DOI:
10.1039/C9RA04578C
(Paper)
RSC Adv., 2019,
9, 24377-24385
Tunable preparation of highly dispersed NixMn-LDO catalysts derived from NixMn-LDHs precursors and application in low-temperature NH3-SCR reactions†
Received
18th June 2019
, Accepted 25th July 2019
First published on 6th August 2019
Abstract
A series of NixMn bimixed metal oxides (NixMn-LDO) were prepared via calcining NixMn layered double hydroxides (NixMn-LDHs) precursors at 400 °C and applied as catalysts in the selective catalytic reduction (SCR) of NOx with NH3. The DeNOx performance of catalysts was optimized by adjusting the Ni/Mn molar ratios of NixMn-LDO precursors, in which Ni5Mn-LDO exhibited above 90% NOx conversion and N2 selectivity at a temperature zone of 180–360 °C. Besides, Ni5Mn-LDO possessed considerable SO2 & H2O resistance and outstanding stability. Multiple characterization techniques were used to analyze the physicochemical properties of the catalysts. The analysis results indicated that all catalysts had the same active species Ni6MnO8, while their particle sizes showed significant differences. Notably, the uniform distribution of active species particles in the Ni5Mn-LDO catalyst provided the rich surface acidity and suitable redox ability which were the primary causes for its desirable DeNOx property.
1. Introduction
During the past decades, nitrogen oxide (NOx) emissions have increased dramatically, which triggered a battery of environmental problems including photochemical smog, acid rain and greenhouse effects, further endangering human health.1,2 With the increasingly severe strictness of environmental regulations, it is necessary to find a valid method to eliminate NOx. Among numerous removal techniques, selective catalytic reduction of NOx with NH3 (NH3-SCR) has been considered as the most effective measure for denitrification (DeNOx).3,4 Currently, commercial catalysts V2O5–WO3/TiO2 have been widely used in the field of industrial stationary sources, which displayed good catalytic performance at the high temperature region of 300–400 °C.5 However, V-based catalysts still possess defects, such as the narrow operation temperature window, low N2 selectivity at higher temperatures and inherent toxicity, which restricted its further application.6 Thus, it is worthwhile to develop non-toxic DeNOx catalysts with good low temperature activity.
Based on the features of environmentally friendly and variable valence of Mn element, MnOx has been deemed as the potential catalyst for NH3-SCR reaction in the low temperature zone.7–9 Whereas there were some inevitably problems caused by its stronger oxidation capacity which easily led to the over-oxidation of NH3 at higher temperature and further reduced SCR activity as well as N2 selectivity.10,11 Recently, it has been reported that the strong oxidation capacity of MnOx catalyst could be adapted by introducing transition metals.12–15 Among all the attempts, Ni was considered to be a functional component with excellent synergistic catalysis. For instance, Wan et al.16 and Chen et al.17 prepared Ni–Mn bi-metal oxides and Ni–Mn spinel oxides catalysts, respectively, the catalytic performance results showed that both catalysts exhibited higher SCR activity and N2 selectivity than single MnOx catalyst. Although some progress has been made, it was unavoidable that the active specie particles of Ni–Mn catalysts prepared by traditional methods were prone to agglomerate, resulting in the decline of catalytic performance.18
As investigated in the previous studies, the dispersion of active specie particles over catalyst could be effectively improved via regulating the morphology and structure.19–21 Lately, the layered double oxides (LDO) catalysts obtained via calcining layered double hydroxides (LDHs) precursor attracted proverbial attention due to the excellent physical and chemical properties.22–24 Moreover, our group have also found that the catalysts prepared by employing LDHs as precursor could achieve better SCR activity than other methods, it might be related with the lattice limiting effect and surface hydrazine structure existed in bulk laminate of LDHs, which were helpful to improve the dispersion and stability of the active specie particles.25
In this work, on account of the synergistic effect of Ni and Mn as well as satisfactory function of LDHs, NixMn-LDHs with different Ni/Mn molar ratios were successfully fabricated with aid of coprecipitation methods. NixMn-LDO obtained by calcining NixMn-LDHs at a proper temperature were hired as DeNOx catalysts in NH3-SCR reaction. In order to optimize physical and chemical properties of NixMn-LDO catalysts, the relative proportions of Ni and Mn of the NixMn-LDHs precursors were explored in detail. A series of characterization techniques were used to reveal the intrinsic reason for the differences in catalytic activities among catalysts. This research might expand a new horizon to develop new catalysts with outstanding efficiency.
2. Experimental section
2.1 Preparation of catalysts
The NixMn-LDHs (“x” represent Ni/Mn molar ratio = 3, 4, 5, 6 and total molar amount Ni + Mn = 0.06 mol) were prepared by co-precipitation method.26 To be specific, for the synthesis of Ni3Mn-LDHs, 0.045 mol (13.09 g) of Ni(NO3)2·6H2O (98.0%) and 0.015 mol (5.37 g) of Mn(NO3)2 (50%) were dissolved in 100 mL deionized water to prepare metal precursor solution. The mixed alkali solution were obtained by dissolving 0.04 mol (1.6 g) of NaOH and 2.8 g of NH3·H2O (25–28%) in 100 mL deionized water. Then, the metal precursor solution and alkali solution were simultaneously added dropwise with magnetic stirring into flask at room temperature. The suspension was crystallized at 80 °C for 24 h, and then the resulting subsidence by filtration, washing and drying at 80 °C for 12 h to obtain the Ni3Mn-LDH precursor. The Ni3Mn-LDO catalyst was obtained by calcining Ni3Mn-LDHs precursor in air at 400 °C for 5 h at a heating rate of 2 °C min−1. Similarly, other samples such as Ni4Mn-LDO, Ni5Mn-LDO and Ni6Mn-LDO were obtained with the same method.
2.2 Characterization of catalysts
X-ray diffraction (XRD) information of catalysts was measured on a Rigaku DX-2700 instrument using Cu/Kα radiation source. The 2θ range of meterage from 5 to 85° and a scanning rate was 8° min−1. The Ni and Mn contents were determined by inductively coupled plasma-atomic (ICP) on Optima 7000 DV instrument. Fourier transform infrared spectrometer (FT-IR) experiment were tested from 400 to 3600 cm−1 on a TENSOR II FT-IR (Bruker). Thermogravimetry (TG) analyzer (Setaram Evolution) equipped with quadrupole mass spectrometry (QMS) real-time monitored the thermal decomposition of LDHs sample with the temperature rising from 50 °C to 800 °C. The surface area and pore information of catalysts were obtained by using Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods on Micromeritics ASAP-2460 apparatus. The test conditions are mainly that the catalyst samples were degassed at 250 °C for 4 h. Ammonia-temperature programmed desorption (NH3-TPD) was performed on FINESORB-3010C chemisorptions analyzer with thermal conductivity detector (TCD). About 100 mg catalysts (40–60 mesh) was pretreated at 300 °C for 1 h under 25 mL min−1 He atmosphere; subsequently, the sample adsorbed NH3 to achieve saturation at 50 °C for 1 h and He was purged at the same temperature to remove weakly adsorbed NH3 on the surface; finally, the desorption curve was recorded via increasing temperature (from 50 °C to 560 °C) with heating rate of 10 °C min−1 under 25 mL min−1 He atmosphere. Hydrogen-temperature programmed reduction (H2-TPR) was tested in the same way. About 80 mg catalysts was pretreated at 300 °C for 1 h under 30 mL min−1 Ar atmosphere; after cooling down to 50 °C, the catalyst samples accomplished the reduction process from 80 °C to 800 °C in a flow of 30 mL min−1 10% H2/Ar. In order to explore the surface morphology and structure of the sample, scanning electron microscope (SEM) and high resolution-transmission electron microscopy (HR-TEM) were conducted on a Hitachi SU8010 apparatus and JEM-2100F (JEOL) with an accelerating voltage of 200 kV, respectively. The EDS equipped with SEM was also used to characterize the surface composition of catalyst.
2.3 Activity measurements of catalysts
The SCR activity measurements of catalysts were carried out on a fixed-bed quartz reactor with the internal diameter of 6 mm. About 350 mg catalyst samples (40–60 mesh) were placed in the reactor. The reaction gas composition was as follows: 600 ppm NO, 600 ppm NH3, 5 vol% O2, 10 vol% H2O (when used), 100 ppm SO2 (when used) and N2 as the balance gas. The total flow rate of gas was 187 mL min−1 and GHSV = 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. The activity performance were tested at range from 90 to 390 °C and the data were collected for every 30 °C, meanwhile, the output gases were monitored by multigas 2000 FTIR. The NOx conversion and N2 selectivity were calculated as follows:
000 h−1. The activity performance were tested at range from 90 to 390 °C and the data were collected for every 30 °C, meanwhile, the output gases were monitored by multigas 2000 FTIR. The NOx conversion and N2 selectivity were calculated as follows:
| NOx conversion = 100% × ([NOx]in − [NOx]out)/[NOx]in |
| N2 selectivity = 100% × ([NOx]in + [NH3]in − [NOx]out − [NH3]out − 2[N2O]out)/([NO]in + [NH3]in − [NOx]out − [NH3]out) |
where “in” represents the gas concentration at the inlet, “out” represents the gas concentration at the output.
