DOI:
10.1039/C9RA04496E
(Paper)
RSC Adv., 2019,
9, 25533-25543
Synthesis of novel benzopyran-connected pyrimidine and pyrazole derivatives via a green method using Cu(II)-tyrosinase enzyme catalyst as potential larvicidal, antifeedant activities
Received
15th June 2019
, Accepted 18th July 2019
First published on 15th August 2019
Abstract
A series of benzopyran-connected pyrimidine (1a–g) and benzopyran-connected pyrazole (2a–i) derivatives were synthesized via Biginelli reaction using a green chemistry approach. Cu(II)-tyrosinase was used as a catalyst in the synthesis of compounds 1a–g and 2a–i via the Biginelli reaction. The as-synthesized compounds were characterized by IR, 1H NMR, 13C NMR, mass spectroscopy, and elemental analysis. The as-synthesized compounds were screened for larvicidal and antifeedant activities. The larvicidal activity was evaluated using the mosquito species Culex quinquefasciatus, and the antifeedant activity was evaluated using the fishes of Oreochromis mossambicus. The compounds 2a–i demonstrated lethal effects, killing 50% of second instar mosquito larvae when their LD50 values were 44.17, 34.96, 45.29, 45.28, 75.96, and 28.99 μg mL−1, respectively. Molecular docking studies were used for analysis based on the binding ability of an odorant binding protein (OBP) of Culex quinquefasciatus with compound 2h (binding energy = −6.12 kcal mol−1) and compound 1g (binding energy = −5.79 kcal mol−1). Therefore, the proposed target compounds were synthesized via a green method using Cu(II)-enzyme as a catalyst to give high yield (94%). In biological screening, benzopyran-connected pyrazole (2h) was highly active compared with benzopyran-connected pyrimidine (1a–g) series in terms of larivicidal activity.
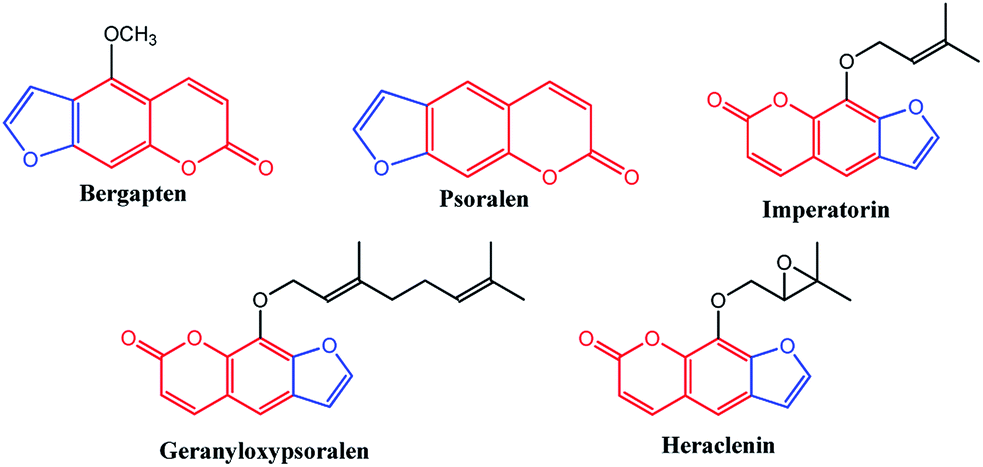
Benzopyrans (coumarins) are an important group of naturally occurring compounds widely distributed in the plant kingdom and have been produced synthetically for many years for commercial uses.1 In addition, these core compounds are used as fragrant additives in food and cosmetics.2 The commercial applications of coumarins include dispersed fluorescent brightening agents and as dyes for tuning lasers.3 Some important biologically active natural benzopyran (coumarin) derivatives are shown in Fig. 1. Mosquitoes are the vectors for a large number of human pathogens compared to other groups of arthropods.4 Their uncontrollable breeding poses a serious threat to the modern humanity. Every year, more than 500 million people are severely affected by malaria. The mosquito larvicide is an insecticide that is specially targeted against the larval life stage of a mosquito. Particularly, the compound bergapten (Fig. 1), which shows the standard of larivicidal activity,5 is commercially available, and it was used as a control in this study for larvicidal screening. Moreover, the antifeedant screening defense mechanism makes it a potential candidate for the development of eco-friendly ichthyocides. Coumarin derivatives exhibit a remarkably broad spectrum of biological activities, including antibacterial,6,7 antifungal,8–10 anticoagulant,11 anti-inflammatory,12 antitumor,13,14 and anti-HIV.15
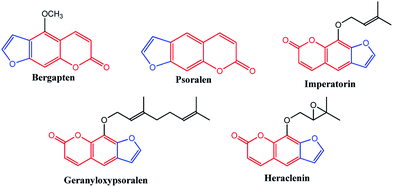 |
| | Fig. 1 Biologically active natural benzopyran compound. | |
Coumarin and its derivatives can be synthesized by various methods, which include the Perkin,16 Knoevenagel,17 Wittig,18 Pechmann,19 and Reformatsky reactions.
Among these reactions, the Pechmann reaction is the most widely used method for the preparation of substituted coumarins since it proceeds from very simple starting materials and gives good yields of variously substituted coumarins. For example, coumarins can be prepared by using various reagents, such as H2SO4, POCl3,20 AlCl3,21 cation exchange resins, trifluoroacetic acid,22 montmorillonite clay,23 solid acid catalysts,24 W/ZrO2 solid acid catalyst,25 chloroaluminate ionic liquid,26 and Nafion-H catalyst.27
Keeping the above literature observations, coumarin derivatives 1a–g and 2a–i are usually prepared with the conventional method involving CuCl2·2H2O catalysis with using HCl additive. This reduces the yield and also increases the reaction time. To overcome this drawback, we used mushroom tyrosinase as a catalyst without any additive, a reaction condition not reported previously. The as-synthesized compounds were used for the biological screening of larvicidal and antifeedant activities (marine fish). In addition, in this study, we considered the molecular docking studies study based on previous studies for performing the binding ability of hydroxy-2-methyl-4H-pyran-4-one (the root extract of Senecio laetus Edgew) with the odorant binding protein (OBP) of Culex quinquefasciatus.28
Results and discussion
Chemistry
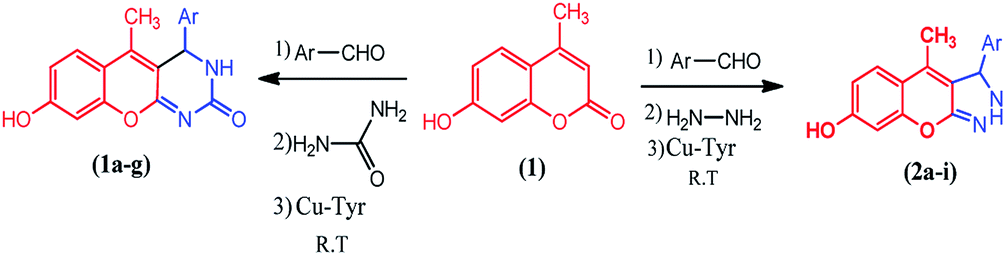
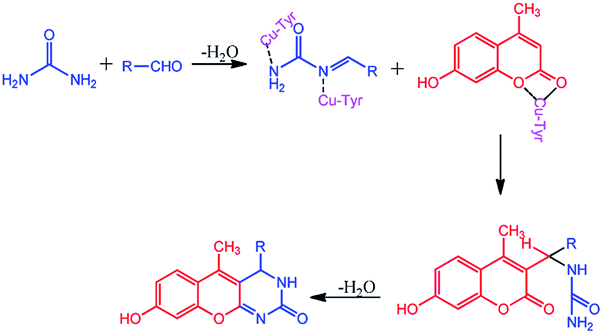
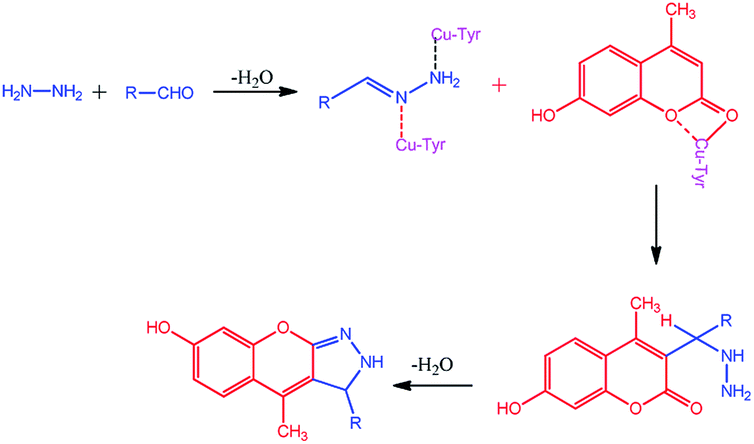
The compounds 1a–g and 2a–i were prepared according to the synthesis sequences illustrated in Scheme 1. The proposed mechanism for the formation of the compounds 1a–g and 2a–i are shown in Schemes 2 and 3. The Cu-tyrosinase enzyme catalysis performed well to afford yields ranging from 82 to 98% within 2 to 4 min of reaction time compared with other catalysts. The catalyst optimization is shown in Table 1, and the optimization of reaction conditions is shown in Table 2.
