
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLong noncoding RNA ZFAS1 enhances adriamycin resistance in pediatric acute myeloid leukemia through the miR-195/Myb axis†
Qun Lia and
Jianmin Wang *b
*b
aDepartment of PICU, First People's Hospital of Shangqiu City, Henan Province, China
bDepartment of Pediatric Medicine, First People's Hospital of Shangqiu City, No. 292, Kaixuan Road, Yuyang District, Shangqiu, Henan Province 476100, China. E-mail: zdedyy@163.com; Tel: +86-370-3255865
First published on 6th September 2019
Abstract
Background: Development of chemoresistance remains a major obstacle for pediatric acute myeloid leukemia (AML) management. Zinc finger antisense 1 (ZFAS1) is a novel tumor-related lncRNA that has been reported as an oncogene involved in the development of pediatric AML. The purpose of the present study was to investigate the role and underlying mechanism of ZFAS1 in AML chemoresistance. Methods: The expression levels of ZFAS1 and miR-195 were assessed by qRT-PCR and Myb expression was detected using western blotting. The CCK-8 assay was used to determine the IC50 value for adriamycin (ADR) and cell proliferation. Cell apoptosis was measured by flow cytometry. The targeted interaction between miR-195 and ZFAS1 or Myb was evaluated by the dual-luciferase reporter assay or RNA immunoprecipitation assay. Results: Our data revealed that ADR treatment induced ZFAS1 expression in pediatric AML. Silencing of ZFAS1 or Myb alleviated AML cell resistance to ADR in vitro. ZFAS1 directly targeted miR-195 and negatively modulated miR-195 expression. Myb was a direct target of miR-195. Moreover, the inhibitory effect of ZFAS1 silencing on ADR resistance of AML cells was mediated by miR-195 in vitro. Myb was involved in the regulation of the ZFAS1/miR-195 axis in ADR resistance of AML cells. Conclusion: Our data indicated that ZFAS1 silencing alleviated ADR resistance of AML cells in vitro through acting as a sponge for miR-195 and regulating Myb expression. Targeting ZFAS1 might be a promising therapeutic strategy for pediatric AML treatment.
1. Introduction
Pediatric acute myeloid leukemia (AML) accounts for 15–20% of the acute leukemias diagnosed in children. It represents approximately 30% of cancer-related deaths in this age range.1,2 Over the past two decades, the prognosis of pediatric AML has remarkably improved with current long-term survival rates of about 70%.3,4 However, development of chemoresistance remains a major obstacle for pediatric AML treatment.5,6 Therefore, a better understanding of the molecular mechanisms of chemoresistant pediatric AML is very important.Long noncoding RNAs (lncRNAs), a novel heterogeneous class of transcripts with >200 nucleotides in length, play crucial roles in various aspects of cellular biological processes.7 They are emerging as new players in the cancer paradigm with potential roles in both oncogenic and tumor suppressive pathways.8 LncRNAs regulate the progression and chemoresistance development of pediatric AML via various mechanisms, including their microRNA (miRNA) binding functions.9–11 Zinc finger antisense 1 (ZFAS1) is a novel tumor-related lncRNA that has been identified as an oncogene in the development of multiple human cancers.12–14 Previous studies had demonstrated that ZFAS1 was abnormally expressed in AML, and ZFAS1 overexpression promoted AML progression through acting as a sponge for miR-150.15,16 A recent document manifested that ZFAS1 promoted cervical cancer cell chemoresistance to cisplatin.17 Nevertheless, the role and underlying mechanism of ZFAS1 on AML chemoresistance remain undefined.
In this study, our data demonstrated that ZFAS1 expression was elevated by adriamycin (ADR) treatment in bone marrow (BM) samples of pediatric AML patients and AML cells. Furthermore, ZFAS1 silencing alleviated ADR resistance of AML cells in vitro through sponging miR-195 and regulating Myb expression.
