
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBovine serum albumin assisted preparation of ultra-stable gold nanoflowers and their selective Raman response to charged dyes†
Xiaoyu Zhanga,
Yandong Hanb,
Zihao Xinga,
Zhenzhen Huang a,
Renguo Xiea and
Wensheng Yang*a
a,
Renguo Xiea and
Wensheng Yang*a
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130022, China. E-mail: wsyang@jlu.edu.cn; Fax: +86 431 85168868; Tel: +86 431 85168185
bInstitute of Molecular Plus, Tianjin University, Tianjin 300072, China
First published on 9th September 2019
Abstract
In this work, we demonstrated a facile, one-pot approach for preparation of gold nanoflowers by using tetrachloroauric acid as a gold precursor, ascorbic acid as a reductant, and bovine serum albumin (BSA) as a ligand. It was found that the morphology of the as-prepared gold nanoparticles (Au NPs) was dependent on the concentration of BSA introduced into the reaction solutions. It is identified that BSA directed the preferential growth along the 〈111〉 direction, which contributed to the anisotropic growth of Au NPs and thus the formation of Au nanoflowers. An increased concentration of BSA reduced the reactivity of the gold precursor, leading to the formation of Au nanoflowers with increased size, which could also be obtained by decreasing the amount of reductant added. The Au nanoflowers were ultra-stable in the presence of chloride ions under acidic pH, making them suitable for selective detection of oppositely charged dyes via surface-enhanced Raman scattering according to the isoelectric point (∼4.7) of BSA capped on their surface.
Introduction
Gold nanoflowers (Au NFs) have attracted extensive attention due to their unique multi-branched structure and abundant “hot spots” localized around their branches and corners,1,2 making them potentially useful in the fields of catalysis,3 biolabeling,4 thermal therapy5 and surface-enhanced Raman scattering (SERS).6–8 A general strategy to synthesize Au NFs is to carry out the growth of Au nanoparticles (NPs) in the presence of shape-directing agents, which can preferentially adsorb onto specific crystal planes and thus direct the anisotropic growth of NPs along the uncapped facets. A variety of agents, such as surfactants,9,10 polymers,11 and biomolecules with active groups that allow their anchoring onto the specific planes,12 have been proven to be effective to direct the growth of Au NFs. However, it is documented that Au NFs are readily to go ripening and become spherical in shape, especially in presence of chloride ions and/or under acidic pH.13–15 Coating the NFs with silica protection layer was known to be effective to suppress the ripening of Au NFs, in which the Au NFs were supported on Al2O3 to improve their dispersing ability and then transferred into the ethanol/ammonia system to carry out the silica coating by conventional Stöber method.15Herein, we demonstrated a facile one-pot approach to prepare ultra-stable Au NFs by using bovine serum albumin (BSA), a globular protein composed of 583 amino acid residues,16 as ligand, tetrachloroauric acid (HAuCl4) as gold precursor and L-ascorbic acid (AA) as reductant. The effect of BSA on the morphology of the resulting Au NPs were investigated. It is identified that BSA promoted the preferential growth of the Au NPs along the 〈111〉 direction and thus the formation of the Au NFs. Increased concentration of BSA resulted in the reduced activity of gold precursor and thus the formation of Au NFs with increased size. The resulting BSA-capped Au NFs were ultra-stable in presence of chloride ions even under low pH (3.0), making them qualified for selective detection of oppositely charged dyes by SERS according to the isoelectric point of the protein capped on their surface.
Experimental
Materials
Tetrachloroauric acid (AR, HAuCl4·4H2O, FW 411.85) was purchased from Shanghai Chemical Reagent Co. Int. Ascorbic acid (≥99%, C6H8O6, AA, FW 176.12) was obtained from Alfa Aesar. Bovine Serum Albumin (≥98%, BSA, FW 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 430), Rhodamine 6G (∼95%, FW 479.01) and sodium hydroxide (99%, NaOH, FW 40.00) were purchased from Sigma-Aldrich. Methyl blue (FW 799.80), 4-aminothiophenol (97%, FW 125.19) and 4-mercaptobenzoic acid (90%, FW 154.19) were purchased from Aladdin. Acetic acid, phosphate acid, boric acid, hydrochloric acid and nitric acid were purchased from Beijing Chemical Work. All of the chemicals were used without further purification. High-purity water (Pall Purelab Plus) with a resistivity of 18.2 MΩ cm was used in all experiments. All glassware used was cleaned in a bath of aqua regia solution (HCl/HNO3, 3:1) and then rinsed thoroughly with high-purity water before use.
430), Rhodamine 6G (∼95%, FW 479.01) and sodium hydroxide (99%, NaOH, FW 40.00) were purchased from Sigma-Aldrich. Methyl blue (FW 799.80), 4-aminothiophenol (97%, FW 125.19) and 4-mercaptobenzoic acid (90%, FW 154.19) were purchased from Aladdin. Acetic acid, phosphate acid, boric acid, hydrochloric acid and nitric acid were purchased from Beijing Chemical Work. All of the chemicals were used without further purification. High-purity water (Pall Purelab Plus) with a resistivity of 18.2 MΩ cm was used in all experiments. All glassware used was cleaned in a bath of aqua regia solution (HCl/HNO3, 3:1) and then rinsed thoroughly with high-purity water before use.
