
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational investigations of the binding mechanism of novel benzophenone imine inhibitors for the treatment of breast cancer†
Amneh Shtaiwi ab,
Rohana Adnan*b,
Melati Khairuddeanb and
Shafi Ullah Khanc
ab,
Rohana Adnan*b,
Melati Khairuddeanb and
Shafi Ullah Khanc
aSchool of Pharmacy, Middle East University, Queen Alia Airport Street, 11118 Amman, Jordan. E-mail: ashtaiwi@meu.edu.jo
bSchool of Chemical Sciences, Universiti Sains Malaysia, 11800 Penang, Malaysia. E-mail: r_adnan@usm.my; Tel: +6046533262
cSchool of Pharmacy, Monash University Malaysia, Jalan Lagoon Selatan, Bandar Sunway, 47500 Subang Jaya, Malaysia. E-mail: shafiullahpharmD@gmail.com
First published on 1st November 2019
Abstract
4-Hydroxytamoxifen (4-OHT), the most common hormone used for the treatment of breast cancer, is a selective estrogen receptor modulator (SERM) inhibitor that acts as an antagonist in breast tissue and a partial agonist in the endometrium. However, the detailed molecular mechanism of 4-OHT structure modification has not been well investigated to date. Herein, molecular docking, molecular dynamics simulations and free energy calculations were performed to explore the mechanisms of the molecular interactions between newly designed benzophenone imines (BIs) and the three forms apo, antagonist and agonist of the human estrogen receptor hERα. The proposed inhibitors were designed by replacing the triarylethylene estrogenic scaffold found in 4-OHT with Schiff base triarylimine derivatives. The antiestrogen scaffold i.e. the O-alkyl side chain in 4-OHT was developed by incorporating an alanine amino acid side chain functionality into the triarylimine scaffold. Docking results reveal that the newly designed BIs bind to the hydrophobic open pocket of the apo and antagonist hERα conformations with higher affinity as compared to the natural and synthetic estrogen estradiol (E2) and 4-OHT. The analysis of the molecular dynamics simulation results based on six different systems of the best docked BI (5c) with hERα receptors demonstrates stable interactions, and the complex undergoes fewer conformational fluctuations in the open apo/antagonist hERα receptors as compared to the case of the closed agonist. In addition, the calculated binding free energies indicate that the main factor that contributes to the stabilization of the receptor–inhibitor complexes is hydrophobic interactions. This study suggests that the development of these Schiff base derivatives may be worth exploring for the preparation of new 4-OHT analogues.
Introduction
Breast cancer is the most common cancer among women worldwide and in the Asia-Pacific region, accounting for 18% of all cancer cases in 2012. Although the incidence rates of breast cancer remain significantly higher in New Zealand and Australia, in recent years, a rapid increase in the cases of breast cancer has been observed in several Asian countries, particularly Malaysia and Thailand.1 The incidence and mortality estimates for Malaysia in 2012 alone were 5410 and 2572 cases, respectively.1,2 The human estrogen receptor hERα protein, which is located on the surface of tumors, can bind to estrogen (17β-estradiol, E2) and enhance the growth and spreading of cancer cells;3 moreover, the estrogen hormone has been identified as a key stimulant in the development of hERα-positive breast cancer, which constitutes around 70–80% of all breast cancers.4,5 Breast cancer involves the generation of tumors through estrogen signalling after the binding of estrogen to the hERα receptors; this activates hormone-responsive genes that promote DNA synthesis and cell reproduction;6 the estrogen receptor is a nuclear receptor, and these receptors have common structural functions, referred to as domains, including the N-terminal (A/B) domain, the DNA binding domain (C), the hinge domain (D) and the ligand binding domain (LBD, E/F) or the C-terminal domain. The activation function 2 (AF-2) in the ligand binding domain has a ligand-dependent transcription factor.7,8 A common treatment of hormone-sensitive breast cancer in the early stage is surgery to remove the tumour, radiotherapy and hormone therapy.9 The hormone therapy can be used to remove or block the action of hormones such as estrogen, which has been recognised as a key molecular driver in breast cancers.9,10 In the ligand–receptor pathway, the binding of E2 to hERα induces a dynamic conformational change that leads to hERα dimerization and association with co-regulatory proteins with the subsequent transcriptional activation of estrogen-responsive genes.11 Selective estrogen receptor modulators (SERMs), such as the benzothiophene derivative 4-hydroxy tamoxifen (4-OHT), act as competitive blockers of the estrogen-hERα binding and have been successfully used in the treatment of hERα-positive breast cancer.12 When the ligands bind to hERα, many changes occur, particularly in the position of helix12 (H12) at the ligand binding site, and these changes differ when different ligands bind to the receptor. Moreover, the transcriptional activity differs when different ligands bind to hERα.13 Upon the binding of E2 to the hERα ligand binding domain, estradiol rests in a binding cavity within the hydrophobic core of the LBD formed by the helices H3, H6, H8, H11 and H12.14 The substrate forms a series of specific hydrogen bonds via the two hydroxyl groups. The phenolic hydroxyl group from the A ring forms direct hydrogen bonds with the carboxylate of a glutamic acid residue in H3 (Glu353) and the arginine residue in H6 (Arg394). On the other hand, the 17β-hydroxyl group in the D ring forms a hydrogen bond with a single histidine residue in H11 (His524) (see Fig. 1). The hydrophobic core of E2 also plays a role in the binding of E2 with the hydrophobic residues of the ER-LBD, which forms close contacts with the alanine and phenylalanine amino acid residues that bind the ligand.14,15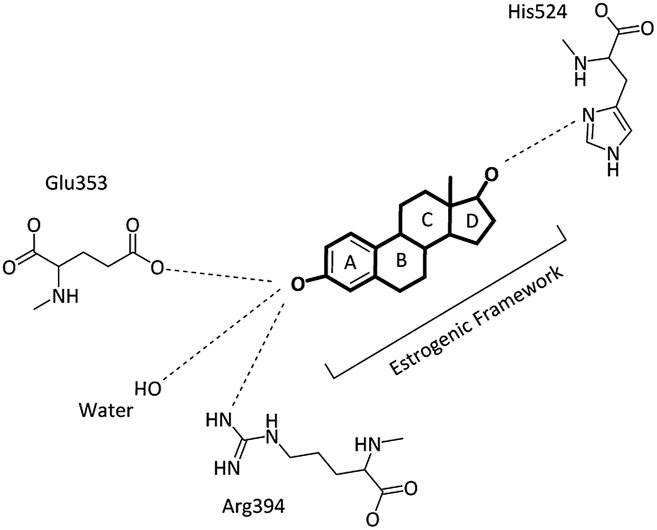 | ||
| Fig. 1 Schematic showing the binding mode of E2 in the hERα-LBD complex. | ||
The result of the hydrophobic and polar binding modes of E2 is the folding of H12 across H3 and H11, leading to the agonist conformation and thus enhancing gene transcription.15,16 When agonists, such as estradiol, bind to ER (see Fig. 2a), the ligand is trapped within a hydrophobic binding cavity formed by the helices H3 (blue), H6 (grey) and H11 (green).17 This allows the inner hydrophobic surface of H12 (red) to fold across H3 and H11 and cap the entrance of the cavity.18 Conversely, antagonists, such as the synthetic antiestrogen 4-OHT, have polar or bulky steric side-chains and occupy the same binding cavity as agonists; however, they force H12 to move towards the open binding site. This allows H12 (red) to overlap with the H3 (blue) and H5 (orange) regions (see Fig. 2b) and occupy the surface where the co-activator protein should bind.13,17,18
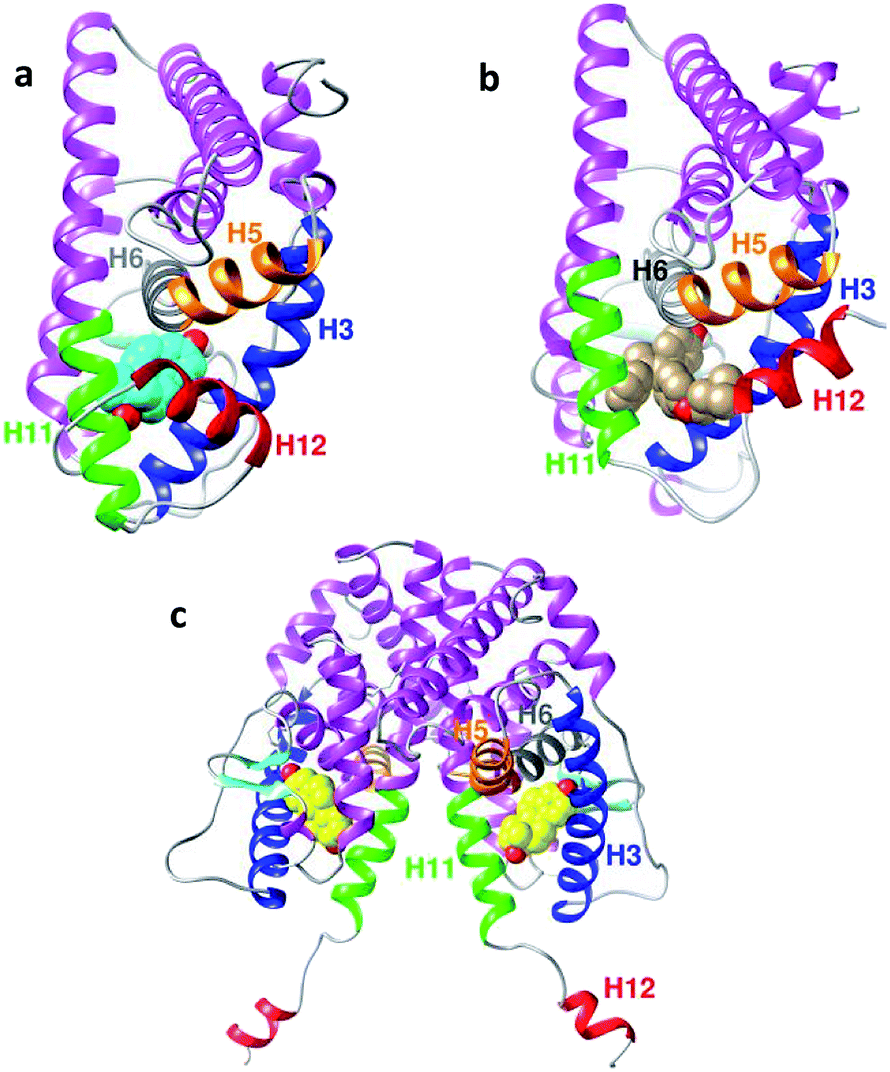 | ||
| Fig. 2 (a) Backbone of the agonist conformation of hERα LBD (PDB ID: 1G50) in the complex with estradiol E2 (cyan). (b) Antagonist conformation of the hERα LBD (PDB ID: 3ERT) in the complex with 4-OHT (grey). (c) Apo conformation (PDB ID: 1A52) in the complex with estradiol E2 (yellow). Important helices are highlighted: H3 (blue), H5 (orange), H6 (grey), H11 (green) and H12 (red). | ||
The extended apo conformation of the NR LBDs was first described in retinoic X receptor-α (RXRα), where H12 was extended away from the surface of the ligand binding core and did not have any hydrophobic interactions with the LBD.19 Similarly, the apo-form of the human estrogen receptor hERα PDB ID: 1A52 (see Fig. 2c) employs this type of extended conformation,20 where H12 interacts with the other monomer of the dimer binding site. Moreover, H11 and H12 in two neighboring monomers bond via disulphide bonds. Therefore, H12 in the first monomer interacts with the binding site of the second monomer to result in these cross-monomer interactions.21,22
SERMs, such as 4-OHT, can deactivate the estrogen signaling pathway via competitive binding to the ER, causing a conformational change in the subsequently formed ER dimer. This involves the shifting of H12 to an adjacent coactivator site (AF-2), thus blocking the binding of the co-activator; this significantly reduces the level of estrogen-regulated gene transcription.16
Antiestrogens
4-OHT has been used as a front-line endocrine therapy for breast cancer in pre-menopausal and post-menopausal women for the last 40 years. Moreover, it is used in the treatment of male breast cancer.23 At the hERα binding site, 4-OHT occupies the same hydrophobic binding pocket as E2, involving the helices H3, H6, H8 and H11 (see Fig. 3). Similar to the A-ring of E2, the phenolic hydroxyl group of 4-OHT interacts with Glu353 and Arg394.24 The dimethylaminoethoxy group, which is the side-chain of 4-OHT, lies in a narrow channel between H3 and H11, and the tertiary amine of the chain is placed near an amino acid residue, Asp351.16 These strong interactions prevent the hydrophobic inner surface of H12 from entering the region and folding over the binding pocket, thereby disrupting the coactivator surface and forcing the H12 orientation towards an open/antagonist conformation. For this reason, the majority of SERMs possess an alkylaminoethoxy side-chain that contributes to the blocking of the transcription of estrogen-dependent genes in breast tissue.25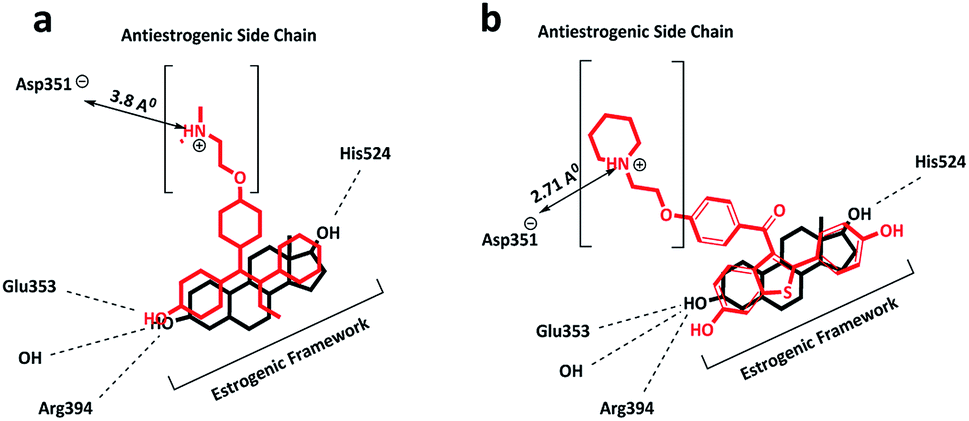 | ||
| Fig. 3 Schematic showing the overlay of the binding modes of (a) E2 with 4-OHT and (b) E2 with raloxifene in the hERα-LBD. The amino acids that interact with E2 and SERMs and the hydrogen bonding formation are shown. | ||
The interactions of the side-chain terminal in 4-OHT with the Asp351 amino acid residue in the LBD differ from those in other SERMs, such as the raloxifene side-chain, as the alkylaminoethoxy side-chain is significantly stronger in raloxifene as compared to that in 4-OHT. The side-chain adopts a position much closer to the Asp351 residue, with a distance of 2.7 Å (as compared to 3.8 Å in other SERMs) and this contributes to the improved shielding of Asp351 from H12 binding and an increased antagonistic effect.
