
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe polysaccharides from Grifola frondosa attenuate CCl4-induced hepatic fibrosis in rats via the TGF-β/Smad signaling pathway
Chao Li†
a,
Meng Meng† a,
Mingzhu Guoa,
Mengyang Wangb,
Aining Juc and
Chunling Wang*a
a,
Mingzhu Guoa,
Mengyang Wangb,
Aining Juc and
Chunling Wang*a
aKey Laboratory of Food Nutrition and Safety, Ministry of Education, College of Food Engineering and Biotechnology, Tianjin University of Science and Technology, No. 29, 13th Avenue, Tianjin Economy Technological Development Area, Tianjin 300457, People Republic of China. E-mail: wangchunling@tust.edu.cn; Fax: +86-022-60912421; Tel: +86-022-60912421
bCangzhou Institutes for Food and Drug Control, Cangzhou, 061000, People Republic of China
cDepartment of Clinical Laboratory, Yantai Affiliated Hospital of Binzhou Medical University, Yantai 264100, People Republic of China
First published on 21st October 2019
Abstract
The TGF-β1/Smad signaling pathway has been linked to hepatic fibrosis. Previous studies have shown that yellow polysaccharide can prevent the development of hepatic fibrosis. However, it is unclear whether the polysaccharide affects the TGF-β1/Smad signaling pathway. In this experiment, 50 experimental rats were randomly divided into a normal control group, model group, low GFP dose group (50 mg kg−1), medium GFP dose group (100 mg kg−1), and high GFP dose group (200 mg kg−1). A cirrhotic portal hypertension rat model was established by a CCl4 compound method. After 12 weeks of intragastric administration, the liver index of the medium dose and high dose group was significantly lower than that of the model group. The hepatic fibrosis lesions of rats in each dose group were improved to different extents, and the effect was most significant in the high dose group. The contents of ALT, AST, TBIL and CIV, PCIII, LN and HA in serum were significantly decreased. The activity of SOD and GSH-Px in the liver tissue of GFP medium and high dose groups was significantly increased and the content of MDA was significantly decreased. The contents of TNF-α, IL-1β and IL-6 were significantly decreased. The western blot results showed that the expressions of p-Smad 2/3, Smad4, PAI-1, Imp7 and Imp8 in medium dose and high dose groups were significantly lower than those in the model group, while the expression of Smad7 was significantly higher than that of the model group. The GFP-treated group was able to reduce the expression level of mi R-154 in liver tissue and increase the expression level of miR-146a. GFP has a significant intervention effect on rat hepatic fibrosis, and its mechanism may inhibit the progression of hepatic fibrosis by inhibiting oxidative stress and inflammatory response and regulating TGF-β1/Smad signaling pathway and mi RNA expression.
Introduction
Hepatic fibrosis refers to the excessive deposition of collagen-based extracellular matrix (ECM) in the liver, which is a common and progressive pathological stage of various chronic liver diseases.1,2 Continuous and repeated inflammation and necrosis of hepatocytes caused by chronic liver disease, which results in a large amount of fibrous connective tissue proliferation, relatively or absolutely inadequate degradation activity, and a large amount of ECM deposition. These are the main pathological characteristics of hepatic fibrosis.3,4 Long-term hepatic fibrosis causes structural changes or remodeling of the liver, and further development of cirrhosis, which ultimately leads to portal hypertension and liver failure, and increases the incidence of liver tumors. Serious liver diseases such as hepatic fibrosis and cirrhosis caused by various causes have brought huge economic loss and burden to patients, families and society.5 It is urgent to study the pathogenesis and prevention measures of hepatic fibrosis and cirrhosis. Hepatic fibrosis is a process of non-self-limiting liver injury of chronic liver disease.6 Therefore, active treatment of hepatic fibrosis stage of chronic liver disease can delay, prevent or even reverse hepatic fibrosis. Hepatic fibrosis is a prelude to the occurrence of cirrhosis and an intermediate link.7,8 In recent years, with the extensive and in-depth study of the pathogenesis of hepatic fibrosis, it is currently considered that hepatic fibrosis is a dynamic process, and early anti-fibrosis can stop its development or even reverse, so there is no objection to its reversibility. Interventional drug therapy for certain pathogenesis of hepatic fibrosis has become a hot topic for scholars.9,10 However, the pathological biochemical mechanism of hepatic fibrosis is quite complicated. The research on anti-fibrosis drugs has not made significant breakthroughs. At present, the commonly used anti-fibrosis drugs are colchicine, interferon-γ (IFN-γ), etc. However, the anti-fibrosis effect of some Western medicines is still uncertain, sometimes even has serious side effects. The discovery of multi-channel, multi-level and multi-target anti-hepatic fibrosis active ingredients or compound preparations from natural plants has great potential for the development of new drugs for preventing and treating hepatic fibrosis, and has been widely concerned at home and abroad.In recent years, the mechanism of liver fibrosis has been extensively studied. Most studies affect and regulate liver fibrosis through signal transduction pathways, thereby improving liver injury and reducing the synthesis of collagen and ECM in liver tissue.11 The main signaling pathways are TGF-β/Smad, JAK/STAT, NF-κB, PI3K/Akt, MAPK and so on. Experiments have shown that TGF-β/Smad signaling pathway is highly expressed in hepatic sinuses, endothelial cells and inflammatory cells, and plays an important regulatory role in the treatment of hepatic fibrosis. Collagen gene expression in hepatic fibrosis is mainly activated by TGF-β/Smads signaling pathway.12 At the same time, studies have shown that TGF-β/Smads signaling pathway can inhibit the expression of inflammatory factors, reduce collagen deposition, and thus play a role in anti-hepatic fibrosis.13
With the improvement of the purification process of fungal polysaccharides and the research on it, the understanding and acceptance of fungal polysaccharides are getting higher and higher. The polysaccharides from ash tree, the polysaccharides from lycium and the polysaccharides from Hericium erinaceus has been used clinically.14,15 Studies have found that the polysaccharides from Grifola frondosa has good clinical protection for the central nervous system, immune system, heart, liver, kidney, etc., and has significant anti-emergency, anti-aging, anti-viral, anti-bacterial and anti-inflammatory effects.16–19 It was found that the polysaccharides from Grifola frondosa could decrease the over-active of CYP2E1, TNF-α and ROS fluorescence induced by CCL4. These results indicated that significant protective effect of Grifola frondosa on hepatic cell (L-02 cell line) injury induced by CC14 in vivo.20 Grifola frondosa is a fungus with beneficial lung function, but the effect of the polysaccharides from Grifola frondosa on hepatic fibrosis has not been reported so far. Our previous work revealed the structural analysis of the water-soluble polysaccharides from Grifola frondosa fruiting body (GFP) and demonstrated it can activate macrophage RAW 264.7.21 HPLC and Monosaccharide analysis showed that the average molecular weight of GFP was 155 kDa and it was mainly composed of rhamnose, xylose, mannose, glucose, molar ratio of 1.00![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.04:1.11:6.21. Structural analysis results revealed that its backbone consisted of (1→4)-linked methylation and Glcp residues as major structural polysaccharide GFP units. Accounting of the polysaccharide backbone hypothesized a GFP every→3)-Glcp-(1→and one→3,4)-Glcp-(1→connected interval with a small amount of 1→, 1→4, 1→6 glycosidic linkages. The structure sketch of GFP was shown in Fig. 1. The aim of this study was to prepare a rat model of hepatic fibrosis caused by biliary obstruction (BDO), observe the mechanism of Grifola frondosa polysaccharide (GFP) in treating hepatic fibrosis, and provide experimental basis and theoretical basis for the treatment of hepatic fibrosis.
