
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron phosphide anchored nanoporous carbon as an efficient electrode for supercapacitors and the oxygen reduction reaction†
Ahmed K. Yousefa,
Yena Kim *bc,
Piyali Bhanja*c,
Peng Meicd,
Malay Pramanikc,
M. M. S. Sanade,
M. M. Rashade,
A. Y. El-Sayeda,
Abdulmohsen Ali Alshehrif,
Yousef Gamaan Alghamdif,
Khalid Ahmed Alzahranif,
Yusuke Idec,
Jianjian Linb and
Yusuke Yamauchi*gh
*bc,
Piyali Bhanja*c,
Peng Meicd,
Malay Pramanikc,
M. M. S. Sanade,
M. M. Rashade,
A. Y. El-Sayeda,
Abdulmohsen Ali Alshehrif,
Yousef Gamaan Alghamdif,
Khalid Ahmed Alzahranif,
Yusuke Idec,
Jianjian Linb and
Yusuke Yamauchi*gh
aChemistry Department, Faculty of Science, Al-Azhar University, Assiut Branch, 71524 Assiut, Egypt
bKey Laboratory of Eco-Chemical Engineering, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, China
cInternational Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: piyalibhanja4@gmail.com; yenakim1227@gmail.com
dSchool of Chemistry and Materials Science, South-Central University for Nationalities, Wuhan 430074, China
eCentral Metallurgical Research & Development Institute, P.O. Box 87, Helwan 11421, Egypt
fDepartment of Chemistry, King Abdulaziz University, P.O. Box. 80203, Jeddah 21589, Saudi Arabia
gSchool of Chemical Engineering, Australian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland Brisbane, QLD 4072, Australia. E-mail: y.yamauchi@uq.edu.au
hDepartment of Plant & Environmental New Resources, Kyung Hee University, 1732 Deogyeong-daero, Giheung-gu, Yongin-si, Gyeonggi-do 446-701, South Korea
First published on 13th August 2019
Abstract
Inspired by their distinctive properties, transition metal phosphides have gained immense attention as promising electrode materials for energy storage and conversion applications. The introduction of a safe and large-scale method of synthesizing a composite of these materials with carbon is of great significance in the fields of electrochemical and materials sciences. In the current effort, we successfully synthesize an iron phosphide/carbon (FeP/C) with a high specific surface area by the pyrolysis of the gel resulting from the hydrothermal treatment of an iron nitrate–phytic acid mixed solution. In comparison with the blank (P/C), the as-synthesized FeP/C appears to be an efficient electrode material for supercapacitor as well as oxygen reduction reaction (ORR) applications in an alkaline medium in a three-electrode system. In the study of supercapacitors, FeP/C shows areal capacitance of 313 mF cm−2 at 1.2 mA cm−2 while retaining 95% of its initial capacitance value after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, while in the ORR, the synthesized material exhibits high electrocatalytic activity with an onset potential of ca. 0.86 V vs. RHE through the preferred four-electron pathway and less than 6% H2O2 production calculated in the potential range of 0.0–0.7 V vs. RHE. The stability is found to be better than those of the benchmark Pt/C (20 wt%) catalyst.
000 cycles, while in the ORR, the synthesized material exhibits high electrocatalytic activity with an onset potential of ca. 0.86 V vs. RHE through the preferred four-electron pathway and less than 6% H2O2 production calculated in the potential range of 0.0–0.7 V vs. RHE. The stability is found to be better than those of the benchmark Pt/C (20 wt%) catalyst.
Introduction
Searching for a new renewable, low-cost, and environmentally friendly source for energy storage and/or conversion has become one of the hottest research challenges, driven by diminishing fossil fuel supplies and climate change in addition to ever-increasing demands for energy sources.1,2 Thus, supercapacitors and oxygen reduction reaction (ORR) have been considered the best way out for the current and near future energy issues.3 Supercapacitor devices can be made using non-toxic materials and show characteristic properties such as a superior power density, long cycle life, and high charge–discharge efficiency as compared with conventional batteries.4–6 The ORR is crucial in our daily life processes, and it is one of the most important reactions in energy storage and conversion applications, where it is the heart of metal–air batteries and fuel cells.7–9The electrical energy in supercapacitors can be stored on the electrode surface in two ways. First, the electrolyte ions are adsorbed at the electrical double layer to produce what is called electric double layer capacitors (EDLCs), and a second one is by redox reactions, which are called pseudocapacitors.10–12 Depending on the nature of the electrode materials, the electrical energy storage mechanism could be controlled. Metal oxides and conductive polymers are the main materials used for pseudocapacitors, while carbon is the conventional electrode material used for EDLCs.13 In general, a high-performance supercapacitor requires materials with high electrical conductivity and a large ion-accessible surface area. In addition, a high ionic transport rate requires short ion diffusion length throughout the electrode materials.14 On the other hand, ORR reacts sluggishly, and it needs to be facilitated to improve its efficiency. Although Pt is the current state-of-the-art electrocatalyst for this reaction, intensive research to modify or replace it is ongoing owing to its high cost and finite resource.15–17
In spite of the recently obtained performance of both supercapacitors and ORR, with the rapid development of electronic devices and the extension of their applications toward large-scale devices, much higher performance is required through the improvement of properties such as energy density, rate capability, cyclability, and safety. The development of new electrode materials plays a vital role in reaching the required performance to promote the commercialization of the two applications mentioned above.18–21 Transition metal compounds are an important class of functional electrode materials that have been widely investigated in several energy applications.22–25 In an inert atmosphere, transition metals combine with phosphorus chemicals to form transition metal phosphides (TMPs), and these materials have notable potential toward energy storage and electrocatalytic applications due to their advantages of low cost, easy large-scale synthesis, and interesting electrical properties.26 Among TMPs, iron phosphide (FeP) has been considered as a promising energy electrode material because Fe is naturally abundant and has low cost, low toxicity, and negligible environmental impact.27 However, some synthetic methods for producing this kind of material are dangerous (for example, the use of H2 to produce PH3 gas).26,28 Moreover, the poor stability of iron-based materials limits their electrochemical properties.29–32 Thus, its combination with carbon is an effective strategy for keeping structural stability, improving electronic conductivity during cycling, and obtaining high surface area materials.33–35 Accordingly, providing a safe synthetic method to obtain FeP/carbon (FeP/C) materials with high surface areas and high stability while exploring their performance as supercapacitors and ORR are highly important for satisfying the current needs for energy.