The effects of side-chain and Asp351 amino acid interactions on the enhanced antagonistic properties were further demonstrated by amino acid substitution experiments.26
The mutation of Asp351 to glutamate results in an increased distance between the piperidine nitrogen and the protein residue, which subsequently results in an increase in the agonist effect.26
On the other hand, SERMs, such as 4-OHT and raloxifene, share the same estrogenic framework binding mode in hERα and act as antagonists in some tissues, whereas they show agonist properties in others, as presented in Table 1.27 Fulvestrant is a selective estrogen receptor down-regulator (SERD), a type of inhibitor with a bulky hydrophobic alkyl-sulfinyl side chain that binds to hERα and causes protein degradation. It is used to treat estrogen receptor-sensitive breast cancer, along with older classes of drugs and aromatase inhibitors.28 Fulvestrant has been found to alter the antagonistic behaviour of full antiestrogen as compared to the previously described SERMs. Fulvestrant causes the complete disruption of the AF2 domain and deactivation of the AF1 domain; moreover, it does not demonstrate agonistic behaviour in any tissue as compared to other SERMs (see Table 1).29
Although 4-OHT is a front-line treatment for breast cancer, resistance and increased risk of endometrial and uterus cancers are important clinical problems that have been associated with 4-OHT.30,31 Therefore, novel small molecule inhibitors with the ability to overcome antiestrogen resistance while limiting the adverse side effects are valuable pharmaceutical targets. This study describes new approaches to obtain these inhibitors through the incorporation of imine derivatives with alanine side chain functionalities into the antiestrogen scaffolds to generate analogues of 4-OHT. In this study, novel benzophenone imines (BIs) were designed by replacing the triarylethylene estrogenic scaffold found in 4-OHT with Schiff base triarylimine derivatives. On the other hand, the antiestrogenic tail O-alkyl side chain in 4-OHT was replaced by incorporating the alanine amino acid side chain functionality into the triarylimine scaffolds.
Recently, bisphenols,32 cyclic amides,33 diphenylamines,34 and Schiff bases35 have been studied as estrogen receptor ligands. Schiff bases were first designed as selective ligands for the estrogen receptors α and β, where they behaved as ERα agonists and ERβ antagonists.35 However, the development of Schiff bases as hERα antagonists through the incorporation of side chains to mimic the 4-OHT backbone has never been reported. The presence of alkyl chains of varying lengths in tamoxifen and various SERMs has been shown to increase the activity of Schiff bases as selective estrogen receptor down-regulators (SERDs), leading to enhanced antagonist effects in MCF-7 cells and anti-breast cancer activity.36–38
In this article, we report the binding interactions of newly designed Schiff bases with hERα using molecular docking and molecular dynamics simulations. In addition, we explored the stability and structural changes of the newly designed ligands following their complexation with three hERα forms, i.e. agonist, antagonist and apo conformations, to provide new information that might be useful in the development of new inhibitors with improved anti-estrogenic properties to treat breast cancer.
Methods
Strategies for the design of new benzophenone imines
The development of new BI hybrids with strong affinity for hERα could be achieved using core scaffolds that mimicked the binding interactions of known antiestrogens. In this study, the non-steroidal SERM triphenylethylene backbone of 4-OHT was chosen as a suitable scaffold due to its high affinity for hERα. The triarylethylene framework in tamoxifen, ospemifene, toremifene and many SERMs forms a backbone, which is responsible for mimicking the effect of natural estrogen, that can bind to the hER as an agonist or antagonist.39 The triarylethylene framework acts as an estrogen agonist, and the two alkyl chains attached to this framework are responsible for its antagonist behavior.39,40 Based on these observations, the contributions of polar imine functional groups in the presence of an alanine amino acid side chain (see Fig. 4) towards the anti-breast cancer activities of the novel triarylimines were examined.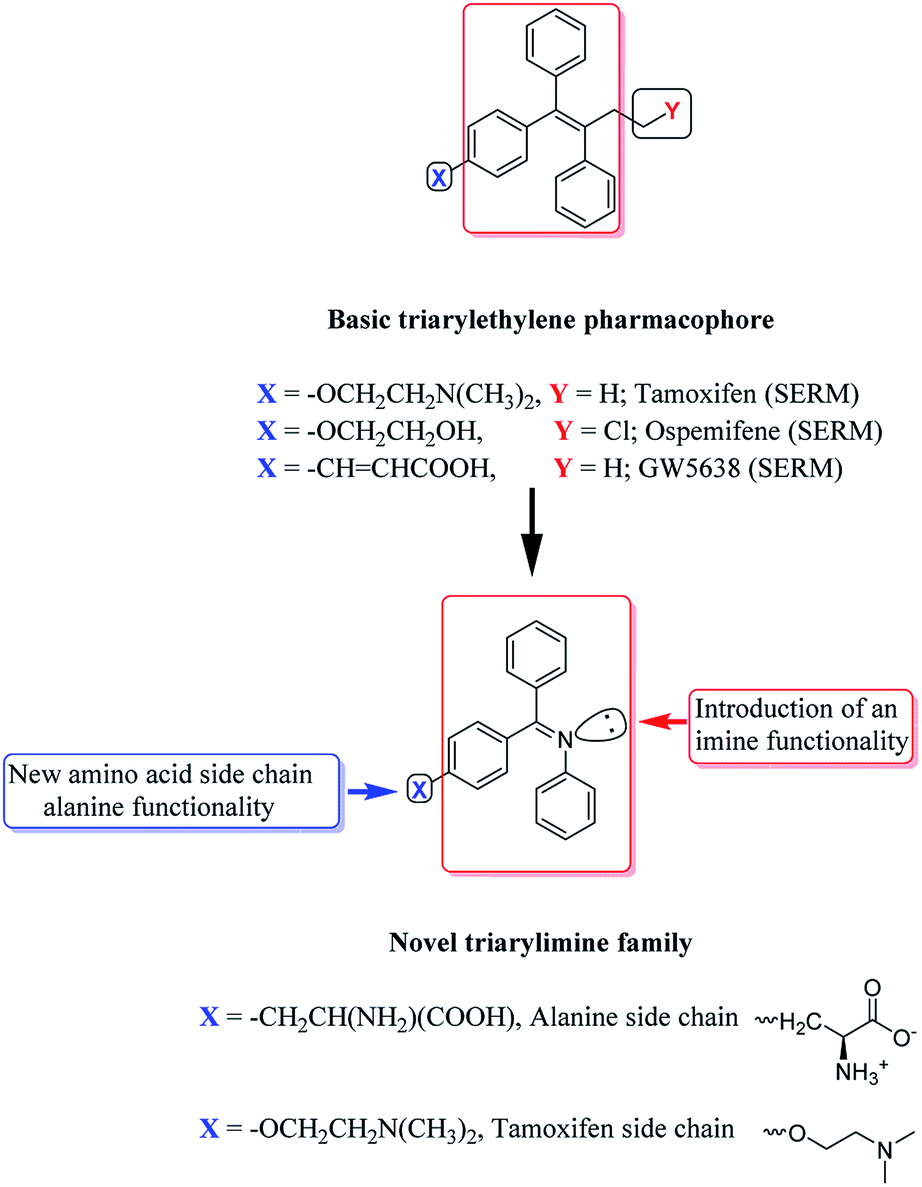 | ||
| Fig. 4 The design of novel benzophenone imine inhibitors. | ||
Molecular preparations of the hERα structures and ligands
The X-ray crystal structures of apo, agonist and antagonist human estrogen receptor (hERα), PDB ID: 1A52, 1G50 and 3ERT were retrieved from the Protein Data Bank (http://www.rcsb.org). In the protein preparation step, all the water molecules were removed except for those involved in the stabilization of the protein–ligand complex. The ligands, i.e. 17β-estradiol (E2) bound to apo 1A52, 4-OHT bound to the antagonist 3ERT and E2 bound to agonist 1G50, were removed from the crystal structures of the host molecules. Prior to conducting the docking calculations using Autodock 4.2.6, hydrogen atoms were added to the protein structures, and the missing residues of the proteins were fixed. Then, the hydrogen bonds were optimized to relax the strains using the AMBER FF99SB-ILDN force field41 program with the steepest descent algorithm for 1000 steps. The 3-D structures of the ligands were built using the Gaussview program,42 and the energy minimizations for the ligands were performed using the semiempirical PM3 method43 available in Gaussian 2009.44 In the second docking protocol, the preparation of proteins was carried out using the protein preparation utilities of Discovery Studio Client 16.1.0. Similarly, all water molecules were removed except for those involved in the stabilization of the protein–ligand complex. Hydrogen atoms were added to the protein structure to obtain the correct ionization and tautomeric states. The active site was selected around the co-crystal ligand within the protein structure using the Make Receptor 3.3.0 tool in OpenEye Suite.45 In the ligand preparation step, the structures of all nine benzophenone imine derivatives were generated using ChemDraw Professional v15, and energy minimizations of the compounds were carried out using the ligand preparation utilities available in Discovery Studio Client 16.1.0; moreover, the maximum conformers were generated using the OMEGA 2.5.1 tools of OpenEye Suite.46Molecular docking
The molecular docking calculations were performed using two approaches. The first approach uses a Lamarckian Genetic Algorithm available in Autodock 4.2.6.47,48 Docking simulations were applied for the three hERα conformations, i.e. 17β-estradiol bound to the apo (1A52.pdb), 4-OHT bound to the antagonist (3ERT.pdb), and 17β-estradiol bound to the agonist (1G50.pdb) structure, to ensure reproducibility in predicting the binding sites, binding conformations and binding affinities. The receptors and the ligands were prepared using the Autodock Tools program. Gasteiger partial charges were computed for each molecule. Rotatable bonds were defined as free for the ligands and rigid for the receptors. The grid maps of the docking simulation for the apo system 1A52 protein were prepared using a grid box of 70 × 70 × 65 Å and centred on the x, y, and z coordinates (107.27, 13.94, and 96.38), respectively. For the antagonist system 3ERT protein, the grid maps were prepared using an 80 × 80 × 80 Å grid box along the axes (30.01, −1.90, and 24.46), respectively. Finally, for the agonist system, the 1G50 protein was centred on the coordinates 105.28, 15.09, and 23.94 with the grid box size of 70 × 70 × 65 Å, respectively. Grid spacings were set at 0.375 Å for all systems. In this study, each docked compound was derived from 100 independent docking runs that were set to terminate after a maximum of 25 × 106 energy evaluations, a maximum number of 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 generations, and a mutation rate of 0.02; moreover, the population size was set to use 300 randomly placed individuals. A total of 100 independent docking runs were carried out for each docking system. For the second comparative docking approach, molecular docking calculations were performed using the two utilities FRED and HYBRID within the OEDocking tool of OpenEye Suite.49 The docking protocol was validated by re-docking the crystal structures of E2 and 4-OHT present in the respective proteins and found to have the RMSD values lower than 2 Å. After the docking validation protocol, all nine benzophenone imines were docked into the active sites of the target proteins, and a minimum 10 poses were generated. After the completion of the docking calculations, the best poses of each compound were selected based on the lowest FRED Chemguass4 and HYBRID Chemguass4 scores. The best possible conformations were then selected and analysed using the LigPlot and Chimera (http://www.cgl.ucsf.edu/chimera) programs.50,51 The newly designed BI ligands including the natural and synthetic substrates used in this study are shown in Fig. 5. The newly designed BIs were verified by the Lipinski's rule of five (RO5, molecular weight less than 500, logP or coefficient partition between −5 and 5, less than five hydrogen bond donors and less than ten hydrogen bond acceptors),52 as presented in Table 2.