:1.04:1.11:6.21. Structural analysis results revealed that its backbone consisted of (1→4)-linked methylation and Glcp residues as major structural polysaccharide GFP units. Accounting of the polysaccharide backbone hypothesized a GFP every→3)-Glcp-(1→and one→3,4)-Glcp-(1→connected interval with a small amount of 1→, 1→4, 1→6 glycosidic linkages. The structure sketch of GFP was shown in Fig. 1. The aim of this study was to prepare a rat model of hepatic fibrosis caused by biliary obstruction (BDO), observe the mechanism of Grifola frondosa polysaccharide (GFP) in treating hepatic fibrosis, and provide experimental basis and theoretical basis for the treatment of hepatic fibrosis.
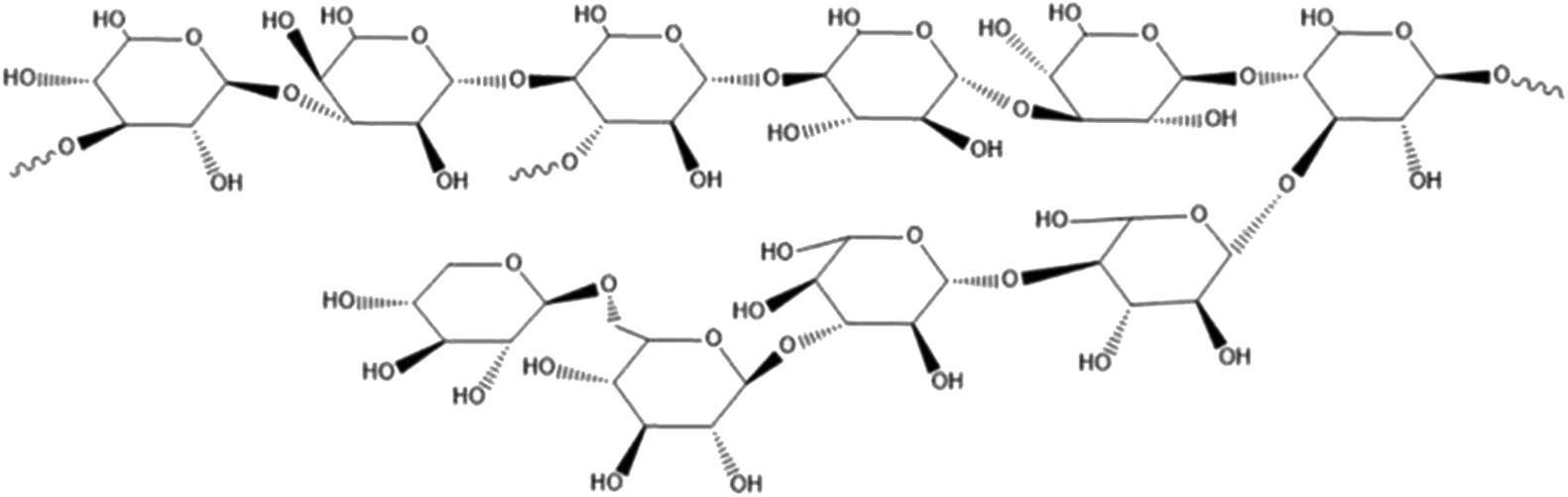 | ||
| Fig. 1 The structure sketch of GFP. | ||
Materials and methods
Materials
The preparation of water-soluble polysaccharides (GFP) followed the procedure described in our previous study.21Animal grouping and processing
Fifty male SD rats aged 55 d were selected and fed with deionized water and standard diet for one week before the experiment. This study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH publication no. 85-23 rev. 1985). All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of “Tianjin University of Science and Technology” University and approved by the Animal Ethics Committee of “Animal Ethical and Welfare Committee (AEWC)”. Then 50 rats were randomly divided into 5 groups: blank control group, model group and low, medium and high dose of GFP (50, 100, 200 mg kg−1) groups. 10 rates were in each group. During the experiment, the rats were given a free diet at a temperature of 18 to 22 °C and natural light. In addition to the blank control group, the rats in each group were intraperitoneally injected with 40% CCl4 peanut oil 3 mL kg−1 for the first time, and then injected 2 mL kg−1 every 3 days for 12 weeks to establish a hepatic fibrosis model. Dosing started on the second day after the first injection. GFP was administered intragastrically, and the rates in the control group and the model group were given 2 mL of normal saline. After 8 weeks, the experiment was finished. After the last administration, the rats were fasted for 12 h and intraperitoneally injected with a concentration of 3 g L−1 sodium pentobarbital physiological saline solution (10 mL kg−1). After anesthetized, blood was taken from the aorta.HE staining
Liver tissue was taken from the fixed position, fixed by neutral formaldehyde, dehydrated, paraffin-embedded and sliced (4 μm thick). After dewaxing and hematoxylin–eosin (HE) staining, the pathological changes of liver tissue were observed under optical microscope.Determination of ALT, AST, TBIL content and hepatic fibrosis four indicators (CIV, PCIII, LN, HA)
The serum levels of ALT, AST and TBIL were determined by biochemical detector according to the kit procedure. The serum levels of CIV, PCIII, LN and HA in each group were determined by microplate reader.Determination of antioxidant enzyme activity and MDA content in liver tissue
The liver tissue was taken, the homogenate was ground, and the supernatant was taken by low temperature centrifugation (4 °C, 12000 rpm, 15 min). The activity of antioxidant enzymes (SOD, GSH-Px) and MDA content in liver tissues of rats in each group were determined by ultraviolet-visible spectrophotometer according to the kit method.
Determination of serum inflammatory cytokine levels
Rat serum was taken and processed according to the procedure of the ELISA kit. Then, the levels of cytokines (TNF-α, IL-1β and IL-6) in the serum of each group were determined by a microplate reader.Real time RT-PCR detection
Total RNA was extracted from 100 mg of liver tissue in 1 m LRNAiso PLus. The expression of miR-154 and miR-146a in each group was detected by reverse transcription kit and fluorescent quantitative SYBR Green/Fluorescein qPCR Master Mixture Kit. U6 was used as the reaction internal reference. U6, miR-154, miR-146a reverse transcription primer and RT-PCR reaction primer were designed and synthesized by Wuhan Buffalo Biotechnology Service Co., Ltd. RT-PCR reaction conditions were: 95 °C, 30 s, 1 cycle; 95 °C, 5 s, 60 °C, 40 s, a total of 40 cycles; 95 °C, 30 s, 60 °C, 1 min; 95 °C, 15 s. The reaction was carried out in a 96-well plate with 3 replicate wells per reaction containing a total of 20 μL of reaction system. At the end of the reaction, the Ct values of the target gene and the internal reference of each group were obtained, and the internal control was standardized. The data obtained by RT-PCR was analyzed by ΔΔCT method, and the relative expression of the target genes miR-154 and miR-146a was calculated according to formula 2−ΔΔCT.Western blot analysis
Rat liver tissue was taken and total protein was extracted according to the kit instructions. BCA protein quantification method was used to measure the concentration of each group of proteins. Each group of proteins was diluted to an equal concentration with cell lysate, mixed with 5× loading buffer at 4:1, and denatured at 98 °C for 5 min. 60 μg of protein in each group was taken for 12% SDS-PAGE gel electrophoresis (100 V, 100 min). After that, it was transferred to PVDF membrane for 100 min. Then the primary antibody (1:1000) was added at 4 °C. 1:5000 labeled anti-rabbit secondary antibody was added. The gray values of the target bands and the internal reference bands were recorded by ECL chemiluminescence. The internal reference GAPDH was used as a control, and the image analysis was performed using Image J1.42q software.