In this study, we report a facile and safe method for the synthesis of FeP/C with a high surface area through a pyrolysis strategy for the gel resulting from the hydrothermal treatment of Fe nitrate–phytic acid mixed solution. When the as-synthesized FeP/C is used as an electrode for a supercapacitor, it exhibits good areal capacitance and shows outstanding cycle performance (retains 95% of its initial capacitance value after 10000 cycles), which indicates the promising practical applications of synthesized FeP/C for supercapacitors. The electrocatalytic performance towards ORR in an alkaline medium is also investigated. The obtained FeP/C electrodes show electrocatalytic activity through a four-electron reaction pathway that was higher than that of P/C samples. In addition, the limiting current density at 0 V vs. RHE and stability are found to be better than those of the benchmark Pt/C (20 wt%) catalyst.
Experimental section
Materials
Iron nitrate nonahydrate (Fe (NO3)3·9H2O), isopropanol, ethanol, methanol, and KOH were purchased from Wako Pure Chemicals Co., Ltd. Phytic acid solution (50 wt% in H2O) and Nafion solution (5 wt%) were purchased from Sigma-Aldrich, and commercial Pt/C (20 wt%) from Alfa Aesar. Carbon Paper TGP-H-090 was obtained from Toray. All chemicals were of analytical grade and were used without further treatment.Synthesis of FeP/C and P/C
In a typical synthesis procedure, 0.5 mmol of Fe (NO3)3·9H2O was added to 10 mmol of phytic acid solution, and it was continuously stirred for one hour. The resultant clear solution was transferred to a 25 mL stainless steel autoclave and heated to 140 °C at a rate of 1 °C min−1 and left for 36 h. The resulting gel was transferred to a tubular furnace and left in a nitrogen atmosphere at room temperature for 1 h; after that, the temperature was increased to 300 °C and maintained for 2 h. Subsequently, the temperature was increased to 900 °C and maintained for 2 h. Finally, the sample was naturally cooled. The heating rate for the entire annealing process was 5 °C min−1. After cooling to room temperature, a black powder material of FeP/C was obtained. P/C was prepared through the same procedure without adding Fe salt with a slight modification, where the gel resulting from the hydrothermal treatment was washed once with methanol and dried at 60 °C for one day before annealing.Characterization
Wide-angle powder X-ray diffraction (XRD) patterns were collected on a Rigaku SmartLab X-ray diffractometer using Cu Kα radiation (40 kV and 30 mA) at a scanning rate of 1° min−1. Raman spectroscopy was acquired with a HORIBA Scientific Lab RAM HR Raman spectrometer system using 532.4 nm laser excitation. Field-emission SEM measurements were performed on a HITACHI SU-8230 to check the morphologies. Investigation of the inner structure and energy-dispersive X-ray spectroscopy (EDS) elemental mapping were carried out using a transmission electron microscope (TEM, JEOL JEM-2100F). The nitrogen adsorption–desorption of the samples was measured on a Micromeritics BK122T-B analyzer. The Brunauer–Emmett–Teller (BET) theory was used for the specific surface area (SSA) determination in the relative pressure range of 0.05–0.30. Pore size distributions were determined from the adsorption isotherms, according to the quenched solid-state density functional theory (QSDFT) assuming a mixed cylinder/sphere pore model. Inductively coupled plasma optical emission spectroscopy (ICP-OES) was done on a Hitachi High-Tech Science Corporation model SPS3520UV-DD. The standard was made by Kanto Chemical Co., Inc., and the qualified standard solution by JCSS. The samples were fused by a mixture of Na2CO3, H3BO3, KNO3 and K2S2O7 salts to digest the material into a dissoluble solid. During this process carbon burns out and removed as CO2, Remaining solid melt was then dissolved in HNO3 for the chemical analysis.Electrochemical measurements
The electrochemical performances of the prepared electrodes were investigated with a three-electrode system.:1) and 20 μL of 5.0 wt% Nafion. After 30 min sonication, 120 μL of the suspension was dropped onto carbon paper (thickness: 1 mm) with an area of 1 × 1 cm2 and dried overnight at 60 °C. The mass loading was 1.2 mg cm−2. All electrochemical measurements were carried out using a CHI 660E instrument; cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) measurements were carried out in 3 M KOH between −0.8 and 0.0 V vs. SCE, with a platinum wire and saturated calomel electrode (SCE) as a counter and a reference electrode, respectively. The test of long-term stability for FeP/C was conducted by cycling between −0.8 and 0.0 V vs. SCE in 3 M KOH at a scan rate of 100 mV s−1.The gravimetric specific capacitances (Cg, F g−1) of the as-synthesized electrodes were calculated from the GCD curves using the following equation:
| Cg = (I × Δt)/(m × ΔV), |
The areal capacitance (CA, mF cm−2) was calculated using the following equation:
| CA = Cg × ml, |