000 generations, and a mutation rate of 0.02; moreover, the population size was set to use 300 randomly placed individuals. A total of 100 independent docking runs were carried out for each docking system. For the second comparative docking approach, molecular docking calculations were performed using the two utilities FRED and HYBRID within the OEDocking tool of OpenEye Suite.49 The docking protocol was validated by re-docking the crystal structures of E2 and 4-OHT present in the respective proteins and found to have the RMSD values lower than 2 Å. After the docking validation protocol, all nine benzophenone imines were docked into the active sites of the target proteins, and a minimum 10 poses were generated. After the completion of the docking calculations, the best poses of each compound were selected based on the lowest FRED Chemguass4 and HYBRID Chemguass4 scores. The best possible conformations were then selected and analysed using the LigPlot and Chimera (http://www.cgl.ucsf.edu/chimera) programs.50,51 The newly designed BI ligands including the natural and synthetic substrates used in this study are shown in Fig. 5. The newly designed BIs were verified by the Lipinski's rule of five (RO5, molecular weight less than 500, logP or coefficient partition between −5 and 5, less than five hydrogen bond donors and less than ten hydrogen bond acceptors),52 as presented in Table 2.
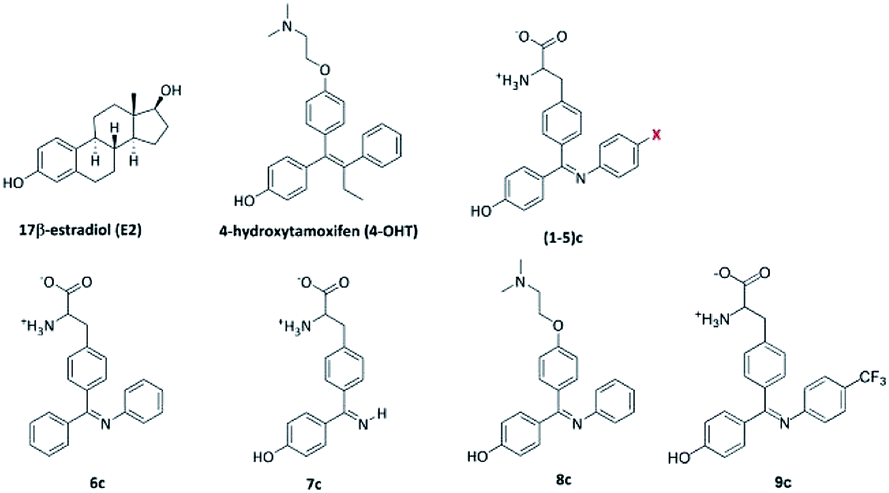 | ||
| Fig. 5 Structures of 17β-estradiol E2, synthetic estrogen 4-OHT and the newly designed ligands used in this study, where X for 1, 2, 3, 4 and 5 is H, F, Cl, CH3 and OH groups, respectively. | ||
| Ligand | Molecular weight | H-Bond donors | H-Bond acceptors | logP |
|---|---|---|---|---|
| 4-OHT | 387 | 1 | 3 | 4.28 |
| 1c | 360 | 4 | 5 | 3.51 |
| 2c | 378 | 4 | 5 | 3.65 |
| 3c | 394 | 4 | 5 | 3.39 |
| 4c | 374 | 4 | 5 | 3.82 |
| 5c | 376 | 5 | 6 | 3.22 |
| 6c | 344 | 3 | 4 | 3.81 |
| 7c | 284 | 5 | 5 | 1.76 |
| 8c | 360 | 1 | 4 | 4.50 |
| 9c | 428 | 4 | 5 | 4.64 |
Molecular dynamics simulations
A 100 ns MD simulation was performed for each of the six models obtained from the best benzophenone imines BIs 5c with the three hERα conformations apo 1A52, antagonist 3ERT and agonist 1G50, as shown in Table 3. All simulations were performed using the GROMACS 5.0.7 software package with the AMBER FF99SB-ILDN force field.41 The starting conformations of the protein–ligand were obtained from the lowest binding energy conformations via the docking study. The topology parameters of the hERα conformations were created using the GROMACS program, whereas the topology of ligand 5c was generated using ACPYPE available in the Amber-Tools package.53 The systems were solvated with the TIP3P water model in a cubic box with the spacing distance of 1.2 nm around the surface.54 To neutralize the systems, counter ions were added to balance the charge of the proteins. Then, the systems were minimized using the steepest descent method for 6000 steps, followed by the Berendsen thermostat equilibration run in the NVT (constant number of particles, volume and temperature) ensemble for 200 ps at 300 K.55 Then, the production runs were performed using the Parrinello–Rahman barostat in the NPT ensemble (constant number of particles, pressure and temperature) for 1 ns at 1 bar and 300 K.56 After the temperature and pressure adjustments, MD simulation runs were performed for the six different models for 100 ns each (see Table 3). The simulations were performed at 1 bar and 300 K without position restraints on the solute. The cutoffs for the Coulomb and van der Waals interactions were set to 12 Å and updated every 2 fs. The particle mesh Ewald (PME) method was used to correct the electrostatic interactions.57 The LINCS algorithm was used to constrain the bonds with hydrogen atoms.58 All simulations were computed with a time step of 2 fs, and the coordinates were obtained every 500 steps. Molecular graphics images were produced using VMD59 and PyMOL.60 The graphs were prepared using the xmgrace software (http://plasma-gate.weizmann.ac.il/Grace/). The results of the MD simulation trajectories were analysed using root mean square deviation (RMSD), root mean square fluctuation (RMSF), structure stability, transition path and hydrogen bond analysis.| Model no. | Initial conformation | Ligand |
|---|---|---|
| Ap | Apo_1A52 | None |
| ap_c | Apo_1A52 | 5c |
| An | Antagonist_3ERT | None |
| an_c | Antagonist_3ERT | 5c |
| Ag | Agonist_1G50 | None |
| ag_c | Agonist_1G50 | 5c |
Free energy calculations
Free energy calculations were performed using the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) method available in the GROMACS software package prepared using the g_mmpbsa tool.61 In this study, the last 20 ns of all the simulations of the complexes were chosen as the equilibrium part of the trajectory for energy analysis. MM-PBSA was applied to predict the average binding free energies using a Python script, MmPbSaStat.py, available in the g_mmpbsa package. Moreover, two output files, summary_energy.dat and full_energy.dat, were obtained; summary_energy.dat contains the average and standard deviations of all energetic components including the binding energy, polar solvation energy, ΔGpolar, solvent-accessible surface area (SASA), ΔGnonpolar, electrostatic interaction, ΔEelec, and van der Waals interaction, EvdW, whereas full_energy.dat contains the values of the energetic terms as a function of time, which have been plotted using the xmgrace software. On the other hand, to calculate the average contribution of the residues to the binding energy, the Python script MmPbSaDecomp.py was used, and the results, including the binding energy for each residue, were plotted using the energyMapIn.dat file with the xmgrace software. Furthermore, the energy2bfac tool was used to visualize the energy contribution of each residue with its structure using the VMD program.Results and discussion
Docking studies
Molecular docking of benzophenone imines (BIs) (1–9)c was studied with the three hERα forms i.e. apo, antagonist and agonist binding sites to compare the binding interactions and free binding energies of the benzophenone imine derivatives with those of the three receptors (see Table 4). It is interesting to observe that the binding affinities of BIs (1–6)c are higher than those of natural E2 and synthetic 4-OHT in the apo and antagonist models except for the diarylimine 7c and the O-alkyl chain scaffold 8c, which have lower binding affinities.| Ligand | Autodock | FRED | HYBRID | ||
|---|---|---|---|---|---|
| Cluster | Binding energy, ΔG | Calculated Ki | Binding energy, ΔG | Binding energy, ΔG | |
| Apo hERα-ligand | |||||
| E2 | 100 | −9.67 | 81.36 | −16.53 | −16.83 |
| 4-OHT | 100 | −10.30 | 28.06 | −15.24 | −14.37 |
| 1c | 100 | −11.67 | 2.81 | −18.52 | −10.34 |
| 2c | 100 | −11.68 | 2.73 | −12.32 | −10.43 |
| 3c | 100 | −11.30 | 5.22 | −12.32 | −13.84 |
| 4c | 100 | −11.59 | 3.21 | −17.32 | −10.06 |
| 5c | 100 | −11.89 | 1.91 | −18.16 | −15.14 |
| 6c | 100 | −11.28 | 5.43 | −16.46 | −10.04 |
| 7c | 92 | −9.39 | 131.2 | −12.80 | −10.72 |
| 8c | 96 | −11.18 | 5.16 | −16.13 | −10.36 |
| 9c | — | — | — | −14.81 | −12.70 |
|
|||||
| Antagonist hERα-ligand | |||||
| E2 | 100 | −10.23 | 27.86 | −14.69 | −14.95 |
| 4-OHT | 100 | −10.84 | 11.40 | −18.02 | −17.72 |
| 1c | 100 | −10.29 | 28.84 | −16.92 | −15.77 |
| 2c | 100 | −10.30 | 28.06 | −13.03 | −9.67 |
| 3c | 100 | −10.64 | 15.77 | −16.74 | −16.18 |
| 4c | 100 | −10.85 | 11.14 | −16.58 | −16.49 |
| 5c | 100 | −10.92 | 9.95 | −17.91 | −17.32 |
| 6c | 100 | −9.81 | 63.99 | −15.18 | −13.75 |
| 7c | 92 | −7.86 | 1730 | −12.92 | −12.12 |
| 8c | 82 | −9.99 | 47.96 | −17.60 | −17.48 |
| 9c | — | — | — | −16.40 | −14.44 |
|
|||||
| Agonist hERα-ligand | |||||
| E2 | 100 | −10.81 | 12.00 | −18.31 | −18.31 |
| 4-OHT | 48 | −7.31 | 4350 | −8.95 | −6.71 |
| 1c | 13 | −6.35 | 22180 |
−10.63 | −9.82 |
| 2c | 6 | −5.52 | 90310 |
−8.75 | −8.87 |
| 3c | 37 | −5.03 | 207180 |
−9.03 | −9.18 |
| 4c | 47 | −5.14 | 170180 |
−9.44 | −9.19 |
| 5c | 19 | −5.00 | 217020 |
−9.57 | −9.59 |
| 6c | 18 | −6.43 | 19210 |
−12.28 | −9.91 |
| 7c | 11 | −7.40 | 3770 | −14.01 | −11.62 |
| 8c | 13 | −8.12 | 1120 | −10.55 | −9.33 |
| 9c | — | — | — | −7.88 | −7.77 |
Table 4 shows that BIs (1–6)c, 4-OHT and E2 display single cluster docking conformations, which means that the ligands are docked in the same orientation. On the other hand, the diarylimine 7c and the o-alkyl chain scaffold 8c showed multiple cluster docking conformations (see Fig. 7). A comparison of the docked poses of 100 conformations of E2 at the apo hERα binding site with its original crystal structure is shown in Fig. 6a. It can be observed that E2 engages in hydrophobic interactions along with hydrogen bonding interactions with Arg394, Glu353 and His524. The docked pose of 4-OHT with apo hERα is shown in Fig. 6b.
