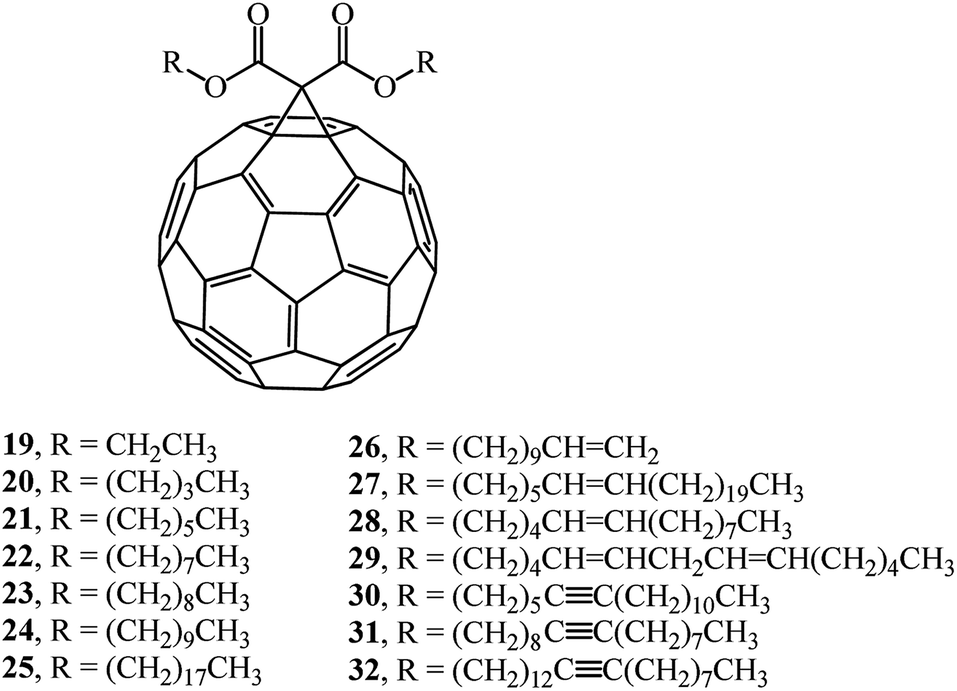 | ||
| Fig. 3 Dialkyl 1,2-[6,6]-methano-[60]-fullerene dicarboxylate derivatives 19–32. | ||
A study of the physical characteristics of the presented dialkyl 1,2-[6,6]-methano-[60]-fullerene dicarboxylates showed that compounds 20, 21 and 23 were dark brown solids with very high melting points. Dioctyl and longer-chain saturated and unsaturated dialkyl 1,2-[6,6]-methano-[60]-fullerene dicarboxylates 22 and 24–32 are viscous liquids that do not crystallize from organic solvents.
The Bingel approach is significantly expanded if bis-tert-butyl- and bis(2-methylhexan-2-yl)2-bromomalonates are used as halogen-activated substrates for synthesizing methanofullerenes to give 33 and 34, which were converted to the corresponding C60 diacids under specific conditions (Scheme 10).34
 | ||
| Scheme 10 | ||
The compounds 33 and 34 synthesized are thermo-cleavable fullerene materials and can be used as buffer layers for increasing the efficiency of inverted organic solar cells. In this case, the efficiency of inverted solar cells increases due to improved dissociation of excitons and electron transfer, along with a reduction in the series resistance.
The principal possibility of using α,β-dibromo derivatives of acid esters in the Bingel reaction was demonstrated by synthesizing methanofullerene 35 (Scheme 11).35
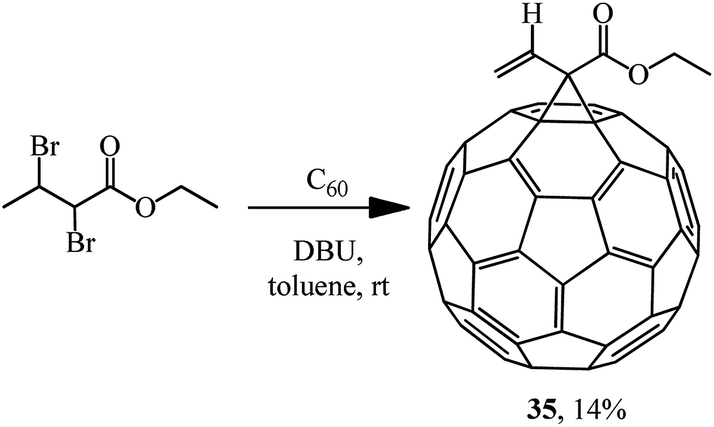 | ||
| Scheme 11 | ||
In a search for highly water-soluble fullerene derivatives, condensation of equimolar amounts of a bromomalonodiamide derivative and C60 gave adduct 36,36 hydrolysis of which resulted in methanofullerene 37 (Scheme 12). However, neither tetraacetate 36 nor even tetraol 37 showed any noticeable water solubility. Therefore, the reaction of C60 with a large excess of the aforementioned bromomalonodiamide (>10 equiv.) was carried out to give a mixture of 4–6 substituted cyclopropanation products, which was then hydrolyzed under similar conditions. The solubility of the resulting mixture of methanofullerenes 37 (n = 4–6) in water was found to be 240 mg mL−1 which, as noted in ref. 36, appears to be a good result for fullerene derivatives.
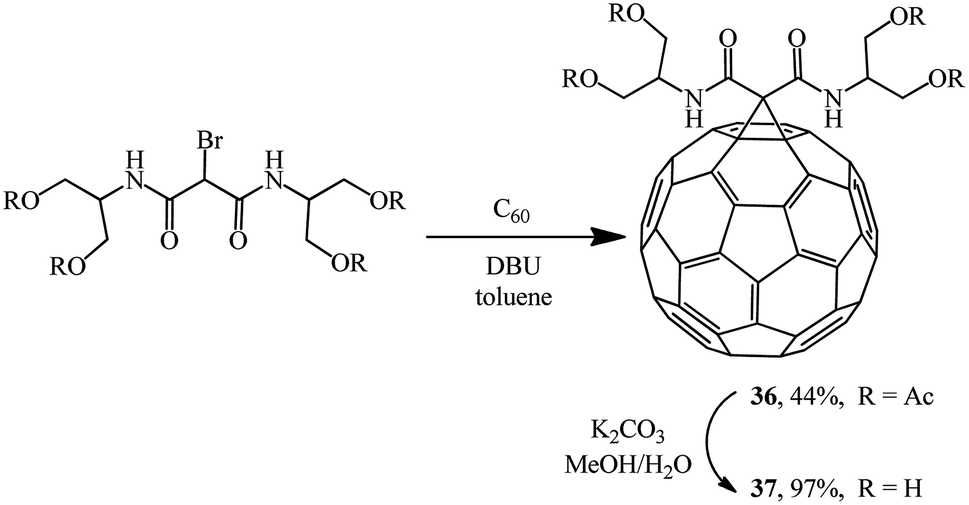 | ||
| Scheme 12 | ||
The reaction of 5-bromobarbituric acid with C60 in toluene:dimethylsulfoxide (DMSO) solution (95:5, v/v) gave monocyclopropanation adduct 38 as a brown powder (Scheme 13). This synthesis is the first example of intermolecular photodimerization of a C60 derivative in homogeneous solution that was made possible by the formation of supramolecular assemblies in which the fullerenes are maintained in close proximity.37
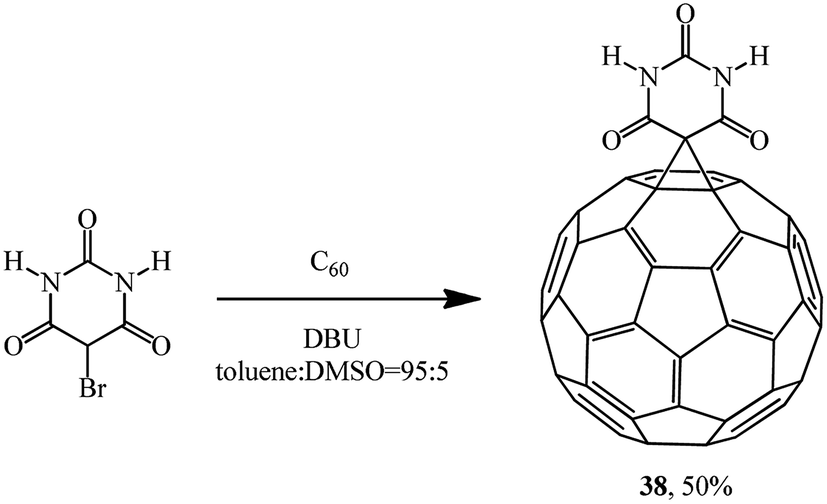 | ||
| Scheme 13 | ||
Cyclopropanation of C60 also smoothly occurs with α-bromoketones. It was used to incorporate a diphenylaminofluorene chromophore group into fullerene structure. Using this scheme, new acceptor–keto–donor assembly diads 39,38 40,39 41 (ref. 40) were obtained in yields of about 70% (Scheme 14). Molecules of this kind are characterized by an increase in the relative intensity of the fluorescent zone. This is explained by direct covalent bonding of diphenylaminofluorene moieties with the fullerene frame, which facilitates efficient intramolecular processes of electron or energy transfer.
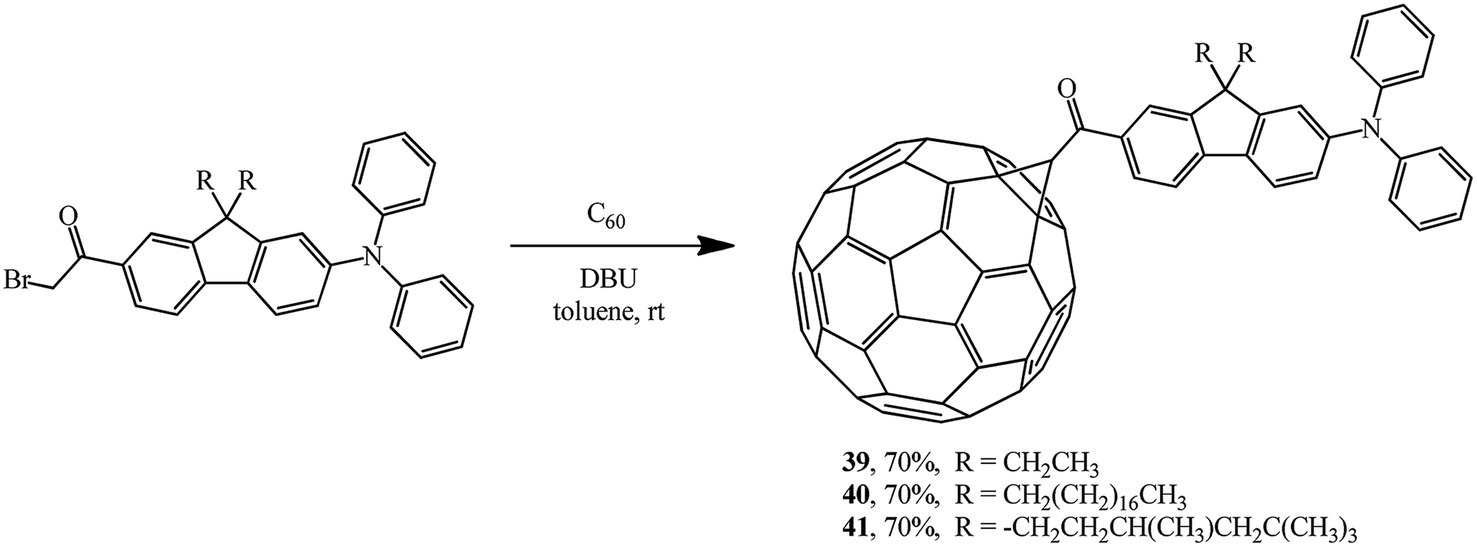 | ||
| Scheme 14 | ||
Methanofullerene 42 containing a phenylazide group was used for grafting C60 to a nanotube via aniline derivative 43 and diazonium salt 44 (Scheme 15).41
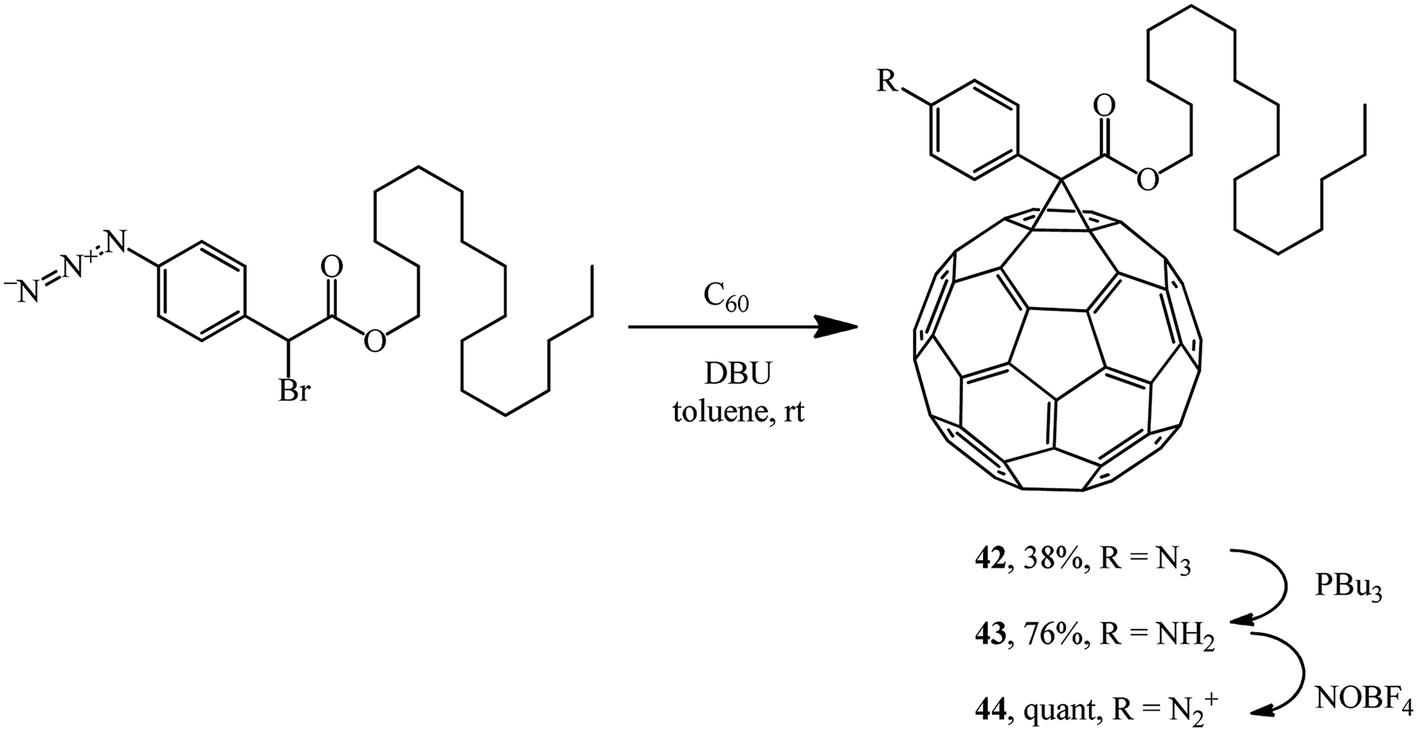 | ||
| Scheme 15 | ||
Phosphonate reagents with a reactive methylene group that underwent monobromination were found to be very reactive agents for C60 cyclopropanation. For example, the reaction of dimethoxybromomethylphosphonate alkyl esters with fullerene gave P-modified methanofullerenes 45–47 (Scheme 16).42
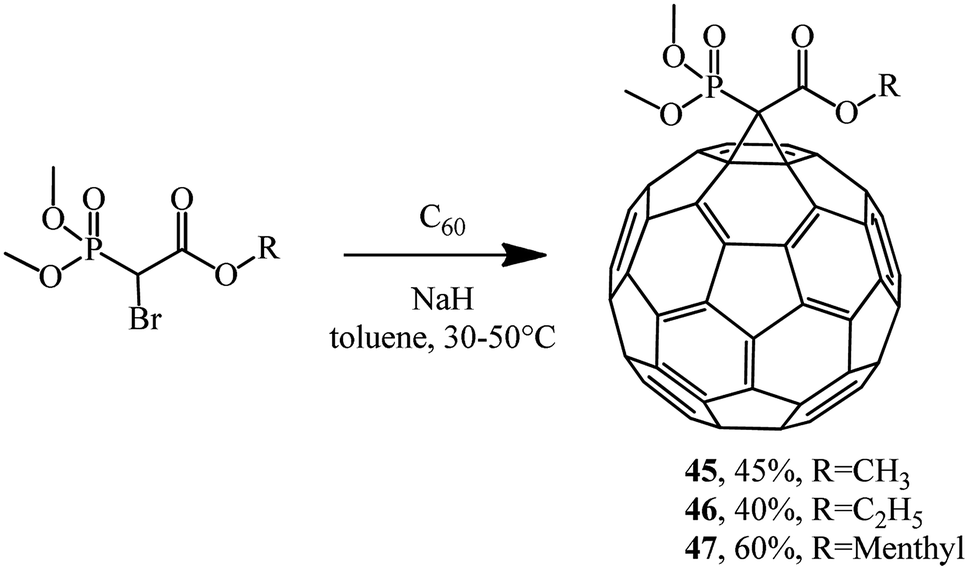 | ||
| Scheme 16 | ||
Using the methodology developed, the same researchers43 expanded this series by synthesizing (NaH, THF) phosphorylated methanofullerene 48, phenyl-containing α-ketoester 49 and diketones 50 (Fig. 4). According to the authors, the latter compounds are interesting objects for studying their biological activity and may be useful in the search for new functional materials.
 | ||
| Fig. 4 Methanofullerenes 48–50. | ||
Bis-methanophosphonates were also used in C60 cyclopropanation to give adducts 51 (ref. 44) and 52,45 which were treated with iodo- and bromotrimethylsilanes to give acid 53 via labile trimethylsilyl ethers easily hydrolyzable on stirring in an organic-aqueous medium (Scheme 17). Acid 53 is unstable in organic solvents and poorly soluble in water.
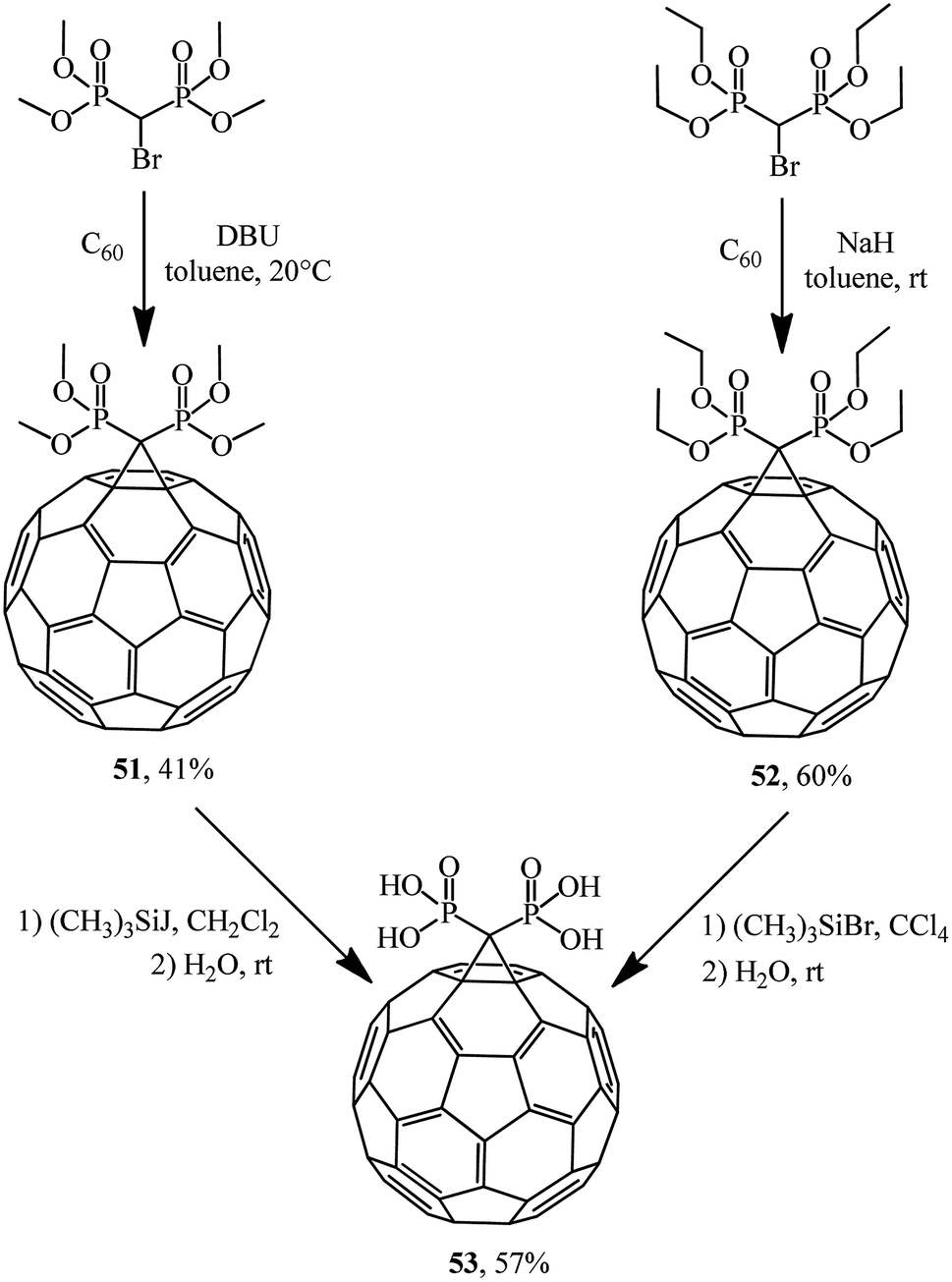 | ||
| Scheme 17 | ||
The multidisciplinary studies by F. Diederich46 also dealt with various methanofullerenes incorporating phosphonate or sulfone groups. In the syntheses of 54–56, the corresponding cyclopropanating agent reacted with C60 in oDCB (Fig. 5).
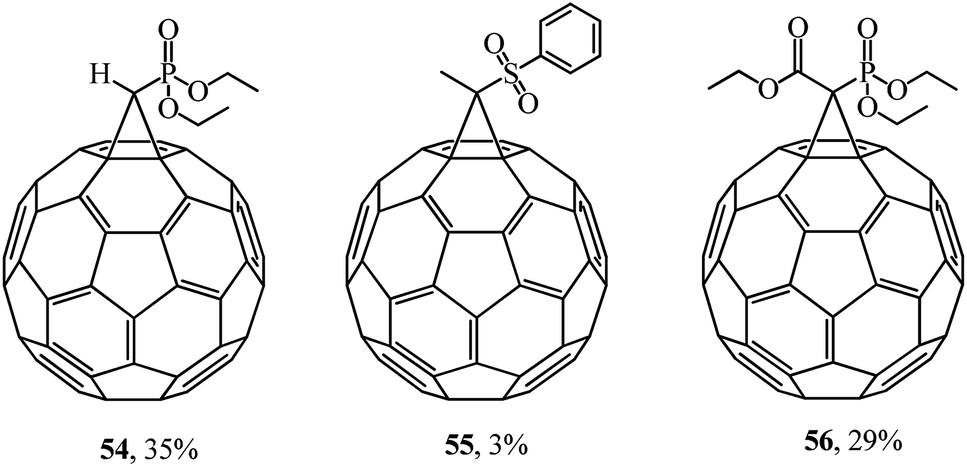 | ||
| Fig. 5 Methanofullerenes 54–56. | ||
By varying the conditions of the reaction of C60 with 2-bromo- or 2-iodo-1,3-diphenylpropane-1,3-dione in the presence of various bases (Scheme 18), the optimal conditions for the process giving the highest possible yield of methanofullerene 57 (45%) were determined.47 It was found that a bromohaloketone:DBU ratio of 1.0:2.4 had to be strictly maintained.
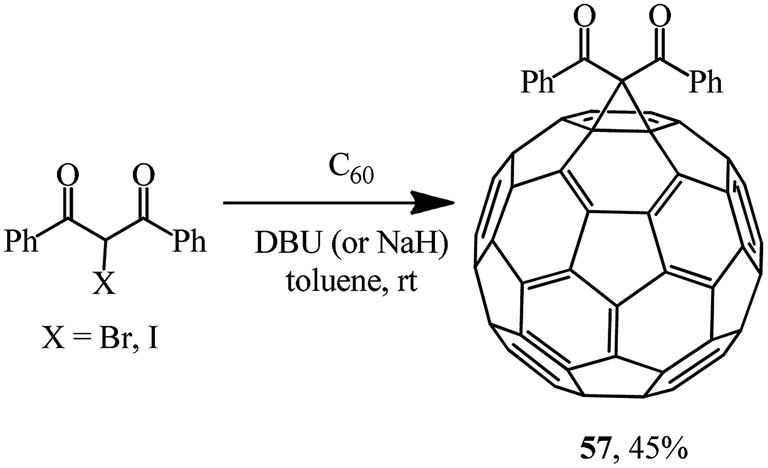 | ||
| Scheme 18 | ||
The approach by M. Miftakhov implemented in a number of publications48,49 that involved the use of dichloroacetic acid derivatives with an activated dichloromethine function capable of forming stabilized carbanions as a new cyclopropanating agent proved to be fruitful. The methine proton in dichloroacetic acid derivatives is strongly activated, therefore a carbanion is readily generated from it, while the readily leaving group (Cl−) ensures the intramolecular cyclization. Examples of aliphatic derivatives of dichloroacetic acid are presented in Scheme 19.
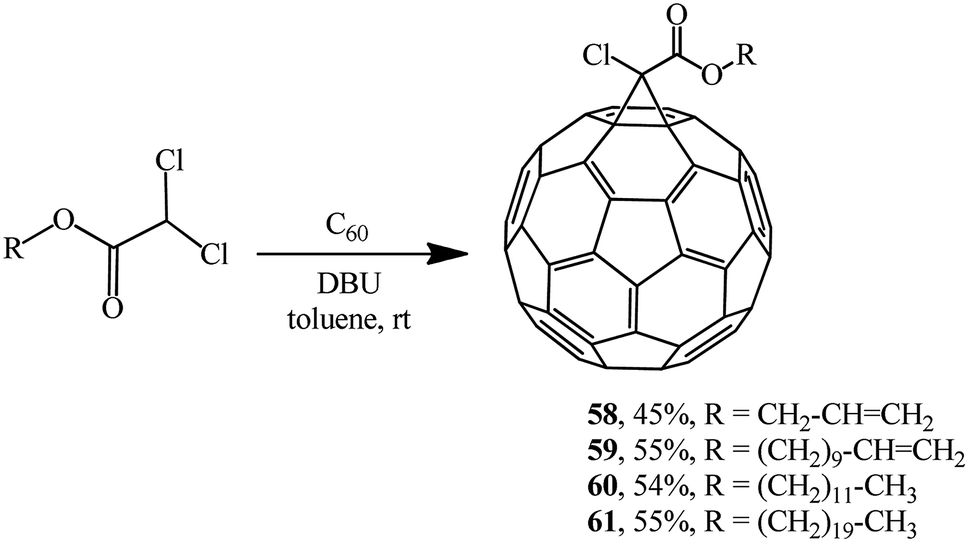 | ||
| Scheme 19 | ||
Using dichloroacetic acid derivatives and the Bingel methodology at the key stages, facile syntheses of fullerene-containing monomers of (meth)acrylic 62 and 63 and norbornene types 64 have been developed, aimed at their subsequent conversion into high-molecular compounds for application in solar cells (Scheme 20).50,51
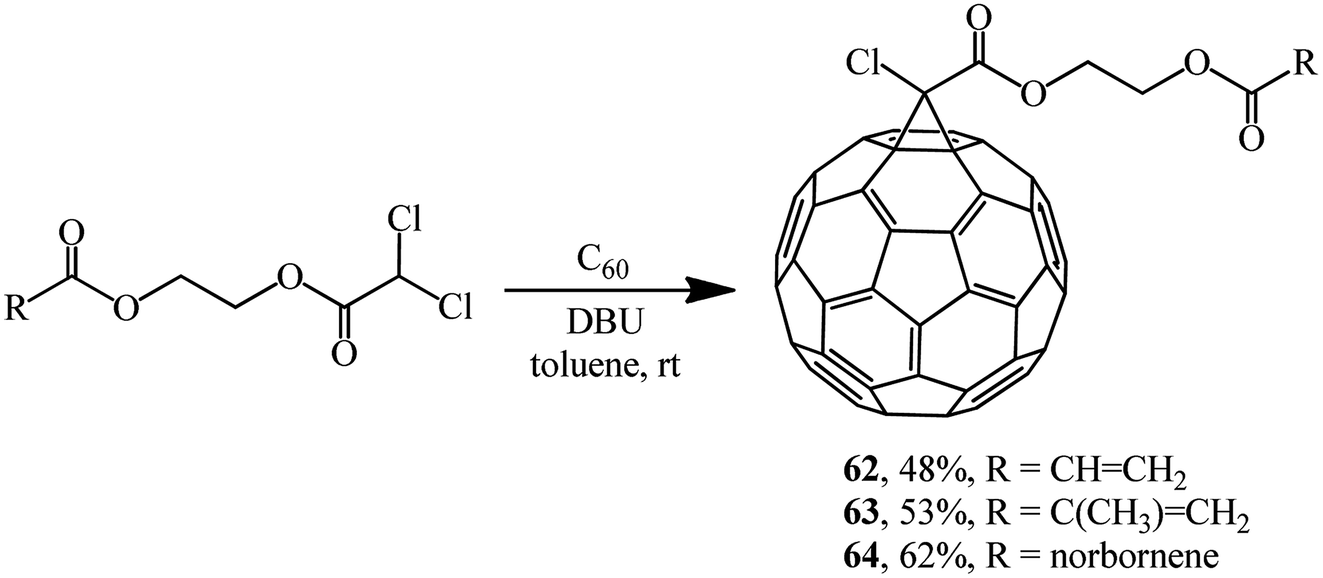 | ||
| Scheme 20 | ||
Keeping in mind that the broad-spectrum antibiotic laevomycetin contains the CHCl2 dichloromethine function that is reactive in the Bingel reaction, a group of researchers52 attempted to obtain its conjugate with fullerene as a continuation of these studies. In an experimental test, laevomycetin reacted with C60 in oDCB solution to give monoadduct 65. In a similar way, the cyclopropanation of fullerene with laevomycetin diacetate in toluene solution to give methanofullerene 66 (Scheme 21) was also successfully implemented.
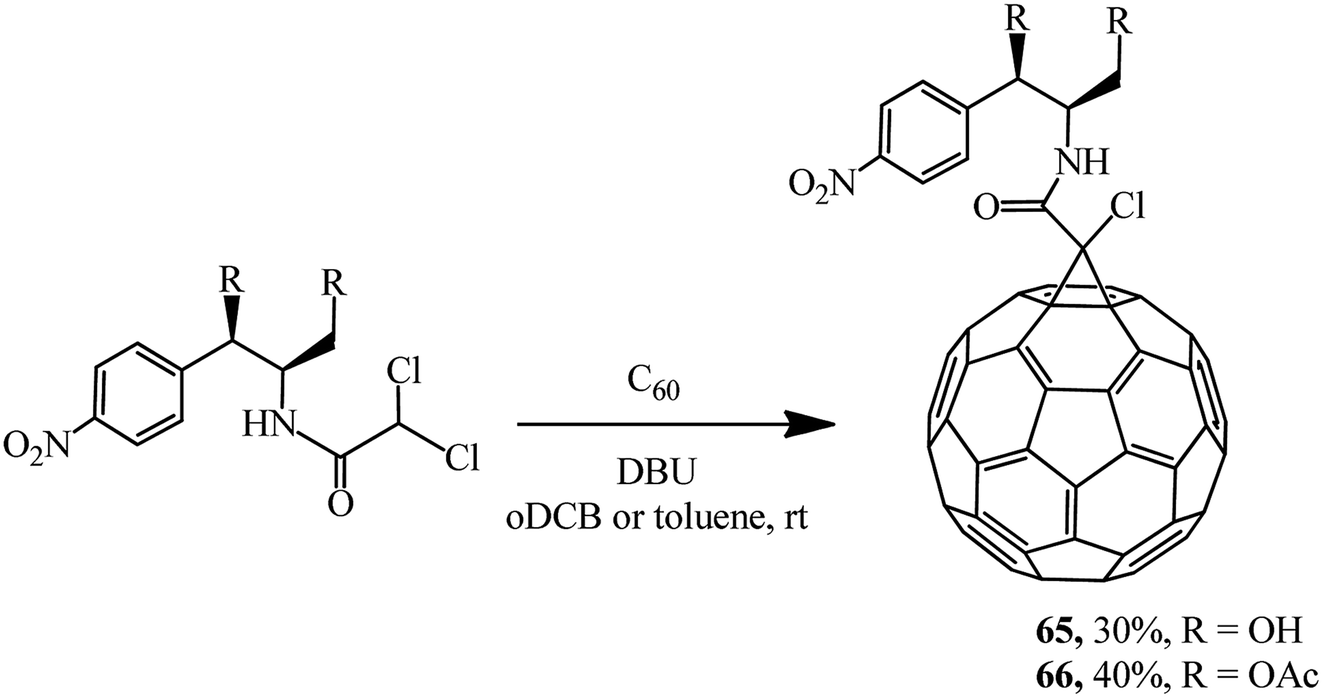 | ||
| Scheme 21 | ||
Unlike dichloroacetic acid esters that were previously used for C60 cyclopropanation, methyl (2Z)-2,4,4-trichloro-3-methoxybut-2-enoate obtained in two stages from hexachlorobutadiene contains no activating α-carboxy function. Nevertheless, it was assumed53 that due to the concerted action of donor (MeO) and acceptor (CO2Me, Cl) substituents in the substrate, the level of CH-acidity is sufficient for quite successful implementation of the Bingel reaction under standard conditions. As one can see from structure 67, the donor and acceptor parts in the target product are effectively cross-conjugated (Scheme 22).
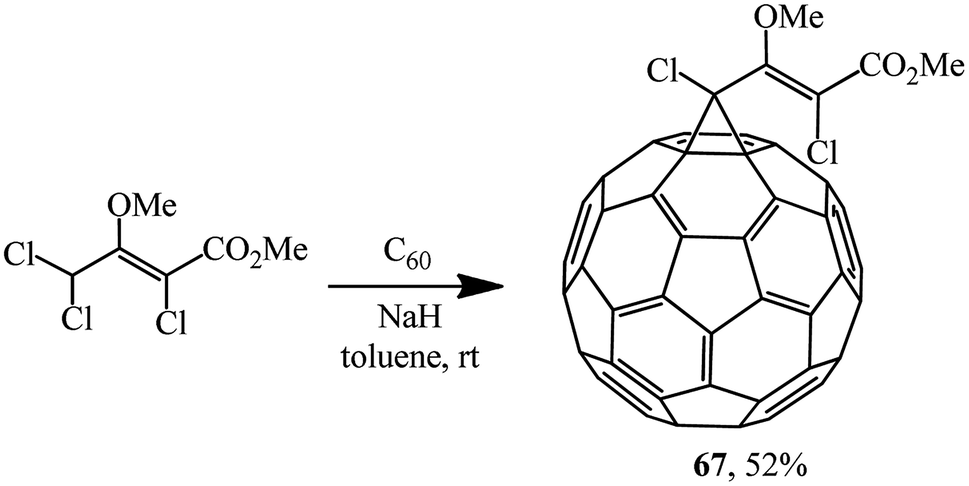 | ||
| Scheme 22 | ||
The literature sources covering the Bingel reaction offer a variety of bases used in the syntheses of methanofullerenes. Examples of the use of alternative activating bases are also provided, for example, the use of triethylamine in a synthesis of nitromethanofullerene 68 from bromonitromethane and pyridine in a synthesis of ethyl cyanocarboxymethanofullerene 69 from ethyl bromocyanoacetate (Scheme 23). According to ref. 54, the methanofullerenes with acceptor substituents (NO2, CO2Et) they isolated are promising in the design of photoconversion devices.
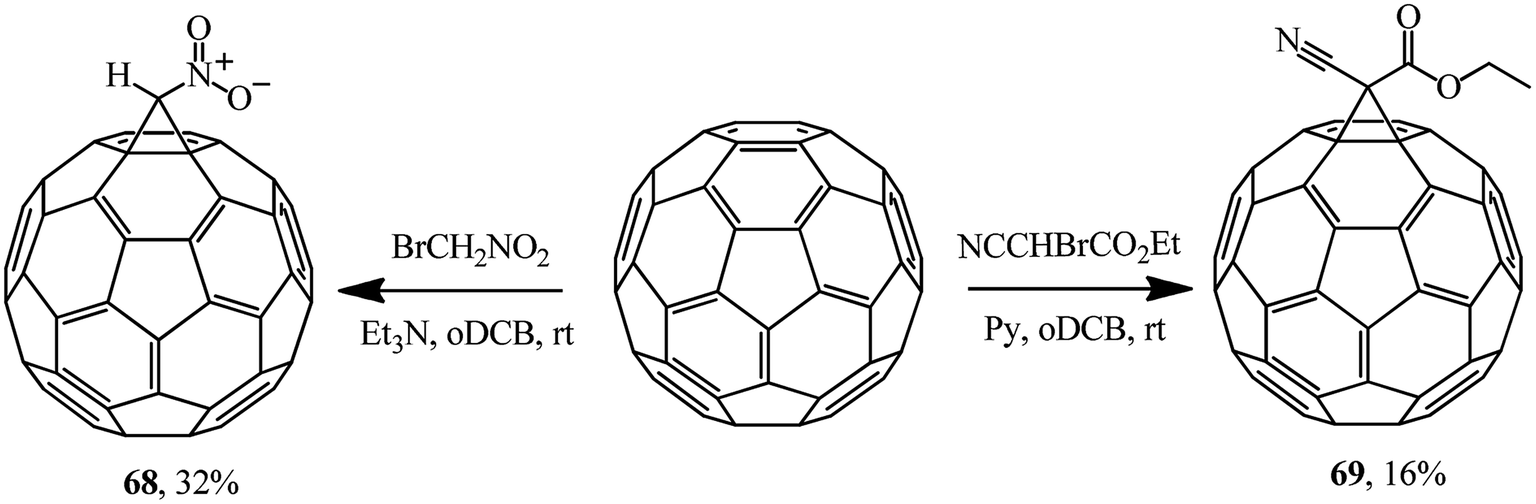 | ||
| Scheme 23 | ||
The reactions of fullerene with bromoacetonitrile and tribromomethane also resulted in 1′-cyano- 70 and 1′,1′-dibromo-1,2-methano[60]fullerenes 71, respectively. Since the reaction in the presence of sodium hydride failed, a stronger base, namely lithium diisopropylamide (LDA), had to be used (Scheme 24).55
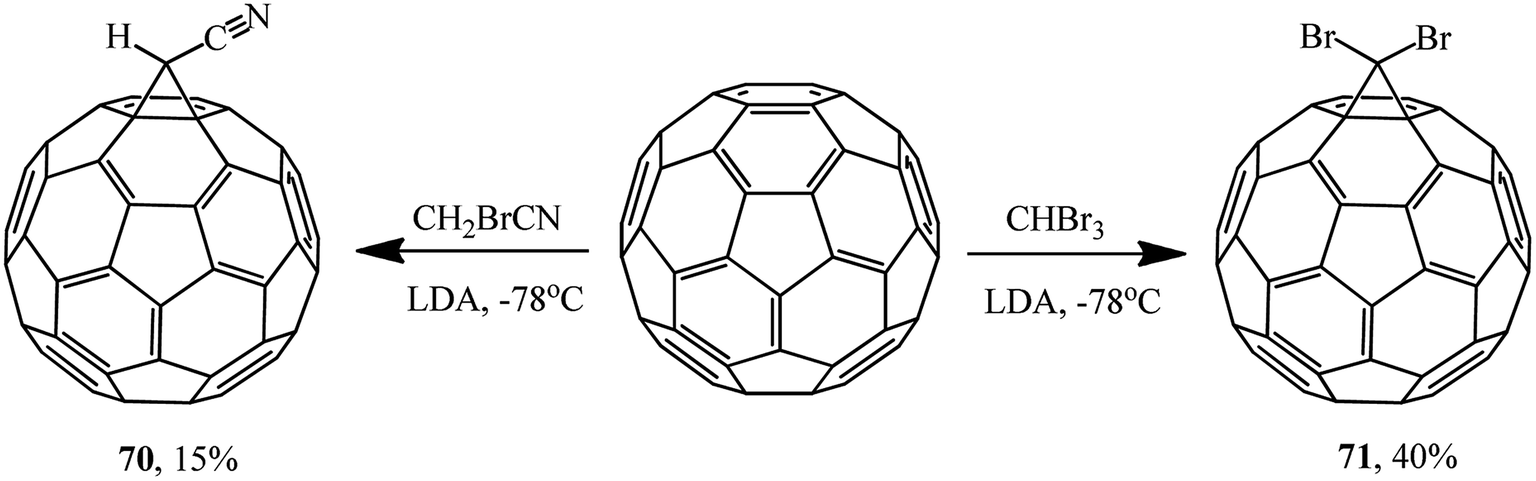 | ||
| Scheme 24 | ||
In order to increase the product yield and simultaneously simplify the process, the reaction of C60 with diethyl bromomalonate under high-speed vibration milling (HSVM) conditions in the absence of solvents was studied.56 As a result, the opinion was formulated that under these conditions methanofullerenes can be obtained in good or even excellent yields in the presence of inorganic bases (Scheme 25).
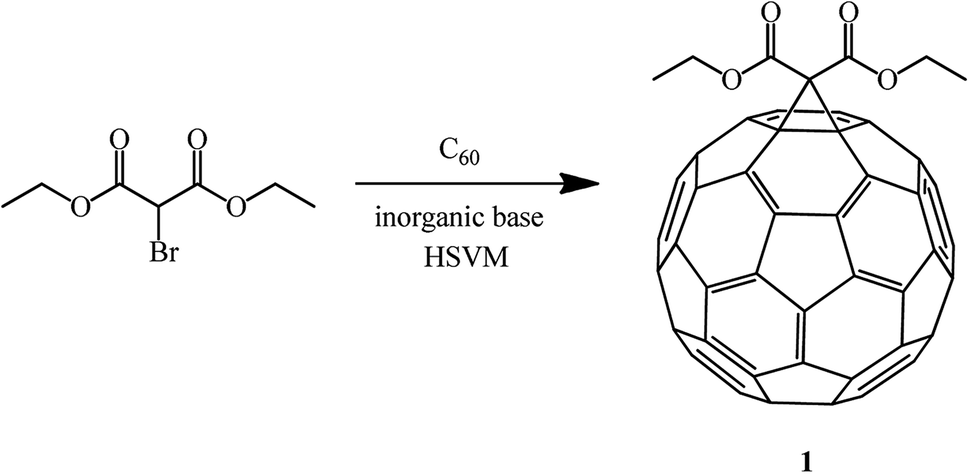 | ||
| Scheme 25 | ||
It has been found that some weak inorganic bases are very effective in Bingel's mechanochemical reactions of C60 under high-speed vibration milling conditions without a solvent. A comparative analysis and study of bases revealed the fact that the presence of sodium acetate in the system provides the maximum yield of the mono-adduct, whereas a significant amount of bis-adducts is formed in the presence of potassium carbonate (Table 1).
| Entry | Base | Yield of 1, % |
|---|---|---|
| 1 | NaOAc | 59 |
| 2 | Na2CO3 | 51 |
| 3 | NaHCO3 | 48 |
| 4 | Ca(OH)2 | 45 |
| 5 | Na2B4O7 × 10H2O | 44 |
| 6 | K2CO3 | 29 |
| 7 | NaOH | 11 |
| 8 | KOH | 17 |
| 9 | Alkaline Al2O3 | 10 |
Recently, a method for fullerene cyclopropanation by bromo-substituted substrates with a reactive methylene function in the presence of amino acids and dimethyl sulfoxide and without a base was suggested. This method was claimed to be efficient.57 Sarcosine, glycine, N-ethylglycine, and N-benzylglycine were used as the catalytic additives. As shown by experimental data for the reaction with diethyl bromomalonate, the yield of products varies from 5 to 42% and depends on the presence of a particular amino acid, stoichiometric ratio of reagents, and nature of solvents (C6H5Cl:DMSO) (Table 2). The process is completed in 1–2 hours at 20 °C (Scheme 26).
| Entry | Catalyst | C6H5Cl:DMSO (v/v) |
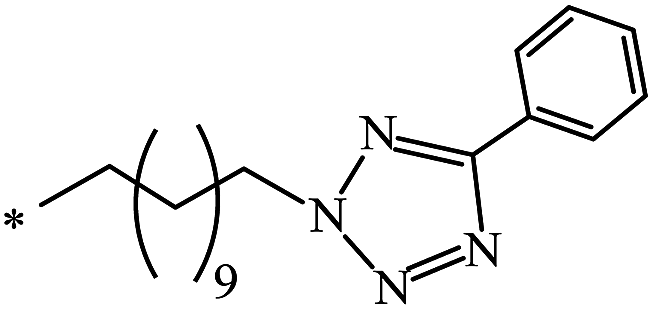
Yield of 1, % |
|---|---|---|---|
| 1 | — | 2:1 |
0 |
| 2 | N-Ethylglycine | 2:1 |
37 |
| 3 | Glycine | 2:1 |
33 |
| 4 | N-Benzylglycine | 2:1 |
38 |
| 5 | Sarcosine | 2:1 |
42 |
| 6 | Sarcosine | 20:1 |
5 |
| 7 | Sarcosine | 20:3 |
8 |
| 8 | Sarcosine | 4:1 |
17 |
| 9 | Sarcosine | 5:2 |
39 |
| 10 | Sarcosine | 5:3 |
41 |
| 11 | Sarcosine | 4:3 |
38 |
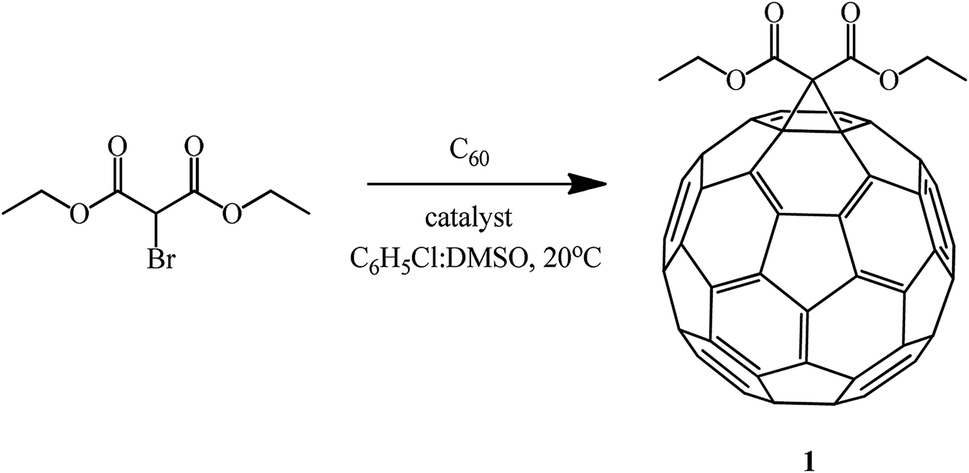 | ||
| Scheme 26 | ||
This method allows the cyclopropanation of fullerene to be performed at room temperature with bromomalonates and bromoacetoacetates that are sensitive to bases. This approach proved to be convenient for involving bromomalonic acid in the cyclopropanation of C60 to give methanofullerene 72, which is impossible to synthesize under “classical” Bingel reaction conditions (Scheme 27).