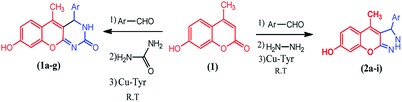 |
| | Scheme 1 Synthesis of coumarin derivatives 1a–g and 2a–i. | |
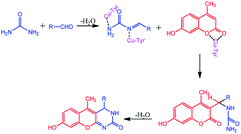 |
| | Scheme 2 Proposed mechanism for the formation of 1a–g. | |
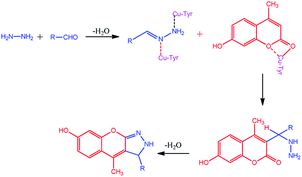 |
| | Scheme 3 Proposed mechanism for the formation of 2a–i. | |
Table 1 Optimization of the catalyst for compounds (1a)
| Entry |
Catalyst |
Additive |
Yield (%) |
| 1 |
ZnCl2 |
HCl |
47 |
| 2 |
SnCl2·2H2O |
HCl |
53 |
| 3 |
ZrOCl2 |
HCl |
68 |
| 4 |
AlCl3 |
HCl |
76 |
| 5 |
CuCl2·2H2O |
HCl |
87 |
| 6 |
Cu-tyrosinase enzyme catalysis |
Without additive |
94 |
Table 2 Optimization of the reaction conditions for compounds 1a–g and 2a–i
| Compd no. |
Ar |
Catalysis activity, time (min)/yield (%) |
| CuCl2 |
Cu-tyrosinase enzyme catalysis |
| 1a |
-Ph |
5/87 |
2/94 |
| 1b |
–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–Ph CH–Ph |
5/78 |
3/88 |
| 1c |
-4-ClC6H4 |
5/83 |
4/87 |
| 1d |
-4-OHC6H4 |
5/87 |
2/89 |
| 1e |
-3-No2C6H4 |
5/75 |
2/85 |
| 1f |
4-OCH3C6H4 |
5/80 |
2/89 |
| 1g |
Furfuryl |
5/83 |
2/89 |
| 2a |
-Ph |
5/72 |
2/82 |
| 2b |
–CHCH–Ph |
5/87 |
2/89 |
| 2c |
-4-ClC6H4 |
5/89 |
2/91 |
| 2d |
-4-OHC6H4 |
5/82 |
2/92 |
| 2e |
-3-No2C6H4 |
5/85 |
2/95 |
| 2f |
-N(CH3)2C6H4 |
5/89 |
2/96 |
| 2g |
4-OCH3C6H4 |
5/71 |
2/98 |
| 2h |
Furfuryl |
5/83 |
3/89 |
| 2i |
Citral |
5/79 |
4/86 |
All the as-synthesized compounds were characterized by IR, 1H NMR, and 13C NMR analyses. Important assignments of the compounds 1a–g using the IR spectroscopy include absorption bands at 3174.54–3175.59, 2596.13–2596.89, and 1648.12–1715.02 cm−1, corresponding to the –NH, –CN and –CO. The 1H NMR spectrum shows signals at δ 9.19–9.27, 3.97–4.17, and 1.21–1.29 ppm, corresponding to the NH, 4CH protons, and CH3. The 13C NMR spectrum shows peaks at δ 160.4, 160.4, and 15.0–15.7 ppm, corresponding to the CO, CN, and CH3 carbon atoms. The important assignments of the compounds 2a–i using the IR spectroscopy include absorption bands at 3174.54, 3034, and 2596.13 cm−1, corresponding to the –NH, Ar–H and –CN. The 1H NMR spectrum shows signals at δ 8.70, 3.99–4.59, and 2.42 ppm, corresponding to the NH, 4CH protons, and CH3. The 13C NMR spectrum shows peaks at δ 150.6, 122.3–123.3, and 15.0–15.7 ppm, corresponding to the CN, CC, and CH3 carbon atoms. All compounds were confirmed via mass spectroscopy and elemental analysis supported the expected target compounds.
Larvicidal activity
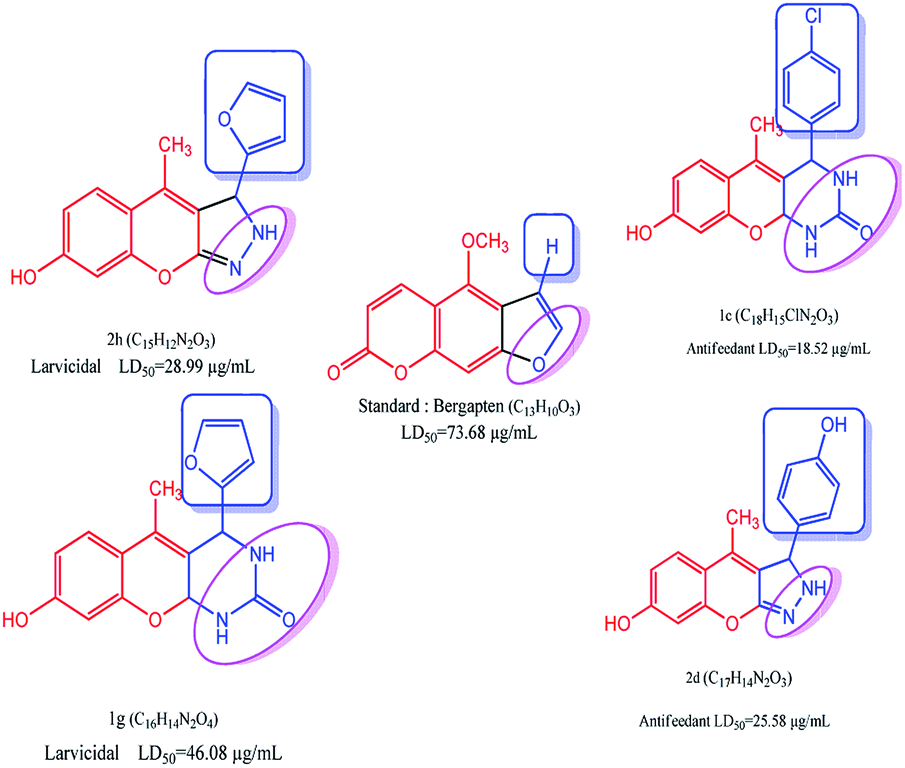
Compounds 1a–g and 2a–i were screened for larvicidal activity. Compounds 1a–g had low activity compared with compounds 2a–i. The experiments were performed at room temperature. The compound 2h produced 100% mortality at 100 μg mL−1. The compound 1g was the only highly active compound with the LD50 value of 46.08 μg mL−1 in the 1a–g series. Overall, compounds 1g, 2a, 2b, 2d, 2e and 2h were highly active against Culex quinquefasciatus with the LD50 values of 46.08, 44.17, 34.96, 45.29, 45.28, and 28.99 μg mL−1, respectively, than the control bergapten with the LD50 value of 73.68 μg mL−1. Among the as-synthesized compounds 1a–g and 2a–i, compound 2h was highly active against Culex quinquefasciatus with the LD50 value of 28.99 μg mL−1 compared with the control bergapten (the LD50 value of 73.68 μg mL−1). The compounds 1g, 2a, 2d, and 2e were equipotently active (range from 44.17 to 46.08 μg mL−1) due to the nature of chemical performance; however, some important functional groups displayed the biological activity in different ways. For example, compound 1g containing pyrimidine with furfuryl gave equipotent activity compared with compound 2a (electron donating group phenyl with pyrazole), compound 2d (electron donating group 4-HO-phenyl with pyrazole), and compound 2e (electron withdrawing group 3-NO2-Ph with pyrazole). The values are summarized in Table 3.
Table 3 Larvicidal activity of title compounds 1a–g and 2a–i
| Compounds |
Concentration (μg mL−1)/mortalitya (%) |
LD50 (μg mL−1) |
| 10 |
25 |
50 |
100 |
| Values are the means of three replicates ± SD. |
| 1a |
— |
0 ± 0.00 |
20 ± 0.07 |
43 ± 0.87 |
>100 |
| 1b |
— |
0 ± 0.00 |
14 ± 0.96 |
33 ± 1.65 |
>100 |
| 1c |
— |
— |
— |
— |
>100 |
| 1d |
— |
— |
0 ± 0.00 |
20 ± 1.61 |
>100 |
| 1e |
— |
— |
0 ± 0.00 |
20 ± 0.88 |
>100 |
| 1f |
— |
— |
— |
— |
>100 |
| 1g |
18 ± 1.87 |
40 ± 1.21 |
60 ± 0.56 |
80 ± 1.54 |
46.08 |
| 2a |
18 ± 1.20 |
40 ± 1.87 |
60 ± 0.67 |
84 ± 0.00 |
44.17 |
| 2b |
16 ± 1.87 |
40 ± 1.61 |
60 ± 1.67 |
80 ± 0.09 |
34.96 |
| 2c |
— |
— |
0 ± 0.00 |
20 ± 1.14 |
>100 |
| 2d |
18 ± 0.95 |
42 ± 1.38 |
60 ± 1.42 |
80 ± 1.23 |
45.29 |
| 2e |
20 ± 1.76 |
40 ± 1.23 |
62 ± 1.94 |
80 ± 1.11 |
45.28 |
| 2f |
0 ± 0.00 |
20 ± 0.67 |
44 ± 1.47 |
60 ± 2.12 |
75.96 |
| 2g |
— |
0 ± 0.00 |
8 ± 1.63 |
20 ± 1.42 |
>100 |
| 2h |
46 ± 1.33 |
62 ± 1.34 |
84 ± 1.35 |
100 ± 0.00 |
28.99 |
| 2i |
— |
0 ± 0.00 |
10 ± 1.34 |
20 ± 1.34 |
>100 |
| Bergapten |
11 ± 2.05 |
25 ± 1.90 |
41 ± 1.65 |
63 ± 0.58 |
73.68 |
Antifeedant activity (ichthyotoxicity activity)
The as-synthesized compounds 1a–g and 2a–i were screened for the antifeedant activity with many of the compounds showing high toxicity. All the as-synthesized compounds had 100% mortality at 100 μg mL−1 except compounds 1a, 1b, and 2a. Among the as-synthesized compounds 1a–g and 2a–i, the compound 1a was the least toxic with the mortality of 0% at 100 μg mL−1. The compound 1c was highly active with the LD50 value of 18.52 μg mL−1 compared with compounds 1a–g and 2a–i. The compounds 1f, 2c, and 2e (range from 35.07 to 35.98 μg mL−1) had equipotent activity due to the presence of different functional groups, such as electron donating groups at 4-CH3O-Ph with pyrimidine in compound 1f, electron donating groups at 4-Cl-Ph with pyrazole in compound 2c, and electron withdrawing group 3-NO2-Ph with pyrazole in compound 2e. The values are summarized in Table 4.