:water mixed solution (volume ratio of 1:3) and 50 μL of 5.0 wt% Nafion. After sonication for 60 min, 5 μL of the suspension was dropped onto a glassy carbon electrode (GCE) of a rotating ring-disk electrode (RRDE) with an area of 0.1256 cm2 and dried under a lamp. The mass loading was 0.2 mg cm−2. To activate the GCE, it was pre-polished with 1 μm diamond, then 0.05 μm alumina powder, and washed with water. All electrochemical measurements were carried out using CHI 842B instrument; cyclic voltammetry (CV), linear sweep voltammetry (LSV) at 10 mV s−1, and chronoamperometry (i–t) measurements were performed in 0.1 M KOH saturated solution. The electrolyte solution was continuously purged with ultra-pure O2 or N2 for at least 1 h before starting the measurements. A platinum wire and silver–silver chloride (Ag/AgCl) were used as the counter and reference electrode, respectively. The potentials were expressed with regard to the reversible hydrogen potential electrode (RHE). The overall electron transfer numbers per oxygen molecule were calculated from the slope of the Koutecky–Levich (K–L) plots according to the following equation:| J−1 = Jk−1 + JL−1 = Jk−1 + B−1ω−1/2 |
| B = 0.2nFCo(Do)2/3ν−1/6, |
485 C mol−1), Co is the concentration of O2 (1.2 × 10−6 mol cm−3), Do is the diffusion coefficient of O2 (1.9 × 10−5 cm2 s−1), ν is the kinematic viscosity of the electrolyte (0.01 cm2 s−1), and ω is the electrode rotating speed in rpm (0.2 is a constant when the rotating speed is expressed in rpm).36,37 n and % H2O2 were calculated from RRDE measurements according to the following equations:| n = 4 × Id/[Id + (Ir/N)] |
| % H2O2 = 200 × (Ir/N)/[Id + (Ir/N)]. |
Results and discussion
FeP/C was synthesized through a two-step process involving hydrothermal solution-phase crystallization and carbonization under inert atmosphere. During the crystallization process, the clear mixtures of the phytic acid and Fe(III) precursor solutions undergo polycondensation from the initial gel through the hydrothermal treatment at 140 °C for 36 h. Under these conditions, the phytic acid molecule, which bears six phosphonic acid moieties, has a strong affinity to bind with Fe(III) species present in the synthesis medium to form a highly cross-linked structure via the Fe–O–P bonds. The cross-linking increased during the hydrothermal treatment, leading to improved mechanical strength of the metal-ligand network structure.38 The multidentate nature of the phytic acid molecule together with the presence of PO4 tetrahedral and FeO6 octahedral units separated by organic fragments could provide high structural robustness, as reflected in the crystalline nanostructure. Finally, the pyrolysis of the resulted cross-linked structure at a high temperature leads to the decomposition of Fe–phytate and the interaction of Fe with P to form FeP nanoclusters, while the phytate rings carbonized to obtain FeP/C as a final product.The XRD patterns of P/C and FeP/C are shown in Fig. 1a. All XRD peaks in the FeP/C sample can be indexed as pure FeP (JCPDS card no. 89-2746) with the presence of two broad characteristic graphitic peaks located at around 23° and 43° in both samples, which can be assigned to (002) and (101) planes, respectively. Raman analysis was used to investigate the graphitic degree of the carbon content in the synthesized samples. As can be seen in Fig. 1b, both samples exhibit typical D and G bands at 1353 and 1596 cm−1, representing the disordered and graphitic phases in carbon, respectively. Therefore, the integrated intensity ratio of D and G bands can reveal the level of ordering and defects in the carbon structure, indicating a higher disorder for a higher ID/IG value.39 ID is higher than IG in the two samples, and the intensity ratio of ID/IG is about 1.02 and 1.07 for P/C and FeP/C, respectively. This indicates that many defects or disordered sites are present in the carbon component, which agrees with the obtained XRD data.
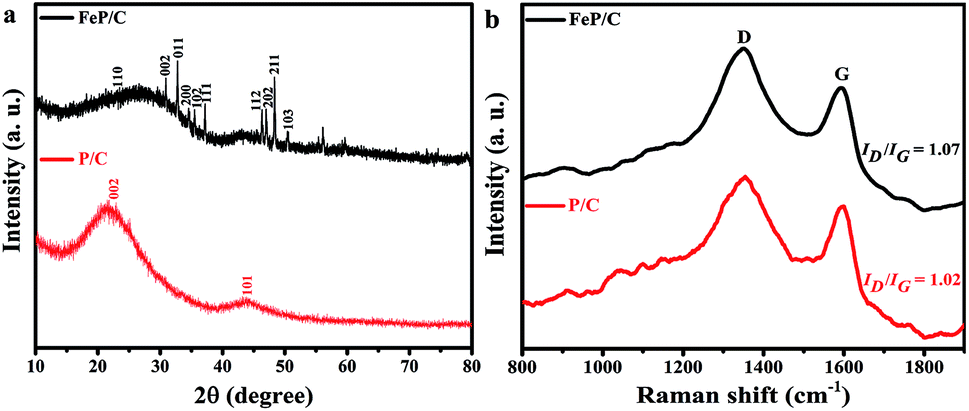 | ||
| Fig. 1 (a) XRD patterns and (b) Raman spectra of P/C and FeP/C. | ||
The morphology of the P/C and FeP/C materials were investigated using scanning electron microscopy (SEM) (Fig. 2a and b). As shown in Fig. 2a, the P/C possesses irregularly shaped particles, with the presence of some spherical particles (as indicated by arrows). However, the FeP/C appears as a stacked layered structure with no distinct particles, as can be seen in its SEM image (Fig. 2b and S2b†). Thus, the morphology has been changed from P/C to FeP/C, which is understandable where the temperature is high enough to cause a reaction of Fe particles with the carbon matrix to form Fe carbide, which, with increasing temperature, dissociates again into graphitic carbon and Fe particles.40 A variety of graphitic carbons of small crystallite size are supposed to be formed, as the graphitization effect of the Fe particle is localized. The high temperature (900 °C) and the presence of the Fe particle enhance the degree of graphitization.41 The amorphous nature of the synthesized materials and the disordered micropores were observed under a high-resolution transmission electron microscope (HRTEM), as shown in Fig. 2c, d and S1.† The HRTEM images indicate the layered-like structure of synthesized FeP/C samples, highlighted by the yellow boxes in Fig. 2d and S2.† This sheet-like structure can improve the electrocatalytic performance of active materials through fast electron transfer and ion diffusion pathways, and can also increase the effective contact area between the electrolytes and active materials.42,43 Elemental mapping of FeP/C shows the uniform distribution of Fe and P elements over the carbon matrix, as can be seen in Fig. S3.† From this elemental mapping, it is noticed that the amount of P element is higher than that of Fe, which is different from the results obtained by XRD (1:1). From ICP analysis, the amount of P in the P/C sample was found to be 20%, while in the FeP/C sample, the amount of P was 15% and of Fe was 7%, indicating the dominant content of C in both samples (80%). That may explain its amorphous appearance, as shown in Fig. 2c and d.
