DOI:
10.1039/C9RA04478G
(Paper)
RSC Adv., 2019,
9, 28548-28553
Doxorubicin loaded ferritin nanoparticles for ferroptosis enhanced targeted killing of cancer cells
Received
14th June 2019
, Accepted 2nd September 2019
First published on 10th September 2019
Abstract
In recent years, ferroptosis has been investigated widely as a new form of cell death. Development of nanodrugs for ferroptosis induction in cancer cells may be a promising approach for cancer treatment. Here, we developed a type of nanoparticle consisting of the antitumor drug doxorubicin and exogenous ferritin. The drug loading process did not change the size of ferritin obviously. And this nanoparticle could induce the accumulation of ROS and cell ferroptosis for transferrin receptor overexpressed tumor cell, HT29. The ferroptosis process was also confirmed using inhibitors for ferroptosis. The cytotoxicity of this nanoparticle is similar to that of free DOX. This study provides a new strategy for targeting and killing transferrin receptor overexpressed tumor cells.
Introduction
Ferroptosis is a new type of cell death proposed by Dixon in 2012.1 Unlike apoptosis, necrosis, autophagy and other cell death mechanisms, Ferroptosis depends on the iron and the accumulation of lipid reactive oxygen species (L-ROS).1–5 This type of cell death can be activated by small molecules (erastin, sulfasalazine, RSL3, etc.) and effectively inhibited by iron ion chelating agents (desferrioxamine, cyclopyramide), and lipophilic antioxidants.1,3,4 It is an adaptive process different from apoptosis and other non-apoptotic cell death pathways.6 Usually, the lethal L-ROS forms iron-dependently on the polyunsaturated fatty acids chain of cell membrane lipids.7,8 However, this ferroptosis process can be held back by the glutathione (GSH)-dependent antioxidant enzyme glutathione peroxidase 4 (GPX4). The biosynthesis of cellular GSH needs transmembrane cystine/glutamate antiporter system xc−, which imports extracellular Cys in exchange for intracellular Glu.1 So removal of cystine,1 inhibition of system xc−,1,9 inhibition of GSH-dependent GPX4 enzyme activity,8 and reduction of intracellular iron1 can lead to L-ROS accumulation and subsequent ferroptosis.
Doxorubicin (DOX) is one of anti-tumor chemotherapeutic drugs, which have been used in clinical. DOX showed powerful therapeutic effects on a wide range of cancer,10 including acute leukemia, lymphoma, breast cancer, and lung cancer. The cytotoxic effects on tumor cells for DOX were exerted mainly by two mechanisms.11 One is inhibiting DNA replication and RNA synthesis in cells by inserting into the base pairs of the DNA strands.12 Another is that DOX can act as an electron acceptor in redox reactions and then be oxidized to semiquinone free radical, which causing oxidative damage to cellular membranes, proteins and DNA by producing ROS.12–14 However, the cytotoxicity of DOX result in some side effects, especially in myocardial lesions and cardiac failure.15–17 To reduce the side effects of DOX in vivo, various formulations of DOX have been investigated in clinical or preclinical fields including encapsulated in nanomaterial, such as liposome, protein and PLGA nanoparticles.18–20
Nanomaterials have shown to great potential in encapsulating and transporting drugs, penetrating cell membranes and releasing drugs in tumor cells through the enhanced permeability and retention (EPR) effect,21 which augment the accumulation of drugs in tumor cells but reduce that in normal cells.22 Ferritin (Fn), a universal intracellular protein that stores iron and releases it in a controlled fashion, was designed to encapsulate DOX for targeting delivery drugs to transferrin receptor 1 (TFR1) overexpressed tumor cells,23,24 human leukemia (HL-60), human colorectal carcinoma (Lovo), human breast adenocarcinoma (MDA-MB-468), human liver carcinoma (Hep G2), human cervical adenocarcinoma (HeLa), human breast (MCF-7), human small cell lung carcinoma (NCI-H69).25,26 Ferritin, with DOX loaded, could directly interact with TFR1 (ref. 27) on the membranes of tumor cells and subsequently internalize and release DOX within the cells. By this way, DOX was released in tumor cells for therapeutic research.24
In this study, DOX was loaded into iron saturated ferritin. The ferritin was designed as a tumor targeting drug carrier. And also, the intracellular iron concentration can be increased by ferritin, which is a promoter for ferroptosis.28 As DOX can cause oxidative damage to cell in the tumor cells, it will produce ROS.29 Combined these two processes, DOX loaded ferritin may have great potential in tumor cell killing.
Experimental section
Materials
Doxorubicin hydrochloride and ferritin from equine spleen were purchased from Sigma Aldrich (Shanghai, China). Erastin and ferrostatin-1 were obtained from Selleck (Shanghai, China). Deferoxamine was obtained from Novartis (Shanghai, China), glutathione reduced form (GSH) was purchased from Tokyo Chemical Industry (Shanghai, China). Fe3+(FeCl3·6H2O) was obtained from Sinopharm Chemical Reagent Co., Ltd, Float-A-Lyzer G2 Dialysis Cassettes (MWCO 3.5 kDa) and Tube-O-DIALYZER™ Micro Dialysis System (MWCO 50 kDa) were purchased from Sangon Biotech (Shanghai, China). PD-10 desalting columns were obtained from GE Healthcare. C11-BODIPY was obtained from Thermo Fisher. The kits for Cell Counting Kit-8 (CCK-8), enhanced BCA protein assay, and reactive oxygen species assay, anti-actin, goat anti-rabbit IgG (H + L)-HRP conjugate and goat anti-mouse IgG (H + L)-HRP conjugate were purchased from Beyotime Institute of Biotechnology (Jiangsu, China). Anti-GPX4 was obtained from Abcam. Anti-GPX4 and anti-actin were used as primary antibodies. Goat anti-rabbit IgG (H + L)-HRP conjugate and goat anti-mouse IgG (H + L)-HRP were used as secondary antibodies. All obtained chemicals were used without further purification.