2. Materials and methods
2.1. Clinical samples
The present study enrolled 30 children (younger than 12 years of age) with newly diagnosed AML, received ADR-based chemotherapy according to the BFM backbone protocol at Department of PICU Section; First People's Hospital, Shangqiu City, Henan Province, China between April 2012 and August 2016. BM samples were collected from these pediatric AML children before and after treatment with ADR-based chemotherapy. All samples were immediately frozen in liquid nitrogen and stored at −80 °C until RNA extraction. Informed consent was signed by all parents, and approval was obtained from the Ethics Committee of Department of PICU Section; First People's Hospital, Shangqiu City, Henan Province, China.2.2. Cell culture and treatment
Human acute leukemia cell lines (HL60 and THP-1) obtained from ATCC (Manassas, VA, USA) were maintained in Iscove's Modified Dulbecco's Medium (IMDM, Gibco, Invitrogen Ltd, Milan, Italy) supplemented with 10% fetal bovine serum (FBS, Cripion, Industria Brasileira, Andradina, SP, Brazil), 1% (v/v) antibiotics (100 U ml−1 penicillin and 100 μg ml−1 streptomycin, Gibco) at 37 °C in a humidified 5% CO2 incubator. ADR-resistant AML cell lines (HL60/ADR and THP-1/ADR) were established as described previously18 as follows: (1) HL60 and THP-1 cells in the logarithmic phase were treated with 0.8 μM of ADR (Sigma-Aldrich, St. Louis, MO, USA) for 12 h, and then fresh ADR-free medium was replaced; (2) cells were allowed to reach the logarithmic phase before another ADR treatment cycle, until the treatment showed no significant influence on cell morphology and proliferation. Additional 0.5 μM of ADR was added into the culture medium to maintain the ADR-resistant phenotype of HL60/ADR and THP-1/ADR.2.3. Cell transfection
All synthetic miR-195 mimic, miRNA inhibitor (anti-miR-195) and corresponding negative control (miR-NC mimic and anti-miR-NC) were purchased from Applied Biosystems (Nieuwerkerk a/d IJssel, Netherlands). ZFAS1 overexpression plasmid (pcDNA-ZFAS1) and negative control plasmid (pcDNA-NC), silencer select ZFAS1 and Myb siRNAs (si-ZFAS1 and si-Myb) and negative control siRNA (si-NC) were obtained from Applied Biosystems. Cells were transfected with the indicated oligonucleotide or plasmid using Lipofectamine 3000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA) following the instructions of manufacturers.2.4. RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from BM samples and cells using Trizol Reagent (Invitrogen) according to the protocols of manufacturers. The quality and quantity of RNA extracts were assessed by a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Montchanin, DE, USA). For ZFAS1 expression analysis, cDNA was synthesized using random primers (Fermentas, St. Leon-Rot, Germany) and M-MLV Reverse Transcriptase (Invitrogen), and the quantification of cDNA was carried out using SYBR Green Master Mix (Applied Biosystems) with GAPDH as a housekeeping gene for normalization. The expression of miR-195 was determined by the Applied Biosystems TaqMan MicroRNA Assay System on an ABI7500 Quantitative PCR Detector (Applied Biosystems). U6 was used as the internal reference for miR-195 expression. The expression levels of ZFAS1 and miR-195 were defined from the threshold cycle (Ct), and relative expression levels were calculated using the 2−ΔΔCt method.2.5. Determination of IC50 values and cell proliferation
Cellular IC50 value for ADR and cell proliferation ability were determined using a Cell Counting Kit-8 (CCK-8, Dojindo Laboratories, Kumamoto, Japan) following the manufacturer's guidance. For the measurement of cell proliferation, at the indicated time points (0, 24, 48 and 72 h) after transfection, cells were seeded into a 96-well plate, and then 10 μl of CCK-8 solution was added into each well at 37 °C for 2 h. For the detection of IC50 value for ADR, transfected cells were treated with various concentrations of ADR (0, 2, 4, 8, 16, 32, 64 and 128 μM) for 48 h, followed by the conduction of CCK-8 assay. Absorbance at the wavelength of 450 nm was measured using a microplate reader (Bio-Rad Laboratories, Richmond, CA, USA).2.6. Flow cytometry
Cell apoptosis was detected by flow cytometry using the Annexin V-FITC Apoptosis Detection Kit (EMD Chemicals, Gibbstown, NJ, USA) in accordance with the instructions of manufacturers. Transfected cells were double stained with Annexin V-FITC conjugate and PI in isotonic solution, followed by the measurement of cell apoptotic rate by a FACS Calibur (BD Biosciences, San Jose, CA, USA).2.7. Western blotting
Total cellular protein was extracted using ice-cold RIPA lysis buffer (50 mM Tris–HCl, pH = 7.4, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 0.5% sodium deoxycholate) containing 1× complete protease inhibitor cocktail (EDTA-free, Sigma-Aldrich). Protein extracts were electrophoresed on a 10% SDS polyacrylamide gel and transferred to the Immobilon-P PVDF membrane (Millipore, Billerica, MA, USA). The following primary antibodies were used: anti-multidrug resistance-related protein 1 (anti-MRP1, Abcam, Cambridge, UK; dilution 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000), anti-multidrug resistance 1 (anti-MDR1, Abcam; dilution 1:2000), anti-Myb (Abcam; dilution 1:1000) and anti-β-actin (Abcam; dilution 1:5000). Horseradish peroxidase-conjugated IgG (Abcam; dilution 1:5000) was used a second antibody. Protein bands were detected using ECL reagent (GE Healthcare Technologies, Waukesha, WI, USA) and analyzed by ImageJ software (National Institutes of Health, Bethesda, MD, USA).
:1000), anti-multidrug resistance 1 (anti-MDR1, Abcam; dilution 1:2000), anti-Myb (Abcam; dilution 1:1000) and anti-β-actin (Abcam; dilution 1:5000). Horseradish peroxidase-conjugated IgG (Abcam; dilution 1:5000) was used a second antibody. Protein bands were detected using ECL reagent (GE Healthcare Technologies, Waukesha, WI, USA) and analyzed by ImageJ software (National Institutes of Health, Bethesda, MD, USA).