3. Results and discussion, experimental
3.1 Characterization of NixMn-LDHs precursors
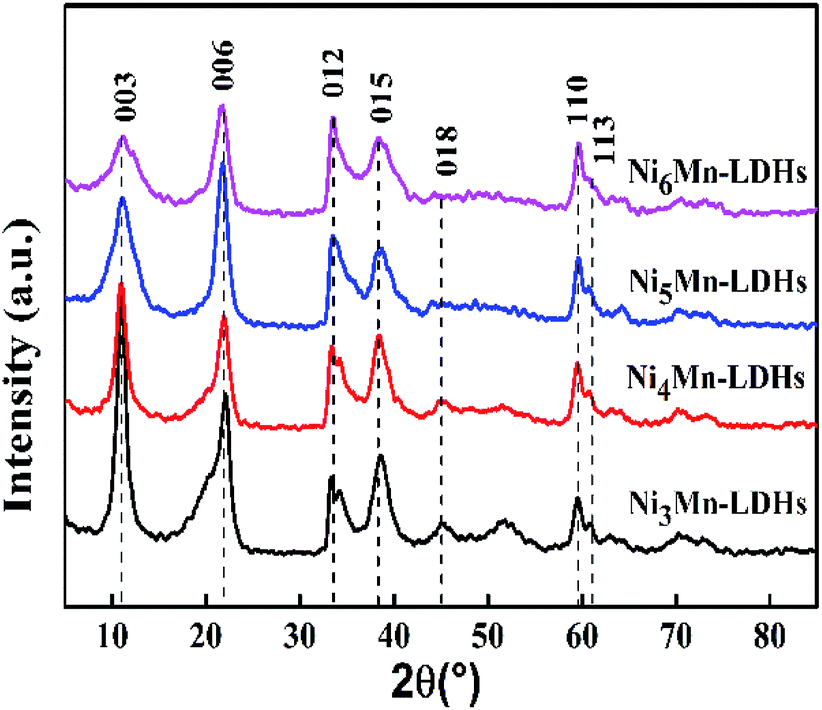
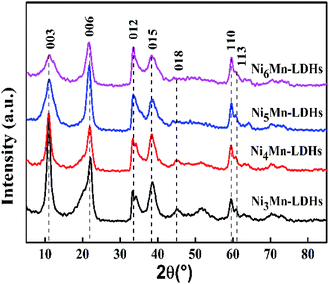
3.1.1 XRD analysis. XRD patterns of the NixMn-LDHs precursors with different Ni/Mn molar ratios were displayed in Fig. 1. As the results presented, all samples showed the typical diffraction peaks at about 11.0°, 22.1°, 34.3°, 38.7°, 45.0°, 60.0° and 61.1°, which can be indexed to (003), (006), (012), (015), (018), (110) and (113) planes of NixMn-LDHs,27–29 and no other miscellaneous peaks were observed. That is to say, NixMn-LDHs with pure phase were synthesized successfully. It can be seen that the (003) diffraction peaks of four samples appear at 11°, further the basal spacings of NixMn-LDHs were calculated as 8.04 Å by Bragg's equation of nλ = 2dsinθ, which indicated that CO32− existed in the interlayers of LDHs.26,30 In addition, with the increase of Ni/Mn molar ratios for NixMn-LDHs, it can be observed that the intensity of the (003) diffraction peaks showed a tendency to reduce, and the peaks width became wider. This implied that Ni5Mn-LDHs and Ni6Mn-LDHs possessed smaller LDHs crystal sizes compared with Ni3Mn-LDHs and Ni4Mn-LDHs (Table 1), which may change the appearance of the LDHs precursors and thus affect the dispersibility of the calcined product. Furthermore, tested Ni/Mn molar ratios were determined by ICP analysis (Table 1), and the result was consistent with feeding ratios. The composition and morphology information of the NixMn-LDHs were further investigated by FT-IR and SEM.
 |
| | Fig. 1 XRD patterns of NixMn-LDHs precursors (x = 3, 4, 5, 6). | |
Table 1 Chemical compositions and XRD parameters of NixMn-LDHs (x = 3, 4, 5, 6) precursors
| Samples |
Weight contenta |
Ni:Mn |
FWHM003 |
D003b (nm) |
| Ni |
Mn |
| Weight content of Ni and Mn was determined by ICP analysis. D003 of samples was calculated by Scherrer equation. |
| Ni3Mn-LDHs |
44.37 |
15.32 |
2.89 |
0.0164 |
8.39 |
| Ni4Mn-LDHs |
47.66 |
12.41 |
3.84 |
0.0170 |
8.09 |
| Ni5Mn-LDHs |
49.01 |
9.55 |
5.13 |
0.0216 |
6.40 |
| Ni6Mn-LDHs |
51.22 |
8.71 |
5.88 |
0.0222 |
6.21 |
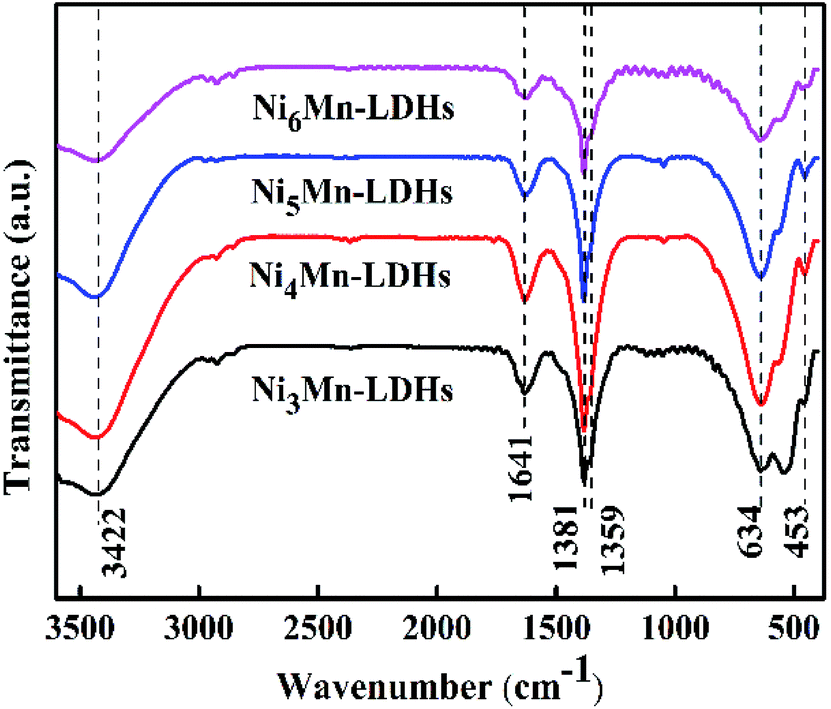
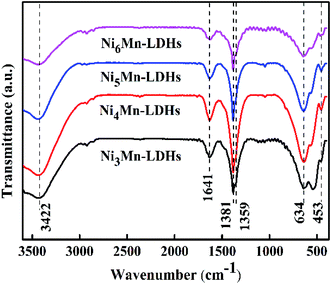
3.1.2 FT-IR analysis. Fig. 2 showed the FT-IR spectra for NixMn-LDHs (x = 3, 4, 5, 6). It could be found that a series of infrared adsorption bands were displayed within the wavenumber range of 400–3600 cm−1. Broad adsorption band at 3422 cm−1 could be assigned to stretching vibration of –OH groups in the NixMn-LDHs layers and interlayer water molecules.31 The adsorption band at 1641 cm−1 might be due to the angular deformation of water.32 The vibration of CO32− and NO3− existed in interlayers could be observed at 1381 and 1359 cm−1, respectively.26 Additionally, the adsorption bands around 634 and 453 cm−1 could be attributed to the cation-oxygen vibrations, such as O–M–O, M–O–M or M–OH.33 These results further indicated that the prepared NixMn-LDHs samples were CO32− and NO3− intercalated.
 |
| | Fig. 2 FT-IR spectra of NixMn-LDHs precursors (x = 3, 4, 5, 6). | |
3.1.3 SEM analysis. The SEM characterization was adopted to probe the microscopic morphology of the precursors, the corresponding results were shown in Fig. 3. As can be seen, the four samples exhibited typical nanosheet structure, which might further witnessed the successful synthesis of LDHs precursors.34,35 Moreover, it could also be observed that surface morphology of four samples has changed obviously with the increase of Ni/Mn molar ratios. For Ni3Mn-LDHs and Ni4Mn-LDHs, the LDHs sheets presented a larger size and seriously stacked, which will greatly affect the exposure of the effective components on the surface of the sample. In contrast, the sheets size of Ni5Mn-LDHs and Ni6Mn-LDHs was significantly reduced and the degree of accumulation was greatly improved. In fact, thus morphology difference of precursor often influence the properties of the calcined product.