Synthesis of ferritin-doxorubicin (Fn-DOX)
Ferritin was diluted into saline solution at a concentration of 5 mg mL−1 first. Then the pH value of the solution was adjusted to 4 approximately by 0.05 N HCl. 2 mL ferritin solution was mixed with 2 mL DOX aqueous solution (0.5 mg mL−1) and incubated for 1 h. After that, the products were run through a PD-10 column to remove free Dox. The pH value of the solution was then adjusted to 7 by 0.05 N NaOH.
The loaded Dox was quantified using a fluorescence spectrometer (LS55, PerkinElmer) by comparing to a standard curve.
Characterization
The size distribution of Fn-DOX were measured by dynamic light scattering (DLS) using a Zetasizer Nano ZSE (Malvern Instruments, Ltd.). The UV-vis absorption of the sample solutions was measured by a PerkinElmer Lambda 25 UV-vis spectrophotometer. High-performance liquid chromatography was performed with Waters 1525 equipped with 2998 photodiode array detector.
Drug release
Fn-DOX was placed into a dialysis tubes (MWCO 50 kDa) and dialysis in 50 mL PBS (pH 7.4 or 5) for 24 h. The solutions outside the dialysis tubes were collected at various time point. The DOX concentrations were determined by HPLC analysis.
Cell culture
Human hepatoma cell line (Hep G2), and Human colorectal adenocarcinoma grade II cell line (HT29) were obtained from the Cell Bank in Shanghai Institute of Cell Biology, China. The Hep G2 cells were cultured in a humidified atmosphere of a 37 °C incubator with 5% CO2 in DMEM medium supplemented with 10% fetal bovine serum and 0.1% penicillin–streptomycin. HT29 cells were cultured in McCoy's 5A medium supplemented with 10% fetal bovine serum and 0.1% penicillin–streptomycin.
Cell uptake for Fn-DOX
The uptake of Fn-DOX was evaluated by flow cytometric in Hep G2 and HT29 cells. Cells with media were added into 6-well plate (3 × 105 cells per well) and incubated for overnight at 37 °C. Cells were then treated with 0.5 μM free DOX for 2 h or Fn-DOX, with the same concentration of free DOX (0.5 μM), for 15, 30, 60, 120 min. All of cells were collected and evaluated by flow cytometer.
Cell viability assay
HT29 cells were seeded into 96-well plate (1 × 104 cells per well) and incubated for overnight at 37 °C. The cells were incubated with Fn-DOX, DOX, DOX-Fe (mixture of DOX and ferric chloride), and ferritin at different concentrations for 24 h at 37 °C, and then analyzed via the standard CCK-8 assay. To confirm Fn-DOX-mediated cell ferroptosis pathways, we selected several typical cell ferroptosis inhibitors for cell viability assays, including DFO, GSH and Fer-1. The cells were incubated with each inhibitor and Fn-DOX for 24 h. The concentrations of the inhibitors were used as follows: DFO, 500 μM; GSH, 5 mM; Fer-1, 10 μM. After the treatment, cell viabilities were assessed using the CCK-8 assay.
Cell lipid peroxide staining
HT29 cells were seeded with a density of 1 × 105 per well in 24-well plates and incubated overnight. Cells were treated with agents for 24 h. After incubation, the culture medium was replaced and cells were washed by PBS for three times. 2.5 mL of fresh medium containing the lipid peroxidation sensor C11-BODIPY was added into each well with the final concentration of 5 μM and incubating cells for another 30 min. After washed by PBS, the cells were observed using a fluorescence microscopy (Olympus).
In vitro ROS detection
The fluorescent probe DCFH-DA was used to detect the intracellular ROS changes. HT29 cells were seeded into 6-well plate (5 × 105 cells per well) and incubated for overnight at 37 °C. Then the cells were treated with DFO, GSH, Fer-1 and Fn-DOX for 24 h. A final concentration of 10 μM DCFH-DA mixed with fresh cell culture medium was added to the wells and the cells were incubated in a 37 °C incubator for 20 min and washed three times by PBS. Finally, cells were digested and dispersed in 500 μL PBS for flow cytometry study.
Western blot analysis
The samples were lysed. Cell lysates were prepared in Western and IP lysis buffer containing 1 mM PMSF, 20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, sodium pyrophosphate, β-glycerophosphate, EDTA, Na3VO4, leupeptin, and other inhibitors. The enhanced BCA protein assay kit was used to quantify protein levels. The protein samples with loading buffer were heated at 70 °C for 10 min and then run on 12% Bis-Tris gels (Invitrogen Novex). After transferred to a polyvinylidene fluoride (PVDF) membrane, proteins were incubated with primary antibodies as follows: anti-GPX4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15000), and anti-actin (1:1000) at 4 °C overnight. On next day, the samples were incubated with goat anti-rabbit-HRP or anti-mouse-HRP secondary antibodies. The relative quantity of proteins was analyzed by quantity one software and normalized to that of loading controls.
:15000), and anti-actin (1:1000) at 4 °C overnight. On next day, the samples were incubated with goat anti-rabbit-HRP or anti-mouse-HRP secondary antibodies. The relative quantity of proteins was analyzed by quantity one software and normalized to that of loading controls.
Statistical analysis
All data were tested at least three times independently. Each experiment in the cell death assay were performed by 3–6 replicates. All data were represented as means ± SD (n ≥ 3). Student's t-test was performed to assess the statistical significance. A p-value < 0.05 was considered statistically significant.