 | ||
| Fig. 2 (a and b) SEM and (c and d) HRTEM images of (a and c) P/C and (b and d) FeP/C, respectively. The white arrows in (a) indicate the presence of spherical particles in the P/C sample. The yellow box in (d) indicates the layered-like structure of FeP/C. | ||
The specific surface area and pore size of P/C and FeP/C were examined by nitrogen adsorption–desorption measurements (Fig. 3). Table S1† summarizes the pore sizes, specific surface areas (SSAs), and total pore volumes (TPVs) of both samples. P/C has possessed a high BET surface area of 596 m2 g−1; after introducing Fe in the synthesis process, it is dramatically increased to 1269 m2 g−1. The high surface area without the use of any template may be due to the pore-generating ability of the starting precursor. At a sufficiently high temperature, phytic acid releases its six molecules of orthophosphate, which are removed from the surface of carbon as CO2 gas and generate mainly micropores in the carbon framework.44,45 The higher surface area of FeP/C can be ascribed to its higher total pore volume. Its pore size distribution (Fig. 3b) indicates the micro-/meso-porous structure of both samples. While FeP/C shows a sharp and strong peak at a pore diameter of 1.5 nm, P/C shows a similar peak at 1.8 nm. Both samples show a peak around 2.9 nm. It is confirmed that the FeP/C is highly porous in nature with a high surface area. The large surface area and abundant micro/meso-pores provide sufficient channels for the efficient diffusion of the electrolyte and facilitate the accumulation of electrons on the electrode surface in the electrochemical system. This is expected to achieve stable and high performance as electrode materials for supercapacitor and ORR applications.46,47
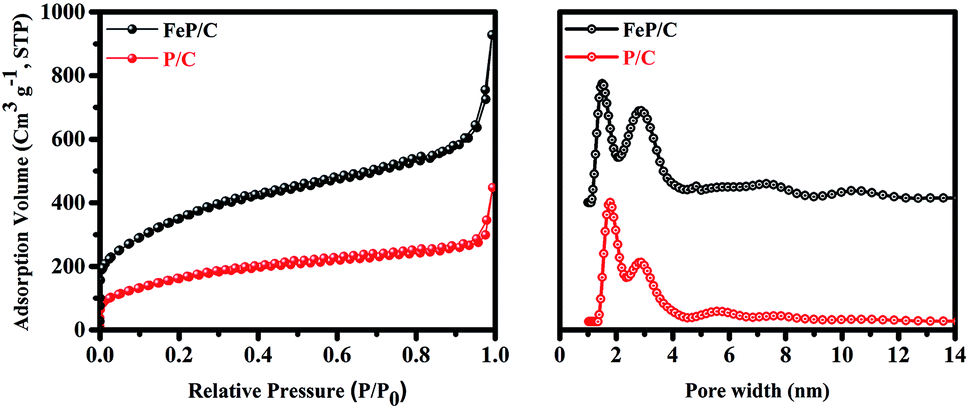 | ||
| Fig. 3 N2 adsorption–desorption isotherms and pore size distribution curves of P/C and FeP/C. | ||
The supercapacitor (SC) performances of P/C and FeP/C samples were investigated by cyclic voltammetry (CV) and galvanostatic charge/discharge (GCD) measurements in a conventional three-electrode configuration at room temperature using 3 M KOH solution as an electrolyte. Fig. 4a shows a comparison of the CV curves of P/C and FeP/C obtained at a scan rate of 100 mV s−1. Both samples show a quasi-rectangular shape, indicating an electric double layer capacitive behavior. The FeP/C electrode shows a considerably higher current density than that of P/C, revealing the higher capacitance of FeP/C based on the directly proportional relationship between the CV curve area and capacitance. The CV curves were also obtained by varying the scan rate for both samples, P/C (Fig. S4a†) and FeP/C (Fig. 4b). The good symmetry shown indicates a good rate capability.