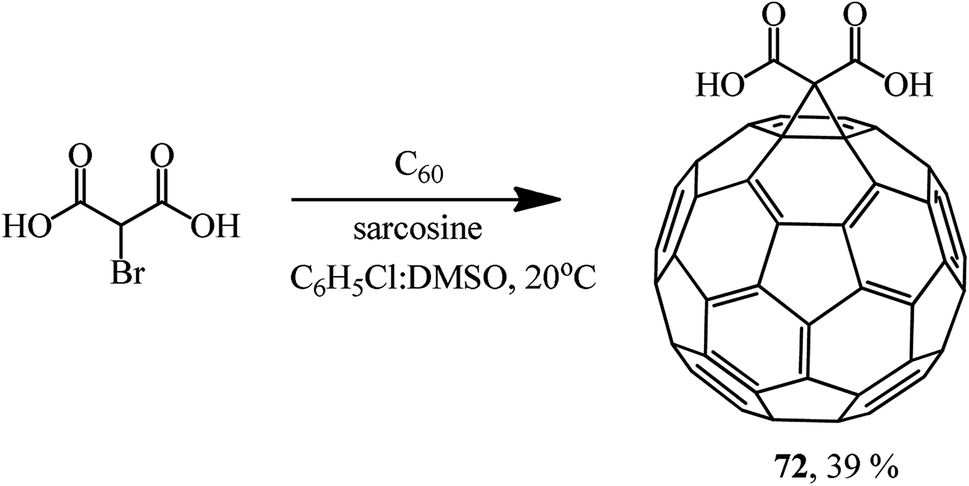 | ||
| Scheme 27 | ||
Using sarcosine as a catalyst, monofunctionalized methanofullerenes 73–78 (Scheme 28) and 79 and 80 (Scheme 29) were isolated in a similar way.57
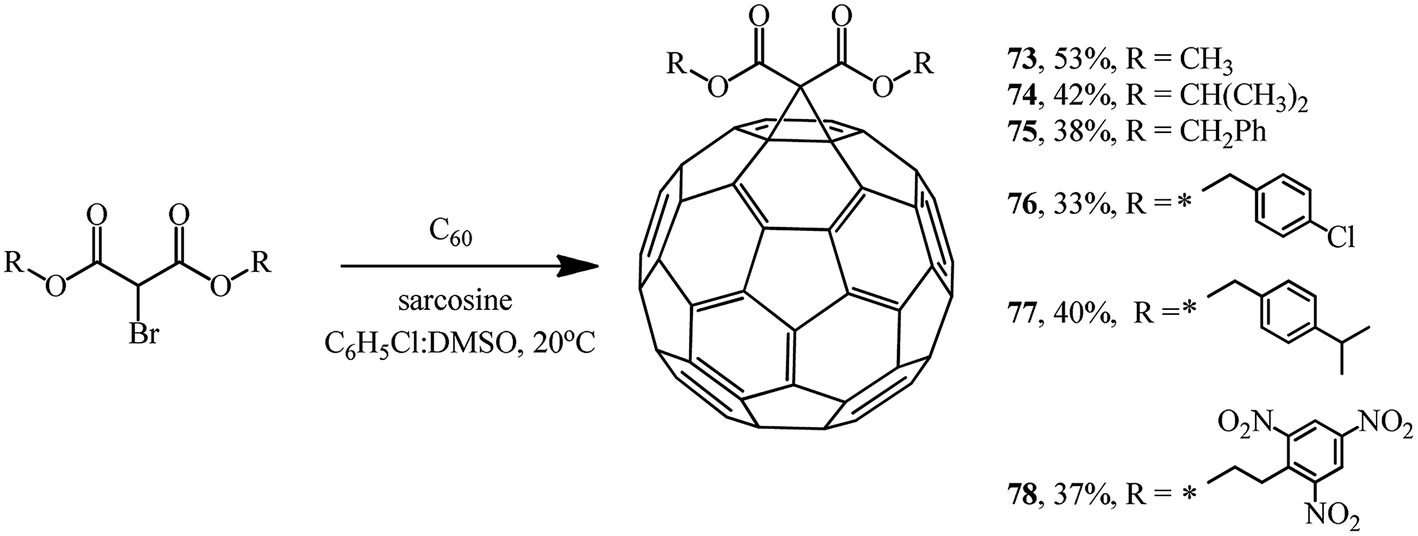 | ||
| Scheme 28 | ||
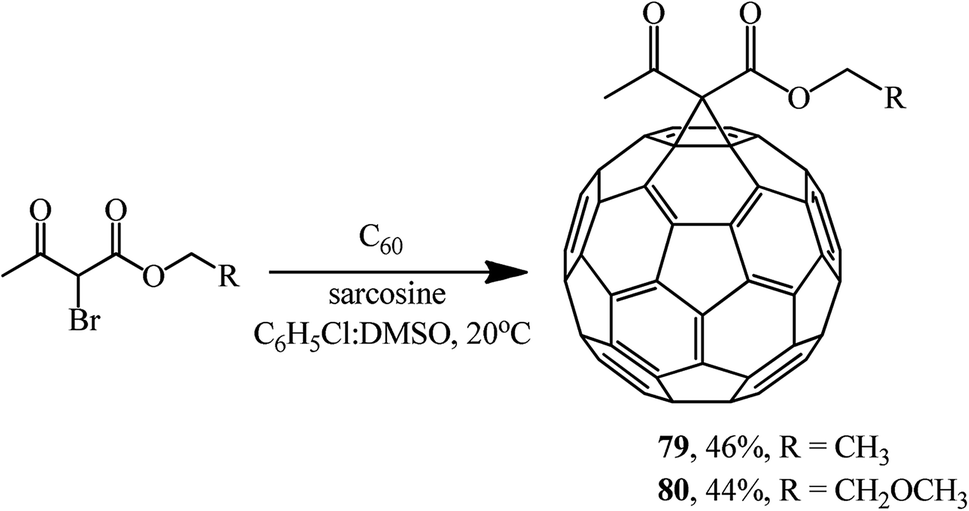 | ||
| Scheme 29 | ||
The assumed process mechanism is presented in Scheme 30. The fullerene anion radical E and amino acid radical cation F are formed in the presence of dimethylsulfoxide due to electron transfer from the amino acid to C60. Subsequently, radical anion E attacks the methylene group of bromomalonate to give radical G with elimination of a bromide ion. At the final stage of cyclization with participation of the amino acid radical ion, methanofullerene H is formed.
 | ||
| Scheme 30 | ||
The new energy-enriched derivative 81 was synthesized by a modified Bingel's reaction of C60 with bromomalonic acid poly (glycidyl nitrate) ester (Scheme 31).58
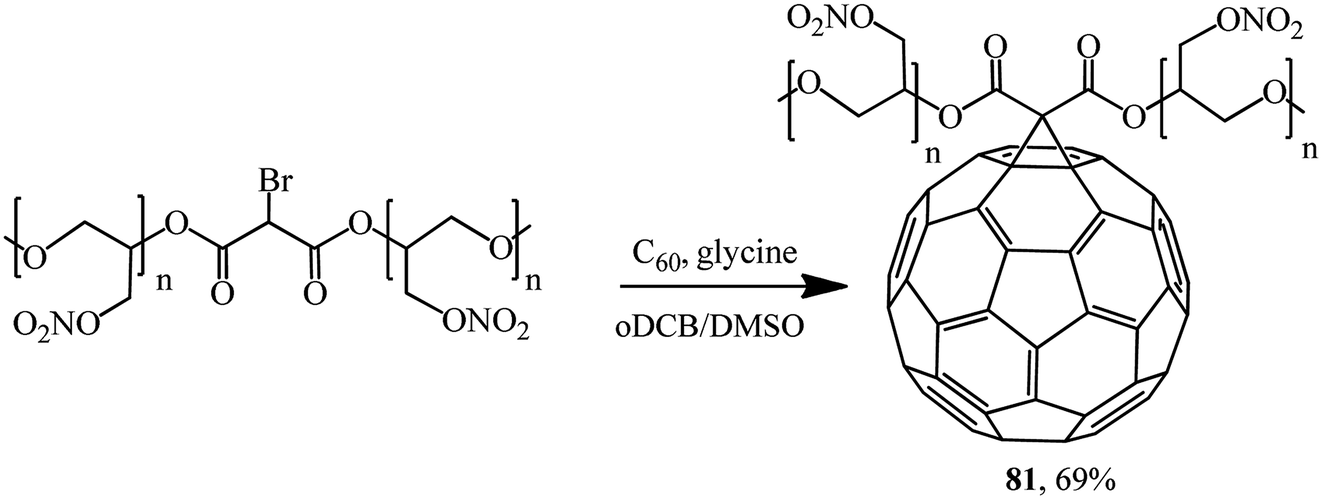 | ||
| Scheme 31 | ||
A promising fullerene-containing polymer 82 based on bromomalonic acid poly (3-azidomethyl-3-methyl oxetane) ester was also obtained by a modified Bingel's reaction (Scheme 32).59
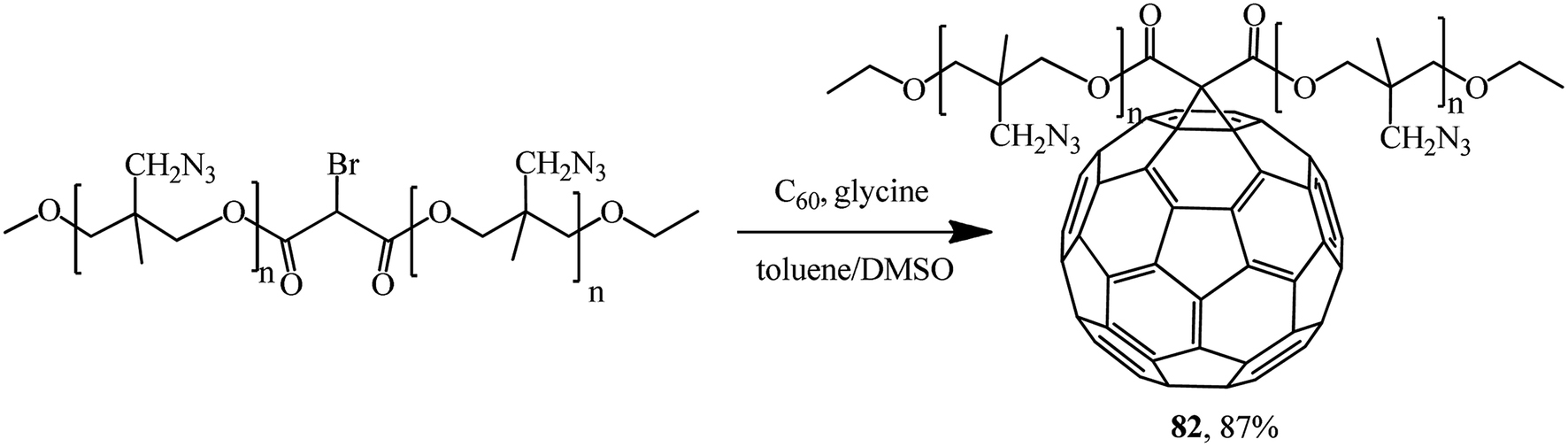 | ||
| Scheme 32 | ||
The compounds with a reactive methylene group used in the Bingel reaction can include not only bromomalonates but also α-bromo-β-ketoesters, 2-bromo-1,3-diketones, and other bromo-substituted reactive substrates. By changing the process technique to some extent and using the DMSO/Na2CO3 system,60 we isolated methanofullerenes in good yields even at 10 °C. In fact, cyclopropanation of C60 with various bromomalonic esters in the presence of DMSO/Na2CO3 (with a 20-fold excess of ether and a 40-fold excess of Na2CO3) in a binary C6H5Cl/DMSO solvent mixture showed the presence of methanofullerenes 73–77 and 83–87 in 29–68% yields even in 5 min after the start of the reaction (Scheme 33).
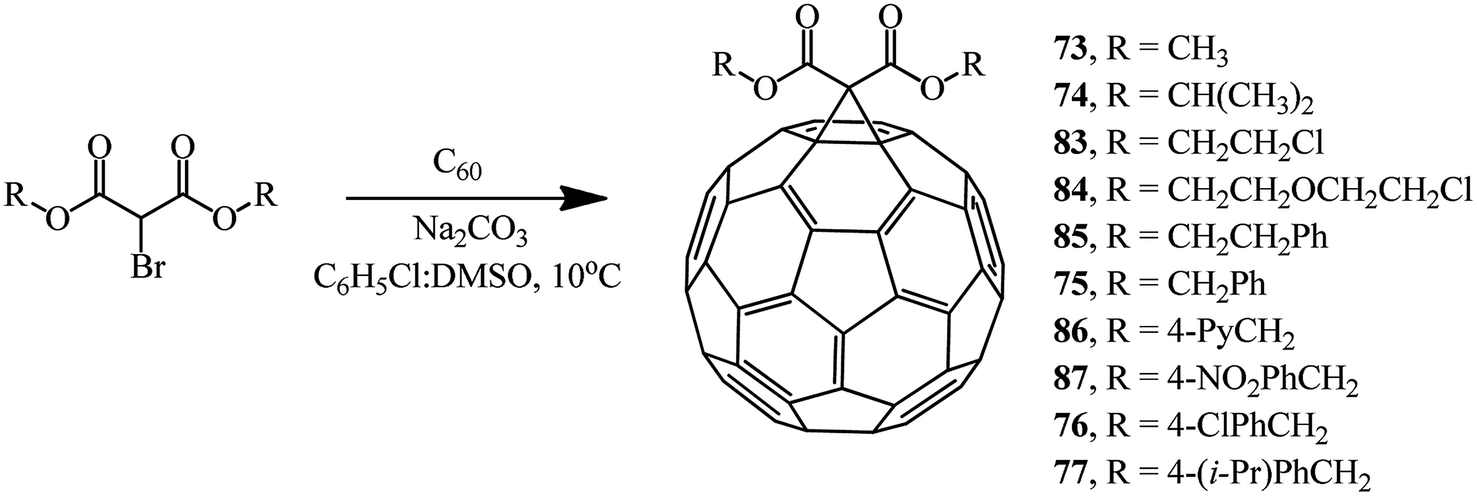 | ||
| Scheme 33 | ||
Various brominated β-ketoesters and aromatic ethyl bromoformyl acetates react with fullerene C60 under identical conditions to give monocyclopropanation adducts 88–93 (34–63%) and 94–100 (43–66%) (Schemes 34 and 35).
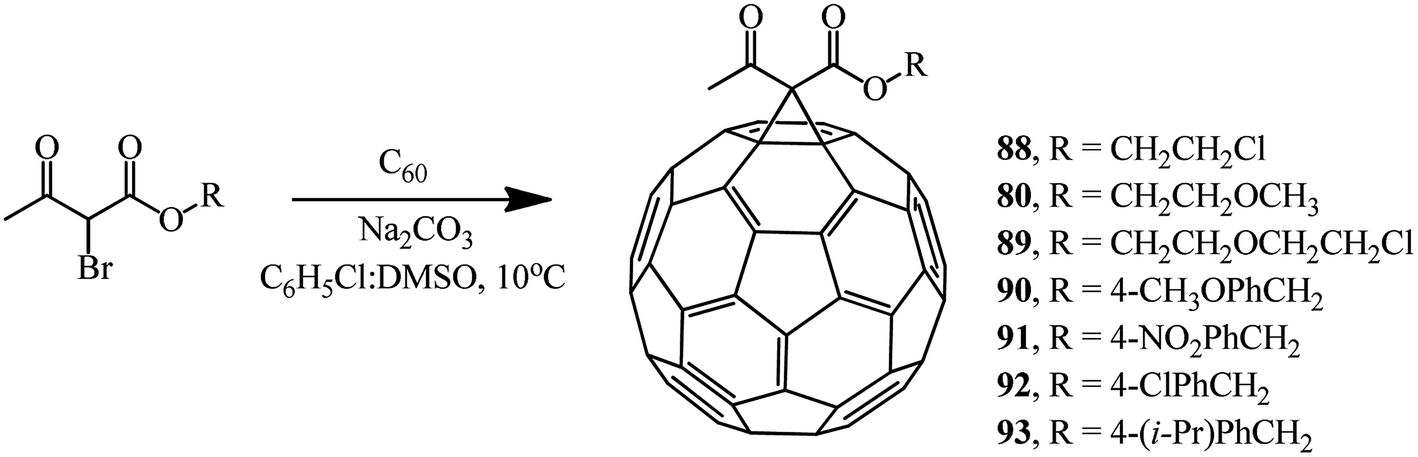 | ||
| Scheme 34 | ||
 | ||
| Scheme 35 | ||
The mechanism of cyclopropanation in the presence of the DMSO/Na2CO3 catalytic system was studied. It is assumed that nucleophilic attack of the dimsil anion on the bromine-containing center of the substrate to give dimethylsulfoxonium intermediate I occurs. At the next stage, I loses a proton in the weakly alkaline environment to give intermediate J. The latter reacts with the fullerene to form an intermediate charged complex K, followed by its intramolecular cyclization with elimination of DMSO and formation of methanofullerene L (Scheme 36).
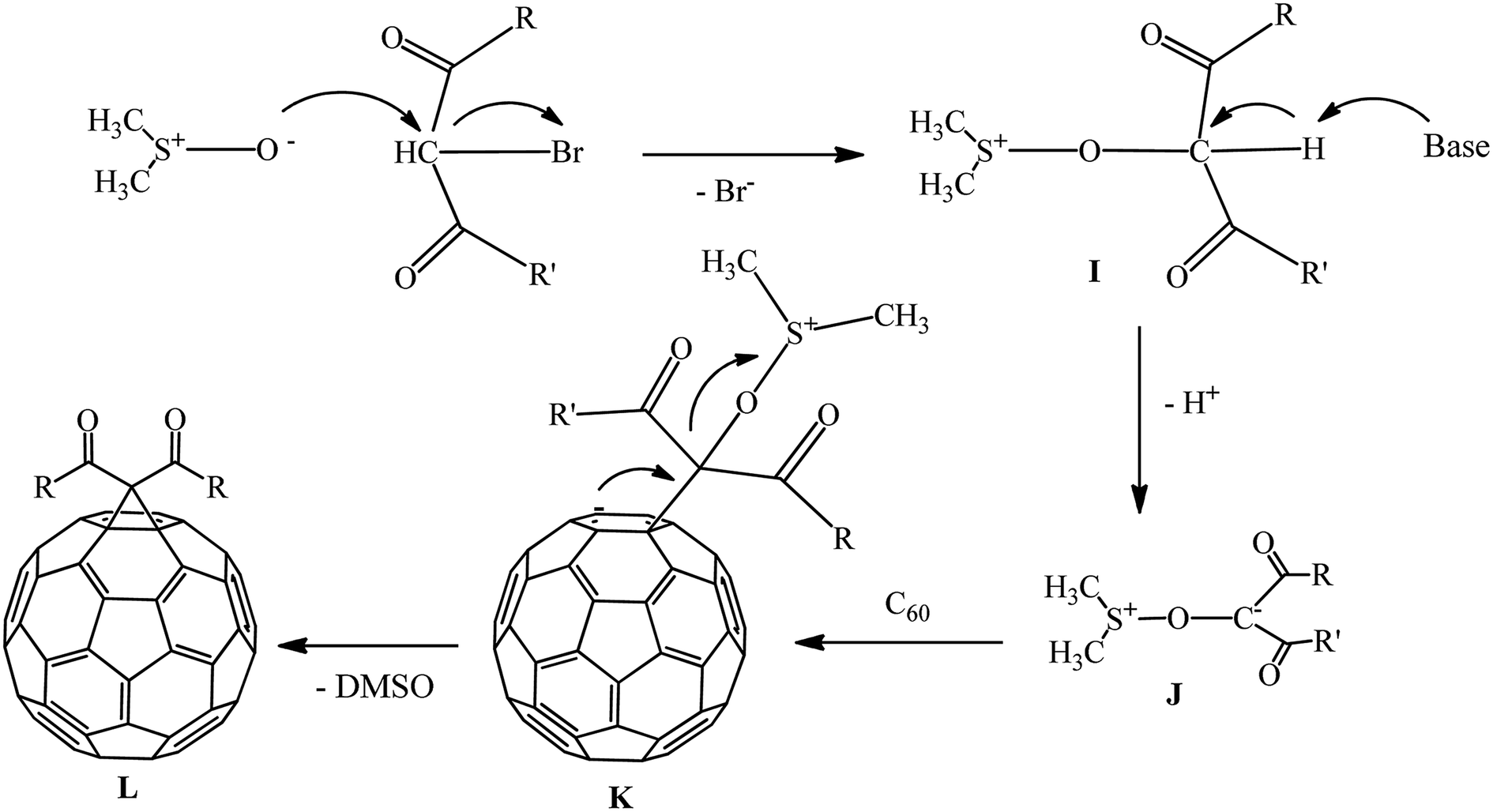 | ||
| Scheme 36 | ||
Cyclopropanation in the presence of DMSO/Na2CO3 is valuable precisely because it allows one to functionalize not only C60 fullerene but also the C70 molecule, as well as single-walled carbon nanotubes and, which is particularly valuable, also graphene.
The nearly comprehensive Bingel method presented above makes it possible to synthesize methanofullerenes predominantly from bromo-substituted substrates with diverse structures containing a reactive methylene function.
2 The Bingel–Hirsch technique in the synthesis of methanofullerenes
In the previous chapter, Bingel's approach was noted as an almost comprehensive method for synthesizing methanofullerenes mainly based on bromo-substituted substrates. After the method was improved by his colleague, A. Hirsch, it was named the Bingel–Hirsch method.Using four different malonates containing chiral and sterically hindered dendritic side chains as examples, X. Camps and A. Hirsch61 developed a method for synthesizing methanofullerenes in a “one-pot” reaction involving tetrabromomethane and DBU. The monoadducts were synthesized by direct treatment of C60 with malonates in the presence of CBr4 and DBU in a molar ratio of 1:1.5:1.5:3, respectively. The cyclopropanation of fullerene under these conditions occurred smoothly even at room temperature to give the corresponding mono adducts 1, 14, 22 and 101. Their yields were found to be comparable to, or even higher than those in the case of the process with bromomalonates (Scheme 37).
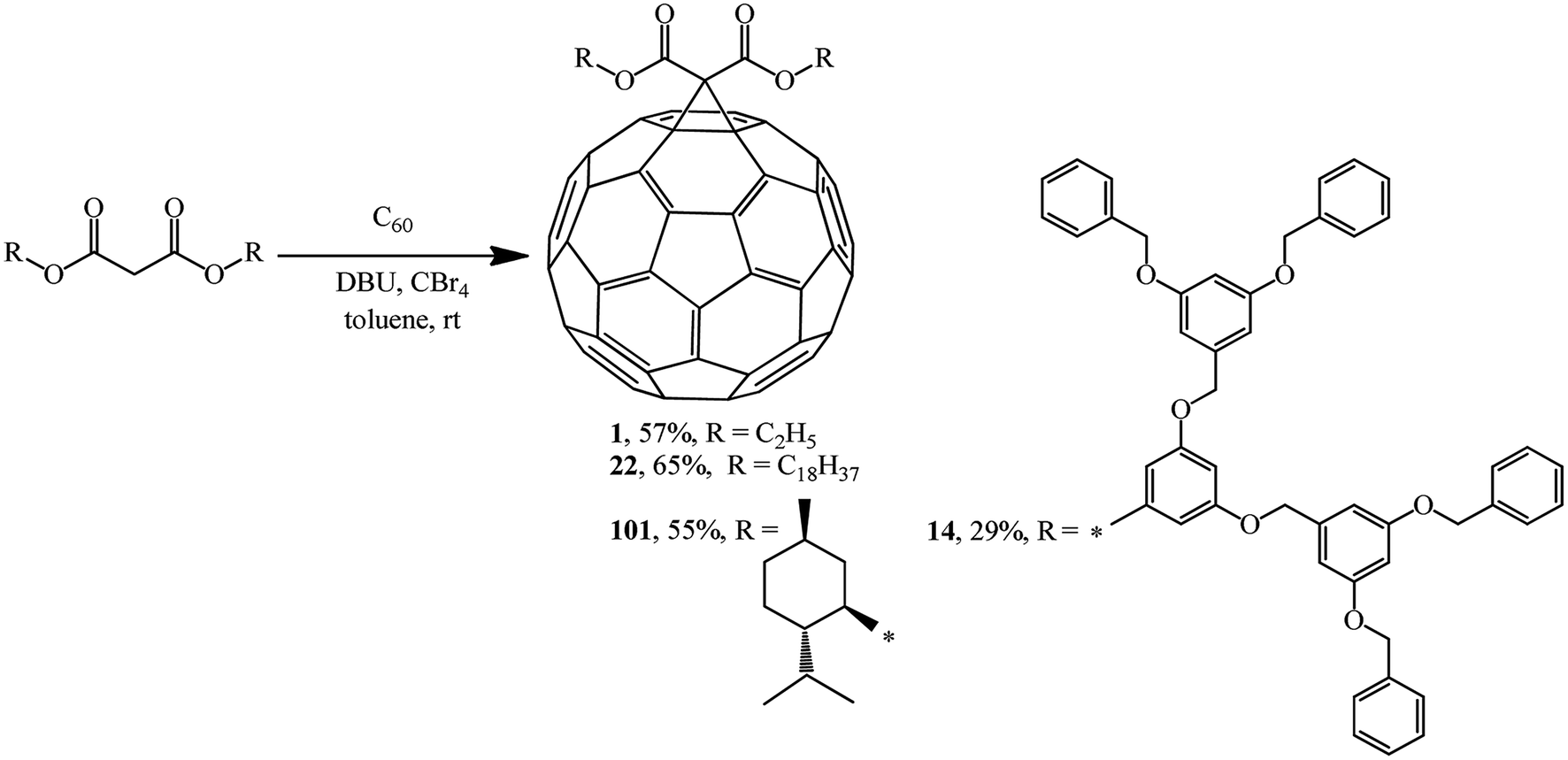 | ||
| Scheme 37 | ||
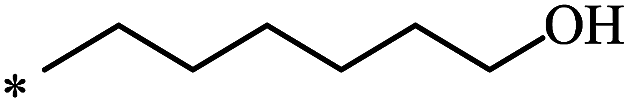
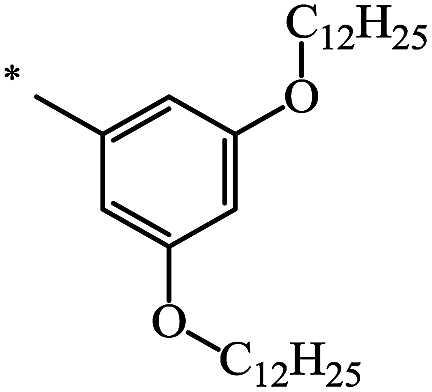
The Bingel–Hirsch approach was further extended to include malonates with various substituents listed in Table 3 (Scheme 38).
| Entry | R1 | R2 | Yield, % | Reference |
|---|---|---|---|---|
| a nr – the yield is not provided. | ||||
| 73 | CH3 | CH3 | 28 | 62 |
| 17 | C4H9 | C4H9 | 35 | |
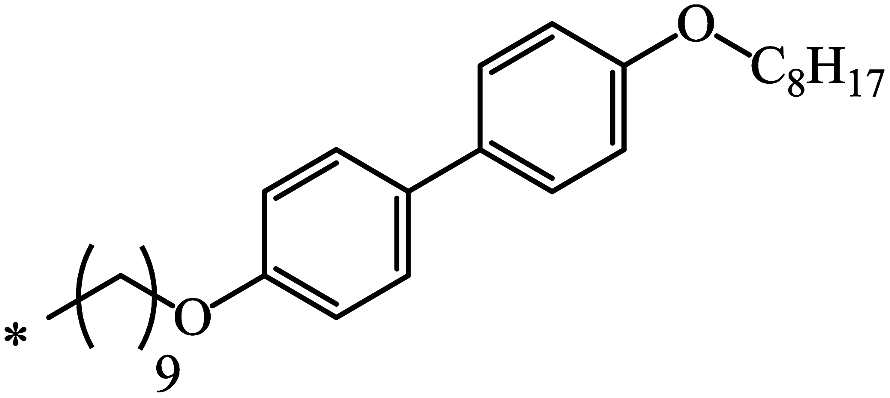
| 19 | C8H17 | C8H17 | 32 | |
| 102 | (CH2)2OCH3 | (CH2)2OCH3 | 21 | |
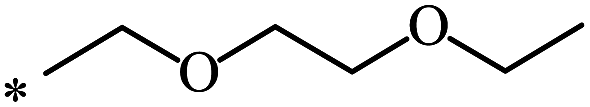
| 85 | 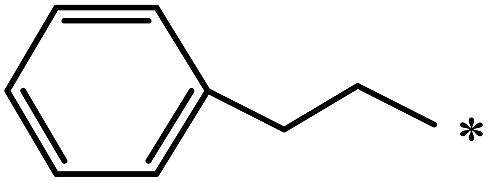 |
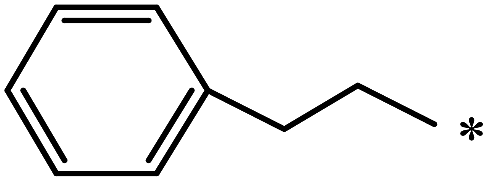 |
37 | |
| 103 | (CH2)11–CH3 | CH3 | 59 | 49 |
| 104 | (CH2)19–CH3 | CH3 | 50 | |
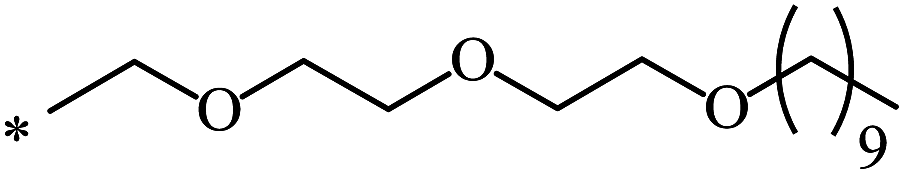
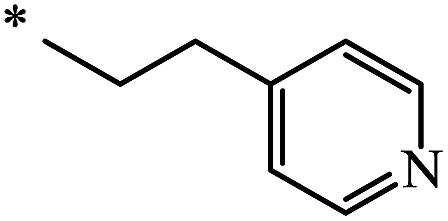
| 105 | (CH2)2–O–CH3 | C2H5 | nra | 63 |
| 106 | CH2–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
CH2–CHCH2 |
32 | 64 |
| 107 | (CH2)9–CHCH2 |
CH3 | 50 | 51 |
| 108 | 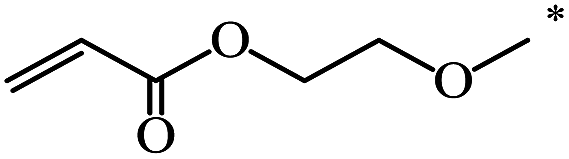 |
CH3 | 59 | |
| 109 | 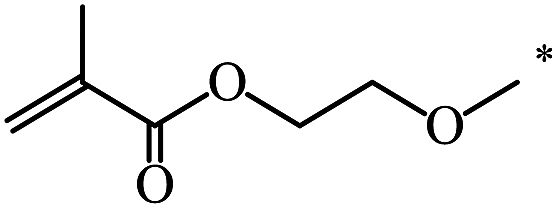 |
CH3 | 46 | |
| 110 | 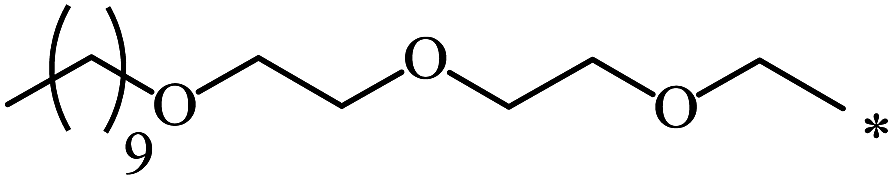 |
 |
48 | 65 |
| 111 |  |
 |
nr | 66 |
| 112 | 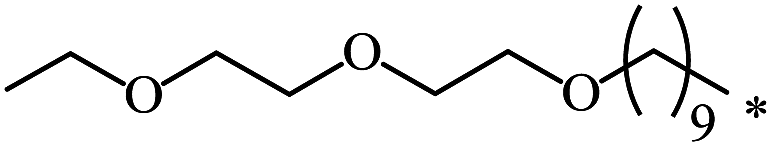 |
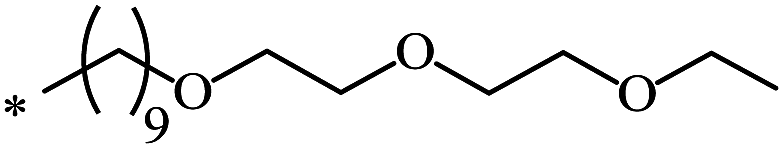 |
nr | 66 |
| 113 | 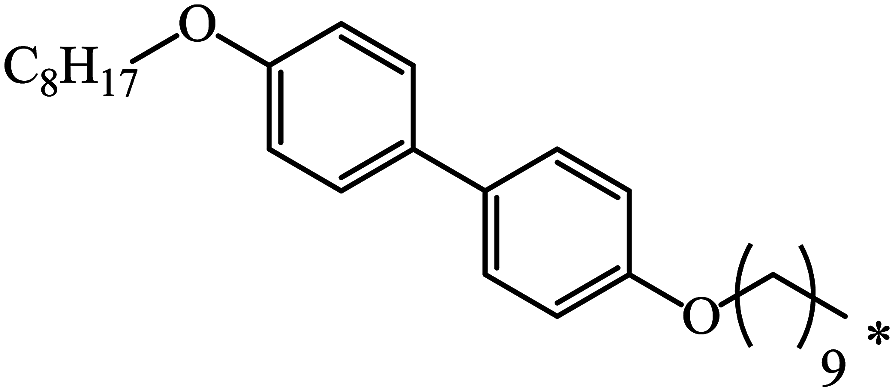 |
 |
51 | 67 |
| 114 | 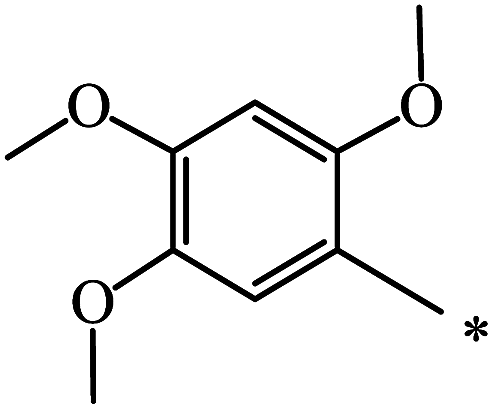 |
CH3 | 51 | 68 |
| 115 | 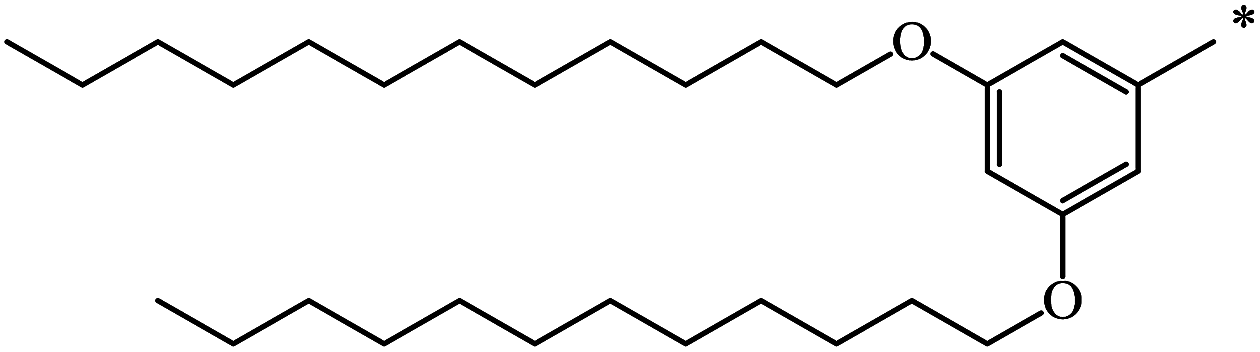 |
 |
47 | 69 |
| 116 | 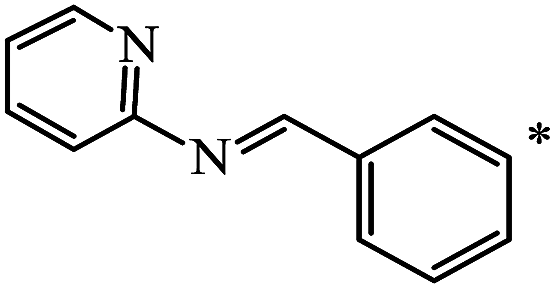 |
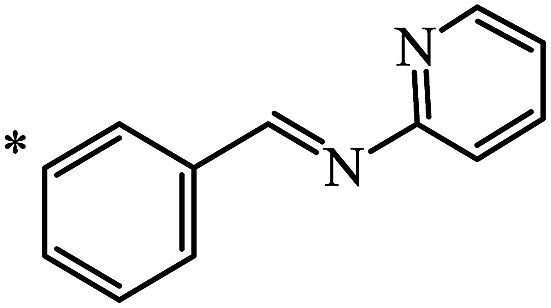 |
33 | 70 |
| 117 | 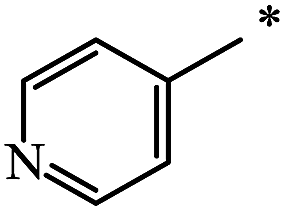 |
 |
23 | 71 |
| 118 |  |
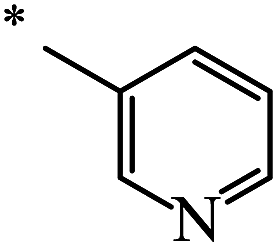 |
21 | |
| 119 |  |
CH3 | 50 | 72 |
| 120 |  |
 |
40 | |
| 121 |  |
 |
50 | |
| 122 | 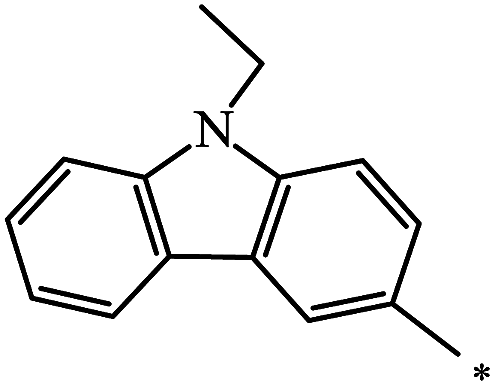 |
C2H5 | 22 | 73 |
| 123 | 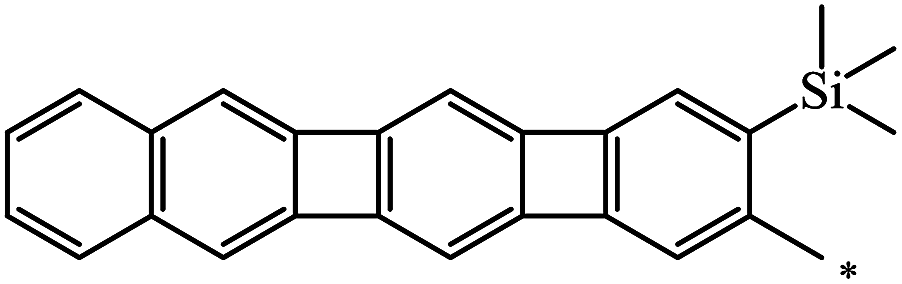 |
 |
52 | 74 |
| 124 | 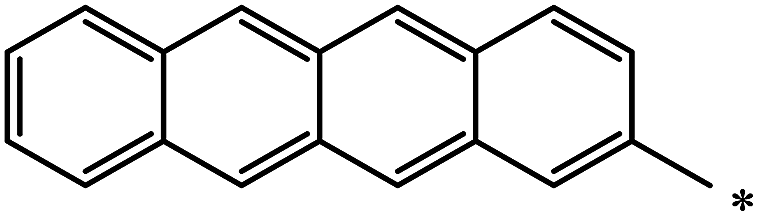 |
 |
27 | 75 |
| 125 |  |
CH3 | 16 | 76 |
| 126 |  |
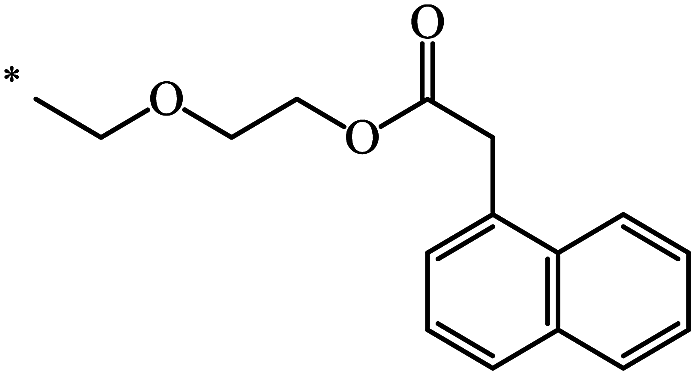 |
39 | |
| 127 |  |
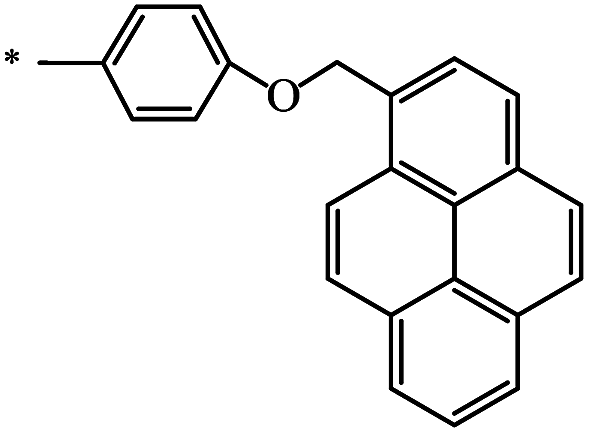 |
48 | 77 |
| 128 |  |
C2H5 | 62 | 78 |
| 129 | 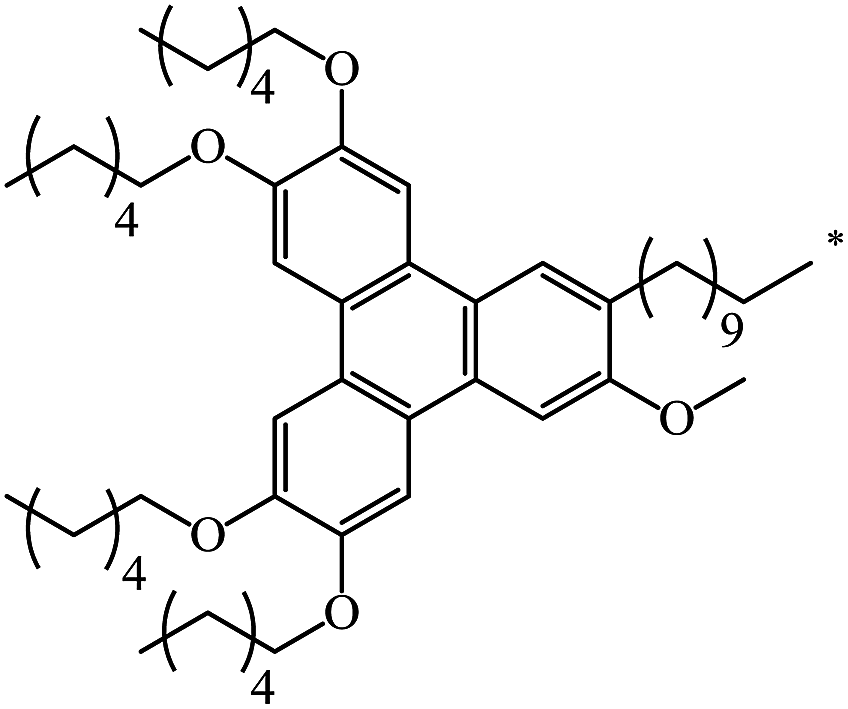 |
 |
62 | |
| 130 |  |
 |
38 | 79 |
| 131 |  |
 |
nr | 80 |
| 132 |  |
 |
||
| 133 |  |
 |
||
| 134 | 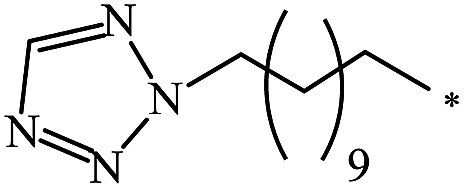 |
 |
||
| 135 |  |
C2H5 | 14 | 81 |
| 136 |  |
C2H5 | 21 | |
| 137 | 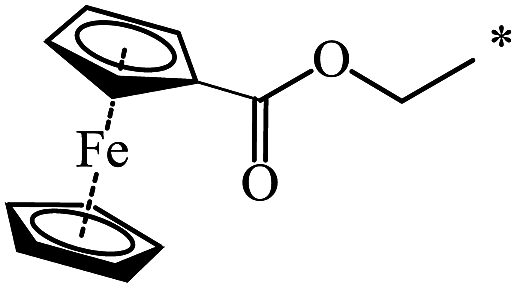 |
 |
45 | 82 |
| 138 | (CH2)3–Br | C18H37 | 70 | 83 |
| 139 | (CH2)12–N3 | C18H37 | 11 | |
 | ||
| Scheme 38 | ||
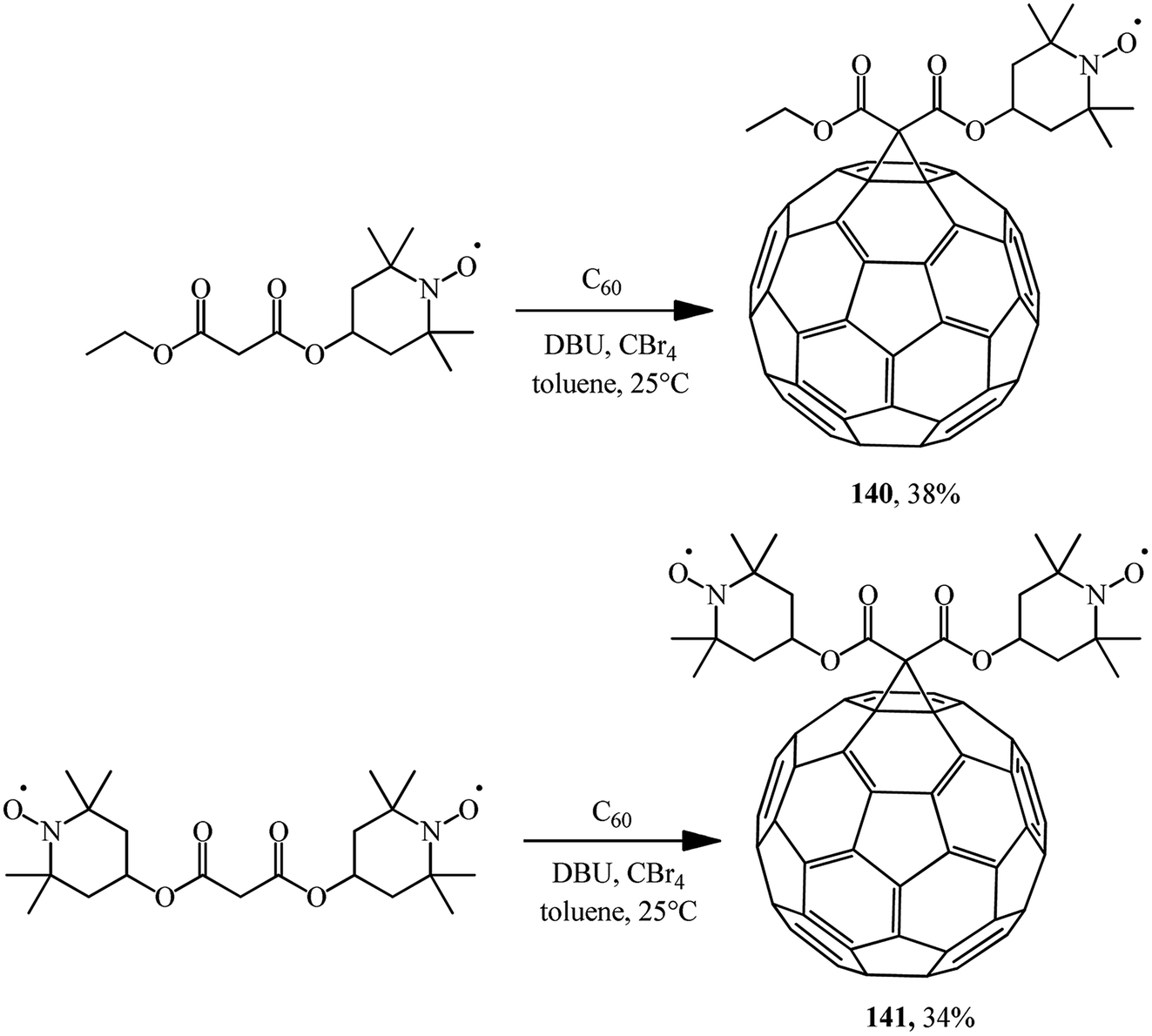
Using the Bingel–Hirsch technique, methanofullerenes 140 and 141 were obtained using malonates with nitroxide-containing radicals (Scheme 39).84 It has been found for the first time that these compounds combined with the known anticancer drug cyclophosphamide show high antitumor activity against P-388 leukemia.
 | ||
| Scheme 39 | ||
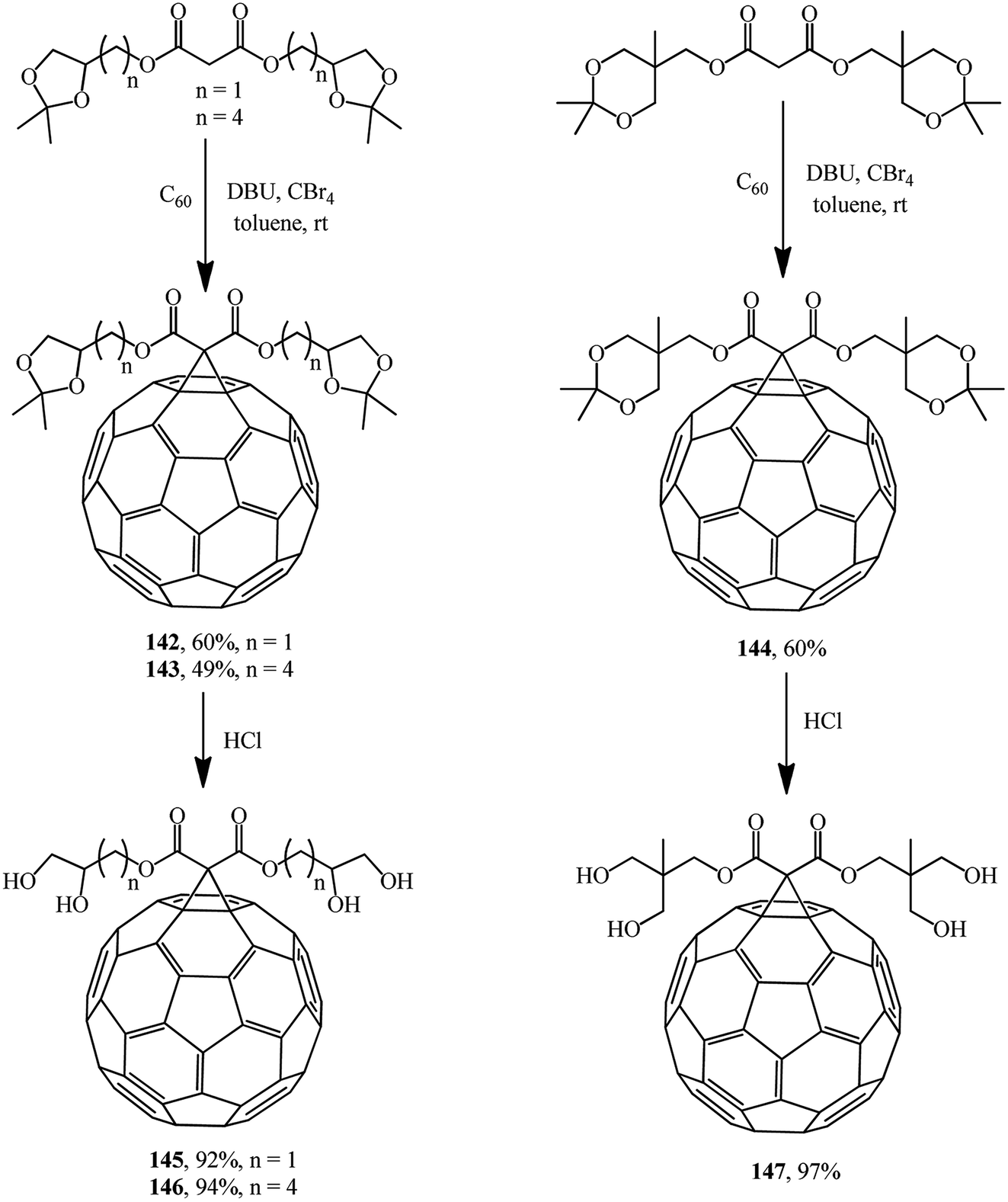
The same researchers isolated and characterized monofunctionalized methanofullerenes differing in the number of acetonide groups, for example, compounds 142–144.85 Removal of acetonide protection results in new chromatographically pure water-soluble polyol methanofullerenes 145–147 in nearly quantitative yields (Scheme 40).
 | ||
| Scheme 40 | ||
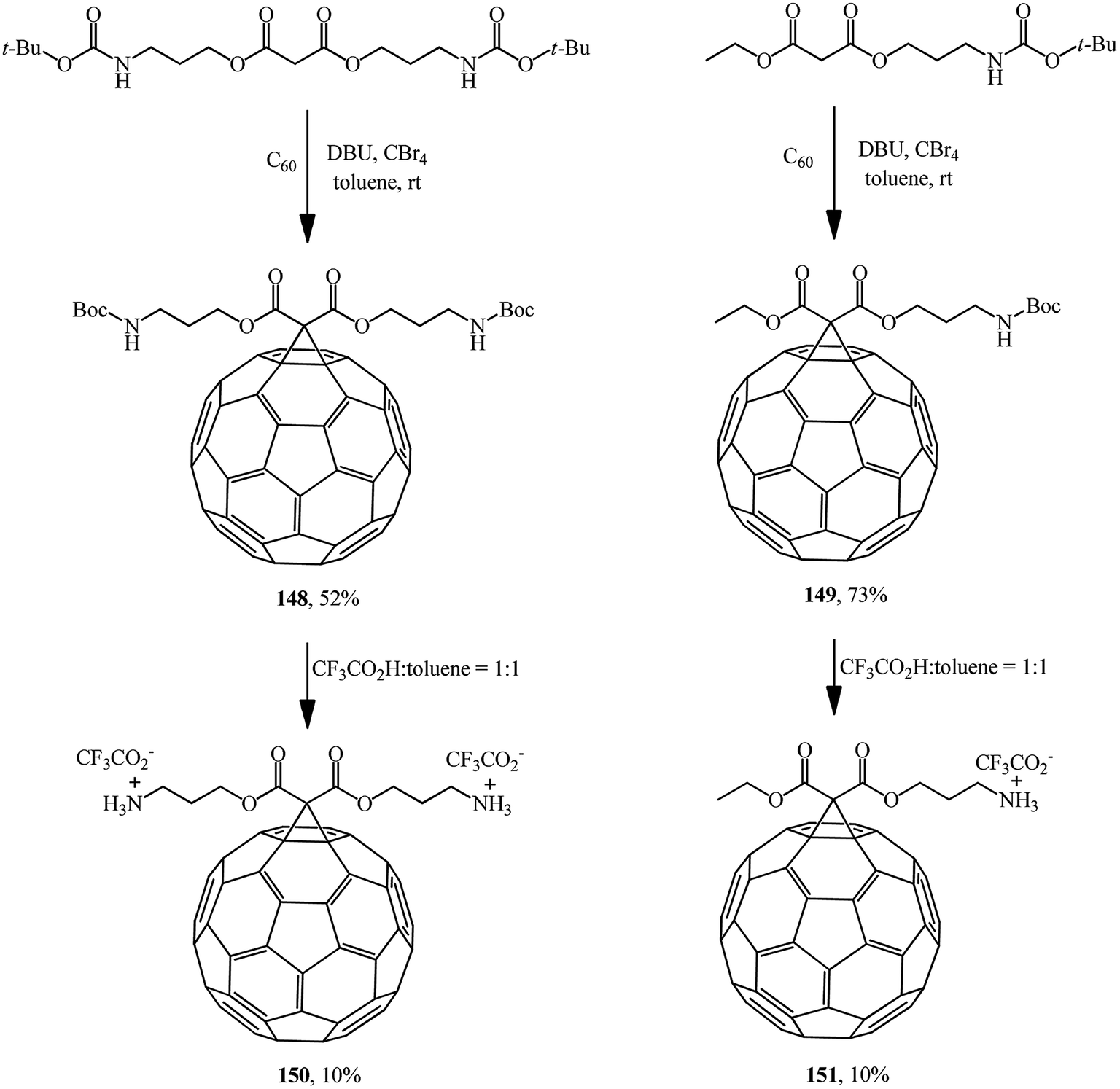
Mono- and diamino-functionalized methanofullerenes 148 and 149 were obtained by the Bingel–Hirsch reaction using N-protected malonic derivatives synthesized by the reaction of tert-butyl N-(3-hydroxypropyl)carbamate with malonyl chloride in the presence of pyridine (Scheme 41).86 Removal of tert-Boc-protected amine groups was performed using trifluoroacetic acid; the methanoaminofullerenes 150 and 151 that were isolated exhibited good solubility in water.
 | ||
| Scheme 41 | ||
Studies on the synthesis of methanofullerenes with practical purposes, primarily for medical purposes, attract considerable attention. For instance, monoadducts 150 and diamine adducts 151 (ref. 86) as well as adduct 152 (ref. 87) (Fig. 6) selectively inactivate neuronal isoforms of nitric oxide synthase by splitting reactive oxygen intermediates from NO and formation of citrulline in the presence of arginine.