2.8. Bioinformatics analysis
LncBase Predicted v.2 software was used to predict the targeted miRNAs of ZFAS1 at http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=lncbasev2/index. Analysis for the targets of miR-195 was performed using TargetScan v.7.1 online software available at http://www.targetscan.org.2.9. Dual-luciferase reporter assay
ZFAS1 wild-type luciferase reporter vector (ZFAS1-WT) containing miR-195 targeted sequence and its mutant in the seeded region (ZFAS1-MUT), Myb luciferase reporter vector (Myb 3′-UTR-WT) which included the 3′-UTR of Myb downstream from the luciferase reporter gene and site-directed mutation of miR-195 binding site (Myb 3′-UTR-MUT) were constructed by Applied Biosystems. To verify the targeted interrelation between ZFAS1 and miR-195, cells were transfected with ZFAS1-WT or ZFAS1-MUT, together with miR-NC mimic or miR-195 mimic using Lipofectamine 3000 Transfection Reagent. To confirm whether Myb was a direct target of miR-195, Myb 3′-UTR-WT or Myb 3′-UTR-MUT was cotransfected into cells and miR-NC mimic or miR-195 mimic. After transfection for 24 h, cells were lysed using the Dual-Luciferase Reporter Assay Protocol (Promega, Madison, WI, USA) and luciferase luminescence was detected by the GloMax® 20/20 luminometer (Promega).2.10. RNA immunoprecipitation (RIP) assay
Cell lysates were prepared in ice-cold RIPA lysis buffer supplemented with 1× protease inhibitor cocktail. Cellular RNA-induced silencing complex (RISC) was isolated as described previously.19 RISC was immunoprecipitated from cell lysates using 1 μg anti-Argonaute 2 (anti-Ago2, Abcam) antibody overnight at 4 °C, with IgG antibody as negative control. Immunoprecipitates were captured with protein A/G-plus agarose beads (Stanta Cruz Biotechnology, Santa Cruz, CA, USA). Total RNA was extracted from the immunoprecipitates using Trizol Reagent, and qRT-PCR was performed to determine the enrichment of ZFAS1.2.11. Statistical analysis
The difference in ZFAS1 expression among different BM samples was analyzed by Mann–Whitney U test, and the differences between two groups were compared using an unpaired Student's t-test. All experimental data were expressed as mean ± standard deviation (SD) from at least three independent repeats. P values at 0.05 or smaller were considered significant.3. Results
3.1. ADR treatment induced the expression of ZFAS1 in pediatric AML
For a preliminary investigation of the involvement of ZFAS1 in ADR resistance of pediatric AML, we firstly evaluated ZFAS1 expression in the BM samples from 30 pediatric AML patients before and after treatment with ADR-based chemotherapy. qRT-PCR results revealed that the expression levels of ZFAS1 were higher in post-treatment group than those in un-treatment group (Fig. 1A). Our data also showed that ADR treatment triggered a significant increase of MRP1 and MDR1 levels in BM samples (ESI Fig. 1†). Subsequently, we established two ADR resistant AML cell lines (HL60/ADR and THP-1/ADR). CCK-8 assay verified that the IC50 values for ADR of HL60/ADR and THP-1/ADR cells were significantly elevated compared with their parents (Fig. 1B and C), indicating a successful establishment for ADR resistant cell lines. Moreover, in comparison to their parents, ZFAS1 expression was significantly increased in HL60/ADR and THP-1/ADR cells (Fig. 1D). These results together suggested that ZFAS1 was involved in the development of ADR resistance in pediatric AML. | ||
| Fig. 1 ADR treatment increased the expression of ZFAS1 in pediatric AML. (A) The expression of ZFAS1 by qRT-PCR in the BM samples from 30 pediatric AML patients before and after treatment with ADR-based chemotherapy. (B and C) The IC50 values for ADR by CCK-8 assay in HL60, HL60/ADR, THP-1 or THP-1/ADR cells. (D) ZFAS1 level by qRT-PCR in HL60, HL60/ADR, THP-1 or THP-1/ADR cells. *P < 0.05. | ||
3.2. Silencing of ZFAS1 attenuated AML cell resistance to ADR in vitro