Syntheses of Au NPs
Typically, aqueous solutions of HAuCl4 and BSA were mixed and then kept at room temperature (25 ± 2 °C). Total volume of the solutions were 5.0 mL. The concentration of HAuCl4 solutions was fixed to be 0.25 mM and that of BSA varied from 0 to 4.0 μM. After 1 h incubation, 20 μL aqueous solution of AA (31.25 mM) was added into the mixtures under magnetic stirring (400 rpm). After 30 min, the resulting Au NPs were collected by centrifugation (4000 rpm, 10 min) and then redispersed in 5 mL pure water for characterizations.Preparation of SERS samples
2.0 mL dispersions of the Au NPs prepared with 3.0 μM BSA were collected by centrifugation (4000 rpm, 10 min) and then redispersed in 2.0 mL pure water with pH 3.0, 5.0 and 8.0, respectively. The pH was adjusted by addition of 1 M HCl or NaOH. 10 μL Rhodamine 6G (1 mM), 10 μL methyl blue (1 mM) and 20 μL of their 1:1 mixture were added into the Au NP dispersions with pH 3.0, 5.0 and 8.0, respectively, and then transferred into 1 cm quartz cuvettes for SERS measurements after 20 min standing. In another series of experiments, 8 μL 4-aminothiophenol (0.5 mM) and 8 μL 4-mercaptobenzoic acid (0.5 mM) were added into 2 mL Au NP dispersions with pH 3.0 and 8.0, and then transferred into 1 cm quartz cuvettes for SERS measurements.
Measurements
UV-Vis spectra were recorded on a Shimadzu UV-1800 UV-Vis spectrophotometer. Transmission electron microscopy (TEM) observations were carried out with a JEOL JEM-2010 electron microscope with an acceleration voltage of 100 kV. High-resolution TEM (HRTEM) images were obtained by a JEOL JEM-3010 electron microscope operated with an acceleration voltage of 300 kV. The dispersions were dropped onto carbon-coated copper grids and then dried in air before the TEM observations. Temporal evolutions of the UV-Vis spectra were acquired by an Ocean Optics HR4000CG-UV-NIR high-resolution spectrophotometer. Zeta-potential was measured on a Brookhaven ZetaPlus apparatus. The pH of samples was adjusted from 2.5 to 8.0 by using Britton–Robinson buffer (mixture of 2 mM acetic acid, phosphate acid and boric acid with different amount of NaOH). Power XRD patterns were recorded on a Rigaku D-Max 2550 diffractometer equipped with a graphite monochromator using Cu Kα radiation (λ = 1.54 Å) at a scanning speed of 5° min−1. The samples for XRD measurements were dried frozenly in vacuo. Raman spectra were obtained by using a Horiba LabRAM UR evolution instrument with 785 nm laser (50 mW) and a holder for cuvette-based liquid measurements. The accumulation time was 60 s for all the data presented.Results and discussion
In all the reactions, the initial concentrations of HAuCl4 and AA were fixed to be 0.25 mM and 1.25 mM (corresponding to a molar ratio of 1:5) unless stated especially. The solution of HAuCl4 presented a pH of ca. 3.3, which decreased slightly to ca. 3.2 after the addition of 5.0 μM BSA (Fig. S1a†). No surface plasmon resonance (SPR) peak of Au particles was observable after the solutions of HAuCl4 and BSA were mixed for 1 h, attributed to the very weak reducing ability of BSA in acidic solution.17 After the addition of AA, Au hydrosols with different colors were obtained, dependent on the amounts of BSA contained in the reaction solutions (Fig. 1a). In absence of BSA, the resulting hydrosol was red in colour, corresponding to a SPR peak at 525 nm, which is the typical character of spherical Au particles. When the concentration of BSA increased to 1.0, 1.5, 2.0, 3.0 and 4.0 μM, color of the hydrosols evolved from pink to purple, deep blue, blue and light blue. Correspondingly, the SPR peak became broad and experienced a red-shift from 546 nm to 565, 589, 610 and 635 nm. It was deduced that the resulting Au particles became larger and/or more anisotropic in shape with the increased concentration of BSA in the reaction solutions.18 It should be mentioned that when the concentration of BSA was 5.0 μM or higher, turbid grey hydrosols were obtained and the particles settled to the bottom of vials within a couple of hours, possibly due to the formation of large particles.