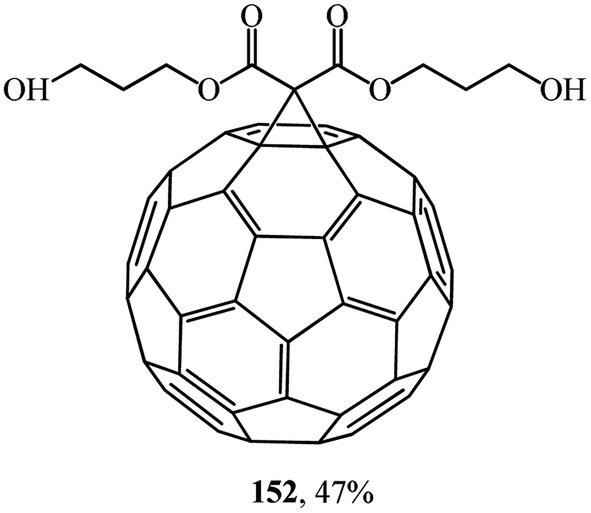 | ||
| Fig. 6 Di(carboxypropan-3-ol)methanofullerene 152. | ||
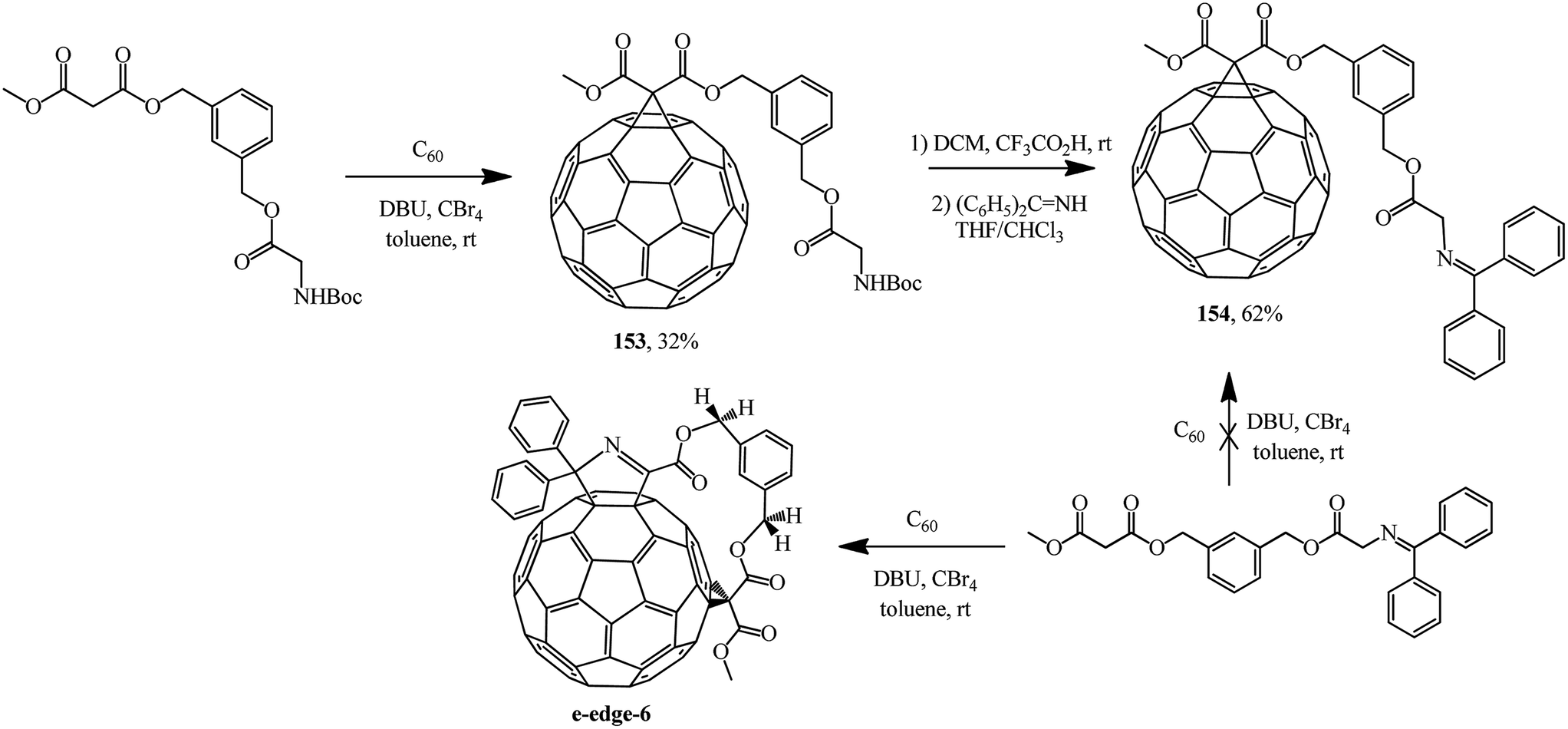
Treatment of spacer-bound derivative of 1,3-bis-(hydroxymethylbenzene)diester with methyl malonate and Boc-ester of glycine with fullerene C60 gave the expected adduct 153.88 Based on the latter, removal of the tert-butyloxycarbonyl protecting group (Boc) with 4-(dicyanomethylene)-2-methyl-6-(4-dimethylaminostyryl)-4H-pyran (DCM) and treatment with benzophenone imine gave block 154 containing an N-(diphenylmethylene)glycine moiety (Scheme 42). Under Bingel–Hirsch conditions, this moiety can act as a cyclopropanating “element”, as shown by the example of a regioselective synthesis of ansa-type e-edge-6 with participation of an acyclic compound with a simpler structure.
 | ||
| Scheme 42 | ||
Due to the availability of malonates with very diverse structures, the Bingel–Hirsch reaction is widely used for the functionalization of the fullerene sphere in targeted syntheses of new compounds with useful properties.
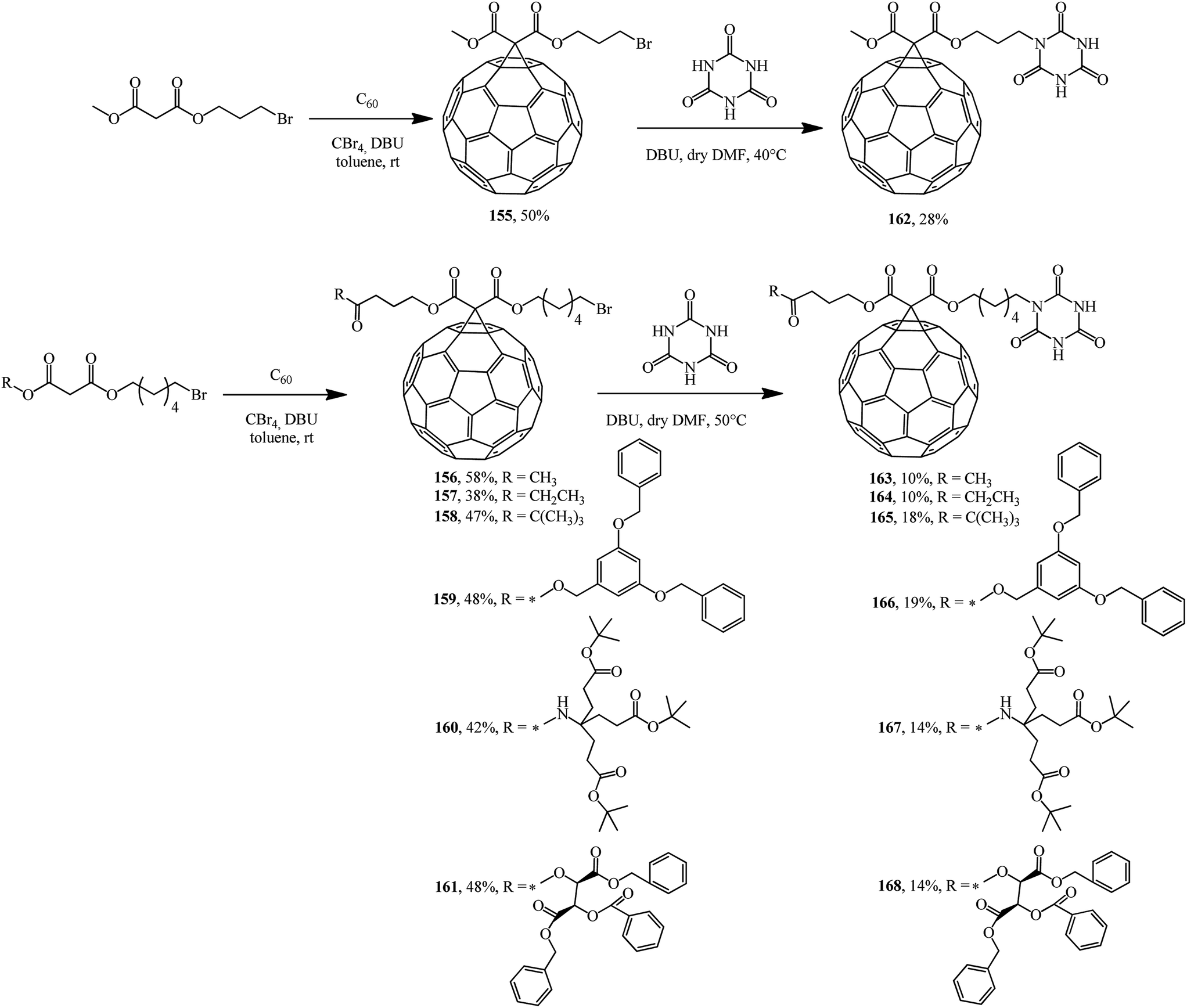
In fact, a series of terminal bromoalkyl fullerene adducts 155–161 were synthesized. Treatment of these adducts with cyanuric acid gave substitution products 162–168 (Scheme 43).89
 | ||
| Scheme 43 | ||
The adducts that were isolated were used in self-assembly and in studies of the photophysical properties of supramolecular donor–acceptor fullerenoporphyrin nanohybrids bound by a hydrogen bond based on a Hamilton receptor.90
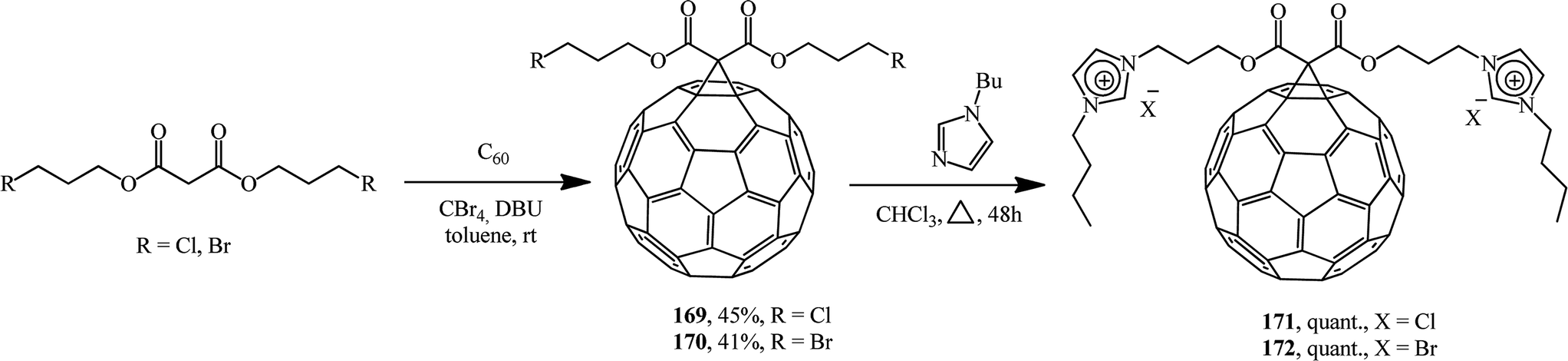
Methanofullerenes can also be obtained using halogenated malonic esters 169 and 170.91 Reactions of the compounds obtained with 1-butylimidazole convert the former into a series of “fullerene-ionic liquid” hybrids 171 and 172 (Scheme 44). Combinations of C60 and ionic liquids give rise to new unique properties of the conjugates such as solubility in water, which was higher than 800 mg mL; moreover, the hybrids were employed for the immobilization of palladium nanoparticles through ion exchange followed by reduction with sodium borohydrie.
 | ||
| Scheme 44 | ||
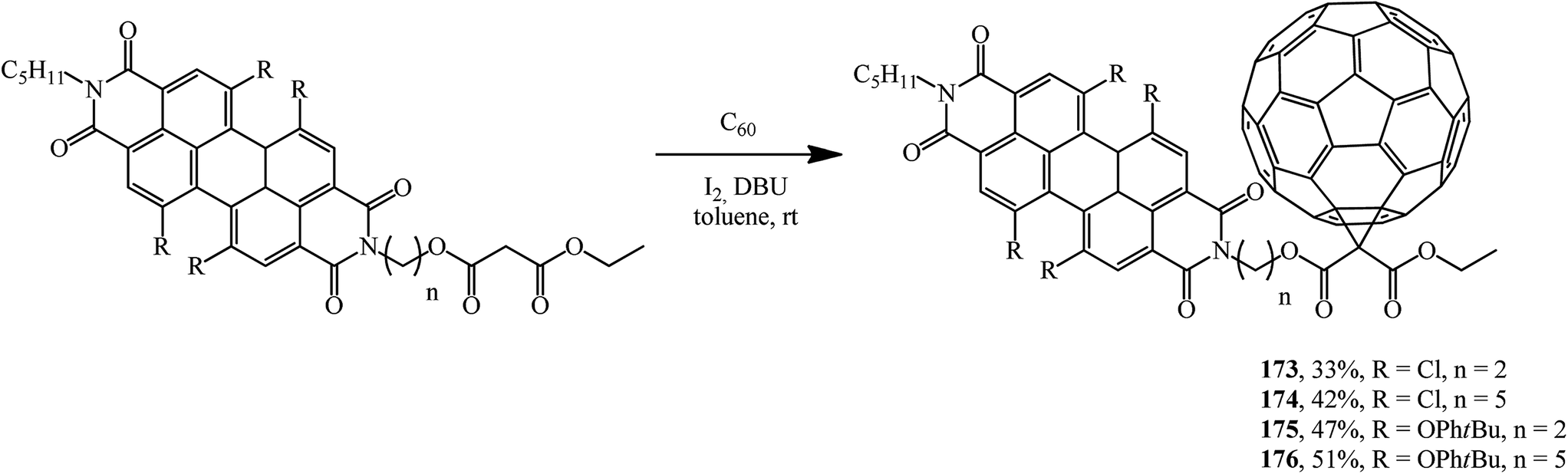
Fullerene conjugates are of considerable interest for practical applications. For example, in order to improve the energy transfer from the dye as a photoactive antenna to the fullerene part in photovoltaic systems, a group of fullerene–perylenediimide diads 173–176 was obtained from the corresponding N-malonyl perylenediimides and characterized (Scheme 45). Their electrochemical and photophysical properties were studied.92–94
 | ||
| Scheme 45 | ||
It was demonstrated using steady-state and time-resolved spectroscopy that quantitative photoinduced energy transfer occurs from the perylenediimide moiety, which acts as a light-harvesting antenna, to the C60 unit playing the role of an energy acceptor.94 At the authors' suggestion, the fullerene was intentionally attached at the bay region of the perylene diimide to achieve close proximity of the two chromophores and to ensure efficient photoinduced electron transfer95 (Fig. 7).
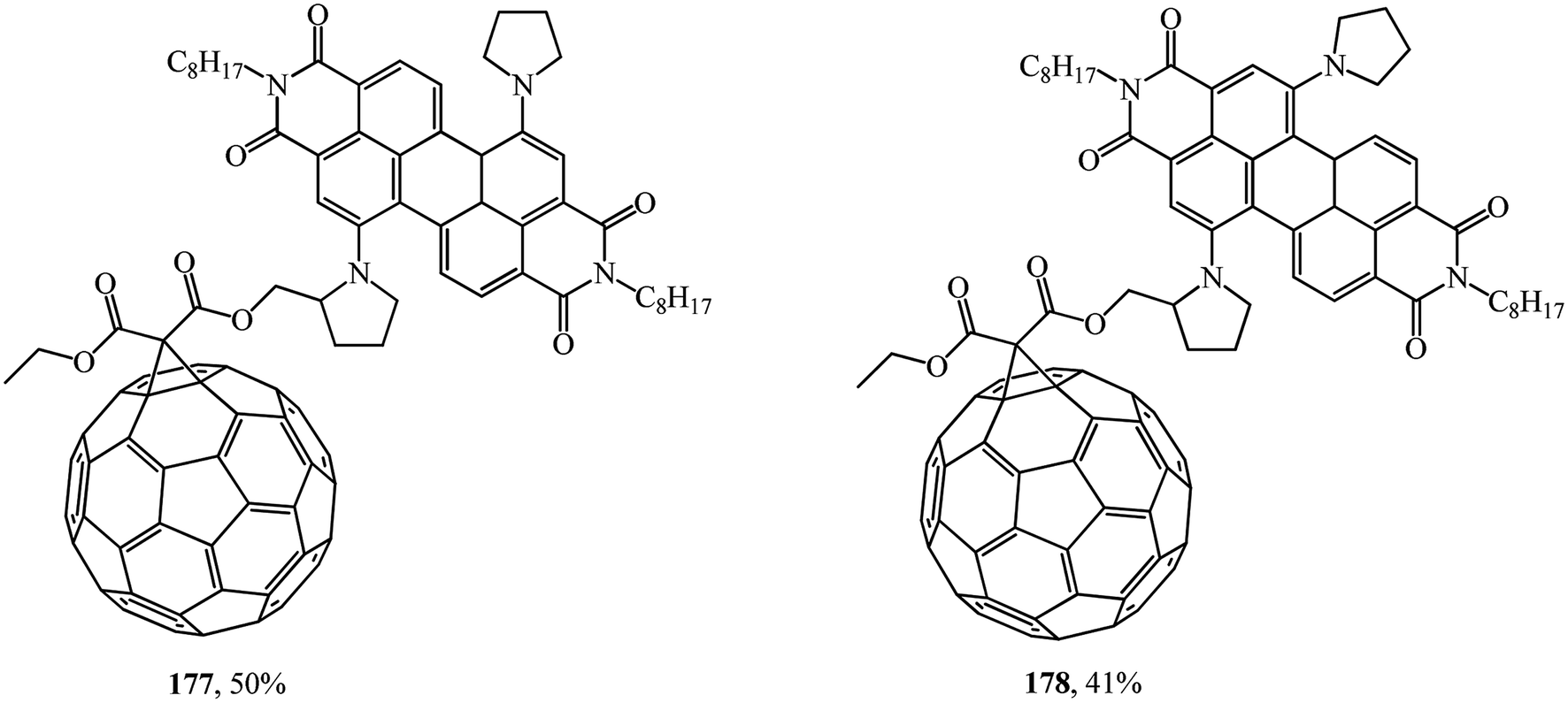 | ||
| Fig. 7 Fullerene–perylenediimide diads 177 and 178. | ||
Fullerene C60 diads can also be formed with 4-amino-1,8-naphthalimide chromophores (Fig. 8). A study of the fluorescent properties of the monoadducts obtained identified the role of naphthalimide chromophore with charge transfer as a light absorbing antenna and an excited fullerene sensitizer. According to ref. 96, conjugates 179 and 180 are of great practical importance.
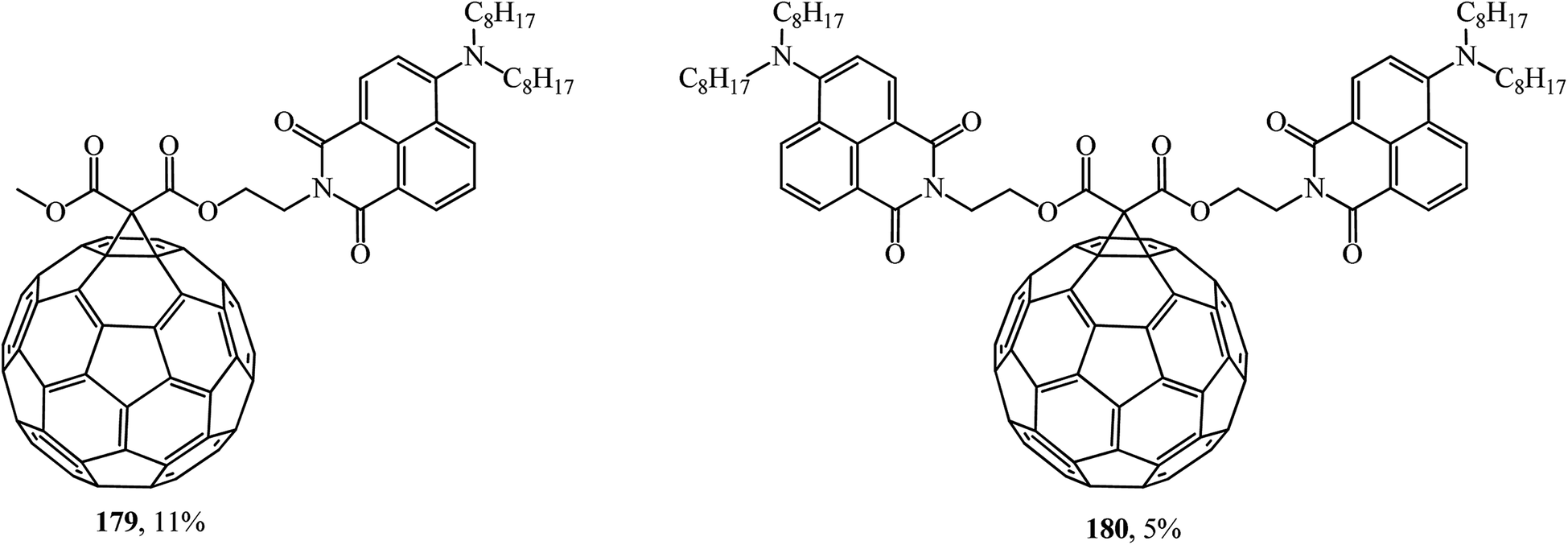 | ||
| Fig. 8 Fullerene–naphthalimide diads 179 and 180. | ||
Fullerene-containing derivatives carrying alkyne functional groups are attractive in terms of various transformations. A considerable number of studies deal with the synthesis of fullerene-containing malonates with terminal acetylene bonds which then easily react with azides by the “click-chemistry” mechanism to give the corresponding triazole derivatives.
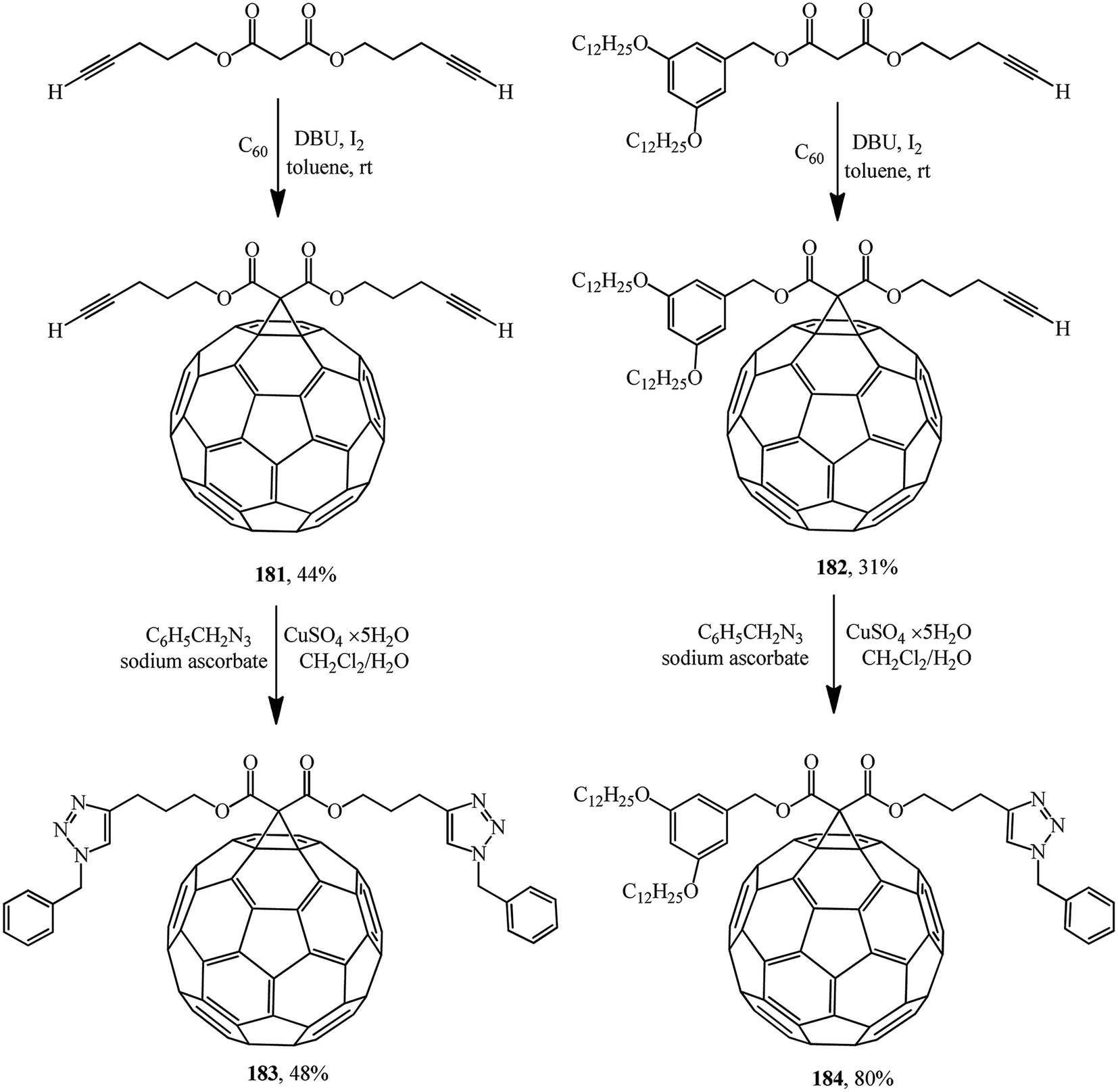
One of the variants was implemented in ref. 97 where, for example, compounds 181 or 182 were synthesized and used as building blocks under copper-mediated Huisgen 1,3-dipolar cycloaddition conditions to afford 1,2,3-triazole derivatives 183 or 184, respectively (Scheme 46).
 | ||
| Scheme 46 | ||
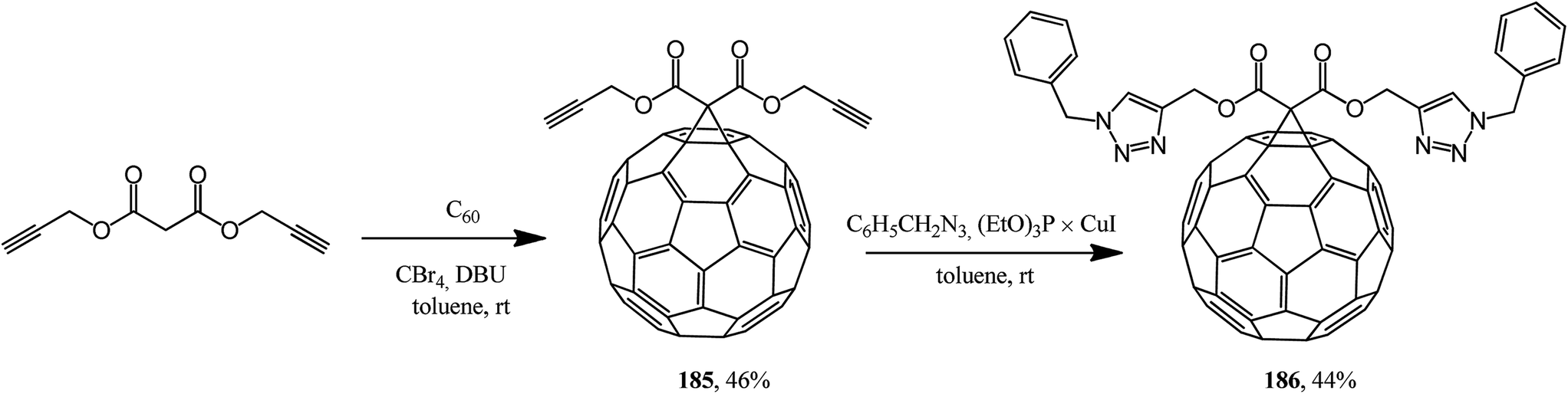
This approach was implemented in ref. 98 where the originally isolated symmetric 61-bis(propargyloxycarbonyl)methano[60]fullerene 185 was converted according to Huisgen into adduct 186 (Scheme 47).
 | ||
| Scheme 47 | ||
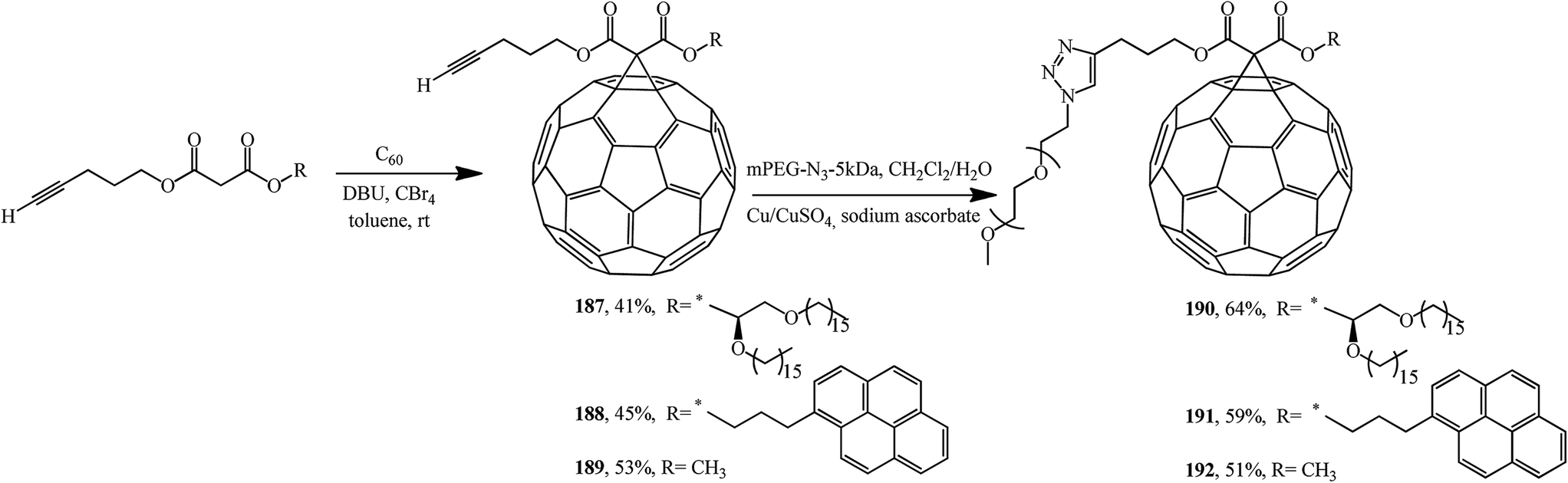
Using malonates containing a terminal-acetylenic bond, the corresponding fullerene-containing adducts 187–189 were synthesized (Scheme 48).99 Treatment of the latter with excess methoxypoly(ethylene glycol)azide in the presence of copper shavings and copper(II) sulfate gave triazoles 190–192 in the yields indicated in Scheme 48.
 | ||
| Scheme 48 | ||
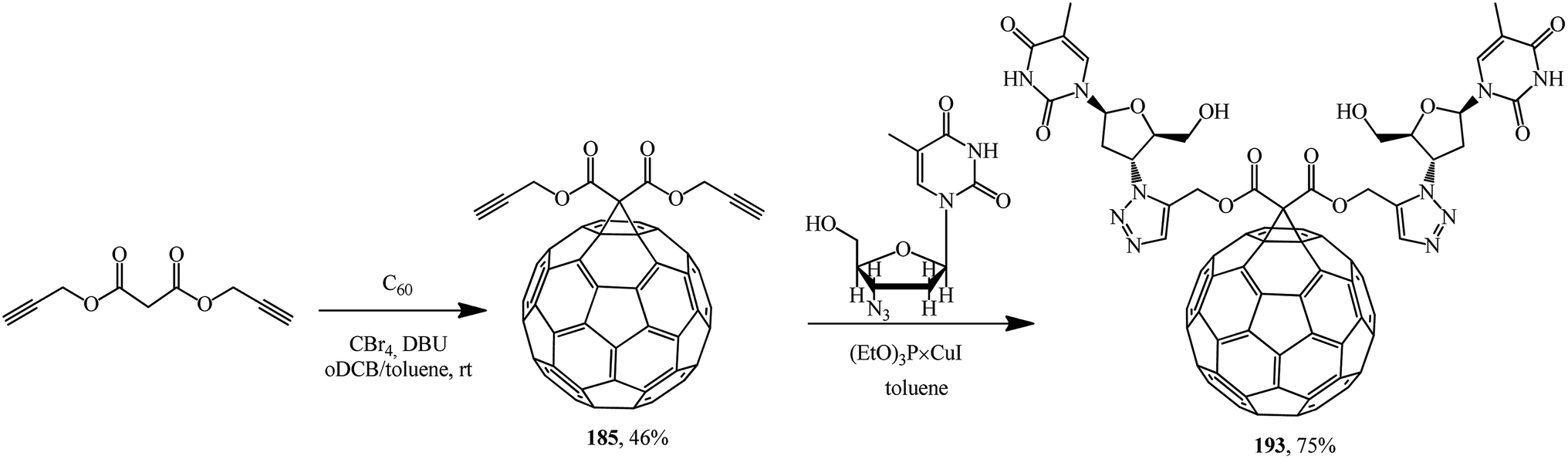
A methanofullerene containing triazole and deoxythymidine moieties that is promising in terms of biological activity was described in ref. 100 For this purpose, diacetylenic precursor 185 was synthesized, which in the presence of catalytic amounts of (EtO)3P × CuI reacted with azidodeoxythymidine to give the desired compound 193 (Scheme 49).
 | ||
| Scheme 49 | ||
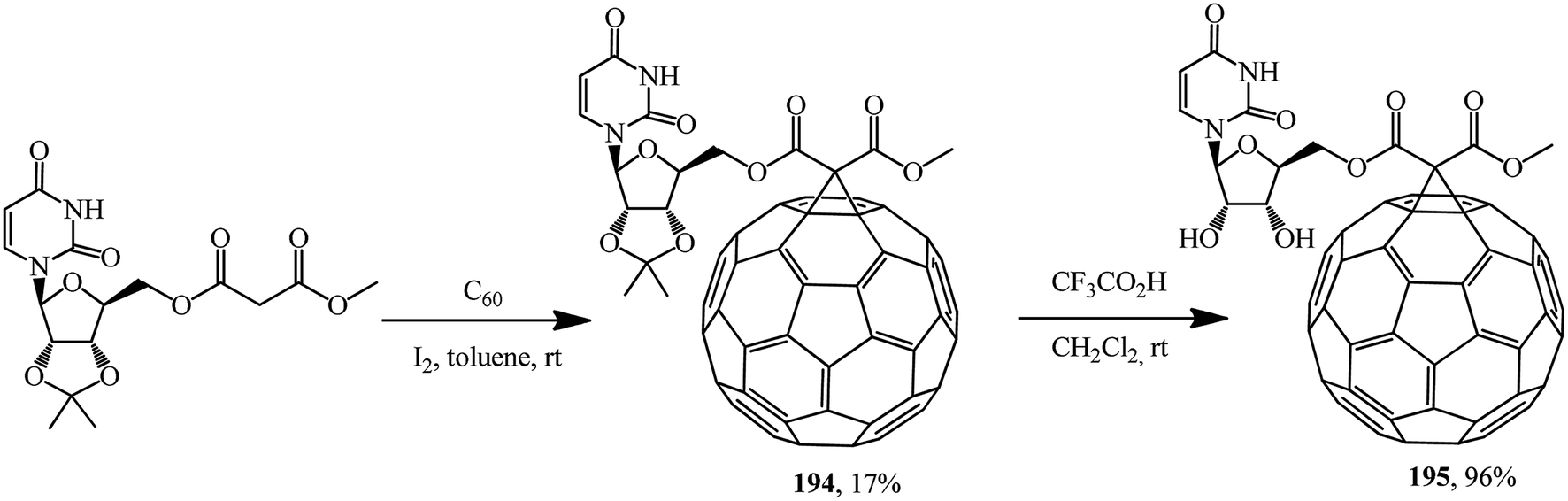
Based on methyl uridinemalonate synthesized, cyclopropanation first gave a fullerene conjugate with uridine 194, deblocking of which gave its analogue 195 in an almost quantitative yield (Scheme 50).101
 | ||
| Scheme 50 | ||
Using the Bingel–Hirsch reaction, the class of methanofullerenes was expanded with adducts containing porphyrin radicals. Numerous publications dealing with the synthesis of porphyrin–fullerene diads already exist, where the subject of the mutual effect of the C60 sphere and porphyrin moiety is also covered. The keen interest in these extremely important aromatic heterocyclic systems underlying natural compounds, such as hemoglobin, chlorophyll and vitamin B12, is quite understandable. This is particularly true for combinations with fullerene where extraordinary results may be expected.
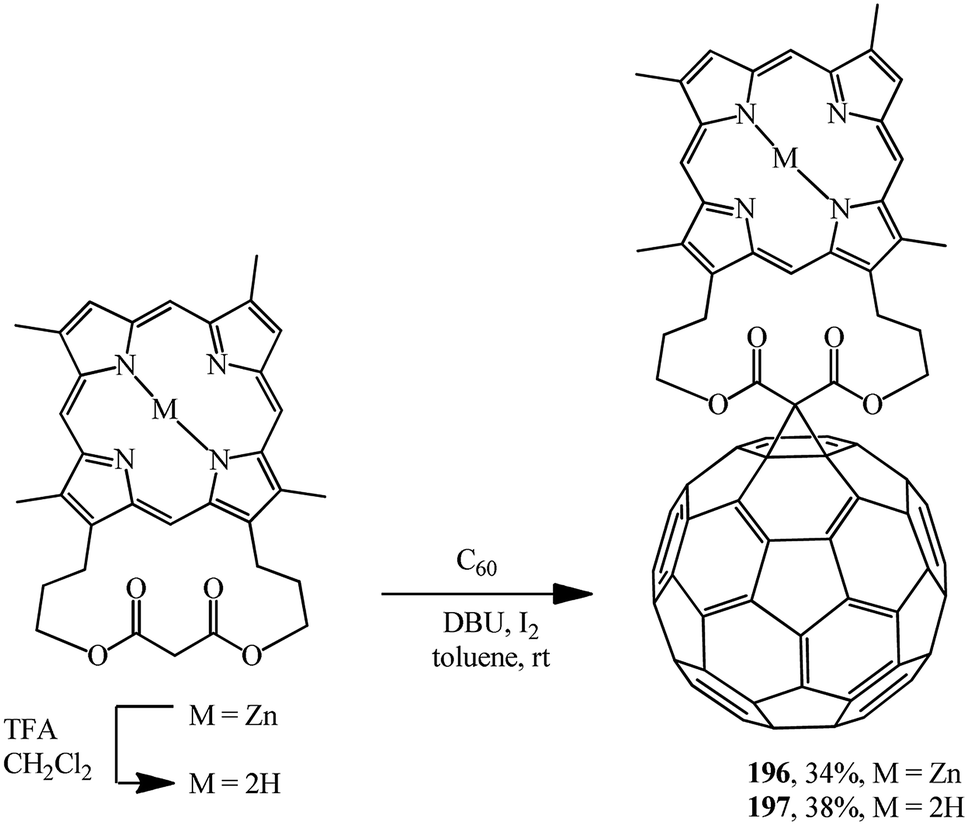
For example, in ref. 102 and 103 the original [3,7,12,17-tetramethyl-2,18-(propanoxypropanoxypropano)-21H,23H-porphine-29,31-dionato-(2-)]-zinc was demetallated with trifluoroacetic acid and these compounds were converted into the corresponding “porphyrin–fullerene” diads 196 and 197 under Bingel–Hirsch reaction conditions (Scheme 51).
 | ||
| Scheme 51 | ||
Porphyrin–fullerene diads 196 and 197 bound with a bislactone show promising photophysical properties in photosynthesis simulation.
Using a similar route, covalent-bound porphyrin–fullerene diads with “parachute” topology 198,104 199 and 200 (ref. 105) were obtained (Scheme 52).
 | ||
| Scheme 52 | ||
In all cases, luminescence quenching was observed upon formation of porphyrin–fullerene diads, which is characteristic of the original porphyrin derivatives.
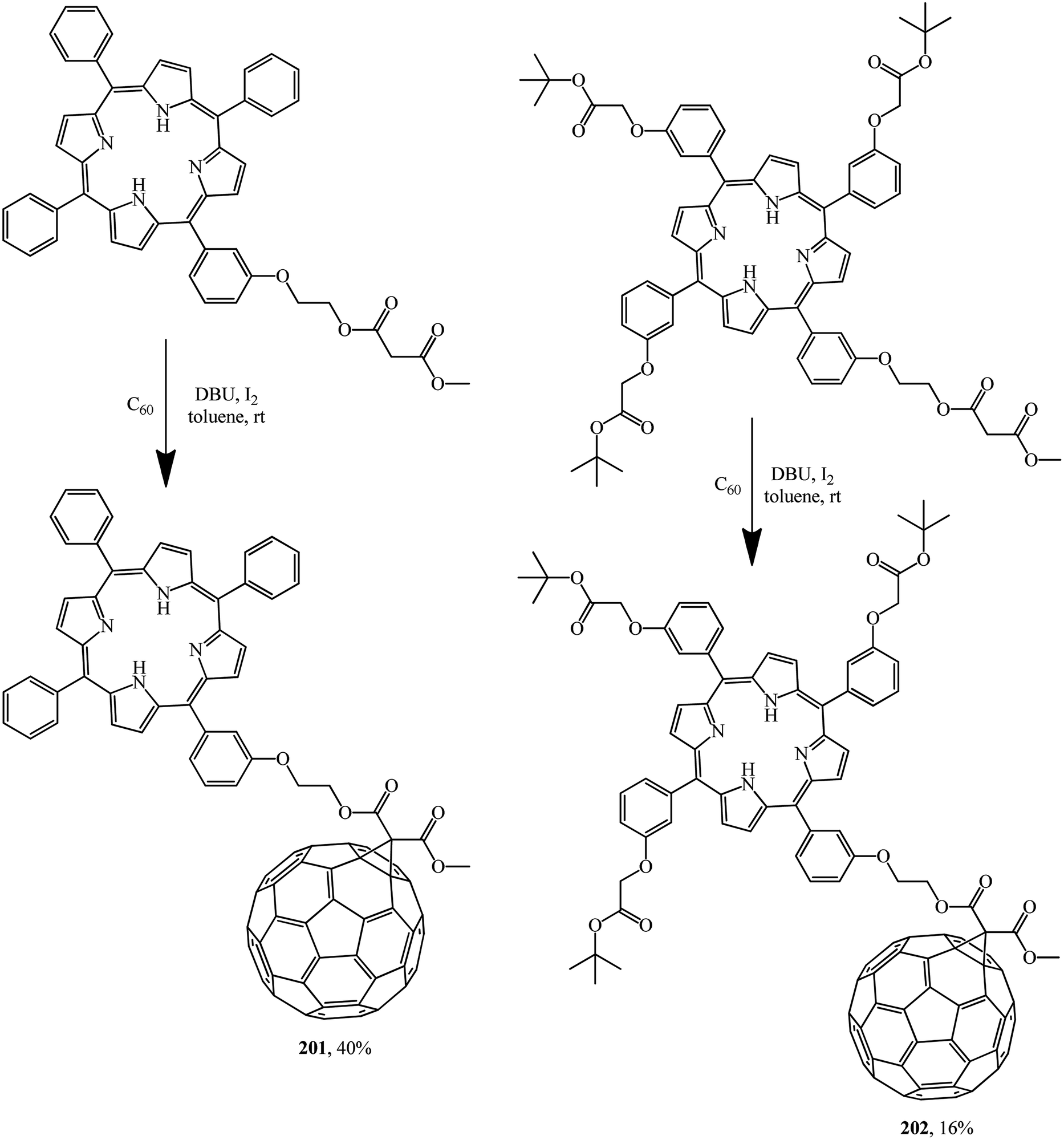
A detailed electrochemical and photophysical study revealed that the porphyrin–fullerene diads 201 and 202 synthesized allow storing a nearly 1.7 eV radical ion pair state with a lifetime of one nanosecond. In this context, the flexible linkage, powering a through space charge transfer, prevents, however, stabilization of the radical ion pair state beyond nanoseconds (Scheme 53).82
 | ||
| Scheme 53 | ||
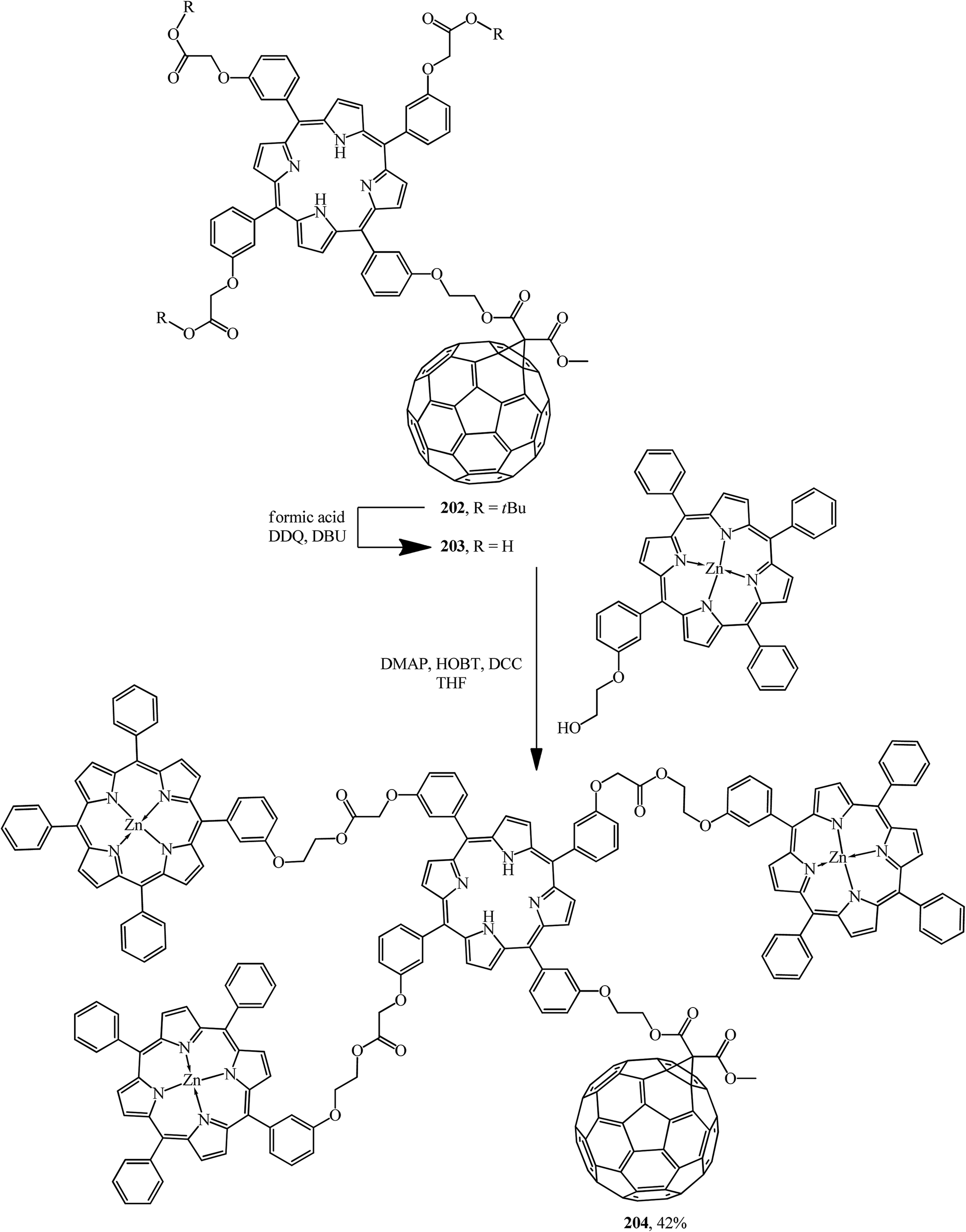
Light harvesting, unidirectional energy transfer, charge transfer, and charge-shift reactions are attainable due to dendritic architectures bound to C60, which gives rise to the successful mimicry of the primary events in photosynthesis. Therefore, as a continuation of studies,106 compound 202 synthesized was quantitatively converted by hydration of its ester moiety to the corresponding triacid 203. Addition of excess porphyrin to the latter in the presence of 4-dimethylaminopyridine (DMAP), 1-hydroxybenzotriazole hydrate (HOBT) and N,N′-dicyclohexylcarbodiimide (DCC) in THF gave dendritic porphyrin–fullerene conjugate 204 (Scheme 54).
 | ||
| Scheme 54 | ||
The interest in incorporation of donor porphyrin moieties into the C60 molecule is due to the search for covalent-bound diads as models of natural electron transfer centers and as potential photochemical molecular devices. Two methods are used to achieve favorable steric conditions favoring the electronic transfers in these diads. The first one involves separating the donor and fullerene moieties by flexible bonds, while the second one involves “face-to-face” aligning the porphyrin plane directly to the C60 sphere. Upon photoexcitation, long-lived states with separated charges are formed in this type of covalent-bound diads. In fact, arranging four bulky chromophore porphyrin groups at C60 periphery enhances the light collection function and the light conversion of the resulting hybrid 204. The authors showed that the latter can demonstrate two main functions of photosynthesis: light assembly and charge transfer; as a result, this approach results in highly efficient solar energy conversion.
