
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural characterization of an amorphous VS4 and its lithiation/delithiation behavior studied by solid-state NMR spectroscopy†
Keiji Shimoda *a,
Kazuto Koganeib,
Tomonari Takeuchib,
Toshiyuki Matsunagaa,
Miwa Murakamia,
Hikari Sakaebeb,
Hironori Kobayashib and
Eiichiro Matsubarac
*a,
Kazuto Koganeib,
Tomonari Takeuchib,
Toshiyuki Matsunagaa,
Miwa Murakamia,
Hikari Sakaebeb,
Hironori Kobayashib and
Eiichiro Matsubarac
aOffice of Society-Academia Collaboration for Innovation, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan. E-mail: k-shimoda@saci.kyoto-u.ac.jp; Fax: +81-774-38-4996; Tel: +81-774-38-4967
bNational Institute of Advanced Industrial Science and Technology, Ikeda, Osaka 563-8577, Japan
cDepartment of Materials Science and Engineering, Kyoto University, Kyoto 606-8501, Japan
First published on 5th August 2019
Abstract
Vanadium sulfide (VS4) is one of the promising positive electrode materials for next-generation rechargeable lithium-ion batteries because of its high theoretical capacity (1196 mA h g−1). Crystalline VS4 has a unique structure, in which the Peierls-distorted one-dimensional chains of V–V bonds along the c axis are loosely connected to each other through van der Waals interactions. In this study, an amorphous VS4 is prepared by mechanical milling of the crystalline material, and its lithiation/delithiation behavior is investigated by solid-state nuclear magnetic resonance (NMR) spectroscopy. The amorphous VS4 shows a chain structure similar to that of crystalline VS4. The amorphous host structure is found to change drastically during the lithiation process to form Li3VS4: the V ions become tetrahedrally coordinated by S ions, in which the valence states of V and S ions simultaneously change from V4+ to V5+ and S− to S2−, respectively. When the Li insertion proceeds further, the valence state of V ions is reduced. After the 1st cycle, the amorphous VS4 recovers to the chain-like structure although it is highly disordered. No conversion to elemental V is observed, and a high capacity of 700 mA h g−1 is reversibly delivered between 1.5 and 2.6 V.
1. Introduction
Elemental sulfur is one of the most promising electrode materials for next-generation rechargeable lithium-ion batteries, because of its high theoretical capacity of 1672 mA h g−1. Its corresponding electrochemical reaction can be expressed as S + 2Li+ + 2e− = Li2S, and involves the S0/S2− redox couple at an average potential of 2.15 V vs. Li/Li+.1 However, two well-known problems have yet to be solved. The first is that both S and Li2S are electronically resistive, which gives rise to relatively poor electrochemical performance in practical applications. The other problem is the dissolution of lithium polysulfide intermediates Li2Sx (2 < x < 8) in nonaqueous electrolyte solutions during the charge–discharge cycles, leading to capacity fading.1–3To enhance the electronic conductivity of S, composite electrodes with carbonaceous materials have been developed, and excellent electrochemical properties were reported for a highly ordered nanostructured carbon–sulfur composite electrode.4–6 As an alternative approach, considerable attention has been paid to transition metal (TM) sulfides, such as TiS2, FeS2, and MoS2,7–16 which have been examined as positive or negative electrode materials for conventional lithium-ion batteries. TM sulfide-based positive electrodes were introduced for Li–S batteries, in which the TM ions were considered to act merely as electron conductive agents. New sulfur-rich electrode materials based on TM sulfides have been developed in recent years.17–23 For example, amorphous TiS4 was synthesized as a cathode material by a mechanochemical process from a TiS2 and S mixture and showed a stable charge–discharge profile in a nonaqueous electrolyte solution.17 An Fe-doped Li2S-based material, Li8FeS5, showed a high discharge capacity close to 730 mA h g−1.21 These studies suggest that the formation of metal–sulfur bonds is important to suppress polysulfide dissolution.
Vanadium sulfide (VS4, mineral name: patronite) has a unique crystalline form, in which infinite chains of V–V bonds along the c axis show Peierls distortion with alternate V–V distances of 2.83 and 3.22 Å, and each V ion is coordinated by 4 disulfide anions [S2]2−.24 Initial discharge and charge capacities of the crystalline VS4 (c-VS4) electrode composite with reduced graphene oxide (VS4-rGO) were reported to be 1669 and 1105 mA h g−1 at a current rate of 0.1C, and an excellent charge capacity retention of 954 mA h g−1 was obtained after 100 cycles.25 Good capacity retention at high current rates was also demonstrated.25,26 The charge–discharge mechanism of the c-VS4 electrode was carefully studied by pair distribution function (PDF) analysis of synchrotron-radiation X-ray scattering data and X-ray absorption near-edge spectroscopy (XANES) as well as X-ray diffraction (XRD):27 in situ XRD measurements showed that c-VS4 started to transform into a highly disordered or amorphous phase from the beginning of lithiation. The results of X-ray PDF and S K-edge XANES analyses suggest the breaking of S–S dimers accompanied by the formation of [VS4]3− tetrahedral units during lithiation, and the subsequent formation of face-centered cubic (fcc) V and Li2S at the end of discharge at 0 V.27 These reactions were proved to be partially reversible, in contrast with the previous speculation that the formed V is inactive and Li2S is reversibly delithiated/lithiated.26 Solid-state nuclear magnetic resonance (NMR) was also successfully applied to understand the structural changes around Li and V ions during the lithiation/delithiation process:27 the 51V signal of c-VS4 disappeared and metallic V and Li2S were observed during the lithiation. Recently, our group examined the charge–discharge properties of the c-VS4 electrode without rGO in a potential window between 1.0 and 3.0 V.28 The conversion to elemental V was not observed within this potential window, and the pristine VS4-like local structure, although non-crystalline, was recovered after the initial charge–discharge cycle. Moreover, it was found that c-VS4 transforms into an amorphous phase by mechanical milling.29 The charge–discharge profile of the amorphous VS4 (a-VS4) resembles that of c-VS4, and its electrochemical properties were moderately improved.29 However, the exact structural information of the pristine a-VS4 and its charge–discharge mechanism are still unclear. As shown in a previous study,27 NMR is a suitable technique to characterize the framework structure of V ions as well as the non-framework environment of labile Li ions. Therefore, in the present study, we investigate the structure of an amorphous VS4 and its changes during the lithiation/delithiation process using solid-state NMR spectroscopy.