 |
| | Fig. 3 SEM images of (a) Ni3Mn-LDHs; (b) Ni4Mn-LDHs; (c) Ni5Mn-LDHs and (d) Ni6Mn-LDHs precursors. | |
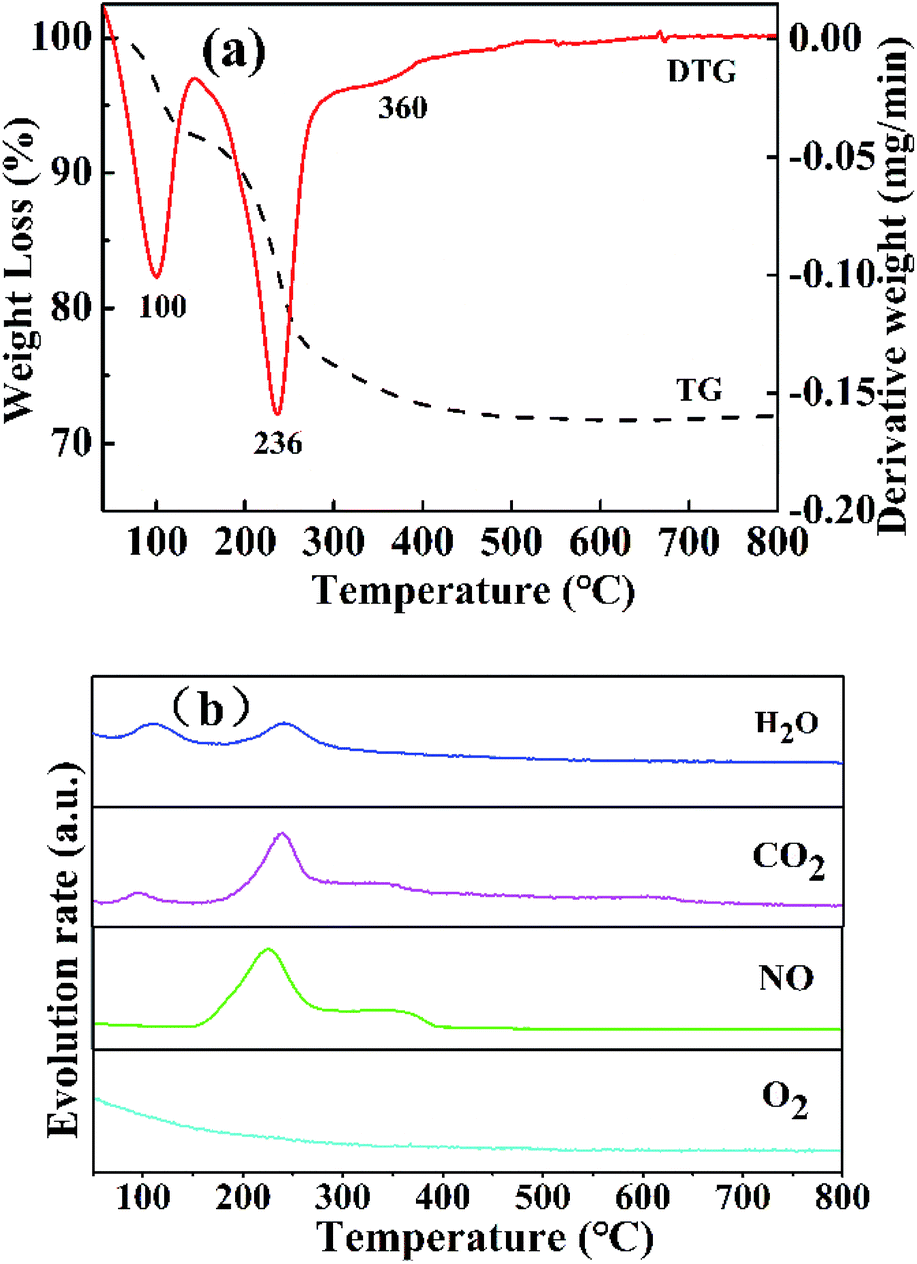
3.1.4 TG-MS analysis. The thermal decomposition of the NixMn-LDHs precursors was clearly explored by TG-MS analysis. As shown in Fig. 4, take Ni5Mn-LDHs as an example. Three distinct weight loss peaks appeared on the curves of TG and DTG (Fig. 4(a)). The first weight loss stage around 80–130 °C was caused by the removal of physically adsorbed water, interlayer water and CO2 molecules, where the release signal of the corresponding gases (H2O and CO2) was detected by MS (Fig. 4(b)).36 The major weight loss process occurred at second stage, which took place at the temperature range of 150–300 °C, mainly arising from the dehydration of –OH group on the LDHs laminate as well as the loss of volatile gases CO2 and NO from the interlayer CO32− and NO3− ions.37 The last weight loss stage displayed a slight exothermic peak at about 360 °C, a small amount of CO2 and NO release can be still observed from MS (Fig. 4(b)), which can be assigned to decomposition of interlayer anions CO32− and NO3− at higher temperature.32 As the temperature further increased (>400 °C), no weight loss peak was found, indicating that the Ni5Mn-LDHs precursor achieved complete decomposition. Based on the TG-MS analysis, it can be concluded that calcining LDHs precursors at 400 °C can form stable mixed metal oxides.
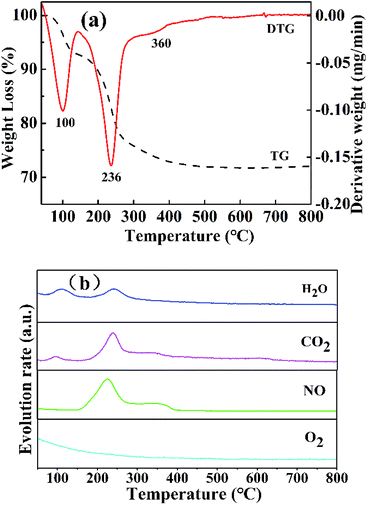 |
| | Fig. 4 Curves of TG and DTG (a) and MS (b) for Ni5Mn-LDHs samples. | |
3.2 Characterization of NixMn-LDO (x = 3, 4, 5, 6) catalysts
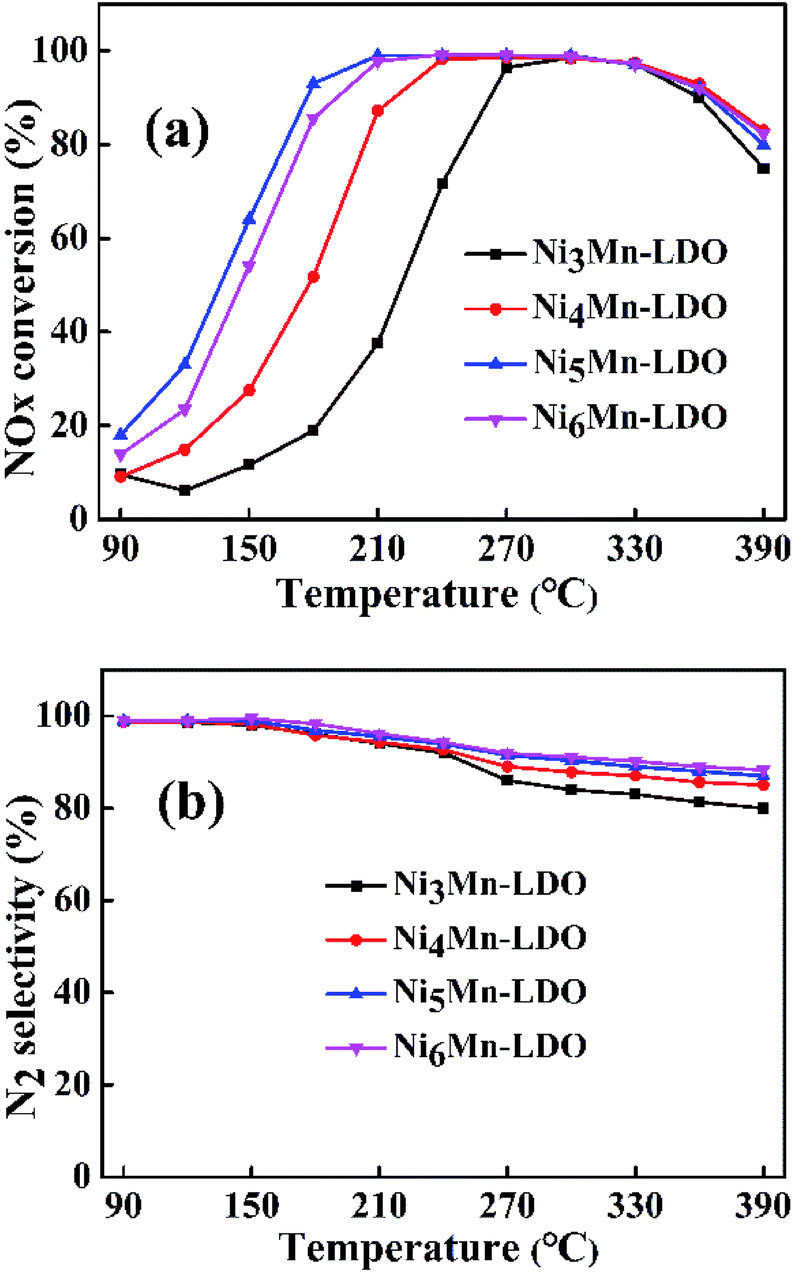
3.2.1 SCR performance of NixMn-LDO (x = 3, 4, 5, 6) catalysts. SCR activity and N2 selectivity of NixMn-LDO catalysts with different metal molar ratios (Ni/Mn = 3, 4, 5, 6) were tested under a GHSV of 45000 h−1. As illustrated in Fig. 5(a), the NOx conversion of all catalysts has experienced three stages at the test temperature range of 90–390 °C. At the first stage, the NOx conversion of NixMn-LDO catalysts increased sharply with increasing temperature and gradually reached the maximum of 98%. Followed by the second stage, the NOx conversion substantially remained at 98% without obvious changes. On the third stage, the NOx conversion presented a declining trend. Notably, it can be observed that the NOx conversion of NixMn-LDO catalysts were significantly influenced by Ni/Mn molar ratios on the first stage. When the Ni/Mn molar ratios rose from 3 to 5, the NOx conversion over NixMn-LDO catalysts displayed an upward trend, Ni5Mn-LDO catalyst afforded the best catalytic activity. However, when the Ni/Mn molar ratios was further increased, Ni6Mn-LDO exhibited lower activity than the Ni5Mn-LDO catalyst, which might be related to the further reduction of the Mn content. Thus, the SCR activity of different catalysts presented as the following order: Ni5Mn-LDO > Ni6Mn-LDO > Ni4Mn-LDO > Ni3Mn-LDO. In order to further understand the catalytic performance of the NixMn-LDO catalysts, the SCR activity comparison of present catalyst and the other Ni–Mn catalysts in literatures was involved in Table S1.† As the result presented that NixMn-LDO catalyst exhibited different activity temperature windows, which may be related to the structural composition of the LDHs precursors.