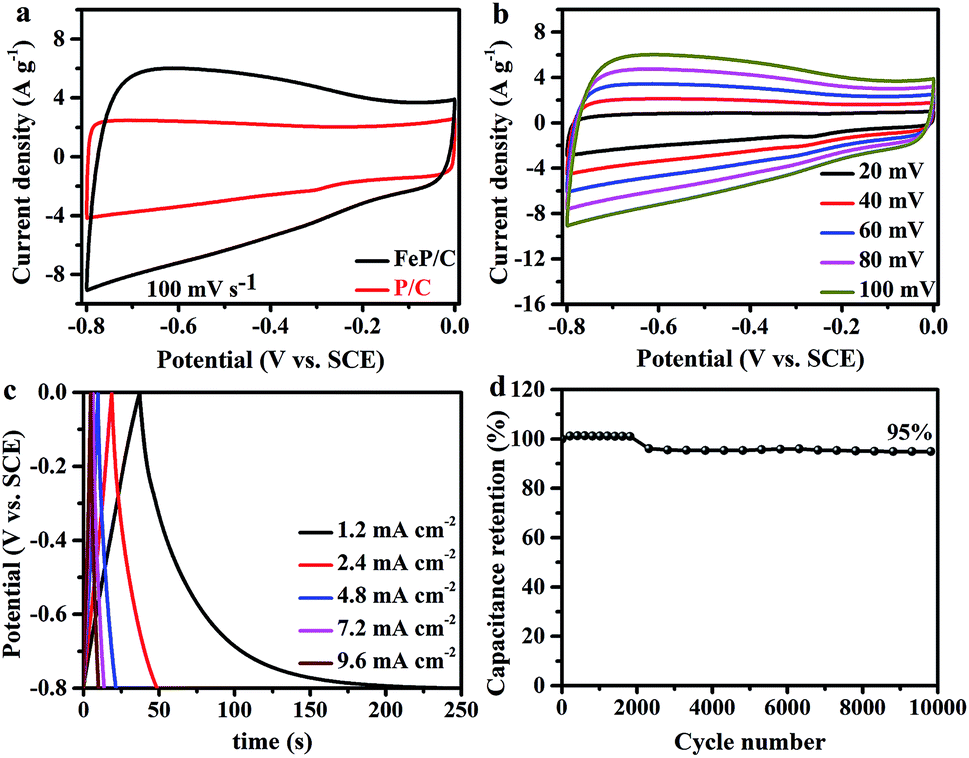 | ||
| Fig. 4 Electrochemical properties measured in a three-electrode system using 3 M KOH as an electrolyte. (a) CV curve comparison of P/C and FeP/C at 100 mV s−1 scan rate, (b) CV curves of FeP/C at various scan rates, (c) GCD curves of FeP/C at different current density, and (d) the cycling performance of FeP/C at 100 mV s−1. | ||
The GCD curves of P/C and FeP/C are depicted in Fig. S4b† and 4c, respectively. Curves of P/C exhibit nearly triangular traces at various current densities, showing the capacitive behavior of the prepared sample. In contrast, the discharging curve in the FeP/C sample shows an initially sharp potential drop, especially at the current density of 1.2 mA cm−2, which returns back to the internal resistance (ohmic resistance). This IR drop mainly arises from the internal resistance of FeP/C due to the migration of ions between the electrode and electrolyte.48 Fig. S5† shows the change of areal capacitance with current density for both samples. The P/C shows better rate capability than that of FeP/C. This can be understood in the light of the pore size of both samples (Table S1†), where the reversible adsorption/desorption of electrolyte ions on the surface of electrodes determines the electric double layer (EDL) capacitance. At low current densities, electrolyte ions can easily access the inner micro- and meso-pores through holes on the P/C and FeP/C. However, at high current densities, the small pores may reduce the ions' movements. Therefore, the capacitance decreases significantly in the FeP/C sample.49
The areal capacitances (CA) of both electrodes were estimated based on the galvanostatic discharge curves. The CA values of P/C and FeP/C achieve 41 and 313 mF cm−2 at a current density of 1.2 mA cm−2, respectively. The specific capacitance values of P/C and FeP/C at an equivalent current density of 1 A g−1 correspond to 34 and 261 F g−1, respectively. The obtained areal capacitance of FeP/C is close to or considerably better than some previously reported iron-based materials. Table S2† shows data comparing its electrochemical performance with those of some previously reported Fe-based materials.
The electrochemical stability of the electrode material was evaluated using long-term cycling performance at a scan rate of 100 mV s−1 for 10000 cycles (Fig. 4d). It is well known that because of structural deformation during the long-time charge/discharge process, iron-based supercapacitor electrode materials often suffer from poor stability.50,51 In previous studies, arrays from FeP nanotubes and nanorods were used as negative electrodes for supercapacitor application and showed only 41% and 24.19% retention after 5000 cycles, respectively.29,31 After coating FeP nanorod arrays with PEDOT, the stability increased to 82.1%.31 Porous N/P co-doped carbon showed capacitance retention of 86.3% after 10000 cycles.52 Interestingly, FeP/C exhibits outstanding cycle performance and retains 95% of its initial capacitance value after 10000 cycles, indicating the synergetic effect of the coupling of FeP and carbon.
The electrochemical activities of the as-synthesized samples toward ORR were studied in O2- and N2-saturated 0.1 M KOH solutions by cyclic voltammetry (CV) at a scan rate of 100 mV s−1 (Fig. 5a and S6†). It is clear in Fig. 5a that the CV curves in the N2-saturated solution did not display any characteristic reduction peaks, while the same electrodes in the O2-saturated solution display characteristic peaks on the cathodic scan at about 0.52 and 0.68 V vs. RHE for P/C and FeP/C, respectively. Moreover, as shown in Fig. S6,† the area under the FeP/C curve is clearly higher than that of P/C, indicating the increase in ORR active sites with the introduction of Fe. The ORR curves of FeP/C are well defined with the most positive onset potential (ca. 0.86 V vs. RHE), which is 180 and 20 mV more positive than those of P/C (Fig. 5a) and a previously reported Fe–phosphate/C,41 respectively.