Table 4 Antifeedant activity of title compounds 1a–g and 2a–i
| Compounds |
Concentration (μg mL−1)/mortalitya (%) |
LD50 (μg mL−1) |
| 10 |
25 |
50 |
100 |
| Values are the means of three replicates ± SD. |
| 1a |
— |
— |
— |
— |
>100 |
| 1b |
0 ± 0.00 |
10 ± 1.23 |
24 ± 0.96 |
40 ± 1.65 |
>100 |
| 1c |
34 ± 1.23 |
68 ± 1.54 |
80 ± 1.42 |
100 ± 0.00 |
18.52 |
| 1d |
14 ± 1.35 |
38 ± 1.42 |
60 ± 1.67 |
100 ± 0.00 |
32.63 |
| 1e |
24 ± 1.56 |
46 ± 1.23 |
80 ± 1.78 |
100 ± 0.00 |
30.28 |
| 1f |
18 ± 1.12 |
40 ± 1.32 |
60 ± 1.42 |
100 ± 0.00 |
35.07 |
| 1g |
20 ± 1.87 |
38 ± 1.21 |
62 ± 0.56 |
100 ± 0.00 |
36.54 |
| 2a |
0 ± 0.00 |
20 ± 1.87 |
42 ± 0.67 |
80 ± 1.78 |
59.97 |
| 2b |
10 ± 1.87 |
32 ± 1.61 |
60 ± 1.67 |
100 ± 0.00 |
41.87 |
| 2c |
20 ± 1.20 |
42 ± 1.42 |
68 ± 1.65 |
100 ± 0.00 |
35.53 |
| 2d |
24 ± 0.95 |
60 ± 1.38 |
80 ± 1.42 |
100 ± 0.00 |
25.58 |
| 2e |
14 ± 1.76 |
32 ± 1.23 |
64 ± 1.94 |
100 ± 0.00 |
35.98 |
| 2f |
28 ± 1.20 |
46 ± 0.67 |
68 ± 1.47 |
100 ± 0.00 |
33.72 |
| 2g |
12 ± 1.42 |
38 ± 1.87 |
70 ± 1.63 |
100 ± 0.00 |
31.56 |
| 2h |
24 ± 1.33 |
40 ± 1.34 |
74 ± 1.35 |
100 ± 0.00 |
33.75 |
| 2i |
20 ± 1.42 |
42 ± 1.23 |
60 ± 1.34 |
100 ± 0.00 |
34.52 |
Mosquito larval growth inhibition activities
Considering the regulation of coumarin analogues on growth and metamorphosis, the effect of the compound 2h after a 72 h treatment on the weight gain of larvae and rate of inhibition (Table 5), the duration of the pupal and adult stages and the eclosion rate of the treated Culex quinquefasciatus were evaluated at 10 μg mL−1 (Table 6). The weight gain of larvae were controlled by the compound 2h, which gave an inhibitory rate of 55.06%. The effect of the compound 2h on the duration of the pupal and adult stages was evident, and the rate of eclosion was only 40% after treatment with the compound 2h. The results show that the compound 2h exhibited potent inhibitory activity against the growth and development of Culex quinquefasciatus.
Table 5 The effect of the title compound 2h on the growth of mosquito larvae
| Compound |
Culex quinquefasciatusa |
| Weight (mg) |
Weight gain (mg) |
Inhibition (%) |
| 0 h |
72 h |
| Values are the means of three replicates ± SD. The concentration of 2h was 10 μg mL−1. Control does not contain the compounds. |
| 2hb |
100.28 ± 1.20 |
102.14 ± 1.33 |
3.86 ± 1.14 |
55.06 ± 1.42 |
| Controlc |
100.06 ± 1.21 |
108.65 ± 0.67 |
8.59 ± 1.11 |
— |
Table 6 The effect of the title compound 2h on the growth and development of mosquito larvae
| Compound |
Culex quinquefasciatusa |
| Duration of pupae (h) |
Duration of adult (h) |
Rate of eclosion (%) |
| Values are the means of three replicates ± SD. The concentration of 2h was 10 μg mL−1. Control does not contain the compounds. |
| 2hb |
70.1 ± 1.32 |
25.1 ± 2.34 |
40 ± 3.10 |
| Controlc |
65.5 ± 1.28 |
24.2 ± 1.98 |
90 ± 2.80 |
Molecular docking
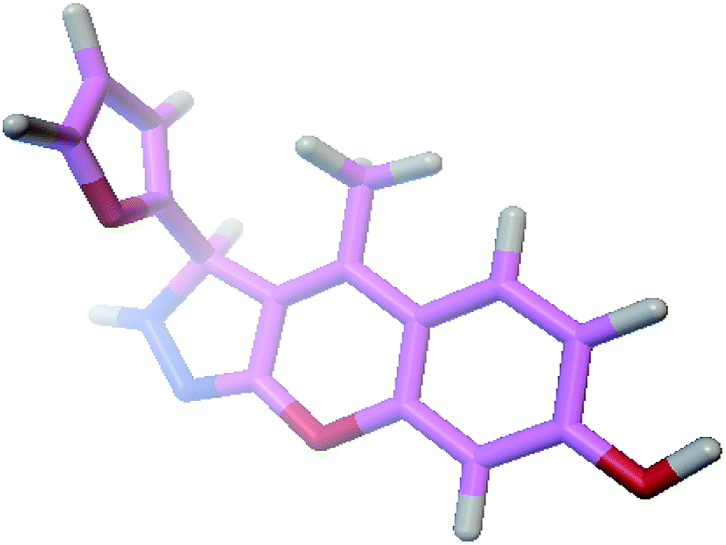
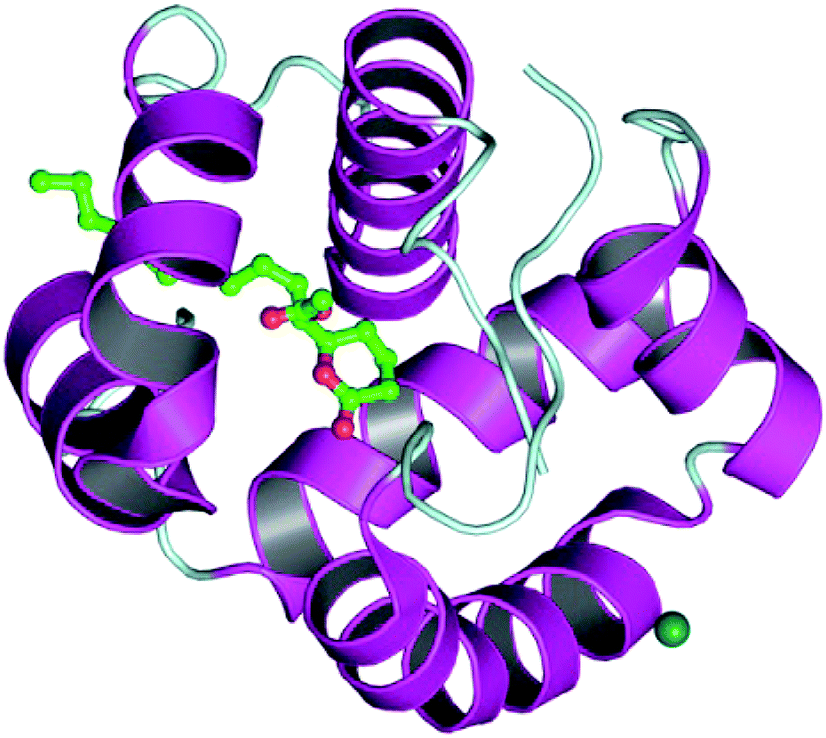
The docking process involves two basic steps: the prediction of the ligand conformation and assessment of the binding affinity. These two steps are related to sampling methods and scoring schemes respectively. Docking studies were accomplished for compounds 1g and 2h in order to calculate their binding affinities against 3OGN protein using the AutoDock4 (version 4.2.6) software. The 3D structure of the ligand 2h is shown in Fig. 2. The 3D structure of the 3OGN protein is shown in Fig. 3. The results were examined based on the binding energies of the docked complexes. AutoDock4 generated 10 conformers for each ligand. The selection of the best conformer was based on the smallest binding energy between the ligand and the protein. After docking, the ligands were ranked according to their binding energies.