2. Experimental
Crystalline VS4 was first synthesized without rGO from a mixture of V2S3 (99%, Kojundo Chemical Laboratory) and S (99.9%, Wako) in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6, which was sealed in a glass tube under vacuum and heated two times at 400 °C for 12 h.28 After cooling to room temperature, excessive sulfur was removed by annealing at 200 °C under vacuum. Then, the amorphous VS4 was prepared from the sintered sample by mechanical milling for 40 h at 270 rpm in a planetary ball mill (Pulverisette 7, Fritsch). The working electrode consisted of a mixture of the active material (a-VS4), Ketjen Black (KB), and polytetrafluoroethylene (PTFE) binder in a weight ratio of 59:29:12 on a Ni mesh current collector. A Li foil (0.2 mm in thickness, >99.9%, Honjo Metal) was used as the negative electrode, and a microporous polyolefin sheet was chosen as the separator. A solution of 1 M LiPF6 dissolved in a 1:1 volume ratio mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) was used as the electrolyte solution (Tomiyama Pure Chemical Industries, battery grade). Coin-type or laminate-type cells were assembled in an Ar-filled glove box. The electrochemical measurements were performed at 30 °C. The cells were galvanostatically cycled between 1.5 and 2.6 V at a current density of 59.8 mA g−1 (0.05C rate) using a TOSCAT-3100 battery-testing system (Toyo System). They were then carefully disassembled at selected discharge/charge states in the glove box and rinsed with DMC to remove the residual electrolyte solution.
:6, which was sealed in a glass tube under vacuum and heated two times at 400 °C for 12 h.28 After cooling to room temperature, excessive sulfur was removed by annealing at 200 °C under vacuum. Then, the amorphous VS4 was prepared from the sintered sample by mechanical milling for 40 h at 270 rpm in a planetary ball mill (Pulverisette 7, Fritsch). The working electrode consisted of a mixture of the active material (a-VS4), Ketjen Black (KB), and polytetrafluoroethylene (PTFE) binder in a weight ratio of 59:29:12 on a Ni mesh current collector. A Li foil (0.2 mm in thickness, >99.9%, Honjo Metal) was used as the negative electrode, and a microporous polyolefin sheet was chosen as the separator. A solution of 1 M LiPF6 dissolved in a 1:1 volume ratio mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) was used as the electrolyte solution (Tomiyama Pure Chemical Industries, battery grade). Coin-type or laminate-type cells were assembled in an Ar-filled glove box. The electrochemical measurements were performed at 30 °C. The cells were galvanostatically cycled between 1.5 and 2.6 V at a current density of 59.8 mA g−1 (0.05C rate) using a TOSCAT-3100 battery-testing system (Toyo System). They were then carefully disassembled at selected discharge/charge states in the glove box and rinsed with DMC to remove the residual electrolyte solution.
XRD measurements were carried out for sealed samples on a D8 ADVANCE X-ray diffractometer (Bruker AXS) with a Cu Kα source and a SmartLab diffractometer (Rigaku) with a Mo Kα source. Raman spectra were acquired using a LabRAM HR-800 spectrometer (Horiba-Jobin Yvon) with a 50× lens, 600 and 1800 grooves per mm grating, and an excitation wavelength of 632.8 nm (He–Ne laser). The electrode samples were sealed in transparent gas-barrier films. The laser power was reduced to less than 1 mW to avoid laser-induced degradation on the targeted particles. The exposure time was 10 s × 10 times on more than ten different particles. XPS measurements were performed on a PHI5000 VersaProbe II (ULVAC-PHI) photoelectron spectrometer with monochromatic Al Kα radiation (1486.6 eV). The samples were transferred to an ultra-high vacuum sample chamber (<4 × 10−7 Pa) without exposing them to air. The pass energy was set to 23.5 eV. Dual-beam charge neutralization (simultaneous irradiation with low-energy electrons and Ar+ ion beams) was applied to suppress sample charging. The spectra were acquired without Ar+ ion sputtering to avoid sample damage. The binding energies were calibrated with respect to the C 1s signal from KB at 284.6 eV.
NMR spectra were acquired on a DD2 600 spectrometer (Agilent Technologies) at a magnetic field of 14.1 T. Operando 7Li NMR measurements were performed with a homemade wide-bore static probe, in which a flat laminate-type cell was placed in the center of a 10 mm-diameter solenoid coil. A Hahn echo pulse sequence was used, with a first pulse width of 4.5 μs and an echo decay of 8 μs. Each spectrum was averaged over 30 min (scan number of 1792 with a relaxation delay of 1 s). 7Li and 51V magic-angle spinning (MAS) NMR spectra were acquired with a wide-bore T3 MAS probe (Agilent Technologies). Powder samples were packed into 1.2 mmϕ MAS ZrO2 rotors with airtight caps in an Ar-filled glove box and spun at a rate of 60 kHz during the measurements. A rotor-synchronized Hahn echo pulse sequence (π/2-τ-π-τ-acq) was used with a π/2 pulse width of 1.0 μs and a relaxation delay of 5 s for 7Li measurements (longer delay for a full signal detection including byproduct components), and 0.5 μs and 0.5 s for 51V measurements, respectively. All spectra were referenced to 1 M LiCl solution at 0.0 ppm and V2O5 at −612 ppm (a secondary solid reference relative to neat VOCl3 at 0.0 ppm) for 7Li and 51V measurements, respectively.