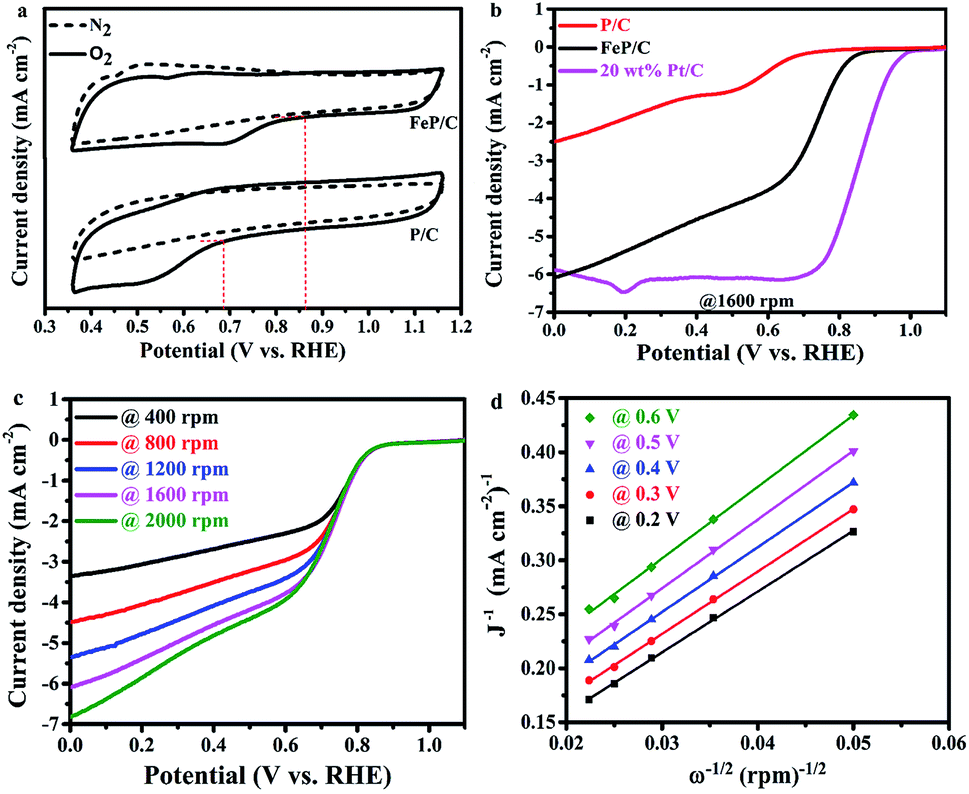 | ||
| Fig. 5 ORR measurements in a three-electrode system using 0.1 M KOH as an electrolyte. (a) CV curve comparison of P/C and FeP/C at 100 mV s−1, (b) LSV curve comparison of P/C, FeP/C, and 20 wt% Pt/C at 1600 rpm, (c) LSV curves of FeP/C at different rotation rates, and (d) Koutecky–Levich plots at different potentials. | ||
The ORR kinetics of P/C and FeP/C have been studied by linear sweep voltammetry (LSV) through RRDE measurements at rotation speeds ranging from 400 to 2000 rpm with a scan rate of 10 mV s−1 (Fig. 5b and c). The LSV curves of as-synthesized FeP/C show a more positive value of onset potential (0.86 V vs. RHE) than that of prepared P/C (0.68 V vs. RHE), as shown in Fig. 5b. Also, the half-wave potential values of FeP/C, P/C, and commercial Pt/C are estimated to be 0.74 V, 0.59 V, and 0.86 V (vs. RHE), respectively. These results indicate the better ORR catalytic activity of prepared FeP/C than that of P/C sample in 0.1 M KOH solution. Interestingly, the limiting current density of our FeP/C electrode at 0 V vs. RHE was a little bit higher than that of the commercial Pt/C electrode. Fig. S7† shows the differences in the potential and limiting current density values of the three electrodes. The higher ORR activity of FeP/C than P/C may be due to the higher surface area, the presence of sufficient P–C interactions which break the electroneutrality of carbon, and the presence of Fe atom which helps to break the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds.51–55
O bonds.51–55
Using the Koutecky–Levich plot, the electrons transferred per O2 molecule (n) during ORR at different potentials (0.2, 0.3, 0.4, 0.5, and 0.6 V vs. RHE) were calculated, as shown in Fig. 5d. The obtained K–L plots show high linearity, suggesting first-order reaction kinetics based on our measurement conditions.56 The average electron number is 4.6 for FeP/C, which reveals the preferred H2O formation mechanism.36,57
In further study, the electron transfer numbers and hydrogen peroxide percentage were calculated by using the currents obtained from disk and ring electrodes through RRDE measurement in the potential range of 0.0–0.7 V vs. RHE (Fig. 6).37 As shown in Fig. 6b, electron transfer number values are around 4 along all selected potential ranges, which is consistent with the results obtained by K–L plot analysis. The hydrogen peroxide yield is between 1.4% and 5.7%, which agrees with a previously reported study of Fe–phosphate/C material,41 which overcomes the reported defects of phosphate-based ORR electrocatalysts.58 Such a high electron transfer number and low H2O2 percentage are very important, according to the operating point of view that requires protection of the catalyst layer and membrane from degradation by crossover H2O2.59
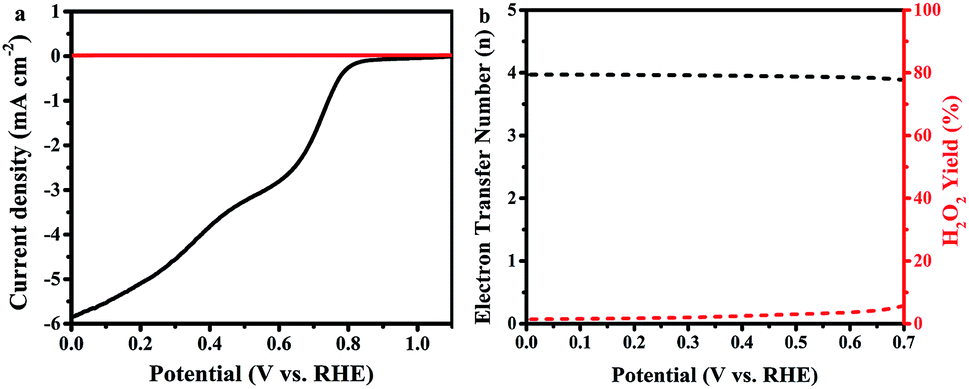 | ||
| Fig. 6 (a) RRDE test of the ORR on FeP/C in an O2-saturated 0.1 M KOH electrolyte at a scan rate of 10 mV s−1 and a rotating rate of 1600 rpm; (b) assessment of electron transfer number and peroxide percentage in ORR catalyzed by FeP/C based on RRDE data. | ||
The stability of the FeP/C electrode was investigated using chronopotentiometry measurements at a constant potential (0.46 V vs. RHE) in O2-saturated 0.1 M KOH at 1600 rpm as a rotation speed and compared with the benchmark Pt/C catalyst (Fig. S8†). The as-synthesized FeP/C electrode retains 84% of its initial current value after 9000 seconds, a bit better than 20 wt% Pt/C, which shows 81% current retention under the same measurement conditions. All of the above results highlight the promising application of FeP/C as an electrode for ORR applications.