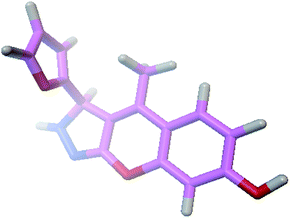 |
| | Fig. 2 3D structure of the compound 2h. | |
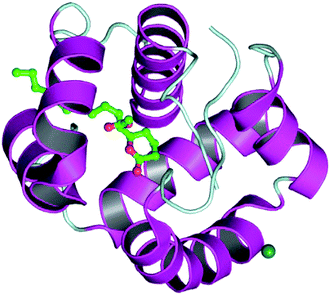 |
| | Fig. 3 3D structure of the 3OGN mosquito odorant binding protein. | |
Docked results using the AutoDock4 software
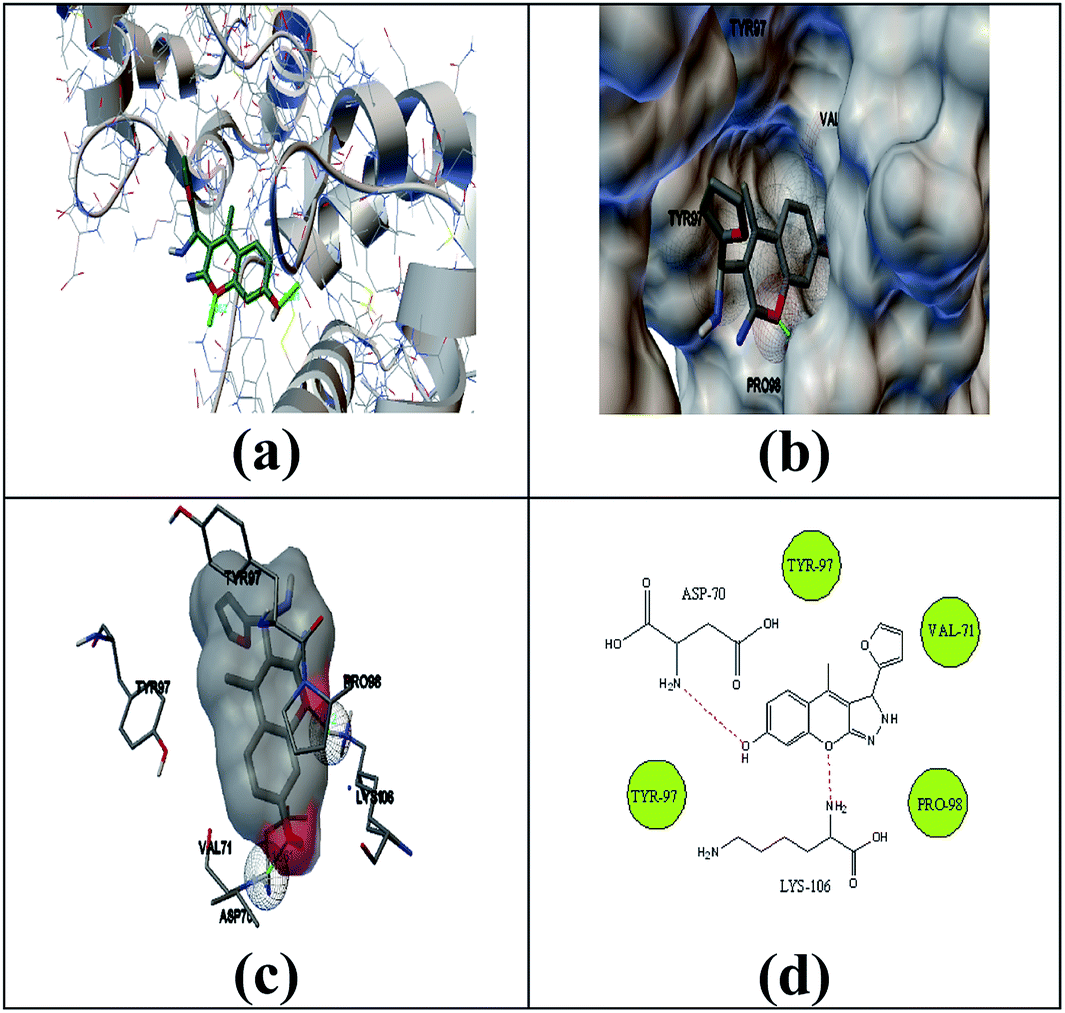
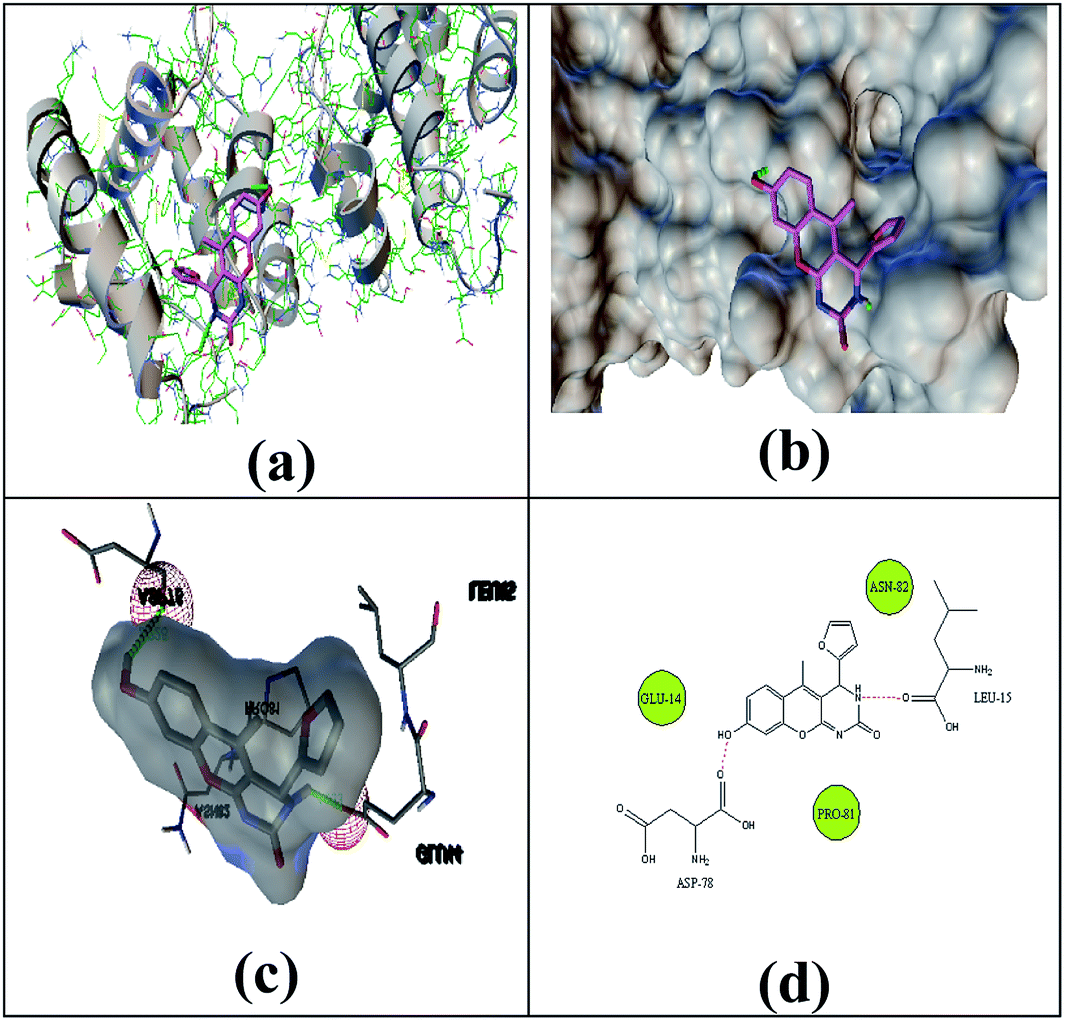
The selected ligand 2h was docked against 3OGN protein using the AutoDock4 software. The ball and stick representation of the 3OGN-2h docked complex is shown in Fig. 4a. The molecular surface representation of the 3OGN-2h docked complex is shown in Fig. 4b. The best-selected pose of the 3OGN-2h docked complex (binding energy −6.12 kcal mol−1) with binding site residues is shown in Fig. 4c. The hydrogen bonds and the types of contacts involved in the 3OGN-2h docked complex are shown in Fig. 4d. It was observed that ASP-70 and LYS-106 are involved in hydrogen bond interactions and the residues VAL-71, TYR-97, TYR-97 and PRO-98 are involved in van der Waals interactions. Then, the selected ligand 1g was docked against the 3OGN. The ball and stick representation of 3OGN-1g docked complex is shown in Fig. 5a. The molecular surface representation of the 3OGN-1g docked complex is shown in Fig. 5b. The best-selected pose of the 3OGN-1g docked complex (binding energy −5.79 kcal mol−1) with binding site residues is shown in Fig. 5c. The hydrogen bonds and the types of contacts involved in the 3OGN-1g docked complex are shown in Fig. 5d. It was observed that ASP-78 and LEU-15 are involved in hydrogen bond interactions and the residues GLU-14, PRO-81 and ASN-82 are involved in van der Waals interactions. The compound 2h has the highest binding affinity with the mosquito odorant binding protein 3OGN compared with compound 1g. The molecular docking characterizations for compound 2h are listed in Table 7, while those for 1g are listed in Table 8.
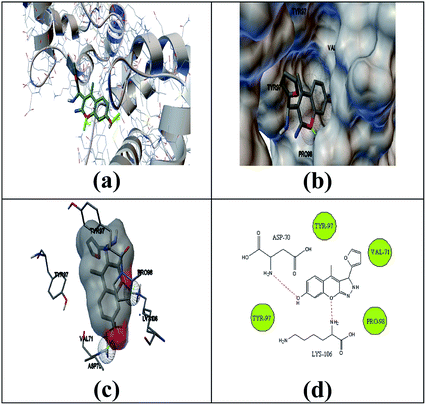 |
| | Fig. 4 Ball and stick (a), molecular surface (b), 3D (c), and 2D (d) interaction modes of compound 2h within the binding site of the mosquito odorant protein 3OGN. | |
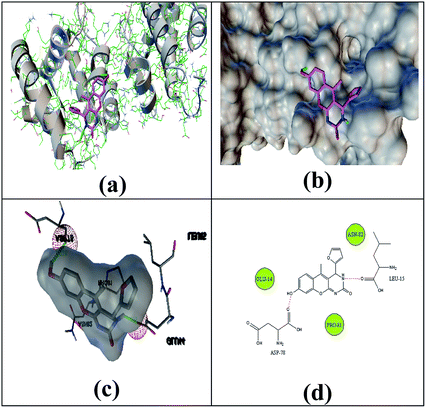 |
| | Fig. 5 Ball and stick (a), molecular surface (b), 3D (c), and 2D (d) interaction modes of compound 1g within the binding site of the mosquito odorant protein 3OGN. | |
Table 7 Molecular docking for highly active compound 2h
| Conformer |
Binding energy (kcal mol−1) |
Inhibition constant (Ki) (μM) |
Intermolecular energy (kcal mol−1) |
Internal energy (kcal mol−1) |
| 1 |
−6.12 |
32.64 |
−6.72 |
−0.48 |
| 2 |
−6.12 |
32.66 |
−6.72 |
−0.48 |
| 3 |
−6.11 |
33.27 |
−6.71 |
−0.47 |
| 4 |
−6.10 |
33.64 |
−6.70 |
−0.48 |
| 5 |
−6.07 |
35.27 |
−6.67 |
−0.47 |
| 6 |
−5.85 |
51.38 |
−6.45 |
−0.48 |
| 7 |
−5.29 |
132.17 |
−5.89 |
−0.47 |
| 8 |
−5.02 |
208.20 |
−5.62 |
−0.47 |
| 9 |
−4.83 |
287.15 |
−5.43 |
−0.38 |
| 10 |
−4.80 |
303.05 |
−5.40 |
−0.47 |
Table 8 Molecular docking for low activity compound 1g
| Conformer |
Binding energy (kcal mol−1) |
Inhibition constant (Ki) (μM) |
Intermolecular energy (kcal mol−1) |
Internal energy (kcal mol−1) |
| 1 |
−5.79 |
57.40 |
−6.38 |
−0.20 |
| 2 |
−5.76 |
60.25 |
−6.36 |
−0.20 |
| 3 |
−5.73 |
63.54 |
−6.32 |
−0.20 |
| 4 |
−5.71 |
65.63 |
−6.30 |
−0.07 |
| 5 |
−5.69 |
69.10 |
−6.28 |
−0.20 |
| 6 |
−5.66 |
71.39 |
−6.25 |
−0.20 |
| 7 |
−5.61 |
77.81 |
−6.20 |
−0.20 |
| 8 |
−5.34 |
121.82 |
−5.94 |
−0.20 |
| 9 |
−4.99 |
220.99 |
−5.58 |
−0.20 |
| 10 |
−4.86 |
272.58 |
−5.46 |
−0.20 |
Structure–activity relationship
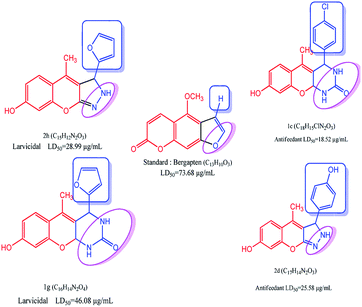
The as-synthesized compounds are compared for the structure and activity relationship, as shown in Fig. 6. The larvicidal activities of the as-synthesized compounds are compared with those of the control bergapten. The larvicidal activity in the presence of pyrazole moiety at the five-membered ring in compound 2h is highly active with the LD50 value of 28.99 μg mL−1 than the five-membered ring at control bergapten with the LD50 value of 73.68 μg mL−1.