Statistical analysis
The monitoring data were analyzed by SPSS19.0 statistical software. The results of data analysis were showed as mean ± standard deviation (mean ± SD). Multigroup data analysis was based on one-way ANOVA. LSD test is used for subsequent analysis. P < 0.05, the difference was significant.Results
Rat liver quality and liver index
Compared with the control group, the body weight of the model group was decreased. However, after treatment, the mean body weight of the 100, 200 mg of GFP treated group was remarkably greater than that of the model group. The results suggested that GFP could improve the symptom of mass loss in CCl4-induced hepatic fibrosis rats.Compared with the control group, the liver index of the treatment group and the model group were significantly higher (P < 0.05). Compared with the control group, the liver index of the model group was significantly higher (P < 0.05). Compared with the model group, the liver index of the middle and high dose groups of GFP was significantly lower (P < 0.05). The results were shown in Table 1.
| Group | Initial weight (g) | Final weight (g) | Liver index |
|---|---|---|---|
| Control | 189.27 ± 9.62 | 452.71 ± 35.28 | 34.67 ± 6.19 |
| Model | 191.62 ± 9.45 | 307.48 ± 25.50## | 69.82 ± 15.81## |
| GFP 50 mg | 185.17 ± 9.96 | 321.75 ± 29.13 | 57.18 ± 11.45 |
| GFP 100 mg | 192.59 ± 9.62 | 381.62 ± 31.14* | 49.23 ± 9.80* |
| GFP 200 mg | 187.63 ± 9.15 | 412.67 ± 33.59** | 41.35 ± 8.47** |
Histopathological analysis in liver tissues
In the control group, the hepatic cell line was neatly arranged, the hepatocyte size was uniform, and there was no degeneration, necrosis, no inflammatory cell infiltration, and no abnormalities in liver tissue. In the model group, the normal structure of the hepatic lobule was destroyed, the hepatic cord arrangement was disordered, and the hepatocytes showed extensive balloon-like changes. The fibrous connective tissue around the portal area and part of the central vein proliferated to form fine strands, a large number of inflammatory cells infiltrated, collagen fibers deposited and fell into the liver tissue to separate and surround the hepatocytes to form fibrous septa, and some seriously damaged areas had appeared pseudolobules of varying sizes separated by collagen fibers. The pathological changes of liver tissue in the GFP treatment group were significantly improved. Among them, the high-dose and medium-dose groups of GFP had better therapeutic effect. The arrangement of hepatic cells was basically clear, the structure of hepatic lobule was basically intact, only slight destruction, collagen fiber proliferation was less, pseudo-lobules did not appear, and a small amount of inflammatory cells infiltrated, as shown in Fig. 2.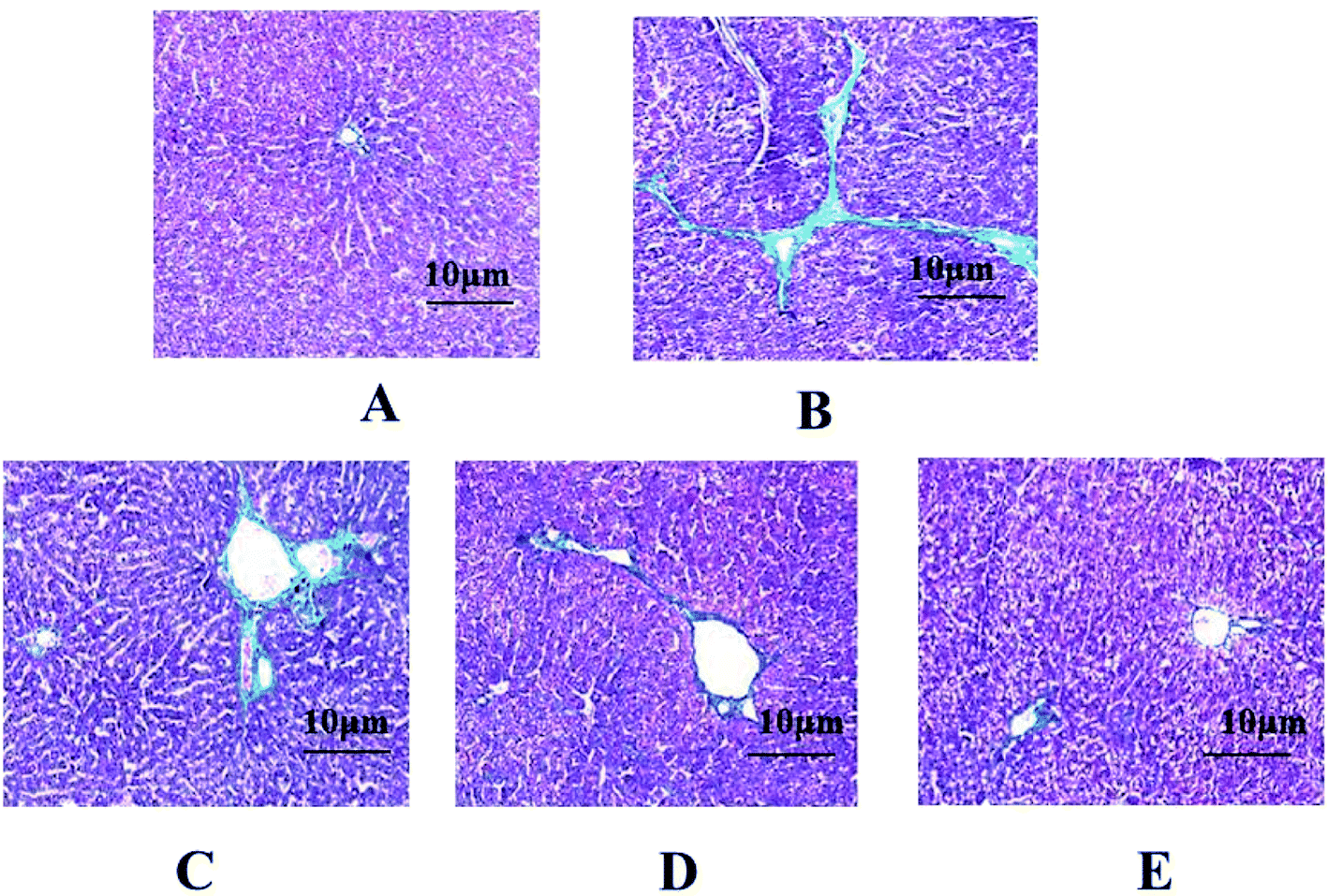 | ||
| Fig. 2 Effect of GFP on HE staining in liver tissues (SP × 400). (A) Control group; (B) model group; (C) GFP 50 mg-treated group; (D) GFP 100 mg-treated group; (E) GFP 200 mg-treated group. | ||
Effect of GFP on serum ALT, AST, TBIL content in liver tissues of rats with hepatic fibrosis.