Results and discussion
Synthesis and characterization of Fn-DOX
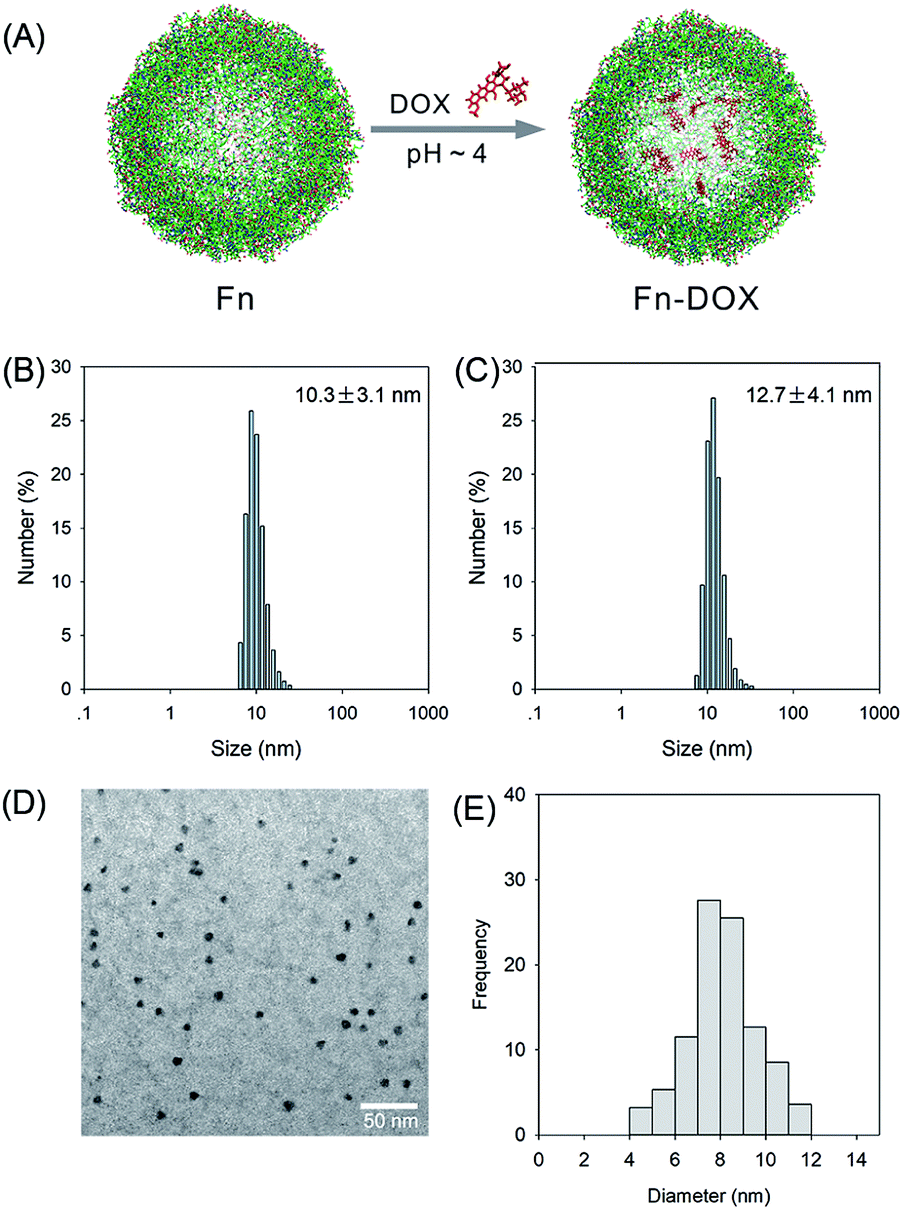
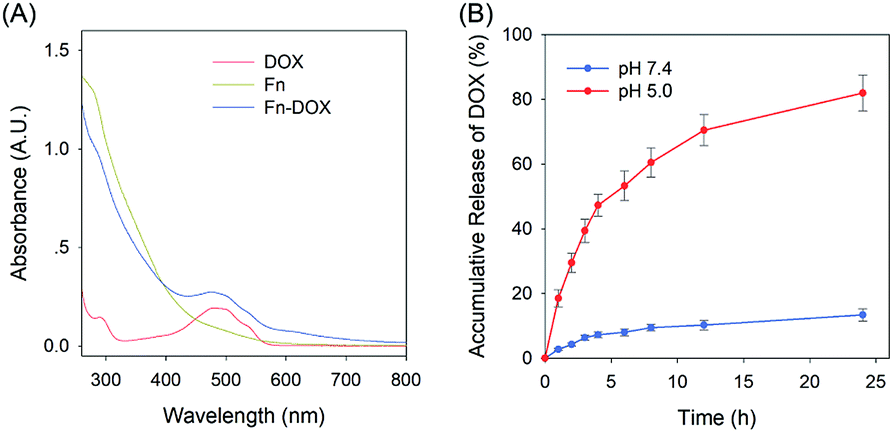
The fabrication process of Fn-DOX was illustrated in Fig. 1A. The hydrodynamic size of Fn-DOX was determined by dynamic light scattering (DLS). This resulted in an average hydrodynamic diameter of 12.7 ± 4.1 nm (Fig. 1C), which is a little larger than that of ferritin (10.3 ± 3.1 nm) (Fig. 1B). These results suggest that the Fn-DOX did not aggregate after drug loading and remained single protein particles. TEM characterization was also performed to determine the size distribution of Fn-DOX (Fig. 1D and E). It showed that the average size of Fn-DOX is around 8 nm with a narrow size distribution. The UV-vis-NIR spectra of Fn-DOX demonstrated a characteristic absorption peak at 490 nm, corresponding for the component of DOX in the Fn-DOX (Fig. 2A). The drug loading efficiency is 5.27% which determined by fluorescence spectrum. To simulate the extracellular and endolysosomal environments, the release of DOX from Fn-DOX was studied at pH 7.4 and pH 5.0, respectively (Fig. 2B). It was found that Fn-DOX was almost stable at pH 7.4 with only less than 10% DOX released after 24 h incubation. And as the pH decreased to pH 5, the DOX loaded in Fn-DOX exhibits rapid dissociation release compared to pH 7.4, the release of DOX reaches about 80% after 24 h. This pH-dependent drug release behavior of Fn-DOX reveal that the DOX can be released in endolysosome.