Conclusions
In summary, we have successfully synthesized FeP/C material using an eco-friendly and simple hydrothermal method followed by carbonization. FeP/C sample shows a high surface area (1269 m2 g−1) that is mainly microporous with the presence of mesopores. FeP/C has been also investigated as an electrode for supercapacitor and ORR applications using a three-electrode system. The electrochemical properties of P/C are significantly improved by incorporating Fe. When FeP/C is used as a supercapacitor electrode, it exhibits a maximum areal capacitance of 313 mF cm−2 at a current density of 1.2 mA cm−2, accompanied by 95% capacitance retention after 10000 cycles. While in use as an electrode material for ORR in an alkaline medium, it illustrated high electrocatalytic activity and exhibited a positive shift value of onset potential (0.86 V vs. RHE) and high limiting current at 0 V vs. RHE with the production of a low percentage from hydrogen peroxide. It also provided a four-electron transfer pathway with 84% current retention after 9000 seconds. The reported results would provide useful information for the development of metal phosphide based electrodes with high performance for supercapacitor and ORR applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was supported by the National Natural Science Foundation of China (NSFC) (61604070) and the National Natural Science Foundation of Jiangsu Province (BK20161000). Ahmed K. Yousef appreciates the financial support by the joint supervision scholarship from the cultural affairs and missions sector, the Egyptian Ministry of Higher Education (MOHE). The authors acknowledge the Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah for technical and financial support. This work was performed in part at the Queensland node of the Australian National Fabrication Facility, a company established under the National Collaborative Research Infrastructure Strategy to provide nano- and microfabrication facilities for Australia's researchers.References
- B. Liu, Y. Sun, L. Liu, J. Chen, B. Yang, S. Xu and X. Yan, Energy Environ. Sci., 2019, 12, 887–922 RSC.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS PubMed.
- K. Jost, D. P. Durkin, L. M. Haverhals, E. K. Brown, M. Langenstein, H. C. De Long, P. C. Trulove, Y. Gogotsi and G. Dion, Adv. Energy Mater., 2015, 5, 1401286 CrossRef.
- Y. Wang and Y. Xia, Adv. Mater., 2013, 25, 5336–5342 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- H.-W. Liang, W. Wei, Z.-S. Wu, X. Feng and K. Müllen, J. Am. Chem. Soc., 2013, 135, 16002–16005 CrossRef CAS PubMed.
- B. E. Conway, Electrochemical supercapacitors: scientific fundamentals and technological applications, Springer Science & Business Media, 2013, pp. 11–31 Search PubMed.
- Z. Lin, E. Goikolea, A. Balducci, K. Naoi, P.-L. Taberna, M. Salanne, G. Yushin and P. Simon, Mater. Today, 2018, 21, 419–436 CrossRef CAS.
- R. Zhao, L. Zhang, C. Wang and L. Yin, J. Power Sources, 2017, 353, 77–84 CrossRef CAS.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- W. Wei, J. Wu, S. Cui, Y. Zhao, W. Chen and L. Mi, Nanoscale, 2019, 11, 6243–6253 RSC.
- Q. Liu, L. Du, G. Fu, Z. Cui, Y. Li, D. Dang, X. Gao, Q. Zheng and J. B. Goodenough, Adv. Energy Mater., 2019, 9, 1803040 CrossRef.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef PubMed.
- L. Yang, J. Shui, L. Du, Y. Shao, J. Liu, L. Dai and Z. Hu, Adv. Mater., 2019, 31, 1804799 CrossRef PubMed.
- W. Wei, S. Cui, L. Ding, L. Mi, W. Chen and X. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 40655–40670 CrossRef CAS PubMed.
- H. S. Kim, M. S. Kang and W. C. Yoo, J. Mater. Chem. A, 2019, 7, 5561–5574 RSC.
- Z. Peng, Y. Hu, J. Wang, S. Liu, C. Li, Q. Jiang, J. Lu, X. Zeng, P. Peng and F. F. Li, Adv. Energy Mater., 2019, 9, 1802928 CrossRef.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- J.-J. Zhou, X. Han, K. Tao, Q. Li, Y.-L. Li, C. Chen and L. Han, Chem. Eng. J., 2018, 354, 875–884 CrossRef CAS.
- D. Kong, C. Cheng, Y. Wang, J. I. Wong, Y. Yang and H. Y. Yang, J. Mater. Chem. A, 2015, 3, 16150–16161 RSC.
- Z. Sun, M. Zhu, X. Lv, Y. Liu, C. Shi, Y. Dai, A. Wang and T. Majima, Appl. Catal., B, 2019, 246, 330–336 CrossRef CAS.
- Y. Yu, J. Ma, C. Chen, Y. Fu, Y. Wang, K. Li, Y. Liao, L. Zheng and X. Zuo, ChemCatChem, 2019, 11, 1722–1731 CrossRef CAS.
- K. N. Dinh, Q. Liang, C.-F. Du, J. Zhao, A. I. Y. Tok, H. Mao and Q. Yan, Nano Today, 2019, 25, 99–121 CrossRef CAS.
- B. C. Martindale and E. Reisner, Adv. Energy Mater., 2016, 6, 1502095 CrossRef.
- T.-Q. Zhang, J. Liu, L.-B. Huang, X.-D. Zhang, Y.-G. Sun, X.-C. Liu, D.-S. Bin, X. Chen, A.-M. Cao and J.-S. Hu, J. Am. Chem. Soc., 2017, 139, 11248–11253 CrossRef CAS PubMed.