3. Results and discussion
3.1. Structural characterization of a-VS4
Fig. 1a compares the XRD profiles of c-VS4 and a-VS4. The diffraction pattern of c-VS4 prepared in this study was indexed to the space group C2/c, which was recently revised against I2/c, as reported previously.24,30 The diffraction peaks disappeared after mechanical milling of c-VS4, and a broad halo was observed at ∼15°. This indicates mechanochemical amorphization of the VS4 crystal structure (i.e., a-VS4). We note that the crystalline component started to decrease after the mechanical milling of 10 h and completely disappeared with the milling time of up to 40 h. Needle-shaped particles characteristic of c-VS4 was completely broken and pulverized on the scanning electron microscope (SEM) images (not shown). The amorphous structure of a-VS4 was examined by X-ray PDF analysis using a Mo Kα source (Fig. 1a, lower panel). The interatomic distances in g(r) functions of c-VS4 and a-VS4 were similar to each other up to 5 Å: a shoulder at ∼2.0 Å and a peak at 2.4 Å are assigned to S–S distance in a disulfide bond and V–S distance, respectively. A longer V–V distance in the Peierls-distorted chain and a second-neighbor S–S distance are merged at 3.3 Å. This indicates that the local structure of a-VS4 resemble that of c-VS4.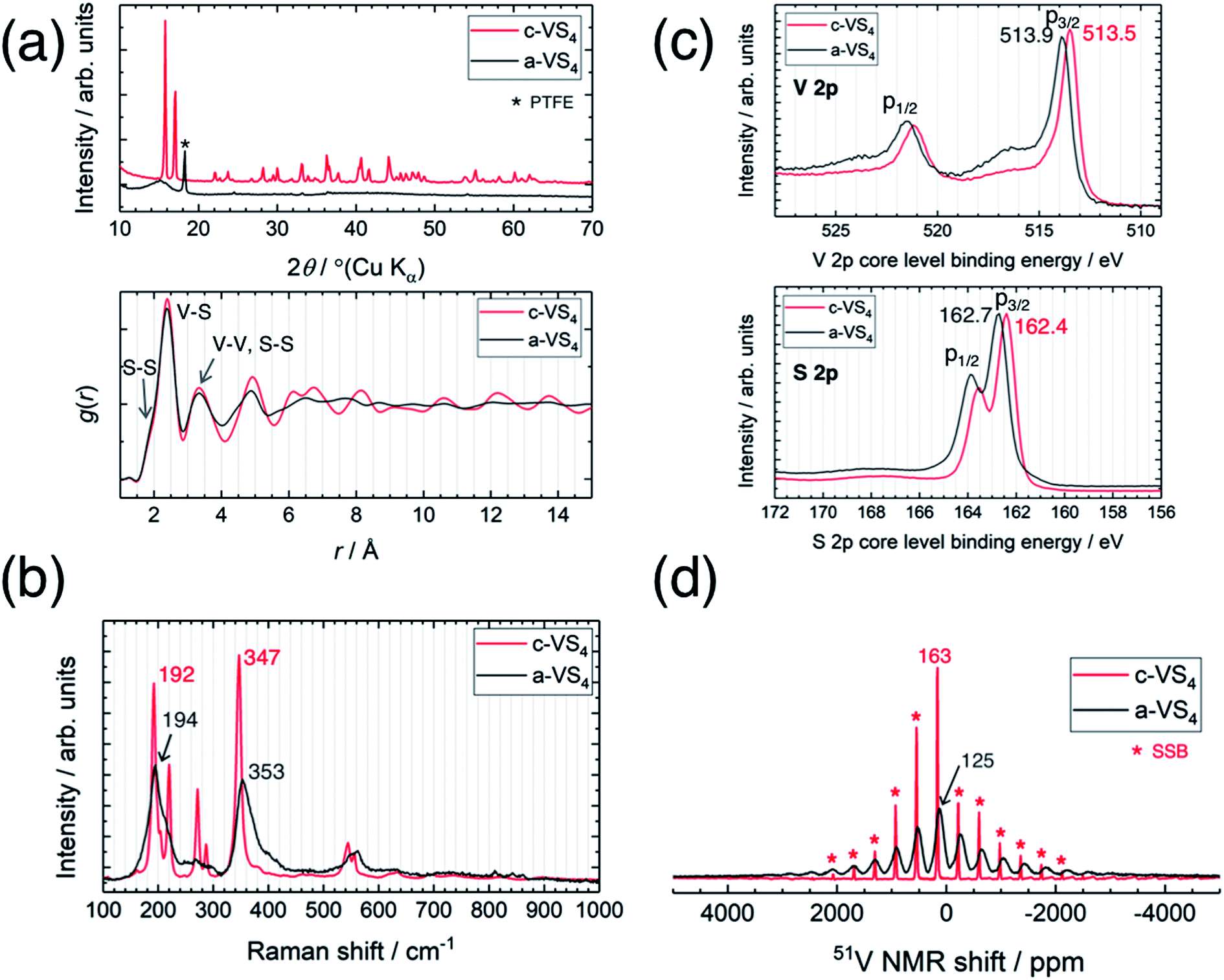 | ||
| Fig. 1 Comparison between crystalline and amorphous VS4. (a) XRD profiles (upper panel) and PDF patterns (lower panel), (b) Raman spectra, (c) XPS spectra, and (d) 51V MAS NMR spectra. | ||
The Raman spectra of c-VS4 and a-VS4 are shown in Fig. 1b. First, we obtained a high-quality and high-resolution spectrum for c-VS4 using reduced laser power. Intense peaks were observed at 192, 220, 272, 287, 347, 544, and 555 cm−1. We confirmed that these vibrational peaks arise from c-VS4 based on the theoretical calculation of Raman-active frequencies and intensities (Fig. S1†). The Raman spectrum of a-VS4 shows broader features than that of c-VS4, proving a similar structural unit with local disorder. Two intense bands at 194 and 353 cm−1 and a weaker band at ∼560 cm−1 are attributed to V–S bond stretching mode, breathing motion of the V2S4 cage, and S–S bond stretching/twisting, respectively.31
The V 2p and S 2p XPS spectra of c-VS4 and a-VS4 are compared in Fig. 1c. The V 2p core-level spectra are split into two components, p3/2 and p1/2, due to spin–orbit coupling. The V 2p3/2 peak position of a-VS4 (513.9 eV) is close to that of c-VS4 (513.5 eV), which is attributed to the V4+ valence state (514.1 eV in ref. 32). Similarly, the S 2p core-level spectra show a doublet peak, p3/2 and p1/2 with an intensity ratio of 2:1, due to spin–orbit coupling. The S 2p3/2 peak position of a-VS4 (162.7 eV) is close to that of c-VS4 (162.4 eV), which is assigned to the S− (or [S2]2−) state.32,33 Therefore, the valence state of a-VS4 is almost the same as that of c-VS4. In addition, the S/V atomic ratios of c- and a-VS4 were estimated to be 4.0(1) and 3.7(1), respectively.