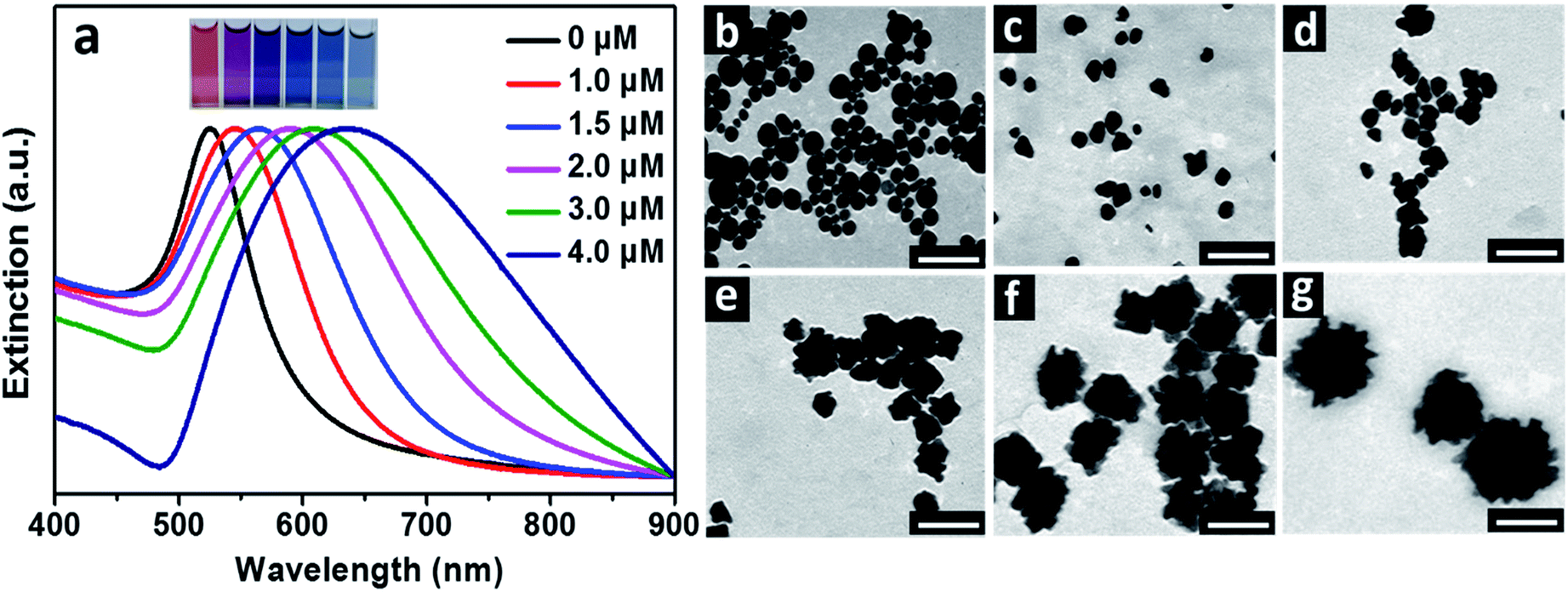 | ||
| Fig. 1 (a) Normalized UV-Vis spectra of the Au particles prepared under different BSA concentrations. Inset gives the photos of the corresponding Au hydrosols prepared under different BSA concentrations (from left to right, 0, 1.0, 1.5, 2.0, 3.0 and 4.0 μM). TEM images of the corresponding Au particles prepared under BSA concentrations of (b) 0, (c) 1.0, (d) 1.5, (e) 2.0, (f) 3.0 and (g) 4.0 μM. The scale bars were 100 nm. | ||
TEM observations were carried out to further illustrate the change in size and morphology of the Au particles with the concentrations of BSA (Fig. 1b–g). Spherical Au particles with average size of 27 nm were obtained in absence of BSA. When 1.0 and 1.5 μM BSA were introduced into the solutions, average sizes of the resulting Au particles were 24 and 28 nm, respectively, and morphology of the particles became less regular in shape. When the concentrations of BSA were 2.0, 3.0 and 4.0 μM, the resulting Au particles became flower-like in shape, with average sizes of 47, 64 and 120 nm respectively. By comparing the variations in sizes and SPR peaks of the Au hydrosols prepared by using BSA as ligand with these of the typical spherical Au particles,19 it was obvious that the red-shifted SPR peaks and color changes of the Au hydrosols were primarily related to the evolution in their morphologies, especially for these prepared in the range of BSA concentrations of 1.0–3.0 μM.
Temporal evolution in UV-Vis spectra of the reaction solutions with different concentrations of BSA were recorded to understand the effect of BSA on growth of the Au particles (Fig. 2). For the reaction proceeded at BSA concentration of 1.0 μM, a SPR peak at 545 nm became observable after 2 s, whose intensity increased rapidly and kept almost unchanged after 30 s of the reaction (Fig. 2a). For the reaction proceeded at BSA concentration of 3.0 μM (Fig. 2b), the SPR peak became observable after 10 s of the reaction, which shifted rapidly from 554 to 592 nm after 20 s of the reaction, whose intensity increased slowly and kept almost unchanged after 10 min of the reaction, suggesting the slowed down reduction of the gold precursor under the high BSA concentration. The evolution in SPR peak position and maximum extinction of the Au hydrosols prepared at different BSA concentrations was summarized as shown in Fig. 2c and d. It was deduced that there existed continuous nucleation in the reaction with 1.0 μM BSA, as suggested by the slight change in position and the rapid increase in maximum extinction of the SPR peak at the early stage of the reaction (0–5 s), which was in consistent with the formation of quasi-spherical Au particles with high polydispersity (25.6%) as shown in Fig. 1c. In comparison, the SPR peak experienced obvious red-shift and gradual increase in intensity in the reaction with 3.0 μM BSA, suggesting the rapid, anisotropic growth of the Au hydrosols at the early stage of the reaction (0–20 s).