 |
| | Fig. 6 Structure–activity relationship studies of coumarin derivative. | |
The larvicidal activity in the presence of pyrimidine moiety with the six-membered ring in compound 1g was highly active with the LD50 value of 46.08 μg mL−1 than the five-membered ring in bergapten with the LD50 value of 73.49 μg mL−1. The presence of natural aldehyde scaffolds in the title compound shows high larvicidal and antifeedant activities. The presence of the furfuraldehyde scaffold in 2h shows high larvicidal activity with the LD50 value of 28.99 μg mL−1 than that in the compounds 1a–g and 2a–i. The presence of 4-Cl-phenyl scaffold in 1c shows high antifeedant activity with the LD50 value of 18.52 μg mL−1 than that in the compounds 1a–g and 2a–i.
Experimental
General
The chemicals 7-hydroxy-4-methylcoumarin, tyrosinase, and bergapten were purchased from Sigma-Aldrich and used without further purification. Melting points were recorded in open capillary tubes and were uncorrected. The IR spectra (KBr) were recorded in KBr on a Shimadzu 8201 pc (4000–400 cm−1). The 1H NMR and 13C NMR spectra were recorded on a Bruker DRX-300 MHz. Elemental analysis (C, H, and N) was performed using an Elemental analyzer model (Varian EL III). The purity of the compounds was checked via thin layer chromatography (TLC) with silica gel plates.
General method for the preparation of 8-hydroxy-5-methyl-4-phenyl-3,4-dihydro-2H-chromeno[2,3-d]pyrimidin-2-one (1a)
The mixture of 7-hydroxy-4-methyl-2H-chromen-2-one (0.001 mol, 0.17 g), substituted aldehyde (0.003 mol), urea (0.003 mol, 0.18 g), and Cu(II)-tyrosinase enzyme (0.5 g) were mixed in a mortar. Then, 2 mL of 50 mM potassium phosphate buffer (pH 6.0) was added and filtered. The insoluble product was washed with excess ice-cold water, and then filtered and dried. The product was confirmed via TLC. The product was washed in ethanol to get pure product. The same method was followed for the synthesis of compounds 1b–g.
White solid; mw: 306.32; mp 164 °C; IR (cm−1): 3174.54 (NH), 3074.57 (Ph-CHstr), 3034 (Ar–H), 2596.43 (CN), 1612.5 (CO); 1H NMR (300 MHz, DMSO-d6): δ 10.5 (1H, s, OH), 8.70 (1H, s, NH), 7.33 (2H, dd, J = 7.33 Hz, J = 7.37 Hz, phenyl), 7.26 (1H, d, J = 6.21 Hz, phenyl), 7.23 (2H, dd, J = 7.31 Hz, J = 7.35 Hz, phenyl), 7.04 (1H, d, J = 6.21 Hz, phenyl), 6.45 (1H, s, phenyl), 6.21 (1H, d, J = 6.21 Hz, phenyl), 5.13 (1H, d, J = 6.21 Hz, –CH), 2.35 (3H, s, –CH3); 13C NMR (300 MHz, DMSO-d6): 160.4 (1C, CO), 160.4 (1C, CN), 159.1 (1C, –C–OH), 155.7 (1C, –C–O–), 143.3, 128.5, 127.2, 126.9, 126.7, 108.4, 107.9, 99.5 (10C, Ar ring), 136.4 (1C, –C–CH3), 112.8 (1C, –CC–CH3), 42.2 (1C, –C–NH), 15.6 (1C, –CH3); EI-MS: 307.10 (M+, 20%); elemental analysis (C18H14N2O3): calculated: C, 70.58; H, 4.61; N, 9.15%; found: C, 70.56; H, 4.62; N, 9.16%.
8-Hydroxy-5-methyl-4-styryl-3,4-dihydro-2H-chromeno[2,3-d]pyrimidin-2-one (1b)
Light yellow solid; mw: 332.35; mp 146 °C; IR (cm−1): 3174.54 (NH), 3074.57 (Ph-CHstr), 3034 (Ar–H), 2596.43 (CN), 1612.5 (CO); 1H NMR (300 MHz, DMSO-d6): δ 10.5 (1H, s, OH), 8.70 (1H, s, NH), 7.40 (2H, dd, J = 7.33 Hz, J = 7.37 Hz, phenyl), 7.33 (1H, d, J = 6.21 Hz, phenyl), 7.24 (2H, dd, J = 7.31 Hz, J = 7.35 Hz, phenyl), 7.04 (1H, d, J = 6.21 Hz, phenyl), 6.56 (1H, d, J = 6.21 Hz, –CH), 6.45 (1H, s, phenyl), 6.21 (1H, d, J = 6.21 Hz, phenyl), 6.19 (1H, d, J = 6.21 Hz, –CH), 4.96 (1H, d, J = 6.21 Hz, –CH), 2.42 (3H, s, –CH3); 13C NMR (300 MHz, DMSO-d6): 160.4 (1C, CO), 160.4 (1C, CN), 159.1 (1C, –C–OH), 155.7 (1C, –C–O–), 136.4 (1C, –C–CH3), 136.4, 128.6, 128.5, 127.9, 127.4, 108.4, 107.9, 99.5 (10C, Ar ring), 129.5 (1C, –CC), 123.3 (1C, –CC), 112.8 (1C, –CC–CH3), 41.6 (1C, –C–NH), 15.7 (1C, –CH3); EI-MS: 333.12 (M+, 22%); elemental analysis (C20H16N2O3): calculated: C, 72.28; H, 4.85; N, 8.43%; found: C, 72.27; H, 4.86; N, 8.45%.
4-(4-chlorophenyl)-8-hydroxy-5-methyl-3,4-dihydro-2H-chromeno[2,3-d]pyrimidin-2-one (1c)
Brown solid; mw: 340.76; mp 178 °C; IR (cm−1): 3174.54 (NH), 3074.57 (Ph-CHstr), 3034 (Ar–H), 2596.43 (CN), 1612.5 (CO); 1H NMR (300 MHz, DMSO-d6): δ 10.5 (1H, s, OH), 8.70 (1H, s, NH), 7.37 (2H, dd, J = 7.33 Hz, J = 7.37 Hz, phenyl), 7.34 (2H, dd, J = 7.31 Hz, J = 7.35 Hz, phenyl), 7.04 (1H, d, J = 6.21 Hz, phenyl), 6.45 (1H, s, phenyl), 6.21 (1H, d, J = 6.21 Hz, phenyl), 5.13 (1H, d, J = 6.21 Hz, –CH), 2.42 (3H, s, –CH3); 13C NMR (300 MHz, DMSO-d6): 160.4 (1C, CO), 160.4 (1C, CN), 159.1 (1C, –C–OH), 155.7 (1C, –C–O–), 141.4, 132.3, 128.6, 127.2, 126.1, 108.4, 107.9, 99.5 (10C, Ar ring), 136.4 (1C, –C–CH3), 112.8 (1C, –CC–CH3), 42.2 (1C, –C–NH), 15.6 (1C, –CH3); EI-MS: 342.06 (M+, 32%); elemental analysis (C18H13ClN2O3): calculated: C, 63.44; H, 3.85; N, 8.22%; found: C, 63.42; H, 3.86; N, 8.23%.