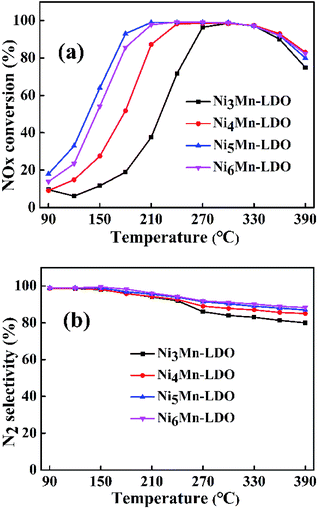 |
| | Fig. 5 (a) NOx conversion and (b) N2 selectivity of NixMn-LDO (x = 3, 4, 5, 6) catalysts. Reaction conditions: [NO] = [NH3] = 600 ppm, [O2] = 5 vol%, N2 as balance gas, GHSV = 45000 h−1. | |
The N2 selectivity of NixMn-LDO catalysts was also considered. As shown in Fig. 5(b), all catalysts exhibited similar N2 selectivity about 85% or higher over the entire temperature range. Whereas, with the temperature raising and exceeding 180 °C, the N2 selectivity of each catalyst decreased to varying degrees, in which Ni5Mn-LDO and Ni6Mn-LDO catalysts displayed better N2 selectivity than the other two. These results indicated that SCR performance of NixMn-LDO could be affected by the Ni/Mn molar ratios.
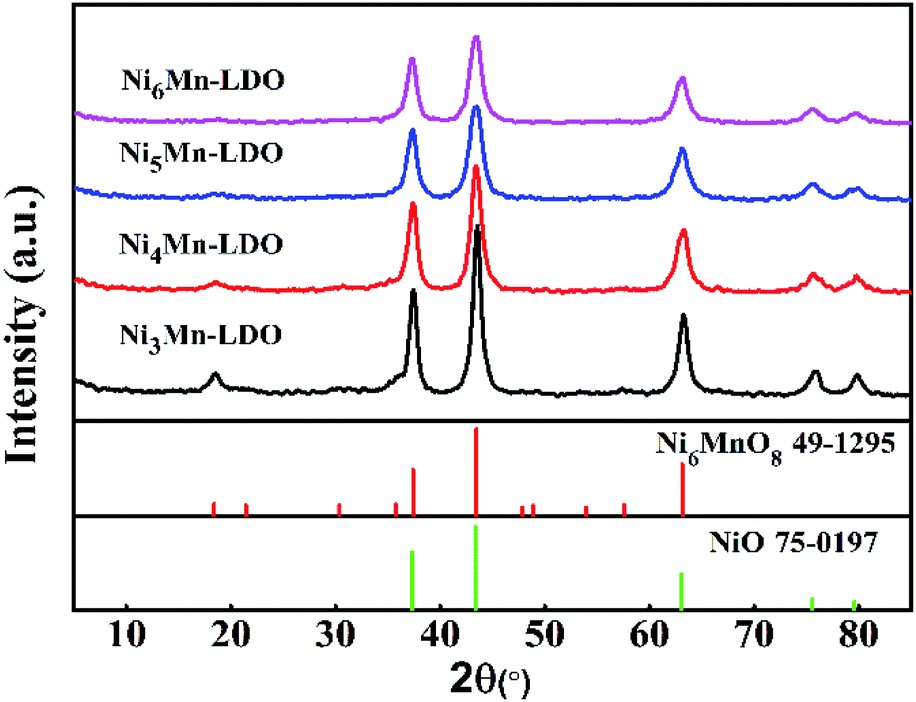
3.2.2 XRD analysis. Fig. 6 gave XRD patterns of NixMn-LDO catalysts. As can be seen from the diagram, all catalysts exhibited a similar XRD characteristic diffraction peaks. The peaks located at 18.7°, 37.3°, 43.4°, 63.1° can be attributed to Ni6MnO8 phase (JCPDS 49-1295).38 The appearance of Ni6MnO8 crystal phase can be related to the formation of Mn–Ni–O solid solution, which is mainly because that part of Mn3+ and Mn4+ can insert the lattice of NiO where the radius of Ni2+ (0.069 nm) is greater than Mn3+ (0.066 nm) and Mn4+ (0.056 nm).39 It was reported that the phase of Ni6MnO8 was conducive to the improvement of SCR activity.20 The peaks at 2θ of 37.3°, 43.4°, 63.1°, 75.5°, 79.7°can be assigned to NiO phase (JCPDS 75-0197),38,39 it was partially overlapped with those of Ni6MnO8 and can be further analyzed by TEM. Meanwhile, the absence of the diffraction peaks for pure MnOx may be due to that a part of Mn participated in the formation of Ni6MnO8 and remained Mn existed in an amorphous state. In addition, with the increase of Ni/Mn molar ratios for NixMn-LDO, diffraction peaks of samples were broadened and its intensity descended, indicating the gradual decreasing of crystal size. Generally speaking, small crystal can expose more crystal defects, which is more beneficial to improving catalytic activity.39 This well explained the difference in catalytic activity for catalysts to some extent.
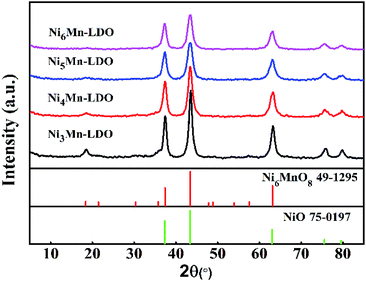 |
| | Fig. 6 XRD patterns of NixMn-LDO (x = 3, 4, 5, 6) catalysts. | |
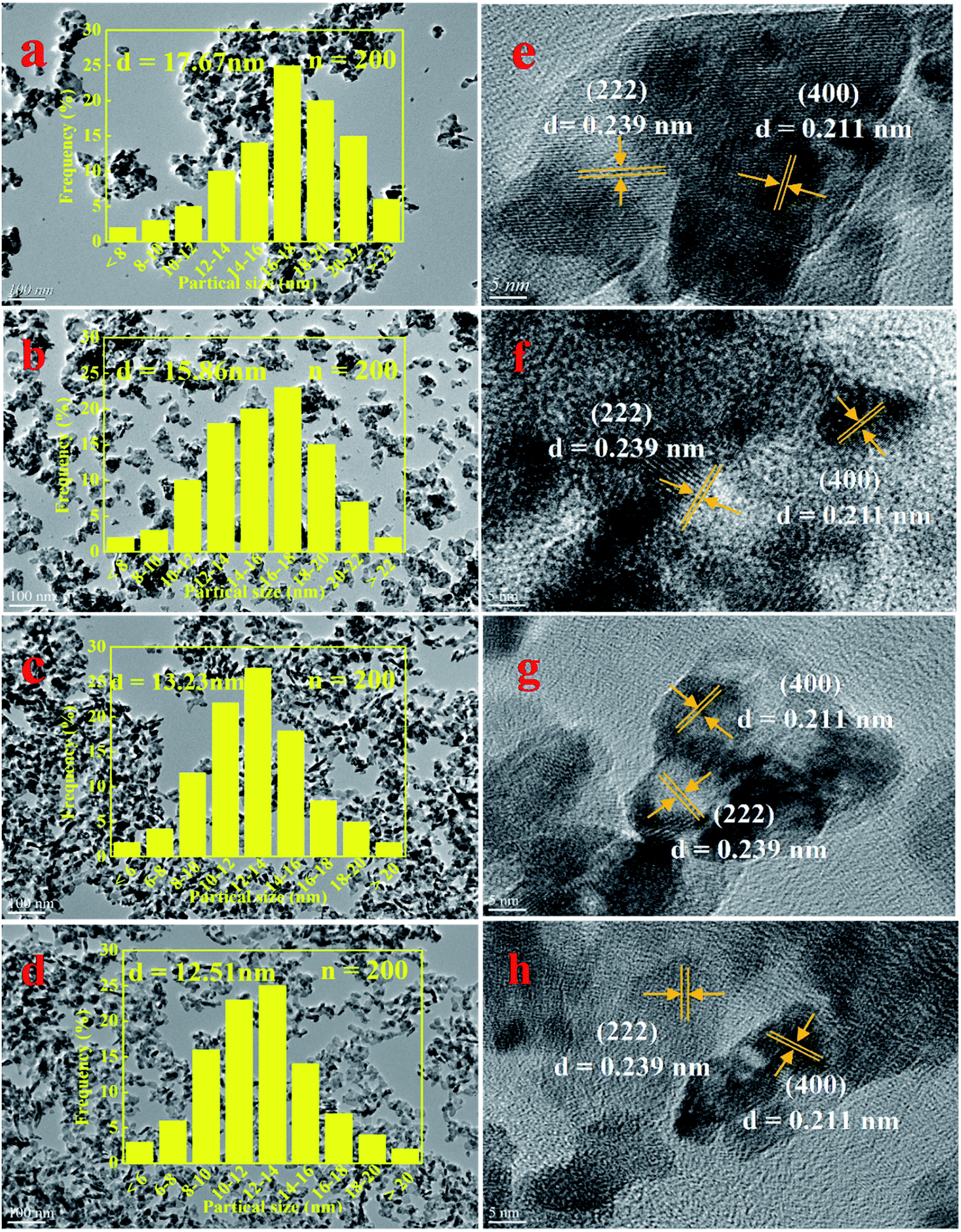
3.2.3 SEM, TEM and HRTEM analysis. The degree of exposure of catalyst surface active species is an important factor affecting catalytic activity.40 Therefore, the surface information of NixMn-LDO catalyst was investigated by SEM and TEM. Above all, the SEM images of four catalysts were shown in Fig. S1–S4.† After 400 °C calcination, all samples exhibited a spherical morphology and the sheet structure disappeared completely, which indicated that the LDHs laminate collapsed and a stable composite metal oxide formed. To further explore the surface element composition of the catalyst, the EDS mapping of NixMn-LDO catalysts was also conducted, as shown in Fig. S1–S4.† It can be seen that the O, Ni and Mn element was well dispersed on the surface of the catalyst. In addition, based on the relative content of Ni and Mn elements of each catalyst, surface Ni/Mn molar ratios of four samples was calculated. For Ni5Mn-LDO catalyst, it was noticed that its Ni/Mn molar ratios (4.68) was much lower than tested value (5.13) determined by ICP analysis. This result suggested that more Mn elements can be fully exposed on the catalyst surface and further enhancing SCR activity, which may be related to the uniform distribution of metal oxide particles in the catalyst.Then the TEM images of