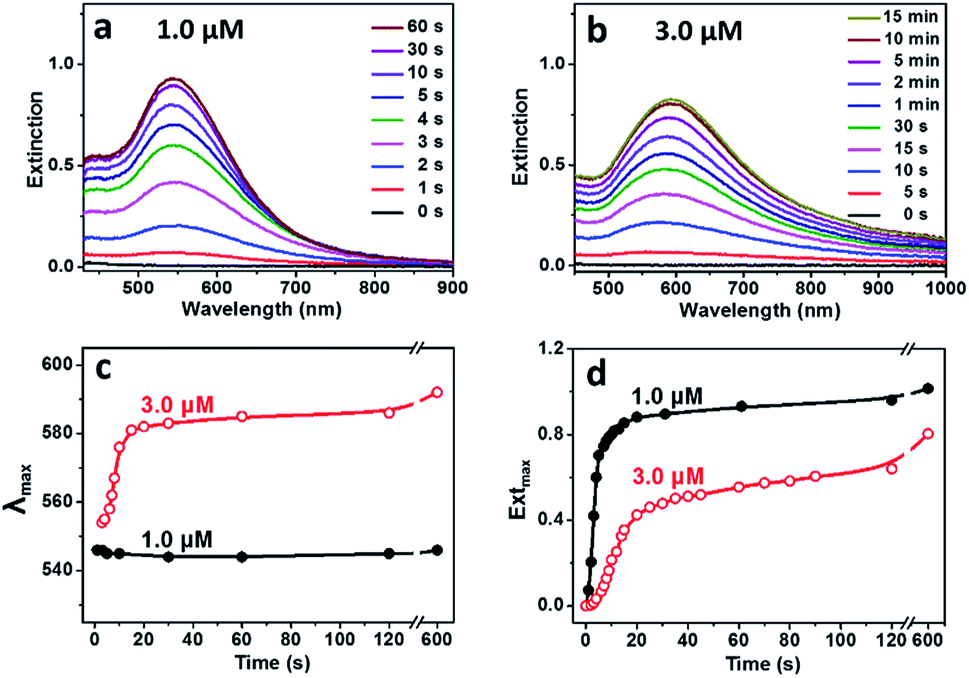 | ||
| Fig. 2 Temporal evolution of UV-Vis spectra of the Au particles grown at BSA concentrations of (a) 1.0 μM and (b) 3.0 μM. Summarized temporal evolution of (c) SPR peak position and (d) maximum extinction of the Au particles grown at 1.0 and 3.0 μM BSA. | ||
It was expected that BSA may act as ligand for both the gold precursor and the gold particles since there are plenty of functional groups, primarily carboxyl and amino ones, on its surface.20 UV-Vis spectrum of HAuCl4 solution presented two bands at 220 and 291 nm in absence of BSA, corresponding to ligand–metal transitions of AuCl4− and AuCl3(OH)−.21 With the increased concentration of BSA added, the band at 220 nm became weaker, and that at 291 nm disappeared gradually (Fig. S1b†), indicating the gradual replacement of chloride ions by the functional groups (mainly carboxyl groups) of BSA.22 These results indicated reduced activity of the gold precursor and thus slowed down growth of Au particles with the increased concentration of BSA contained in the reaction solutions, which may be profitable for the formation of the gold nanoflowers. Control experiments were designed and carried out to illustrate this assumption. At the BSA concentration of 3.0 μM, the resulting hydrosols prepared with AA concentrations of 0.65, 1.50 and 2.50 mM presented SPR peaks around 618 nm (Fig. 3a), accompanied by decreased absorbance in the 900–1100 nm window, suggesting the decreased size and/or anisotropic character of the resulting Au particles with the increased concentration of AA and thus promoted reduction of the gold precursor. TEM observations show that all the particles were flower-like in shape, and their average sizes decreased from 113 nm to 60 and 51 nm when the AA concentration increased from 0.65 mM to 1.50 and 2.50 mM (Fig. 3b–d). At the BSA concentration of 1.0 μM, the Au particles prepared at different AA concentrations were quasi-spherical in shape, and their average size decreased from 28 to 23 nm when the AA concentration increased from 0.65 to 1.50 mM (Fig. S2†). These results suggested that the evolution in shape of the Au particles is primarily dependent on concentration of BSA contained in the reaction solution and the promoted reduction of the gold precursor is profitable for the formation of Au nanoflowers with decreased size.