8-Hydroxy-4-(4-hydroxyphenyl)-5-methyl-3,4-dihydro-2H-chromeno[2,3-d]pyrimidin-2-one (1d)
Brown solid; mw: 322.31; mp 172 °C; IR (cm−1): 3174.54 (NH), 3074.57 (Ph-CHstr), 3034 (Ar–H), 2596.43 (CN), 1612.5 (CO); 1H NMR (300 MHz, DMSO-d6): δ 10.5 (2H, s, OH), 8.70 (1H, s, NH), 7.06 (2H, dd, J = 7.33 Hz, J = 7.37 Hz, phenyl), 7.04 (1H, d, J = 6.21 Hz, phenyl), 6.63 (2H, dd, J = 7.31 Hz, J = 7.35 Hz, phenyl), 6.45 (1H, s, phenyl), 6.21 (1H, d, J = 6.21 Hz, phenyl), 5.13 (1H, d, J = 6.21 Hz, –CH), 2.42 (3H, s, –CH3); 13C NMR (300 MHz, DMSO-d6): 160.4 (1C, CO), 160.4 (1C, CN), 159.1 (1C, –C–OH), 155.7 (1C, –C–O–), 156.5, 135.9, 126.1, 127.2, 115.7, 108.4, 107.9, 99.5 (10C, Ar ring), 136.4 (1C, –C–CH3), 112.8 (1C, –CC–CH3), 42.2 (1C, –C–NH), 15.6 (1C, –CH3); EI-MS: 323.10 (M+, 20%); elemental analysis (C18H14N2O4): calculated: C, 67.07; H, 4.38; N, 8.69%; found: C, 67.06; H, 4.40; N, 8.68%.
8-Hydroxy-5-methyl-4-(3-nitrophenyl)-3,4-dihydro-2H-chromeno[2,3-d]pyrimidin-2-one (1e)
White solid; mw: 351.31; mp 108 °C; IR (cm−1): 3174.54 (NH), 3074.57 (Ph-CHstr), 3034 (Ar–H), 2596.43 (CN), 1612.5 (CO); 1H NMR (300 MHz, DMSO-d6): δ 10.5 (1H, s, OH), 8.70 (1H, s, NH), 8.12 (1H, s, phenyl), 8.07 (1H, d, J = 7.33 Hz, phenyl), 7.62 (1H, d, J = 6.21 Hz, phenyl), 7.59 (1H, dd, J = 7.31 Hz, J = 7.35 Hz, phenyl), 7.04 (1H, d, J = 6.21 Hz, phenyl), 6.45 (1H, s, phenyl), 6.21 (1H, d, J = 6.21 Hz, phenyl), 5.13 (1H, d, J = 6.21 Hz, –CH), 2.42 (3H, s, –CH3); 13C NMR (300 MHz, DMSO-d6): 160.4 (1C, CO), 160.4 (1C, CN), 159.1 (1C, –C–OH), 155.7 (1C, –C–O–), 147.7, 144.2, 133.0, 129.4, 121.9, 120.7, 127.2, 108.4, 107.9, 99.5 (10C, Ar ring), 136.4 (1C, –C–CH3), 112.8 (1C, –CC–CH3), 41.2 (1C, –C–NH), 15.6 (1C, –CH3); EI-MS: 352.09 (M+, 20%); elemental analysis (C18H13N3O5): calculated: C, 61.54; H, 3.73; N, 11.96%; found: C, 61.56; H, 3.72; N, 11.97%.
8-Hydroxy-4-(4-methoxyphenyl)-5-methyl-3,4-dihydro-2H-chromeno[2,3-d]pyrimidin-2-one (1f)
Brown solid; mw: 336.34; mp 180 °C; IR (cm−1): 3174.54 (NH), 3074.57 (Ph-CHstr), 3034 (Ar–H), 2596.43 (CN), 1612.5 (CO); 1H NMR (300 MHz, DMSO-d6): δ 10.5 (1H, s, OH), 8.70 (1H, s, NH), 7.12 (2H, dd, J = 7.33 Hz, J = 7.37 Hz, phenyl), 7.04 (1H, d, J = 6.21 Hz, phenyl), 6.87 (2H, dd, J = 7.31 Hz, J = 7.35 Hz, phenyl), 6.45 (1H, s, phenyl), 6.21 (1H, d, J = 6.21 Hz, phenyl), 5.13 (1H, d, J = 6.21 Hz, –CH), 3.83 (3H, s, –OCH3), 2.42 (3H, s, –CH3); 13C NMR (300 MHz, DMSO-d6): 160.4 (1C, CO), 160.4 (1C, CN), 159.1 (1C, –C–OH), 155.7 (1C, –C–O–), 158.6, 135.6, 125.7, 127.2, 114.1, 108.4, 107.9, 99.5 (10C, Ar ring), 136.4 (1C, –C–CH3), 112.8 (1C, –CC–CH3), 55.8 (1C, –OCH3), 42.2 (1C, –C–NH), 15.6 (1C, –CH3); EI-MS: 337.11 (M+, 21%); elemental analysis (C19H16N2O4): calculated: C, 67.85; H, 4.79; N, 8.33%; found: C, 67.84; H, 4.78; N, 8.35%.
4-(Furan-2-yl)-8-hydroxy-5-methyl-3,4-dihydro-2H-chromeno[2,3-d]pyrimidin-2-one (1g)
Black solid; mw: 296.28; mp 208 °C; IR (cm−1): 3174.54 (NH), 3074.57 (Ph-CHstr), 3034 (Ar–H), 2596.43 (CN), 1612.5 (CO); 1H NMR (300 MHz, DMSO-d6): δ 10.5 (1H, s, OH), 8.70 (1H, s, NH), 7.65 (1H, d, J = 6.21 Hz, furyl), 7.04 (1H, d, J = 6.21 Hz, phenyl), 6.46 (1H, dd, J = 7.33 Hz, J = 7.37 Hz, furyl), 6.45 (1H, s, phenyl), 6.26 (1H, d, J = 6.21 Hz, furyl), 6.21 (1H, d, J = 6.21 Hz, phenyl), 5.13 (1H, d, J = 6.21 Hz, –CH), 2.42 (3H, s, –CH3); 13C NMR (300 MHz, DMSO-d6): 160.4 (1C, CO), 160.4 (1C, CN), 159.1 (1C, –C–OH), 155.7 (1C, –C–O–), 152.5, 142.1, 127.2, 110.6, 108.4, 107.9, 106.7, 99.5 (8C, Ar ring), 136.4 (1C, –C–CH3), 112.8 (1C, –CC–CH3), 43.4 (1C, –C–NH), 15.0 (1C, –CH3); EI-MS: 297.08 (M+, 18%); elemental analysis (C16H12N2O4): calculated: C, 64.86; H, 4.08; N, 9.46%; found: C, 64.87; H, 4.10; N, 9.43%.
General method for preparation of 4-methyl-3-phenyl-2,3-dihydrochromeno[2,3-c]pyrazol-7-ol (2a)
The mixture of 7-hydroxy-4-methyl-2H-chromen-2-one (0.001 mol, 0.17 g), substituted aldehyde (0.003 mol, 0.3 mL), hydrazine hydrate (0.003 mol, 0.1 mL), and Cu(II)-tyrosinase enzyme (0.5 mL) is mixed together in a mortar. Then 2 mL of 50 mM potassium phosphate buffer (pH 6.0) was added and filtered. The insoluble product was washed with excess of ice-cold water filtered and dried. The product was confirmed by TLC. The product was washed in ethanol to get pure product. The same method was followed to the synthesis of other compounds 2b–i.
Black solid; mw: 278.31; mp 112 °C; IR (cm−1): 3174.54 (NH), 3074.57 (Ph-CHstr), 3034 (Ar–H), 2596.43 (CN); 1H NMR (300 MHz, DMSO-d6): δ 10.5 (1H, s, OH), 8.70 (1H, s, NH), 7.33 (2H, dd, J = 7.33 Hz, J = 7.37 Hz, phenyl), 7.26 (1H, d, J = 6.21 Hz, phenyl), 7.23 (2H, dd, J = 7.31 Hz, J = 7.35 Hz, phenyl), 7.04 (1H, d, J = 6.21 Hz, phenyl), 6.45 (1H, s, phenyl), 6.21 (1H, d, J = 6.21 Hz, phenyl), 4.59 (1H, d, J = 6.21 Hz, –CH), 2.42 (3H, s, –CH3); 13C NMR (300 MHz, DMSO-d6): 159.1 (1C, –C–OH), 155.7 (1C, –C–O–), 150.6 (1C, CN), 143.3, 128.5, 127.2, 126.9, 126.7, 108.4, 107.9, 99.5 (10C, Ar ring), 137.2 (1C, –C–CH3), 122.3 (1C, –CC–CH3), 45.8 (1C, –C–NH), 15.6 (1C, –CH3); EI-MS: 279.11 (M+, 19%); elemental analysis (C17H14N2O2): calculated: C, 73.37; H, 5.07; N, 10.07%; found: C, 73.36; H, 5.06; N, 10.09%.
4-Methyl-3-styryl-2,3-dihydrochromeno[2,3-c]pyrazol-7-ol (2b)
Red solid; mw: 304.34; mp 194 °C; IR (cm−1): 3174.54 (NH), 3074.57 (Ph-CHstr), 3034 (Ar–H), 2596.43 (CN); 1H NMR (300 MHz, DMSO-d6): δ 10.5 (1H, s, OH), 8.70 (1H, s, NH), 7.40 (2H, dd, J = 7.33 Hz, J = 7.37 Hz, phenyl), 7.33 (1H, d, J = 6.21 Hz, phenyl), 7.24 (2H, dd, J = 7.31 Hz, J = 7.35 Hz, phenyl), 7.04 (1H, d, J = 6.21 Hz, phenyl), 6.56 (1H, d, J = 6.21 Hz, –CH), 6.45 (1H, s, phenyl), 6.21 (1H, d, J = 6.21 Hz, phenyl), 6.19 (1H, d, J = 6.21 Hz, –CH), 3.99 (1H, d, J = 6.21 Hz, –CH), 2.42 (3H, s, –CH3); 13C NMR (300 MHz, DMSO-d6): 159.1 (1C, –C–OH), 155.7 (1C, –C–O–), 150.6 (1C, CN), 137.2 (1C, –C–CH3), 136.4, 128.6, 128.5, 127.9, 127.2, 108.4, 107.9, 99.5 (10C, Ar ring), 129.5 (1C, –CC), 123.3 (1C, –CC), 122.3 (1C, –CC–CH3), 45.4 (1C, –C–NH), 15.7 (1C, –CH3); EI-MS: 305.12 (M+, 21%); elemental analysis (C19H16N2O2): calculated: C, 74.98; H, 5.30; N, 9.20%; found: C, 74.97; H, 5.31; N, 9.18%.