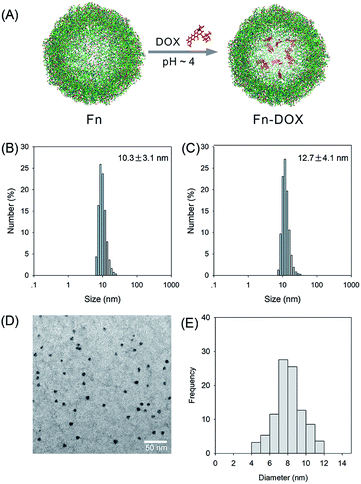 |
| | Fig. 1 (A) The schematic synthesis route of Fn-DOX nanoparticle by self-assembly. Size distribution of Fn (B) and Fn-DOX (C) determined by DLS. (D) TEM image of Fn-DOX. (E) Size distribution of Fn-DOX determined by TEM image. | |
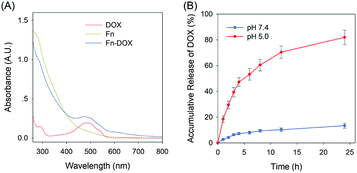 |
| | Fig. 2 (A) UV-Vis spectra of DOX, Fn and Fn-DOX in water. (B) Accumulation release of DOX from Fn-DOX under different pH conditions. | |
Selective uptake of Fn-DOX
Although varies of nanodrugs have been explored to kill cancer cells through ferroptosis, such as cisplatin loaded iron oxide,30 lipid hydroperoxide nanoparticles,31 and metal–organic network encapsulated with p53 plasmid,32 the targeting ability was been ignored. The transferrin receptor is present on the surface of many cells and mediates iron-containing ferritin from outside the cell. In order to verify the targeting capacity of Fn-DOX on tumor cells, we selected HT29 cells (human colon adenocarcinoma cell line) with high expression of transferrin receptor and Hep G2 cells (human liver hepatocyte carcinoma cell line) with low expression of transferrin receptor. The intracellular uptake of Fn-DOX and free DOX in both cell lines were compared by flow cytometry. As shown in Fig. 3, cellular uptake of free DOX is almost identical in these two cells, this result can be verified from the mean fluorescence intensity (MFI). Whereas the uptake of Fn-DOX in these two cells showed significant differences. Since HT29 cells have highly expressed transferrin receptors, more Fn-DOX can be transferred into the cells (Fig. 3C). In other words, high uptake of Fn-DOX is dependent on intracellularly high expression of transferrin receptors and exhibits time-dependent uptake. As shown in Fig. 3D, when Fn-DOX was incubated with HT29 for 120 min, the mean fluorescence intensity of the cells was approximately 16 times that of the control group. These results showed the great potential of Fn-DOX for targeting killing of cancer cells.
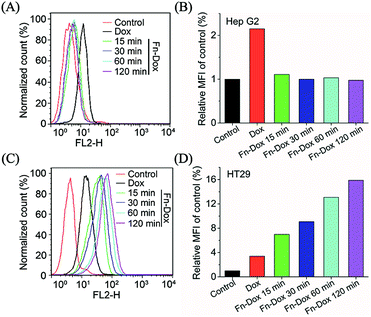 |
| | Fig. 3 (A) Uptake of DOX and Fn-DOX on Hep G2 (low expression of TFR) cells. (B) Relative MFI of control in (A) analyzed by flow cytometry. (C) Uptake of DOX and Fn-DOX on HT29 (high expression of TFR) cells. (D) Relative MFI of control in (C) analyzed by flow cytometry. | |
Cytotoxicity in HT29 cells
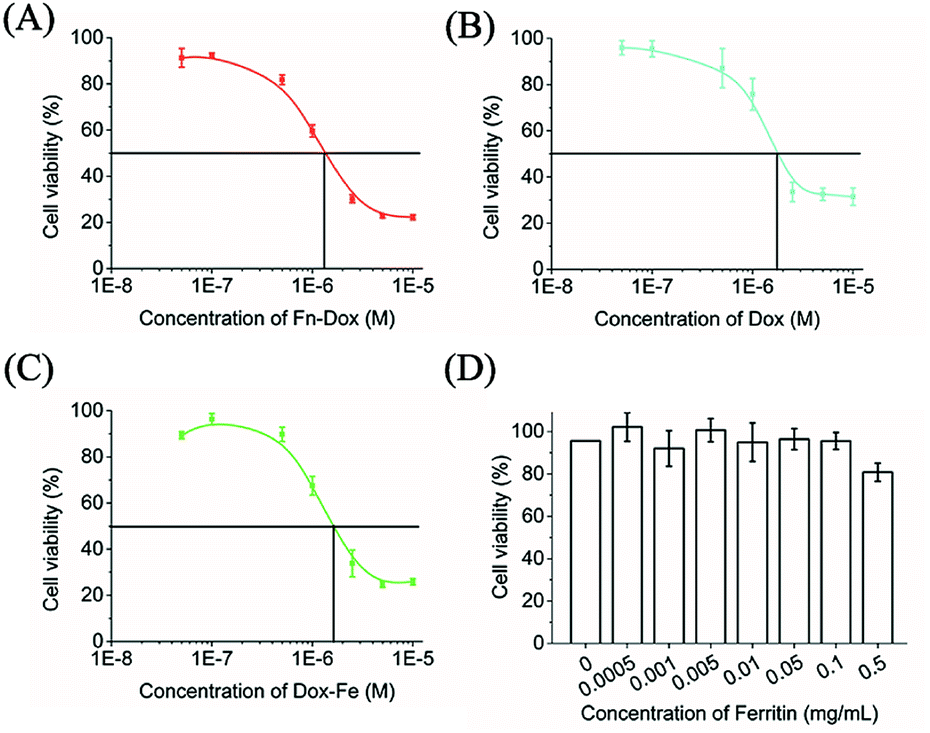
The cytotoxicity of Fn-DOX, DOX, DOX-Fe, and ferritin was evaluated in HT29 cells (Fig. 3A–D). Ferritin without DOX has almost no toxicity to HT29 cells within 0.5 mg mL−1, and the half maximal inhibitory concentration (IC50) were 1.683, 1.330 μM for DOX, and DOX-Fe, respectively. The IC50 of Fn-DOX was 1.349 μM, which was closed to DOX-Fe. This may be due to the dissociation of Fn-DOX in the acidic environment in endosome and lysosome of tumor cells and then DOX released from ferritin. And the transferrin receptors targeting ability of Fn-DOX may also promote the cytotoxicity. These results suggest Fn-DOX got a strong cancer killing ability just like the free DOX (Fig. 4).