The results showed that the serum levels of ALT, AST, TBIL in the model group were significantly higher than in the control group (P < 0.01). After GFP treatment, the contents of ALT, AST, TBIL were significantly decreased (P < 0.05, P < 0.01), and the results were shown in Table 2.| Group | ALT (IU L−1) | AST (IU L−1) | TBIL (pg mL−1) |
|---|---|---|---|
| Control | 46.81 ± 8.06 | 91.46 ± 19.82 | 40.31 ± 7.25 |
| Model | 492.13 ± 112.68## | 752.90 ± 115.94## | 71.68 ± 8.68## |
| GFP 50 mg | 371.54 ± 92.31 | 613.68 ± 82.46 | 62.82 ± 9.27 |
| GFP 100 mg | 311.46 ± 71.35* | 482.92 ± 52.19** | 54.68 ± 9.80* |
| GFP 200 mg | 205.32 ± 52.34** | 335.17 ± 36.85** | 41.35 ± 8.47** |
Effect of GFP on the contents of CIV, PCIII, LN, HA content in liver tissues of rats with hepatic fibrosis
The results showed that the levels of CIV, PCIII, LN, HA in the serum of the model group were significantly higher than those in the control group (P < 0.01). After treatment with GFP, the serum CIV and the contents of PCIII, LN, HA were significantly decreased (P < 0.05, P < 0.01), and the results were shown in Table 3.| Group | CIV (μg L−1) | PCIII (μg L−1) | LN (μg L−1) | HA (μg L−1) |
|---|---|---|---|---|
| Control | 35.82 ± 7.92 | 73.62 ± 15.67 | 24.27 ± 11.35 | 23.68 ± 7.64 |
| Model | 79.45 ± 13.72## | 253.68 ± 38.69## | 125.92 ± 39.02## | 93.64 ± 27.53## |
| GFP 50 mg | 60.92 ± 11.27 | 182.75 ± 20.32 | 81.25 ± 22.69 | 75.51 ± 21.69 |
| GFP 100 mg | 51.69 ± 9.45* | 120.73 ± 19.52** | 54.91 ± 18.47** | 46.82 ± 13.58** |
| GFP 200 mg | 42.78 ± 8.62** | 85.72 ± 17.92** | 36.70 ± 14.73** | 31.42 ± 10.03** |
Antioxidant activity of GFP in liver tissues of rats with hepatic fibrosis
The results showed that the activity of SOD and GSH-Px in the liver tissue of the model group was significantly lower than that of the control group, and the MDA content was significantly increased (P < 0.01). After GFP treatment, the activity of SOD and GSH-Px was significantly increased, and MDA content was significantly decreased (P < 0.05, P < 0.01). The results were shown in Fig. 3.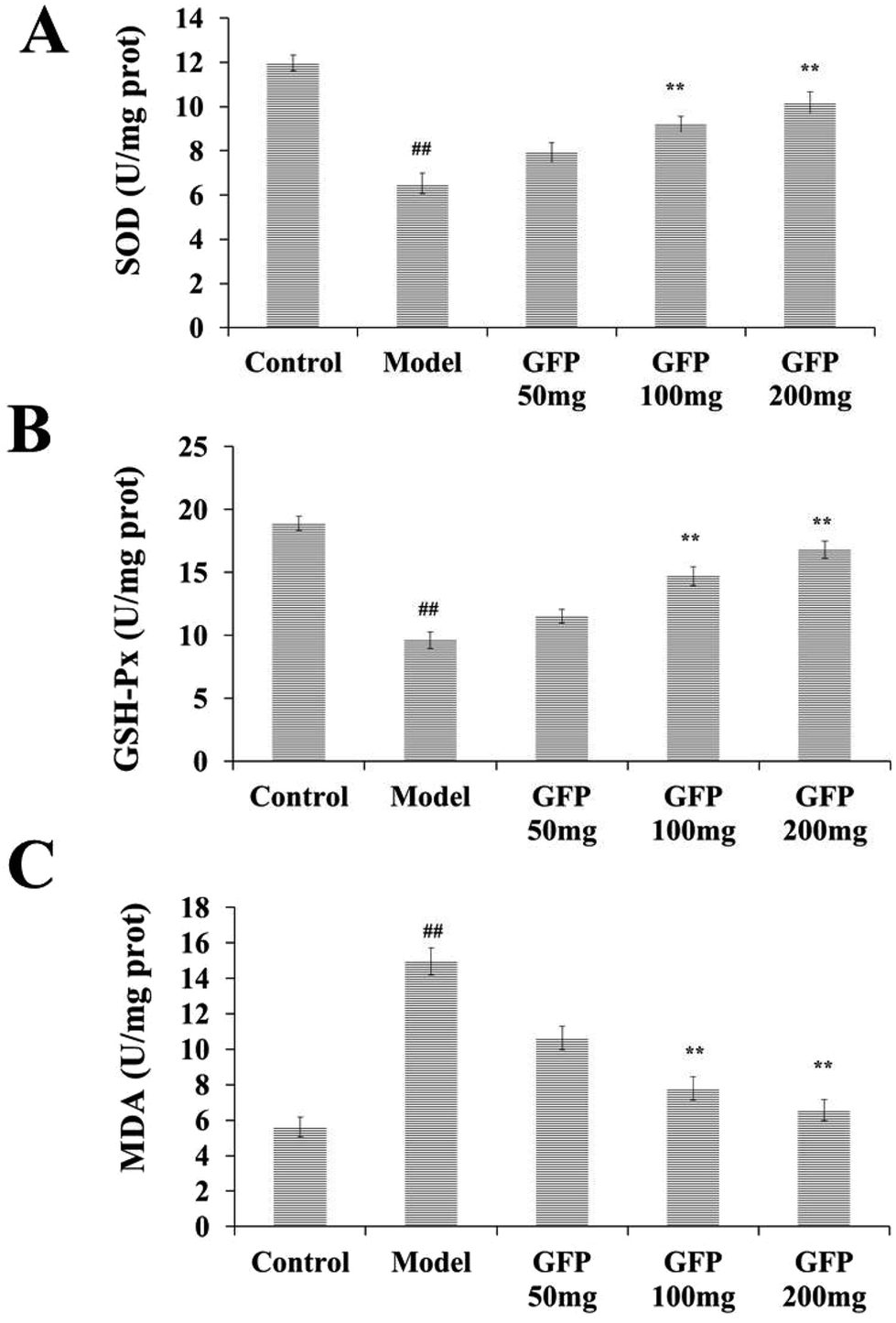 | ||
| Fig. 3 Effects of GFP on SOD activity (A) GSH-Px activity (B) MDA level (C) in liver tissues of rats with hepatic fibrosis. #P < 0.05, ##P < 0.01 vs. control group, *P < 0.05, **P < 0.01 vs. model group, respectively. | ||
Effect of GFP on inflammatory factors TNF-α, IL-1β and IL-6 in liver tissue of rats with hepatic fibrosis