Fig. 1d shows the 51V MAS NMR spectra of c-VS4 and a-VS4. The spectrum of c-VS4 shows a sharp signal at 163 ppm with significant spinning sideband manifolds (SSBs). It is important to note that the valence state of V4+ in c-VS4 is expected to be paramagnetic (the 3d1 electronic configuration). However, if it is indeed paramagnetic, the 51V signal would be almost undetectable due to ultrafast relaxation of free induction decay (FID). The one-dimensional linear chain structure of c-VS4 is stabilized by Peierls distortion, which gives rise to alternating short and long V–V distances.27 Electronic spin-pairing occurs within the short V–V bond, resulting in a diamagnetic shift in the 51V spectrum.27 The spectrum of a-VS4 shows a relatively broad signal at 125 ppm with significant SSBs. The isotropic shift of a-VS4 is close (but not equal) in position to that of c-VS4, suggesting that a-VS4 also has the Peierls-distorted chain structure with some configuration disordering.
3.2. Structural changes during charge–discharge cycling
Fig. 2 shows the charge–discharge profile of the Li//a-VS4 cell. The initial discharge capacity at 1.5 V was ∼750 mA h g−1 (Fig. S2†), corresponding to the average composition of Li5VS4. It should be noted that the theoretical capacity of 1196 mA h g−1 (VS4 + 8Li+ + 8e− = V + 4Li2S) is achieved at a voltage below 0.5 V.25–27 Within the potential window in the present study, a plateau-like feature was observed. The XRD profiles showed that the lithiated/delithiated a-VS4 samples preserved the amorphous structure (Fig. S3†). Structural evaluation of these materials is currently in progress using synchrotron-radiation X-ray PDF analysis.29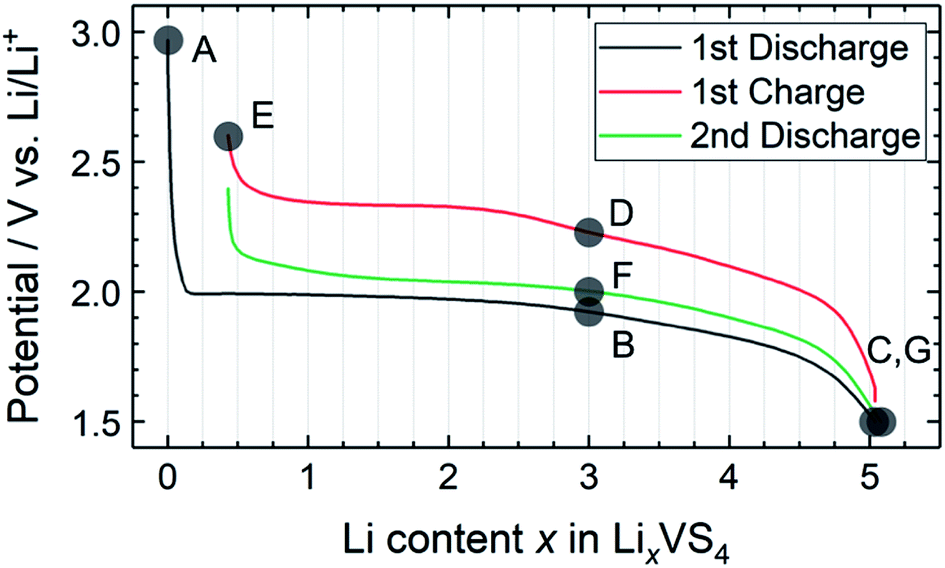 | ||
| Fig. 2 Charge–discharge profile of the Li//a-VS4 cell between 1.5 and 2.6 V. Sampling points are marked on the figure. | ||
To understand the lithiation/delithiation behavior of a-VS4, operando 7Li NMR measurements were carried out (Fig. 3). Two signals were observed before the electrochemical test. The sharp peaks at ∼0 and 275 ppm originate from the LiPF6 salt in the electrolyte solution and the Li metal used as the counter electrode, respectively. During the lithiation process, the 7Li signal from the Li ions inserted in a-VS4 (i.e., a-LixVS4) increased in intensity, which gives rise to a broad signal centered at ∼25 ppm. The spectral evolution changed with an increase in Li content above Li∼3.6VS4: the a-LixVS4 signal shifted to higher frequencies with a significant increase in width. This is followed by a decrease in the integrated intensity of the a-LixVS4 signal after the measurement time of 15 h. The intensity loss may be attributed to the incomplete detection of the broader signal component. These results indicate an increasing dipolar interaction between the 7Li nuclei and the unpaired electrons on the V ions during the latter half of the discharge process. Similar behavior was observed in the previous study.34 On the subsequent charge process, the integrated intensity of the a-LixVS4 signal first increased and then decreased. The spectral changes were basically reversible (see also Fig. S4†).