Methanofullerenocarboxylates are also considered as candidates in the creation of rotaxanes, non-covalent bound self-organizing Sauvage-type systems that are promising for creating photochemical molecular devices. In these systems, electro- and photoactive components are incorporated into the linear part of the system or are as if threaded on the latter. Electron or energy transfers occur in these systems upon external impact. The presence of charged moieties in the molecules does not change the electrophilicity of rotaxane spheres in comparison with that of the non-functionalized methanofullerene.
Using standard techniques, macrocycles were developed onto which fullerene was then grafted. For example, fullerene-based compound 205 was obtained from a macrocyclic precursor by the Bingel–Hirsch reaction. Complexation of 205 with a molecule containing the second phenanthroline group with zinc porphyrin on one end is then performed in the presence of Cu(I) in order to isolate tetrahedral complex 206. Addition of a porphyrin carboxylic acid to the other end of complex 206 gives the target rotaxane 207 (Scheme 55).107,108
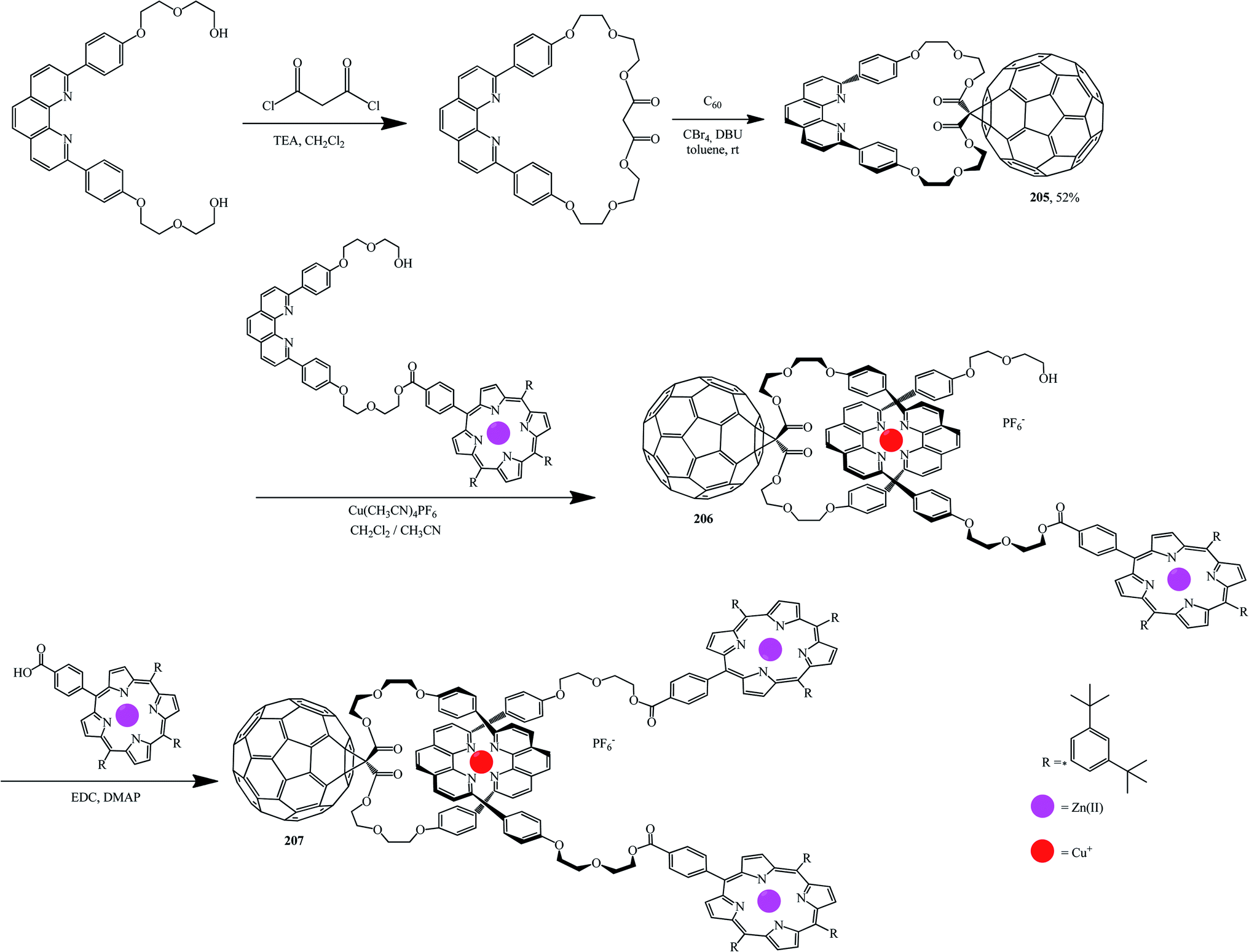 | ||
| Scheme 55 | ||
Titration of porphyrin–fullerene rotaxanes with 1,4-diazabicyclo-[2.2.2]octane (DABCO) or 4,4′-bipyridine led to photo- and redoxactive catenanic architectures M and N, which upon photoexcitation undergo a sequence of short-range energy and electron transfer events to give a long-lived charge-separated radical-pair state (Fig. 9).108
 | ||
| Fig. 9 Formation of 1:1 sandwich complexes. | ||
Using the methodology reported in ref. 108 and 109 a series of porphyrinorotaxanes included in the structure and containing fullerene stoppers109 were synthesized by a convergent route. First, a macrocyclic structure with a 1,10-phenanthroline moiety attached to the porphyrin part was formed. This process was accompanied by the formation of a tetrahedral Cu(I) complex with a linear phenanthroline group and hydroxyls at both ends (Scheme 56). At the final stage, esterification of this diol with fullerene–carboxylic acid 208 at room temperature gave rotaxane 209.
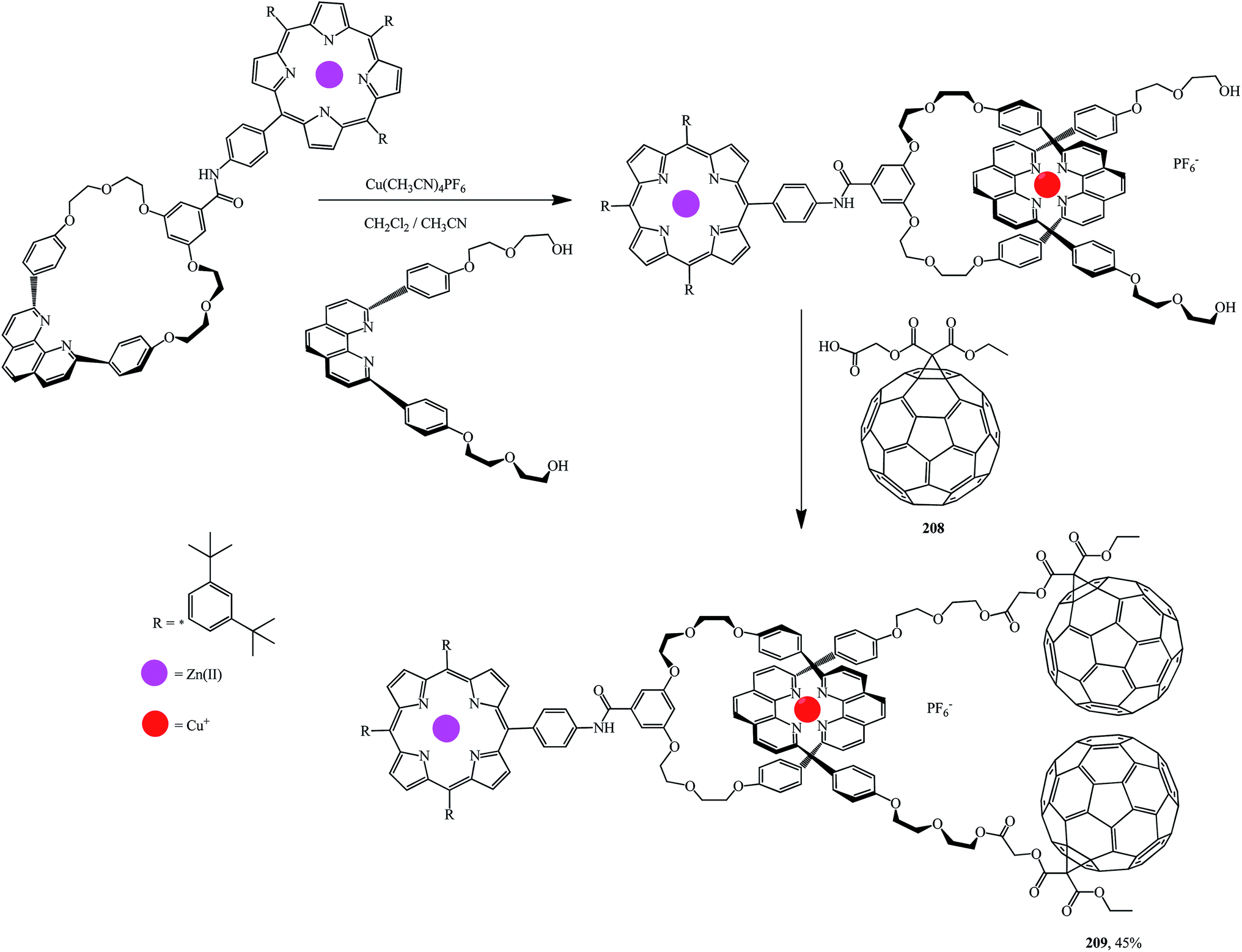 | ||
| Scheme 56 | ||
Fullerene–porphyrin rotaxanes 210 and 211 were synthesized in a similar way (Fig. 10).
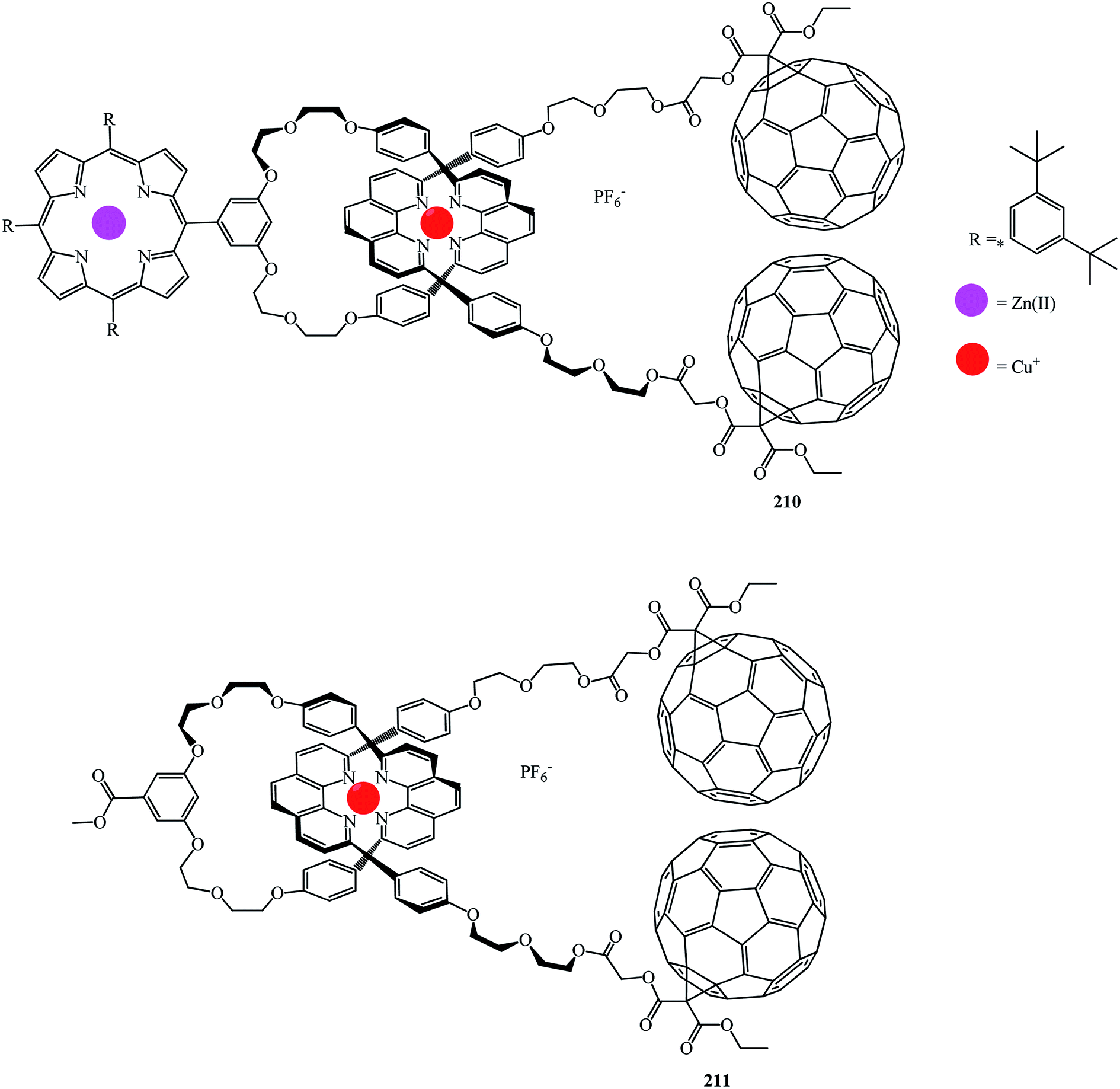 | ||
| Fig. 10 Fullerene–porphyrin rotaxanes 210 and 211. | ||
A synthesis of the first [2]catenaes with porphyrin and fullerene subunits 212 and 213, which combined the advantages of both “click-chemistry” and Sauvage's methods, was performed in yields that are very good for Bingel's reaction (Fig. 11).110 This new electron donor–acceptor catenane family was extensively studied by spectroscopic, computational, electrochemical and photophysical methods.
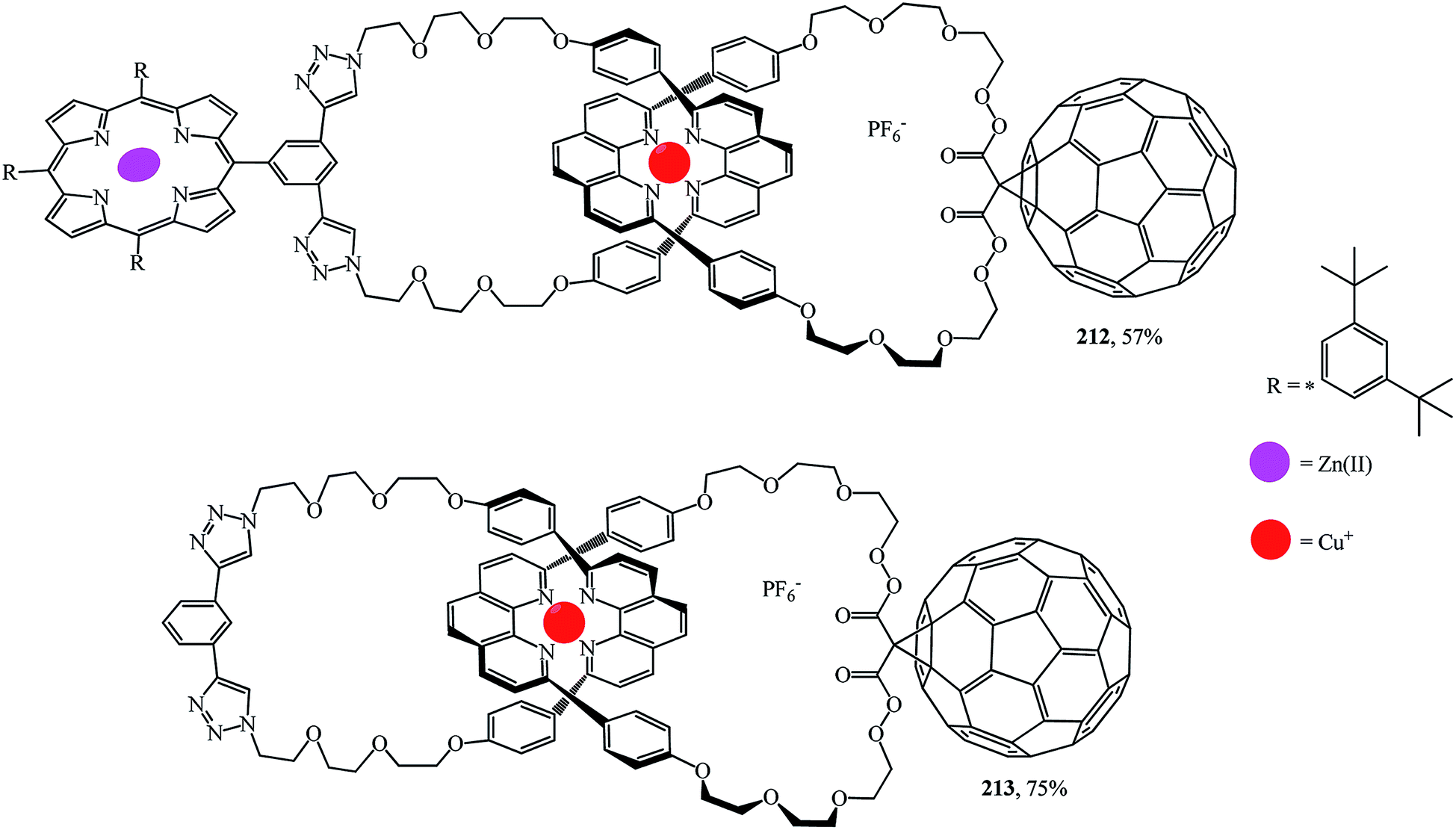 | ||
| Fig. 11 Porphyrin–C60-[2]catenanes 212 and 213. | ||
Thus, the use of C60 as an electron acceptor in catenanes simultaneously ensures the transfer of electrons over long distances and reduces the rate of reverse electron transfer. It has been shown110 that supramolecular methods can be used to create high-complexity 3D nanostructures containing electron donors and acceptors that generate long-lived charged separated states upon photoexcitation. The use of these materials in light energy conversion systems, in photovoltaic devices in particular, will be the subject of intense fundamental studies in the near future.
Fullerene–crown ether conjugates have been the subject of several investigations in view of possible supramolecular modulation of the electronic properties of fullerenes.111–115
One of the first references to monofunctionalized fullerene–crown ether conjugates obtained by the Bingel–Hirsch method can be found in ref. 116. This paper reported a synthesis of a C60 adduct with dibenzo-24-crown-8 macrocyclic polyether (DB24C8) 214 (Scheme 57).
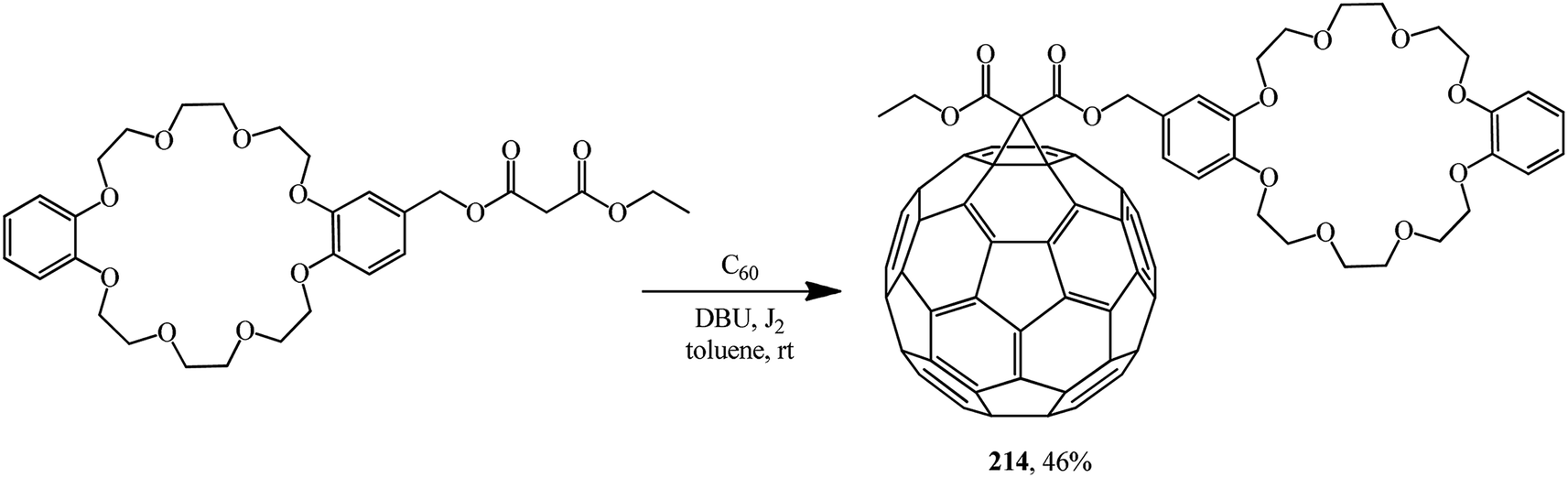 | ||
| Scheme 57 | ||
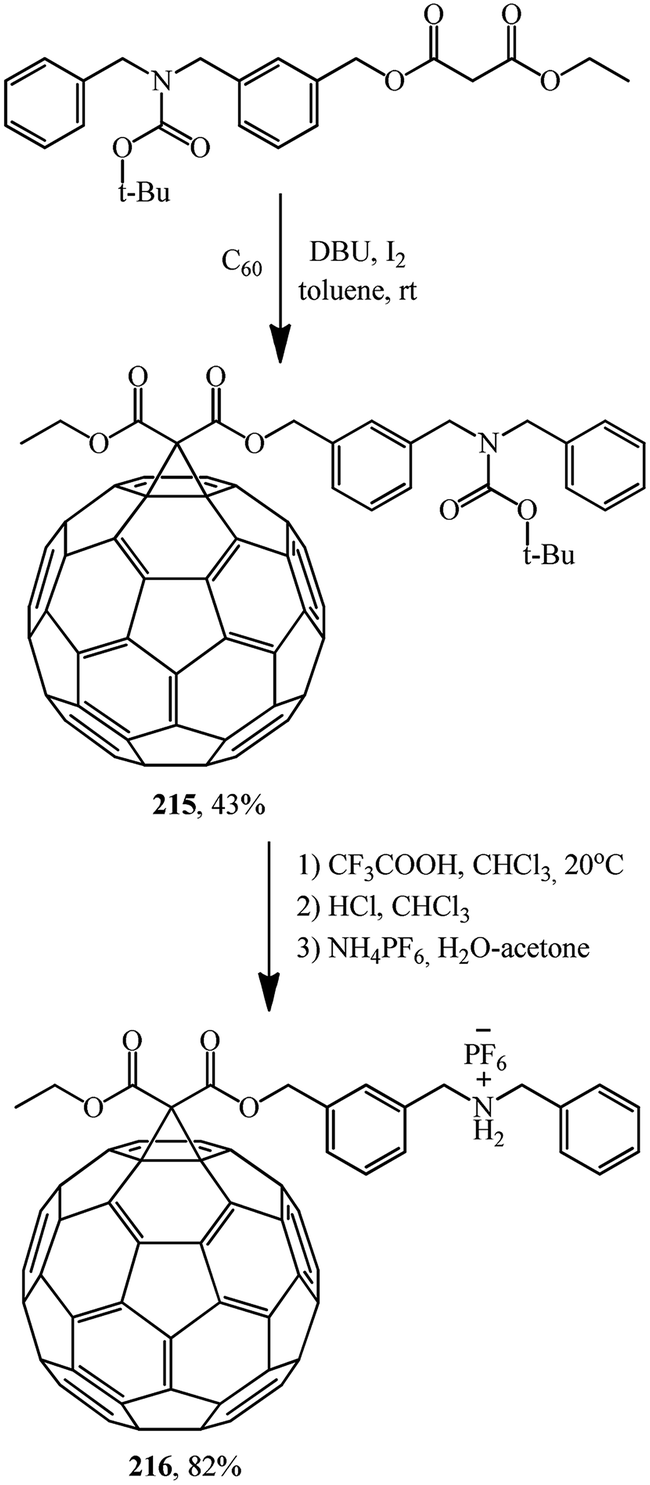
In addition, article116 reports a synthesis, via intermediate 215, of an adduct of dibenzylammonium salt 216 covalent-bound to carbon allotrope C60 (Scheme 58).
 | ||
| Scheme 58 | ||
Conjugate 214 forms a stable, pseudorotaxane-like 1:1 complex with dibenzylammonium hexafluorophosphate. Equally, the C60 dibenzylammonium conjugate 216 threads through the cavity of DB24C8 to form a 1:1 complex with a pseudorotaxane-like geometry. Furthermore, the C60-DB24C8 adduct 214 and the C60 dibenzylammonium conjugate 215 interact with each other by hydrogen bonding and ion-dipole interactions to form the first supramolecular C60 dimer 217 (Fig. 12). In all the three cases, the dibenzylammonium component is threaded through the cavity of the crown ether macrocycle.
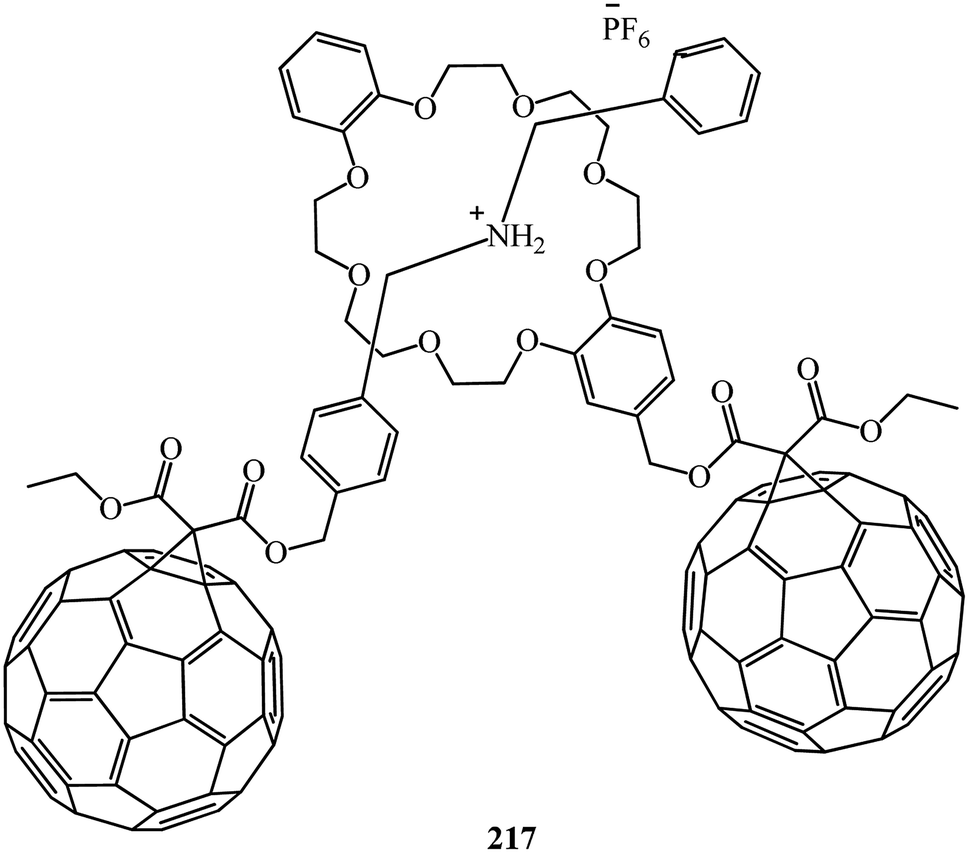 | ||
| Fig. 12 Supramolecular C60 dimer 217. | ||
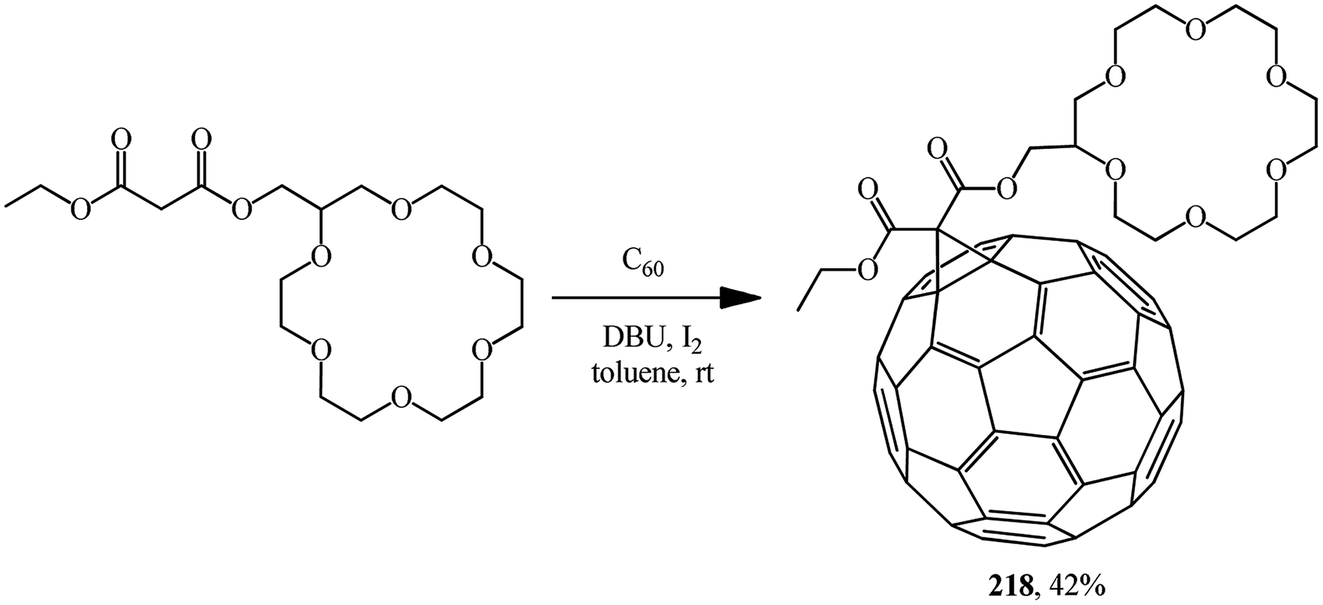
A time resolved EPR spectrum of a radical-triplet pair (RTP) in the quartet excited state was recorded by irradiating a solution of a fullerene–crown ether conjugate 218 (Scheme 59) and an ammonium aminoxyl radical with visible laser light. This is the first direct observation of an RTP in liquid solution for a stable host–guest complex between a macrocyclic ligand bearing a triplet precursor and an aminoxyl ammonium cation.
 | ||
| Scheme 59 | ||
E. Sartori et al.117 performed a comparative analysis of the complexing properties of several methanofullerenes incorporating a macrocyclic crown ether bound to C60. Studies of the complexing properties of the methanofullerenes with respect to ammonium ions with various degrees of substitution made it possible to identify the particularly high complexing ability toward monosubstituted ammonium ions. The first observation of a RTP is reported which occurs upon photoexcitation of a host–guest complex between [60]fullerene–crown ether conjugate 218 and the benzoate ammonium salt of 3-aminomethyl-[2,2,5,5-tetramethylpyrrolidin-1-oxyl] with formation of non-covalent 219. The electrochemical behavior of the compounds obtained and the results of their analysis by ESR method were considered (Scheme 60).
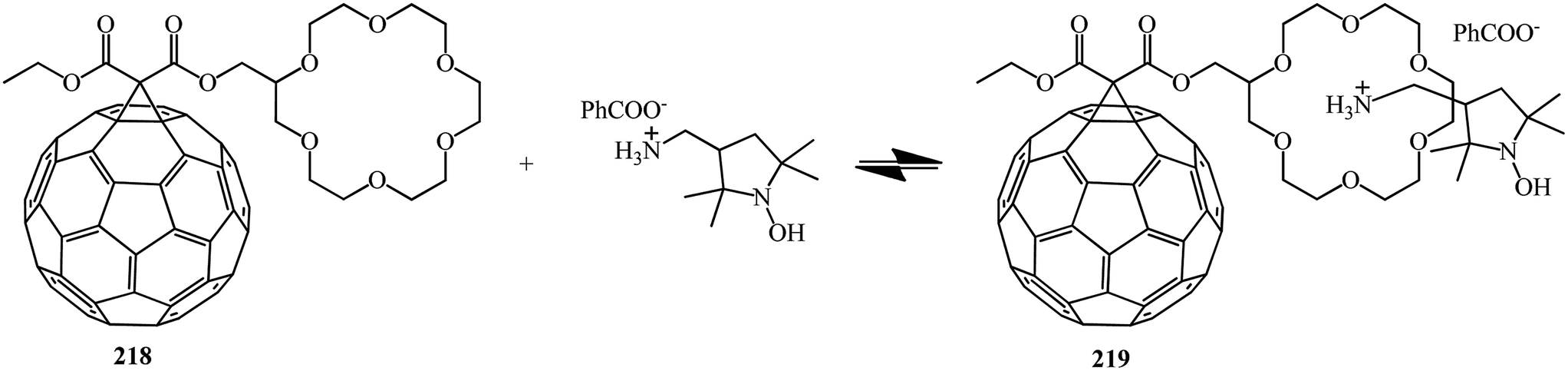 | ||
| Scheme 60 | ||
Methanofullerenes in which the malonate part is a linker between C60 and the macrocyclic crown ether were reported by Pasini. Based on malonate type crown ethers, C60 conjugates with symmetric polyether groups 220–223 (ref. 118) and 224–226 (ref. 119) were synthesized using iodine as the halogenating agent (Scheme 61).
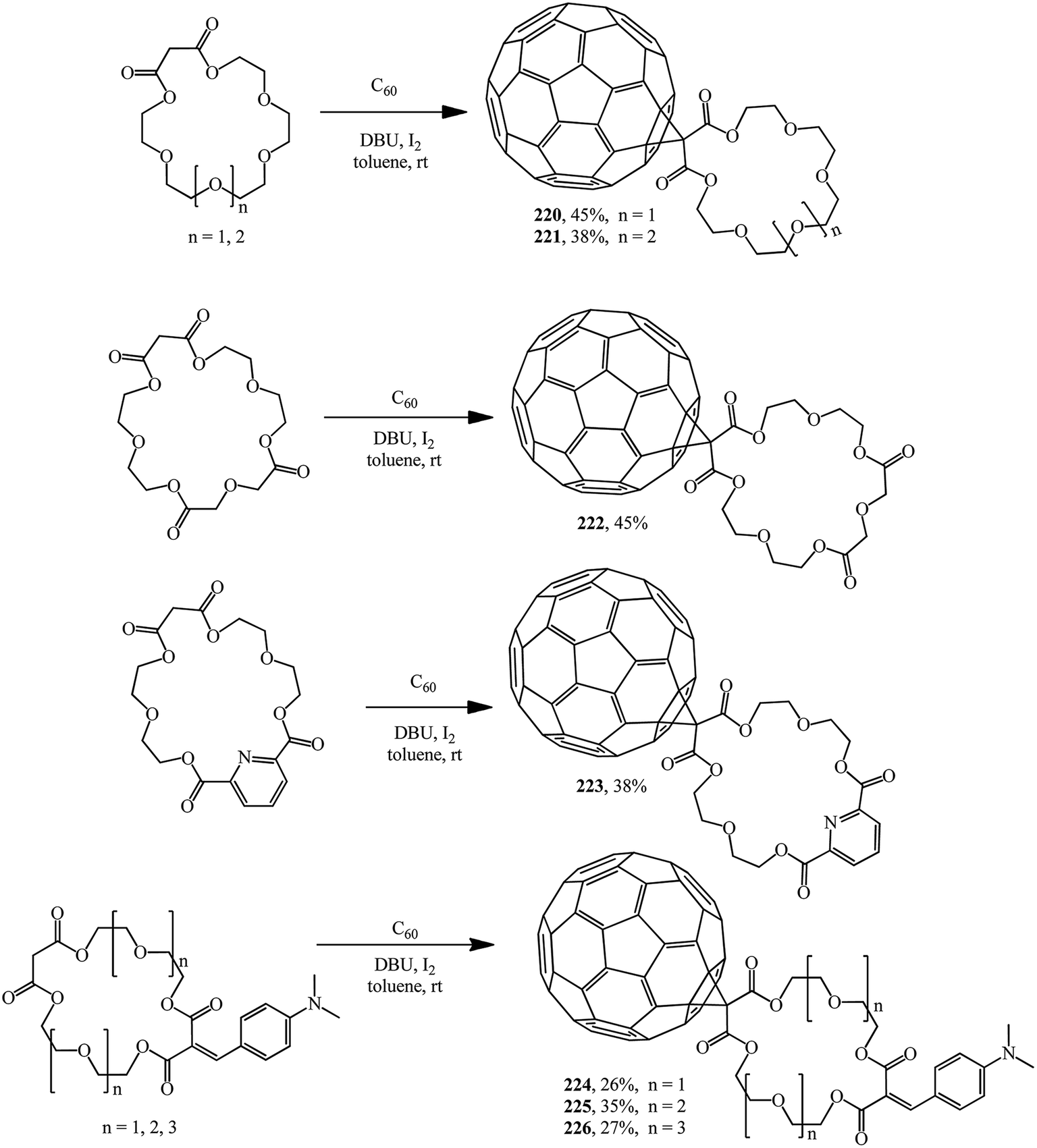 | ||
| Scheme 61 | ||
Based on the readily available adduct 227, a synthesis of derivative 228 with an ammonium subunit capable of forming a supramolecular complex with oligo(p-phenylenevinylene) (OPV) similarly to the well-known π-binding in ammonium-crown ether 229 was developed. Preliminary luminescence studies show that an intramolecular photoinduced process appears in this new supramolecular assembly C60–OPV (Scheme 62).120
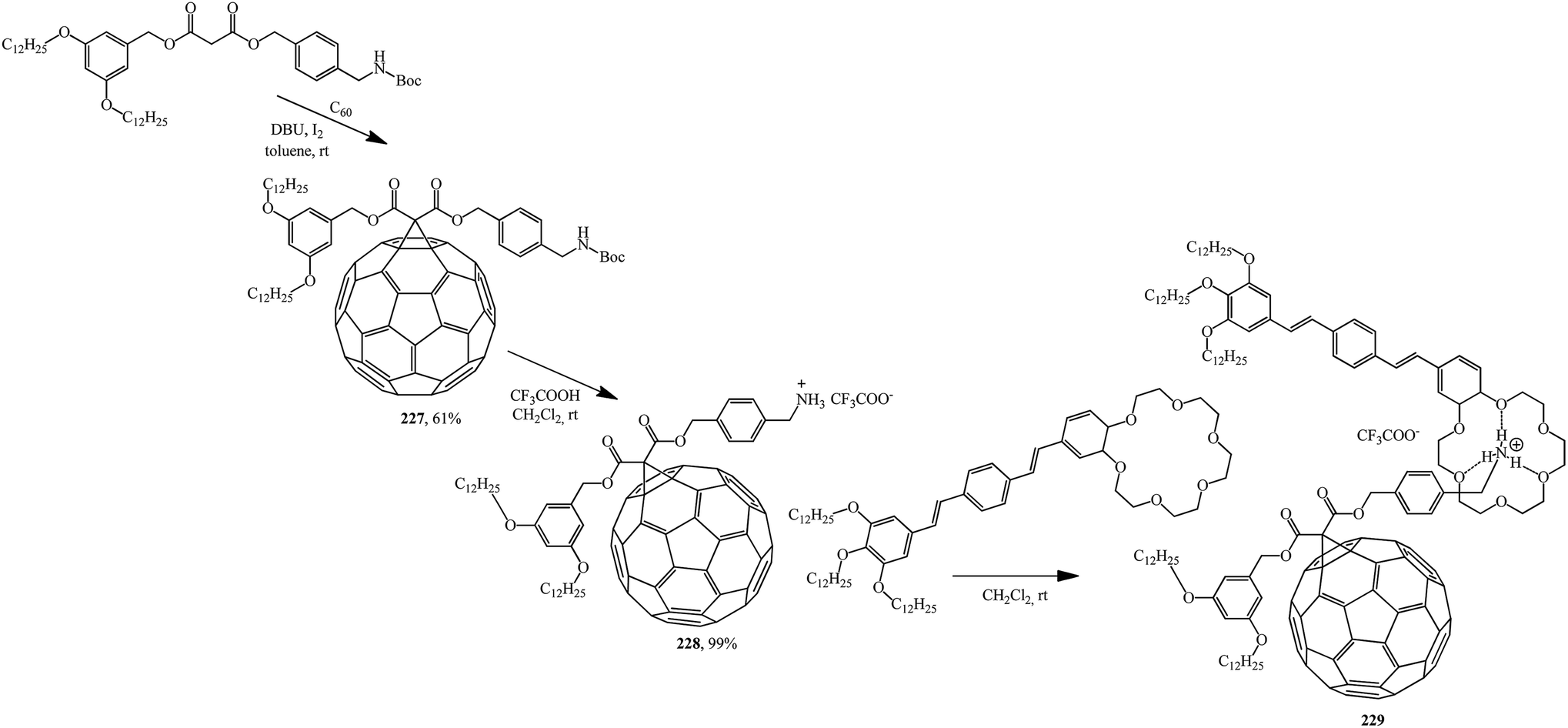 | ||
| Scheme 62 | ||
Using the same principle and methanofullerene 228 with an ammonium subunit derivative synthesized by authors,120 the formation of supramolecular complex 230 with dibenzo-24-crown-6 was proved121 (Scheme 63).
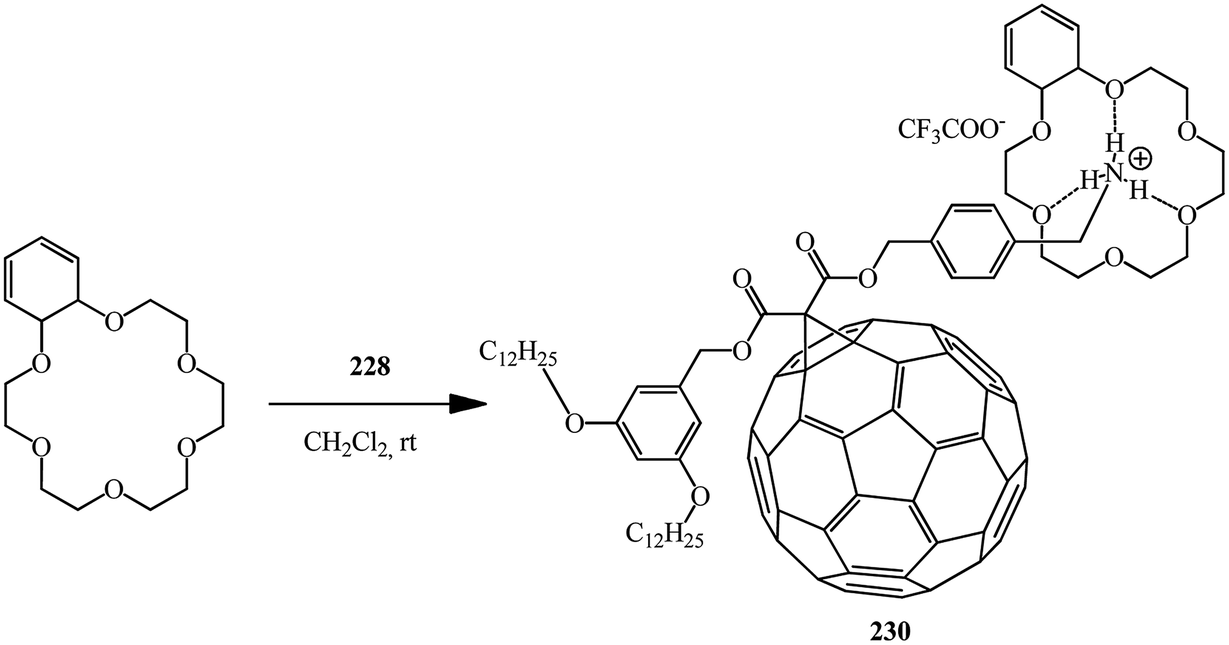 | ||
| Scheme 63 | ||
Publications of this kind open a path for creating stable non-covalent supramolecular assemblies with combined crown ether and fullerene units that are not available in literature. Using successfully chosen molecular components, new supramolecular architectures with various photoinduced intercomponent processes can be constructed.
Examples of cycloaddition to buckminsterfullerene C60 where high-molecular objects are involved rather than low-molecular ones are of appreciable interest.
For instance, oligomers are used as starting materials in the Bingel–Hirsch reaction: disymmetrically substituted oligo(phenyleneethynediyl) (OPE) derivatives were prepared from 2,5-bis(octyloxy)-4-[(triisopropylsilyl)ethynyl]benzaldehyde by an iterative approach.122 The starting dimalonic esters in disymmetrically substituted OPE derivatives undergo cyclopropanation to give the corresponding C60–OPE conjugates 231 and 232 (Scheme 64).
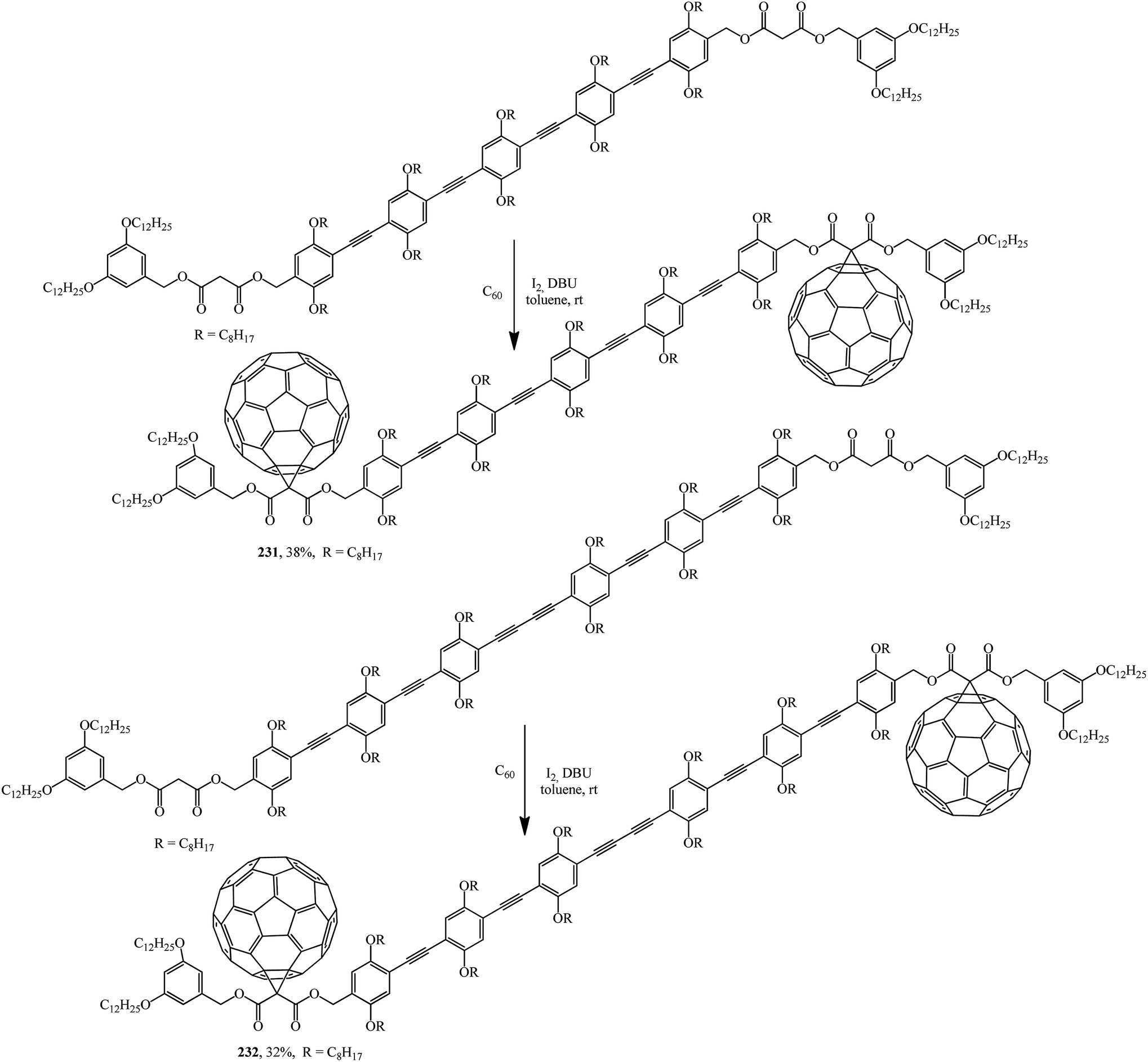 | ||
| Scheme 64 | ||
Derivatives 231 and 232, the structure of which includes a long conjugation chain, were tested as active materials in photovoltaic devices. The performance of devices prepared from these products shows that the efficiency of devices can be significantly improved by increasing the donor ability of the OPE moiety.
Later, the same group of scientists isolated acid 234 itself from fullerene carboxylic acid tert-butyl ether derivative 233. The reaction of 234 with oligophenylenevinyleneylene heptamers containing one or two hydroxy groups gave C60–OPV conjugates incorporating one 235 (ref. 123) or two 236 (ref. 124) fullerene subunits (Scheme 65).
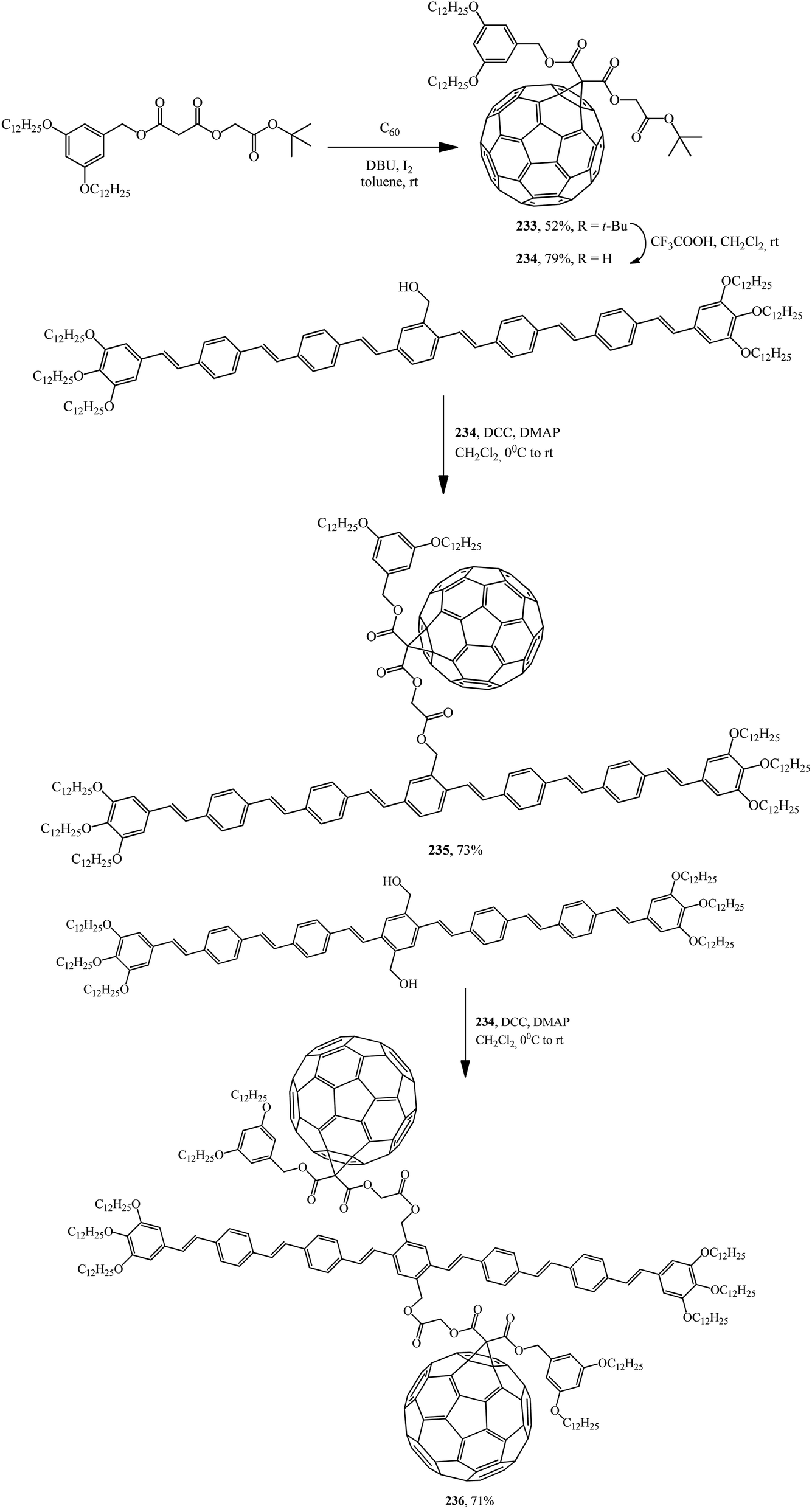 | ||
| Scheme 65 | ||
The electrochemical properties of the resulting C60–OPV derivatives were studied by cyclic voltammetry. While the first reduction of both C60–OPV conjugates is centered on the C60 unit, the oxidation is centered on the OPV rod. Photophysical studies revealed that intramolecular photo-induced processes of energy and electron transfer from the OPV central unit to the attached fullerene moieties appeared. The charge separation process shows a significant dependence on the solvent nature.