Compared with the control group, the levels of TNF-α, IL-1β and IL-6 in the liver of the model group were increased significantly (P < 0.01). After GFP treatment, the contents of TNF-α, IL- 1β and IL-6 were significantly increased (P < 0.01), as shown in Fig. 4.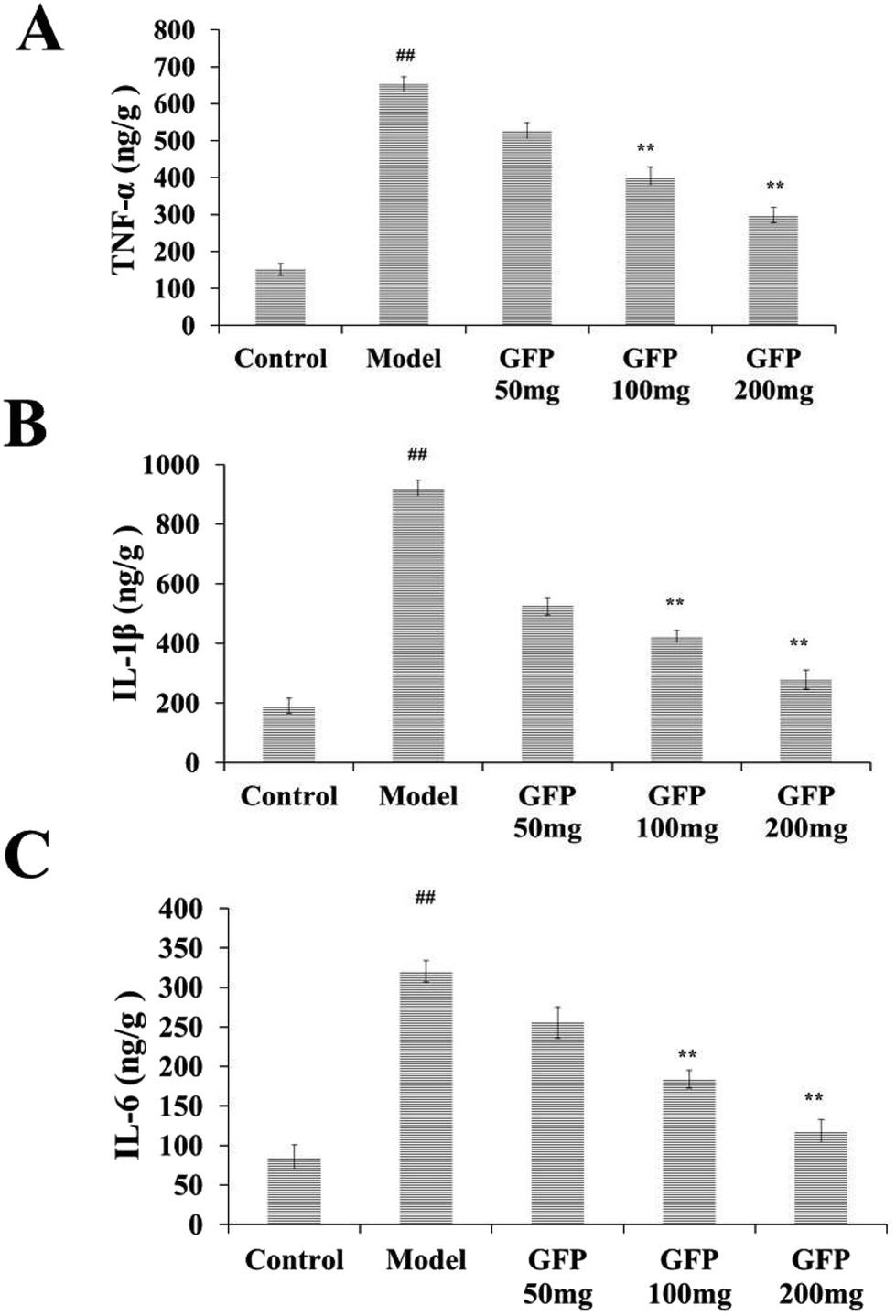 | ||
| Fig. 4 Effects of GFP on inflammatory factors (A) TNF-α, (B) IL-1β and (C) IL-6 in liver tissue of rats with hepatic fibrosis. #P < 0.05, ##P < 0.01 vs. control group, *P < 0.05, **P < 0.01 vs. model group, respectively. | ||
Effect of GFP on TGF-β/Smad pathways in liver tissues of rats with hepatic fibrosis
Compared with the control group, the content of p-Smad 2/3, Smad4 and PAI-1 in liver tissue was significantly increased in the model group, the content of Smad7 protein was significantly decreased in the model group (P < 0.01), and the total Smad 2/3 in liver tissue was is no significant difference in the model group. The expression of Smad4 and PAI-1 in liver tissue of medium dose and high dose group was significantly lower than that of model group, and the expression of Smad7 was significantly higher than that of model group (P < 0.01) (Fig. 5A and B). These results suggested that medium and high dose of GFP can significantly inhibit the expression of Smad4 and PAI-1 in CCL4-induced hepatic fibrosis and induce the expression of Smad7.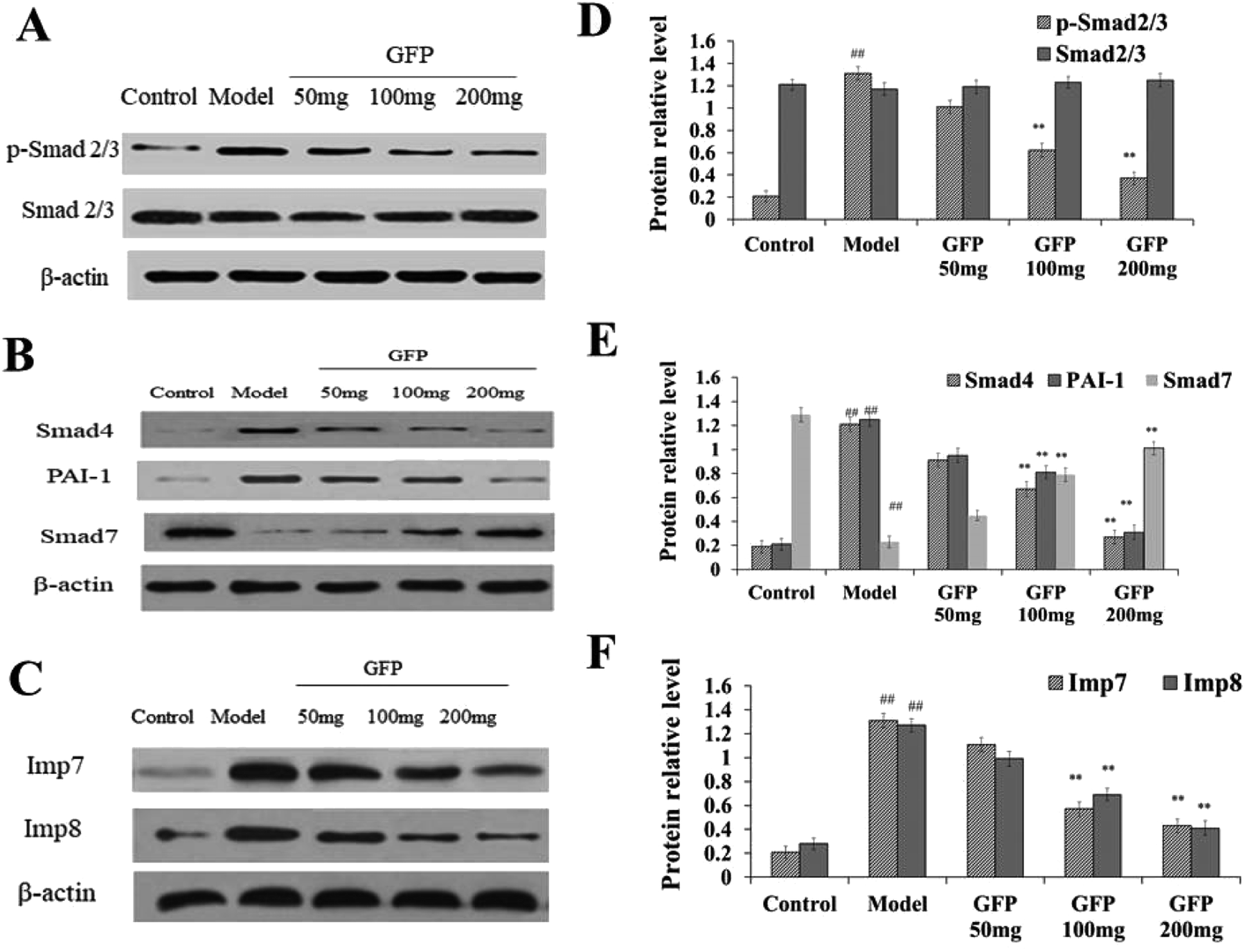 | ||
| Fig. 5 (A) Effect of GFP on the protein expression level of p-Smad 2/3 and Smad 2/3 in liver tissue. (B) Effect of GFP on the protein expression level of Smad4,PAI-1and Smad7 in liver tissue. (C) Effect of GFP on the protein expression level of Imp7 and Imp8 in liver tissue. Histogram represents quantification of protein expression levels using ImageJ software. (D) Levels of p-Smad 2/3 and Smad 2/3. (E) Levels of Smad4, PAI-1and Smad7. (F) Levels of Imp7 and Imp8. The expression of protein was analyzed by western bolt. β-Actin was used as an equal loading control. #P < 0.05, ##P < 0.01 vs. control group, *P < 0.05, **P < 0.01 vs. model group, respectively. | ||
Western blot analysis showed (Fig. 5B) that the expression of Imp7 and Imp8 was significantly increased in liver tissue compared with that in the control group (P < 0.01). The expression of Imp7 and Imp8 in liver tissue of GFP medium and high dose group was significantly lower compared with that in the model group (P < 0.01), suggesting that GFP can down-regulate the expression of Imp7 and Imp8 during hepatic fibrosis.