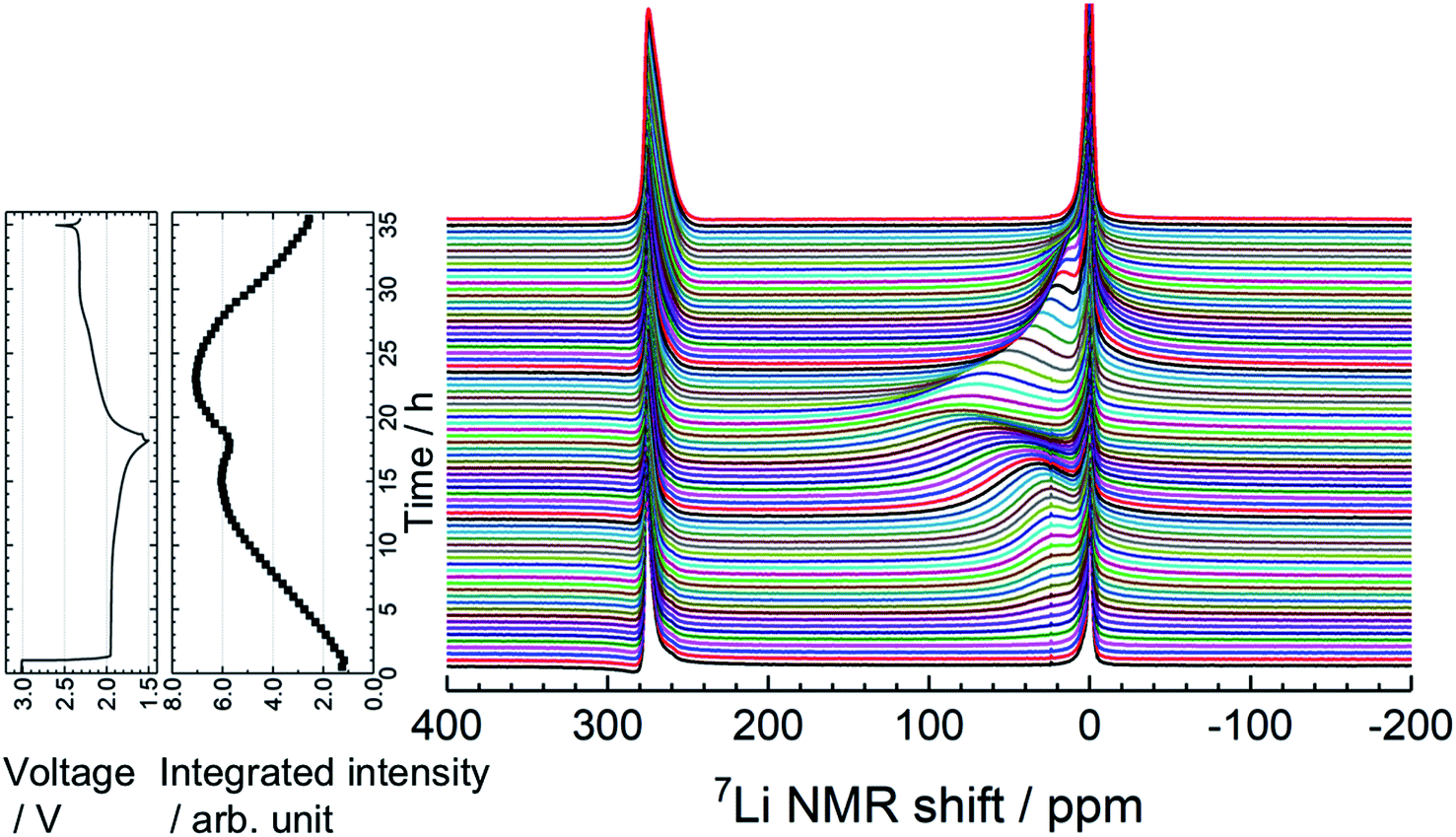 | ||
| Fig. 3 Time evolution of operando 7Li NMR spectra of the Li//a-VS4 cell, along with the corresponding charge–discharge profile and integrated intensity plot of the a-LixVS4 signal (including electrolyte signal). The spectra were accumulated for a flat laminate cell placed vertically in the center of coil. | ||
Fig. 4 shows high-resolution 7Li and 51V MAS NMR spectra for the electrode samples disassembled at the points shown in Fig. 2. The spectra were normalized by the scan number and sample weight in the rotors. The pristine a-VS4 has no signal in the 7Li MAS spectrum as expected (spectrum A in Fig. 4a), but the lithiated samples showed a sharp peak at 0 ppm, indicating the diamagnetic nature of the a-LixVS4 signal (B, C, D, F, G). Based on the operando 7Li spectral evolution and 51V MAS NMR spectra discussed later, the signal at 0 ppm was attributed to a diamagnetic V5+-containing material, most probably a-Li3VS4, as had been suggested in a previous study.27 Another shoulder peak at ∼5 ppm for the samples at the midpoint of the charge–discharge process (B, D, F) may be attributed to the a-Li3+dVS4 intermediate material.27 A broad signal centered at ∼20 ppm was observed at 1.5 V (C), which is clearly affected by the hyperfine interactions between Li ions and paramagnetic ions, indicating the formation of V4+ and/or V3+ species in a-Li3+d′VS4.27 This corresponds to the signal broadening and intensity loss observed in the operando measurements. Small peaks were also observed at 2.0 and 2.6 ppm at 1.5 V (C, G), one of which may be associated with the formation of Li2S.27 The spectral changes were reversible.