NixMn-LDO catalysts were shown in Fig. 7(a–d), it can be clearly observed that the active nanoparticles are anchoring on the catalyst surface, but there are some difference in dispersion with Ni/Mn molar ratios change of catalysts. As for Ni3Mn-LDO and Ni4Mn-LDO catalysts, a portion of active species particles have undergone significant agglomeration, with the corresponding average particle size being 17.67 nm and 15.68 nm, respectively. In contrast, the aggregation degree of active nanoparticles for Ni5Mn-LDO and Ni6Mn-LDO catalysts was improved significantly with the average particle size being 13.23 nm and 12.51 nm, respectively. Combined with the SEM analysis of the NixMn-LDHs samples, it can be concluded that the morphological difference of the precursors can seriously affect the exposure opportunities of catalyst surface active species. This result revealed that higher catalytic activity was achieved to Ni5Mn-LDO and Ni6Mn-LDO catalysts. In order to further investigate the phase composition of the catalyst, HRTEM analysis of NixMn-LDO was carried out in this part. As compiled in Fig. 7(e–h), all catalysts exhibited two different types of lattice fringes, the lattice spacings of 0.239 nm can be assigned to the (222) plane of NiO, and the lattice spacings of 0.211 nm correspond to Ni6MnO8 (400) plane,39 which is in good accordance with the XRD analysis results.
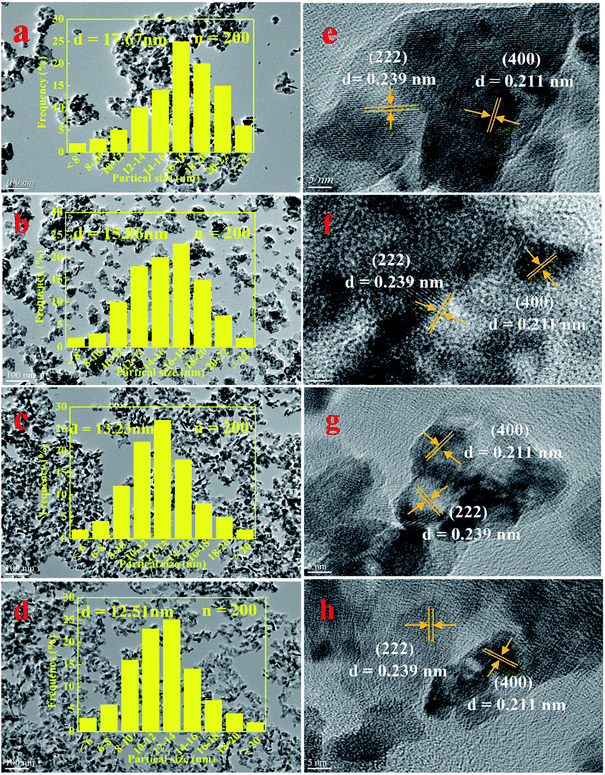 |
| | Fig. 7 (a–d) TEM and (e–h) HRTEM images of NixMn-LDO catalysts: (a, e) Ni3Mn-LDO; (b, f) Ni4Mn-LDO; (c, g) Ni5Mn-LDO; (d, h) Ni6Mn-LDO. | |
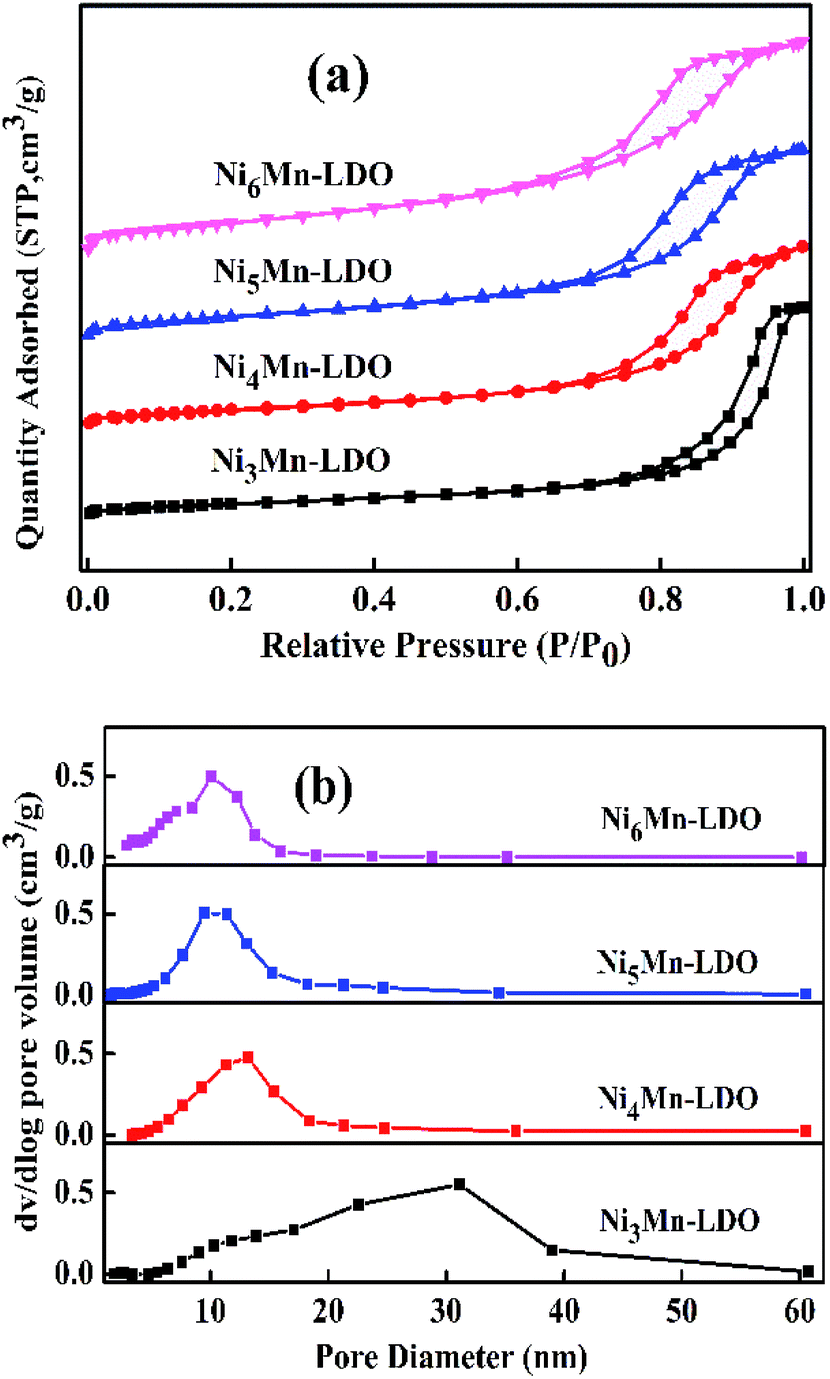
3.2.4 N2 adsorption–desorption analysis. The N2 adsorption–desorption characterization was used to test the surface area and pore structure distribution of the NixMn-LDO catalysts. Related results were shown in Fig. 8(a and b) and Table 2. As illustrated in Fig. 8(a), four catalysts exhibit the same type of desorption curves belonging to type IV isotherms with type H2 hysteresis loops, which fully indicated the existence of mesoporous structure.34,41,42 The corresponding pore diameter distribution was considered in Fig. 8(b). It can be observed that the sample of Ni3Mn-LDO has a wide pore size distribution with 10–40 nm. Comparatively, the pore size distribution range of NixMn-LDO (x = 4, 5, 6) was greatly reduced and more concentrated around 5–20 nm, which implies that the latter had a more uniform pore size structure.43 Moreover, the surface area was obtained in Table 2 by calculating with BET method. Obviously, the surface area value of samples was sensitive to the proportion of Ni/Mn and displayed an upward trend from 41 to 83 m2 g−1 with enhancement of Ni/Mn molar ratios. Combined with TEM analysis, thus may be related to the uniform distribution of the active species particles. Generally, a large specific surface area can provide maximum active and adsorption sites, which might be favorable to the improvement of catalytic activity.44 The Ni5Mn-LDO catalyst presented a large specific surface area (70 m2 g−1), suitable average pore diameter (9.35 nm) and pore volume (0.17 cm3 g−1), resulting in the desirable SCR activity.43,45
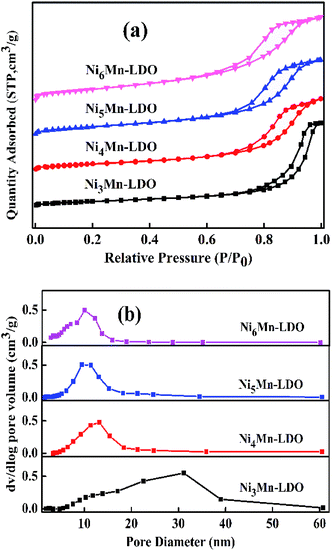 |
| | Fig. 8 (a) N2 adsorption–desorption isotherms and (b) pore diameter distribution of NixMn-LDO (x = 3, 4, 5, 6) catalysts. | |
Table 2 The information of surface area and pore structure distribution of NixMn-LDO (x = 3, 4, 5, 6) catalysts
| Samples |
SBET (m2 g−1) |
DBJH (nm) |
Vtotal (cm3 g−1) |
| Ni3Mn-LDO |
41 |
17.71 |
0.23 |
| Ni4Mn-LDO |
53 |
11.19 |
0.15 |
| Ni5Mn-LDO |
70 |
9.35 |
0.17 |
| Ni6Mn-LDO |
83 |
7.73 |
0.19 |