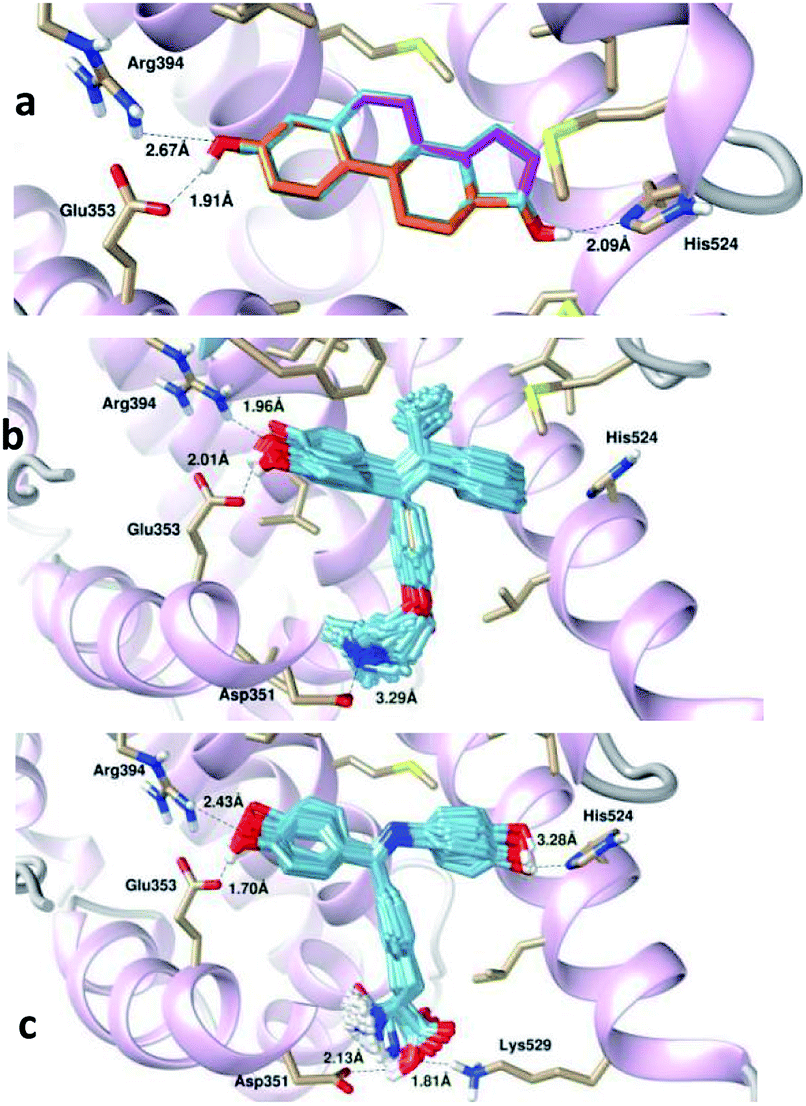 | ||
| Fig. 6 Superimpositions of the 100 conformations of (a) E2, (b) 4-OHT and (c) 5c docked in the binding pocket of the apo hERα complex. The important bond lengths involved in the hydrogen bond formation are highlighted. | ||
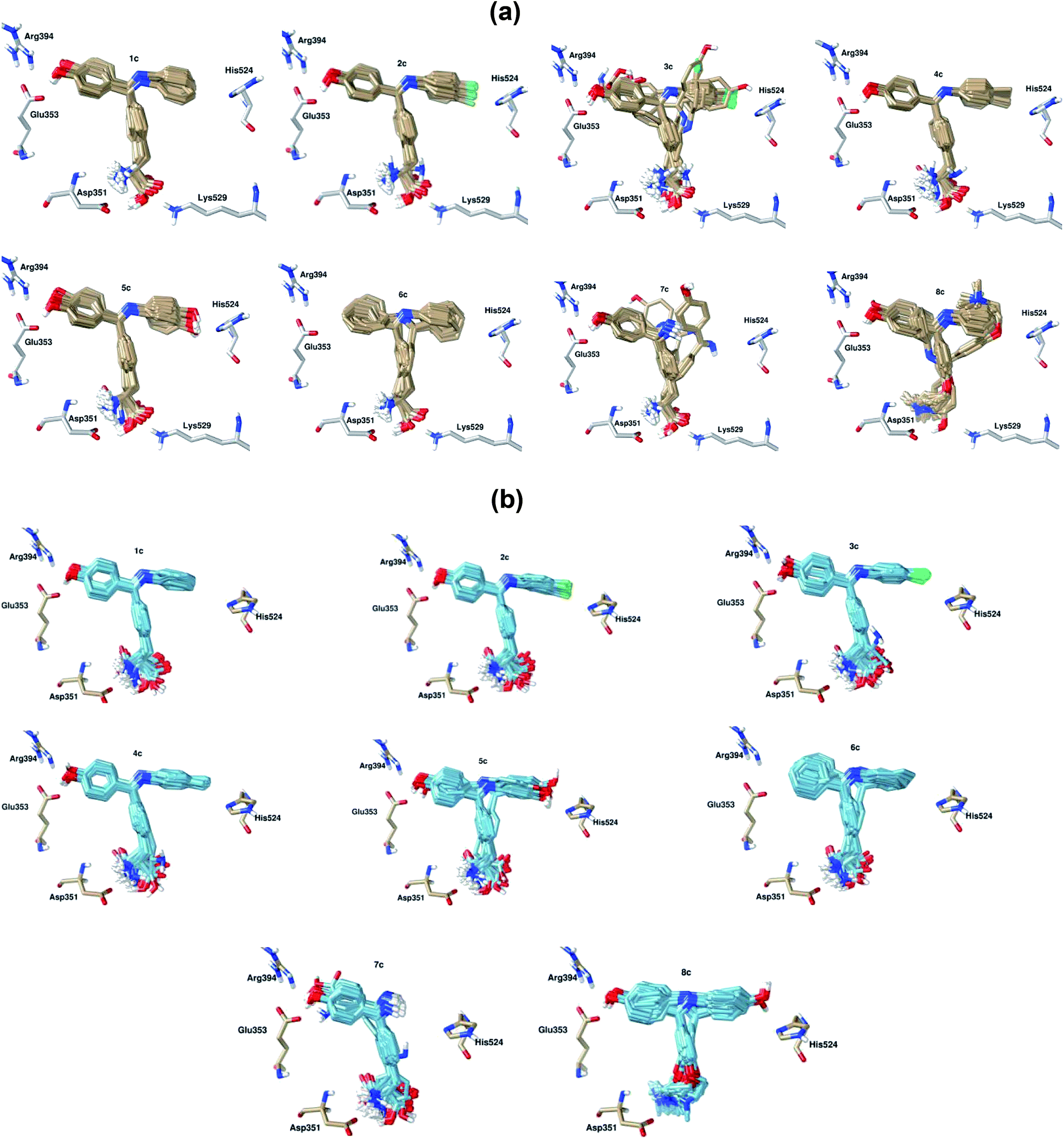 | ||
| Fig. 7 Superimpositions of 100 conformations for BIs 1–6c docked in the same orientation in (a) apo hERα–BIs and (b) antagonist hERα–BIs. 6c and 7c show multiple cluster docking conformations in both the apo and the antagonist models. Important amino acid residues are highlighted. | ||
It has been observed that the estrogenic scaffold in 4-OHT has the same binding mode as that of E2; moreover, the O-alkyl side chain in 4-OHT is responsible for the interaction with Asp351, which is responsible for the antiestrogen properties. For the newly designed Schiff bases, the hydroxyl groups in all BI derivatives except 6c form hydrogen bonding interactions with the Glu353 and/or Arg394 amino acid residues, thus mimicking the binding modes of E2 and 4-OHT. Moreover, the hydroxyl group in the imine aromatic ring forms a hydrogen bond with His524 at the other site of the LBD. On the other hand, the carboxylic acid groups in the 1–7c side chains, along with the N-alkyl side chain in 8c, form hydrogen bonds with Asp351. The best docked conformer, 5c, formed a hydrogen bond with Asp351 that was shorter (2.13 Å) than the bond observed in 4-OHT (3.29 Å, see Fig. 6c); this indicated that the BI derivatives might form stronger hydrogen bonds with Asp351 and decrease the agonistic activity.
This means that triarylimines in the 1–9c framework along with triarylethylene in 4-OHT act as agonists mimicking the effect of the natural estrogen E2, which can bind to ER as an agonist or antagonist. On the other hand, the two side chains, alanine in 1–7c and the O-alkyl chain in 8c, attached to triarylimine along with the O-alkyl side chain in 4-OHT are responsible for its antagonist behaviour. In addition, the docking studies of the BI derivatives revealed that polar (Glu353, Arg394, His524, Asp351 and Lys529), aromatic (Trp383 and Phe404) and non-polar (Leu346, Leu349, Leu384, Leu391, Leu525, Met388, Ala350, Thr347, Trp383, Phe404 and Gly521) amino acid residues played important roles in the stabilization of the hERα–BIs complexes, as presented in Table 5. Interestingly, the docking poses for BIs 1–6c display a single mode interaction with the antagonist hERα-LBD, as shown in Fig. 7b, which confirms the selectivity of the apo hERα–1–6c clusters. This can be attributed to fact that the newly designed triarylimines with alanine side chains display significant structural fitting to the apo and antagonist binding sites, thus mimicking the effects of the triarylethylene and O-alkyl side chain frameworks in 4-OHT.
| Ligand | Hydrophobic interactions | ||
|---|---|---|---|
| Apo hERα-ligand | Antagonist hERα-ligand | ||
| E2 | Phe404, Met388, Ile424, Gly521, Leu384, Leu387, Leu391, Leu428, Leu525 | 4-OHT | Leu346, Leu384, Leu387, Leu391, Leu525, Met343, Met421, Thr347, Ala350, Trp383, Gly521, Phe404 |
| 1c | Leu346, Leu384, Leu387, Leu391, Leu525, Trp383, Gly521, Met388, Met421, Ala350, Thr347, His524 | Leu346, Leu384, Leu387, Leu391, Leu525, Met343, Met421, Thr347, Ala350, Trp383, Gly521 | |
| 2c | Leu346, Leu384, Leu387, Leu391, Leu525, Trp383, Gly521, Met388, Met421, Ala350, Thr347, His524, Phe404 | Leu346, Leu384, Leu387, Leu391, Leu525, Met343, Met421, Thr347, Ala350, Trp383, Gly521 | |
| 3c | Leu346, Leu349, Leu384, Leu525, Gly521, Met388, Met421, Ala350, Thr347, Phe404 | Leu346, Leu384, Leu391, Ile424, Met421, Thr347, Ala350, Trp383, Gly521 | |
| 4c | Leu346, Leu349, Leu384, Leu525, Ile424, Met388, Met421, Ala350, Thr347, Trp383, Phe404, His524, Gly521 | Leu346, Leu384, Leu391, Leu525, Ile424, Met343, Met388, Met421, Thr347, Ala350, Trp383, Gly420, Gly521, His524 | |
| 5c | Leu346, Leu349, Leu384, Leu391, Leu525, Met388, Ala350, Thr347, Trp383, Phe404, Gly521 | Leu346, Leu349, Leu525, Met343, Ala350, Thr347, Trp383, Gly521 | |
| 6c | Leu346, Leu387, Leu384, Leu391, Leu525, Ala350, Gly521, Met388, Met421, Trp383 | Leu346, Leu349, Leu391, Ala350, Thr347, Trp383, Gly521, Met343, Met421, Phe404 | |
| 7c | Leu346, Leu349, Leu384, Leu387, Leu391, Leu525, Ala350, Thr347, Trp383, Phe404 | Leu346, Leu384, Leu387, Ala350, Thr347, Trp383 | |
| 8c | Leu346, Leu354, Leu384, Leu387, Leu391, Leu525, Met388, Met421, Thr347, Ala350, Gly521, Trp383 | Leu346, Leu384, Leu387, Leu391, Leu525, Met343, Met421, Thr347, Ala350, Trp383, Gly521 | |
| 9c | Ala350, Asp351, Glu353, Leu387, Leu391, Ile424, Met421, Gly521, His524, Leu525 | Met343, Leu346, Thr347, Ala350, Glu353, Leu387, Leu391, Leu384, Glu421, Ile424, Met421, Gly521, His524, Leu525 | |
The docking results of the antagonist model show that the binding free energies of BIs 1–6c to antagonist hERα are in the same range as that of 4-OHT, −10.84 kcal mol−1, whereas 5c has the lowest binding free energy of −10.92 kcal mol−1. The antagonist hERα–7c shows a higher binding energy of −7.86 kcal mol−1, and this is in agreement with the apo hERα–7c complex, which also shows the highest binding energy of −9.39 kcal mol−1 but with multiple clusters.
The results were also compared with those of other docking protocols, i.e. FRED and HYBRID Chemgauss4, and the results are tabulated in Table 4. Generally, the results obtained from the three different docking protocols showed different trends for the binding affinities of all the investigated ligands. However, the three docking protocols agree that the antagonist hERα–5c has highest binding affinities. FRED performs a systematic and non-stochastic examination of all possible protein–ligand poses and filters for shape complementarity and chemical feature alignment before selecting and optimizing the poses using the Chemgauss4 scoring function.49 The HYBRID program, on the other hand, uses the information present in both the structure of the protein and the bound ligand to enhance the docking performance. The results are also consistent with the calculated binding energies obtained using Autodock, which show that the binding affinities of the apo and antagonist hERα complexes are stronger than that of the agonist complex.
It has been observed that 4-OHT is engaged in hydrogen bonding interactions with the Glu353, Arg394 and Asp351 amino acid residues (see Fig. 8a), whereas 5c, which has two hydroxyl groups, forms four hydrogen bonds with Glu353, Arg394, His524 and Asp351, as shown in Fig. 8b. Previously, Celik and his co-workers highlighted the importance of hydrogen bond formation with the Glu353 and Arg394 amino acid residues in their docking study on E2 and 4-OHT.62 It was observed that BIs formed hydrophobic contacts mainly with leucines (Leu346, Leu384, Leu387, Leu391 and Leu525) and methionines (Met343, Met388 and Met421). Other amino acid residues, such as Thr347, Ala350, Trp383, Gly521 and Phe404, also form hydrophobic contacts with 5c, as shown in Table 5. The ligand forms four hydrogen bonds with Glu353, Arg394, His524 and Asp351. In both the apo and the antagonist models, 5c and 7c reveal the lowest hydrophobic interactions due to the hydrophilic nature of 5c, whereas the small diaryl ligand 7c contributes to fewer interactions with amino acid residues at the binding site. On the other hand, 4c has the highest hydrophobic interactions due to the hydrophobic methyl group in the imine aryl ring.