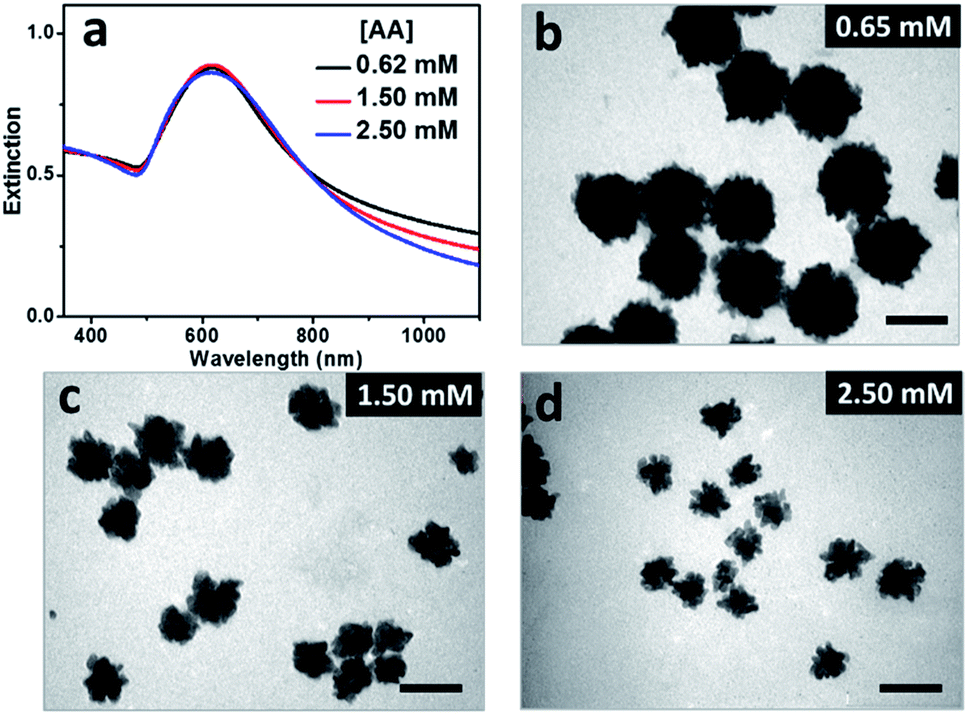 | ||
| Fig. 3 (a) UV-Vis spectra of Au hydrosols prepared at different AA concentrations. The concentration of BSA was fixed to be 3.0 μM in all the reactions. (b–d) TEM images of the Au particles prepared at AA concentrations of (b) 0.65, (c) 1.50 and (d) 2.50 mM. The scale bars were 100 nm. | ||
X-ray diffraction (XRD) patterns were recorded to further characterize the crystal structures of the Au particles prepared at 1.0 and 3.0 μM BSA. Both the particles presented four peaks at 2θ angles of 38.2°, 44.4°, 64.8° and 77.9°, corresponding to (111), (200), (220) and (311) planes of face-centered-cubic (fcc) lattice of gold (Fig. 4a).10 It is identified the intensity ratios of the (200) and (111) planes, I(200)/I(111), were 0.39 and 0.23 respectively for the particles prepared at 1.0 and 3.0 μM BSA, suggesting the (111) is the predominant lattice plane to direct the growth of Au nanoflowers.11 Enlarged TEM image indicated that the surface of the Au particle prepared with 1.0 μM BSA was quite smooth (Fig. 4b). High-resolution TEM image of the edge presented (111) lattice planes with random orientations (Fig. 4b, inset). However, there exit many branches on surface of the Au particle prepared with 3.0 μM BSA (Fig. 4c), which is expected to be related to the decreased intensity ratio of I(200)/I(111) in the XRD pattern. High-resolution TEM images of a typical branch in the Au NF prepared with 3.0 μM BSA showed the preferential growth of the branch along the 〈111〉 direction (Fig. 4d), which was consistent with the results of XRD analysis. Thus it was deduced that the role of BSA in formation of Au nanoflowers was to direct the preferential growth along 〈111〉 direction of the Au particles by anchoring onto other planes. The coordination of the functional groups of BSA, primarily carboxylic ones, with the Au ions resulted in reduced activity of the gold precursor and thus decreased number of Au nuclei, contributing to the formation of gold flowers with increased size with the increased BSA concentrations as shown in Fig. 1e and f, which can also be acquired by decreasing the concentration of the reductant (AA) used in the reactions (Fig. 3b–d).