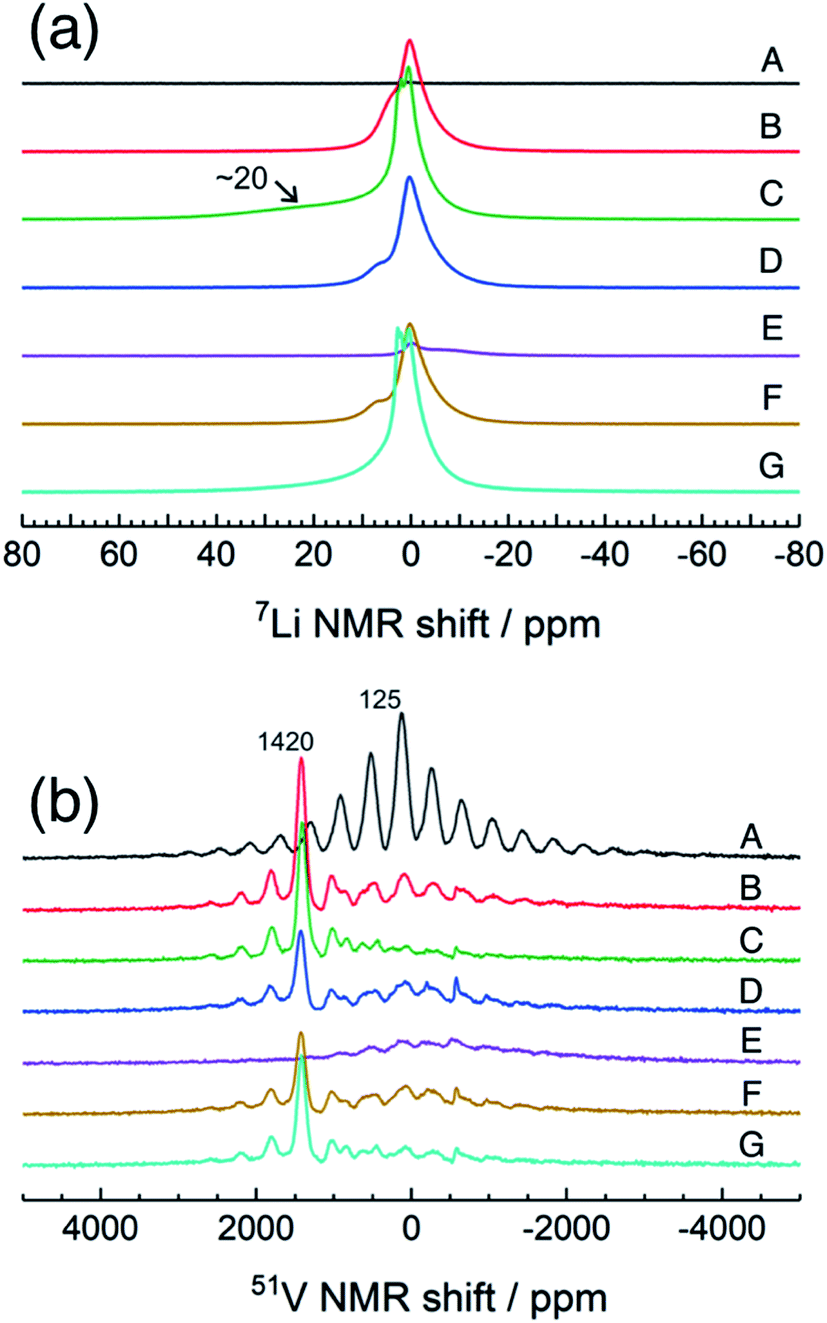 | ||
| Fig. 4 7Li and 51V MAS NMR spectra of a-VS4 electrode samples disassembled at the points marked in Fig. 2. | ||
The pristine a-VS4 shows an isotropic 51V signal at 125 ppm with significant SSBs (A in Fig. 4b). A new peak was clearly observed at 1420 ppm for the a-LixVS4 samples (B, C, D, F, G). This chemical shift is close to that of K3VS4 (1375 ppm),27 in which the V5+ ions are tetrahedrally coordinated by S2− ions.35 Therefore, it is ascribed to a-Li3VS4. We emphasize that the formation of the [VS4]3− tetrahedral structure was explicitly demonstrated in the present 51V MAS NMR spectra compared to the previous study.27 Both the signals from a-VS4 and a-Li3VS4 were observed for the samples at the midpoint of the charge–discharge process (B, D, F). This suggests a two-phase reaction process between a-VS4 and a-Li3VS4, in agreement with the flat voltage in this region (Fig. 2). At 1.5 V (C, G), the a-VS4 signal almost disappeared, and a weak signal at ∼830 ppm was identified. The latter signal may come from the V5+ ions close to the paramagnetic V4+ and/or V3+ ions in a-Li3+d′VS4, which is affected by the hyperfine interactions. We did not find the 51V signal from metallic V at ∼5500 ppm with a carrier frequency at 5500 ppm, in contrast to the previous study.27 Therefore, we believe that the conversion reaction to elemental V starts at a potential below 1.5 V. The a-Li3VS4 signal disappeared, and a broad signal at ∼120 ppm was recovered after full delithiation (E). The latter signal was attributed to a-VS4, but its intensity was significantly reduced compared to the pristine material (A). This intensity reduction is probably due to the increasing local structural disorder caused by back-transformation from the [VS4]3− tetrahedral monomer to the one-dimensional chain-like structure, in which V–V spin-pairing may be partly ineffective and, in that case, the V4+ ions will be partially invisible in the 51V MAS NMR spectrum. The recovery of the pristine structure was also confirmed by Raman spectra (Fig. S5†). In addition, a small peak was shown at −580 ppm in some spectra. A similar peak was identified in the previous study and tentatively ascribed to the V5+ ions with nearby V4+ ions.27 However, we here attribute this signal to the LiVO3 impurity phase (−573 ppm).36
Examination of these results gives essential information on the structure of pristine a-VS4 and its changes during the lithiation/delithiation process. The local structure of a-VS4 resembles that of c-VS4: the pristine a-VS4 consists of a one-dimensional chain-like structure with alternate short and long V–V distances, but long-range ordering is lost due to the positional disorder introduced by high-energy ball milling. The first Li insertion into a-VS4 proceeds as a two-phase reaction. The lithiation of a-VS4 to a-Li3VS4 involves drastic changes in local structure: the one-dimensional chains of V–V bonds and S–S disulfide bonds are broken, and [VS4]3− tetrahedral monomers are formed. This reaction involves the oxidation of V ions as well as the reduction of S ions: the valence states of V4+ and S− (or [S2]2−) in a-VS4 changes to V5+ and S2− in a-Li3VS4, respectively.27 Further lithiation of a-Li3VS4 to a-Li3+d′VS4 causes partial reduction of the V valence state. The amorphous phase at 1.5 V (Li5VS4 composition) still includes a large amount of [VS4]3− tetrahedral units. Information on the local structure around the paramagnetic V4+ and/or V3+ ions is not available from the 51V NMR spectra. Theoretical calculations of the Li3+dVO4 model structures proposed the coexistence of tetrahedral and octahedral V sites in Li5VO4.37 Recently, the V3+ ion in an octahedral environment was suggested in Li3+dVO4 based on the V K-edge XANES spectra.38 Similar coordination change may be considered in a-Li3+d′VS4, and the V K-edge XANES spectrum of a-LixVS4 provides supplementary information.29 The conversion to elemental V does not occur within the potential window used in this study. The structure of a-Li3+d′VS4 changes reversibly to that of a-VS4 during the delithiation process, but it necessitates drastic changes in the local environments of the V and S ions, leading to apparent voltage hysteresis in the charge–discharge profile. The structure of a-VS4 after the 1st cycle delithiation is similar to that of the pristine material, but with significant local disorder. The tetrahedral structure phase completely disappeared and, therefore, the irreversible capacity of 65 mA h g−1 may be associated with the formation of solid-electrolyte interphases (SEI) involving electrolyte decomposition. The XPS spectra of the lithiated and delithiated samples showed the deposition of organic components on the electrode surface, which makes it difficult to evaluate the valence states of V and S ions in these samples (not shown). The present study