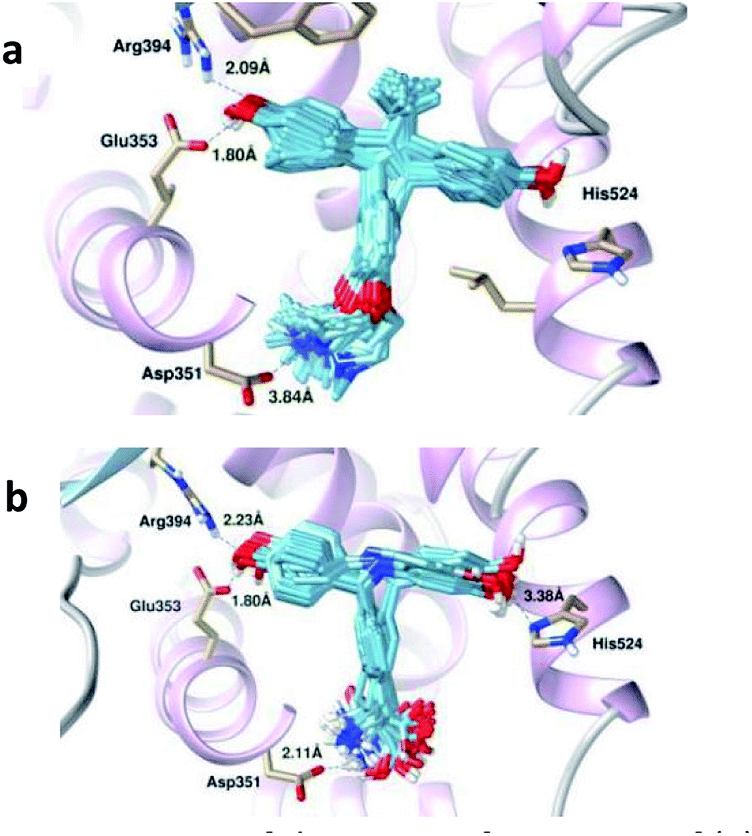 | ||
| Fig. 8 Superimpositions of the 100 conformations of (a) 4-OHT and (b) 5c docked in the binding pocket of the antagonist hERα complex. The important bond lengths involved in the hydrogen bond formation are highlighted. | ||
Furthermore, the side chain tails in the BIs 1–7c ligands are highly flexible, as noted from the docking poses with the antagonist hERα-LBD as compared to the case of the apo system, as shown in Fig. 7. The NH2 groups in the side chains of BIs 1–7c form hydrogen bonds with the Asp351 amino acid residue in both the apo and the antagonist systems. Moreover, the carboxylic acid group (COOH) forms a hydrogen bond with the Lys529 amino acid residue in the apo system only. This is due to the difference in the Lys529 amino acid positions in the apo and antagonist systems. Lys529 is located at the end of H11 in the apo system and faces the binding site. However, Lys529 in the antagonist system points out of the binding pocket, thus preventing the formation of hydrogen bonds with the COOH group in the BI ligands. The agonist hERα forms stable complexes with small ligands such as E2, and this allows H12 to cover the binding site and restricts the movement of the ligand. The natural and synthetic substrates E2 and 4-OHT were redocked to the hERα agonist binding site to compare the interaction energies and cluster distributions in the close LBD towards the bulky BI SERMs-like ligands, and the results are tabulated in Table 4. By comparing the binding energies between E2, 4-OHT and the BIs, we have found that E2 forms single clusters in the LBDs of all three hERα forms (see Fig. 9a). Moreover, both 4-OHT and BIs exist in multiple clusters in the agonist form and generally have higher binding energies as compared to E2. In addition, the interactions of the BIs and 4-OHT in the hERα binding pocket involve hydrophobic interactions with the helices H3 (blue), H6 (grey) and H11 (green) (see Fig. 9b). On the other hand, the synthetic antagonist 4-OHT and the newly designed BIs interact with the open/apo and open/antagonist hERα with higher binding affinities. This suggests that the binding poses of the newly designed BIs adopt 4-OHT-like modes in the binding pocket of the antagonist hERα.
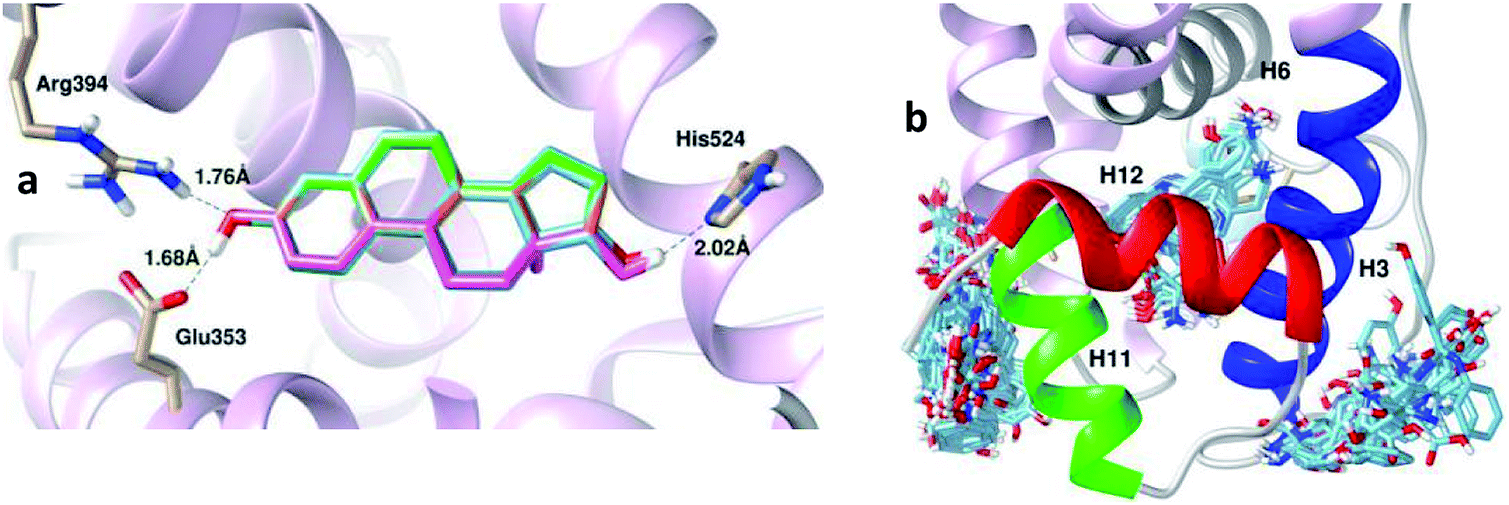 | ||
| Fig. 9 Superimpositions of the 100 conformations of (a) E2 and (b) 5c docked in the binding pocket of the hERα agonist complex. | ||
Molecular dynamics studies
The flexibility of the hERα binding site is an important factor to investigate the conformational changes of the apo, antagonist and agonist forms of hERα upon its binding with BIs. Molecular dynamics (MD) simulations have provided insights into the dynamic fluctuations of the hERα apo, antagonist and agonist forms upon binding with natural E2 or synthetic 4-OHT.62–65 Furthermore, the dynamics of H12 at the binding site in the apo and antagonist systems have been reported to be profoundly different.63,65Simulations were conducted on each of the six different models to determine the best candidate among the benzophenone imines, 5c, with the three hERα conformations. Previously, Fratev investigated the transition of the apo form to either the agonist or the antagonist state,63 whereas Celik and co-workers investigated the binding of E2 in the presence and absence of co-activator proteins.62
Herein, we report the results of the simulations of the apo state to study its behavior as an open binding site towards 4-OHT-like ligands and compare their stabilities with the antagonist and agonist forms. The RMSD values showed that the antagonist and agonist models did not reach stable RMSD values until 20 ns. This is in agreement with previous MD simulations involving the agonist and antagonist forms of hERα, which shows stable RMSD values only after 20 ns.65 The average and maximum RMSDs of 2.8 and 4.0 Å for the antagonist form were larger than those for the agonist form, which had the values of 1.8 and 2.8 Å, respectively (see Fig. 10). Moreover, relatively large fluctuations of the antagonist form as compared to those of the agonist form were observed in the longest MD simulations reported to date65 and in the 5 ns MD simulations reported by Celik and co-workers.62 The larger RMSD value fluctuation from 0 to 35 ns in the free and bound apo forms appears to have occurred due to the large fluctuation in the extended H12 region, and the values started to stabilize after 35 ns until the end of the simulation. These results demonstrate that 5c promotes the stability of the apo hERα complex as compared to the free apo protein. This is also in agreement with a previous study, which states that the apo ERα monomer exhibits high conformational flexibility with respect to H12, thus affecting the stability of the overall hERα structure as compared to the folding of the H12 conformation in the antagonist and agonist forms.63 Upon comparing the RMSD values for the three systems, the results suggest that the complexation of 5c to the apo and antagonist hERα is more favourable and provides stability to the protein structure. On the contrary, the close/agonist hER–5c complex has higher RMSD values as compared to its free agonist form due to the position of H12 that covers the binding site and restricts the movement of the ligand. Variation in the gyration radii (Rg) values of the three systems was expected due to the differences in the folding structure of H12 (ESI Fig. S1†). H12 in the apo free and complex forms is unfolding and extending away from the binding site. However, in the antagonist conformation, H12 shifts to an adjacent coactivator site. In the agonist form, H12 covers the ligand binding site with the highest level of folding as compared to the apo and antagonist forms.
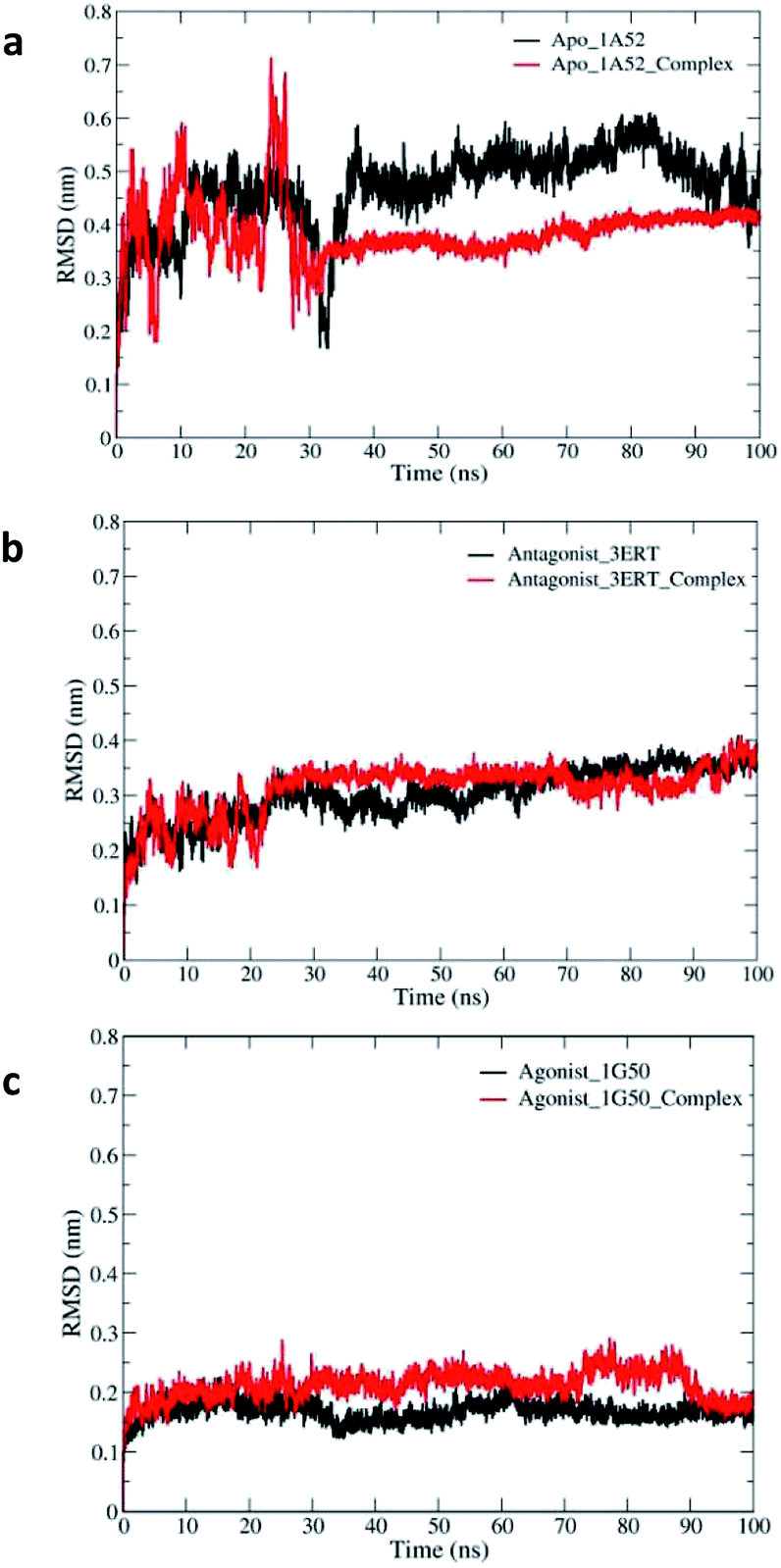 | ||
| Fig. 10 The RMSDs of the backbone atoms of free and bound hERα throughout the simulation times for the (a) apo, (b) antagonist and (c) agonist models. | ||
Therefore, Rg of the agonist was stable from the beginning to the end of the simulation. Moreover, the Rg values for the antagonist model continued to fluctuate until 60 ns.