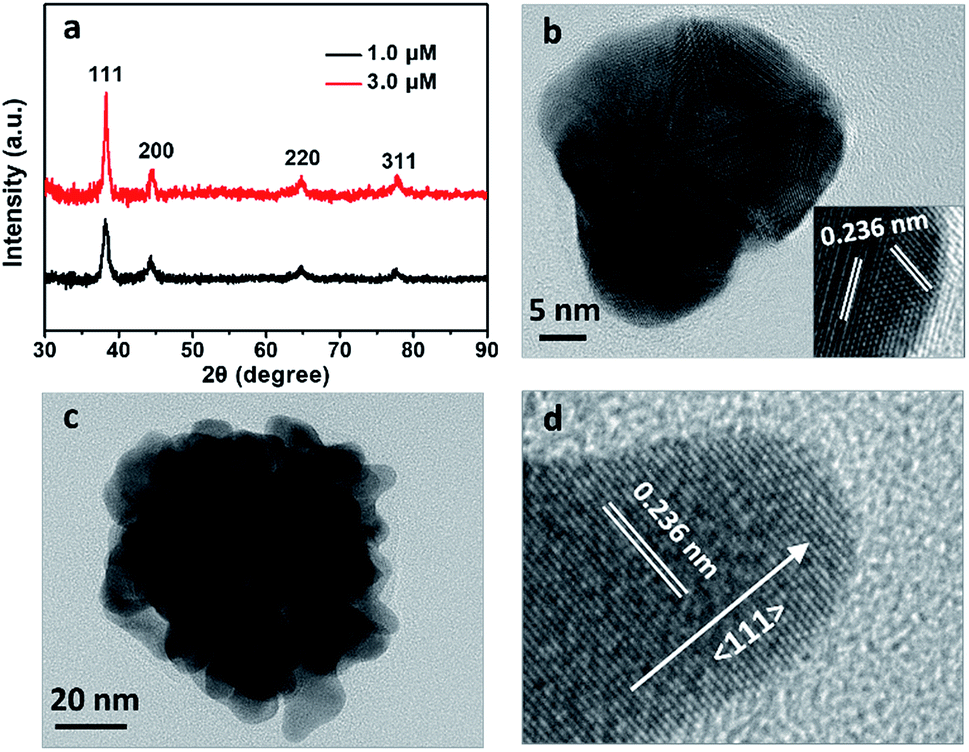 | ||
| Fig. 4 (a) XRD patterns of the Au particles prepared with 1.0 and 3.0 μM BSA. (b) Enlarged TEM images of a single spherical-like Au nanoparticle prepared with 1.0 μM BSA. Inset of (b) was the high-resolution TEM image of the edge. (c) Enlarged TEM images of a single Au NF prepared with 3.0 μM BSA. (d) High-resolution TEM image of a typical branch of the Au NF obtained at 3.0 μM BSA. The lattice spacing of 0.236 nm is attributed to (111) plane of fcc gold. | ||
It is known that Au NFs are readily to go ripening and become spherical in shape, especially under acidic condition and in presence of chloride ions, which greatly limit their performance. It is surprising to see that the Au NFs synthesized by using BSA as ligand present superior stability in the acidic solution and in presence of chloride ions. As shown in Fig. 5a, UV-Vis spectra of the Au NFs kept almost unchanged even after 120 h when pH of the solution was lowered to 3.0 in presence of 5 mM sodium chloride, much stable than the citrate-capped Au NFs, which evolved into spherical in shape within 5 min under the acidic solution (pH 4.2) in presence of 1.3 mM sodium chloride.13 Zeta-potential measurements indicated that the Au nanoflowers presented an isoelectric point around 4.7 (Fig. 5b), the same as that of BSA, suggesting that the surface properties of the Au NFs are dominated by BSA. TEM observations show that the resulting Au NFs were coated by a thin layer of BSA with thickness ca. 3 nm (inset of Fig. 5b), which contributed to the superior stability of the Au NFs against ripening. Based on above results and discussion, we can conclude that BSA plays multiple roles in preparation of the Au particles. Firstly, it acts as ligand to tune the activity of the gold precursor via coordination of its surface functional groups, such as carboxyl and amino ones, with the gold ions. As such, increase in concentration of BSA will contribute to the formation of gold precursor with reduced activity and thus the growth of Au particles with increased size. Secondly, it acts as shape-directing agent to promote the preferential growth of the gold particles along the 〈111〉 direction by anchoring onto other planes of the fcc gold, contributing to the formation of Au NFs under optimized BSA concentration. Thirdly, it acts as ligand to endow the resulting Au NFs with excellent stability and reversible surface charge switchable according to its isoelectric point. At present stage, such BSA assisted approaches still lack the ability to tune the length and number of branches of the resulting Au nanoflowers precisely, which remains a great challenge in the field of solution-based synthesis of branched nanocrystals.