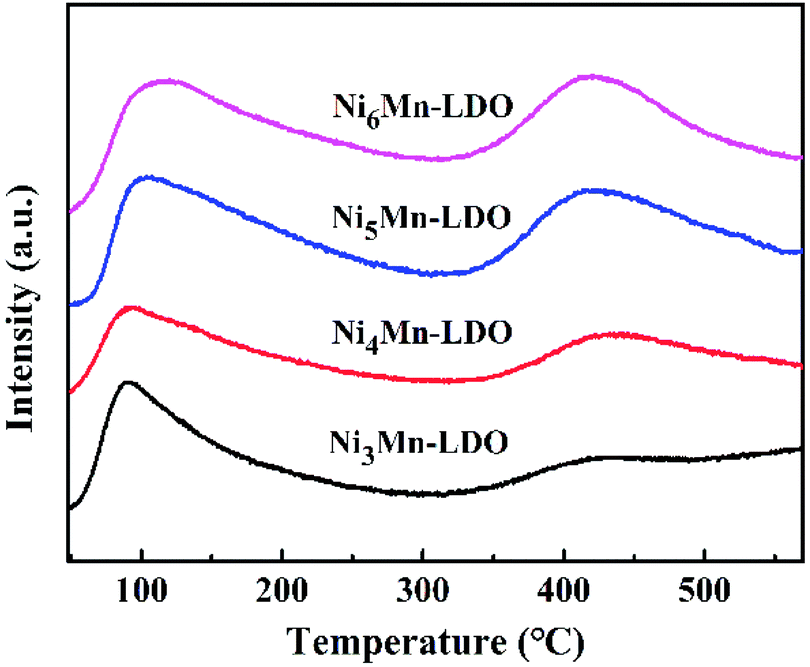
3.2.5 NH3-TPD analysis. In NH3-SCR reaction, the adsorption and activation of NH3 by catalyst is an important factor affecting the DeNOx action.41,46 Therefore, surface acidity of all catalysts was measured by NH3-TPD experiment (Fig. 9). As can be seen, the four catalysts exhibited two similar NH3 desorption peaks in the test temperature range with 50–560 °C. The peak at lower temperature with 50–300 °C was considered to be the desorption of weak acid sites on the catalyst surface, which can be attributed to NH4+ ions bound to the Brønsted acid sites.41,47,48 The desorption peak at high temperature with 350–550 °C was usually caused by coordinated NH3 molecular originating from the Lewis acid sites with stronger acidity, which might be because that the complex formed by the NH3 lone pair electron coordination possess stronger thermal stability than the NH4+ ions.49–52 Since the desorption peak positions of the four catalysts were similar, so the relative acid amount of catalysts were normalized by the peak area of the desorbed peak and shown in Table 3. Noticeably, the amount of acid of NixMn-LDO catalysts at the weak acid sites decreased and increased at strong acid sites gradually with the increase of Ni/Mn molar ratios, which suggest that the acid sites on the surface of catalyst are related to the relative distribution of Ni and Mn elements. In addition, Ni5Mn-LDO catalyst displayed more amount of acid than others, which implied that more NH3 can be adsorbed on the catalyst surface and further promote the low-temperature NH3-SCR reaction. What's more, the acid amount distribution of the four catalysts has a similar order to the size of the metal oxide particles. This results suggest that the uniform distribution of the active species particles is an crucial factor on the full exposure of the acid sites on the catalyst surface.
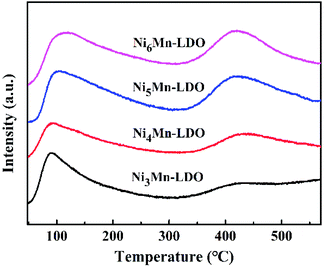 |
| | Fig. 9 NH3-TPD curves of NixMn-LDO (x = 3, 4, 5, 6) catalysts. | |
Table 3 Relative amount and proportion of NH3 desorption for NixMn-LDO (x = 3, 4, 5, 6) catalysts
| Samples |
Total amount of NH3 desorption |
Proportion of NH3 desorption (%) |
| Weak acid site (50–300 °C) |
Strong acid site (350–550 °C) |
| Ni3Mn-LDO |
1.00 |
88.4 |
11.6 |
| Ni4Mn-LDO |
1.08 |
67.4 |
32.6 |
| Ni5Mn-LDO |
2.10 |
62.2 |
37.8 |
| Ni6Mn-LDO |
1.91 |
59.6 |
40.4 |
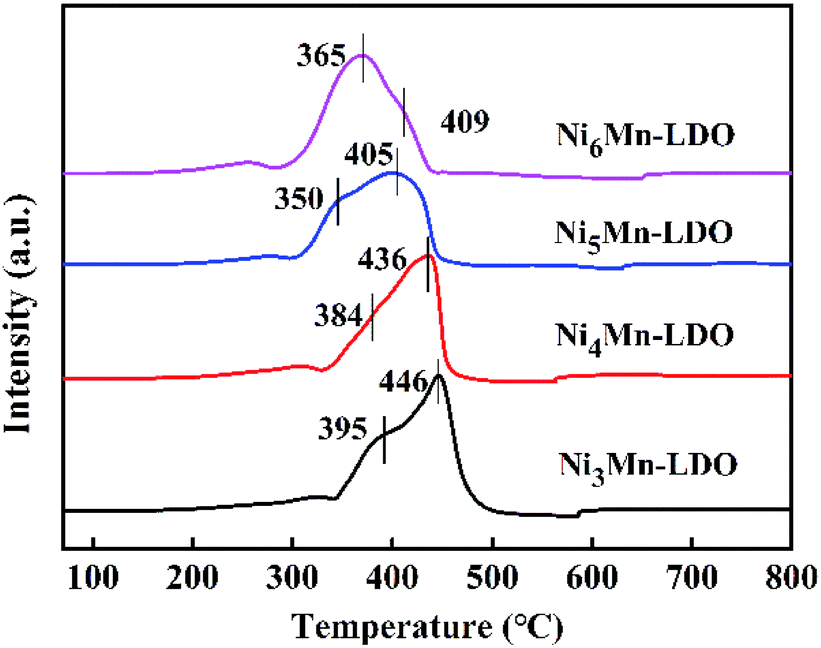
3.2.6 H2-TPR analysis. The redox capability of the catalysts is another indispensable indicator for evaluating catalytic activity in the NH3-SCR reaction. The H2-TPR results of four catalysts was demonstrated in Fig. 10. There are two reduction peaks on the curves, the first peak appears in the temperature range of 350–395 °C, corresponding to the process: Ni6MnO8 + H2 → MnO–NiO + H2O, which represents the reduction from MnOx species to MnO; the second step of the reduction takes place at 405–446 °C and can be attributed to: MnO–NiO + H2 → Ni + MnO + H2O, indicating the transformation of NiO species to Ni.20,39,53,54 Likewise, it can be observed that the positions of the reduction peaks of four catalysts have changed significantly. Obviously, with the increase of Ni/Mn molar ratios, the peak position of NixMn-LDO catalysts shifted to the lower temperature, which suggests that the reduction process is becoming easier. Especially, Ni5Mn-LDO catalyst shows the lowest reduction peak temperature and correspondingly the highest catalytic activity. The reason may be related to the uniform dispersion of the reducible species on the surface of catalyst, which can be confirmed by BET and TEM analysis results.