A symmetrical fullerene–bis(styryl)benzene diad 237 was synthesized in practice-oriented studies into single-component organic solar cells. In this case, the oxyethylene macrocyclic spacer attached to bis(styryl)benzene served as the cyclopropanating agent (Scheme 66).125
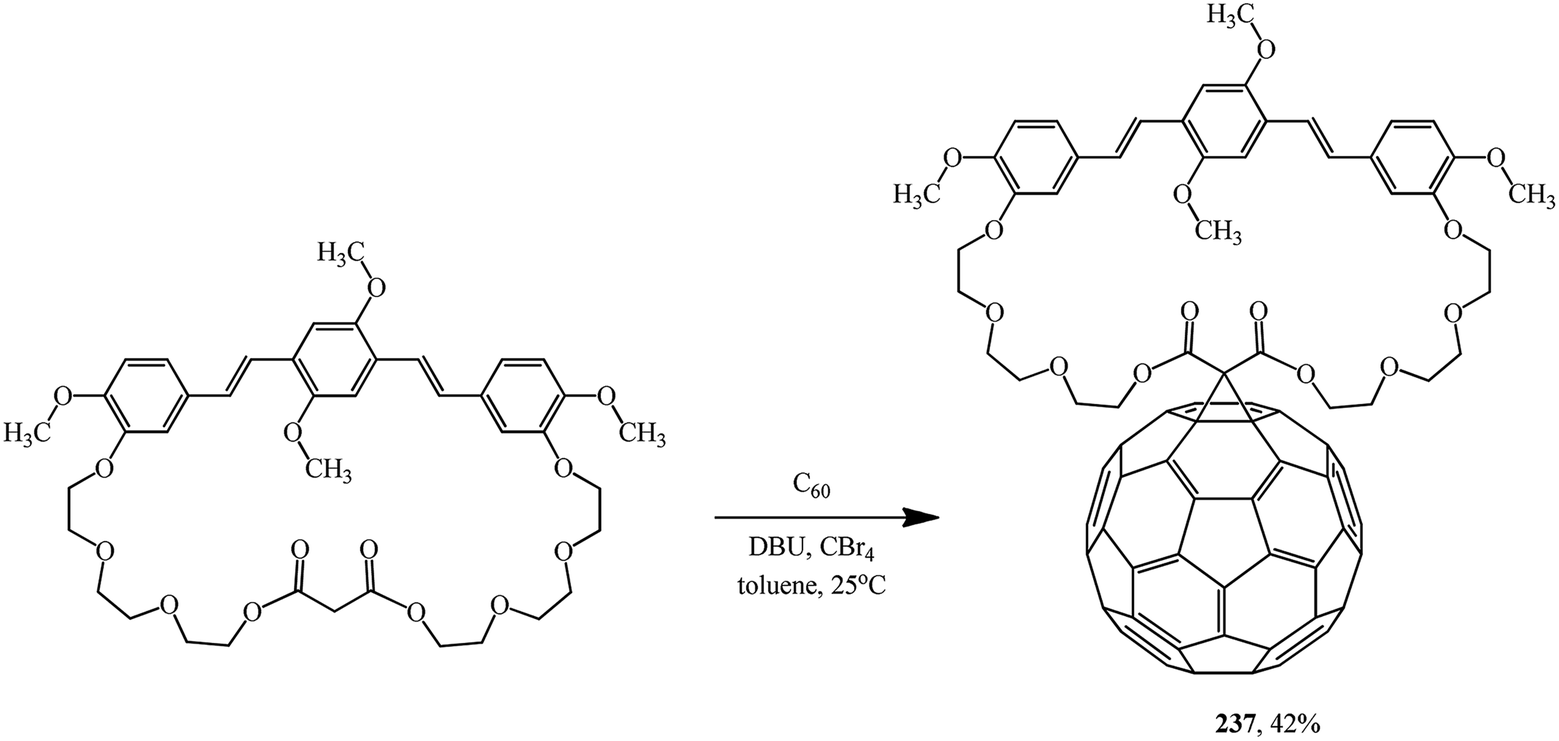 | ||
| Scheme 66 | ||
The overall power conversion efficiency calculated for solar cells made of the bis(styryl)benzene–fullerene diad was as low as 0.022%, which was the highest value reported at the time, under high-level irradiation conditions, for solar cells based on diads in which a fullerene is tethered to a phenylenevinylene structure.
The separately prepared hydroxy-containing fullerene block 238 was fused by the carbodiimide method with the homooxacalix[3]arene moiety to give compound 239, which exhibits interesting selfcomplexation–decomplexation properties in response to the solvent polarity (Scheme 67).126
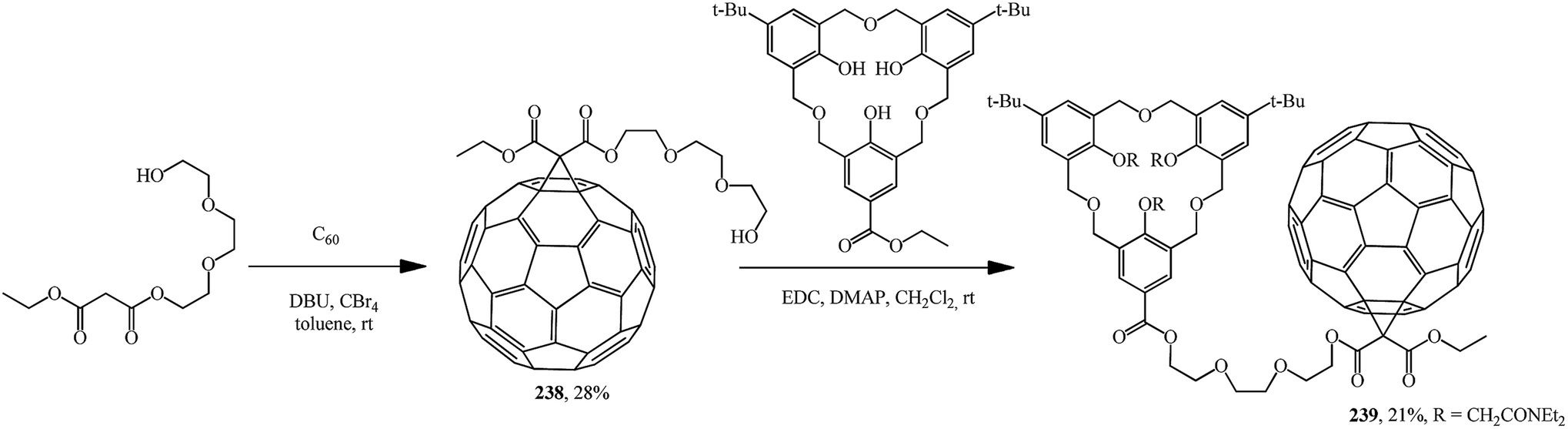 | ||
| Scheme 67 | ||
A macrocycle having two isosteviol moieties and malonate fragment was synthesized and its ability to inhibit the growth of Mycobacterium tuberculosis H37Rv in vitro at a minimum concentration of 1 mg × cm−3 was demonstrated. The macrocycle and the acyclic analogue add fullerene C60 at the activated methylene group to furnish the corresponding methanofullerene conjugates 240 and 241 (Scheme 68).127
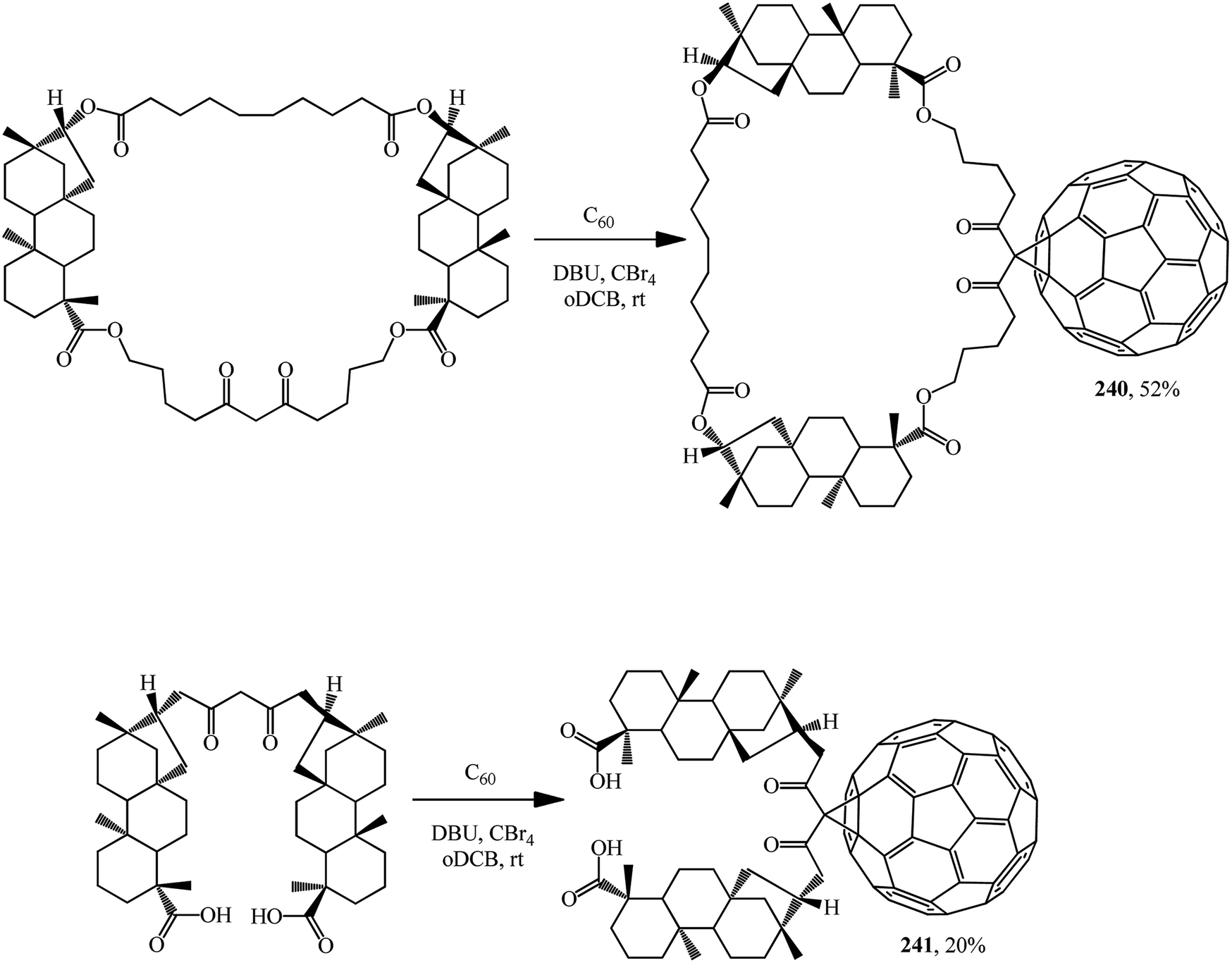 | ||
| Scheme 68 | ||
A number of methanofullerenes containing natural flavonoids were synthesized by A. C. Tome's team.128 Starting from quercetin, a natural flavonol with high antioxidant activity, three novel fullerene–flavone derivatives 242–244 were synthesized (Scheme 69).
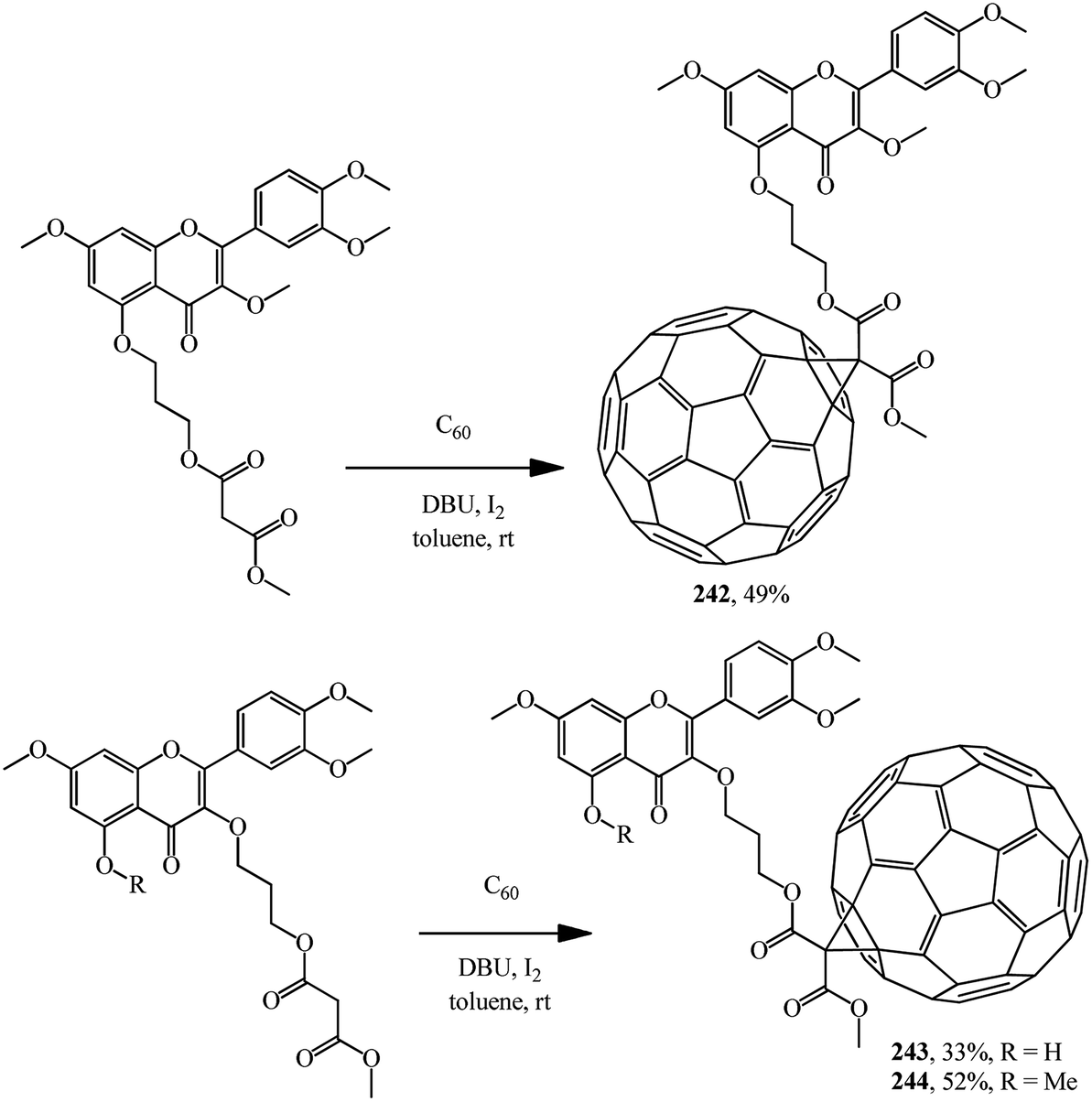 | ||
| Scheme 69 | ||
Similarly, fullerene–flavonoid derivatives 245, 246 were subsequently obtained from methyl(4′-flavonylmethyl)- and bis(4′-flavonylmethyl)malonates (Fig. 13).129
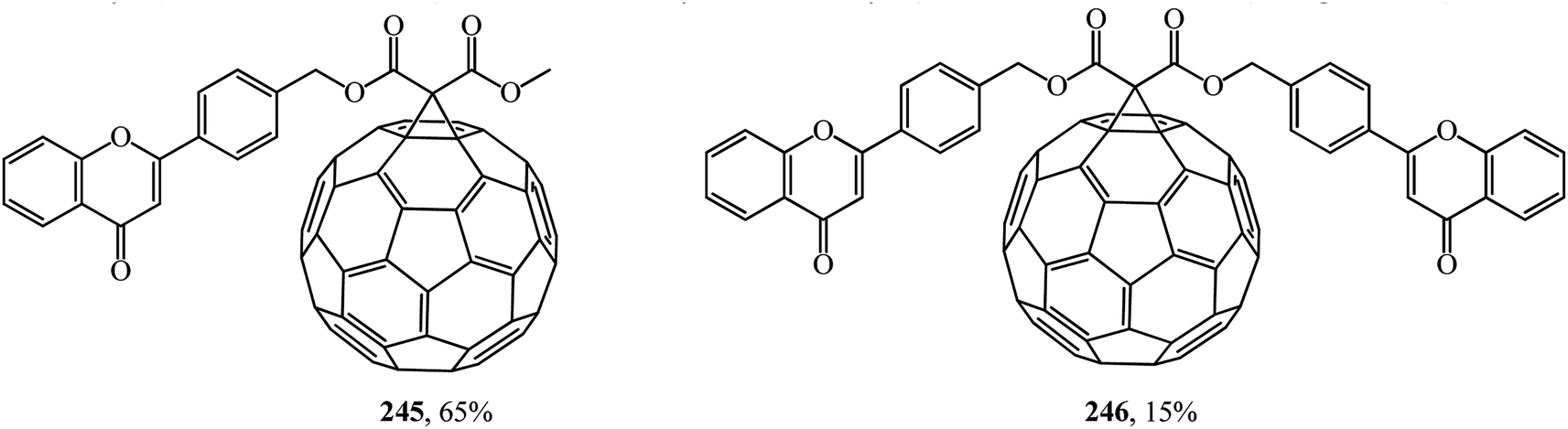 | ||
| Fig. 13 Fullerene–flavonoid conjugates 245–246. | ||
The scope of fullerene–flavonoid conjugates 247–249 bearing one or two 3,5-di-tert-butyl-4-hydroxyphenyl groups was successfully expanded in ref. 130 using chalcone, flavone and flavanone as the starting materials (Scheme 70).
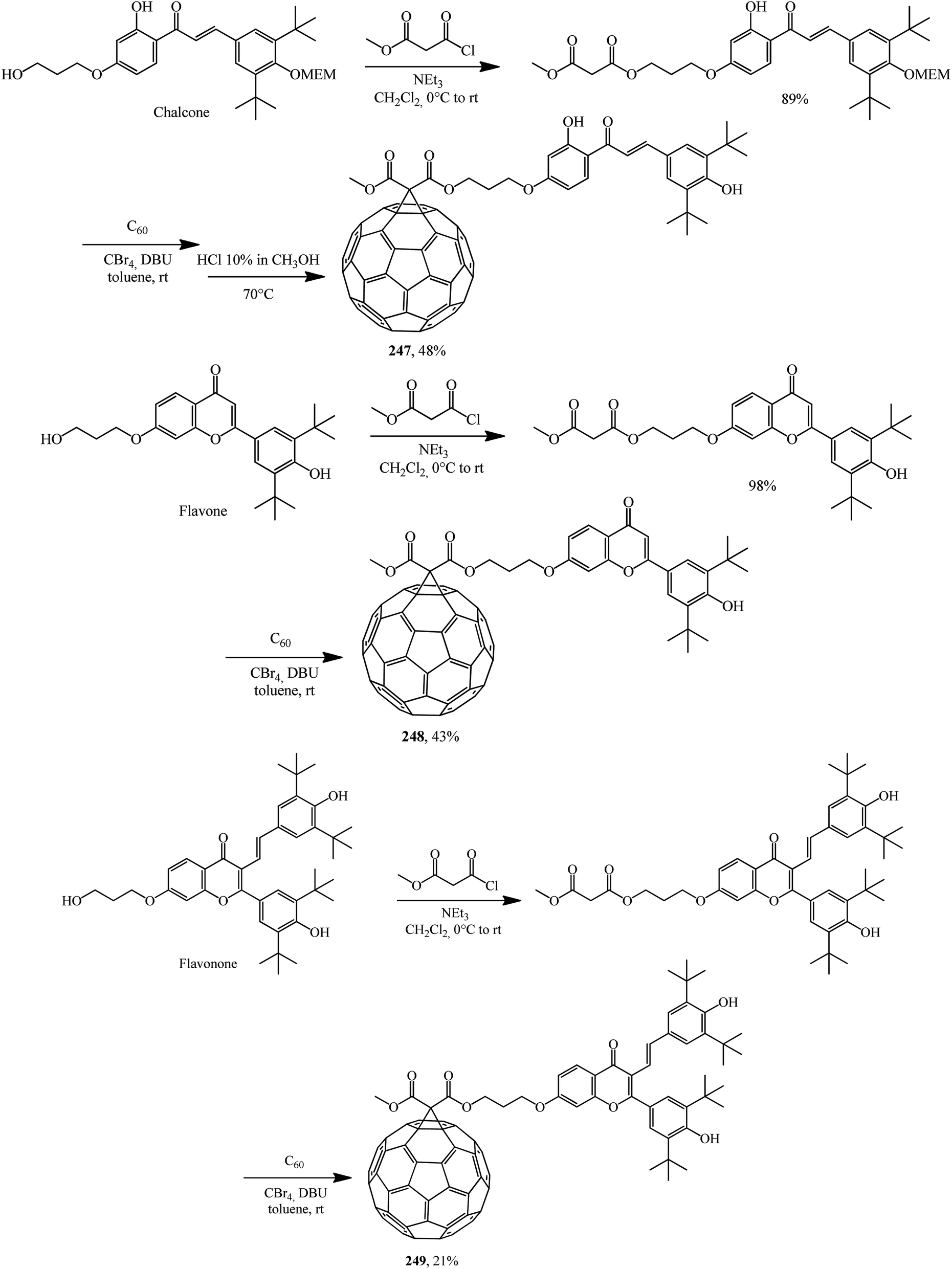 | ||
| Scheme 70 | ||
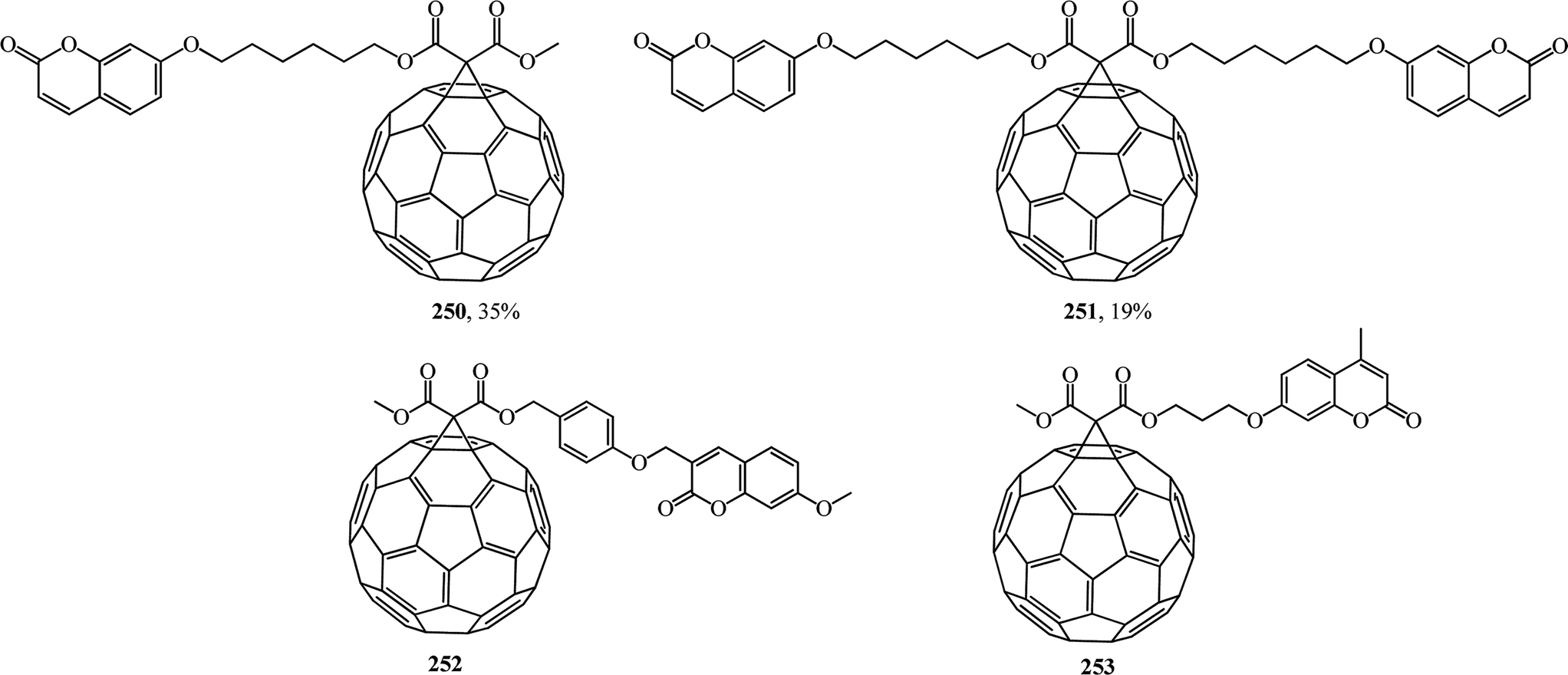
Coumarins are attractive synthetic objects due to their high quantum yields of radiation and photostability. Moreover, coumarin derivatives are often used as signal elements in sensors and in complex photophysical systems. Therefore, studies aimed at the synthesis of new covalent-bound fullerene–coumarin diads 250, 251 (ref. 131) and 252, 253,132 as well as studies of their photophysical properties (Fig. 14) are quite timely.
 | ||
| Fig. 14 Fullerene–coumarin diads 250–253. | ||
Fluorescence quenching of the coumarin fluorophore in the diads results from efficient dipole–dipole resonance energy transfer from the coumarin moiety to the fullerene moiety.
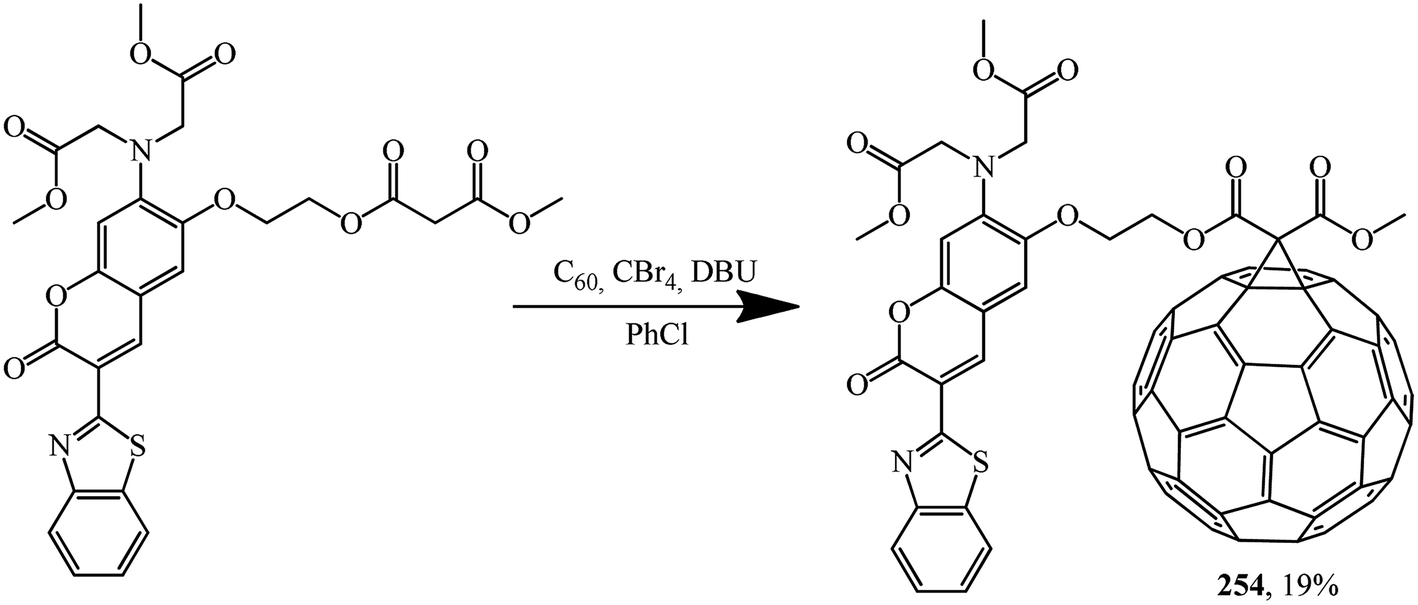
The malonate derivative of coumarin that was synthesized, which contains a benzothiazole chromophore block as a continuation of the π-conjugated system of the coumarin skeleton, undergoes a Bingel–Hirsch reaction to give fullerene–coumarin diad 254 (Scheme 71):133
 | ||
| Scheme 71 | ||
The electron interactions between the two components of diad 254 were studied by UV/Vis spectroscopy, fluorescence emission and electrochemical measurements. Studies clearly show the existence of electronic interactions between C60 and modified coumarin in the ground state; efficient electron-transfer quenching of the singlet excited state of the coumarin moiety by the appended fullerene sphere was also observed. As a result of intramolecular transfer of electrons from coumarin to C60, quenching of the characteristic radiation of coumarin in diad 254 occurs, as demonstrated in photoluminescence experiments performed in non-polar and polar solvents.
A considerable number of studies deal with dendrons functionalized with fullerene. This direction is headed by J.-F. Nierengarten. Fullerene-functionalized dendrons, where C60 serves as the branching core or fullerene is located in each branching unit, became the subject of their studies. 1,3,5-Substituted benzenes are convenient building blocks in the creation of such dendrimeric structures.
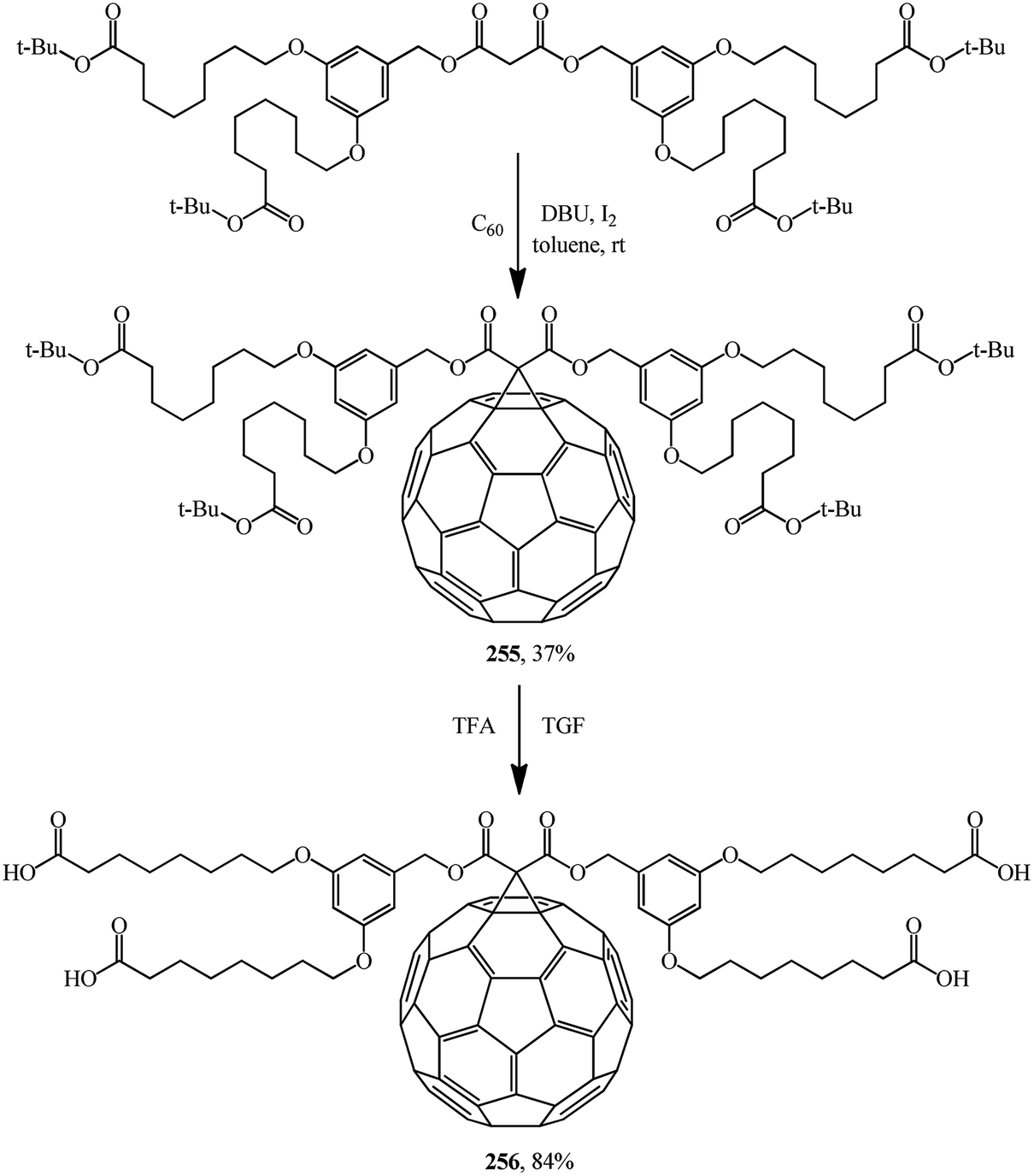
Bearing in mind that the solubility of C60 in polar solvents, in water in particular, is extremely low, derivatives of methanofullerenes that are free from this drawback are particularly promising as the possible range of product applications expands significantly. For example, a group of scientists134 reported a hitherto unknown amphiphilic methanofullerene derivative 256 obtained via its intermediate tert-butyl adduct 255. Owing to the high solubility of 256 in polar solvents widely used in sol–gel treatment, it was successfully included into a sol–gel (Scheme 72).
 | ||
| Scheme 72 | ||
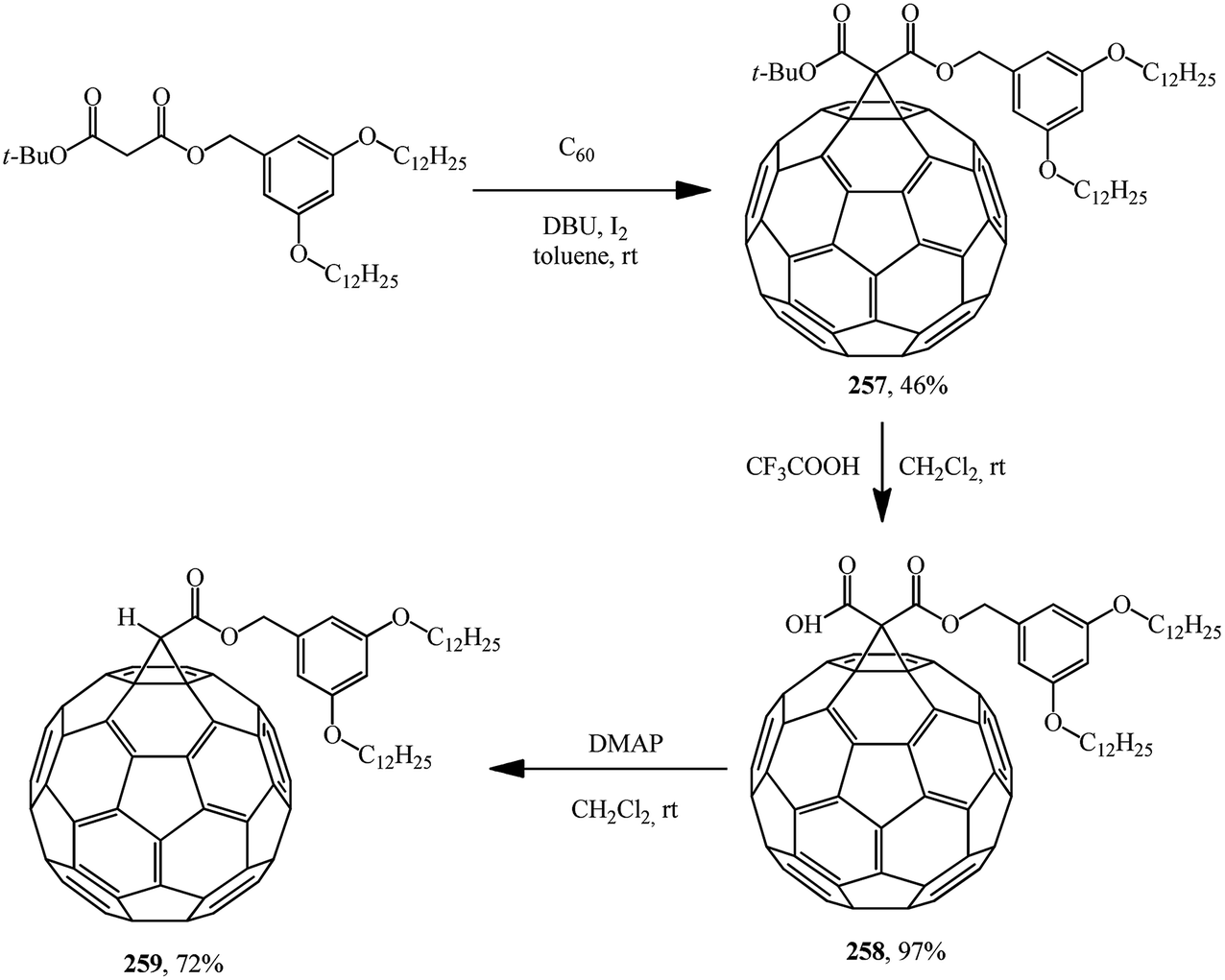
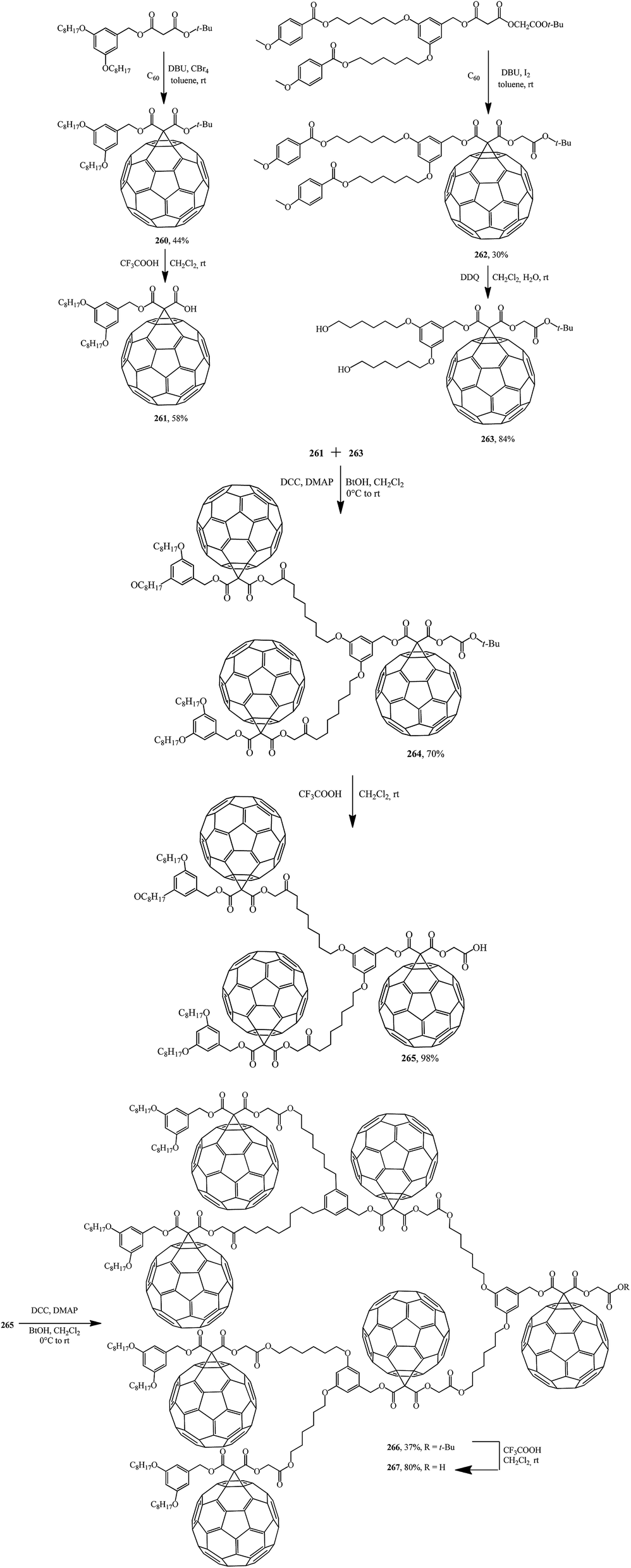
Similarly to the synthesis via methanofullerene–dicarboxylic acid mono-ester 258 reported in (Scheme 73),135 a related derivative 261 was obtained (Scheme 74)136 The authors135 also showed that the reaction of compound 258 with a catalytic amount of DMAP in CH2Cl2 at room temperature afforded 259 in a good yield (72%). In this case, the unstable carbanion resulting from the decarboxylation reaction is immediately quenched by the more acidic DMAP-H+ resulting from the reaction of DMAP with the carboxylic acid function of 258; therefore, the conversion of 258 into 259 occurs smoothly under these conditions.
 | ||
| Scheme 73 | ||
 | ||
| Scheme 74 | ||
Further, fullerene-functionalized dendrons (265, 267) containing a C60 sphere at each branching unit were prepared by a convergent approach using successive DCC-mediated esterification followed by elimination of a tert-butyl ester moiety under acidic conditions (Scheme 74).
In was found in a study on the electrochemical properties of methanofullerenes that their cyclic voltammograms differed only a little from those of the C60 molecule.137–139 It was concluded on this basis that methanofullerenes maintain the unique electrochemical properties of the original C60. This conclusion prompted the creation of covalently bound donor–acceptor diads based on fullerenes by attaching donor fragments to the C60 frame. The significance of this direction is determined by the fact that, as compared to planar acceptors, the fullerene moieties in such diads provide a low value of reorganization energy in photocatalytic processes. As a result of this advantage, covalent-bound fullerene-containing diads became irreplaceable models for studying the key points of photosynthesis and for creating photovoltaic cells and optoelectronic materials. Taking the above into consideration, there is a need for fundamental studies on the development of fullerene-containing dendrimer structures. Studies on the synthesis of dendrimer structures with one fullerene-containing center were carried out under the direction of R. Deschenaux and were reported in ref. 140. Addition reactions of mesomorphic malonate-based dendrimers (up to the fourth generation) to C60 gave liquid-crystal symmetrical fullerene derivatives 268–273 (Fig. 15) and non-symmetrical ones 274, 275 (Fig. 16). The supramolecular organization of fullerene-based molecular units was studied by X-ray diffraction. Two structural modes were identified. The supramolecular organization of low generation dendrimers is determined by steric factors. For high generation dendrimers, the mesogenic groups impose a microphase organization.
 | ||
| Fig. 15 Symmetrical fullerene-containing mesomorphic dendrimers 268–273. | ||
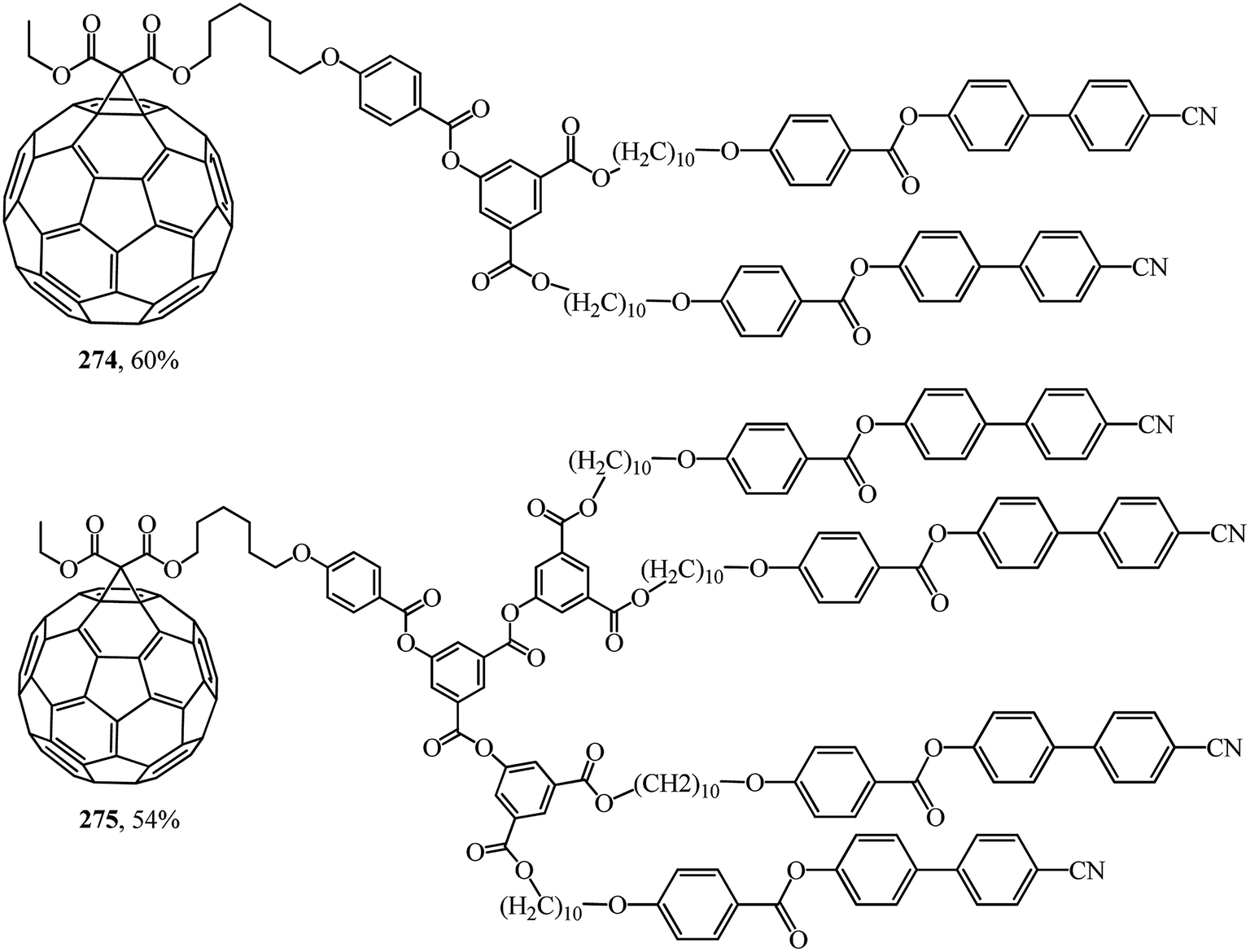 | ||
| Fig. 16 Non-symmetrical fullerene-containing mesomorphic dendrimers 274 and 275. | ||
Structures containing both the C60 core and moieties that provide liquid crystal properties should not be ignored. In these cases, not only are existing properties improved but also new ones often appear. The synthesis of methanofullerene 276, the first C60-functionalised liquid crystal dendrimer possessing a chiral mesophase (Scheme 75),141 can serve as an example.
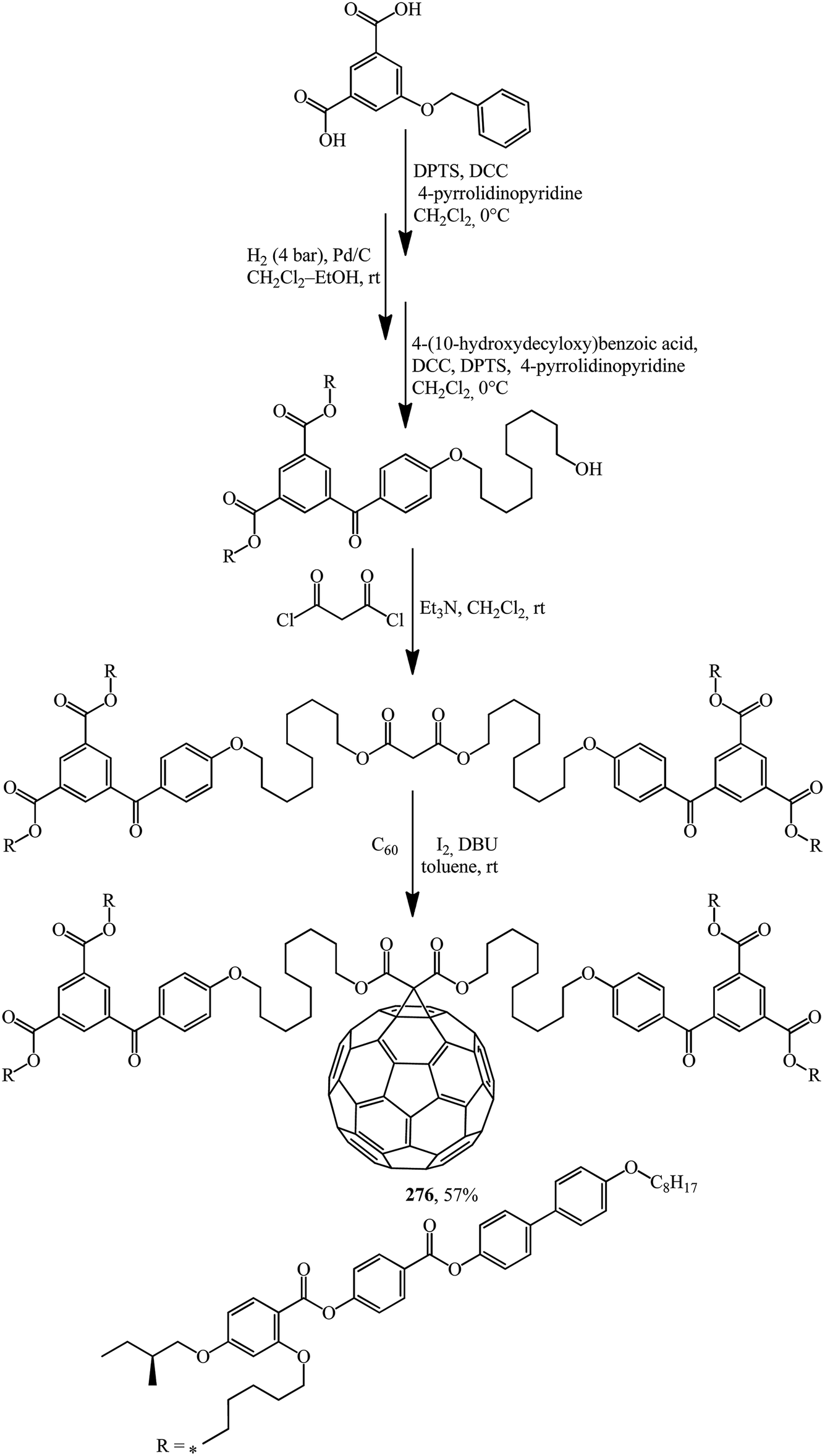 | ||
| Scheme 75 | ||
In continuation of studies on this subject, B. Dardel et al.142 isolated various symmetrical 277 and non-symmetrical 278 fullerene-containing liquid crystal dendrimers (Fig. 17). They were characterized by the method based on the electro-optical Kerr effect, by determination of the permanent dipole moment, which is sensitive to the molecular polar ordering and anisotropy of polarizability, and by hydrodynamic methods in solution.