For the extended apo state, the Rg values started to fluctuate between 2.03 and 1.85 nm during the simulation time. The fluctuations of the free and complex systems were high at the beginning of the simulation time and then started to decrease to around 1.9 ± 0.05 nm. This result supports the changes observed in the RMSD plot, which show high fluctuations at the beginning of the simulation time.
To illustrate and compare the flexibilities and conformational changes of the three free and bound models, the RMSFs were analyzed, and the results are presented in Fig. 11. The RMSFs of the free and complex antagonist systems are similar to those of the apo system. However, lower flexibility regions in the beginning for H4 and H10 were observed in the antagonist system. On the other hand, the higher regions of the antagonist systems as compared to those of the apo system involve the Asp531 amino acid residue in the end of H11. In general, the fluctuations in the free and complex forms of the apo system are quite similar to each other and to those in the antagonist systems, in which the fluctuation profile of the free antagonist form is similar to that of its complex system.
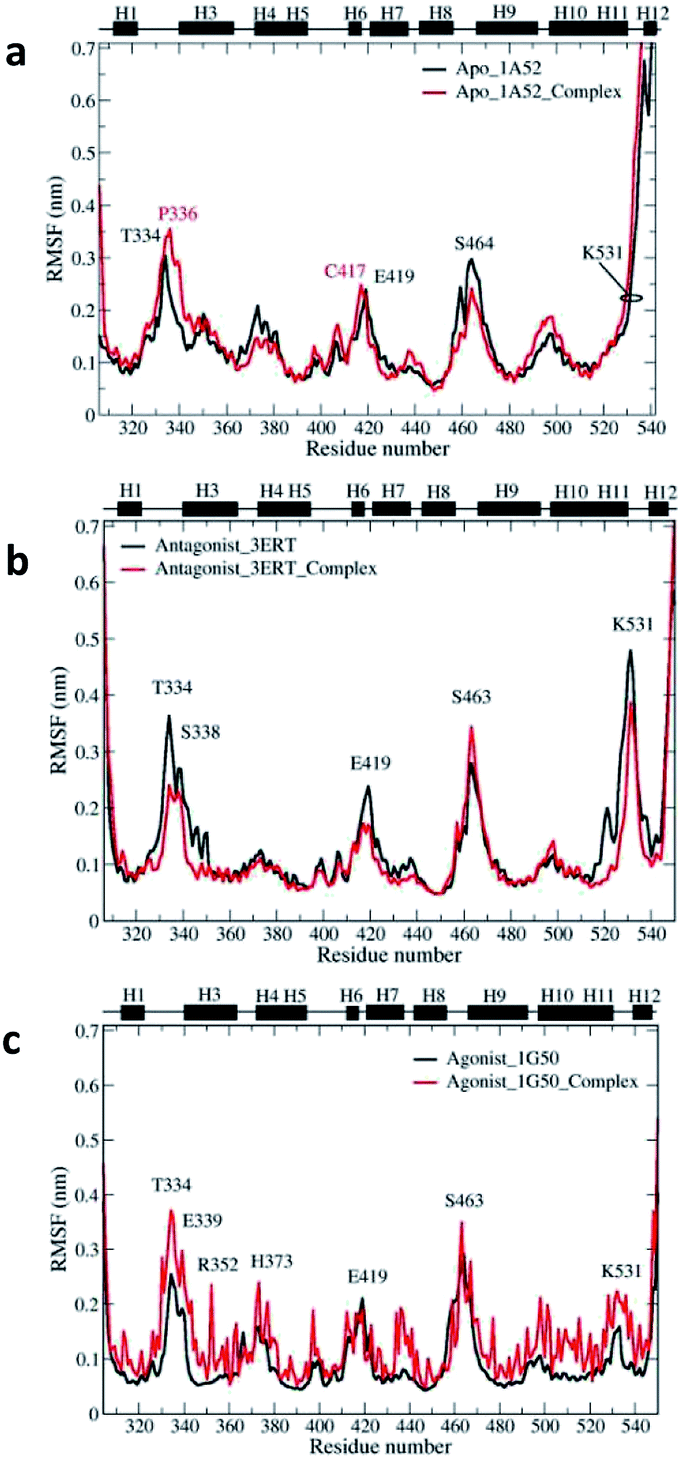 | ||
| Fig. 11 RMSF profiles of the free and complex hERα throughout the simulation period for the (a) apo, (b) antagonist and (c) agonist models. | ||
However, the agonist complex differs greatly from the apo and antagonist open systems. Further analysis reveals that most of the amino acid residues in the agonist complex have highest RMSF values throughout the simulation period. This can be explained by the close LBD of the agonist estrogen receptor conformation with H12, which restricts the ligand movement.
As a whole, amino acid fluctuations in the antagonist forms, i.e., free and complex systems, are located in the residue number 526–535 and 545–550, corresponding to the loops that are attached to both H12 terminals. These fluctuations in the loops around H12 were also observed in the hERα–4-OHT simulation study.64 A high dynamic region was also observed in a loop preceding the H9 involving residues 460–466. The dynamic behavior observed in this region has also been reported in many agonist/antagonist simulation studies.66,67 Simulations of the apo systems show slightly different RMSF values in the same region as compared to those of the antagonist forms. Moreover, the free agonist systems have similar dynamic regions as those of the apo and antagonist systems, with decreased fluctuations in a loop preceding H9 (residues 460–466), and this agrees well with a previous simulation study involving hERα.65
To study the conformational flexibility and binding of the 5c ligand at the three binding sites of the apo, antagonist and agonist forms of hERα, the RMSDs for 5c in the three systems were analyzed and are presented in Fig. 12a. The calculated ligand RMSDs reached stable values after approximately 20–100 ns and stabilized at 0.1 ± 0.05 nm in both the apo and the antagonist hERα complexes.
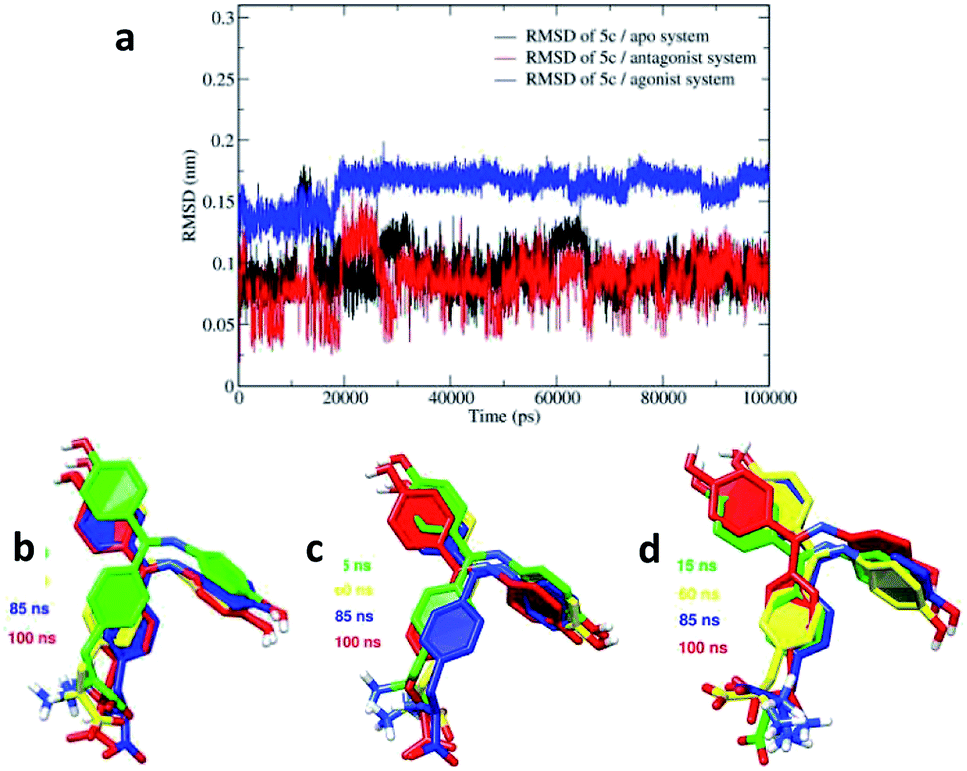 | ||
| Fig. 12 Conformational changes of the 5c ligand throughout the simulation period. (a) The RMSDs of 5c in the binding pockets of the apo, antagonist and agonist forms. Ligand dynamics images in the binding pockets obtained at different simulation times for the (b) apo, (c) antagonist and (d) agonist systems. | ||
However, in the agonist hERα complex, higher RMSDs of 0.17 ± 0.02 nm were observed for the ligand. This high variation for the ligand in the agonist system is in agreement with the increasing conformational fluctuations of the agonist receptor amino acid residues in the RMSF plot. The images showing the dynamics of 5c in the binding pocket of the apo, antagonist and agonist systems are shown in Fig. 12. The binding orientation of 5c shows relatively similar variations in the open LBD apo and antagonist forms. The amino group in the alanine side chain binds with the Asp351 amino acid residue, whereas the carboxylic acid functional group in 5c interacts with the Lys529 amino acid residue. In contrast, relatively higher variation in the alanine side chain was observed in the agonist system, as shown in Fig. 12d. In addition, the amino group in the alanine side chain along with the carboxylic acid functional group in 5c bind in opposite orientations in the agonist system as compared to that in the apo and antagonist systems. This prevents the hydrogen bond formation between 5c and the Lys529 amino acid residue in the agonist system.