- B. Liang, Z. Zheng, M. Retana, K. Lu, T. Wood, Y. Ai, X. Zu and W. Zhou, Nanotechnology, 2019, 30, 295401 CrossRef PubMed.
- C. Guan, J. Liu, Y. Wang, L. Mao, Z. Fan, Z. Shen, H. Zhang and J. Wang, ACS Nano, 2015, 9, 5198–5207 CrossRef CAS PubMed.
- J. Luo, Z. Zheng, A. Kumamoto, W. I. Unah, S. Yan, Y. H. Ikuhara, X. Xiang, X. Zu and W. Zhou, Chem. Commun., 2018, 54, 794–797 RSC.
- V. D. Nithya and N. S. Arul, J. Power Sources, 2016, 327, 297–318 CrossRef CAS.
- X. Wang, K. Chen, G. Wang, X. Liu and H. Wang, ACS Nano, 2017, 11, 11602–11616 CrossRef CAS PubMed.
- H. Fan, H. Liu, X. Hu, G. Lv, Y. Zheng, F. He, D. Ma, Q. Liu, Y. Lu and W. Shen, J. Mater. Chem. A, 2019, 7, 11321–11330 RSC.
- A. M. Zardkhoshoui and S. S. H. Davarani, J. Alloys Compd., 2019, 773, 527–536 CrossRef CAS.
- J. Duan, S. Chen, S. Dai and S. Z. Qiao, Adv. Funct. Mater., 2014, 24, 2072–2078 CrossRef CAS.
- R. Zhou, Y. Zheng, M. Jaroniec and S.-Z. Qiao, ACS Catal., 2016, 6, 4720–4728 CrossRef CAS.
- H. B. Aiyappa, S. Saha, P. Wadge, R. Banerjee and S. Kurungot, Chem. Sci., 2015, 6, 603–607 RSC.
- S. Dou, L. Tao, J. Huo, S. Wang and L. Dai, Energy Environ. Sci., 2016, 9, 1320–1326 RSC.
- W. Weisweiler, N. Subramanian and B. Terwiesch, Carbon, 1971, 9, 755–761 CrossRef CAS.
- K. P. Singh, E. J. Bae and J.-S. Yu, J. Am. Chem. Soc., 2015, 137, 3165–3168 CrossRef CAS PubMed.
- G. B. B. Varadwaj and V. O. Nyamori, Nano Res., 2016, 9, 3598–3621 CrossRef CAS.
- W. Wei, W. Chen, L. Ding, S. Cui and L. Mi, Nano Res., 2017, 10, 3726–3742 CrossRef CAS.
- A. M. Puziy, O. I. Poddubnaya, A. Martínez-Alonso, F. Suárez-García and J. M. Tascón, Carbon, 2005, 43, 2857–2868 CrossRef CAS.
- M. A. Nahil and P. T. Williams, Biomass Bioenergy, 2012, 37, 142–149 CrossRef CAS.
- A. E. Allah, Y. Yamauchi, J. Wang, Y. Bando, H. Tan, A. A. Farghali, M. H. Khedr, A. Alshehri, Y. G. Alghamdi, D. Martin, M. A. Wahab, M.-d. A. Hossain and A. K. Nanjundan, ChemElectroChem, 2019, 6, 1–8 CrossRef.
- S. Liu, H. Zhang, Q. Zhao, X. Zhang, R. Liu, X. Ge, G. Wang, H. Zhao and W. Cai, Carbon, 2016, 106, 74–83 CrossRef CAS.
- A. Pendashteh, M. S. Rahmanifar, R. B. Kaner and M. F. Mousavi, Chem. Commun., 2014, 50, 1972–1975 RSC.
- J. Wang, T. Park, J. W. Yi, B. Ding, J. Henzie, Z. Chang, H. Dou, X. Zhang and Y. Yamauchi, Nanoscale Horiz., 2019, 4, 526–530 RSC.
- R. Li, Y. Wang, C. Zhou, C. Wang, X. Ba, Y. Li, X. Huang and J. Liu, Adv. Funct. Mater., 2015, 25, 5384–5394 CrossRef CAS.
- Y. Zeng, M. Yu, Y. Meng, P. Fang, X. Lu and Y. Tong, Adv. Energy Mater., 2016, 6, 1601053 CrossRef.
- Y. Zhang, Q. Sun, K. Xia, B. Han, C. Zhou, Q. Gao, H. Wang, S. Pu and J. Wu, ACS Sustainable Chem. Eng., 2019, 7, 5717–5726 CrossRef CAS.
- H. Tan, Y. Li, J. Kim, T. Takei, Z. Wang, X. Xu, J. Wang, Y. Bando, Y.-M. Kang and J. Tang, Adv. Sci., 2018, 5, 1800120 CrossRef PubMed.
- L. Yang, S. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Wang, Q. Wu, J. Ma, Y. Ma and Z. Hu, Angew. Chem., Int. Ed., 2011, 50, 7132–7135 CrossRef CAS PubMed.
- C. H. Choi, S. H. Park and S. I. Woo, ACS Nano, 2012, 6, 7084–7091 CrossRef CAS PubMed.
- X. Fan, Y. Liu, Z. Peng, Z. Zhang, H. Zhou, X. Zhang, B. I. Yakobson, W. A. Goddard III, X. Guo and R. H. Hauge, ACS Nano, 2017, 11, 384–394 CrossRef CAS PubMed.
- L. Zhang, J. Zhang, D. P. Wilkinson and H. Wang, J. Power Sources, 2006, 156, 171–182 CrossRef CAS.
- M. Pramanik, C. Li, Y. V. Kaneti and Y. Yamauchi, Chem. Commun., 2017, 53, 5721–5724 RSC.
- X.-Z. Yuan and H. Wang, PEM fuel cell electrocatalysts and catalyst layers: fundamentals and applications, Springer Science & Business Media, 2008, pp. 1–87 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04326h |
| This journal is © The Royal Society of Chemistry 2019 |