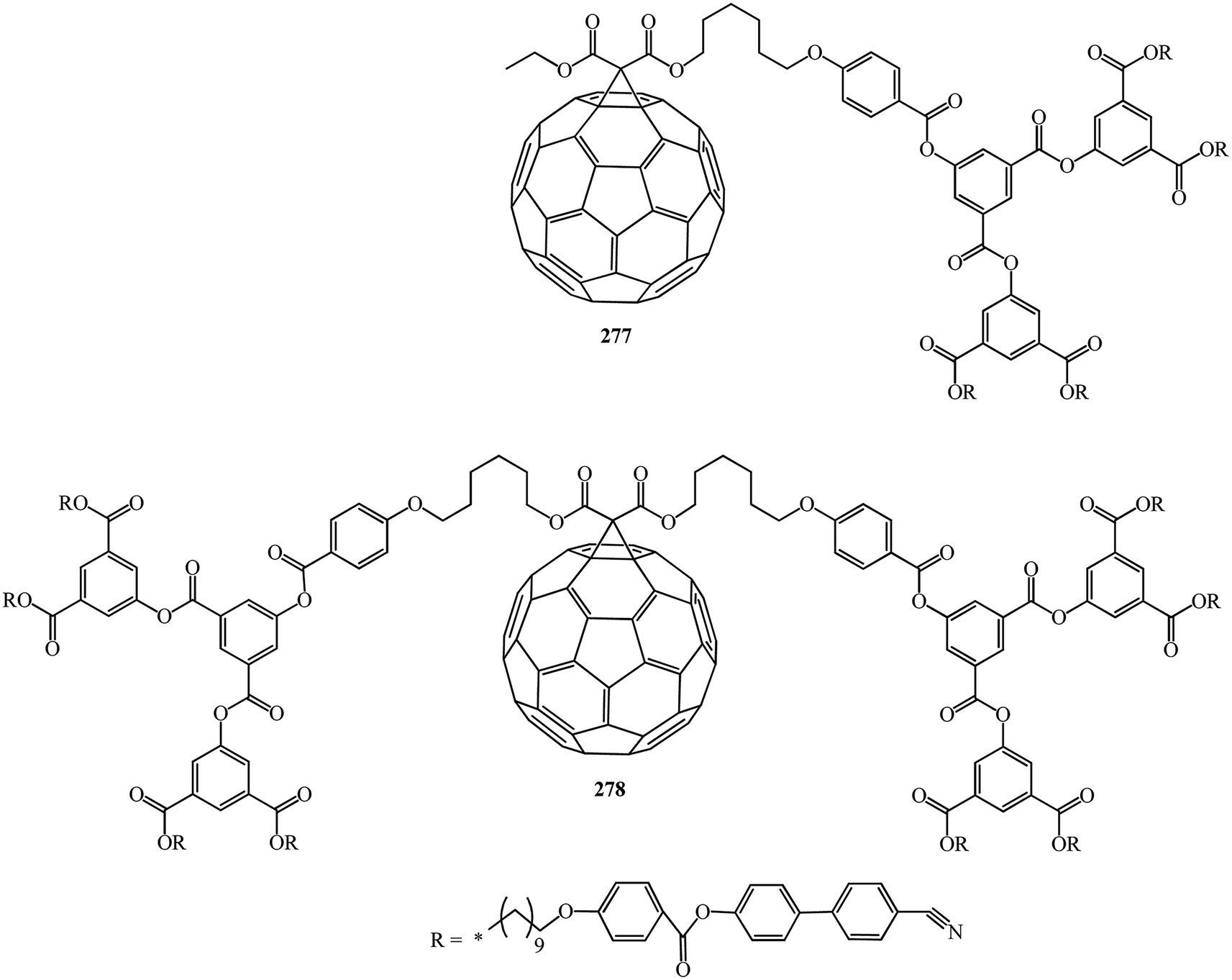 | ||
| Fig. 17 Fullerene-containing liquid crystalline dendrimers 277 and 278. | ||
The effect of covalent-bound fullerene on the polarity, electro-optical, and viscometric properties of fullerene derivatives was identified and analyzed. It was concluded that fullerene produces a change in the original ordering in dendrite molecules, which mainly resulted in a correction of the shape asymmetry of these molecules as a whole.
Using convergent and modular synthetic methodology, symmetrical 279, 280 (Fig. 18), and non-symmetrical 281–283 (Fig. 19) methanofullerodendrimers were obtained.143 The rather low yield of the target product (14–28%) is explained by participation of mesomorphic malonate derivatives with substituents that create spatial hindrance in this process. The liquid-crystal properties of malonates and fullerodendrimers were studied by polarized optical microscopy, differential scanning calorimetry, and X-ray diffraction.
 | ||
| Fig. 18 Symmetrical fullerene-containing liquid crystal dendrimers 279 and 280. | ||
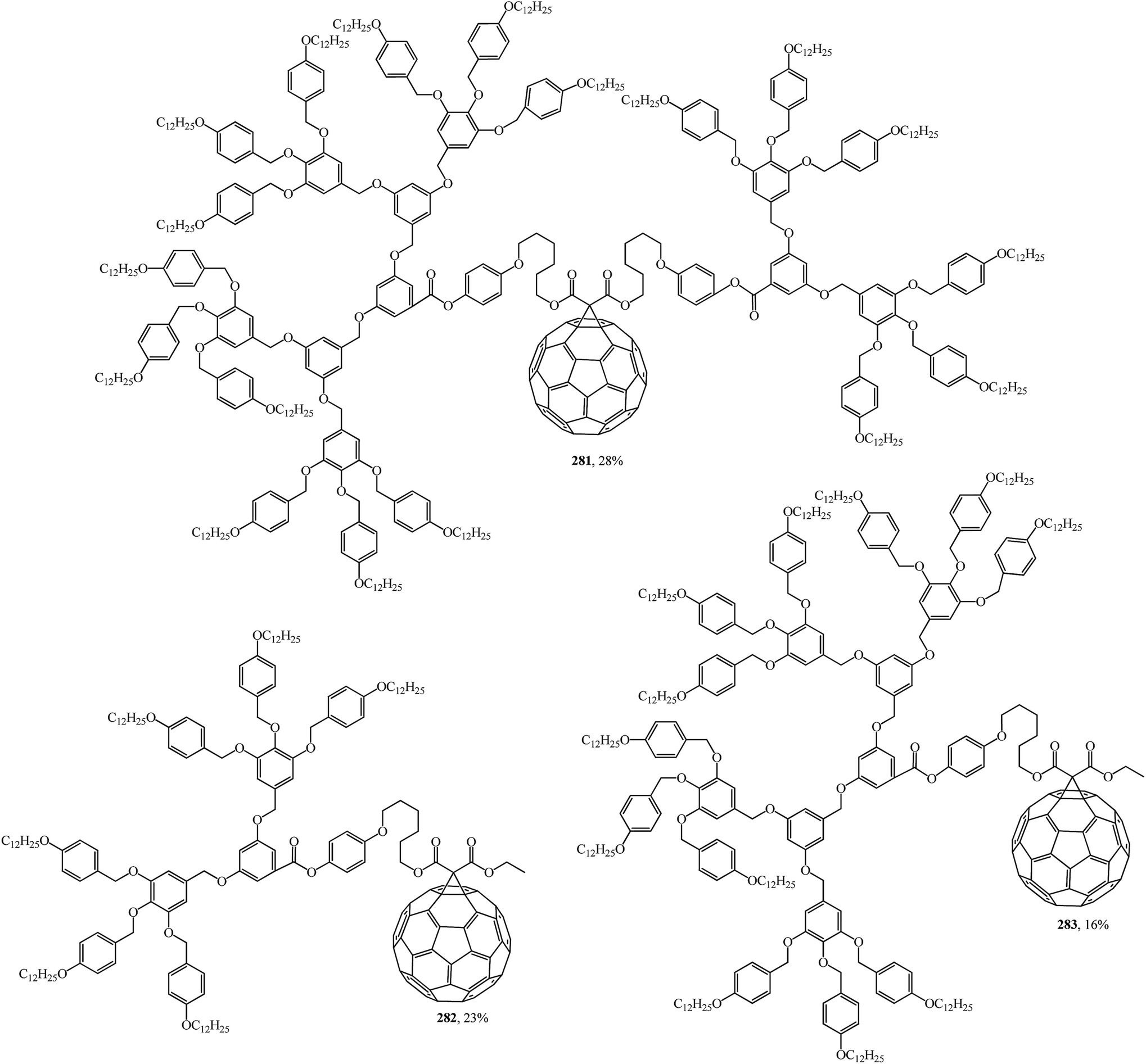 | ||
| Fig. 19 Non-symmetrical fullerene-containing liquid crystal dendrimers 281–283. | ||
The design, synthesis and liquid-crystal properties of hybrid compound 284 built of rod-like OPV and disc-like poly(benzyl ether) units were reported (Fig. 20).144
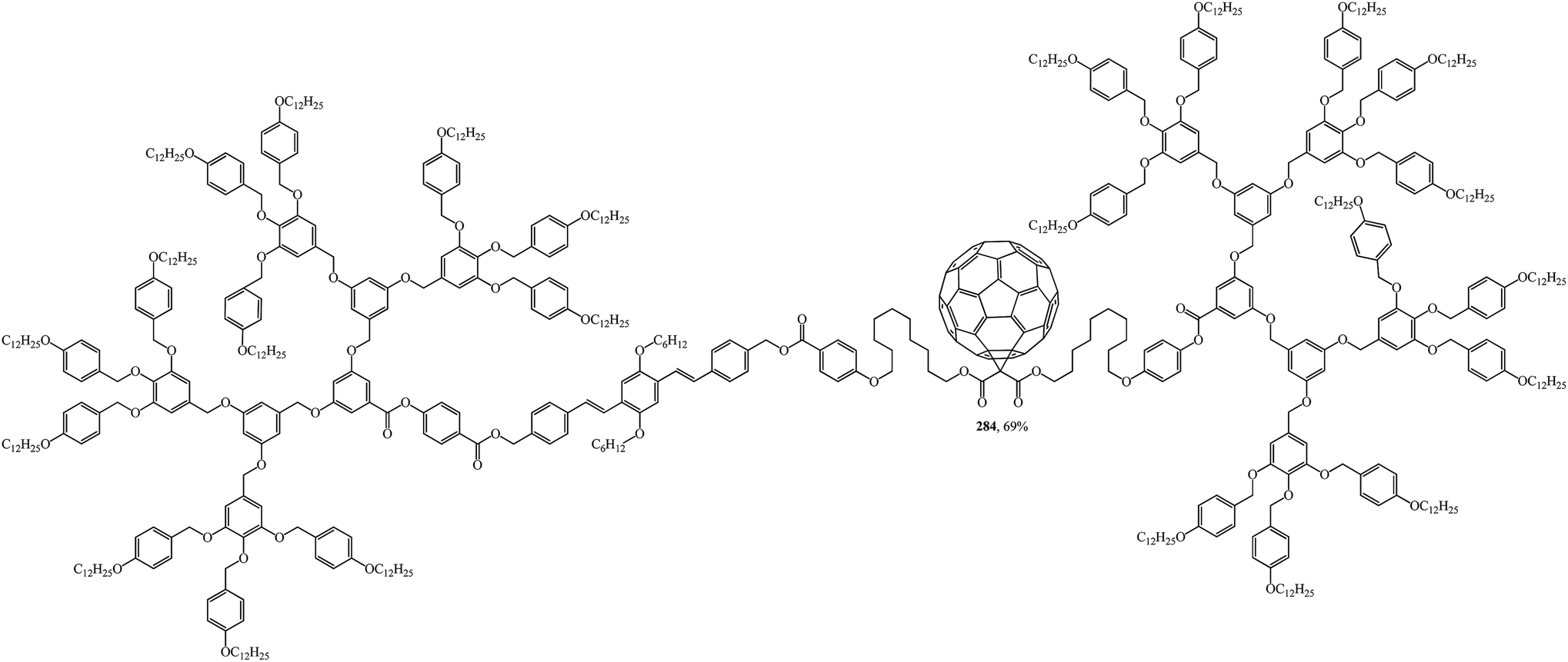 | ||
| Fig. 20 Fullerene-containing liquid crystal dendrimer 284. | ||
An unusual way to obtain bis[60]fullerodendrimers was chosen by Deschenaux et al.145 The synthetic modular approach developed in that study allowed new substances to be isolated by assembling type I methanofullerenes 285, 286 containing a terminal olefin, and type II methanofullerenes 287, 288 containing an α,β-unsaturated carbonyl olefin (Fig. 21). Both mono[60]fullerodendrimers and their fullerene-free analogues were synthesized in this study.
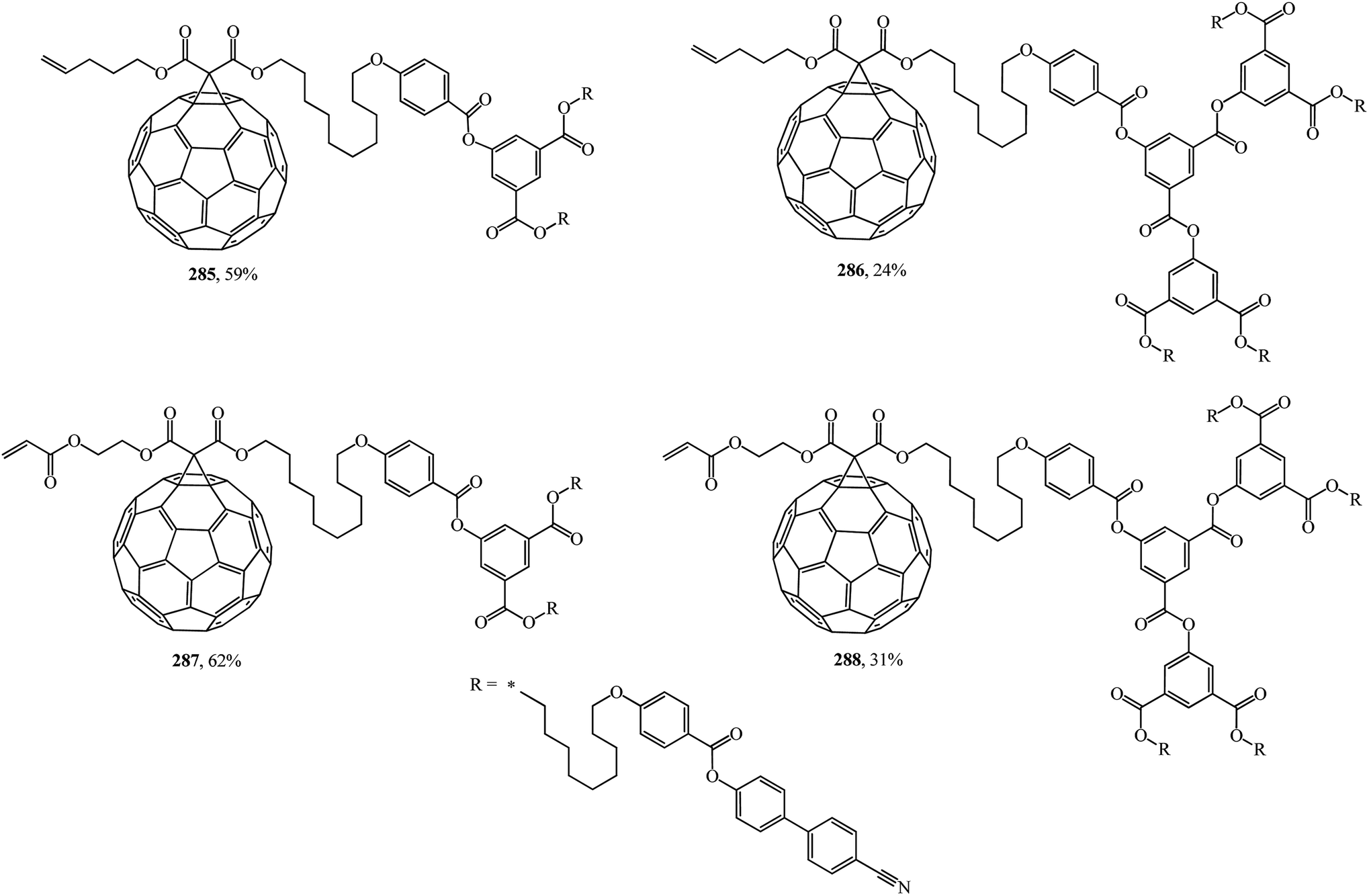 | ||
| Fig. 21 Fullerene-containing dendrimers 285–288. | ||
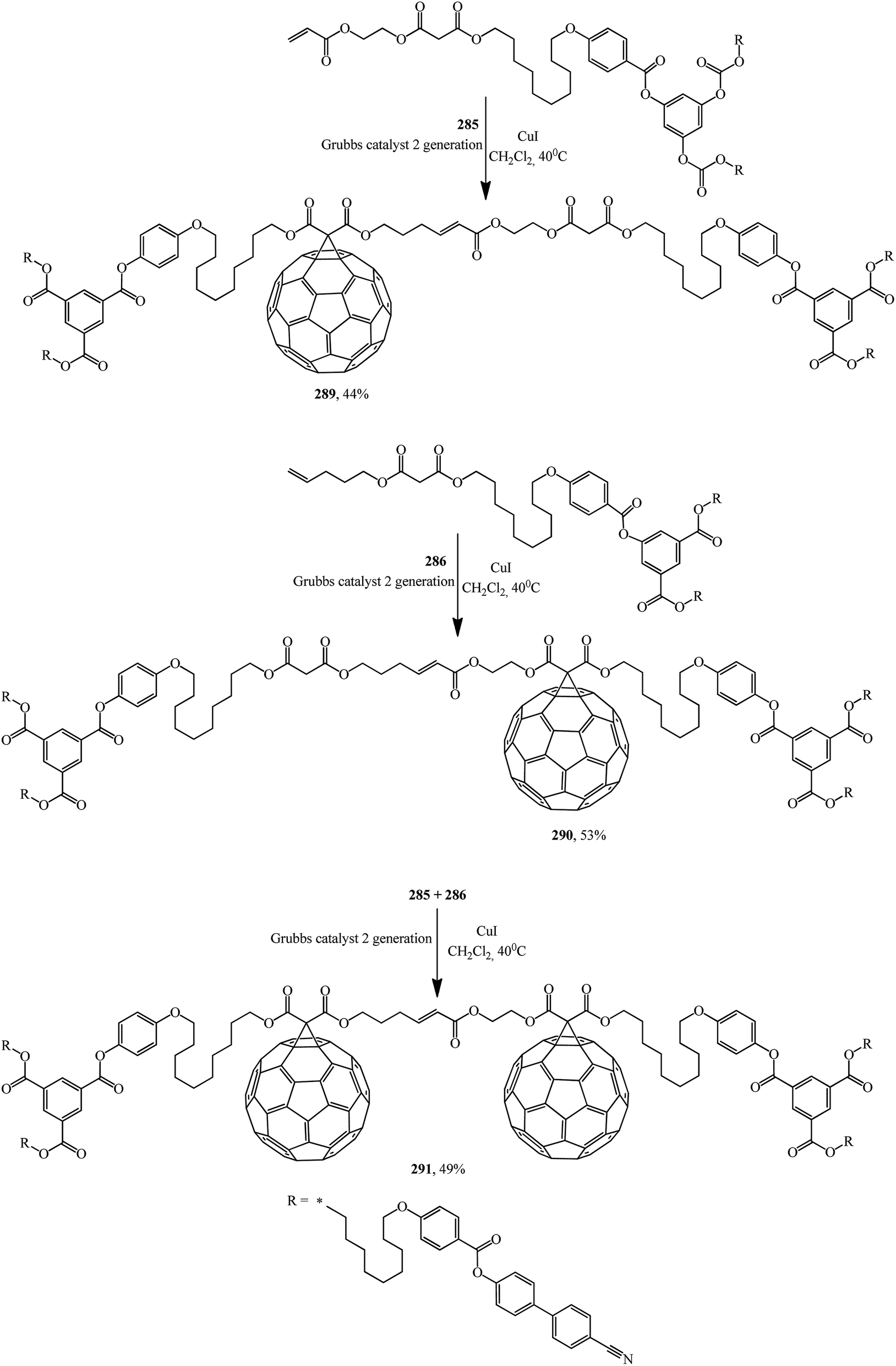
First- and second-generation poly(aryl ester) dendrons carrying cyanobiphenyl mesogens were used as liquid-crystalline promoters. First-generation mono- 289, 290 and bis-[60]fullerodendrimers 291 were obtained from mono-[60]fullerene derivatives 285 and 287 (Scheme 77) under olefin cross metathesis reaction conditions aided by the very efficient second-generation Grubbs catalyst according to general Scheme 76.
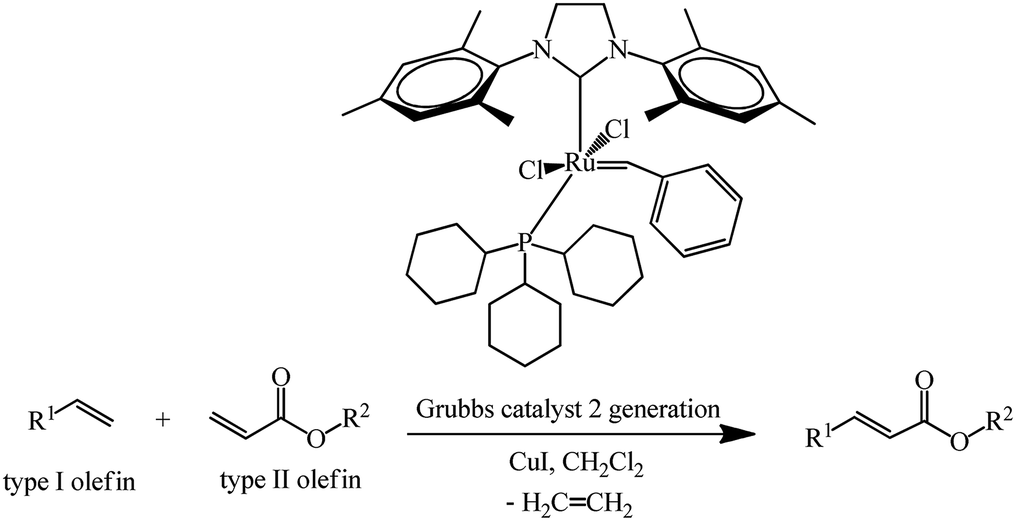 | ||
| Scheme 76 | ||
 | ||
| Scheme 77 | ||
The second generation of bis-[60]fullerodendrimer 292 based on mono-[60]fullerenes 286, 288 containing a terminal double bond was also implemented by the proven technique and is shown in Scheme 78.
 | ||
| Scheme 78 | ||
The liquid-crystal properties were studied by polarized optical microscopy, differential scanning calorimetry, and small-angle X-ray scattering. Nematic, smectic, and multisegregated lamellar phases were observed in agreement with the nature and structure of dendrimers.145 Owing to its versatility and tolerance towards many functional groups, olefin cross-metathesis proved to be the reaction of choice for the elaboration of molecular materials with complex architectures.
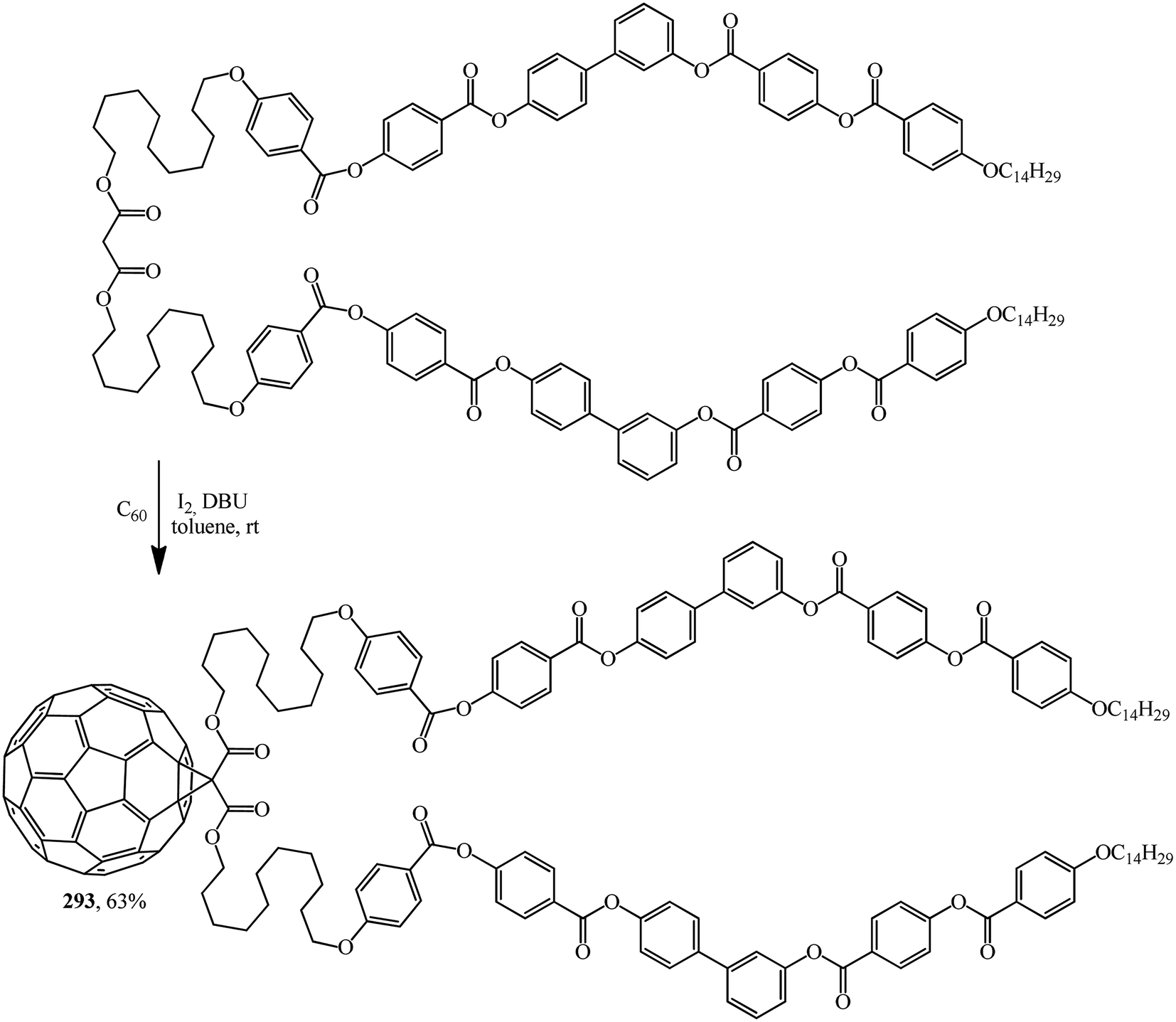
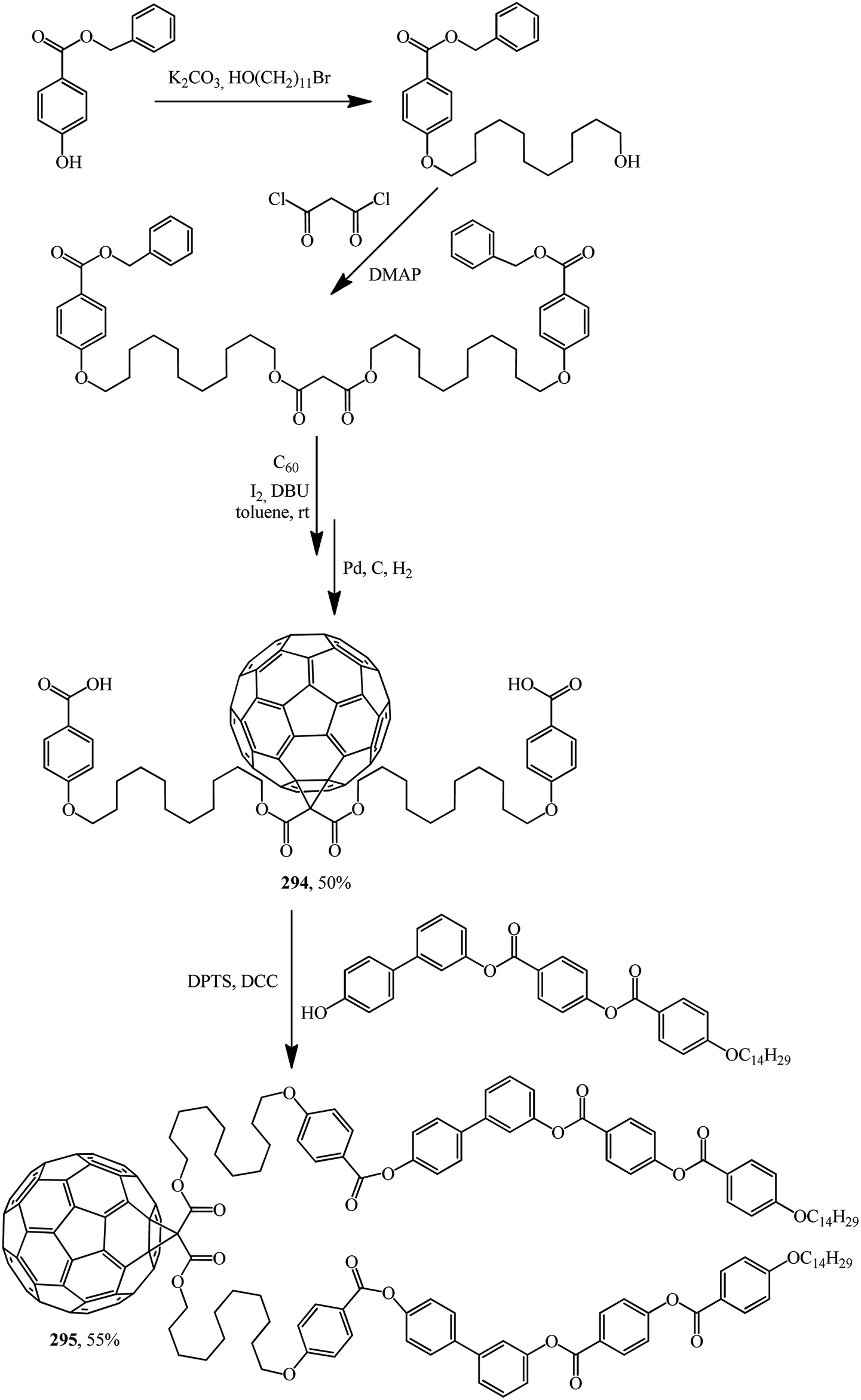
Excellent candidates for organizing fullerenes into supramolecular ordered structures were reported in ref. 146 Moreover, a new class of nondendritic hybrid C60-containing bended molecules corresponds to liquid crystals. In this case, the goal is achieved using two approaches: in the first one, a fullerene is incorporated by the Bingel–Hirsch method into the center of a bended malonate to give 293 (Scheme 79). In the second one, two alcohol groups are grafted onto a fullerene-containing malonate with simplified structure 294 in the presence of 4-(dimethylamino)pyridinium 4-toluenesulfonate (DPTS)/DCC and a similar product 295 is isolated (Scheme 80).
 | ||
| Scheme 79 | ||
 | ||
| Scheme 80 | ||
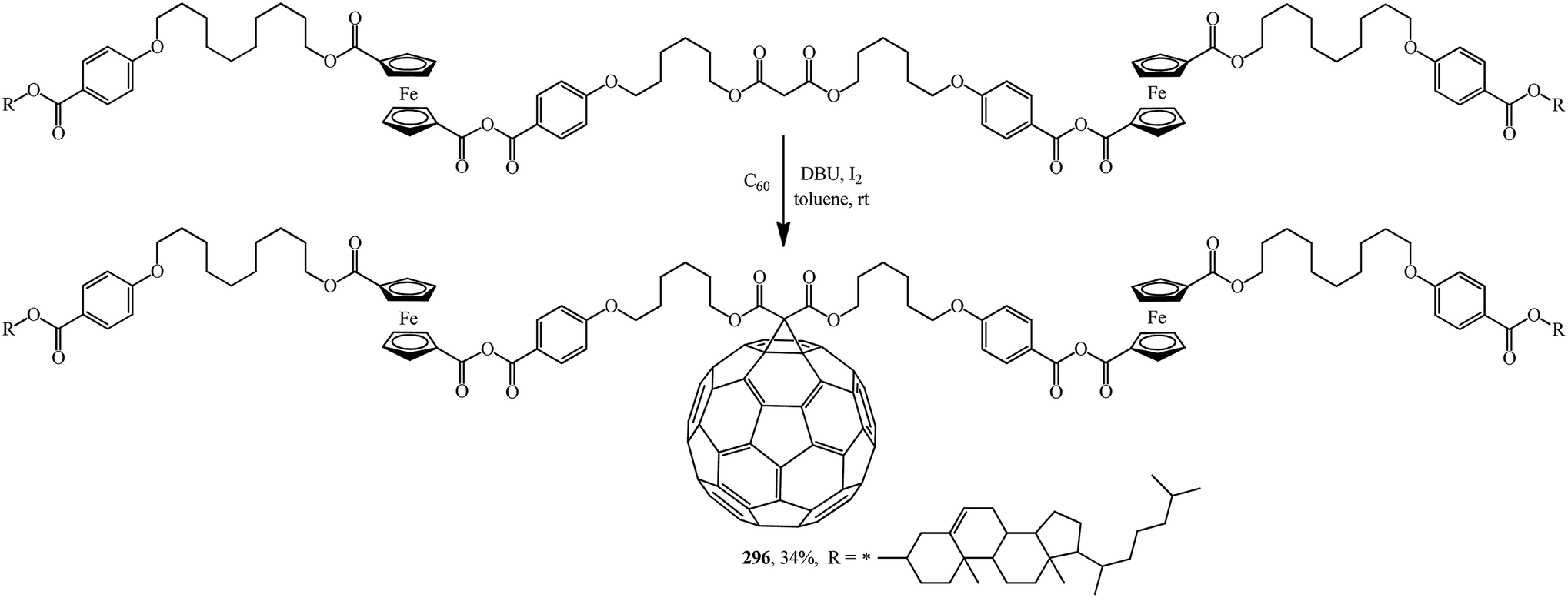
A fruitful idea exists to use malonates with a combination of various functions in the Bingel–Hirsch process, which is a path for generating target products with planned characteristics. In fact, grafting of a ferrocene-containing liquid-crystal malonate derivative (1-[10-{4-[(cholest-5-en-3b-yloxy)carbonyl]phenoxy}-decyloxycarbonyl]-1′-[4-(hydroxyphenoxy)carbonyl]ferrocene) to C60 led to a mixed fullerene–ferrocene adduct 296 that was found to contain smectic phase A (Scheme 81).147 Cholesterol can be used as a liquid-crystal promoter. X-ray diffraction experiments and volumetric measurements indicated that 296 is organized in double layered structures. The corresponding supramolecular organization within the mesomorphic lamellar phase is characterized by a microsegregation of the different units (ferrocene, fullerene, and cholesterol) in distinct sublayers. In such a smectic A phase, C60 imposes the arrangement of the other molecular moieties.
 | ||
| Scheme 81 | ||
Previously we noted the extremely low solubility of C60 and the high need for water-soluble compounds based on it to study its biological properties. It appears that one of the approaches to solve this problem may involve synthesizing dendrimers with hydrophilic substituents at the fullerene frame.
The authors148 describe an ingenious enantioselective synthesis of a large series of optically active dendri-adducts of fullerenes, whose chirality is solely due to their architecture.
2-(tert-Butoxy)-2-oxoethyl ethyl propanedioate was used as the starting malonate. It was converted to adduct 297 under Bingel–Hirsch reaction conditions. Treatment of the latter with TsOH × H2O resulted in 2-(tert-butoxy)-2-oxoethyl ethyl-1,2-methano[60]fullerene-61,61-dicarboxylate 298 (Scheme 82).30
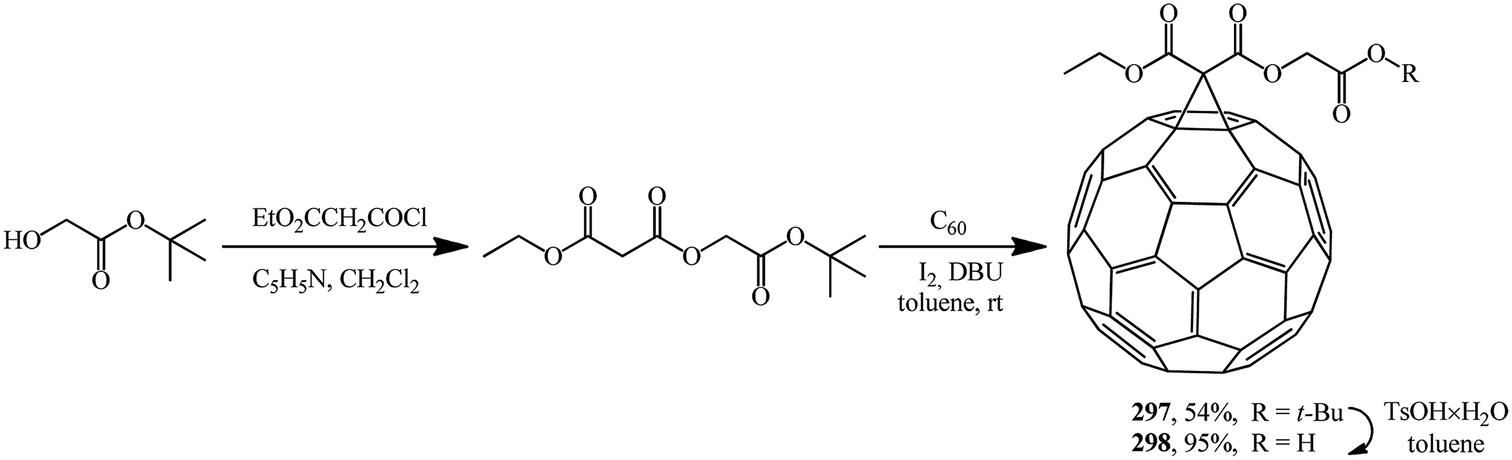 | ||
| Scheme 82 | ||
Methanofullerene 298 thus obtained was used as the base adduct in the coupling of first-, second- and third-generation dendrons, which led to the expected dendrite fullerene derivatives of the first 299, second 300 and third 301 generations, respectively, that are characterized by fairly good solubility in water (Scheme 83).148
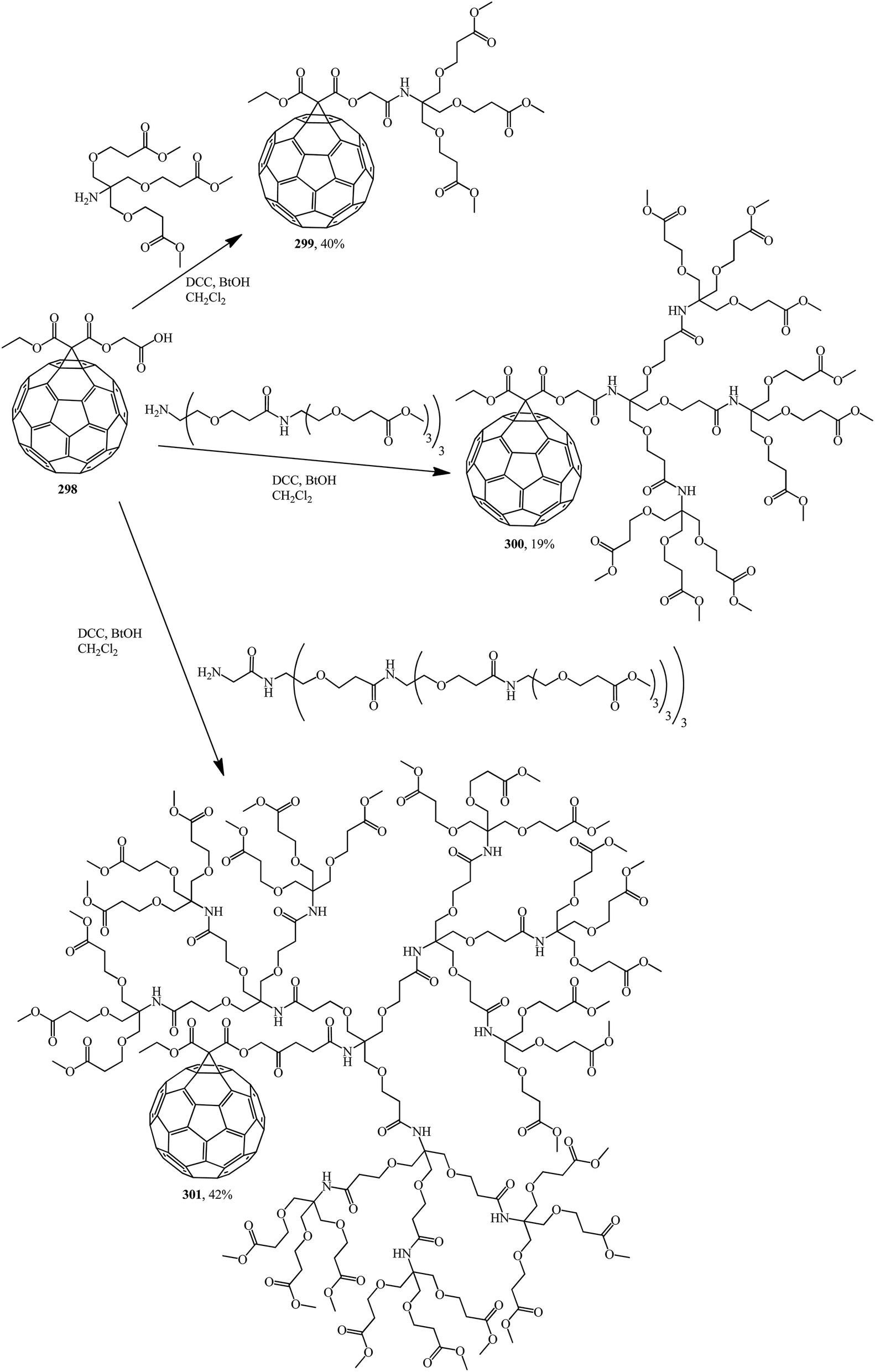 | ||
| Scheme 83 | ||
Various drugs are currently actively developed on the basis of methanofullerenes. In particular, there is much hope to find an anti-AIDS drug. HIV-1 protease, the protein responsible for virus penetration into blood cells, has a spherical cavity with a diameter of 10 Å whose shape remains unchanged upon all mutations. This size almost coincides with the diameter of the C60 molecule. Methanofullerenes were found to inhibit the HIV-1 protease. Apparently taking this information into account, the team headed by Professor Hirsch performed the nucleophilic cyclopropanation of C60 with a second-generation bis(polyamide)-malonate dendrimer in the presence of CBr4 and DBU to give methanofullerene 302 (Scheme 84).149 Removal of the terminal tert-butyl groups provides dendro[60]fullerene 303 with 18 carboxylic groups at the periphery. Methanofullerene 303 showed antiviral properties in biological tests. This is explained by the fact that the fullerene frame is close in size to the active center of the HIV-1 protease enzyme. As a result, some methanofullerenes can penetrate into the hydrophobic cavity of the enzyme and inhibit it.
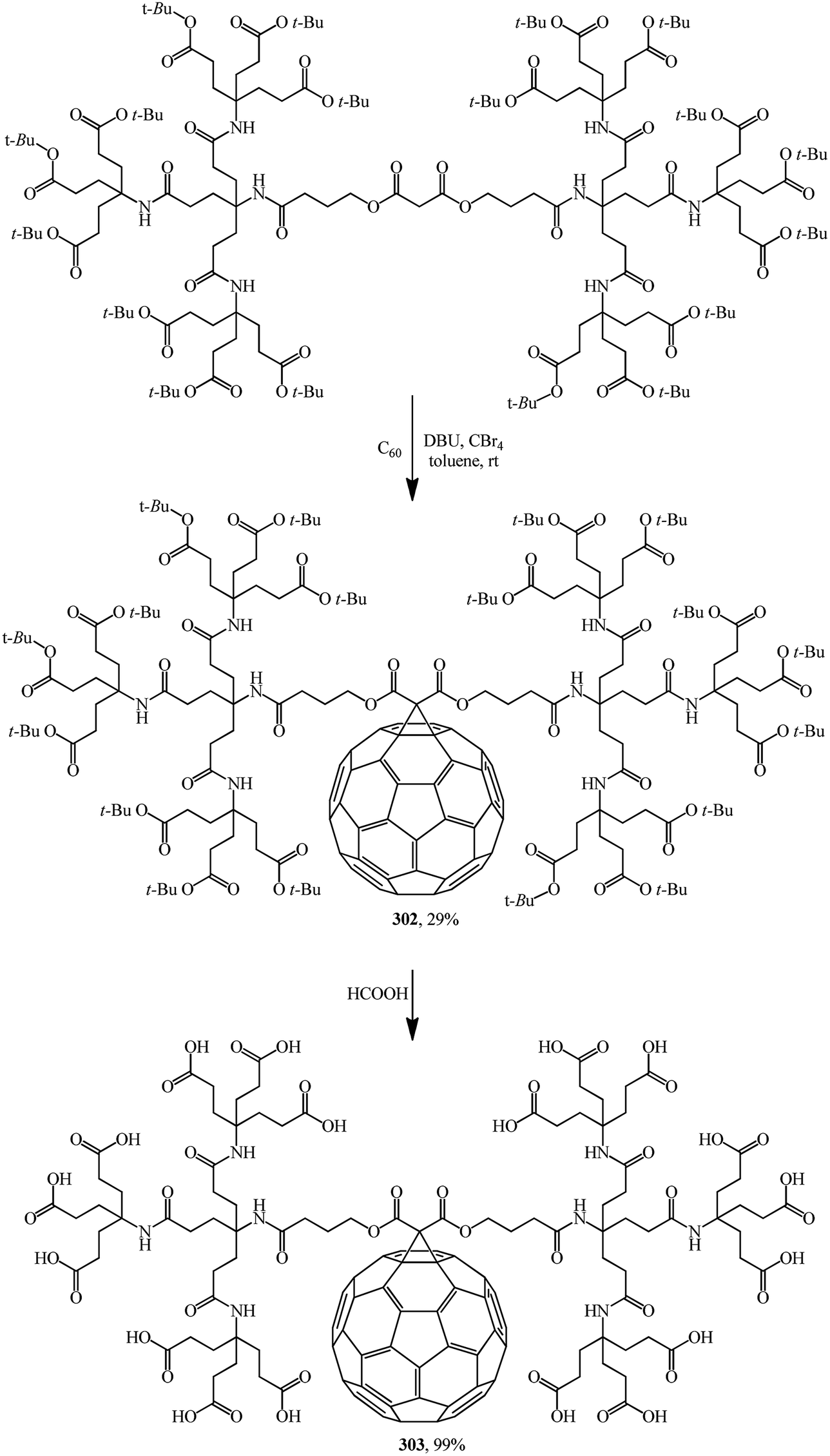 | ||
| Scheme 84 | ||
The same team performed a synthesis of non-symmetrical fullerene-containing dendrite diads terminated by a metallo-tetraphenylporphyrin (M = ZnII, CoII, CoIII) on one end and by Newkome-type amide dendrons on the other end.150 Cyclopropanation of C60 with second-generation dendrons zinc(II)–porphyrin and cobalt(II/III)–porphyrin gives fullerene monoadducts 304 and 305 that represent a new type of dendritic diads (Scheme 85).
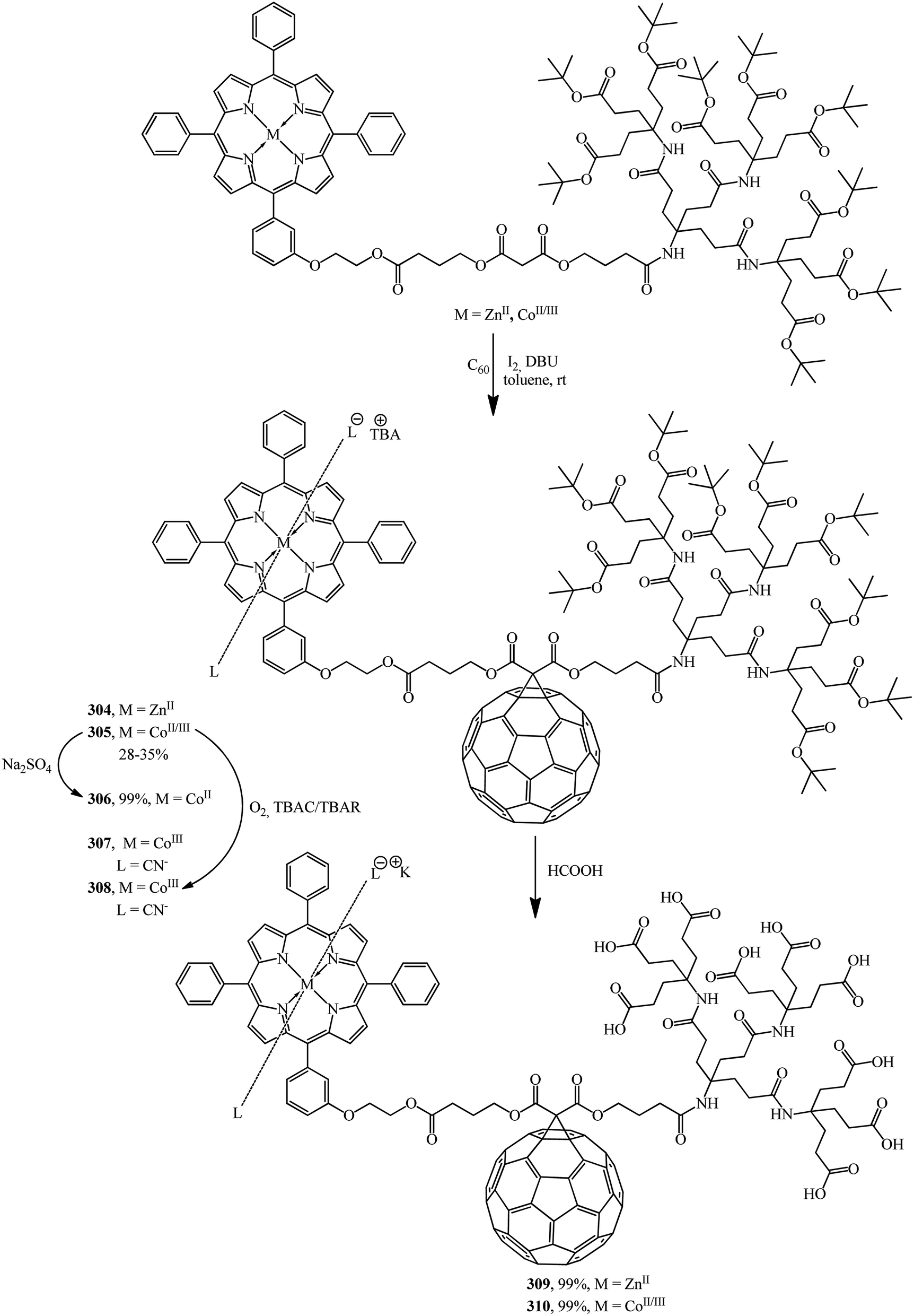 | ||
| Scheme 85 | ||
The formation of Co in oxidation states +II and +III was achieved by treatment with Na2S2O4 or with O2 in the presence of CN− and SCN− ions, respectively, to give a paramagnetic complex cobalt(II)–porphyrin–dendrimer–fullerene 306 and diamagnetic axially coordinated complexes cobalt(III)–porphyrin–dendrimer–fullerene 307 and 308. Deprotection of the tert-butyl groups terminating the dendritic branch of diads 304 and 305 generates the corresponding water-soluble micellar systems zinc(II)–porphyrin–[60]fullerene 309 and cobalt(II/III)–porphyrin–[60]fullerene 310 in quantitative yields. Due to their amphiphilic character, aggregates are formed in water.