3-(4-chlorophenyl)-4-methyl-2,3-dihydrochromeno[2,3-c]pyrazol-7-ol (2c)
Yellow solid; mw: 312.75; mp 174 °C; IR (cm−1): 3174.54 (NH), 3074.57 (Ph-CHstr), 3034 (Ar–H), 2596.43 (CN); 1H NMR (300 MHz, DMSO-d6): δ 10.5 (1H, s, OH), 8.70 (1H, s, NH), 7.37 (2H, dd, J = 7.33 Hz, J = 7.37 Hz, phenyl), 7.34 (2H, dd, J = 7.31 Hz, J = 7.35 Hz, phenyl), 7.04 (1H, d, J = 6.21 Hz, phenyl), 6.45 (1H, s, phenyl), 6.21 (1H, d, J = 6.21 Hz, phenyl), 4.59 (1H, d, J = 6.21 Hz, –CH), 2.42 (3H, s, –CH3); 13C NMR (300 MHz, DMSO-d6): 159.1 (1C, –C–OH), 155.7 (1C, –C–O–), 150.6 (1C, CN), 141.4, 132.3, 128.6, 127.2, 126.1, 108.4, 107.9, 99.5 (10C, Ar ring), 137.2 (1C, –C–CH3), 122.3 (1C, –CC–CH3), 45.8 (1C, –C–NH), 15.6 (1C, –CH3); EI-MS: 314.06 (M+, 32%); elemental analysis (C17H13ClN2O2): calculated: C, 65.29; H, 4.19; N, 8.96%; found: C, 65.27; H, 4.20; N, 8.97%.
3-(4-hydroxyphenyl)-4-methyl-2,3-dihydrochromeno[2,3-c]pyrazol-7-ol (2d)
Yellow solid; mw: 294.30; mp 232 °C; IR (cm−1): 3174.54 (NH), 3074.57 (Ph-CHstr), 3034 (Ar–H), 2596.43 (CN); 1H NMR (300 MHz, DMSO-d6): δ 10.5 (2H, s, OH), 8.70 (1H, s, NH), 7.06 (2H, dd, J = 7.33 Hz, J = 7.37 Hz, phenyl), 7.04 (1H, d, J = 6.21 Hz, phenyl), 6.63 (2H, dd, J = 7.31 Hz, J = 7.35 Hz, phenyl), 6.45 (1H, s, phenyl), 6.21 (1H, d, J = 6.21 Hz, phenyl), 4.59 (1H, d, J = 6.21 Hz, –CH), 2.42 (3H, s, –CH3); 13C NMR (300 MHz, DMSO-d6): 159.1 (1C, –C–OH), 155.7 (1C, –C–O–), 150.6 (1C, CN), 156.5, 135.9, 126.1, 127.2, 115.7, 108.4, 107.9, 99.5 (10C, Ar ring), 137.2 (1C, –C–CH3), 122.3 (1C, –CC–CH3), 45.8 (1C, –C–NH), 15.6 (1C, –CH3); EI-MS: 295.10 (M+, 19%); elemental analysis (C17H14N2O3): calculated: C, 69.38; H, 4.79; N, 9.52%; found: C, 69.39; H, 4.81; N, 9.51%.
4-Methyl-3-(3-nitrophenyl)-2,3-dihydrochromeno[2,3-c]pyrazol-7-ol (2e)
Yellow solid; mw: 323.30; mp 198 °C; IR (cm−1): 3174.54 (NH), 3074.57 (Ph-CHstr), 3034 (Ar–H), 2596.43 (CN); 1H NMR (300 MHz, DMSO-d6): δ 10.5 (1H, s, OH), 8.70 (1H, s, NH), 8.12 (1H, s, phenyl), 8.07 (1H, d, J = 7.33 Hz, phenyl), 7.62 (1H, d, J = 6.21 Hz, phenyl), 7.59 (1H, dd, J = 7.31 Hz, J = 7.35 Hz, phenyl), 7.04 (1H, d, J = 6.21 Hz, phenyl), 6.45 (1H, s, phenyl), 6.21 (1H, d, J = 6.21 Hz, phenyl), 4.59 (1H, d, J = 6.21 Hz, –CH), 2.42 (3H, s, –CH3); 13C NMR (300 MHz, DMSO-d6): 159.1 (1C, –C–OH), 155.7 (1C, –C–O–), 150.6 (1C, CN), 147.7, 144.2, 133.0, 129.4, 121.9, 120.7, 127.2, 108.4, 107.9, 99.5 (10C, Ar ring), 137.2 (1C, –C–CH3), 122.3 (1C, –CC–CH3), 44.8 (1C, –C–NH), 15.6 (1C, –CH3); EI-MS: 324.09 (M+, 20%); elemental analysis (C17H13N3O4): calculated: C, 63.16; H, 4.05; N, 13.00%; found: C, 63.15; H, 4.03; N, 13.01%.
3-(4-(dimethylamino)phenyl)-4-methyl-2,3-dihydrochromeno[2,3-c]pyrazol-7-ol (2f)
Red solid; mw: 321.37; mp 78 °C; IR (cm−1): 3174.54 (NH), 3074.57 (Ph-CHstr), 3034 (Ar–H), 2596.43 (CN); 1H NMR (300 MHz, DMSO-d6): δ 10.5 (1H, s, OH), 8.70 (1H, s, NH), 7.05 (2H, dd, J = 7.33 Hz, J = 7.37 Hz, phenyl), 7.04 (1H, d, J = 6.21 Hz, phenyl), 6.65 (2H, dd, J = 7.31 Hz, J = 7.35 Hz, phenyl), 6.45 (1H, s, phenyl), 6.21 (1H, d, J = 6.21 Hz, phenyl), 4.59 (1H, d, J = 6.21 Hz, –CH), 3.06 (6H, s, –(CH3)2), 2.42 (3H, s, –CH3); 13C NMR (300 MHz, DMSO-d6): 159.1 (1C, –C–OH), 155.7 (1C, –C–O–), 150.6 (1C, CN), 149.1, 132.8, 127.2, 126.1, 112.7, 108.4, 107.9, 99.5 (10C, Ar ring), 137.2 (1C, –C–CH3), 122.3 (1C, –CC–CH3), 45.8 (1C, –C–NH), 41.3 (2C, –(CH3)2), 15.6 (1C, –CH3); EI-MS: 322.15 (M+, 21%); elemental analysis (C19H19N3O2): calculated: C, 71.01; H, 5.96; N, 13.08%; found: C, 71.23; H, 5.95; N, 13.07%.
3-(4-methoxyphenyl)-4-methyl-2,3-dihydrochromeno[2,3-c]pyrazol-7-ol (2g)
Yellow solid; mw: 308.33; mp 144 °C; IR (cm−1): 3174.54 (NH), 3074.57 (Ph-CHstr), 3034 (Ar–H), 2596.43 (CN); 1H NMR (300 MHz, DMSO-d6): δ 10.5 (1H, s, OH), 8.70 (1H, s, NH), 7.12 (2H, dd, J = 7.33 Hz, J = 7.37 Hz, phenyl), 7.04 (1H, d, J = 6.21 Hz, phenyl), 6.87 (2H, dd, J = 7.31 Hz, J = 7.35 Hz, phenyl), 6.45 (1H, s, phenyl), 6.21 (1H, d, J = 6.21 Hz, phenyl), 4.59 (1H, d, J = 6.21 Hz, –CH), 3.83 (3H, s, –OCH3), 2.42 (3H, s, –CH3); 13C NMR (300 MHz, DMSO-d6): 159.1 (1C, –C–OH), 155.7 (1C, –C–O–), 150.6 (1C, CN), 158.6, 135.6, 125.7, 127.2, 114.1, 108.4, 107.9, 99.5 (10C, Ar ring), 137.2 (1C, –C–CH3), 122.3 (1C, –CC–CH3), 55.8 (1C, –OCH3), 45.8 (1C, –C–NH), 15.6 (1C, –CH3); EI-MS: 309.12 (M+, 20%); elemental analysis (C18H16N2O3): calculated: C, 70.12; H, 5.23; N, 9.09%; found: C, 70.13; H, 5.21; N, 9.08%.
3-(Furan-2-yl)-4-methyl-2,3-dihydrochromeno[2,3-c]pyrazol-7-ol (2h)
Brown solid; mw: 268.27; mp 104 °C; IR (cm−1): 3174.54 (NH), 3074.57 (Ph-CHstr), 3034 (Ar–H), 2596.43 (CN); 1H NMR (300 MHz, DMSO-d6): δ 10.5 (1H, s, OH), 8.70 (1H, s, NH), 7.65 (1H, d, J = 6.21 Hz, furyl), 7.04 (1H, d, J = 6.21 Hz, phenyl), 6.46 (1H, dd, J = 7.33 Hz, J = 7.37 Hz, furyl), 6.45 (1H, s, phenyl), 6.26 (1H, d, J = 6.21 Hz, furyl), 6.21 (1H, d, J = 6.21 Hz, phenyl), 4.82 (1H, d, J = 6.21 Hz, –CH), 2.42 (3H, s, –CH3); 13C NMR (300 MHz, DMSO-d6): 159.1 (1C, –C–OH), 155.7 (1C, –C–O–), 150.6 (1C, CN), 152.5, 142.1, 127.2, 110.6, 108.4, 107.9, 106.7, 99.5 (8C, Ar ring), 137.2 (1C, –C–CH3), 122.3 (1C, –CC–CH3), 45.5 (1C, –C–NH), 15.0 (1C, –CH3); EI-MS: 269.09 (M+, 16%); elemental analysis (C15H12N2O3): calculated: C, 67.16; H, 5.51; N, 10.44%; found: C, 67.17; H, 5.50; N, 10.45%.