Effect of GFP on the expression of miR-154 and miR-146a in liver tissues of rats with hepatic fibrosis
As shown in Fig. 6, the expressions of miR-154 and miR-146a in the control group were set to 1. Compared with the control group, the model group miR-154 was significantly increased, and miR-146a was significantly lower (P < 0.01). Compared with the model group, the miR-154 in the liver tissues of GFP-treated groups was significantly lower, and miR-146a was significantly higher (P < 0.01). It indicated that GFP could decrease the expression of miR-54 and increase the expression of miR-146a.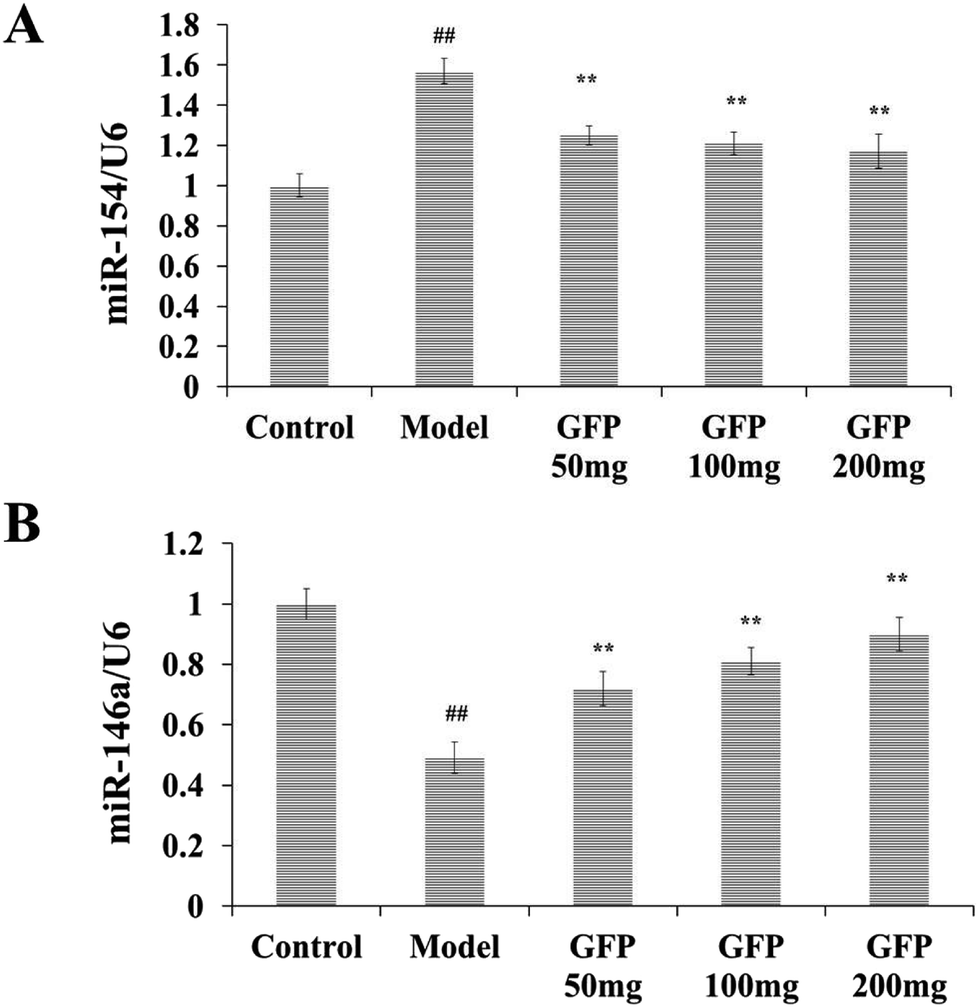 | ||
| Fig. 6 Effects of GFP on miR-154 and miR-146a expression in liver tissues of rats with hepatic fibrosis. (A) Effects of GFP on miR-154 expression in liver tissue. (B) Effects of GFP on miR-146a expression in liver tissue. #P < 0.05, ##P < 0.01 vs. control group, *P < 0.05, **P < 0.01 vs. model group, respectively. | ||
Discussion
Hepatic fibrosis is a necessary stage in liver cirrhosis, which has a serious impact on the prognosis of liver disease.22,23 Its occurrence and development seriously affect the function of the liver. Some related studies have shown that active treatment and control can progress or cure hepatic fibrosis if the disease progresses to the stage of hepatic fibrosis.24 Since the mechanism of hepatic fibrosis has not been specifically clarified, various drugs appear to have limited therapeutic effects on liver fibers. The mechanism of occurrence and development of hepatic fibrosis is extremely complicated, but the current therapeutic effect of western medicine is not very satisfactory, and its drug price is relatively expensive, and the side effects are relatively large, which is not conducive to the treatment of hepatic fibrosis to some extent. Studies have shown that polysaccharides have obvious advantages in the treatment of liver fibers. Polysaccharides can play a role through a network of various links, multiple pathways, a variety of intracellular molecules and cytokines.25–27Grifola frondosa is a precious combination of food and medicine, and its polysaccharide is its main physiologically active substance.16 Previous studies have focused on the anti-tumor effect of the polysaccharides from Grifola frondosa.28 Recent studies have shown that the polysaccharides from Grifola frondosa can significantly reduce serum AST and ALT levels in CC14 acute liver injury model rats, and down-regulate CCL4-induced CYP2E1, TNF-α protein expression, which suggests that the polysaccharides from Grifola frondosa may have a good anti-fibrosis effect.29,30
In this study, a rat model of hepatic fibrosis induced by CCl4 was used to study the intervention effect and mechanism of the polysaccharides from Grifola frondosa (GFP) on hepatic fibrosis. Since the self-healing tendency of fibrosis induced by CCl4 is obvious, a preventive administration method is employed. After 12 weeks of modeling, the rats in the model group showed typical changes in hepatic fibrosis according to the external manifestations, liver status and biochemical indicators of the model group, which confirmed the successful modeling of the experimental animals.