indicates that the lithiation/delithiation process of a-VS4 is basically the same as that previously reported for c-VS4:27 lithium insertion into c-VS4 results in amorphous phases such as a-Li3VS4 and a-Li3+d′VS4, which then revert to a-VS4 after the initial charge–discharge cycling. Recently, a similar lithium insertion/extraction behavior has been proposed for amorphous TiS4 (a-TiS4):39 the structural changes of a-TiS4 toward a-Li4TiS4 involve the breaking of S–S disulfide bonds and a gradual decrease in the average coordination number of Ti ions. These results indicate that the structural changes of sulfur-rich TM sulfides during Li insertion/extraction are related to the changes in both the bonding nature of sulfide species and the coordination number of TM ions. The amorphous host structure can accommodate these modifications, which is not topotactic as in crystalline electrode materials. From the viewpoint of the charge compensation mechanism, stable cation and anion redox reactions are easily available for amorphous sulfur-rich TM sulfide electrodes, which is a key factor for stable high capacity batteries.40
4. Conclusions
The amorphous VS4 (a-VS4) was mechanochemically prepared from crystalline VS4 (c-VS4), and its structural changes during the lithiation/delithiation process were investigated using solid-state NMR spectroscopy. 51V MAS NMR measurements gave extensive information on the amorphous host structures and valence states of the pristine a-VS4 and its lithiated materials. The local structure of a-VS4 resembles that of c-VS4, which consists of a one-dimensional chain structure with Peierls distortion. The lithiation/delithiation mechanism of a-VS4 is expected to be the same as that of c-VS4, and lithiated a-LixVS4 materials are amorphous during charge–discharge cycling. The local structure of a-VS4 changes drastically when Li ions are inserted. In the amorphous Li3VS4 phase (a-Li3VS4), the one-dimensional chain structure is decomposed to an isolated [VS4]3− structure with interstitial Li ions, which involves a unique internal redox process between V and S ions. Further Li insertion into a-Li3+d′VS4 is associated with the reduction of V ions. After the 1st cycle, a-VS4 recovers to a highly disordered chain-like structure. Stable Li insertion/extraction is feasible in between a-VS4 and a-Li5VS4 with a high capacity of ∼700 mA h g−1 using a narrow potential window between 1.5 and 2.6 V. Amorphous sulfur-rich TM sulfides can accommodate reversible changes in both the bonding nature of sulfide species and the coordination number of TM ions, which is a source of stable cation and anion redox reactions, leading to high charge–discharge capacities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Research and Development Initiative for Scientific Innovation of New Generation Batteries 2 (RISING2) funded by the New Energy and Industrial Technology Development Organization (NEDO), Japan (Project code: P16001). The authors thank Mr Takashi Moroishi for his support in the sample preparation and NMR measurements.References
- X. Ji and L. F. Nazar, J. Mater. Chem., 2010, 20, 9821–9826 RSC.
- R. D. Rauh, F. S. Shuker, J. M. Marston and S. B. Brummer, J. Inorg. Nucl. Chem., 1977, 39, 1761–1766 CrossRef CAS.
- Q. Liu, D. Mu, B. Wu, L. Wang, L. Gai and F. Wu, RSC Adv., 2017, 7, 33373–33377 RSC.
- C. Liang, N. J. Dudney and J. Y. Howe, Chem. Mater., 2009, 21, 4724–4730 CrossRef CAS.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- G. Zheng, Q. Zhang, J. J. Cha, Y. Yang, W. Li, Z. W. She and Y. Cui, Nano Lett., 2013, 13, 1265–1270 CrossRef CAS PubMed.
- J. R. Dahn and R. R. Haering, Mater. Res. Bull., 1979, 14, 1259–1262 CrossRef CAS.
- J. R. Dahn, W. R. McKinnon, R. R. Haering, W. J. L. Buyers and B. M. Powell, Can. J. Phys., 1980, 58, 207–213 CrossRef CAS.
- S. Kostov, M. denBoer, E. Strauss, D. Golodnitsky, S. G. Greenbaum and E. Peled, J. Power Sources, 1999, 81–82, 709–714 CrossRef CAS.
- Y. Shao-Horn, S. Osmialowski and Q. C. Horn, J. Electrochem. Soc., 2002, 149, A1547–A1555 CrossRef CAS.
- C. Feng, J. Ma, H. Li, R. Zeng, Z. Guo and H. Liu, Mater. Res. Bull., 2009, 44, 1811–1815 CrossRef CAS.
- G. Du, Z. Guo, S. Wang, R. Zeng, Z. Chen and H. Liu, Chem. Commun., 2010, 46, 1106–1108 RSC.
- N. Sharma, G. Du, A. J. Studer, Z. Guo and V. K. Peterson, Solid State Ionics, 2011, 199–200, 37–43 CrossRef CAS.
- X. Xu, W. Liu, Y. Kim and J. Cho, Nano Today, 2014, 9, 604–630 CrossRef CAS.
- M. M. Butala, M. Mayo, V. V. T. Doan-Nguyen, M. A. Lumley, C. Göbel, K. M. Wiaderek, O. J. Borkiewicz, K. W. Chapman, P. J. Chupas, M. Balasubramanian, G. Laurita, S. Britto, A. J. Morris, C. P. Grey and R. Seshadri, Chem. Mater., 2017, 29, 3070–3082 CrossRef CAS.
- L. Zhang, D. Sun, J. Kang, H.-T. Wang, S.-H. Hsieh, W.-F. Pong, H. A. Bechtel, J. Feng, L.-W. Wang, E. J. Cairns and J. Guo, Nano Lett., 2018, 18, 4506–4515 CrossRef CAS PubMed.