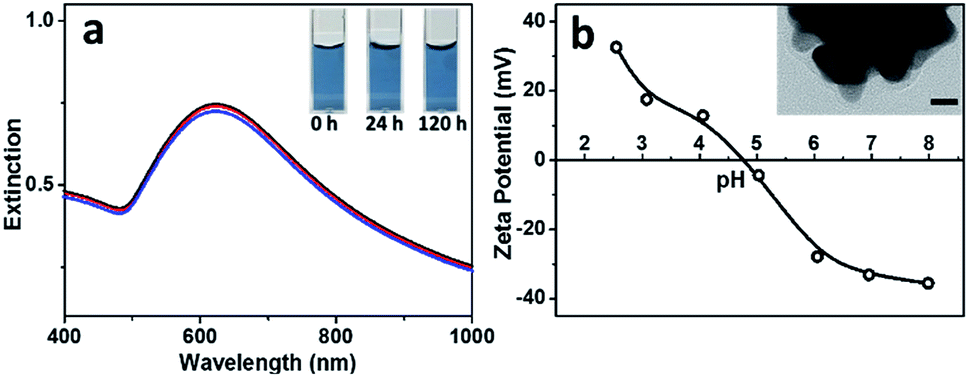 | ||
| Fig. 5 (a) UV-Vis spectra of the Au NF hydrosol prepared with 3 μM BSA (pH 3.0) after the addition of 5 mM sodium chloride for 0 (black line), 24 (red line) and 120 h (blue line). Inset gives photos of the hydrosol taken at 0, 24 and 120 h after the addition of 5 mM sodium chloride. (b) Variation in zeta potentials of the Au NF hydrosol with pH. Inset gives high-resolution TEM image of a single Au nanoflower. Scale bar was 10 nm. | ||
Surface properties of the Au nanoflowers were further illustrated by pH-dependent SERS experiments. As shown in Fig. 6a, the positively charged dye, Rhodamine 6G, only presented strong SERS signal at pH (8.0) higher than the isoelectric point (4.7). In contrast, the negatively charged dye, methyl blue, only presented SERS signals at pH (3.0) lower than the isoelectric point (Fig. 6b). This provides us the opportunity for selective detection of charged dyes by optimizing the electrostatic attraction and repulsion between the Au nanoflowers and dye molecules. When the Au nanoflowers were added into the mixtures of Rhodamine 6G and methyl blue, only the signal from positively charged Rhodamine 6G was observed when the surface of the Au nanoflowers were negatively charged (pH 8.0), and only that from the negatively charged methyl blue were observed when the nanoflowers were positively charged (pH 3.0). However, both the signals from Rhodamine 6G and methyl blue were greatly weakened when pH of the solution (5.0) was around the isoelectric point of the Au nanoflowers (see Fig. S3† for molecular structures of Rhodamine 6G and methyl blue, and Tables S1 and S2† for detailed assignments of the Raman signals). It was noted that when there exist functional groups with strong affinity to gold particles in the analytes, for example, 4-aminothiophenol and 4-mercaptobenzoic acid, the charge selective Raman responses will be suppressed to some extent since the electrostatic repulsion is not enough to push the analytes away from the Au NF surface at this time (see Fig. S4†).
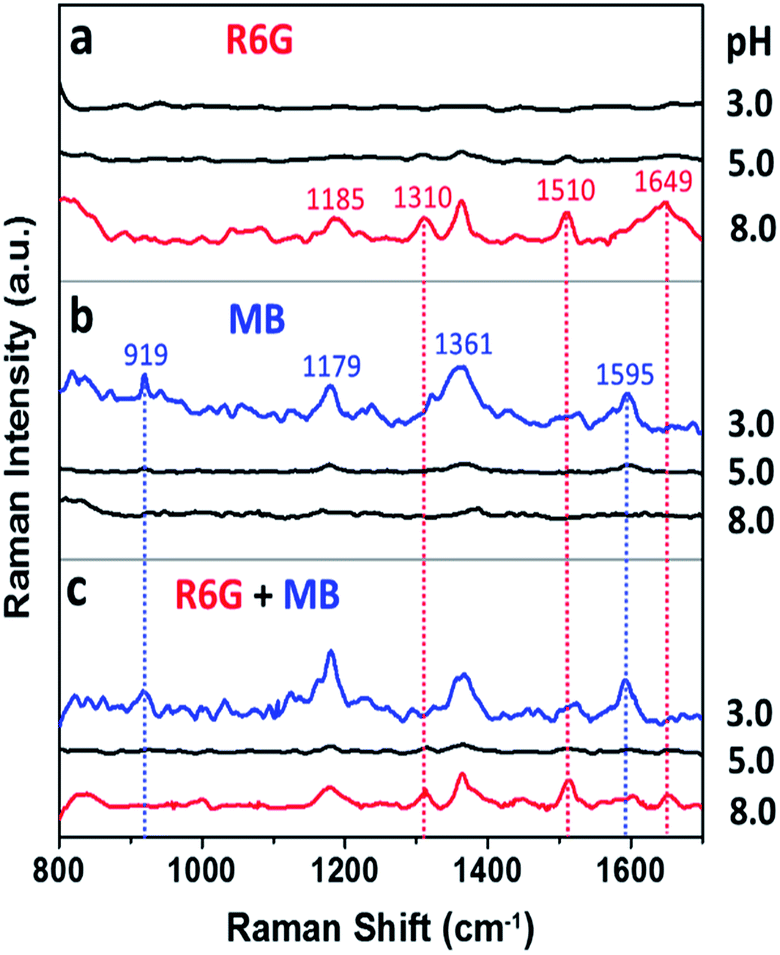 | ||
| Fig. 6 SERS spectra of (a) Rhodamine 6G (R6G), (b) methyl blue (MB) and (c) their 1:1 mixture (R6G + MB) at pH 3.0, 5.0 and 8.0, respectively, recorded after being mixed with the Au nanoflower hydrosols prepared with 3.0 μM BSA for 20 min. | ||
Conclusions
In summary, we developed a facile approach for preparation of ultra-stable Au nanoflowers by using BSA as ligand at room temperature. Preferential growth along the 〈111〉 direction of gold contributed to the anisotropic growth of Au particles and thus the formation of gold nanoflowers. Increased concentration of BSA or decreased concentration of the reductant contributed to reduced activity or reduced reduction of the gold precursor, leading to the formation of Au nanoflowers with increased size. The BSA capped on surface of the gold nanoflowers not only contributed to the superior stability of the Au nanoflowers against ripening, but also endowed them with the ability to recognize oppositely charged dyes by SERS measurements according to the isoelectric point of the protein.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21773089, 51372097).References
- L. Shao, A. S. Susha, L. S. Cheung, T. K. Sau, A. L. Rogach and J. Wang, Langmuir, 2012, 28, 8979–8984 CrossRef CAS PubMed.