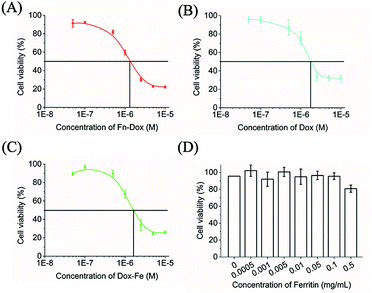 |
| | Fig. 4 In vitro cytotoxicity of Fn-DOX (A), DOX (B), DOX-Fe (C), and Fn (D) in HT29 cells for 24 h. | |
Ferroptosis induced by Fn-DOX
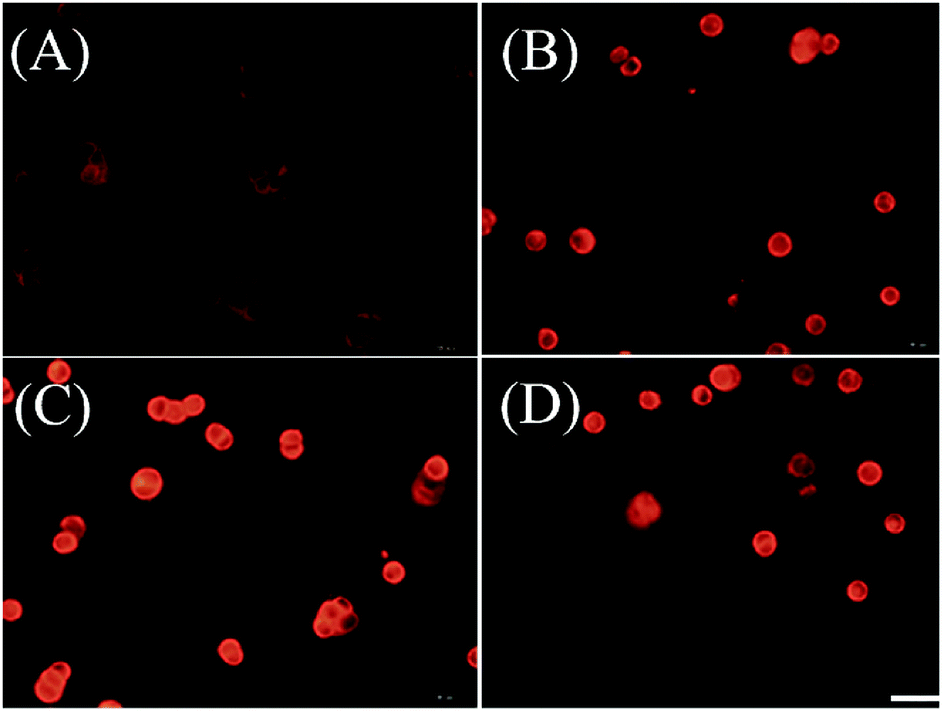
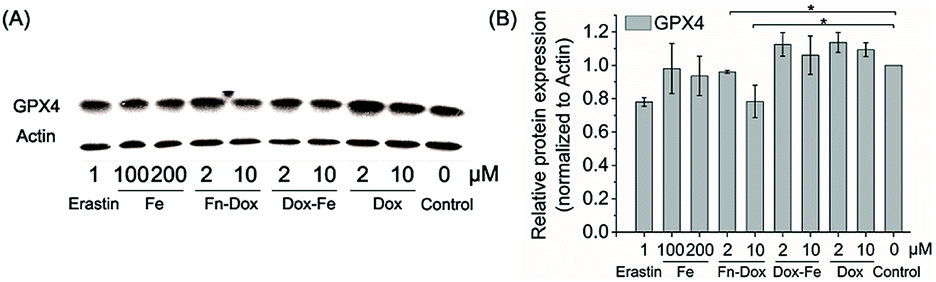
We further examined if cell death was occurring by ferroptosis pathway. Membrane lipid oxidation is one of the hallmarks of cell ferroptosis.1,3 The level of intracellular membrane lipid oxidation was measured by fluorescence microscopy. C11-BODIPY as a membrane lipid oxidation detection probe, was used to incubate with the HT29 cells which was first treated by Fn-DOX, DOX, or DOX-Fe. As shown in Fig. 5, the Fn-DOX group showed highest fluorescence intensity compared with the control group, DOX, and DOX-Fe which means highest membrane lipid oxidation level in cells induced by Fn-DOX. In addition, detection of changes in protein levels is also necessary to determine the form of cell death. GPX4 is a phospholipid hydroperoxidase that protects cells against membrane lipid peroxidation.2 Erastin caused GPX4 reduced synthesis in HT29 cells, suggesting that the protein reduction is involved in ferroptosis. Similarly, GPX4 expression level was reduced in Fn-DOX-treated cells, approximately to 70% of the control group. However, cells treated with DOX, Fe ions alone, or mixed with DOX and Fe, GPX4 levels were not significantly decreased (Fig. 6). These results indicate that ferroptosis can be induced by Fn-DOX.