The content of hepatic fibrosis (CIV, PCIII, LN, HA) in liver tissue is a common indicator used to monitor the progress of hepatic fibrosis, and during the development of hepatic fibrosis, collagen fibers proliferate in large quantities.31 The main component of collagen fibers is collagen, so the content of liver fiber can also reflect the degree of hepatic fibrosis.32 In this study, a rat model of hepatic fibrosis was prepared by carbon tetrachloride (CCl4) composite method, and GFP was given to intervene treatment. It was found that GFP can effectively reduce the serum levels of CIV, PCIII, LN, HA, ALT, AST and TBIL in rats with hepatic fibrosis, improve the pathological changes of liver tissue, inhibit the necrosis of hepatocytes and the proliferation of hepatic fibrosis tissue, and improve the fibrosis status of liver tissue, suggesting that GFP has protective effect on experimental hepatic fibrosis rats.
In recent years, pathophysiological studies have found that oxidative stress and inflammatory response are important pathological mechanisms for the development of hepatic fibrosis and transformation into cirrhosis.33 Superoxide dismutase (SOD) can reduce oxygen free radicals to H2O by providing hydrogen atom ligands. Under the catalysis of glutathione peroxidase (GSH-Px), SOD can be further reduced to form harmless hydrogen dioxide and oxygen, thus eliminating oxidative stress damage.34 The content of malondialdehyde (MDA), which was the final product of lipid peroxidation, can also indirectly reflect the degree of oxidative stress damage.35 TNF-α, IL-1β and IL-6 have played key roles in the regulation of inflammatory response and the occurrence of hepatic fibrosis in vivo.36 This experimental study found that GFP can effectively improve liver tissue in rats with hepatic fibrosis SOD and GSH-Px activities, reduce MDA content and decrease serum levels of TNF-α, IL-1β and IL-6, suggesting that GFP can inhibit oxidative stress and inflammatory response in liver tissue of rats with hepatic fibrosis.
The TGF-β/Smad signaling pathway is closely related to the pathogenesis of hepatic fibrosis-HCC, and PAI-1 is one of the most important target genes regulated by this pathway.37,38 Experimental and clinical studies have shown that PAI-1 plays an important role in the formation of hepatic fibrosis, which is related to its regulation of ECM deposition.39 Smad4 is an integral part of the TGF-β/Smad signaling pathway. Studies have shown that conditional knockout of Smad4 inhibits hepatic fibrosis and HCC, suggesting that Smad4 is one of the factors that promote hepatic fibrosis. Studies have found that Smad4 mRNA expression is increased in experimental hepatic fibrosis, and Smad4 overexpression promotes HBV-related hepatic fibrosis.40 Smad7 is an inhibitory Smad that inhibits the TGF-β/Smad signaling pathway through a negative feedback mechanism. Studies have shown that knockout of Smad7 promotes progression of hepatic fibrosis and HSC activation. Smad7 expression was significantly down-regulated in the CCl4-induced hepatic fibrosis model. The profibrotic signal transmitted by p Smad2L/C and the pro-cancer signal of p Smad3L are required to regulate the transcription of target genes such as PAI-1 from the cytoplasm into the nucleus.41 The entry of Smad2, Smad3 and Smad4 is regulated by Imp7 and Imp8 in the importin family, and knockdown of Imp7 and Imp8 strongly inhibits Smad2/3 entry into the nucleus.42 This study found that GFP can down-regulate the expression of PAI-1, Smad4, Imp7 and Imp8 and up-regulate the expression of Smad4, which may be the molecular mechanism of GFP against hepatic fibrosis.
In addition, the TGF-β/Smad signaling pathway regulates the occurrence and development of hepatic fibrosis through interaction with micro RNA.43,44 After transcription of microRNAs, the expression of genes can be negatively regulated. A target gene can be regulated by multiple different microRNAs at the same time. MicroRNAs can also regulate multiple different genes. It has been found that many MicroRNAs play an important role in the regulation of liver fibrosis.45 With the development of research, it has been reported that microRNAs affect fibrosis by regulating the TGF-β/Smad signal transduction pathway.46 It has been found that microRNA-199a can inhibit the transmission of TGF-β/Smad signaling pathway by inhibiting the expression of TGF-β.47 MicroRNA is a kind of small-molecule single-stranded non-coding RNA, which has been widely concerned in recent years by regulating gene expression and participating in biological processes such as cell proliferation, apoptosis, development, differentiation and metabolism.48,49 This study found that miR-154 was up-regulated in the liver tissue of rats with pulmonary fibrosis, and miR-146a was also down-regulated to a greater extent than that of the control group. After treatment with GFP, the expression level of miR-154 was significantly decreased, and the expression level of miR-146a was significantly increased. The results of this study suggested that miR-154, miR-146a and TGF-β/Smad signaling pathways play an important role in the inhibition of hepatic fibrosis by GFP. However, the relationship between miRNA-154, miRNA-146a and hepatic fibrosis, whether miRNA-154 and miRNA-146a participated in hepatic fibrosis through regulating the TGF-β/Smad signaling pathway remained to be further studied.
Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This study was supported by National “13th Five-Year” Plan for Science & Technology (grant number 2016YFNC010104; 2016YFD0400505); the Innovative Research Team of Tianjin Municipal Education Commission (grant number TD13-5013); National “12th Five-Year” Plan for Science & Technology (grant number 2012BAD33B04); State Key Laboratory of Food Nutrition and Safety, Tianjin University of Science & Technology (grant number 17YDLJNC00130; 17PTSYJC00080; 18ZYPTJC00020); the Project program of Key Laboratory of Food Nutrition and Safety, Ministry of Education, China (No. 2018013); The Open Project Program of State Key Laboratory of Food Nutrition and Safety, Tianjin University of Science & Technology (No. SKLFNS-KF-201830).References
- V. Hernandezgea and S. L. Friedman, Annu. Rev. Pathol.: Mech. Dis., 2011, 6, 425–456 CrossRef CAS.
- F. Liu, X. Wang, G. Wu, L. Chen, P. Hu, H. Ren and H. Hu, PLoS One, 2015, 10, e0142457 CrossRef.
- D. A. Brenner, Trans. Am. Clin. Climatol. Assoc., 2009, 120, 361–368 Search PubMed.
- J. Wiegand and T. Berg, Deutsches Ärzteblatt International, 2013, 110, 85–91 Search PubMed.
- J. Wattacheril, D. Issa and A. J. Sanyal, Annu. Rev. Pharmacol. Toxicol., 2018, 58, 649–662 CrossRef CAS PubMed.
- A. M. Elsharkawy, F. Oakley and D. A. Mann, Apoptosis, 2005, 10, 927–939 CrossRef CAS.
- H. Lam, C. H. Yeh, K. Cheng, C. Hsu and J. Cheng, Neurosci. Lett., 2008, 438, 90–95 CrossRef CAS.
- Y. A. Lee, M. C. Wallace and S. L. Friedman, Gut, 2015, 64, 830–841 CrossRef CAS.
- Y. H. Wang, C. X. Zhao, B. M. Chen, M. He, L. Q. Liu, C. Y. Li and X. Chen, China J. Chin. Mater. Med., 2014, 39, 1473 Search PubMed.
- K. S. Hung, T. H. Lee, W. Y. Chou, C. L. Wu, C. L. Cho, C. N. Lu, B. Jawan and C. H. Wang, Biochem. Biophys. Res. Commun., 2005, 336, 324–331 CrossRef CAS.