The idea to create polyamide malonic dendrofullerenes, which correlates with the studies of the Hirsch team, is provided in ref. 151 A synthesis of water-soluble fullerene derivatives bearing a poly(amidoamine) dendron with peripheral carboxylic groups on one side and an alkyne moiety on the other side was also reported.
Fullerodendrimers with tert-butyl ester groups at the periphery (311, 313) were first prepared by treating C60 with unsymmetrical malonates under Bingel–Hirsch reaction conditions (Schemes 86 and 87). The tert-butyl esters were then cleaved using formic acid to give the corresponding first- and second-generation carboxyfullerene derivatives (312, 314) (Schemes 86 and 87). Second-generation fullerodendrimer 314 with eight carboxylic groups at the periphery is easily soluble in water, whereas first-generation compound 312 with its four carboxylic groups can only be solubilized in a basic medium. These compounds self-assemble into micelle-like aggregates probably composed of a C60 cluster surrounded by a poly(amidoamine) shell.
 | ||
| Scheme 86 | ||
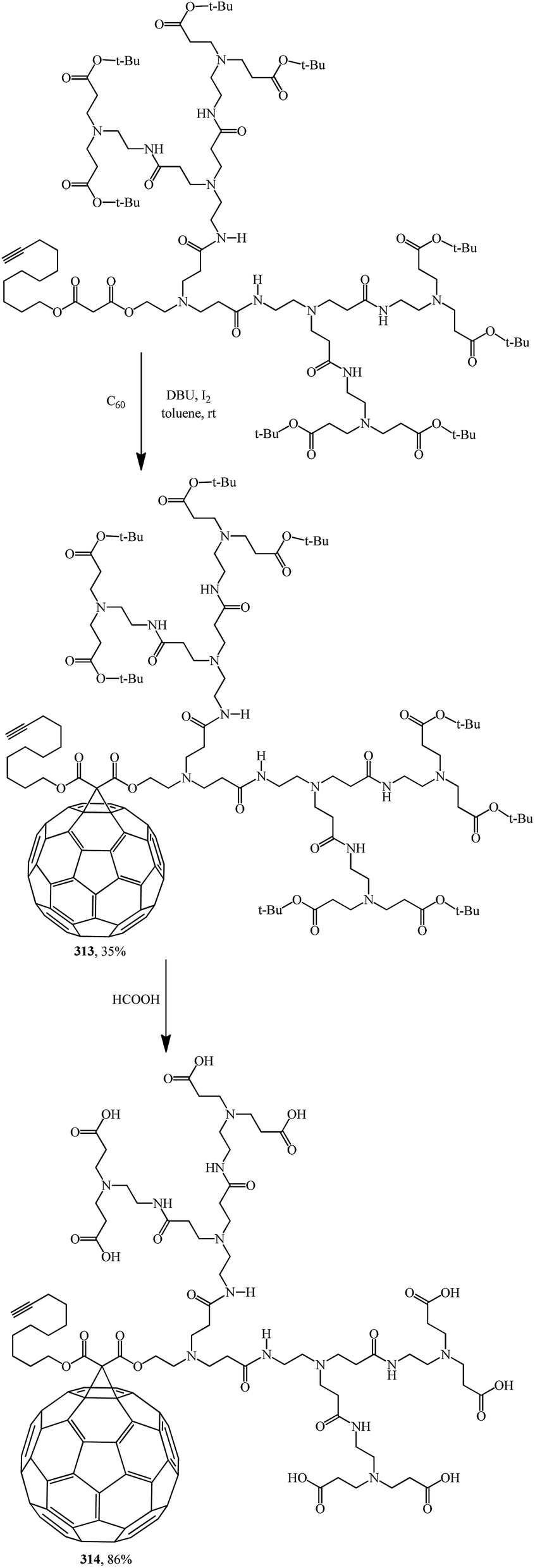 | ||
| Scheme 87 | ||
The alkyne moiety was then used as a chemical anchor to immobilize in water fullerodendrimers on the surface of azido-coated polymer nanoparticles by means of the copper(I)-catalyzed azide and alkyne cycloaddition reaction. At room temperature, this reaction competes with azido cycloaddition onto the fullerene core. Given the high density of anchoring azide groups on the nanoparticle surface and the size of the fullerodendrimers, the unreacted azides are still reactive and are available for subsequent functional arrangements. This strategy paves the way for the design of functional fullerene-rich nanomaterials that could be of interest in the field of materials science.
A wide range of electroactive donor–acceptor systems based on C60, in particular, for light-induced charge separation in artificial photosynthetic systems and photovoltaic devices, was suggested by authors.152 They synthesized novel C60/π-extended tetrathiafulvalene diads 315–320 (Fig. 22) and triads 321–327 (Fig. 23).
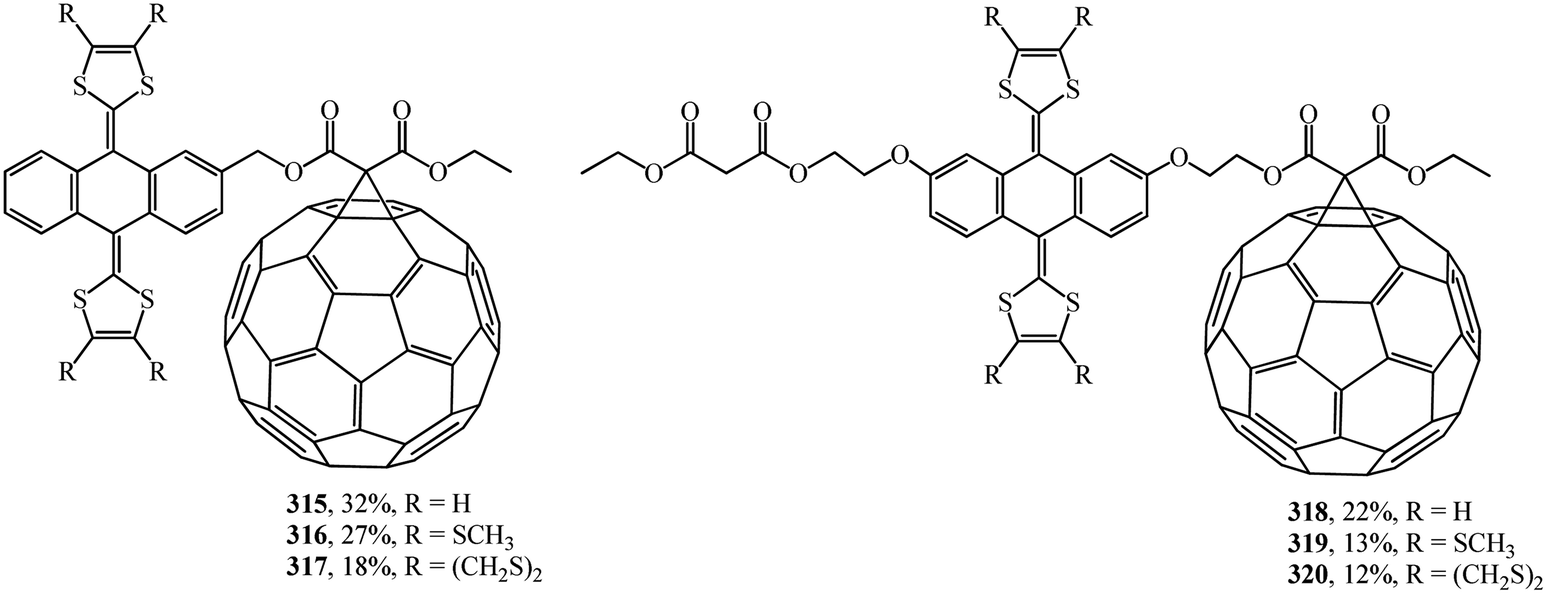 | ||
| Fig. 22 C60/π-extended tetrathiafulvalene diads 315–320. | ||
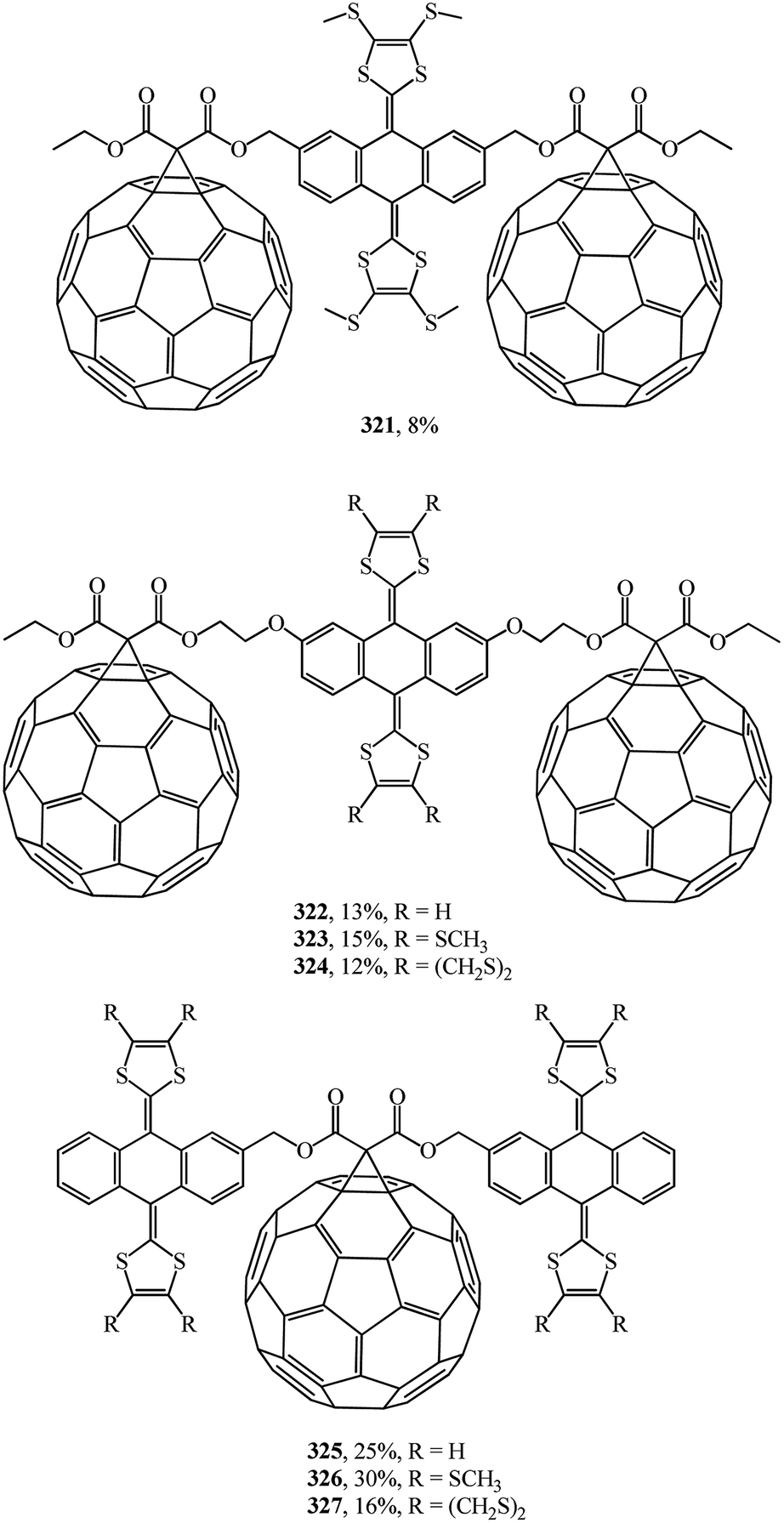 | ||
| Fig. 23 C60/π-extended tetrathiafulvalene triads 321–327. | ||
The oxidative properties of the new compounds were determined by cyclic voltammetry at room temperature. Semi-empirical (PM3) theoretical calculations were carried out to better understand the geometric and electron properties of the systems obtained. Additionally, photophysical measurements (fluorescence and transient absorption spectra) were carried out.
In ref. 153, the synthetic methodology, liquid crystal properties, supramolecular organization and electrochemical behavior of mesomorphic C60–tetrathiafulvalene derivative 328, which is the first liquid crystal representative of this family, are reported (Fig. 24).
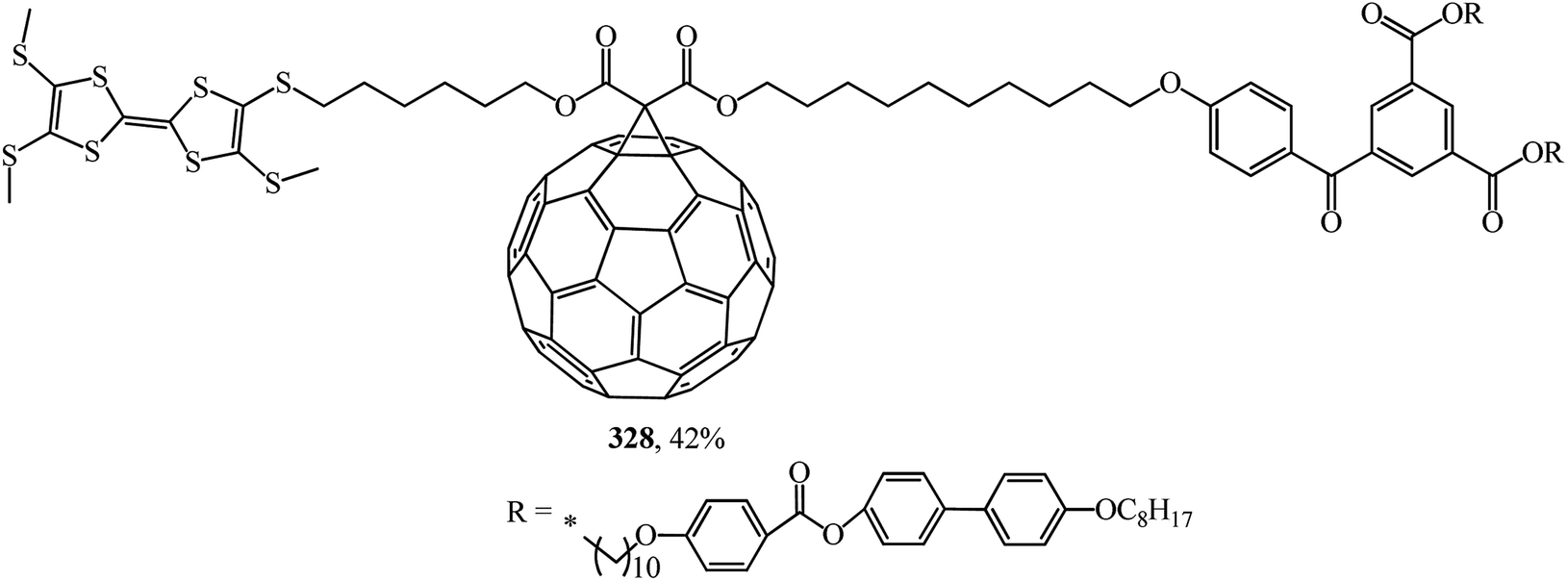 | ||
| Fig. 24 C60–tetrathiafulvalene 328. | ||
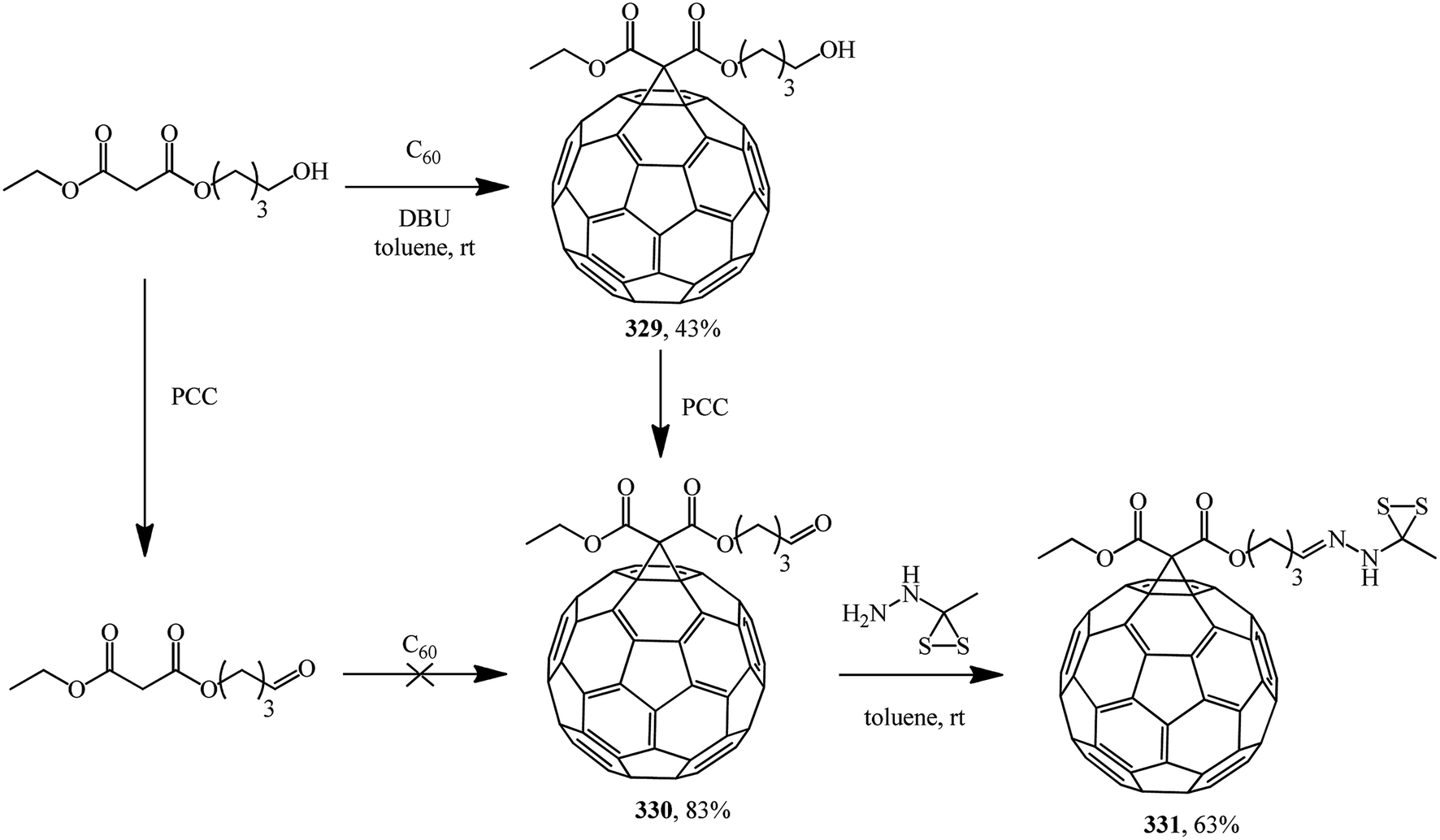
The following standalone studies were performed. Publication154 describes a synthesis of methanofullerene 329, which on treatment with pyridinium chlorochromate (PCC) gives compound 330 (fullerene grafting to a related aldehyde failed, probably due to side reactions with the carbonyl function under basic conditions), which reacts with S-methyl dithiocarbazate to give 331 (Scheme 88).
 | ||
| Scheme 88 | ||
Fullerene-based Schiff bases are prepared and used to stabilise the [ReO]3+ core. To this end, the authors conducted ligand exchange reactions of [NBu4][ReOCl4] with bidentate SN Schiff bases to produce mixed-ligand C60/Re complexes for radiopharmaceutical purposes.
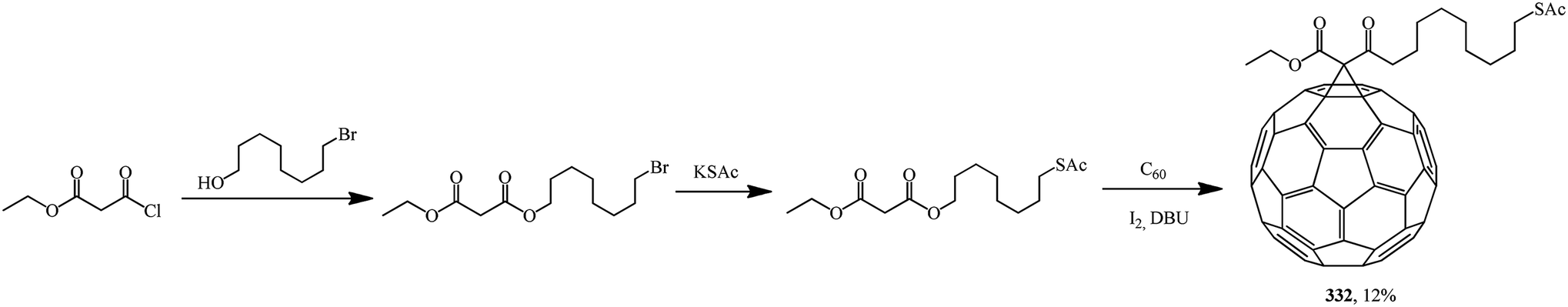
Based on 8-(acetylthio)octyl ethyl malonate, 61-ethyloxycarbonyl-61-[8-(acetylthio)octyl-1-oxycarbonyl]-1,2-methano[60]fullerene 332 was synthesized. The latter was converted to a hexakis adduct by adding a malonate containing the 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) moiety (Scheme 89). The catalytic composite C60TEMPO10@Au allowed for the oxidation of various primary and secondary alcohols under mild conditions to the corresponding aldehyde and ketone analogues with efficiencies as high as 79–98%, thus giving values typical of homogeneous catalysis, while retaining at the same time all the advantages of heterogeneous catalysis, e.g., easy separation by filtration from the reaction mixture.155
 | ||
| Scheme 89 | ||
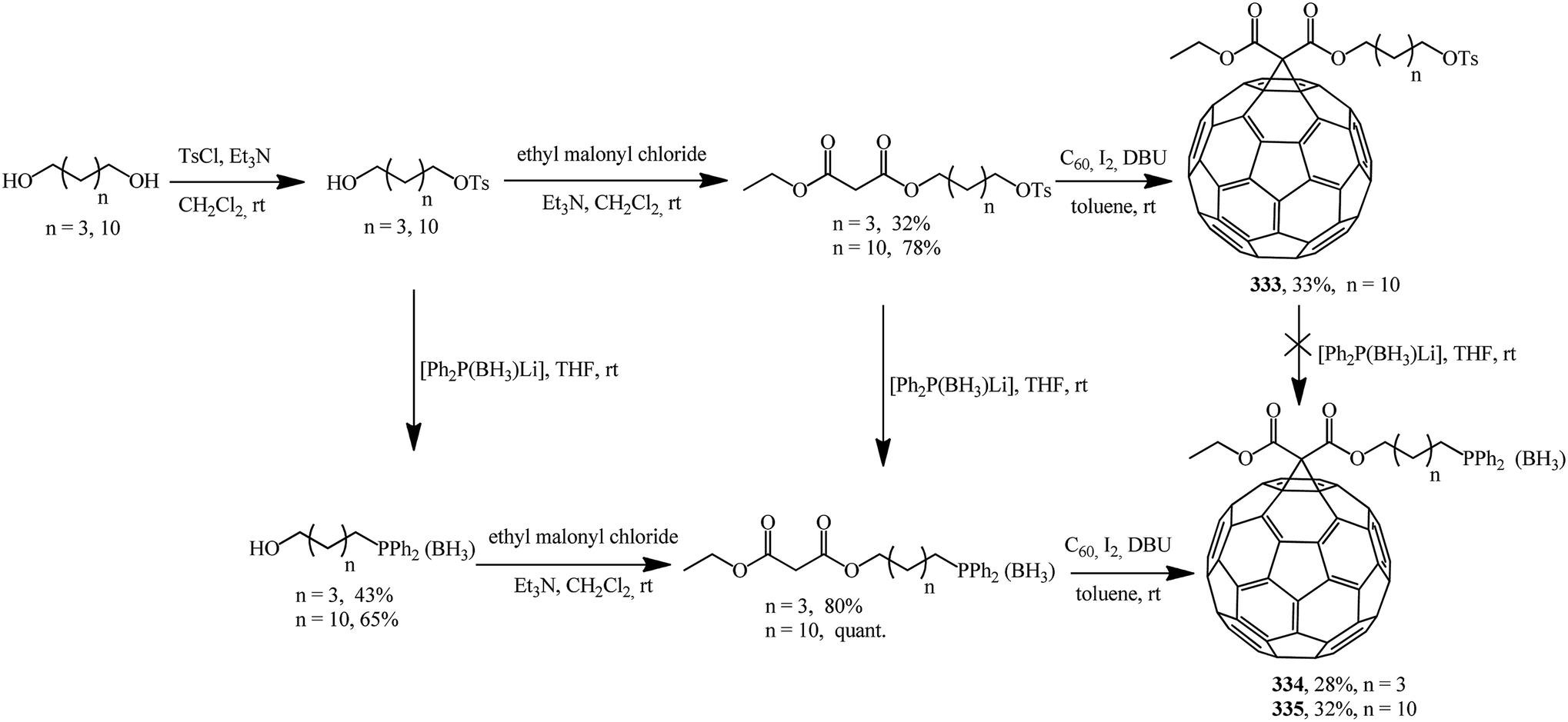
Using asymmetric phosphorus-containing malonates in fullerene cyclopropanation, two series of new phosphine derivatives based on C60 protected by borane 334, 335 were synthesized and characterized (Scheme 90).156
 | ||
| Scheme 90 | ||
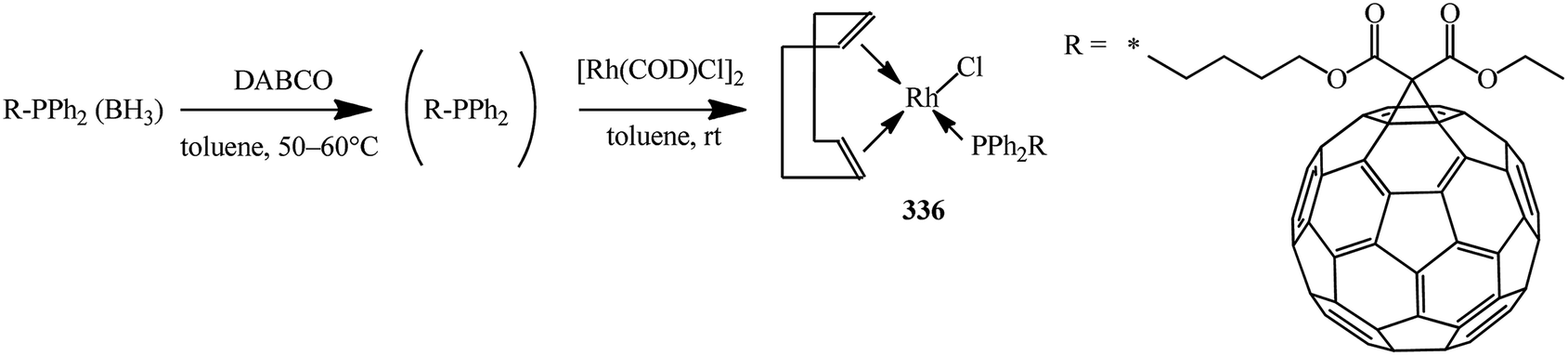
Deprotection of the phosphine–borane complexes was performed using tertiary amines such as DABCO in dry toluene at 50–60 °C under argon atmosphere (Scheme 91). Preliminary complexation experiments with rhodium were performed using chloro(1,5-cyclooctadiene)rhodium(I) dimer, whereas complexation with rhenium was carried out using bis(perthiobenzoato)(dithiobenzoato)rhenium(III) 336.
 | ||
| Scheme 91 | ||
The presence of fullerene covalent-bound to the polymer chain results in a completely new type of macromolecules whose repeating structure contains carbon nanoparticles with sizes comparable to the correlation distances in high-molecular compounds. As a result, fullerene-containing polymers acquire unique physical properties that are significantly different from the polymers containing no C60.157–159 Incorporation of fullerene into polymer chains in the role of photo- and electroactive blocks (a subject of intense competing research in recent years) should afford new materials with outstanding structural, electrochemical and photophysical properties. It is the high content of C60 in macromolecules that explains the appearance of extraordinary physical properties in such polymers, which can be achieved upon conversion of a fullerene-containing low-molecular compound into a high-molecular one. It should be noted that, despite the existing variety of methods for fullerene incorporation into polymer chains, the most effective way to obtain macromolecules with the highest possible C60 content involves the polymerization of a fullerene-containing monomer.
Scientific literature presents examples of synthesizing low-molecular functionalized C60 methano derivatives. As a rule, these are compounds containing functional groups intended for involvement in polymer analogous reactions and polycondensation processes.
A prepolymer pre-synthesized from polyethylene glycol methyl ether and malonyl chloride was treated with C60 under Bingel–Hirsch conditions in order to obtain fullerene-containing high-molecular compound 337 (Scheme 92).160
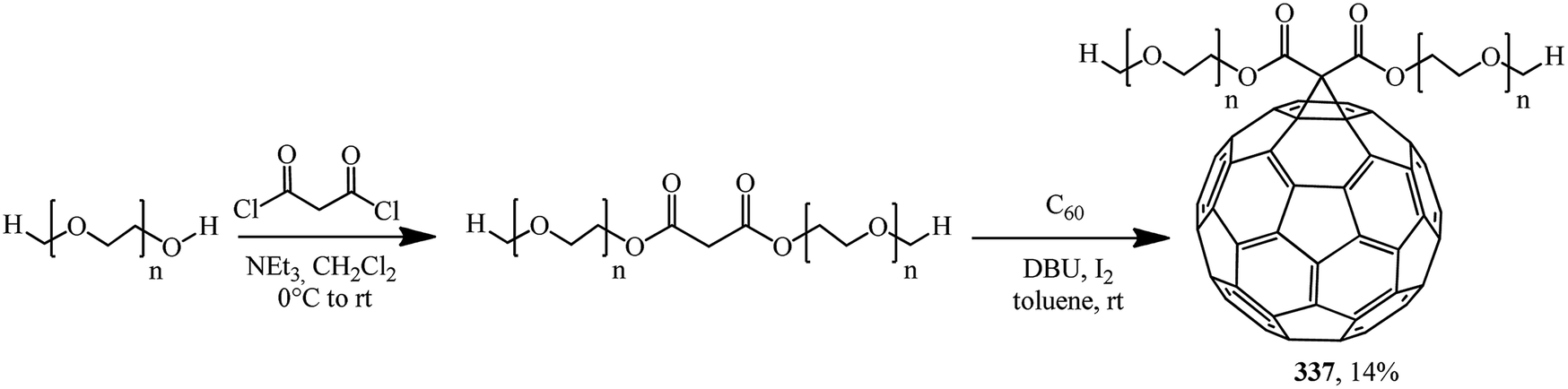 | ||
| Scheme 92 | ||
The low yield of the product can be easily explained by steric hindrance. As a rule, this kind of polymer analogous reactions produce polymers containing various units, where not each repeating unit of the macromolecule contains the C60 “framing group”. As a result, the regular structure of the polymer is lost and hence the performance characteristics of the target product are affected.
Some samples from the series of fullerenes covalent-functionalized with polyethylene glycol were included as additives to CH3NH3PbI3 (∼10−5 mol mol−1) to the most common organic–inorganic perovskite used in perovskite solar cells. A photovoltaic study performed using these materials shows the following beneficial effects: reduced hysteresis and high moisture resistance, owing to which solar cells retain up to 97% of their initial power conversion efficiency. The observed effect is most likely due to the hygroscopicity of polyethylene glycol macromolecular chains, which helps water to be retained, thereby preventing perovskite degradation.
Yet another example of polymer analogous cycloaddition is reported in ref. 83 It is suggested to bind the fullerene frame located in the side chain to the main chain through a 1,2,3-triazole derivative. Preliminarily, radical polymerization is used to obtain a block copolymer of styrene and propargyloxy styrene with a malonate group in the side chain, namely, 3-azidopropyl octadecylmalonate. The latter is then used in cycloaddition to C60 (Bingel–Hirsch) to give fullerene-containing functionalized copolymer 338 with good solubility in common organic solvents and a number-average molecular mass of 71000 g mol−1 (Scheme 93).
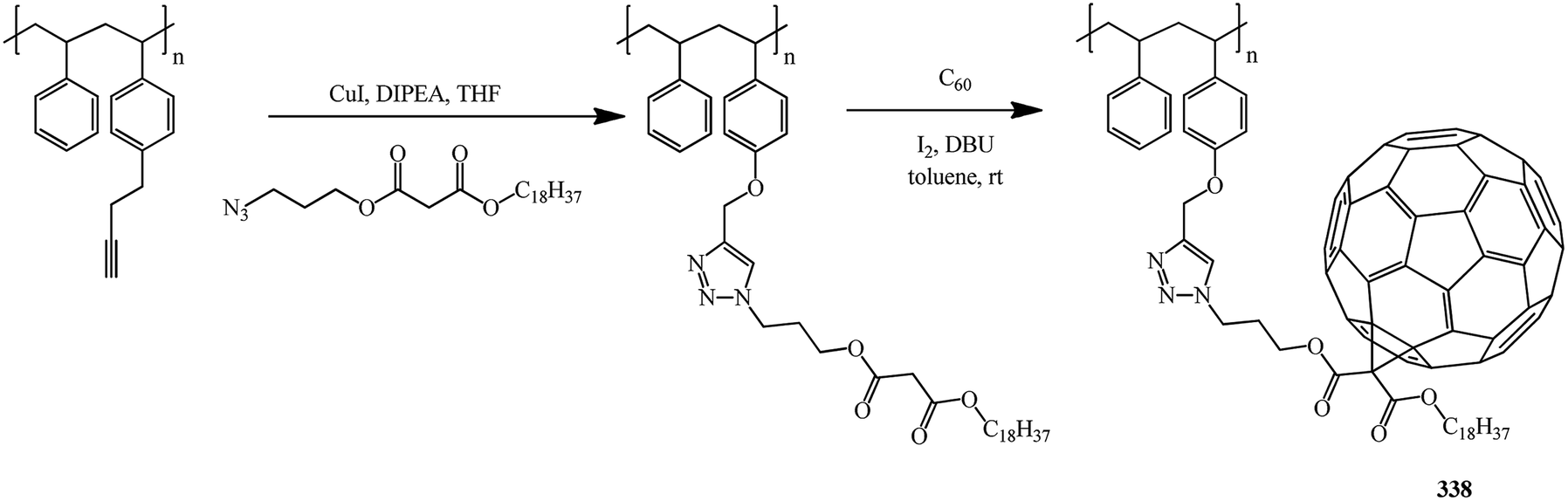 | ||
| Scheme 93 | ||
A multistep synthetic procedure for preparing novel C60-anchored two-armed poly(tert-butyl acrylate) was developed. For this purpose, two-armed poly(tert-butyl acrylate) bearing a malonate ester core with well-controlled molecular weight (number-average molecular mass: 14700 g mol−1) was synthesized by atom transfer radical polymerization. An effective Bingel reaction between C60 and the well-defined polymer was then carried out to yield C60-anchored polymer 339 (Scheme 94). A related dibromo-functionalized methanofullerene 340 was also synthesized.161
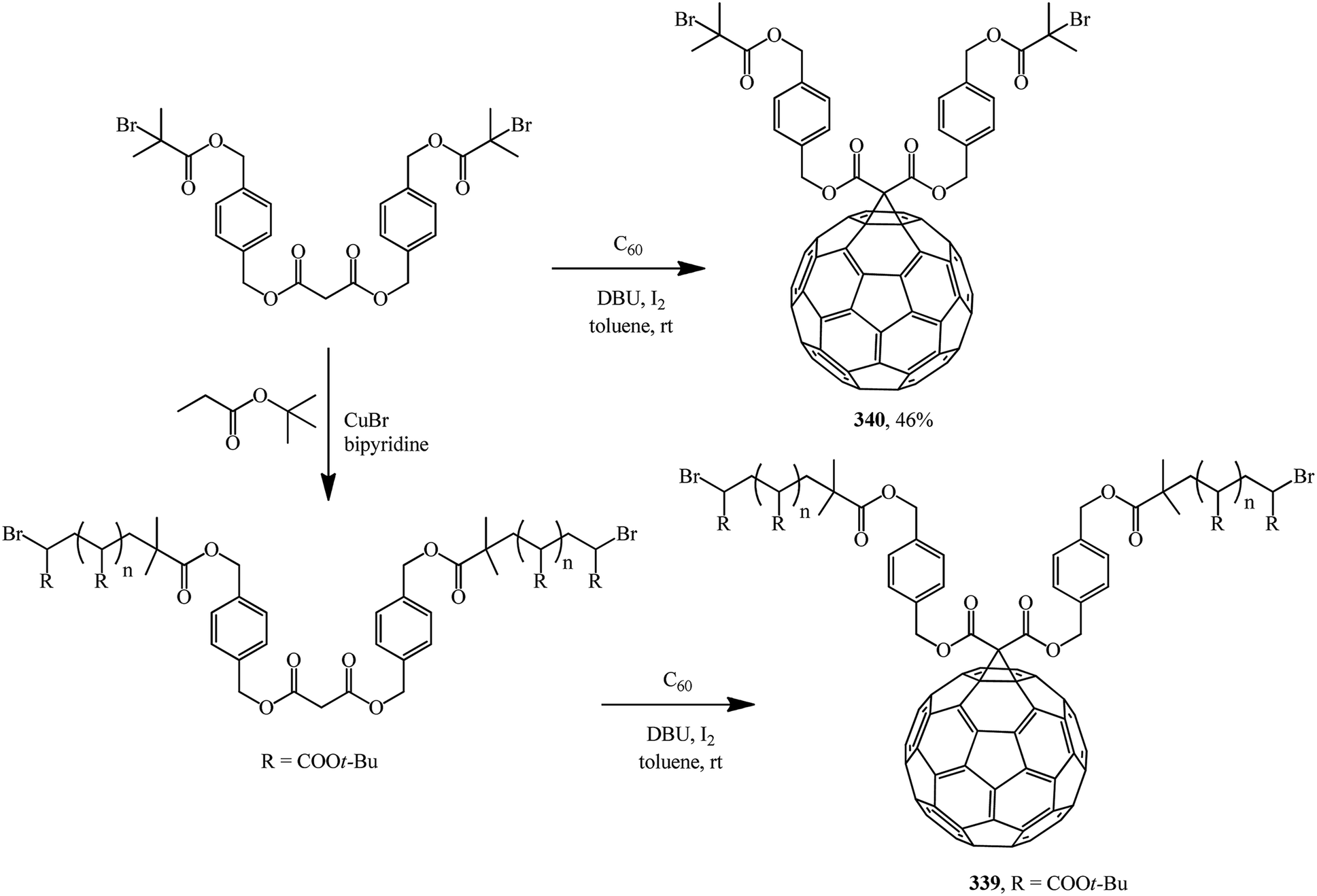 | ||
| Scheme 94 | ||
Scientific literature contains very interesting examples of substrates that can be used as cyclopropanating agents as alternatives to malonic acid derivatives. For example, information is available that spiromethanofullerenes containing a reactive free formyl 341 and diacetyl 342 groups were synthesized by the Bingel–Hirsch process (Scheme 95).162
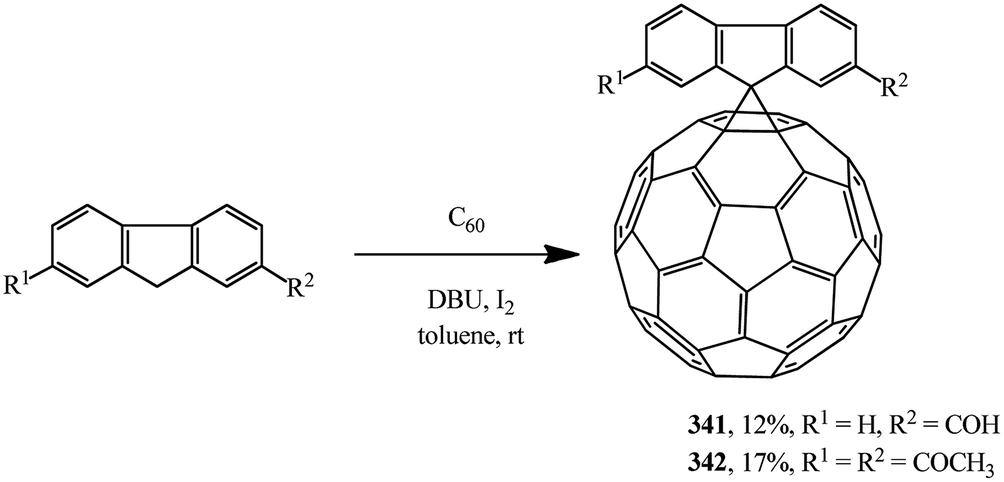 | ||
| Scheme 95 | ||
Meldrum's acid derivatives are also original cyclopropanating agents. Under Bingel–Hirsch reaction conditions, they gave a number of methano derivatives 343–353 in moderate yields (Scheme 96).163
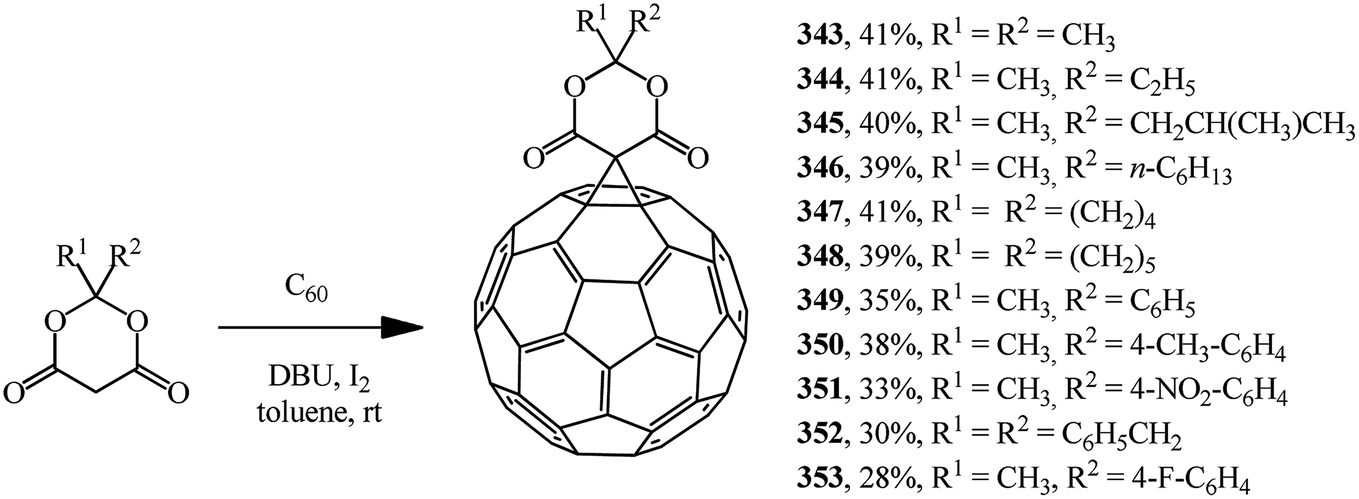 | ||
| Scheme 96 | ||
According to the authors, the carbene intermediate mechanism is most acceptable for this reaction (Scheme 97).
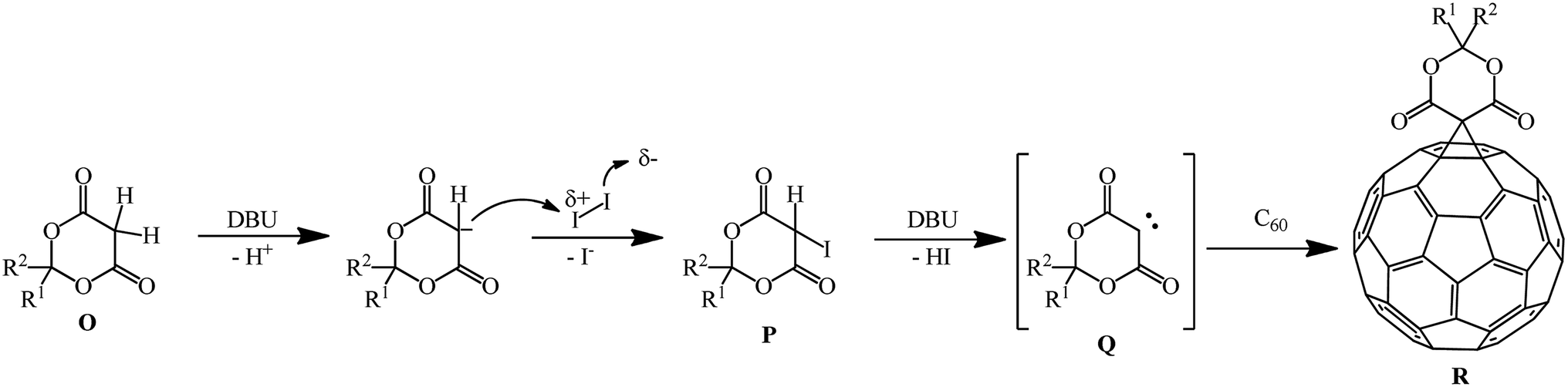 | ||
| Scheme 97 | ||
Meldrum's acid O reacts with I2 under basic reaction conditions in the presence of DBU to give α-iodine-substituted product P, which eliminated HI to give carbene intermediate Q. Then it added to C60 to form the mono-addition product Q or a double addition product, depending on the concentration of carbene intermediate R in the reaction system.
N-(Diphenylmethylene)glycinate esters were suggested as non-traditional cyclopropanating agents. Under Bingel–Hirsch reaction conditions, they were converted to methanofullerene iminoesters 354, 355,164 356, 357 (ref. 165) (Scheme 98).
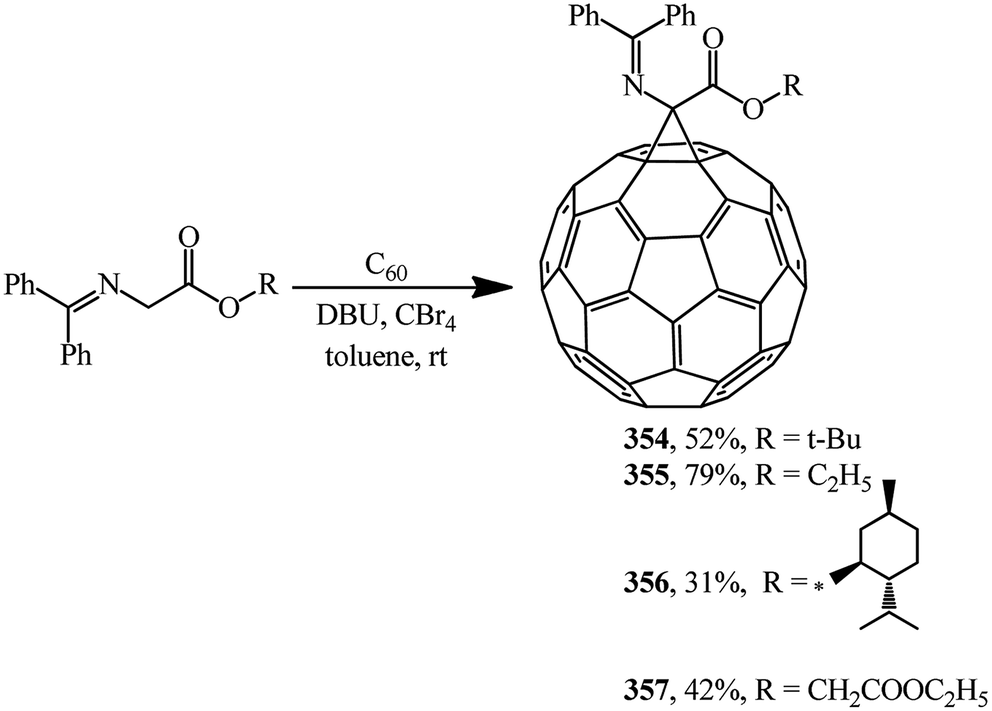 | ||
| Scheme 98 | ||
Based on a number of thiophene-containing ketones and C60 in the Bingel–Hirch process, mono-ketone modified methanofullerene derivatives 358–361 were synthesized (Scheme 99).166
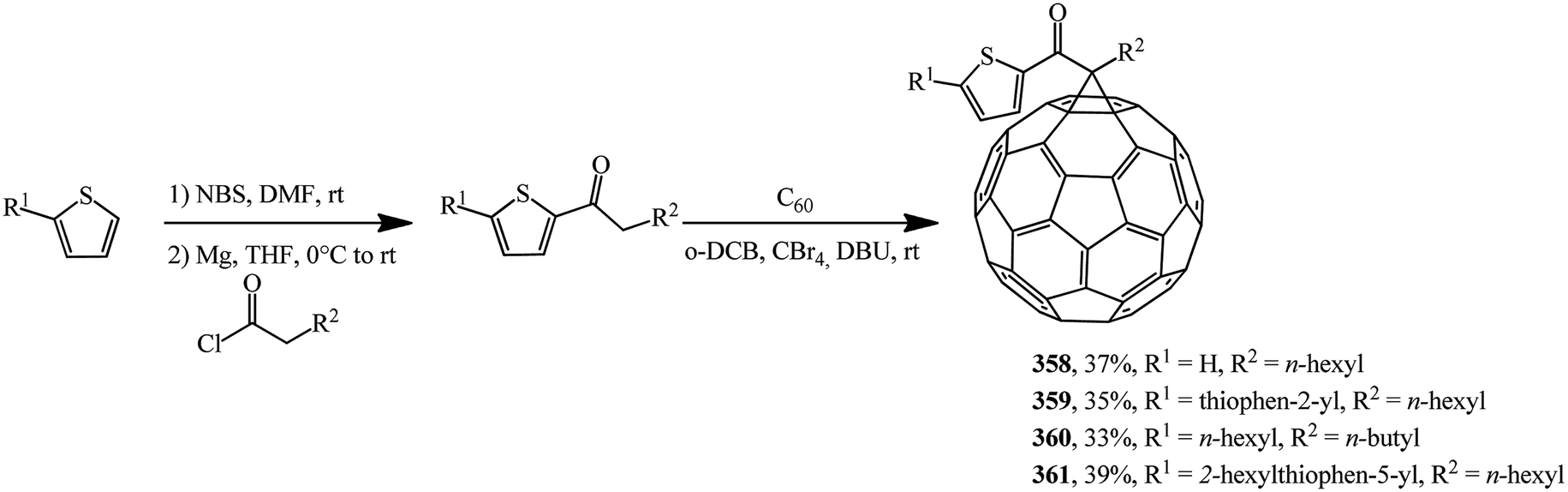 | ||
| Scheme 99 | ||
The authors note that compound 361 is the best one in this series. It shows the highest efficiency of 1.27% under pristine conditions. Methanofullerene 361 has the highest solubility and the lowest unoccupied molecular orbital level among newly synthesized derivatives in this paper and it can be affect to modify the donor–acceptor bulk-heterojunction and improve electron attraction ability from P3HT donor.