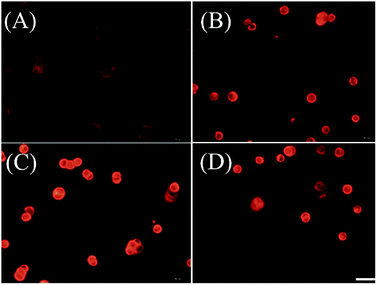 |
| | Fig. 5 Lipid peroxide in HT29 cells which were stained by C11-BODIPY. (A) Control, (B) DOX, (C) Fn-DOX, and (D) DOX-Fe. The DOX concentration in (B)–(D) is 2 μM. Scale bar is 20 μm. | |
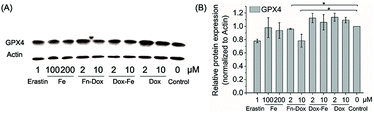 |
| | Fig. 6 (A) The expression of GPX4 in HT29 cells treated by different agents determined by western blot. (B) Quantitation of GPX4 proteins normalized to actin in by using Image J software. Values were presented as mean ± SD, each analysis was repeated three times, *p < 0.05. | |
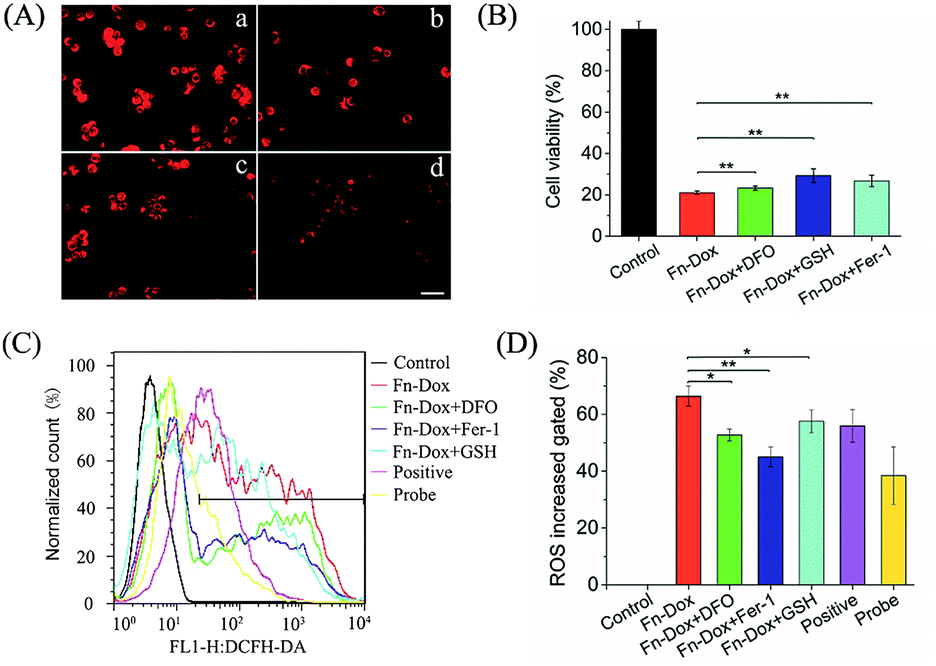
Inhibition of Fn-DOX induced ferroptosis
Further studies found that cells treated with Fn-DOX while adding ferroptosis inhibitor, such as DFO, GSH and Fer-1, the increased red fluorescence of C11-BODIPY can be inhibited (Fig. 7A). Next, we incubated the cells with a medium containing ferroptosis inhibitor and Fn-DOX to investigate whether the cytotoxicity of Fn-DOX was induced by ferroptosis. As shown in the Fig. 7B, DFO, GSH, and Fer-1 reduced the cytotoxicity which means part of the cytotoxicity was induced by ferroptosis process. On the other hand, a large number of reactive oxygen species (ROS) accumulate during the process of ferroptosis in cells,1 and therefore we tested it. The dichlorofluorescein diacetate (DCFH-DA) probe is a non-labeled oxidation-sensitive fluorescent probe that can be used to determine the level of intracellular ROS.33 The levels of ROS in Fn-DOX-treated cells showed a significant increase which was detected by flow cytometry (Fig. 7C). The mean fluorescence intensity of Fn-DOX-treated cells is about 65-fold higher than that of the control group. However, with the presence of DFO, GSH and Fer-1, the fluorescence intensity decreased to 50, 58, and 47-fold higher than that of the control group in Fig. 5D. In general, Fn-DOX induces cell death dependent on ferroptosis which caused by accumulation of lipid peroxidation. These results suggest that ROS generation was also caused by iron of ferritin. And this has a critical role in Fn-DOX-induced ferroptosis. Fn-DOX induces cells to undergo a ferroptosis process that is dependent on ROS accumulation and GPX4 decline. And combined the apoptosis induced by doxorubicin, the tumor cells can be killed efficiently by Fn-DOX.