3-(2,6-Dimethylhepta-1,5-dien-1-yl)-4-methyl-2,3-dihydrochromeno[2,3-c]pyrazol-7-ol (2i)
Brown solid; mw: 324.42; mp 68 °C; IR (cm−1): 3174.54 (NH), 3074.57 (Ph-CHstr), 3034 (Ar–H), 2596.43 (CN); 1H NMR (300 MHz, DMSO-d6): δ 10.5 (1H, s, OH), 8.70 (1H, s, NH), 7.04 (1H, d, J = 6.21 Hz, phenyl), 6.45 (1H, s, phenyl), 6.21 (1H, d, J = 6.21 Hz, phenyl), 5.33 (1H, d, J = 6.21 Hz, –CHC), 5.20 (1H, d, J = 6.21 Hz, –CHC), 3.99 (1H, d, J = 6.21 Hz, –CH), 2.42 (3H, s, –CH3), 2.00 (4H, d, J = 6.21 Hz, –(CH2)2), 1.82 (6H, d, J = 6.21 Hz, –(CH3)2), 1.70 (3H, d, J = 6.21 Hz, –CH3); 13C NMR (300 MHz, DMSO-d6): 159.1 (1C, –C–OH), 155.7 (1C, –C–O–), 150.6 (1C, CN), 137.2 (1C, –C–CH3), 135.5 (1C, –CC–), 132.0 (1C, –CC–), 123.5 (1C, –CC–), 116.7 (1C, –CC–), 127.2, 108.4, 107.9, 99.5 (4C, Ar ring), 122.3 (1C, –CC–CH3), 39.8 (1C, –CH2–), 39.3 (1C, –C–NH), 26.4 (1C, –CH2–), 24.6 (1C, –CH3), 18.6 (1C, –CH3), 16.5 (1C, –CH3), 15.7 (1C, –CH3); EI-MS: 325.19 (M+, 20%); elemental analysis (C20H24N2O2): calculated: C, 74.04; H, 7.46; N, 8.64%; found: C, 74.05; H, 7.47; N, 8.62%.
Larvicidal activity
The larvicidal screenings were performed according to our previously reported methods.29 The LD50 values of several active compounds were evaluated using probit analysis. The results were analyzed using the statistical software SPSS version 16.0. The larval growth inhibition and regulation against Culex quinquefasciatus were determined using the water immersion method30.
Antifeedant activity
Fingerlings (1.5–2.0 cm) of marine-acclimated Oreochromis mossambicus were used for evaluating ichthyotoxic potential.31
Protein preparation for docking
The mosquito odorant binding protein 3OGN was obtained from the protein data bank (PDB).32 For docking, all water molecules were removed from protein, and hydrogen atoms were added to the refined model using AutoDock Tools (ADT), and finally merging the non-polar hydrogens. The as-prepared protein was saved in PDB, partial charge (Q), and atom type (T) (PDBQT) formats.
Ligand preparation for docking
The ligand was drawn in the Chem 3D pro software and converted into a protein data bank (PDB) file format using the Open Babel software.33 The ligands were prepared using AutoDock Tools (ADT). Gasteiger charges were assigned to the ligands. The as-prepared ligands were saved in a PDBQT format. Fig. 2 shows the chemical structure of the ligand 2h.
Molecular docking
AutoDock4 (version 4.2.6) was used for the molecular docking studies. AutoGrid program supplied with AutoDock4 was used for the preparation of grid maps. The grid box size was set at 76, 94, and 76 Å for x, y, and z respectively. The spacing between the grid points was 1.0 Å. The grid center was set at 8.639, 35.906, and 8.905 Å for x, y, and z, respectively. The Lamarckian genetic algorithm (LGA) was chosen from the ADT tool to search for the best conformers. During the docking process, a maximum of 10 conformers was considered for the ligand. The docking processes were performed with the default parameters of AutoDock 4.34 Population size was set to 150, maximum number of evaluations 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500000, the maximum number of generations 27000, the maximum number of a top individual that automatically survived 1, gene mutation rate 0.02 and crossover rate 0.8 AutoDock4 was produced and run under the windows 7 operating system. All figures with structure representations were generated using a python molecule viewer.
500000, the maximum number of generations 27000, the maximum number of a top individual that automatically survived 1, gene mutation rate 0.02 and crossover rate 0.8 AutoDock4 was produced and run under the windows 7 operating system. All figures with structure representations were generated using a python molecule viewer.
Conclusions
From this present study, as-synthesized compounds showed significant activity in the larvicidal and antifeedant screenings. Cu(II)-tyrosinase enzyme was used as a catalyst for Biginelli reaction to give good yield. Compound 2h showed significant activity against mosquito larvae and low toxic with eco-friendly ichthyocides against antifeedant screening when compared with other compounds. Molecular docking studies were well supported for compound 2h (binding energy −6.12 kcal mol−1) with good binding ability (OBP) of Culex quinquefasciatus. Therefore, these compounds might be a potential source for developing ecologically significant bioactive compounds, including biodegradable pesticides and biopharmaceuticals.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors extend their appreciation to the Deanship Scientific Research at King Saud University for funding the work through the Research Group Project No RGP-1438-90.
Notes and references
- K. Othmer, Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, New York, 5th edn, 2007 Search PubMed.
- R. O. Kennedy and R. D. Thornes, Coumarins: Biology, applications and mode of action, John Wiley & Sons, New York, 1997 Search PubMed.
- M. Zabradnik, The Production and Application of Fluorescent Brightening Agents, John Wiley and Sons, New York, 1992 Search PubMed.
- E. A. El-Hag, A. H. Nadi and A. A. Zaitoon, Phytother. Res., 1999, 13, 388–392 CrossRef CAS PubMed.
- Z. Wang, J. R. Kim, M. Wang, S. Shu and Y. J. Ahu, Pest Manage. Sci., 2012, 68, 1041–1047 CrossRef CAS PubMed.
- A. M. El-Agrody, M. S. Abd El-Latif, N. A. El-Hady, A. H. Fakery and A. H. Bedair, Molecules, 2001, 6, 519–527 CrossRef CAS.
- S. M. De Souza, F. Delle Monache and A. Z. Smânia, Z. Naturforsch., C: J. Biosci., 2005, 60, 693–700 CAS.
- H. C. Marcondes, T. T. De Oliveira, J. G. Taylor, M. Hamoy, A. L. Neto, V. J. De Mello and T. J. J. Nagem, J. Chem., 2015, 241243 Search PubMed.
- C. Montagner, S. M. De Souza, C. Groposoa, F. Delle Monache, E. F. Smânia and A. Smânia, Z. Naturforsch., C: J. Biosci., 2008, 63, 21–28 CAS.
- A. F. El-Farargy, Egypt. J. Pharm. Sci., 1991, 32, 625–628 CAS.
- I. Manolov and N. D. Danchev, Eur. J. Med. Chem., 1995, 30, 531–535 CrossRef CAS.
- A. A. Emmanuel-Giota, K. C. Fylaktakidou, D. J. Hadjipavlou-Litina, K. E. Litinas and D. N. Nicolaides, Heterocycl. Chem., 2001, 38, 717–722 CrossRef CAS.
- L. Raev, E. Voinov, I. Ivanov and D. Popov, Pharmazie, 1990, 45, 696–702 CAS.
- Z. M. Nofal, M. El-Zahar and S. Abd El-Karim, Molecules, 2000, 5, 99–113 CrossRef CAS.
- L. Xie, Y. Tukeuchi, L. M. Consetino and K. Lee, J. Med. Chem., 1999, 42, 2662–2672 CrossRef CAS PubMed.
- B. J. Donnelly, D. M. X. Donnelly and A. M. O. Sullivan, Tetrahedron, 1968, 24, 2617–2622 CrossRef CAS.
- F. Bigi, L. Chesini, R. Maggi and G. Sartori, J. Org. Chem., 1999, 64, 1033–1035 CrossRef CAS PubMed.
- I. Yavari, R. Hekmat-shoar and A. Zonouzi, Tetrahedron Lett., 1998, 39, 2391–2392 CrossRef CAS.
- E. V. O. Jhon and S. S. Israelstam, J. Org. Chem., 1961, 26, 240–242 CrossRef.
- S. Z. Ahmed and R. D. Desai, Proc. Indian Acad. Sci. Sect. A, 1937, 5, 277–284 CrossRef.
- A. K. Das Gupta, R. M. Chatterje and K. R. Das, J. Chem. Soc. C, 1969, 1, 29–33 RSC.
- L. L. Woods and J. Sapp, J. Org. Chem., 1962, 27, 3703–3705 CrossRef CAS.
- G. K. Biswas, K. Basu, A. K. Barua and P. Bhattacharya, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1992, 31, 628–672 Search PubMed.
- A. J. Hoefnagel, E. A. Gunnewegh, R. S. Downing and H. V. Bekkum, J. Chem. Soc., Chem. Commun., 1995, 2, 225–228 RSC.
- B. M. Reddy, V. R. Reddy and D. Giridhar, Synth. Commun., 2001, 31, 3603–3607 CrossRef CAS.
- A. C. Khandekar and B. M. Khadilkar, Synlett, 2002, 152–154 CrossRef CAS.
- D. D. Chaudhari, Chem. Ind., 1983, 569–572 Search PubMed.
- S. I. Ali and V. Venugopalan, Nat. Prod. Res., 2019 DOI:10.1080/14786419.2019.1634712.
- I. A. Arif, A. Ahamed, R. S. Kumar, A. Idhayadhulla and A. Manilal, Saudi J. Biol. Sci., 2019, 26, 673–680 CrossRef CAS PubMed.
- G. P. Song, D. K. Hu, H. Tian, Y. S. Li, Y. S. Cao, H. W. Jin and Z. N. Cui, Sci. Rep., 2016, 6, 22977 CrossRef PubMed.
- A. A. Ibrahim Anis, R. Surendra Kumar, A. Idhayadhulla and A. Manilal, Saudi J. Biol. Sci., 2019, 26, 673–680 CrossRef PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 1639–1662 CrossRef PubMed.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33–47 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b