- A. Sakuda, N. Taguchi, T. Takeuchi, H. Kobayashi, H. Sakaebe, K. Tatsumi and Z. Ogumi, Electrochem. Commun., 2013, 31, 71–75 CrossRef CAS.
- A. Sakuda, T. Takeuchi, K. Okamura, H. Kobayashi, H. Sakaebe, K. Tatsumi and Z. Ogumi, Sci. Rep., 2014, 4, 4883 CrossRef CAS PubMed.
- A. Sakuda, N. Taguchi, T. Takeuchi, H. Kobayashi, H. Sakaebe, K. Tatsumi and Z. Ogumi, ECS Electrochem. Lett., 2014, 3, A79–A81 CrossRef CAS.
- T. Matsuyama, A. Hayashi, T. Ozaki, S. Mori and M. Tatsumisago, J. Mater. Chem. A, 2015, 3, 14142–14147 RSC.
- T. Takeuchi, H. Kageyama, K. Nakanishi, M. Ogawa, T. Ohta, A. Sakuda, H. Sakaebe, H. Kobayashi and Z. Ogumi, J. Electrochem. Soc., 2015, 162, A1745–A1750 CrossRef CAS.
- A. Sakuda, T. Takeuchi, M. Shikano, K. Ohara, K. Fukuda, Y. Uchimoto, Z. Ogumi, H. Kobayashi and H. Sakaebe, J. Ceram. Soc. Jpn., 2017, 125, 268–271 CrossRef CAS.
- X. Wang, K. Du, C. Wang, L. Ma, B. Zhao, J. Yang, M. Li, X.-X. Zhang, M. Xue and J. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 38606–38611 CrossRef CAS PubMed.
- R. Allmann, I. Baumann, A. Kutoglu, H. Rosch and E. Hellner, Sci. Nat., 1964, 51, 263–264 CrossRef CAS.
- C. S. Rout, B.-H. Kim, X. Xu, J. Yang, H. Y. Jeong, D. Odkhuu, N. Park, J. Cho and H. S. Shin, J. Am. Chem. Soc., 2013, 135, 8720–8725 CrossRef CAS PubMed.
- X. Xu, S. Jeong, C. S. Rout, P. Oh, M. Ko, H. Kim, M. G. Kim, R. Cao, H. S. Shin and J. Cho, J. Mater. Chem. A, 2014, 2, 10847–10853 RSC.
- S. Britto, M. Leskes, X. Hua, C.-A. Herbert, H. S. Shin, S. Clarke, O. Borkiewicz, K. W. Chapman, R. Seshadri, J. Cho and C. P. Grey, J. Am. Chem. Soc., 2015, 137, 8499–8508 CrossRef CAS PubMed.
- K. K. A. Sakuda, T. Takeuchi, H. Sakaebe, H. Kobayashi, H. Kageyama, T. Kawaguchi, H. Kiuchi, K. Nakanishi, M. Yoshimura, T. Ohta, T. Fukunaga and E. Matsubara, Solid State Ionics, 2018, 323, 32–36 CrossRef.
- K. Koganei, A. Sakuda, T. Takeuchi, H. Sakaebe, H. Kobayashi, H. Kiuchi, T. Fukunaga and E. Matsubara, 69th Annual Meeting of the International Society of Electrochemistry, S06a-028 (abstract), 2018 Search PubMed.
- M. N. Kozlova, Y. V. Mironov, E. D. Grayfer, A. I. Smolentsev, V. I. Zaikovskii, N. A. Nebogatikova, T. Y. Podlipskaya and V. E. Fedorov, Chem.–Eur. J., 2015, 21, 4639–4645 CrossRef CAS PubMed.
- M. S. Weimer, R. F. McCarthy, J. D. Emery, M. J. Bedzyk, F. G. Sen, A. Kinaci, M. K. Y. Chan, A. S. Hock and A. B. F. Martinson, Chem. Mater., 2017, 29, 2864–2873 CrossRef CAS.
- M. Wang, L. Fan, Y. Qiu, D. Chen, X. Wu, C. Zhao, J. Cheng, Y. Wang, N. Zhang and K. Sun, J. Mater. Chem. A, 2018, 6, 11694–11699 RSC.
- R. St, C. Smart, W. M. Skinner and A. R. Gerson, Surf. Interface Anal., 1999, 28, 101–105 CrossRef.
- K. Shimoda, M. Murakami, T. Takeuchi, T. Matsunaga, Y. Ukyo, H. Sakaebe, H. Kobayashi and E. Matsubara, J. Power Sources, 2018, 398, 67–74 CrossRef CAS.
- P. Duerichen and W. Bensch, Eur. J. Solid State Inorg. Chem., 1996, 33, 309–320 CAS.
- J. Skibsted, C. J. H. Jacobsen and H. J. Jakobsen, Inorg. Chem., 1998, 37, 3083–3092 CrossRef CAS.
- M. E. Arroyo-de Dompablo, P. Tartaj, J. M. Amarilla and U. Amador, Chem. Mater., 2016, 28, 5643–5651 CrossRef CAS.
- P. Rozier, E. Iwama, N. Nishio, K. Baba, K. Matsumura, K. Kisu, J. Miyamoto, W. Naoi, Y. Orikasa, P. Simon and K. Naoi, Chem. Mater., 2018, 30, 4926–4934 CrossRef CAS.
- A. Sakuda, K. Ohara, K. Fukuda, K. Nakanishi, T. Kawaguchi, H. Arai, Y. Uchimoto, T. Ohta, E. Matsubara, Z. Ogumi, T. Okumura, H. Kobayashi, H. Kageyama, M. Shikano, H. Sakaebe and T. Takeuchi, J. Am. Chem. Soc., 2017, 139, 8796–8799 CrossRef CAS PubMed.
- E. D. Grayfer, E. M. Pazhetnov, M. N. Kozlova, S. B. Artemkina and V. E. Fedorov, ChemSusChem, 2017, 10, 4805–4811 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04338a |
| This journal is © The Royal Society of Chemistry 2019 |