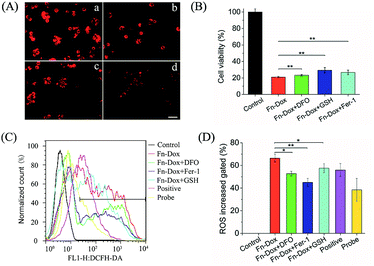 |
| | Fig. 7 (A) Lipid peroxide in HT29 cells exposed to Fn-DOX in the presence of ferroptosis inhibitors, including DFO, GSH, and Fer-1 which were stained by C11-BODIPY. (a) Fn-DOX, (b) Fn-DOX + DFO, (c) Fn-DOX + GSH, (d) Fn-DOX + Fer-1. Scale bar is 20 μm. (B) In vitro cytotoxicity of Fn-DOX in HT29 cells for 24 h in the presence of ferroptosis inhibitors, including DFO, GSH, and Fer-1. (C) ROS stained by DCFH-DA in HT29 cells treated with Fn-DOX in the presence of ferroptosis inhibitors, then were analyzed by flow cytometry. (D) Relative MFI of control in (A) analyzed by flow cytometry. The concentrations of DFO, GSH, Fer-1 were 500 μM, 5 mM, and 10 μM. Values represented as means ± SD (n = 5). *p < 0.05, **p < 0.01. | |
Conclusions
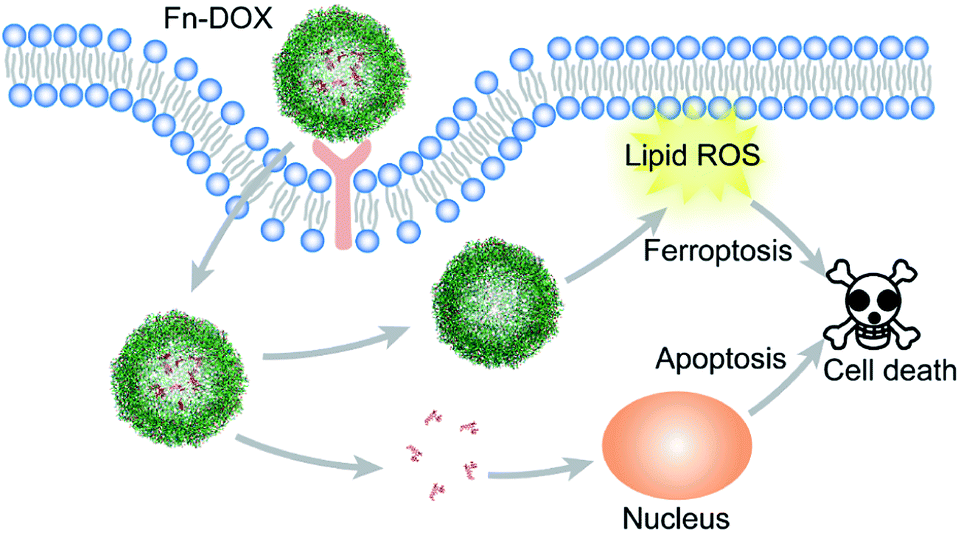
In summary, we designed DOX loaded iron saturated ferritin for cancer treatment (Scheme 1). The small size nanodrug with 12.7 nm hydrodynamic radius showed pH responsive drug release behavior. The HT29 cell targeting capability of Fn-DOX was confirmed by FACS measurement of the instinct fluorescence of DOX. The ferroptosis induction process by Fn-DOX was proved by fluorescent sensor and western blot. Furthermore, it was also confirmed by inhibitors of ferroptosis. And the cytotoxicity of Fn-DOX was similar to free DOX. This ferroptosis enhanced targeting killing of cancer cell by Fn-DOX provides a new insight into ferritin-based drug delivery system.
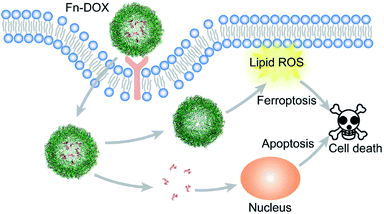 |
| | Scheme 1 Schematic illustration of cell death induction by Fn-DOX. | |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support was from the National Natural Science Foundation of China (31671035, 51803082, 31971316), the National Significant New Drugs Creation Program (2017ZX09304021), the Jiangsu Province Foundation (BK20170204, BK20161137), the Jiangsu Provincial Medical Innovation Team (CXTDA2017024), the Jiangsu Provincial Commission of Health and Family Planning Foundation (LGY2017088, H2018069, H2017031), the Jiangsu Provincial Key Medical Discipline (ZDXKA2016017), Wuxi Science and Technology Development Foundation (WX18IIAN046), the Wuxi Commission of Health and Family Planning Foundation (Q201729) and the Innovation Capacity Development Plan of Jiangsu Province (BM2018023).
Notes and references
- S. J. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley and W. S. Yang, Cell, 2012, 149, 1060–1072 CrossRef CAS PubMed.
- W. S. Yang, R. Sriramaratnam, M. E. Welsch, K. Shimada, R. Skouta, V. S. Viswanathan, J. H. Cheah, P. A. Clemons, A. F. Shamji and C. B. Clish, Cell, 2014, 156, 317–331 CrossRef CAS PubMed.
- N. Yagoda, M. V. Rechenberg, E. Zaganjor, A. J. Bauer, S. Y. Wan, D. J. Fridman, A. J. Wolpaw, I. Smukste, J. M. Peltier and J. J. Boniface, Nature, 2007, 447, 864–868 CrossRef PubMed.
- S. Y. Wan and B. R. Stockwell, Chem. Biol., 2008, 15, 234–245 CrossRef PubMed.