Transition path analysis
The dynamics of the apo, antagonist and agonist hERα complexes at different simulation times were studied and are presented in Fig. 13. Further images were analyzed for the apo complex with an extended H12 conformation, as shown in Fig. 13a. The largest changes occur very quickly, involving H12, which moves from the unfolded form at the beginning of the simulation time to the folded form, and this is actually in agreement with the high fluctuation observed in the RMSD plot during the first 30 ns. Moreover, no transitions to either the agonist or the antagonist form were observed for the folding of H12 until the end of the simulation time; this was also in agreement with a previous study reported by Fratev.63 The author claimed that the apo hERα monomer did not show any transition from the unfolded conformation to either an agonist or an antagonist state, and the transition could only be achieved in the dimer form.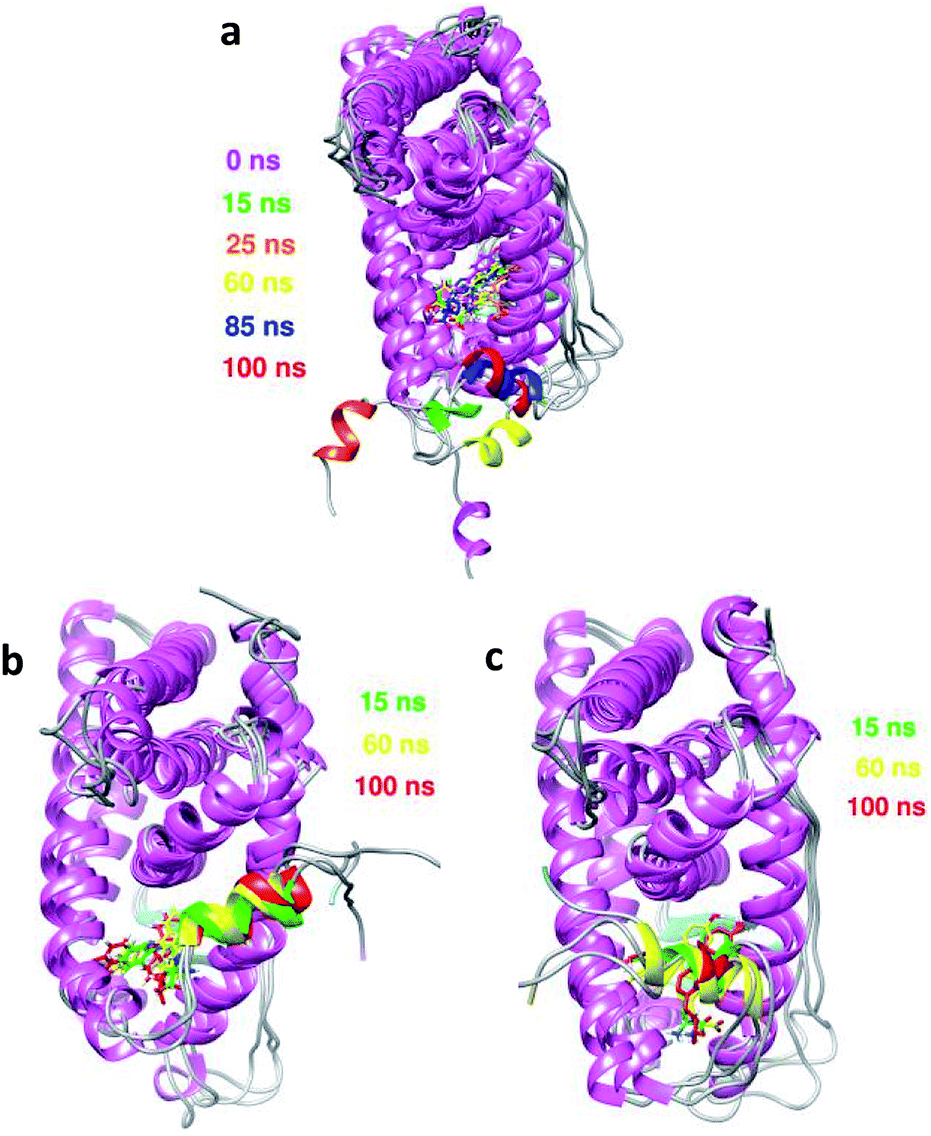 | ||
| Fig. 13 Overlays of the images of the conformational dynamics taken at different simulation times for (a) apo, (b) antagonist and (c) agonist hERα bound to 5c. | ||
Hydrogen bond analysis
The percentages of hydrogen bond occupancy between the three hERα forms and 5c during the simulation period were calculated using the Python script readHBmap.py and the gmx h-bond program in the GROMACS database. The hydrogen bonds were determined based on the acceptor–donor atom distances of less than 3.5 Å and the acceptor–H–donor angles greater than 120°.68Table 6 shows the percentages of the occupancy of H-bonds between 5c and the amino acid residues of the hERα receptor in the three systems. The labelling of the atoms involved in the hydrogen bond formation for the three systems is shown in the ESI Fig. S2.† The highest number of amino acid residues participating in the hydrogen bond formation among the three systems was in the agonist hERα–5c complex. It was observed that the hydrogen bond formation in the agonist complex involved His524, Glu353 and Thr347 with more than 50% occupancy. Sporadically, hydrogen bond formation with less than 50% occupancy was observed with Leu346, Met342, Met343, Gly344 and Val533. This result is expected for the close binding site of the agonist system, which restricts the motion of the ligand, allowing high numbers of amino acids to form hydrogen bonds with 5c, as shown in Table 6. In the apo and agonist cases, hydrogen bonding interactions with Glu353 and His524 were observed with high percentages of occupancy (96.8% and 97.3%, respectively).
| Complex | Donor and acceptor | Occupation (%) |
|---|---|---|
| Ap_c | Glu353@OE2–5c@H3 | 45.6 |
| Glu353@OE1–5c@H3 | 51.2 | |
| His524@ND1–5c@H | 97.3 | |
| Thr347@OG1–5c@H1 | 7.9 | |
| 5c@OXT–Lys529@HZ1 | 22.0 | |
| 5c@O–Lys529@HZ1 | 22.7 | |
| 5c@O2–Arg394@H21 | 3.7 | |
| 5c@OXT–Thr374@HG1 | 1.6 | |
| 5c@O–Thr374@HG1 | 1.0 | |
| An_c | Glu353@OE2–5c@H3 | 52.0 |
| Glu353@OE1–5c@H3 | 47.8 | |
| 5c@O2–Arg394@H21 | 2.0 | |
| Ag_c | Glu353@OE2–5c@H3 | 46.0 |
| Glu353@OE1–5c@H3 | 30.1 | |
| His524@ND1–5c@H | 93.2 | |
| 5c@OXT–Thr347@HG1 | 25.4 | |
| 5c@O–Thr347@HG1 | 58.5 | |
| 5c@OXT–Thr347@H | 17.0 | |
| 5c@O–Thr347@H | 41.3 | |
| 5c@OXT–Leu346@H | 33.2 | |
| 5c@O–Leu346@H | 8.1 | |
| Met342@O–5c@H1 | 31.7 | |
| 5c@OXT–Gly344@H | 26.2 | |
| 5c@O–Gly344@H | 17.8 |
In contrast, the His524 amino acid residue in the antagonist receptor repositioned to the opposite site during the simulation period, and the percentage of occupancy decreased to 2.0%. The MD results further confirmed that the hydrogen bond formation between Glu353 and 5c played a key role in the ligand–receptor interaction and was found in all three systems.
Free energy calculations
To gain an insight into the contributions of the amino acid residues to the binding energy and predict the average binding energies for the apo, antagonist and agonist complexes, free energy calculations were performed, and the obtained binding free energy components and the results are shown in Table 7. 5c is bound to the apo and antagonist receptors with the strong binding affinities of −68.68 kJ mol−1 and −59.96 kJ mol−1, respectively. The calculated free energies, ΔGcalc, of 5c towards the different hERα forms are in the order of agonist > antagonist > apo. These results are consistent with the calculated binding energies obtained from the docking results in this study, which show that the binding affinities of the apo and antagonist complexes are stronger than that of the agonist complex. From the contribution of the calculated energy components of the binding free energies shown in Table 7, the main driving force for hERα–5c binding is hydrophobic nonpolar interactions. The polar interactions contributed unfavorably to the binding of the ligand to hERα.| Energy components | Apo | Antagonist | Agonist |
|---|---|---|---|
| van der Waals | −177.21 | −145.19 | −108.12 |
| Electrostatic | −139.07 | −100.61 | −73.37 |
| Polar solvation | 267.86 | 201.35 | 148.62 |
| SASA | −20.25 | −15.51 | −12.37 |
| Total binding energy | −68.68 | −59.96 | −45.23 |
Indeed, the electrostatic and van der Waals interactions contribute favourably towards the binding of 5c to hERα and are compensated by the large polar solvation energy.
Table 8 summarizes the calculated ΔGcalc values along with the experimental ΔGexp values of the natural estradiol E2 and synthetic 4-OHT obtained from previous studies.69,70 Compared to the natural estrogen E2 and synthetic 4-OHT inhibitor, 5c is bound to the hERα by strong interactions. The calculated binding affinities of the three complexes were in good agreement with the experimental values of E2 and 4-OHT. Surprisingly, the calculated binding affinity of the antagonist complex, −59.96 kJ mol−1, is in excellent agreement with the experimental value of the antagonist hERα–4-OHT (−59.70 kJ mol−1) obtained by Liu and his co-workers.70 Although the calculated binding affinities of E2 and 4OHT with hERα obtained by Liu and his co-workers were high as compared to the experimental ΔGexp values,70 our calculated binding free energies, ΔGcalc, are in very good agreement with the experimental data, especially the calculated value for the antagonist complex hERα–4-OHT (−59.70 kJ mol−1). ΔGcalc of the agonist hERα–5c complex (−45.23 kJ mol−1) is consistent with the calculated and experimental binding energies, −51.21 kJ mol−1 and −51.88 kJ mol−1, respectively, reported by Lipzig and his co-workers.69 The fact that the total binding energies, ΔGcalc, for the different hERα conformations are comparable to the experimental ΔGexp values reported in the literature reflects the similarity in terms of the chemical and physical properties of newly designed Schiff base ligands, such as 5c.
| From previous studiesa,b | In this study | |||
|---|---|---|---|---|
| Complex | ΔGcalc | ΔGexp | Complex | ΔGcalc |
| a a,69 b,70 and ΔGcalc values were calculated using the linear interaction energy (LIE) approximation method.b a,69 b,70 and ΔGcalc values were calculated using the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) method. | ||||
| hERα–E2 | −51.21 (ref. 65) | −51.88 (ref. 65) | Apo | −68.68 |
| hERα–E2 | −127.61 (ref. 64) | −57.44 (ref. 64) | Antagonist | −59.96 |
| hERα–4-OHT | −166.94 (ref. 64) | −59.70 (ref. 64) | Agonist | −45.23 |
A detailed profile of the binding energy contributions was analysed using the MM-PBSA method. Fig. 14 shows the mapping of the energy contributions and the intermolecular ligand–receptor per-residue interaction spectra of the three complexes. During the initial transition to the stable hERα–5c forms, the energy arose mainly from the binding with residues H3, H5 and H11 in the three receptors. The hydrophobic interactions arose from the Leu525, Leu346, Leu387, Leu384, Met343, Met421 and Ala350 amino acid residues in both the apo and antagonist complexes. On the other hand, the nonpolar amino acid residues of the agonist hERα–5c complex interactions mainly arose from Leu346, Leu525, Leu391, Met388, Met421 and Ile424, with the strongest interactions from the hydrophobic residue Leu525. In addition, amino acid residues Glu353, Arg394, Lys529 and Asp531 made obvious polar contributions to 5c with high binding energy values. This indicates that the polar amino acid residues and the hydrogen bond interactions destabilize the ligand–receptor interactions during the simulation. On the other hand, hydrophobic residues, especially Leu525, form strong interactions with 5c, which forms face-to-face interactions with the imine ring; also, Leu387 forms interactions with the second hydroxyl aromatic ring. This suggests that the stability of the 5c ligand in hERα is achieved via hydrophobic interactions.
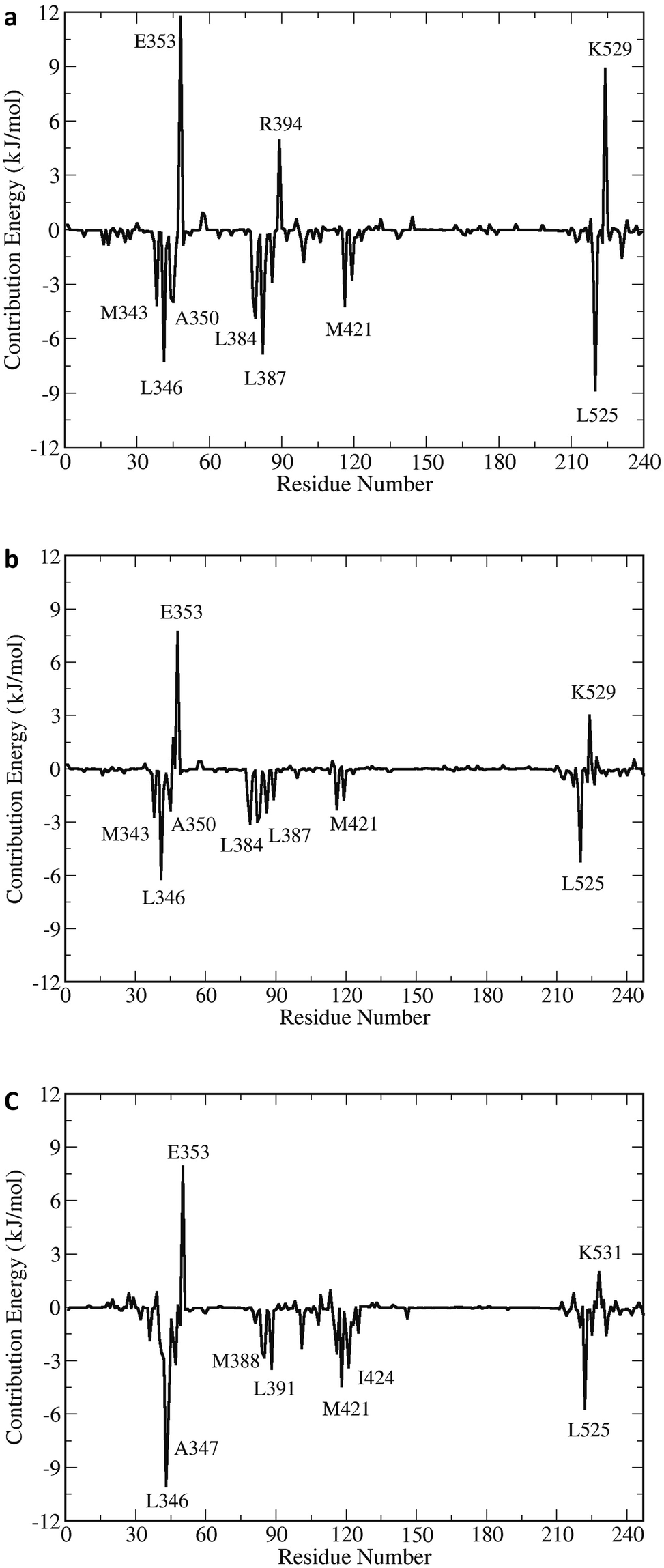 | ||
| Fig. 14 The mapping of the energy contributions on the structure of hERα–5c and the intermolecular ligand–receptor spectra of the (a) apo, (b) antagonist and (c) agonist forms. | ||
Overall, the obtained results show that BIs can form stable complexes with hERα through the open H12 binding pockets of apo and antagonist hERα. Meanwhile, the open binding pocket of the agonist form restricts the movement of the bulky BIs, such that 5c binds to the receptor with lower affinity and selectivity.