- E. Hao, R. C. Bailey, G. C. Schatz, J. T. Hupp and S. Li, Nano Lett., 2004, 4, 327–330 CrossRef CAS.
- K. Mao, Y. Chen, Z. Wu, X. Zhou, A. Shen and J. Hu, J. Agric. Food Chem., 2014, 62, 10638–10645 CrossRef CAS PubMed.
- B. Liu, D. Tang, J. Tang, B. Su, Q. Li and G. Chen, Analyst, 2011, 136, 2218–2220 RSC.
- P. Huang, O. Pandoli, X. Wang, Z. Wang, Z. Li, C. Zhang, F. Chen, J. Lin, D. Cui and X. Chen, Nano Res., 2012, 5, 630–639 CrossRef CAS.
- B. K. Jena and C. R. Raj, Chem. Mater., 2008, 20, 3546–3548 CrossRef CAS.
- Q. Li, Y. Jiang, R. Han, X. Zhong, S. Liu, Z.-Y. Li, Y. Sha and D. Xu, Small, 2013, 9, 927–932 CrossRef CAS PubMed.
- J. Xie, Q. Zhang, J. Y. Lee and D. I. C. Wang, ACS Nano, 2008, 2, 2473–2480 CrossRef CAS PubMed.
- P. Vijayaraghavan, C.-H. Liu and K. C. Hwang, ACS Appl. Mater. Interfaces, 2016, 8, 23909–23919 CrossRef CAS PubMed.
- C. Y. Song, N. Zhou, B. Y. Yang, Y. J. Yang and L. H. Wang, Nanoscale, 2015, 7, 17004–17011 RSC.
- V. M. Kariuki, J. C. Hoffmeier, I. Yazgan and O. A. Sadik, Nanoscale, 2017, 9, 8330–8340 RSC.
- Z. Wang, J. Zhang, J. M. Ekman, P. J. A. Kenis and Y. Lu, Nano Lett., 2010, 10, 1886–1891 CrossRef CAS PubMed.
- L. Zhao, X. Ji, X. Sun, J. Li, W. Yang and X. Peng, J. Phys. Chem. C, 2009, 113, 16645–16651 CrossRef CAS.
- Z. Li, W. Li, P. H. C. Camargo and Y. Xia, Angew. Chem., Int. Ed., 2008, 47, 9653–9656 CrossRef CAS PubMed.
- Y. Imura, S. Koizumi, R. Akiyama, C. Morita-Imura and T. Kawai, Langmuir, 2017, 33, 4313–4318 CrossRef CAS PubMed.
- J. Xie, J. Y. Lee and D. I. C. Wang, J. Phys. Chem. C, 2007, 111, 10226–10232 CrossRef CAS.
- P. Murawala, S. M. Phadnis, R. R. Bhonde and B. L. V. Prasad, Colloids Surf., B, 2009, 73, 224–228 CrossRef CAS PubMed.
- T. K. Sau, A. L. Rogach, F. Jäckel, T. A. Klar and J. Feldmann, Adv. Mater., 2010, 22, 1805–1825 CrossRef CAS PubMed.
- N. G. Bastús, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098–11105 CrossRef PubMed.
- L. Au, B. Lim, P. Colletti, Y. S. Jun and Y. Xia, Chem.–Asian J., 2010, 5, 123–129 CrossRef CAS PubMed.
- D. V. Goia and E. Matijević, Colloids Surf., A, 1999, 146, 139–152 CrossRef CAS.
- X. Ji, X. Song, J. Li, Y. Bai, W. Yang and X. Peng, J. Am. Chem. Soc., 2007, 129, 13939–13948 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04852a |
| This journal is © The Royal Society of Chemistry 2019 |