To observe the effect of ZFAS1 on ADR resistance of pediatric AML in vitro, we performed “phenocopy” silencing by siRNA targeting ZFAS1 (si-ZFAS1). Transient introduction of si-ZFAS1, but not a scrambled control sequence, significantly reduced the expression of ZFAS1 in HL60/ADR and THP-1/ADR cells (Fig. 2A). CCK-8 assay showed that in comparison to negative control, ZFAS1 silencing significantly decreased the IC50 values for ADR in the two cells (Fig. 2B). Moreover, ZFAS1 silencing resulted in a repression of cell proliferation and an enhancement of cell apoptosis in HL60/ADR and THP-1/ADR cells (Fig. 2C–E). The expression levels of MRP1 and MDR1 have been manifested to be closely relevant to chemotherapy resistance of AML.20,21 Herein, we determined the levels of MRP1 and MDR1 proteins in HL60/ADR and THP-1/ADR cells when ZFAS1 silencing. In contrast to negative control, ZFAS1 silencing triggered a significant reduction of MRP1 and MDR1 protein levels (Fig. 2F). All these data indicated that ZFAS1 silencing attenuated ADR resistance of AML cells in vitro.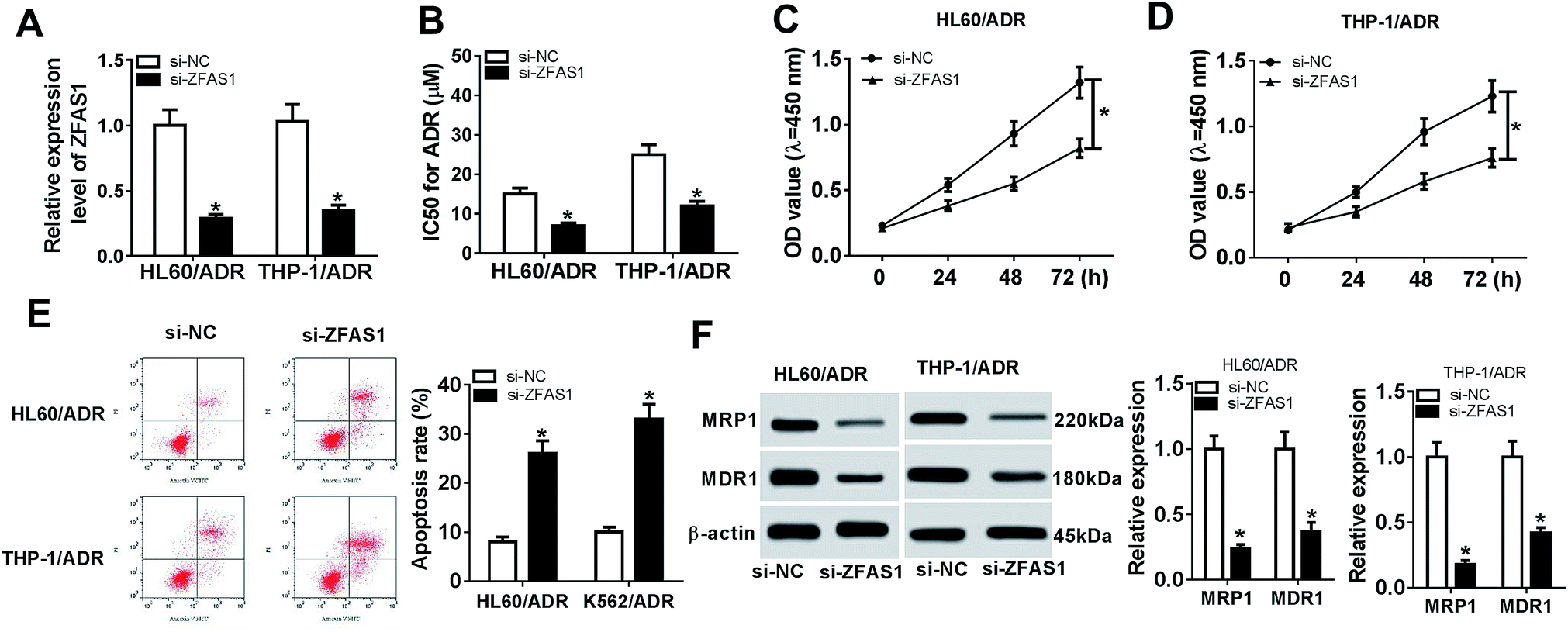 | ||
| Fig. 2 ZFAS1 silencing alleviated ADR resistance of AML cells in vitro. HL60/ADR and THP-1/ADR cells were transfected with si-ZFAS1 or si-NC, followed by the determination of ZFAS1 expression by qRT-PCR (A), IC50 value for ADR by CCK-8 assay (B), cell proliferation by CCK-8 assay (C and D), cell apoptosis by flow cytometry (E), the levels of MRP1 and MDR1 proteins by western blotting (F). *P < 0.05. | ||
3.3. ZFAS1 directly interacted with miR-195 and negatively modulated miR-195 expression
Our above results indicated that ZFAS1 silencing alleviated ADR resistance of AML cells, and then we explored how ZFAS1 achieved it. Of interest, using LncBase Predicted v.2 software, the predicted data showed a putative binding site for miR-195 in ZFAS1 (Fig. 3A). To validate this, dual-luciferase reporter assays were performed by ZFAS1 luciferase reporter (ZFAS1-WT) containing miR-195 targeted sequence and its mutant in the seeded region (ZFAS1-MUT). In comparison to negative control, transfection of miR-195 mimic significantly reduced the luciferase activity of ZFAS1-WT (about 68% in HL60/ADR cells and 75% in THP-1/ADR cells, Fig. 3B and C). However, little change was observed in the activity of ZFAS1-MUT in response to miR-195 overexpression (Fig. 3B and C). Ago2, a key component of RISC, has been identified as a key regulator involved in the mature processes of miRNAs.22 Hence, RIP assays were carried out using anti-Ago2 antibody. These data revealed that compared with negative control, the enrichment levels of ZFAS1 in HL60/ADR and THP-1/ADR cells were highly abundant by miR-195 overexpression (Fig. 3D), providing evidence for the endogenous interaction between ZFAS1 and miR-195 in AML cells. Additionally, our results demonstrated a significant downregulation of miR-195 expression in HL60/ADR and THP-1/ADR cells compared to their parents (Fig. 3E). | ||
| Fig. 3 ZFAS1 functioned as a molecular sponge of miR-195. (A) Schematic of the predicted miR-195 binding site in ZFAS1 and the mutated miR-195 binding sequence. (B and C) ZFAS1 luciferase reporter (ZFAS1-WT) or its mutation in the seeded region (ZFAS1-MUT) was transfected into HL60/ADR and THP-1/ADR cells together with miR-NC mimic or miR-195 mimic, and then the luciferase activity was detected. (D) The enrichment of ZFAS1 was determined in cells transfected with miR-NC mimic or miR-195 mimic. (E) The expression of miR-195 by qRT-PCR in HL60, THP-1, HL60/ADR or THP-1/ADR cells. (F) ZFAS1 expression by qRT-PCR in cells transfected with pcDNA-NC or pcDNA-ZFAS1. (G) MiR-195 expression by qRT-PCR in cells transfected with pcDNA-NC, pcDNA-ZFAS1, si-NC or si-ZFAS1. *P < 0.05. | ||
Next, we assessed whether, if so, how ZFAS1 modulated miR-195 expression in AML cells. In contrast to negative control, transfection of pcDNA-ZFAS1 resulted in about a 2.8-fold increase of ZEAF1 expression in HL60/ADR cells and a 3-fold increase in THP-1/ADR (Fig. 3F). Moreover, miR-195 expression was strikingly repressed by ZFAS1 overexpression, while it was highly elevated following ZFAS1 silencing in HL60/ADR and THP-1/ADR cells (Fig. 3G). Together, these data suggested that ZFAS1 functioned as a molecular sponge of miR-195.