- L. Zou, S. Chen, L. Li and T. Wu, Exp. Toxicol. Pathol., 2017, 69, 451 CrossRef CAS.
- J. H. Lee, H. Lee, Y. K. Joung, K. H. Jung, J. H. Choi, D. H. Lee, K. D. Park and S. S. Hong, Biomaterials, 2011, 32, 1438–1445 CrossRef CAS PubMed.
- T. Wu, J. M. Chen, T. G. Xiao, X. B. Shu, H. C. Xu, L. L. Yang, L. J. Xing, P. Y. Zheng and G. Ji, World J. Gastroenterol., 2016, 22, 4695 CrossRef CAS PubMed.
- G. Gong, T. Dang, Y. Deng, J. Han, Z. Zou, S. Jing, Y. Zhang, Q. Liu, L. Huang and Z. Wang, Int. J. Biol. Macromol., 2018, 109, 611–618 CrossRef CAS.
- Z. Zhang, G. Lv, H. Pan, A. Pandey, W. He and L. Fan, Int. J. Biol. Macromol., 2012, 51, 1140–1146 CrossRef CAS.
- B. C. Lee, J. T. Bae, H. B. Pyo, T. B. Choe, S. W. Kim, H. J. Hwang and J. W. Yun, Enzyme Microb. Technol., 2003, 32, 574–581 CrossRef CAS.
- C. Hsieh, H. Wang, C. Chen, T. Hsu and M. Tseng, Biochem. Eng. J., 2008, 38, 198–205 CrossRef CAS.
- C. Zhuang, T. Mizuno, H. Ito, K. Shimura, T. Sumiya and M. Kawade, J. Food Sci. Technol., 1994, 41, 724–732 CrossRef CAS.
- H. Xu, J. Liu, Z. Shen, Y. Fei and X. Chen, Carbohydr. Polym., 2010, 82, 687–691 CrossRef CAS.
- X. Ma, C. Li, W. Qi, X. Li, S. Wang, X. Cao and C. Wang, Bioact. Carbohydr. Diet. Fibre, 2015, 6, 7–14 CrossRef CAS.
- M. Meng, D. Cheng, L. Han, Y. Chen and C. Wang, Carbohydr. Polym., 2017, 157, 1134–1143 CrossRef CAS.
- Z. Zheng, X. Zhang, J. Wang, A. Dandekar, H. Kim, Y. Qiu, X. Xu, Y. Cui, A. Wang and L. C. Chen, J. Hepatol., 2015, 63, 1397–1404 CrossRef CAS.
- H. J. F. Hodgson, D. R. Davies and R. P. H. Thompson, J. Clin. Pathol., 1976, 29, 11–16 CrossRef CAS.
- E. Seki and R. F. Schwabe, Hepatology, 2015, 61, 1066–1079 CrossRef.
- F. Gan, Q. Liu, Y. Liu, D. Huang, C. Pan, S. Song and K. Huang, Life Sci., 2018, 192, 205–212 CrossRef CAS.
- K. Zhang, Y. Gao, M. Zhong, Y. Xu, J. Li, Y. Chen, X. Duan and H. Zhu, J. Ethnopharmacol., 2016, 179, 38–44 CrossRef CAS.
- Y. Wang, M. Guan, X. Zhao and X. Li, Pharm. Biol., 2018, 56, 325–332 CrossRef CAS.
- S. Konno, S. Aynehchi, D. J. Dolin, A. M. Schwartz, M. Choudhury and H. Tazaki, Int. J. Med. Mushrooms, 2002, 4, 11 CrossRef.
- L. U. Meifang, Nat. Prod. Res. Dev., 2010, p777 Search PubMed.
- X. Ma, C. Li, W. Qi, X. Li, S. Wang, X. Cao and C. Wang, Bioact. Carbohydr. Diet. Fibre, 2015, 6, 7–14 CrossRef CAS.
- Y. Wei, M. Huang, X. Liu, Z. Yuan, Y. Peng, Z. Huang, X. Duan and T. Zhao, Cell. Physiol. Biochem., 2015, 35, 1599–1608 CrossRef CAS.
- M. V. Machado and H. Cortezpinto, J. Hepatol., 2013, 58, 1007–1019 CrossRef.
- V. Sanchezvalle, N. C. Chaveztapia, M. Uribe and N. Mendezsanchez, Curr. Med. Chem., 2012, 19, 4850–4860 CrossRef CAS.
- L. Yuan, F. Chen, L. Ling, P. Dou, H. Bo, M. Zhong and L. Xia, J. Ethnopharmacol., 2008, 116, 539–546 CrossRef CAS.
- H. Wang, W. Wei, N. Wang, C. Wu, S. Yan, L. Yue, L. Zhang and S. Xu, World J. Gastroenterol., 2005, 11, 2124–2129 CrossRef CAS.
- M. M. Mutengo, T. Mduluza, P. Kelly, J. Mwansa, G. Kwenda, P. Musonda and J. Chipeta, J. Parasitol. Res., 2018, 2018, 1–8 CrossRef.
- L. Zhang, C. Liu, X. Meng, C. Huang, F. Xu and J. Li, Mol. Cell. Biochem., 2015, 400, 17–28 CrossRef CAS.
- H. Wang, Y. Zhang and R. O. Heuckeroth, FEBS Lett., 2007, 581, 3098–3104 CrossRef CAS.
- S. Inuzuka, T. Ueno, T. Torimura, S. Tamaki, H. Sugawara, R. Sakata, N. Kusaba, M. Sata and K. Tanikawa, Scand. J. Gastroenterol., 1997, 32, 1052–1060 CrossRef CAS PubMed.
- M. Moussa, N. Helal and M. M. Youssef, APMIS, 2018, 126, 477–485 CrossRef CAS.
- S. Dooley, J. Hamzavi, K. Breitkopf, E. Wiercinska, H. M. Said, J. Lorenzen, P. T. Dijke and A. M. Gressner, Gastroenterology, 2003, 125, 178–191 CrossRef CAS.
- X. Yao, X. Chen, C. L. Cottonham and L. Xu, J. Biol. Chem., 2008, 283, 22867–22874 CrossRef CAS.
- J. Li, B. Cen, S. Chen and Y. He, Mol. Med. Rep., 2016, 13, 4229–4237 CrossRef CAS.
- M. T. Blahna and A. Hata, FEBS Lett., 2012, 586, 1906–1912 CrossRef CAS.
- T. Ogawa, M. Enomoto, H. Fujii, Y. Sekiya, K. Yoshizato, K. Ikeda and N. Kawada, Gut, 2012, 61, 1600–1609 CrossRef CAS.
- W. Qin, A. C. Chung, X. R. Huang, X. M. Meng, D. S. Hui, C. M. Yu, J. J. Sung and H. Y. Lan, J. Am. Soc. Nephrol., 2011, 22, 1462–1474 CrossRef CAS.
- E. A. Lin, L. Kong, X. H. Bai, Y. Luan and C. Liu, J. Biol. Chem., 2009, 284, 11326–11335 CrossRef CAS.
- J. Lu, G. Getz, E. A. Miska, E. Alvarezsaavedra, J. Lamb, D. Peck, A. Sweetcordero, B. L. Ebert, R. H. Mak and A. A. Ferrando, Nature, 2005, 435, 834–838 CrossRef CAS.
- L. He and G. J. Hannon, Nat. Rev. Genet., 2004, 5, 522–531 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2019 |