Conclusions
Herein, the mechanisms of the binding of the newly designed benzophenone imine (BI) Schiff base ligands to the apo, antagonist and agonist forms of human estrogen receptor (hERα) were investigated using molecular docking, molecular dynamics simulations and molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) energy calculations. A total of nine proposed ligands were docked, and the results demonstrated that the newly designed Schiff bases could bind to the hydrophobic open pockets of the apo and antagonist hERα conformations with high affinity, mimicking the behavior of the synthetic inhibitor 4-hydroxytamoxifen (4-OHT). Furthermore, the docking poses of the newly designed Schiff base ligands display single modes of interaction with both the open apo and antagonist hERα-LBD, as denoted by single clusters similar to the natural estradiol (E2) and the synthetic inhibitor 4-OHT. The molecular dynamics simulation results of the best docked benzophenone imine derivatives 5c with the three receptors (apo, antagonist and agonist hERα) demonstrate that 5c binds well to the hydrophobic pockets of the three systems. The flexibility and conformational change analysis based on the RMSD, RMSF and ligand RMSD values revealed stable interactions with fewer conformational fluctuations for 5c with the open apo/antagonist hERα receptors as compared to the closed agonist binding site. These results are consistent with the calculated binding energies obtained from the docking results, which show that the binding affinities of the apo and antagonist hERα–5c complexes are higher than that of the agonist hERα–5c complex. The results also prove that the newly designed Schiff base mimics the behavior of the synthetic antagonist 4-OHT. The occupancy of hydrogen bonds obtained from the MD results further confirmed that the hydrogen bond formation between Glu353 and 5c played a key role in the ligand–receptor interactions and displayed highest hydrogen bond formation among the three systems. The MM-PBSA binding free energy calculation results further confirmed the stability of the hERα–5c systems in the order of apo > antagonist > agonist. These results are consistent with the calculated binding energies obtained from the docking results. In addition, based on the contribution of the calculated energy components of the total binding free energies highlighted herein, the main driving force for the hERα–5c binding is hydrophobic nonpolar interactions. Polar interactions contributed unfavourably to the binding of the ligand. Finally, the high binding affinity for the newly designed Schiff base ligand with hERα suggests that this Schiff base T-shaped C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N family may be worth exploring in the development of new drugs for breast cancer treatment.
N family may be worth exploring in the development of new drugs for breast cancer treatment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support received from the Universiti Sains Malaysia through the USM Fellowship Scheme under the Institute of Postgraduate Studies and Ministry of Higher Education through FRGS Grant No. 203/PKIMIA/6711558. Khan thanks OpenEye Scientific Software, Inc., for a free academic license of the OpenEye Toolkits.References
- D. R. Youlden, S. M. Cramb, C. H. Yip and P. D. Baade, Cancer Biol. Med., 2014, 11, 101–115 Search PubMed.
- G. Agarwal, P. Pradeep, V. Aggarwal, C.-H. Yip and P. S. Cheung, World J. Surg., 2007, 31, 1031–1040 CrossRef.
- G. M. Clark, Breast Cancer, 1995, 2, 79–89 CrossRef.
- W. F. Anderson, N. Chatterjee, W. B. Ershler and O. W. Brawley, Breast Cancer Res. Treat., 2002, 76, 27–36 CrossRef CAS PubMed.
- S. R. Johnston and M. Dowsett, Nat. Rev. Cancer, 2003, 3, 821–831 CrossRef CAS PubMed.
- N. Platet, A. M. Cathiard, M. Gleizes and M. Garcia, Crit. Rev. Oncol. Hematol., 2004, 51, 55–67 CrossRef.
- J.-F. Arnal, C. Fontaine, A. Abot, M.-C. Valera, H. Laurell, P. Gourdy and F. Lenfant, Steroids, 2013, 78, 576–582 CrossRef CAS.
- M. Parker, Biochem. Soc. Symp., 1998, 63, 45–50 CAS.
- L. Downey, R. Livingston and A. Stopeck, J. Am. Geriatr. Soc., 2007, 55, 1636–1644 CrossRef.
- R. Blamey, Eur. J. Cancer, 2003, 39, 286–294 CrossRef CAS.
- Q. Zhou and N. E. Davidson, Cancer Biol. Ther., 2006, 5, 848–849 CrossRef CAS.
- B. L. Riggs and L. C. Hartmann, N. Engl. J. Med., 2003, 348, 618–629 CrossRef.
- W. Bourguet, P. Germain and H. Gronemeyer, Trends Pharmacol. Sci., 2000, 21, 381–388 CrossRef CAS.
- G. M. Anstead, K. E. Carlson and J. A. Katzenellenbogen, Steroids, 1997, 62, 268–303 CrossRef CAS.
- A. M. Brzozowski, A. C. Pike, Z. Dauter, R. E. Hubbard, T. Bonn, O. Engström, L. Öhman, G. L. Greene, J.-Å. Gustafsson and M. Carlquist, Nature, 1997, 389, 753–758 CrossRef CAS PubMed.
- S. Saha Roy and R. K. Vadlamudi, Int. J. Breast Cancer, 2012, 2012, 1–9 CrossRef.
- A. C. Pike, A. M. Brzozowski, R. E. Hubbard, T. Bonn, A. G. Thorsell, O. Engström, J. Ljunggren, J. Å. Gustafsson and M. Carlquist, EMBO J., 1999, 18, 4608–4618 CrossRef CAS.
- U. Egner, N. Heinrich, M. Ruff, M. Gangloff, A. Mueller-Fahrnow and J. M. Wurtz, Med. Res. Rev., 2001, 21, 523–539 CrossRef CAS.
- W. Bourguet, M. Ruff, P. Chambon, H. Gronemeyer and D. Moras, Nature, 1995, 375, 377–382 CrossRef CAS.
- D. M. Tanenbaum, Y. Wang, S. P. Williams and P. B. Sigler, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 5998–6003 CrossRef CAS.
- M. R. Batista and L. Martínez, Biophys. J., 2013, 105, 1670–1680 CrossRef CAS.
- C. Watanabe, H. Watanabe and S. Tanaka, Biochim. Biophys. Acta Protein Proteonomics, 2010, 1804, 1832–1840 CrossRef CAS.
- W.-C. Park and V. C. Jordan, Trends Mol. Med., 2002, 8, 82–88 CrossRef CAS.
- A. K. Shiau, D. Barstad, P. M. Loria, L. Cheng, P. J. Kushner, D. A. Agard and G. L. Greene, Cell, 1998, 95, 927–937 CrossRef CAS.
- V. C. Jordan, J. Med. Chem., 2003, 46, 1081–1111 CrossRef CAS.
- H. Liu, W.-C. Park, D. J. Bentrem, K. P. McKian, A. De Los Reyes, J. A. Loweth, J. M. Schafer, J. W. Zapf and V. C. Jordan, J. Biol. Chem., 2002, 277, 9189–9198 CrossRef CAS.
- S. Fuqua, J. Russo, S. Shackney and M. Stearns, Postgrad. Med. J., 2001, 3–10 CAS.
- C. I. Lee, A. Goodwin and N. Wilcken, Cochrane Database Syst. Rev., 2017, 1–59 Search PubMed.
- H. U. Bryant and W. H. Dere, Proc. Soc. Exp. Biol. Med., 1998, 217, 45–52 CrossRef CAS.
- A. Ring and M. Dowsett, Endocr. Relat. Cancer, 2004, 11, 643–658 CAS.
- S. Sommer and S. A. Fuqua, Semin. Cancer Biol., 2001, 11, 339–352 CrossRef CAS PubMed.
- L. Li, Q. Wang, Y. Zhang, Y. Niu, X. Yao and H. Liu, PLoS One, 2015, 10, e0120330 CrossRef.
- S. R. Stauffer, J. Sun, B. S. Katzenellenbogen and J. A. Katzenellenbogen, Bioorg. Med. Chem., 2000, 8, 1293–1316 CrossRef CAS.
- K. Ohta, Y. Chiba, T. Ogawa and Y. Endo, Bioorg. Med. Chem. Lett., 2008, 18, 5050–5053 CrossRef CAS PubMed.
- Z.-Q. Liao, C. Dong, K. E. Carlson, S. Srinivasan, J. C. Nwachukwu, R. W. Chesnut, A. Sharma, K. W. Nettles, J. A. Katzenellenbogen and H.-B. Zhou, J. Med. Chem., 2014, 57, 3532–3545 CrossRef CAS.
- G. Kaur, M. P. Mahajan, M. K. Pandey, P. Singh, S. R. Ramisetti and A. K. Sharma, Eur. J. Med. Chem., 2014, 86, 211–218 CrossRef CAS.
- G. Kaur, M. P. Mahajan, M. K. Pandey, P. Singh, S. R. Ramisetti and A. K. Sharma, Bioorg. Med. Chem. Lett., 2016, 26, 1963–1969 CrossRef CAS.
- T. Shoda, M. Kato, R. Harada, T. Fujisato, K. Okuhira, Y. Demizu, H. Inoue, M. Naito and M. Kurihara, Bioorg. Med. Chem., 2015, 23, 3091–3096 CrossRef CAS PubMed.
- S. Ray, Drugs Future, 2004, 29, 185–203 CrossRef CAS.
- M. R. Schneider, R. W. Hartmann, F. Sinowatz and W. Amselgruber, J. Cancer Res. Clin. Oncol., 1986, 112, 258–265 CrossRef CAS.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950–1958 CAS.
- R. Dennington, T. Keith and J. Millam, GaussView, version 5, 2009 Search PubMed.
- J. J. Stewart, J. Comput. Chem., 1989, 10, 221–264 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- OpenEye Scientific Software, Santa Fe, NM, http://www.eyesopen.com.
- P. C. Hawkins, A. G. Skillman, G. L. Warren, B. A. Ellingson and M. T. Stahl, J. Chem. Inf. Model., 2010, 50, 572–584 CrossRef CAS.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS.
- OEDocking. 3.2.2, OpenEye Scientific Software, Santa Fe, NM, 2017, http://www.eyesopen.com Search PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS.
- A. C. Wallace, R. A. Laskowski and J. M. Thornton, Protein Eng. Des. Sel., 1995, 8, 127–134 CrossRef CAS.
- C. A. Lipinski, Drug Discov. Today Technol., 2004, 1, 337–341 CrossRef CAS.
- A. W. S. da Silva and W. F. Vranken, BMC Res. Notes, 2012, 5, 367–374 CrossRef.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Phys. Chem., 1983, 79, 926–935 CrossRef CAS.
- H. J. Berendsen, J. P. Postma, W. F. van Gunsteren and J. Hermans, Intermol. Forces, 1981, 14, 331–342 CAS.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- M. J. Abraham and J. E. Gready, J. Comput. Chem., 2011, 32, 2031–2040 CrossRef CAS.
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- W. L. DeLano, CCP4 Newsletter On Protein Crystallography, 2002, vol. 40, pp. 82–92 Search PubMed.
- R. Kumari, R. Kumar, O. S. D. D. Consortium and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS PubMed.
- L. Celik, J. D. D. Lund and B. Schiøtt, Biochemistry, 2007, 46, 1743–1758 CrossRef CAS.
- F. Fratev, Phys. Chem. Chem. Phys., 2015, 17, 13403–13420 RSC.
- T. D. McGee, J. Edwards and A. E. Roitberg, Int. J. Environ. Res. Public Health, 2008, 5, 111–114 CrossRef CAS.
- H. L. Ng, J. Mol. Graphics Modell., 2016, 69, 72–77 CrossRef CAS.
- S. Chakraborty, A. S. Levenson and P. K. Biswas, BMC Struct. Biol., 2013, 13, 27–38 CrossRef CAS.
- J. Zeng, W. Li, Y. Zhao, G. Liu, Y. Tang and H. Jiang, J. Phys. Chem. B, 2008, 112, 2719–2726 CrossRef CAS.
- L. M. Gregoret, S. D. Rader, R. J. Fletterick and F. E. Cohen, Proteins, 1991, 9, 99–107 CrossRef CAS.
- M. M. Van Lipzig, A. M. Ter Laak, A. Jongejan, N. P. Vermeulen, M. Wamelink, D. Geerke and J. H. Meerman, J. Med. Chem., 2004, 47, 1018–1030 CrossRef CAS.
- J.-Y. Liu and S. D. Mooney, Int. J. Biochem. Mol. Biol., 2011, 2, 190–198 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04759j |
| This journal is © The Royal Society of Chemistry 2019 |