3.4. Inhibitory effect of ZFAS1 silencing on ADR resistance of AML cells was mediated by miR-195 in vitro
Then, we further investigated whether ZFAS1 silencing exerted its inhibitory effect on ADR resistance of AML cells by miR-195. Cotransfection of in-miR-195, but not a scrambled control sequence, significantly abrogated the increased effect of ZFAS1 silencing on miR-195 expression in HL60/ADR and THP-1/ADR cells (Fig. 4A). Moreover, the restoration of miR-195 expression significantly antagonized the changes of IC50 value for ADR, cell proliferation and apoptosis, which induced by ZFAS1 silencing (Fig. 4B–E and ESI 2A†). Additionally, as demonstrated by western blotting, the decreased levels of MRP1 and MDR1 proteins triggered by ZFAS1 silencing were highly abolished by miR-195 expression restoration in HL60/ADR and THP-1/ADR cells (Fig. 4F and G). These data together established that miR-195 mediated the inhibitory effect of ZFAS1 silencing on ADR resistance in AML cells in vitro. | ||
| Fig. 4 MiR-195 mediated the inhibitory effect of ZFAS1 silencing on ADR resistance of AML cells in vitro. HL60/ADR and THP-1/ADR cells were transfected with si-NC, si-ZFAS1, si-ZFAS1 + in-miR-NC or si-ZFAS1 + in-miR-195, followed by the detection of miR-195 expression by qRT-PCR (A), IC50 value for ADR by CCK-8 assay (B), cell proliferation by CCK-8 assay (C and D), cell apoptosis by flow cytometry (E), the levels of MRP1 and MDR1 proteins by western blotting (F and G). *P < 0.05. | ||
3.5. Myb was a direct target of miR-195
miRNAs exert biological function through regulating the expression of their target mRNAs. Therefore, we carried out a detailed analysis for the molecular targets of miR-195. In silico prediction by TargetScan v.7.1 online software revealed that Myb harbored a putative target sequence for miR-195 in its 3′-untranslated region (3′-UTR) (Fig. 5A). To determine whether Myb was a target of miR-195, the luciferase reporter assays were performed using a wild-type vector which included the 3′-UTR of Myb downstream from the luciferase reporter gene (Myb 3′-UTR-WT). With Myb 3′-UTR-WT and miR-195 overexpression caused a significant reduction in luciferase activity (Fig. 5B and C). When miR-195-5p binding sequence was partly mutated (Myb 3′-UTR-MUT), no reduction in luciferase activity was observed with miR-195 overexpression (Fig. 5B and C). Our data also demonstrated that Myb expression was significantly increased in HL60/ADR and THP-1/ADR cells compared with their parents (Fig. 5D). Moreover, in contrast to their counterparts, the expression of Myb was markedly decreased by miR-195 overexpression, while it was strikingly increased when miR-195 knockdown in HL60/ADR and THP-1/ADR cells (Fig. 5E), indicating the negative regulation of miR-195 on Myb expression. All these results strongly pointed a notion that Myb was a direct target of miR-195. | ||
| Fig. 5 Myb was a direct target of miR-195. (A) Schematic of Myb 3′-UTR illustrating position of the miR-195 binding site and the mutation in the seed region. (B and C) The luciferase activity in HL60/ADR and THP-1/ADR cells cotransfected with Myb 3′-UTR-WT or Myb 3′-UTR-MUT and miR-NC mimic or miR-195 mimic. (D) Myb expression by qRT-PCR in HL60, THP-1, HL60/ADR or THP-1/ADR cells. (E) Myb expression in HL60/ADR and THP-1/ADR cells transfected with miR-NC mimic, miR-195 mimic, in-miR-NC or in-miR-195. *P < 0.05. | ||
3.6. Myb was involved in the regulation of ZFAS1/miR-195 axis on ADR resistance of AML cells
Given our data that Myb was a direct target of miR-195, we further explored the detailed role of Myb on ADR resistance of AML cells by transfection of si-Myb. As shown by western blotting, transfection of si-Myb resulted in decreased Myb expression (about 65% in HL60/ADR cells and 70% in THP-1/ADR cells) compared with negative control (Fig. 6A). Moreover, Myb silencing led to a decrease of IC50 value for ADR and a repression of cell proliferation, as well as an enhancement of cell apoptosis in HL60/ADR and THP-1/ADR cells (Fig. 6B–E and ESI 2B†). Additionally, in contrast to negative control, the levels of MRP1 and MDR1 proteins were significantly weakened by Myb silencing (Fig. 6F and H), indicating the inhibitory effect of Myb silencing on AML cell resistance to ADR. All these data suggested that Myb silencing attenuated ADR resistance of AML cells. | ||