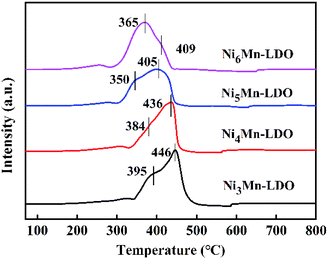 |
| | Fig. 10 H2-TPR curves of NixMn-LDO (x = 3, 4, 5, 6) catalysts. | |
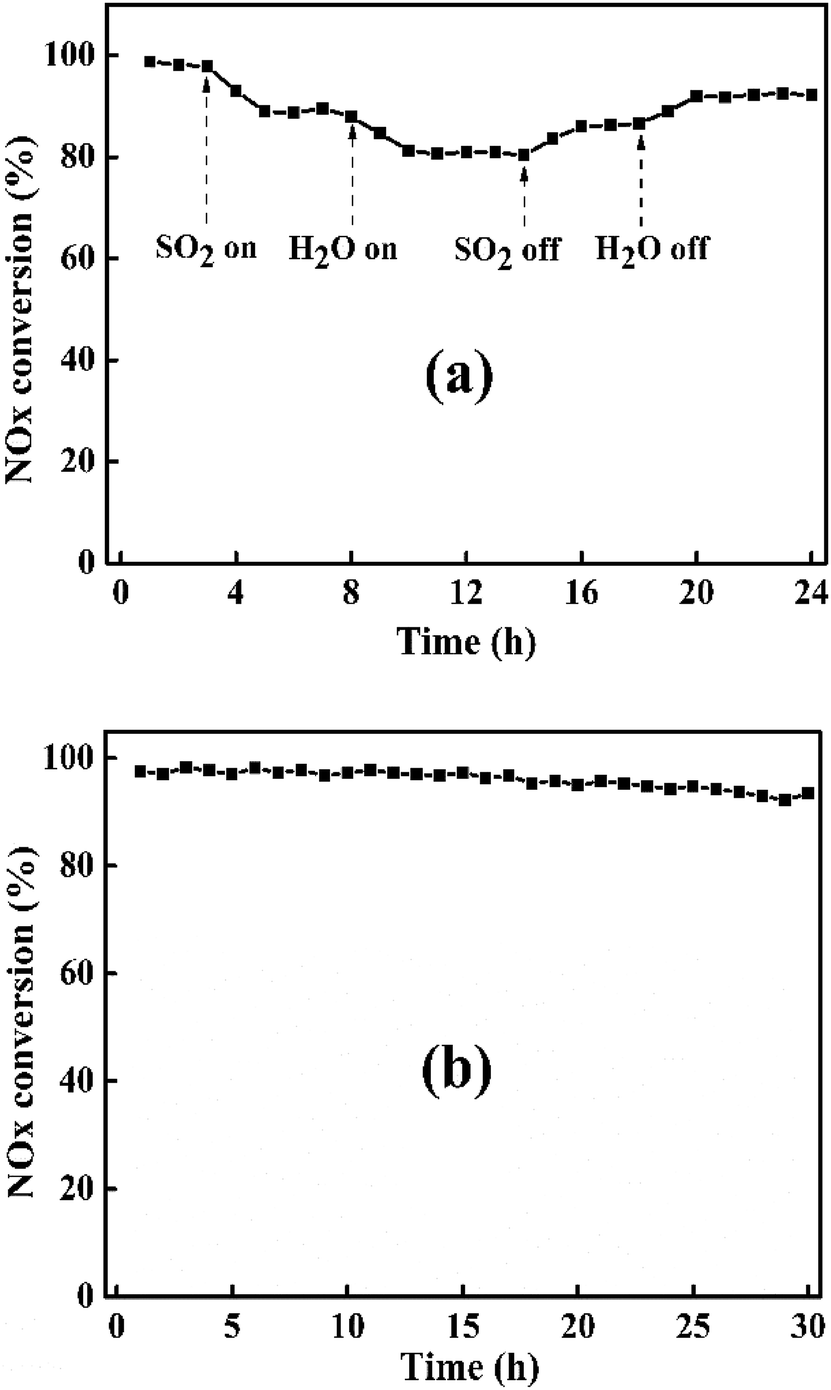
3.2.7 SO2 & H2O resistance and stability tests. In actual NH3-SCR reaction conditions, the flue gas inevitably contains trace amount of SO2 and H2O, which can greatly affect the DeNOx performance of catalyst at low temperature.55 So, it is necessary to study the SO2 & H2O resistance of catalyst. Fig. 11(a) illustrates SO2 & H2O resistance tests of Ni5Mn-LDO catalyst at 240 °C under a GHSV of 45000 h−1. When SO2 was introduced into the mixture gases, NOx conversion of catalyst started to decrease from the original 98% and gradually tended to a smooth value of 88%. And then adding 10 vol% H2O into reaction system, where SO2 and H2O coexisted, DeNOx performance of this catalyst was further suppressed. Upon removing SO2, NOx conversion has risen and remained at around 86%. After further closing H2O, NOx conversion can be restored to 92%, but still slightly lower than the initial value, which may be due to the deposition of ammonium sulfate on the surface of the catalyst. In general, Ni5Mn-LDO catalyst presented considerable SO2 & H2O resistance.
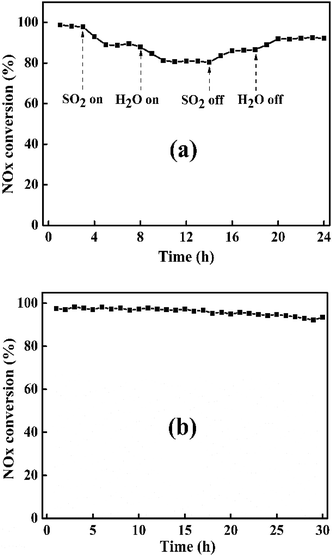 |
| | Fig. 11 (a) SO2 & H2O resistance test and (b) stability test of Ni5Mn-LDO catalyst at 240 °C. Reaction conditions: [NO] = [NH3] = 600 ppm, [O2] = 5 vol%, SO2 = 100 ppm (when used), 10 vol% H2O (when used), N2 as balance gas, GHSV = 45000 h−1. | |
It is also an important challenge for the catalysts to maintain high DeNOx efficiency for a long time in the NH3-SCR reaction. The stability test of Ni5Mn-LDO catalyst was depicted in Fig. 11(b). As can be seen, NOx conversion has only a slight decrease with the extension of the test time throughout the test range, which suggest that the catalyst has good stability.
4. Conclusions
A series of NixMn-LDO (x = 3, 4, 5, 6) metal oxides were obtained by calcining NixMn-LDHs (x = 3, 4, 5, 6) precursors at 400 °C and were employed as NH3-SCR catalysts. The catalytic results presented that all catalysts exhibited good SCR activity and N2 selectivity, which might be attributed to the formation of Ni6MnO8 active species. In addition, catalytic performance of NixMn-LDO catalysts were sensitive to the Ni/Mn molar ratios, which were related to the redox capacity and surface acidity caused by dispersion. In fact, it was demonstrated that the distribution of active species particles on the surface of catalyst could be improved by optimizing the morphology of the NixMn-LDHs precursors. This study provides a new method to prepare catalyst with good dispersion properties by calcining LDHs precursors.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The project was supported by National Natural Science Foundation of the China (No. 21073131), Natural Science Foundation of Shanxi Province, China (No. 201601D102007).
Notes and references
- K. Liu, H. He, Y. B. Yu, Z. D. Yan, W. W. Yang and W. P. Shan, J. Catal., 2019, 369, 372–381 CrossRef CAS.
- D. Damma, P. R. Ettireddy, B. M. Reddy and P. G. Smirniotis, Catalysts, 2019, 9, 349 CrossRef CAS.
- P. Forzatti, I. Nova and E. Tronconi, Angew. Chem., Int. Ed., 2009, 121, 8516–8518 CrossRef.
- Z. B. Zhi, X. Ning, F. Zhou, B. Yang, Y. W. Tu, J. Jin, W. Lu and Z. H. Liu, RSC Adv., 2018, 8, 21915–21925 RSC.
- Z. H. Chen, Q. Yang, H. Li, X. H. Li, L. F. Wang and S. C. Tsang, J. Catal., 2010, 276, 56–65 CrossRef CAS.
- R. H. Gao, D. S. Zhang, X. G. Liu, L. Y. Shi, P. Maitarad, H. R. Li, J. P. Zhang and W. G. Cao, Catal. Sci. Technol., 2013, 3, 191–199 RSC.
- F. Kapteijn, L. Singoredjo, A. Andreini and J. A. Moulijn, Appl. Catal., B, 1994, 3, 173–189 CrossRef CAS.