- A. J. Wolpaw, K. Shimada, R. Skouta, M. E. Welsch, U. D. Akavia, D. Pe'Er, F. Shaik, J. C. Bulinski and B. R. Stockwell, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16151–16152 CrossRef CAS PubMed.
- S. J. Dixon, Immunol. Rev., 2017, 277, 150–157 CrossRef CAS PubMed.
- Z. Cheng and Y. Li, ChemInform, 2010, 107, 748–766 Search PubMed.
- A. Seiler, M. Schneider, H. Förster, S. Roth, E. K. Wirth, C. Culmsee, N. Plesnila, E. Kremmer, O. Rådmark and W. Wurst, Cell Metab., 2008, 8, 237–248 CrossRef CAS PubMed.
- S. J. Dixon, D. N. Patel, M. Welsch, R. Skouta, E. D. Lee, M. Hayano, A. G. Thomas, C. E. Gleason, N. P. Tatonetti and B. S. Slusher, eLife, 2014, 3, e02523 CrossRef PubMed.
- P. Mohan and N. Rapoport, Mol. Pharmaceutics, 2010, 7, 1959–1973 CrossRef CAS PubMed.
- M. Cagel, E. Grotz, E. Bernabeu, M. A. Moretton and D. A. Chiappetta, Drug discovery today, 2017, 22, 270–281 CrossRef CAS PubMed.
- C. F. Thorn, C. Oshiro, S. Marsh, T. Hernandezboussard, H. Mcleod, T. E. Klein and R. B. Altman, Pharmacogenetics Genom., 2011, 21, 440–446 CrossRef CAS PubMed.
- D. A. Gewirtz, Biochem. Pharmacol., 1999, 57, 727–741 CrossRef CAS PubMed.
- J. H. Doroshow, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 4514–4518 CrossRef CAS PubMed.
- M. J. Aiken, V. Suhag, C. A. Garcia, E. Acio, S. Moreau, D. A. Priebat, S. P. Chennupati and N. D. Van, Clin. Nucl. Med., 2009, 34, 762–767 CrossRef PubMed.
- Y. Ichikawa, M. Ghanefar, M. Bayeva, R. Wu, A. Khechaduri, S. V. Naga Prasad, R. K. Mutharasan, T. J. Naik and H. Ardehali, J. Clin. Invest., 2014, 124, 617–630 CrossRef CAS PubMed.
- A. M. Rahman, S. W. Yusuf and M. S. Ewer, Int. J. Nanomed., 2007, 2, 567–583 CAS.
- A. Chanan-Khan, J. Szebeni, S. Savay, L. Liebes, N. M. Rafique, C. R. Alving and F. M. Muggia, Ann. Oncol., 2003, 14, 1430–1437 CrossRef CAS PubMed.
- K. Schmidt, B. R. Gardill, A. Kern, P. Kirchweger, M. Börsch and Y. A. Muller, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5744–5749 CrossRef CAS PubMed.
- S. Sengupta, D. Eavarone, I. Capila, G. Zhao, N. Watson, T. Kiziltepe and R. Sasisekharan, Nature, 2005, 436, 568–572 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, R. Margalit, O. C. Farokhzad and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- Y. Yuan, T. Cai, X. Xia, R. Zhang, P. Chiba and Y. Cai, Drug Delivery, 2016, 23, 3350–3357 CrossRef CAS PubMed.
- M. Liang, K. Fan, M. Zhou, D. Duan, J. Zheng, D. Yang, J. Feng and X. Yan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14900–14905 CrossRef CAS PubMed.
- V. Damiani, E. Falvo, G. Fracasso, L. Federici, M. Pitea, V. D. Laurenzi, G. Sala and P. Ceci, Int. J. Mol. Sci., 2017, 18, 1555 CrossRef CAS PubMed.
- T. R. Daniels, T. Delgado, G. Helguera and M. L. Penichet, Clin. Immunol., 2006, 121, 159–176 CrossRef CAS PubMed.
- T. R. Daniels, T. Delgado, J. A. Rodriguez, G. Helguera and M. L. Penichet, Clin. Immunol., 2006, 121, 144–158 CrossRef CAS PubMed.
- K. Fan, C. Cao, Y. Pan, D. Lu, D. Yang, J. Feng, L. Song, M. Liang and X. Yan, Nat. Nanotechnol., 2012, 7, 459–464 CrossRef CAS PubMed.
- G. O. Latunde-Dada, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 1893–1900 CrossRef CAS PubMed.
- S. Y. Kim, S. J. Kim, B. J. Kim, S. Y. Rah, S. M. Chung, M. J. Im and U. H. Kim, Exp. Mol. Med., 2006, 38, 535–545 CrossRef CAS PubMed.
- P. Ma, H. Xiao, C. Yu, J. Liu, Z. Cheng, H. Song, X. Zhang, C. Li, J. Wang and Z. Gu, Nano Lett., 2017, 17, 928–937 CrossRef CAS PubMed.
- Z. Zhou, J. Song, R. Tian, Z. Yang, G. Yu, L. Lin, G. Zhang, W. Fan, F. Zhang and G. Niu, Angew. Chem., Int. Ed. Engl., 2017, 129, 6492–6496 CrossRef PubMed.
- D. W. Zheng, Q. Lei, J. Y. Zhu, J. X. Fan, C. X. Li, C. Li, Z. Xu, S. X. Cheng and X. Z. Zhang, Nano Lett., 2017, 17, 284–291 CrossRef CAS PubMed.
- S. Ortelli, A. L. Costa, P. Matteucci, M. R. Miller, M. Blosi, D. Gardini, S. A. M. Tofail, L. Tran, D. Tonelli and C. A. Poland, RSC Adv., 2018, 8, 40369–40377 RSC.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Yaoqi Li†,
Xinyu Wang
,
Yaoqi Li†,
Xinyu Wang