| Fig. 6 Myb was involved in the regulation of ZFAS1/miR-195 axis on ADR resistance of AML cells. HL60/ADR and THP-1/ADR cells were transfected with si-NC or si-Myb, followed by the measurement of Myb expression by western blotting (A), the IC50 value for ADR by CCK-8 assay (B), cell proliferation by CCK-8 assay (C and D), cell apoptosis by flow cytometry (E), the levels of MRP1 and MDR1 proteins by western blotting (F and G). (H) Myb expression by western blotting in HL60/ADR and THP-1/ADR cells transfected with si-NC, si-ZFAS1, si-ZFAS1 + in-miR-NC or si-ZFAS1 + in-miR-195. *P < 0.05. | ||
Lastly, we explored whether ZFAS1 functioned as a molecular sponge of miR-195 to regulate the expression of Myb in AML cells. As expected, Myb expression was significantly reduced by ZFAS1 silencing, while this effect was highly abolished in response to in-miR-195 cotransfection in HL60/ADR and THP-1/ADR cells (Fig. 6H). Taken together, these results implied that Myb was involved in the regulation of ZFAS1/miR-195 axis on ADR resistance of AML cells.
4. Discussion
Chemoresistance is a frequent and serious problem in the pharmacological treatment of patients with pediatric AML. Increasing evidence has showed that lncRNAs are related to AML chemoresistance, providing a possibility of lncRNAs as therapeutic targets for AML management. For instance, Li et al. reported that taurine upregulated gene 1 (TUG1) silencing weakened AML cell resistance to ADR through epigenetically elevating miR-34a.23 Dong et al. illuminated that the knockdown of HOXA cluster antisense RNA2 (HOXA-AS2) repressed cell proliferation, enhanced the apoptosis and weakened ADR resistance in ADR-resistant AML cells by targeting miR-520c-3p/S100A4 axis.24 Additionally, coagulation factor IX (FIX) regulated ADR resistance of AML cells by regulating miR-342/ALG3 axis.25 In the present study, we aimed to explore whether or how ZFAS1 influenced ADR resistance of AML cells in vitro. Our data suggested that ZFAS1 silencing alleviated AML cell resistance to ADR through sponging miR-195 and regulating Myb expression.ZFAS1 has been identified as a novel oncogene in a large number of human cancers.12–14 On the other hand, ZFAS1 repressed cellular proliferation and differentiation in human breast cancer, suggesting its tumor suppressive property.26 These contradictory functions may be attributed to different characteristic and type of human cancers. Considering the oncogenic role in AML and chemoresistance-related effect of ZFAS1,15–17 the present study started from the hypothesis that ZFAS1 might enhance the development of AML chemoresistance. To confirm this, we firstly determined ZFAS1 expression in the BM samples of patients with pediatric AML and AML cells in response to ADR treatment. Our data revealed that ADR induced ZFAS1 expression in pediatric AML, suggesting the involvement of ZFAS1 in the development of ADR resistance. Subsequently, we observed how ZFAS1 influenced AML cell resistance to ADR by loss-of-function experiments, and we illuminated that ZFAS1 silencing weakened ADR resistance of AML cells in vitro. A previous study demonstrated that ZFAS1 implicated in the development of multidrug resistance in T-cell acute lymphoblastic leukemia by regulating ADR-resistant cell growth and apoptosis.27
One of the mechanisms by which lncRNAs affect cancer development is to sequester specific miRNAs. Thus, online LncBase v.2 software was used to predict the targeted miRNAs of ZFAS1. Among these candidates, miR-195 was of interest in this research owing to its tumor suppressive role in a series of human cancers, such as breast cancer, bladder cancer and prostate cancer.28–30 Subsequently, we firstly validated that ZFAS1 directly targeted miR-195 and inhibited miR-195 expression. Previous researches reported that miR-195 significantly alleviated cancer cell resistance to chemotherapy in colorectal cancer and gastric cancer.31,32 Moreover, aberrant expression of miR-195 was found in the BM samples of AML patients and low serum level of miR-195 might serve as a potential marker for the supervision of occurrence and prognostic of clinical outcome in pediatric AML.33,34 In the present study, we found that miR-195 expression was highly downregulated in ADR-resistant AML cells. More importantly, our data firstly indicated that miR-195 mediated the inhibitory effect of ZFAS1 silencing on ADR resistance of AML cells in vitro.
It is widely accepted that miRNAs exert biological function by controlling expression of their target mRNAs. Then, we carried out a detailed analysis for the targets of miR-195 using TargetScan v.7.1 software. Of interest, the 3′-UTR of Myb mRNA harbored a putative targeted sequence for miR-195. Further, we confirmed that Myb was a direct target of miR-195, in accordance with former work.35 Additionally, ADR induced the expression of Myb in AML cells. Myb, a human oncogene, functions as a key regulator of stem and progenitor cells in the BM and a neurogenic region of human brain, thereby implicating in a range of leukaemias.36,37 In the present study, we firstly demonstrated that Myb silencing weakened AML cell resistance to ADR, suggesting the involvement of Myb in the development of AML chemoresistance. This effect was similar with the inhibitory function of ZFAS1 silencing on AML chemoresistance. LncRNAs regulate gene expression by acting as a molecular sponge of miRNA. Considering the targeted association between miR-195 and ZFAS1 or Myb and their regulatory effects on AML cell resistance to ADR, we further investigated whether ZFAS1 modulated Myb expression by sponging miR-195 in AML cells. As expected, our data suggested that Myb was involved in the regulation of ZFAS1/miR-195 axis on ADR resistance of AML cells. Similar with our finding, ZFAS1 suppressed AML progression by sponging miR-150 and regulating Sp1 and Myb expression.16 More researches about the regulatory relationship between Myb and ZFAS1/miR-195 axis in AML chemoresistances will be performed in further work.
In conclusion, our study suggested that ZFAS1 silencing attenuated AML cell resistance to ADR in vitro at least partly through acting as a sponge of miR-195 and modulating Myb expression. Targeting ZFAS1 might be a promising therapeutic target for pediatric AML management.
Conflicts of interest
There is no conflict of interest regarding the publication of this paper.References
- G. J. Kaspers, Expert Rev. Anticancer Ther., 2012, 12, 405–413 CrossRef CAS.
- D. A. Arber, A. Orazi, R. Hasserjian, J. Thiele, M. J. Borowitz, M. M. Le Beau, C. D. Bloomfield, M. Cazzola and J. W. Vardiman, Blood, 2016, 127, 2391–2405 CrossRef CAS.
- U. Creutzig, M. Zimmermann, J.-P. Bourquin, M. N. Dworzak, G. Fleischhack, N. Graf, T. Klingebiel, B. Kremens, T. Lehrnbecher and C. von Neuhoff, Blood, 2013, 122, 37–43 CrossRef CAS.
- M. Rasche, M. Zimmermann, L. Borschel, J. P. Bourquin, M. Dworzak, T. Klingebiel, T. Lehrnbecher, U. Creutzig, J. H. Klusmann and D. Reinhardt, Leukemia, 2018, 32, 2167–2177 CrossRef.
- J. D. de Rooij, C. M. Zwaan and M. van den Heuvel-Eibrink, J. Clin. Med., 2015, 4, 127–149 CrossRef CAS.
- F. C. Brown, P. Cifani, E. Drill, J. He, E. Still, S. Zhong, S. Balasubramanian, D. Pavlick, B. Yilmazel, K. M. Knapp, T. A. Alonzo, S. Meshinchi, R. M. Stone, S. M. Kornblau, G. Marcucci, A. S. Gamis, J. C. Byrd, M. Gonen, R. L. Levine and A. Kentsis, Br. J. Haematol., 2017, 176, 86–91 CrossRef CAS.
- A. Fatica and I. Bozzoni, Nat. Rev. Genet., 2014, 15, 7–21 CrossRef CAS.
- E. A. Gibb, C. J. Brown and W. L. Lam, Mol. Cancer, 2011, 10, 38 CrossRef CAS.
- C. Y. Xing, X. Q. Hu, F. Y. Xie, Z. J. Yu, H. Y. Li, Z. Bin, J. B. Wu, L. Y. Tang and S. M. Gao, FEBS Lett., 2015, 589, 1981–1987 CrossRef CAS.
- Y. Zhang, Y. Liu and X. Xu, J. Cell. Biochem., 2018, 119, 6296–6308 CrossRef CAS.
- B. Liu, X. Ma, Q. Liu, Y. Xiao, S. Pan and L. Jia, Cell Death Dis., 2018, 9, 688 CrossRef.
- T. Li, J. Xie, C. Shen, D. Cheng, Y. Shi, Z. Wu, X. Deng, H. Chen, B. Shen and C. Peng, Cancer Res., 2015, 75, 3181–3191 CrossRef CAS.
- L. Pan, W. Liang, M. Fu, Z.-h. Huang, X. Li, W. Zhang, P. Zhang, H. Qian, P.-C. Jiang and W.-R. Xu, J. Cancer Res. Clin. Oncol., 2017, 143, 991–1004 CrossRef CAS.
- S. Xie, Q. Ge, X. Wang, X. Sun and Y. Kang, Cell Cycle, 2018, 17, 154–161 CrossRef CAS.
- H. Guo, L. Wu, P. Zhao and A. Feng, Exp. Ther. Med., 2017, 14, 647–651 CrossRef CAS.
- S. Gan, P. Ma, J. Ma, W. Wang, H. Han, L. Chen, X. Li, F. Wu and H. Sun, Eur. J. Pharmacol., 2019, 844, 38–48 CrossRef CAS.
- L. L. Feng, F. R. Shen, J. H. Zhou and Y. G. Chen, Gene, 2019, 696, 105–112 CrossRef CAS.
- L. Wan, Y. Tian, R. Zhang, Z. Peng, J. Sun and W. Zhang, J. Cell. Biochem., 2018, 119, 3843–3852 CrossRef CAS.
- X. H. Liu, M. Sun, F. Q. Nie, Y. B. Ge, E. B. Zhang, D. D. Yin, R. Kong, R. Xia, K. H. Lu, J. H. Li, W. De, K. M. Wang and Z. X. Wang, Mol. Cancer, 2014, 13, 92 CrossRef.
- S. H. Kweon, J. H. Song and T. S. Kim, Biochem. Biophys. Res. Commun., 2010, 395, 104–110 CrossRef CAS.
- Z. Benderra, A. M. Faussat, L. Sayada, J. Y. Perrot, R. Tang, D. Chaoui, H. Morjani, C. Marzac, J. P. Marie and O. Legrand, Clin. Cancer Res., 2005, 11, 7764–7772 CrossRef CAS.
- R. I. Gregory, T. P. Chendrimada, N. Cooch and R. Shiekhattar, Cell, 2005, 123, 631–640 CrossRef CAS.
- Q. Li, W. Song and J. Wang, Biomed. Pharmacother., 2019, 109, 1793–1801 CrossRef CAS.
- X. Dong, Z. Fang, M. Yu, L. Zhang, R. Xiao, X. Li, G. Pan and J. Liu, Cell. Physiol. Biochem., 2018, 51, 886–896 CrossRef CAS.
- B. Liu, X. Ma, Q. Liu, Y. Xiao, S. Pan and L. Jia, Cell Death Dis., 2018, 9, 688 CrossRef.
- M. E. Askarian-Amiri, J. Crawford, J. D. French, C. E. Smart, M. A. Smith, M. B. Clark, K. Ru, T. R. Mercer, E. R. Thompson and S. R. Lakhani, RNA, 2011, 17, 878–891 CrossRef CAS.
- Q. Liu, H. Ma, X. Sun, B. Liu, Y. Xiao, S. Pan, H. Zhou, W. Dong and L. Jia, J. Exp. Clin. Cancer Res., 2019, 38, 199 CrossRef.
- A. Qattan, H. Intabli, W. Alkhayal, C. Eltabache, T. Tweigieri and S. B. Amer, BMC Cancer, 2017, 17, 799 CrossRef.
- H. J. Li, X. M. Sun, Z. K. Li, Q. W. Yin, H. Pang, J. J. Pan, X. Li and W. Chen, Cell. Physiol. Biochem., 2017, 43, 2548–2561 CrossRef CAS.
- C. Cai, Q. B. Chen, Z. D. Han, Y. Q. Zhang, H. C. He, J. H. Chen, Y. R. Chen, S. B. Yang, Y. D. Wu, Y. R. Zeng, G. Q. Qin, Y. X. Liang, Q. S. Dai, F. N. Jiang, S. L. Wu, G. H. Zeng, W. D. Zhong and C. L. Wu, Clin. Cancer Res., 2015, 21, 4922–4934 CrossRef CAS.
- Y. Jin, M. Wang, H. Hu, Q. Huang, Y. Chen and G. Wang, Int. J. Biol. Macromol., 2018, 117, 445–453 CrossRef CAS.
- R. Ye, B. Wei, S. Li, W. Liu, J. Liu, L. Qiu, X. Wu, Z. Zhao and J. Li, OncoTargets Ther., 2017, 8, 97260–97272 Search PubMed.
- G. Cammarata, L. Augugliaro, D. Salemi, C. Agueli, M. La Rosa, L. Dagnino, G. Civiletto, F. Messana, A. Marfia, M. G. Bica, L. Cascio, P. M. Floridia, A. M. Mineo, M. Russo, F. Fabbiano and A. Santoro, Am. J. Hematol., 2010, 85, 331–339 CAS.
- Z. Hong, R. Zhang and H. Qi, Cancer Biomarkers, 2018, 21, 269–275 CAS.
- Y. C. Zhou, L. W. Tian, X. C. Wang, L. H. Ye, G. Q. Zhao, M. Yu, G. J. Li, Y. J. Lei and Y. C. Huang, Cancer Lett., 2014, 347, 65–74 CrossRef.
- D. R. Pattabiraman and T. J. Gonda, Leukemia, 2013, 27, 269–277 CrossRef CAS.
- D. R. Pattabiraman, C. McGirr, K. Shakhbazov, V. Barbier, K. Krishnan, P. Mukhopadhyay, P. Hawthorne, A. Trezise, J. Ding, S. M. Grimmond, P. Papathanasiou, W. S. Alexander, A. C. Perkins, J. P. Levesque, I. G. Winkler and T. J. Gonda, Blood, 2014, 123, 2682–2690 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04843j |
| This journal is © The Royal Society of Chemistry 2019 |