- X. L. Tang, J. M. Hao, W. G. Xu and J. H. Li, Catal. Commun., 2007, 8, 329–334 CrossRef CAS.
- X. F. Tang, J. H. Li, L. Sun and J. M. Hao, Appl. Catal., B, 2010, 99, 156–162 CrossRef CAS.
- J. H. Li, H. Z. Chang, L. Ma, J. M. Hao and R. T. Yang, Catal. Today, 2011, 175, 147–156 CrossRef CAS.
- C. Liu, J. W. Shi, C. Gao and C. M. Niu, Appl. Catal., A, 2016, 522, 54–69 CrossRef CAS.
- M. Casapu, O. Kröcher and M. Elsener, Appl. Catal., B, 2009, 88, 413–419 CrossRef CAS.
- M. Kang, E. D. Park, J. M. Kim and J. E. Yie, Catal. Today, 2006, 111, 236–241 CrossRef CAS.
- Z. H. Chen, X. H. Li, X. Gao, Y. B. Jiang, Y. X. Lü, F. R. Wang and L. F. Wang, J. Catal., 2009, 30, 4–6 CrossRef.
- Z. H. Lian, F. D. Liu, H. He, X. Y. Shi, J. S. Mo and Z. B. Wu, Chem. Eng. J., 2014, 250, 390–398 CrossRef CAS.
- Y. Wan, W. R. Zhao, Y. Tang, L. Li, H. J. Wang, Y. L. Cui, J. L. Gu, Y. S. Li and J. L. Shi, Appl. Catal., B, 2014, 148, 114–122 CrossRef.
- L. Q. Chen, X. Y. Niu, Z. B. Li, Y. L. Dong, Z. P. Zhang, F. L. Yuan and Y. J. Zhu, Catal. Commun., 2016, 85, 48–51 CrossRef CAS.
- H. R. Li, D. S. Zhang, P. P. Maitarad, L. Y. Shi, R. H. Gao, J. P. Zhang and W. G. Cao, Chem. Commun., 2012, 48, 10645–10647 RSC.
- Y. L. Han, J. C. Mu, X. Y. Li, J. S. Gao, S. Y. Fan, F. Tan and Q. D. Zhao, Chem. Commun., 2018, 54, 9797–9800 RSC.
- S. X. Cai, D. S. Zhang, L. Y. Shi, J. Xu, L. Zhang, L. Huang, H. R. Li and J. P. Zhang, Nanoscale, 2014, 6, 7346 RSC.
- J. Xiong, Q. Q. Wu, X. L. Mei, J. Liu, Y. C. Wei, Z. Zhao, D. Wu and J. M. Li, ACS Catal., 2018, 8, 7915–7930 CrossRef CAS.
- Y. F. Zhao, S. He, M. Wei, D. G. Evans and X. Duan, Chem. Commun., 2010, 46, 3031–3033 RSC.
- H. Yan, M. Wei, J. Ma, F. Li, D. G. Evans and X. Duan, J. Phys. Chem. A, 2009, 113, 6133–6141 CrossRef CAS PubMed.
- Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- X. Wu, R. N. Wang, Y. L. Du, C. L. Zhou, H. Meng and X. M. Xie, Mol. Catal., 2019, 467, 150–160 CrossRef CAS.
- W. Y. Zhou, Q. Y. Tao, F. Sun, X. B. Cao, J. F. Qian, J. Xu, M. Y. He, Q. Chen and J. L. Xiao, J. Catal., 2018, 361, 1–11 CrossRef CAS.
- J. W. Zhao, J. L. Chen, S. M. Xu, M. F. Shao, Q. Zhang, F. Wei, J. Ma, M. Wei, D. G. Evans and X. Duan, Adv. Funct. Mater., 2014, 24, 2938–2946 CrossRef CAS.
- X. J. Li, M. Y. Xin, S. Guo, T. H. Cai, D. F. Du, W. Xing, L. M. Zhao, W. Y. Guo, Q. Z. Xue and Z. F. Yan, Electrochim. Acta, 2017, 253, 302–310 CrossRef CAS.
- W. Ma, R. Z. Ma, J. H. Wu, P. Z. Sun, X. H. Liu, K. C. Zhou and T. Sasaki, Nanoscale, 2016, 8, 10425–10432 RSC.
- S. Miyata, Clays Clay Miner., 1975, 23, 369–375 CrossRef CAS.
- Q. H. Yan, S. N. Chen, L. Qiu, Y. S. Gao, D. O'Hare and Q. Wang, Dalton Trans., 2017, 47, 2992–3004 RSC.
- R. N. Wang, X. Wu, C. L. Zhou, X. J. Li and Y. L. Du, Catalysts, 2018, 8, 384 CrossRef.
- Q. H. Yan, S. N. Chen, C. Zhang, D. O'Hare and Q. Wang, J. Colloid Interface Sci., 2018, 526, 63–74 CrossRef CAS PubMed.
- S. P. Mo, S. D. Li, W. H. Li, J. Q. Li, J. Y. Chen and Y. F. Chen, J. Mater. Chem. A, 2016, 4, 8113–8122 RSC.
- Z. P. Xu, L. Li, C. Y. Cheng, R. G. Ding and C. H. Zhou, Appl. Clay Sci., 2013, 74, 102–108 CrossRef CAS.
- X. Xiang, H. I. Hima, H. Wang and F. Li, Chem. Mater., 2007, 20, 1173–1182 CrossRef.
- X. Wu, Y. L. Feng, Y. L. Du, X. Z. Liu, C. L. Zou and Z. Li, Appl. Surf. Sci., 2019, 467, 802–810 CrossRef.
- J. J. Zhang, R. R. Hu, P. Dai, Z. M. Bai, X. X. Yu, M. Z. Wu and G. Li, J. Mater. Sci.: Mater. Electron., 2018, 29, 7510–7518 CrossRef CAS.
- Y. G. Zhang, Z. F. Qin, G. F. Wang, H. Q. Zhu, M. Dong, S. N. Li, Z. W. Wu, Z. K. Li, Z. H. Wu, J. Zhang, T. D. Hu, W. B. Fan and J. G. Wang, Appl. Catal., B, 2013, 129, 172–181 CrossRef CAS.
- H. Meng, J. N. Liu, Y. L. Du, B. H. Hou, X. Wu and X. M. Xie, Appl. Catal., A, 2019, 119, 101–105 CAS.
- L. Zhang, L. Y. Shi, L. Huang, J. P. Zhang, R. H. Gao and D. S. Zhang, ACS Catal., 2014, 4, 1753–1763 CrossRef CAS.
- Q. Liu, J. J. Gao, F. N. Gu, X. P. Lu, Y. J. Liu, H. F. Li, Z. Y. Zhong, B. Liu, G. W. Xu and F. B. Su, J. Catal., 2015, 326, 127–138 CrossRef CAS.
- X. Wu, X. J. Li, Y. L. Du, R. N. Wang, X. M. Guo and B. H. Hou, ChemCatChem, 2019, 11, 1993–2003 CrossRef CAS.
- S. Ali, L. Q. Chen, Z. B. Li, T. R. Zhang, R. Li, S. U. H. Bakhtiar, X. S. Leng, F. L. Yuan, X. Y. Niu and Y. J. Zhu, Appl. Catal., B, 2018, 236, 25–35 CrossRef CAS.
- Y. Li, G. Q. Li, Y. Lu, W. X. Hao, Z. Wei, J. Liu and Y. F. Zhang, Mol. Catal., 2018, 445, 21–28 CrossRef CAS.
- L. Lietti, I. Nova, G. Ramis, L. D. Acqua, G. Busca, E. Giamello, P. Forzatti and F. Bregani, J. Catal., 1999, 187, 419–435 CrossRef CAS.
- Z. B. Wu, B. Q. Jiang, Y. Liu, H. Q. Wang and R. B. Jin, Environ. Sci. Technol., 2007, 41, 5812–5817 CrossRef CAS PubMed.
- C. Fang, D. S. Zhang, L. Y. Shi, R. H. Gao, H. R. Li, L. P. Ye and J. P. Zhang, Catal. Sci. Technol., 2013, 3, 803–811 RSC.
- S. Roy, B. Viswanath, M. S. Hegde and G. Madras, J. Phys. Chem. C, 2008, 112, 6002–6012 CrossRef CAS.
- Y. Peng, W. W. Yu, W. K. Su, X. Huang and J. H. Li, Catal. Today, 2015, 242, 300–307 CrossRef CAS.
- M. Mhamdi, S. Khaddar-Zine and A. Ghorbel, Appl. Catal., A, 2009, 357, 42–50 CrossRef CAS.
- L. Zhang, D. S. Zhang, J. P. Zhang, S. X. Cai, C. Fang, L. Huang, H. R. Li, R. H. Gao and L. Y. Shi, Nanoscale, 2013, 5, 9821–9829 RSC.
- L. Christel, A. Pierre and D. A.-M. R. Abel, Thermochim. Acta, 1997, 306, 51–59 CrossRef CAS.
- C. Laberty, J. Pielaszek, P. Alphonse and A. Rousset, Solid State Ionics, 1998, 110, 293–302 CrossRef CAS.
- K. Skalska, J. S. Miller and S. Ledakowicz, Sci. Total Environ., 2010, 408, 3976–3989 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04578c |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Xianmei Xie*a
*a and
Xianmei Xie*a