3. Other methods for the synthesis of methanofullerenes by the addition–elimination mechanism
Methanofullerenes can be obtained in low yields (15–25%) by reactions of phosphorous ylides with C60. The first derivatives of [6,6]-closed methanofullerenes 362–368 obtained by this scheme were described in ref. 167 in 1994 (Scheme 100).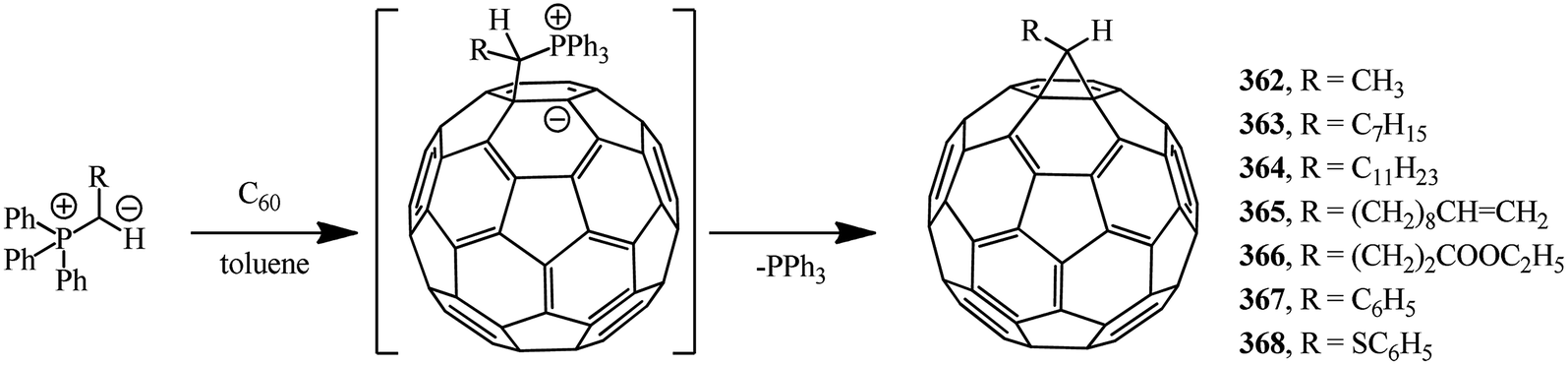 | ||
| Scheme 100 | ||
Wilson et al.168 used sulfonium ylides instead of phosphorus ylides. In this case, [6,6]-methanofullerenes were obtained from stabilized sulfonium ylides 369–374, which in turn were obtained by treatment of sulfonium salts with a base (NaH or NaOH). It was noted that the reaction rates depended on the ylide nucleophilicity, while the yield of the target product reached 58% (Scheme 101).
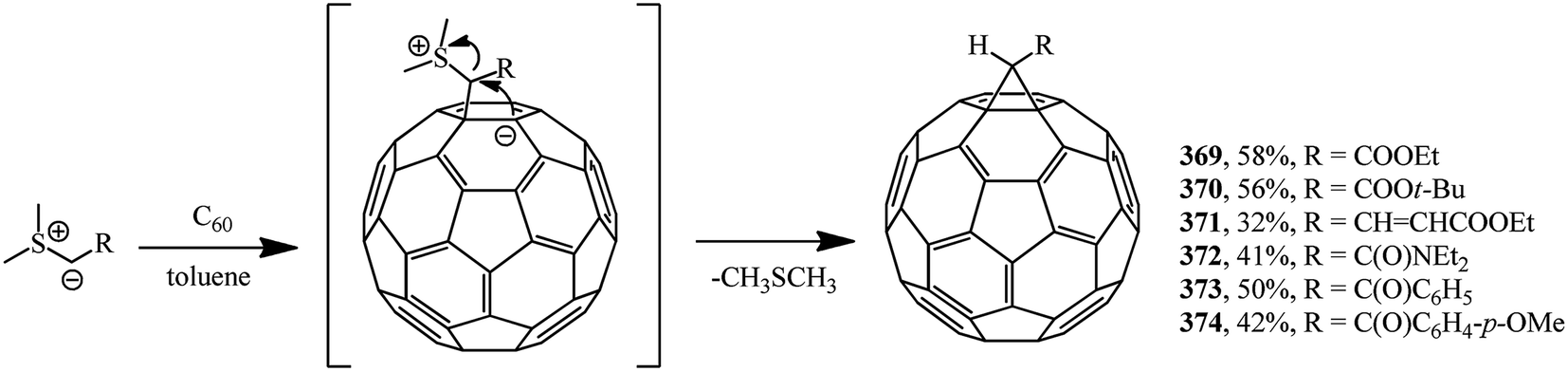 | ||
| Scheme 101 | ||
Examples of the participation of sulfur ylides in the synthesis of methanofullerenes are given in ref. 169 The reaction of fullerene with dimethylsulfonium α-formylalkylide gave novel 2,2-[60]fulleroalkanal 375 having a formyl group at the bridge-head carbon. This derivative 375 was readily converted into a variety of N-aryl-2,2-[60]fulleroaldimines 376–380 by condensation with aromatic amines (Scheme 102). One of them showed efficient quenching of the fullerene core fluorescence due to photoinduced intramolecular electron transfer.
 | ||
| Scheme 102 | ||
A new fullerene reaction with silylated nucleophiles was described.170 The cyclopropanation of C60 with silylated nucleophiles, such as silyl ketene acetals, silyl ketene thioacetals, and silyl enol ethers derived from α-halo carbonyl compounds, smoothly occurred in the presence of KF/18-crown-6 to give the corresponding methanofullerene derivatives 381–389 in moderate to good yields (Scheme 103).
 | ||
| Scheme 103 | ||
Further, the same team171 synthesized 2,2-[60]fullerenoalkanals 390–394 by a fluoride ion mediated reaction of fullerene with 2-bromoenol silyl ethers that were easily prepared by 2-bromination of the corresponding enol silyl ethers (Scheme 104). The reactivity differed significantly depending on the stability of the 2-bromoenol silyl ethers. High yields and selectivities were achieved in the reaction of 2-bromoenol trimethylsilyl ethers under mild conditions (KF/18-crown-6-ether). On the other hand, the reaction of stable 2-bromoenol tert-butyldimethylsilyl ethers required the use of more reactive tetrabutylammonium fluoride as a fluoride ion source.
 | ||
| Scheme 104 | ||
This rearrangement method allows various substituents, including large bulky groups, to be incorporated in C60. An additional advantage of the method is that the Bingel reaction is performed in an almost neutral medium. Reactions of C60 with similar silylketene acetals under photochemical conditions and without F− result in dihydrofullerene acetate rather than methanofullerenes.172
An original synthetic method was suggested: hard-to-access aminomethanofullerenes were synthesized by the reaction of carbon clusters with cyanoacrylates in the presence of ethylmagnesium bromide and Ti(Oi-Pr)4 (catalyst).173 Cyanoacrylates containing cyclic or symmetrical substituents are used to obtain primary methanofullerenes 359–398 (Scheme 105) and 399–402 (Fig. 25).
 | ||
| Scheme 105 | ||
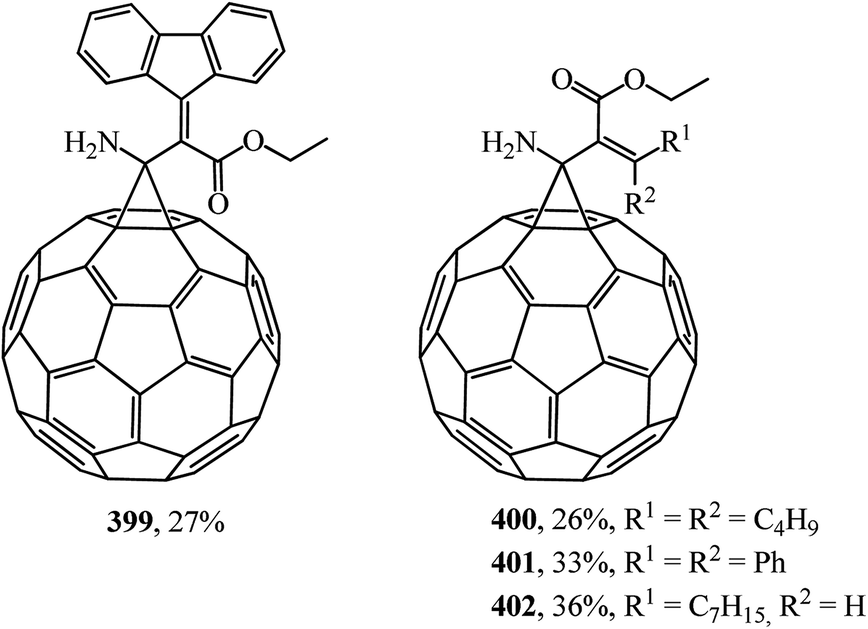 | ||
| Fig. 25 Aminomethanofullerenes 399–402. | ||
A possible process mechanism involves the following. In the first step, the reaction of Ti(Oi-Pr)4 with EtMgBr affords dialkoxytitanocyclopropane S, which exists in equilibrium with the ethylene complex. The presence of fullerene in the reaction mixture causes a displacement of the ethylene molecule from complex S to give the key intermediate of this reaction, i.e., fullerotitanacyclopropane T. The subsequent reaction of intermediate T with cyanoacrylate produces azatitanacyclic C60 adduct U, which reacts with EtMgBr to regenerate dialkoxytitanacyclopropane S, thus completing the catalytic cycle. Upon completion of the reaction, complex U undergoes hydrolysis to give aminomethanofullerenes V (Scheme 106).
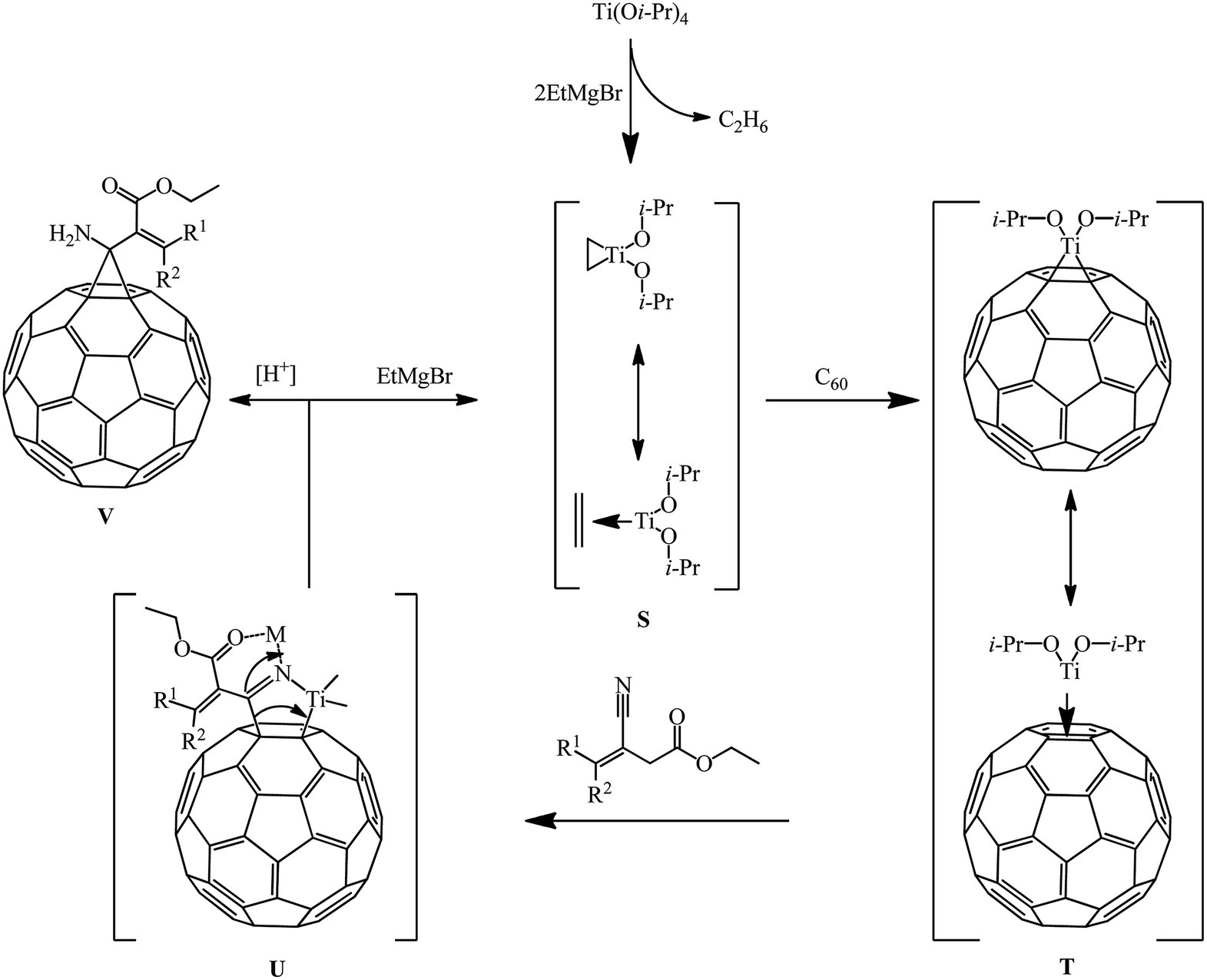 | ||
| Scheme 106 | ||
To synthesize secondary aminofullerenes within the described approach, isonitriles are used instead of cyanoacrylates to give N-substituted amino methanofullerenes 403–409 (Fig. 26).174
 | ||
| Fig. 26 Structures of N-substituted amino methanofullerenes 403–409. | ||
O. Sinyashin's team performed a series of studies by to synthesize methanofullerenes with involvement of phosphorus-containing compounds. Reactions of cyclic β-diketones (acenaphthenequinone, aceanthrenequinone, and N-alkylisatins) with hexaethyltriaminophosphine in the presence of fullerene C60 under mild conditions were studied. This process occurs via deoxygenation of a dicarbonyl compound by a P(III) derivative and is likely to involve the intermediate formation of β-ketocarbenes to give the corresponding spirobicycles 410–415 (ref. 175) and 416 (ref. 176) (Scheme 107). Studies on the electrochemical behavior of the methanofullerenes obtained made it possible to recommend them as acceptor components in organic solar cells.177
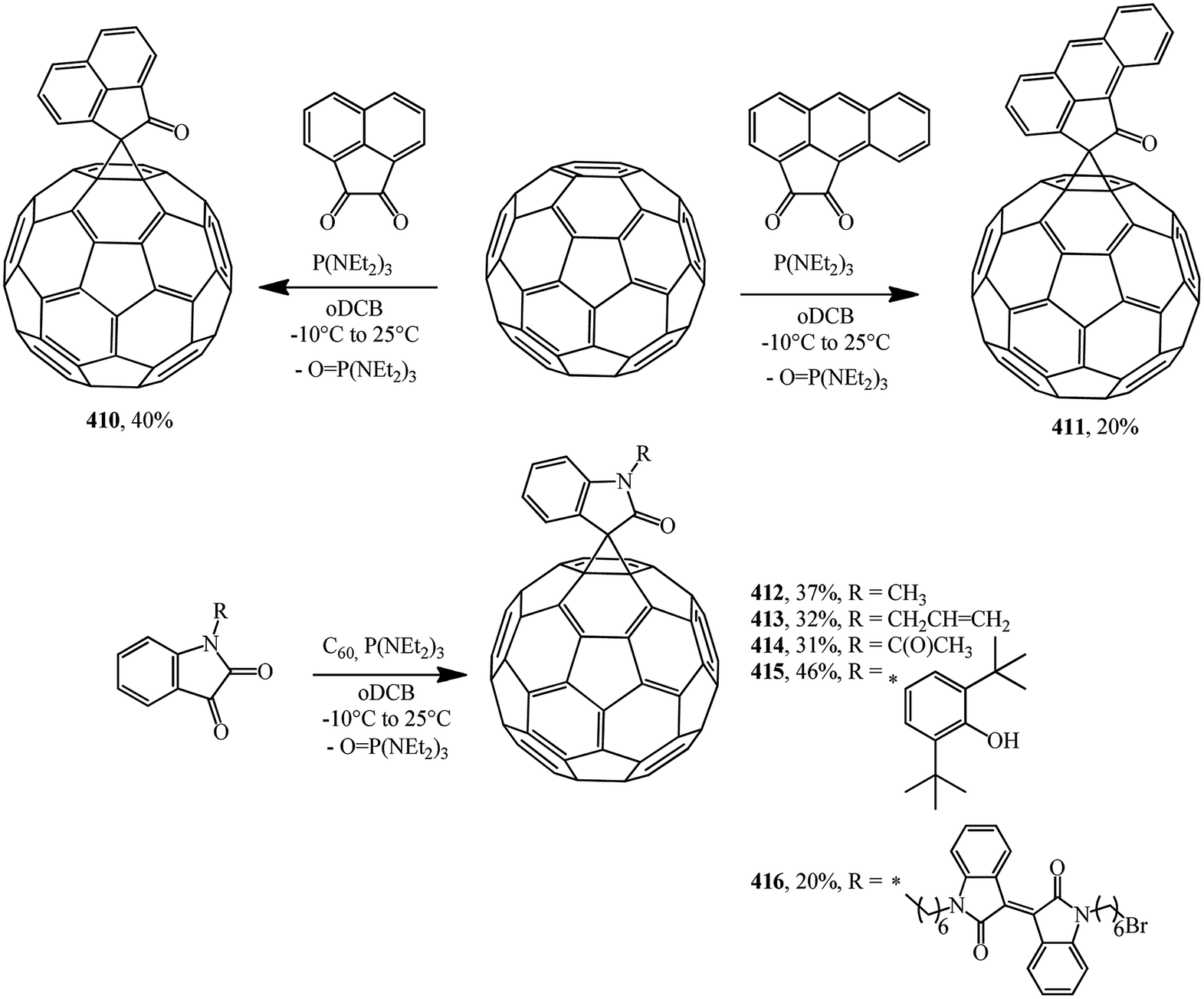 | ||
| Scheme 107 | ||
Conclusions
Thus, our analysis of literature sources dealing with the nucleophilic cyclopropanation of fullerene with stabilized carbanions by the addition–elimination nucleophilic cyclopropanation mechanism led to the following conclusions. The Bingel reaction is among the most efficient methods for synthesizing methanofullerenes. It is almost universal and allows one to synthesize C60 methano derivatives from halo-substituted substrates containing reactive methylene functions with various structures. The advantages of this method for fullerene functionalization include simple and mild reaction conditions that provide good yields of the target products. However, this technique assumes the presence of an additional stage, i.e., preliminary halogenation of a cyclopropanating agent.The Bingel approach modified by Hirsch allows one to obtain methanofullerenes by a “one-pot” reaction: a malonate substrate, CBr4 (or I2) and DBU as a base are used simultaneously. In this case, the synthesis of adducts is performed by direct treatment of C60 with malonates and their yields increase somewhat, as the authors note.
The synthesis of methanofullerenes can be performed using sulfur or phosphorus ydides, silyl derivatives, isonitriles, cyanoacrylates, cyclic β-diketones, etc. as alternative nucleophilic cyclopropanating agents. The base needs to be selected individually in such cases.
In all the above examples of fullerene C60 functionalization, selective formation of [6,6]-bridging adducts occurs. That is why the nucleophilic cyclopropanation of fullerene that occurs by the addition–elimination mechanism is currently used most widely by organic chemists in order to synthesize various methanofullerenes.
Conflicts of interest
Authors declare that they have no conflicts of interest.Acknowledgements
This study was performed in the framework of state assignment (project No. AAAA-A19-119020890014-7).References
- F. Diederich and C. Thilgen, Science, 1996, 271, 317–324 CrossRef CAS.
- S. Jennepalli, S. G. Pyne and P. A. Keller, RSC Adv., 2014, 4, 46383–46398 RSC.
- R. Ganesamoorthy, G. Sathiyan and P. Sakthivel, Sol. Energy Mater. Sol. Cells, 2017, 161, 102–148 CrossRef CAS.
- S. Zhu, F. Li and G. Wang, Chem. Soc. Rev., 2013, 42, 7535–7570 RSC.
- K. Winkler, A. L. Balch and W. Kutner, J. Solid State Electrochem., 2006, 10, 761–784 CrossRef CAS.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS.
- M. A. Yurovskaya and I. V. Trushkov, Russ. Chem. Bull., 2002, 51, 367–443 CrossRef CAS.
- M. Nazario, S. Luis, I. Beatriz and P. Ignacio, Chem. Rev., 1998, 98, 2527–2548 CrossRef.
- D. Manolis and M. Orfanopoulos, Chem. Rev., 2013, 113, 5262–5321 CrossRef.
- C. Li, H. Yip and A. K.-Y. Jen, J. Mater. Chem., 2012, 22, 4161 RSC.
- C. Kästner, C. Ulbricht, D. A. M. Egbe and H. Hoppe, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1562–1566 CrossRef.
- Y. He and Y. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- B. C. Thompson and M. J. Fréchet Jean, Angew. Chem., Int. Ed. Engl., 2008, 47, 58–77 CrossRef CAS.
- F. Diederich, L. Isaacs and D. Philp, Chem. Soc. Rev., 1994, 23, 243–255 RSC.
- C. Brabec, U. Scherf and V. Dyakonov, Organic Photovoltaics: Materials, Device Physics, and Manufacturing Technologies, Wiley-VCH Verlag GmbH & Co. KGaA, 2nd edn, 2014 Search PubMed.
- A. Hirsch and M. Brettreich, Fullerenes. Chemistry and Reactions, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005, p. 445 Search PubMed.
- N. Martín and F. Giacalone, Fullerene Polymers: Synthesis, Properties and Applications, 2009, p. 320 Search PubMed.
- R. F. Verner and C. Benvegnu, Handbook on fullerene: Synthesis, properties, and applications, Nova Science Publishers, Inc, United States, 2012, p. 548 Search PubMed.
- A. Hirsch, Synthesis, 1995, 8, 895–913 CrossRef.
- K. Raghavachari and C. Sosa, Chem. Phys. Lett., 1993, 209, 223–228 CrossRef CAS.
- C. Bingel, Chem. Ber., 1993, 126, 1957–1959 CrossRef CAS.
- H. Hauptmann, Tetrahedron Lett., 1974, 15, 3587–3588 CrossRef.
- H. L. Anderson, R. Faust, Y. Rubin and F. Diederich, Angew. Chem., 1994, 106, 1427–1429 CrossRef CAS.
- H. L. Anderson, R. Faust, Y. Rubin and F. Diederich, Angew. Chem., Int. Ed. Engl., 1994, 33, 1366–1368 CrossRef.
- P. Timmerman, H. L. Anderson, R. Faust, J.-F. Nierengarten, T. Habicher, P. Seiler and F. Diederich, Tetrahedron, 1996, 52, 4925–4947 CrossRef CAS.
- T. Habicher, J.-F. Nierengarten, V. Gramlich and F. Diederich, Angew. Chem., Int. Ed. Engl., 1998, 37, 1916–1919 CrossRef CAS.
- D. Armspach, E. C. Constable, F. Diederich, C. E. Housecroff and J.-F. Nierengarten, Chem. Commun., 1996, 2009–2010 RSC.
- E. C. Constable, A. M. W. C. Thompson, N. Armaroli, V. Balzani and M. Maestri, Polyhedron, 1992, 11, 2707–2709 CrossRef CAS.
- E. C. Constable and M. D. Ward, J. Chem. Soc., Dalton Trans., 1990, 1405–1409 RSC.
- J.-F. Nierengarten, A. Herrmann, R. R. Tykwinski, M. Riittimann and F. Diederich, Helv. Chim. Acta, 1997, 80, 293–316 CrossRef CAS.
- T. Chuard and R. Deschenaux, Helv. Chim. Acta, 1996, 79, 736–741 CrossRef CAS.
- X. Camps, H. Schönberger and A. Hirsch, Chem.–Eur. J., 1997, 3, 561–567 CrossRef CAS.
- S. F. Marcel, K. J. Lie and S. Cheung, Lipids, 1998, 33, 729–732 CrossRef.
- S. Chen, X. Du, G. Ye, J. Cao, H. Sun, Z. Xiao and L. Ding, J. Mater. Chem., 2013, 1, 11170–11176 RSC.
- Y.-P. Sun, R. Guduru, G. E. Lawson, J. E. Mullins, Z. Guo, J. Quinlan, C. E. Bunker and J. R. Gord, J. Phys. Chem. A, 2000, 104, 4625–4632 CrossRef CAS.
- T. Wharton, V. U. Kini, R. A. Mortis and L. J. Wilson, Tetrahedron Lett., 2001, 42, 5159–5162 CrossRef CAS.
- N. D. McClenaghan, C. Absalon and D. M. Bassani, J. Am. Chem. Soc., 2003, 125, 13004–13005 CrossRef CAS.
- P. A. Padmawar, T. Canteenwala, S. Verma, L.-S. Tan and L. Y. Chiang, J. Macromol. Sci., Part A: Pure Appl.Chem., 2004, 41, 1387–1400 CrossRef.
- P. A. Padmawar, T. Canteenwala, S. Verma, L.-S. Tan, G. S. He, P. N. Prasad and L. Y. Chiang, Synth. Met., 2005, 154, 185–188 CrossRef CAS.
- P. A. Padmawar, T. Canteenwala, S. Verma, L.-S. Tan and L. Y. Chiang, J. Mater. Chem., 2006, 16, 1366–1378 RSC.
- N. Mackiewicz, T. Bark, B. Cao, J. A. Delaire, D. Riehl, W. L. Ling, S. Foillard and E. Doris, Carbon, 2011, 49, 3998–4003 CrossRef CAS.
- I. A. Nuretdinov, V. P. Gubskaya, L. Sh. Berezhnaya, A. V. Il'yasov and N. M. Azancheev, Russ. Chem. Bull., 2000, 49, 2048–2050 CrossRef CAS.
- V. P. Gubskaya, F. G. Sibgatullina, V. V. Yanilkin, V. I. Morozov, A. V. Toropchina, V. V. Zverev, N. M. Azancheev and I. A. Nuretdinov, Russ. Chem. Bull., 2005, 54, 1467–1472 CrossRef CAS.
- R. Pellicciari, B. Natalini, L. Amori, M. Marinozzi and R. Seraglia, Synlett, 2000, 12, 1816–1818 Search PubMed.
- P. Damlin, M. Hätönen, S. E. Domínguez, T. Ääritalo, H. Kivelä and C. Kvarnström, RSC Adv., 2014, 4, 8391–8401 RSC.
- M. Oçafrain, M. Á. Herranz, L. Marx, C. Thilgen, F. Diederich and L. Echegoyen, Chem.–Eur. J., 2003, 9, 4811–4919 CrossRef.
- A. Giovannitti, S. M. Seifermann, A. Bihlmeier, T. Muller, F. Topic, K. Rissanen, M. Nieger, W. Klopper and S. Bräse, Eur. J. Org. Chem., 2013, 7907–7913 CrossRef CAS.
- S. A. Torosyan, F. A. Gimalova, Y. N. Biglova, V. V. Mikheev and M. S. Miftakhov, Russ. J. Org. Chem., 2012, 48, 736–738 CrossRef CAS.
- S. A. Torosyan, V. V. Mikheev, Y. N. Biglova and M. S. Miftakhov, Russ. J. Org. Chem., 2015, 51, 1057–1060 CrossRef CAS.
- S. A. Torosyan, Y. N. Biglova, V. V. Mikheev, Z. T. Khalitova, F. A. Gimalova and M. S. Miftakhov, Mendeleev Commun., 2012, 22, 199–200 CrossRef CAS.
- S. A. Torosyan, Y. N. Biglova, V. V. Mikheev, F. A. Gimalova, A. G. Mustafin and M. S. Miftakhov, Russ. J. Org. Chem., 2014, 50, 179–182 CrossRef CAS.
- S. A. Torosyan, V. V. Mikheev, Y. N. Biglova and M. S. Miftakhov, Russ. J. Org. Chem., 2016, 52, 587–589 CrossRef CAS.
- S. A. Torosyan, V. V. Mikheev, Y. N. Biglova, V. A. Egorov and M. S. Miftakhov, Russ. J. Org. Chem., 2016, 52, 456–457 CrossRef CAS.
- K. M. Keshavarz, B. Knight, R. C. Haddon and F. Wudl, Tetrahedron, 1996, 52, 5149–5159 CrossRef.
- A. M. Benito, A. D. Darwish, H. W. Kroto, M. F. Meidine, R. Taylor and D. R. Walton, Tetrahedron Lett., 1996, 37, 1085–1086 CrossRef CAS.
- R.-F. Peng, G.-W. Wang, Y.-B. Shen, Y.-J. Li, T.-H. Zhang, Y.-C. Liu, Y. Murata and K. Komatsu, Synth. Commun., 2004, 34, 2117–2126 CrossRef CAS.
- B. Jin, J. Shen, R. Peng, R. Zheng and S. Chu, Tetrahedron Lett., 2014, 55, 5007–5010 CrossRef CAS.
- W. Gong, B. Jin, R. Peng, N. Deng, R. Zheng and S. Chu, Ind. Eng. Chem. Res., 2015, 54, 2613–2618 CrossRef CAS.
- J. Zhao, B. Jin, R. Peng, N. Deng, W. Gong, Q. Liu and S. Chu, RSC Adv., 2015, 5, 90422–90427 RSC.
- B. Jin, J. Shen, R. Peng, C. Chen, Q. Zhang, X. Wang and S. Chu, Ind. Eng. Chem. Res., 2015, 54, 2879–2885 CrossRef CAS.
- X. Camps and A. Hirsch, J. Chem. Soc., 1997, 1595–1596 CAS.
- L. Zheng, Q. Zhou, X. Deng, W. Fei, N. Bin, Z.-X. Guo, G. Yu and Y. Cao, Thin Solid Films, 2005, 489, 251–256 CrossRef CAS.
- P. A. Troshin, H. Hoppe, J. Renz, M. Egginger, J. Yu. Mayorova, A. E. Goryachev, A. S. Peregudov, R. N. Lyubovskaya, G. Gobsch, N. S. Sariciftci and V. F. Razumov, Adv. Funct. Mater., 2009, 19, 779–788 CrossRef CAS.
- S. A. Torosyan, F. A. Gimalova, V. V. Mikheev, A. A. Fatykhov, Y. N. Biglova and M. S. Miftakhov, Russ. J. Org. Chem., 2011, 47, 1807–1810 CrossRef CAS.
- N. Sun, Y. Wang, Y. Song, Z. Guo, L. Dai and D. Zhu, Chem. Phys. Lett., 2001, 344, 277–282 CrossRef CAS.
- J. Li, N. Sun, Z.-X. Guo, C. Li, Y. Li, L. Dai, D. Zhu, D. Sun, Y. Cao and L. Fan, J. Phys. Chem. B, 2002, 106, 11509–11514 CrossRef CAS.
- S. Gottis, C. Kopp, E. Allard and R. Deschenaux, Helv. Chim. Acta, 2007, 90, 957–962 CrossRef CAS.
- S. A. Torosyan, V. V. Mikheev, Y. N. Biglova and M. S. Miftakhov, Russ. J. Org. Chem., 2015, 51, 940–942 CrossRef CAS.
- A. Trabolsi, M. Urbani, J. L. Delgado, F. Ajamaa, M. Elhabiri, N. Solladié, J. Nierengarten and A. Albrecht-Gary, New J. Chem., 2008, 32, 159–165 RSC.
- A. Kumar, B. Patel, G. Patel and S. K. Menon, Fullerenes, Nanotubes, Carbon Nanostruct., 2010, 18, 186–197 CrossRef CAS.
- J. Fan, Y. Wang, A. J. Blake, C. Wilson, E. S. Davies, A. N. Khlobystov and M. Schröder, Angew. Chem., Int. Ed., 2007, 46, 8013–8016 CrossRef CAS.
- L. J. Santos, D. C. Da-Silva, J. S. Rebouc, M. R. A. Alves, Y. M. Idemori, T. Matencio, R. P. Freitas and R. B. Alves, Tetrahedron, 2011, 67, 228–235 CrossRef CAS.
- Y. Nakamura, M. Suzuki, Y. Imai and J. Nishimura, Org. Lett., 2004, 6, 2797–2799 CrossRef CAS.
- S. Taillemite, C. Aubert, D. Fichou and M. Malacria, Tetrahedron Lett., 2005, 46, 8325–8328 CrossRef CAS.
- G. Dennler, S. Bereznev, D. Fichou, K. Holl, D. Ilic, R. Koeppe, M. Krebs, A. Labouret, C. Lungenschmied, A. Marchenko, D. Meissner, E. Mellikov, J. Méot, A. Meyer, T. Meyer, H. Neugebauer, A. Öpik, N. S. Sariciftci, S. Taillemite and T. Wöhrle, J. Sol. Energy, 2007, 81, 947–957 CrossRef CAS.
- Y. Gao, Z. Ou, G. Yang, L. Liu, M. Jin, X. Wang, B. Zhang and L. Wang, J. Photochem. Photobiol., A, 2009, 203, 105–111 CrossRef CAS.
- R. B. Martin, K. Fu and Y.-P. Sun, Chem. Phys. Lett., 2003, 375, 5–6 CrossRef.
- R. J. Bushby, I. W. Hamley, Q. Liu, O. R. Lozman and J. E. Lydon, J. Mater. Chem., 2005, 15, 4429–4434 RSC.
- L. J. Santos, R. B. Alves, R. P. Freitas, J.-F. Nierengarten, L. E. F. Magalhães, K. Krambrock and M. V. B. Pinheiro, J. Photochem. Photobiol., A, 2008, 200, 277–281 CrossRef.
- L. J. Santos, A. S. P. Gonçalves, K. Krambrock, M. V. B. Pinheiro, M. N. Eberlin, B. G. Vaz, R. P. Freitas and R. B. Alves, J. Photochem. Photobiol., A, 2011, 217, 184–190 CrossRef CAS.
- B. Floris, P. Galloni, R. Seraglia and P. Tagliatesta, J. Organomet. Chem., 2003, 679, 202–207 CrossRef CAS.
- F. Spanig, C. Kovacs, F. Hauke, K. Ohkubo, S. Fukuzumi, D. M. Guldi and A. Hirsch, J. Am. Chem. Soc., 2009, 131, 8180–8195 CrossRef.
- M. Heuken, H. Komber and B. Voit, Macromol. Chem. Phys., 2012, 213, 97–107 CrossRef CAS.
- V. P. Gubskaya, L. Sh. Berezhnaya, A. T. Gubaidullin, I. I. Faingold, R. A. Kotelnikova, N. P. Konovalova, V. I. Morozov, I. A. Litvinov and I. A. Nuretdinov, Org. Biomol. Chem., 2007, 5, 976–981 RSC.
- A. A. Gilmutdinova, V. P. Gubskaya, G. M. Fazleeva, S. K. Latypov, T. A. Zhelonkina, D. R. Sharafutdinova, I. A. Nuretdinov and O. G. Sinyashin, Tetrahedron, 2014, 70, 5947–5953 CrossRef CAS.
- C. F. Richardson, D. I. Schuster and S. R. Wilson, Org. Lett., 2000, 2, 1011–1014 CrossRef CAS.
- D. J. Wolff, K. Mialkowski, C. F. Richardson and S. R. Wilson, Biochemistry, 2001, 40, 37–45 CrossRef CAS.
- L. Chaker, G. E. Ball, J. R. Williams, G. A. Burley, B. C. Hawkins, P. A. Keller and S. G. Pyne, Eur. J. Org. Chem., 2005, 5158–5162 CrossRef CAS.
- G. Pagona, G. Rotas and N. Tagmatarchis, Fullerenes, Nanotubes, Carbon Nanostruct., 2014, 22, 88–98 CrossRef CAS.
- F. Wessendorf, J.-F. Gnichwitz, G. H. Sarova, K. Hager, U. Hartnagel, D. M. Guldi and A. Hirsch, J. Am. Chem. Soc., 2007, 129, 16057–16071 CrossRef CAS.
- V. Campisciano, V. La Parola, L. F. Liotta, F. Giacalone and M. Gruttadauria, Chem.–Eur. J., 2015, 21, 3327–3334 CrossRef CAS.
- J. Baffreau, L. Perrin, S. Leroy-Lhez and P. Hudhomme, Tetrahedron Lett., 2005, 46, 4599–4603 CrossRef CAS.
- A. Łapiński, A. Graja, I. Olejniczak, A. Bogucki, M. Połomska, J. Baffreau, L. Perrin and S. Leroy-Lhez, Mol. Cryst. Liq. Cryst., 2006, 447, 87–103 Search PubMed.
- J. Baffreau, S. Leroy-Lhez, A. N. Vân, R. M. Williams and P. Hudhomme, Chem.–Eur. J., 2008, 14, 4974–4992 CrossRef CAS.
- R. K. Dubey, M. Niemi, K. Kaunisto, A. Efimov, N. V. Tkachenko and H. Lemmetyinen, Chem.–Eur. J., 2013, 19, 6791–6806 CrossRef CAS.
- P. Remón, C. P. Carvalho, C. Baleizão, M. N. Berberan-Santos and U. Pischel, ChemPhysChem, 2013, 14, 2717–2724 CrossRef.
- J. Iehl, R. P. Freitas and J.-F. Nierengarten, Tetrahedron Lett., 2008, 49, 4063–4066 CrossRef CAS.
- V. P. Gubskaya, S. K. Latypov, F. G. Sibgatullina, A. A. Balandina, T. A. Zhelonkina, G. M. Fazleeva, L. N. Islamova, D. R. Sharafutdinova, I. A. Nuretdinov and O. G. Sinyashin, Russ. Chem. Bull., Int. Ed., 2012, 61, 1169–1175 CrossRef CAS.
- A. J. Hilmer, K. Tvrdy, J. Zhang and M. S. Strano, J. Am. Chem. Soc., 2013, 135, 11901–11910 CrossRef CAS.
- L. N. Islamova, V. P. Gubskaya, G. M. Fazleeva, T. A. Zhelonkina, S. K. Latypov, D. R. Sharafutdinova, I. A. Nuretdinov and O. G. Sinyashin, Mendeleev Commun., 2017, 27, 204–206 CrossRef CAS.
- C. I. C. Jordão, A. Farinha, R. F. Enes, A. Tomé, A. Silva, J. Cavaleiro, C. Ramos, M. G. Santana-Marques, F. A. A. Paz, J. de la Torre, M. D. L. de la Torre and M. Nogueras, Tetrahedron, 2008, 64, 4427–4437 CrossRef.
- M. Wedel and F.-P. Montforts, Tetrahedron Lett., 1999, 40, 7071–7074 CrossRef CAS.
- F.-P. Montforts, I. Vlassiouk, S. Smirnov and M. Wedel, J. Porphyrins Phthalocyanines, 2003, 7, 651–666 CrossRef CAS.
- P. Cheng, S. R. Wilson and D. I. Schuster, Chem. Commun., 1999, 89–90 RSC.
- M. A. Fazio, A. Durandin, N. V. Tkachenko, M. Niemi, H. Lemmetyinen and D. I. Schuster, Chem.–Eur. J., 2009, 15, 7698–7705 CrossRef CAS.
- S. Schlundt, G. Kuzmanich, F. Spänig, R. G. deMiguel, C. Kovacs, M. A. Garcia-Garibay, D. M. Guldi and A. Hirsch, Chem.–Eur. J., 2009, 15, 12223–12233 CrossRef CAS PubMed.
- K. Li, D. I. Schuster, D. M. Guldi, M. Á. Herranz and L. Echegoyen, J. Am. Chem. Soc., 2004, 126, 3388–3389 CrossRef CAS.
- D. I. Schuster, K. Li, D. M. Guldi and J. Ramey, Org. Lett., 2004, 6, 1919–1922 CrossRef CAS.
- K. Li, P. J. Bracher, D. M. Guldi, M. Á. Herranz, L. E. David and D. I. Schuster, J. Am. Chem. Soc., 2004, 126, 9156–9157 CrossRef CAS.
- J. D. Megiatto Jr, D. I. Schuster, S. Abwandner, G. de Miguel and D. M. Guldi, J. Am. Chem. Soc., 2010, 132, 3847–3861 CrossRef.
- J. Osterodt, A. Zett and F. Vogtle, Tetrahedron, 1996, 52, 4949–4962 CrossRef CAS.
- S. R. Wilson and Y. Wu, J. Chem. Soc., Chem. Commun., 1993, 784–786 RSC.
- F. Diederich, U. Jonas, V. Gramlich, A. Hermann, H. Ringsdorf and C. Thilgen, Helv. Chim. Acta, 1993, 76, 2445–2453 CrossRef CAS.
- J.-P. Burgeois, L. Echegoyen, M. Fibbioli, E. Pretsch and F. Diederich, Angew. Chem., Int. Ed., 1998, 37, 2118–2121 CrossRef.
- F. Diederich and M. Gómez-López, Chem. Soc. Rev., 1999, 28, 263–277 RSC.
- F. Diederich, L. Echegoyen, M. Gómez-López, R. Kessinger and J. Fraser, J. Chem. Soc., Perkin Trans. 2, 1999, 1577–1586 RSC.
- E. Sartori, L. Garlaschelli, A. Toffoletti, C. Corvaja, M. Maggini and G. Scorranoc, Chem. Commun., 2001, 311–312 RSC.
- M. Carano, C. Corvaja, L. Garlaschelli, M. Maggini, M. Marcaccio, F. Paolucci, D. Pasini, P. P. Righetti, E. Sartori and A. Toffoletti, Eur. J. Org. Chem., 2003, 374–384 CrossRef CAS.
- L. Garlaschelli, I. Messina, D. Pasini and P. P. Righetti, Eur. J. Org. Chem., 2002, 3385–3392 CrossRef CAS.
- M. Gutierrez-Nava, H. Nierengarten, P. Masson, A. Van Dorsselaer and J.-F. Nierengarten, Tetrahedron Lett., 2003, 44, 3043–3046 CrossRef CAS.
- N. Solladié, M. E. Walther, H. Herschbach, E. Leize, A. Van Dorsselaer, T. M. F. Duarte and J.-F. Nierengarten, Tetrahedron, 2006, 62, 1979–1987 CrossRef.
- J.-F. Nierengarten, T. Gu, G. Hadziioannou, D. Tsamouras and V. Krasnikov, Helv. Chim. Acta, 2004, 87, 2948–2966 CrossRef CAS.
- A. Gégout, M. Holler, T. M. Figueira-Duarte and J.-F. Nierengarten, Eur. J. Org. Chem., 2008, 3627–3634 CrossRef.
- A. Gégout, T. M. Figueira-Duarte, J.-F. Nierengarten, A. Listorti and N. Armaroli, Synlett, 2006, 18, 3095–3099 Search PubMed.
- N. Camaioni, G. Fabbrini, E. Menna, M. Maggini, G. Ridolfi and A. Zanelli, New J. Chem., 2006, 30, 335–342 RSC.
- A. Ikeda, S. Nobukuni, H. Udzu, Z. Zhong and S. Shinkai, Eur. J. Org. Chem., 2000, 3287–3293 CrossRef CAS.
- R. N. Khaybullin, I. Y. Strobykina, V. P. Gubskaya, G. M. Fazleeva, S. K. Latypov and V. E. Kataev, Mendeleev Commun., 2011, 21, 134–136 CrossRef CAS.
- M. D. Torre, A. C. Tome, A. M. S. Silva and J. A. S. Cavaleiro, Tetrahedron Lett., 2002, 43, 4617–4620 CrossRef.
- M. D. Torre, A. G. Rodrigues, A. C. Tomé, A. M. Silva and J. A. Cavaleiro, Tetrahedron, 2004, 60, 3581–3592 CrossRef.
- R. F. Enes, A. S. F. Farinha, A. C. Tomé, J. A. S. Cavaleiro, R. Amorati, S. Petrucci and G. F. Pedulli, Tetrahedron, 2009, 65, 253–262 CrossRef CAS.
- M. J. Brites, C. Santos, S. Nascimento, B. Gigante and M. N. Berberan-Santos, Tetrahedron Lett., 2004, 45, 6927–6930 CrossRef CAS.
- M. J. Brites, C. Santos, S. Nascimento, B. Gigante, H. Luftmann, A. Fedorov and M. N. Berberan-Santos, New J. Chem., 2006, 30, 1036–1045 RSC.
- G. Pagona, S. P. Economopoulos, G. K. Tsikalas, H. E. Katerinopoulos and N. Tagmatarchis, Chem.–Eur. J., 2010, 16, 11969–11976 CrossRef CAS.
- D. Felder, D. Guillon, R. Levy, A. Mathis, J.-F. Nicoud, J.-F. Nierengarten, J.-L. Rehspringerd and J. Schel, J. Mater. Chem., 2000, 10, 887–892 RSC.
- J.-F. Nierengarten and J.-F. Nicoud, Tetrahedron Lett., 1997, 38, 7737–7740 CrossRef CAS.
- J.-F. Nierengarten, D. Felder and J.-F. Nicoud, Tetrahedron Lett., 2000, 41, 41–44 CrossRef CAS.
- F. Wudl, Acc. Chem. Res., 1992, 25, 157–161 CrossRef CAS.
- T. Suzuki, Y. Maruyama, T. Akasaka, W. Ando, K. Kobayashi and S. Naguse, J. Am. Chem. Soc., 1994, 116, 1359–1363 CrossRef CAS.
- F. Arias, Q. Xie, Y. Wu, Q. Lu, S. R. Wilson and L. Echegoyen, J. Am. Chem. Soc., 1994, 116, 6388–6394 CrossRef CAS.
- B. Dardel, D. Guillon, B. Heinrich and R. Deschenaux, J. Mater. Chem., 2001, 11, 2814–2831 RSC.
- S. Campidelli, C. Eng, I. M. Saez, J. W. Goodby and R. Deschenaux, Chem. Commun., 2003, 13, 1520–1521 RSC.
- R. Deschenaux, N. Yevlampieva, T. Dmitrieva, B. Dardel and P. Lavrenko, Fullerenes, Nanotubes, Carbon Nanostruct., 2004, 12, 193–199 CrossRef CAS.
- N. Maringa, J. Lenoble, B. Donnio, D. Guillon and R. Deschenaux, J. Mater. Chem., 2008, 18, 1524–1534 RSC.
- T. N. Y. Hoang, D. Pociecha, M. Salamonczyk, E. Goreckab and R. A. Deschenaux, Soft Matter, 2011, 7, 4948–4953 RSC.
- V. Russo, P. Pieper, B. Heinrich, B. Donnio and R. Deschenaux, Chem.–Eur. J., 2016, 22, 17366–17376 CrossRef CAS.
- J. Vergara, J. Barberá, J. L. Serrano, M. B. Ros, N. Sebastián, R. de la Fuente, D. O. López, G. Fernández, L. Sánchez and N. Martín, Angew. Chem., Int. Ed., 2011, 50, 12523–12528 CrossRef CAS.
- M. Even, B. Heinrich, D. Guillon, D. M. Guldi, M. Prato and R. A. Deschenaux, Chem.–Eur. J., 2001, 7, 2595–2604 CrossRef CAS.
- J.-F. Nierengarten, T. Habicher, R. Kessinger, F. Cardullo, F. Diederich, V. Gramlich, J.-P. Gisselbrecht, C. Boudon and M. Gross, Helv. Chim. Acta, 1997, 80, 2238–2276 CrossRef.
- M. Brettreich and A. A. Hirsch, Tetrahedron Lett., 1998, 39, 2731–2734 CrossRef CAS.
- C. Kovacs and A. A. Hirsch, Eur. J. Org. Chem., 2006, 3348–3357 CrossRef CAS.
- G. Rousseau, H. Fensterbank, K. Baczko, M. Cano, I. Stenger, C. Larpent and E. Allard, ChemPlusChem, 2013, 78, 352–363 CrossRef CAS.
- S. González, N. Martín and D. M. Guldi, J. Org. Chem. Res., 2003, 68, 779–791 CrossRef.
- E. Allard, F. Oswald, B. Donnio, D. Guillon, J. L. Delgado, F. Langa and R. Deschenaux, Org. Lett., 2005, 7, 383–386 CrossRef CAS.
- S. Ballot and N. Noiret, J. Organomet. Chem., 2002, 664, 208–213 CrossRef CAS.
- P. Piotrowski, J. Pawłowska, J. G. Sadło, R. Bilewicz and A. Kaim, J. Nanopart. Res., 2017, 19, 161 CrossRef.
- S. Ballot and N. Noiret, Tetrahedron Lett., 2003, 44, 8811 CrossRef CAS.
- Y.-C. Chen, C.-Y. Hsu, R. Y.-Y. Lin, K.-C. Ho and J. T. Lin, ChemSusChem, 2013, 6, 20–35 CrossRef CAS.
- D. Jariwala, V. K. Sangwan, L. J. Lauhon, T. J. Marks and M. C. Hersam, Chem. Soc. Rev., 2013, 42, 2824–2860 RSC.
- A. Bianco and T. D. Ros, Fullerenes: Principles and Applications, Royal Society of Chemistry, 2011, ch. 14, vol. 2, pp. 507–545 Search PubMed.
- S. Collavini, M. Saliba, W. R. Tress, P. J. Holzhey, S. F. Völker, K. Domanski, S. H. Turren-Cruz, A. Ummadisingu, S. M. Zakeeruddin, A. Hagfeldt, M. Grätzel and J. L. Delgado, ChemSusChem, 2018, 11, 1032–1039 CrossRef CAS.
- C.-C. Chu, L. Wang and T.-I. Ho, Macromol. Rapid Commun., 2005, 26, 1179–1184 CrossRef CAS.
- V. P. Gubskaya, K. L. Nodov, F. G. Sibgatullina, G. M. Fazleeva, I. E. Ismaev, S. K. Latypov, Y. Y. Efremov and I. A. Nuretdinov, Russ. Chem. Bull., Int. Ed., 2007, 56, 1843–1848 CrossRef CAS.
- H. Jiang, J.-M. Zhang, W.-Q. Du and S.-Z. Zhu, Chin. J. Chem., 2007, 25, 86–89 CrossRef CAS.
- G. A. Burley, P. A. Keller, S. G. Pyne and G. E. Ball, J. Org. Chem., 2002, 67, 8316–8330 CrossRef CAS.
- G. A. Burley, P. A. Keller, S. G. Pynea and G. E. Ball, Chem. Commun., 1998, 2539–2540 RSC.
- H. S. Lee, S. C. Yoon, J. Lim, M. Lee and C. Lee, Mol. Cryst. Liq. Cryst., 2008, 492, 293–302 CAS.
- H. J. Bestmann, D. Hadawi, T. Röder and C. Moll, Tetrahedron Lett., 1994, 35, 9017–9020 CrossRef CAS.
- Y. Wang, J. Cao, D. I. Schuster and S. R. Wilson, Tetrahedron Lett., 1995, 36, 6843–6846 CrossRef CAS.
- M. Hamada, T. Hino, K. Kinbara and K. Saigo, Tetrahedron Lett., 2001, 42, 5069–5071 CrossRef CAS.
- T. Hino, K. Kinbara and K. Saigo, Tetrahedron Lett., 2001, 42, 5065–5067 CrossRef CAS.
- H. Ito, Y. Kishi, Y. Nishikawa, T. Tada, Y. Ishida and K. Saigo, Synlett, 2010, 12, 1811–1814 Search PubMed.
- P. D. W. Boyd, P. Bhyrappa, P. Paul, J. Stinchcombe, R. Bolskar, Y. Sun and C. A. Reed, J. Am. Chem. Soc., 1995, 117, 2907–2914 CrossRef CAS.
- A. R. Tuktarov, Z. R. Shakirova, A. A. Khuzin and U. M. Dzhemilev, Synthesis, 2016, 48, 136–140 CrossRef CAS.
- A. R. Tuktarov, Z. R. Shakirova, A. A. Khuzin, A. R. Tulyabaev, I. R. Ramazanov and U. M. Dzhemilev, Tetrahedron Lett., 2016, 57, 4314–4317 CrossRef CAS.
- I. P. Romanova, A. V. Bogdanov, V. F. Mironov, G. R. Shaikhutdinova, O. A. Larionova, S. K. Latypov, A. A. Balandina, D. G. Yakhvarov, A. T. Gubaidullin, A. F. Saifina and O. G. Sinyashin, J. Org. Chem., 2011, 76, 2548–2557 CrossRef CAS.
- A. V. Bogdanov, G. G. Yusupova, I. P. Romanova, S. K. Latypov, D. B. Krivolapov, V. F. Mironov and O. G. Sinyashin, Synthesis, 2013, 45, 0668–0672 CrossRef CAS.
- M. I. Valitov, I. P. Romanova, A. A. Gromchenko, G. R. Shaikhutdinova, D. G. Yakhvarov, V. V. Bruevich, V. A. Dyakov, O. G. Sinyashin and D. Y. Paraschuk, Sol. Energy Mater. Sol. Cells, 2012, 103, 48–52 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |