
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHomocysteine induces human mesangial cell apoptosis via the involvement of autophagy and endoplasmic reticulum stress
Shanshan
Liang†
,
Hua
Liu†
,
Sixiu
Liu†
,
Meng
Wei
,
Fanfan
Gao
,
Jinhong
Xue
,
Lingshuang
Sun
,
Meng
Wang
,
Hongli
Jiang
* and
Lei
Chen
 *
*
Dialysis Department of Nephrology Hospital, The First Affiliated Hospital of Xi'an Jiaotong University, West Yanta Road 277, Xi'an 710061, Shaanxi, China. E-mail: chl1221@hotmail.com; j92106@sina.com; Fax: +86 29 85324729; Tel: +86 29 85324729
First published on 7th October 2019
Abstract
Homocysteine (Hcy) level characterizes a progressive increase in chronic kidney disease (CKD). In fact, Hcy accumulation is considered to be a crucial biochemical culprit in CKD progression, but the mechanism underlying this remains poorly understood. This study investigated the role of Hcy in glomerular mesangial cell (MC) apoptosis and the potential involvement of autophagy and endoplasmic reticulum (ER) stress in this process, shedding light on Hcy toxicity in kidney disease. Human mesangial cells (HMCs) were incubated with different concentrations of Hcy for different times. Flow cytometry was used to determine the proportion of apoptotic cells and western blotting was used to analyze protein levels after the administration of Hcy, endoplasmic reticulum inhibitor 4-phenylbutyric acid (4-PBA), and Atg5 siRNA. The results demonstrated that the cell viability gradually decreased and the proportion of HMCs undergoing apoptosis increased with increasing Hcy concentration and prolonged incubation time. Meanwhile, levels of the apoptosis-related proteins Bax and cleaved caspase-3 were significantly increased, while ER stress-related proteins such as ATF4, CHOP, GRP78, and phospho-eIF2α significantly increased. Levels of cleaved LC3, and beclin1 and Atg5 proteins also increased, accompanied by p62 degradation, indicating autophagy activation. 4-PBA effectively inhibited ER stress and reversed Hcy-induced apoptosis and autophagy. Moreover, Atg5 siRNA alleviated Hcy-induced apoptosis. Taken together, these results suggest that Hcy induces HMC apoptosis in a dose- and time-dependent manner via the activation of Atg5-dependent autophagy triggered by ER stress. This study suggests a novel strategy against Hcy toxicity in kidney injury and should help in clarifying the pathogenesis of CKD.
Introduction
Homocysteine (Hcy), a sulfur-containing amino acid derived from methionine metabolism, is mainly metabolized in the kidneys. The accumulation of Hcy, namely, hyperhomocysteinemia (HHcy), characterizes all forms of chronic kidney disease (CKD), and is considered to be a crucial biochemical culprit in CKD development and progression.1–3 Elevated plasma Hcy levels can cause vasoconstriction and impairment of the renal microvasculature, and decrease glomerular filtration, finally leading to glomerulosclerosis and irreversible deterioration of renal function.4 Glomerular mesangial cells (MCs), which are the main factor responsible for regulating glomerular capillary blood flow and modulating glomerular filtration, may be a key component involved in HHcy-induced renal injury.5As the core regulator of mesangial homeostasis, disruption of the balance between the proliferation and apoptosis of MCs results in the progression of kidney disease.6 Enhanced apoptosis is a universal finding in both clinical and experimental kidney diseases that involve the mesangium, which irreversibly affects the pool of MCs and leads to their disappearance from mesangium.7 On the other hand, previous studies showed that Hcy could induce the apoptosis of neuronal cells,8,9 cardiomyocytes,10,11 vascular endothelial cells,12,13 and smooth muscle cells (SMCs).14 Interestingly, there is a similarity in the mechanism that Hcy could induce the apoptosis of these cell types by activating endoplasmic reticulum stress (ERS) and autophagy. As the predominant MCs display morphological and functional characteristics akin to SMCs, Hcy may induce MCs apoptosis. However, the role of Hcy in MC apoptosis and the underlying mechanism is far from elucidation.15,16
Endoplasmic reticulum (ER) is a membranous organelle present in almost every mammalian cell; its main functions include protein synthesis, modification, and folding.17 The capacity of ER to maintain appropriate protein folding is vital to the functional integrity of cells, including MCs. If protein folding is disturbed in the ER, unfolded proteins will accumulate and induce ER stress (ERS) and an adaptive response: the unfolded protein response (UPR).18,19 UPR can protect cells under mild or short-term stress; however, when the stress exceeds the ability of UPR to confer tolerance of it, ERS leads to apoptosis.20 Hcy-triggered ERS-mediated endothelial cell apoptosis is one example in which this occurs.21 Recent studies have shown the significant role of ERS-induced apoptosis in kidney diseases, especially proteinuric kidney diseases caused by podocyte injury.22 Nevertheless, whether ERS is involved in Hcy-induced MC injury remains to be clarified.
Autophagy is another adaptive response in cells under stress, in which autophagosomes form and deliver the cell contents to lysosomes for degradation.23 It has become increasingly clear that ERS stimulates accumulation of the precursors of autophagosomes, inducing autophagy.24 Autophagy is closely related to the apoptosis induced by ERS. Generally, autophagy attenuates apoptosis by inactivating caspases as well as inducing mitophagy. However, in special cases, autophagy or autophagy-relevant proteins may help to induce apoptosis.25 Since autophagy can both promote and inhibit apoptosis in ERS-induced cell injury, the relationship between autophagy and apoptosis in Hcy-treated MCs remains to be explored.
In summary, considering the damage to kidneys caused by Hcy and the potential links among Hcy, ERS, autophagy, and apoptosis, we speculated that ERS and autophagy may be involved in the Hcy-induced damage to MCs. Therefore, in the current study, we tested the hypothesis that elevated Hcy levels could induce MCs apoptosis by modulating ERS and autophagy.
Materials and methods
Cell culture and homocysteine preparation
Human mesangial cells (HMCs) (ScienCell Research Laboratories, CA, USA) were cultured in low-glucose Dulbecco's Modified Eagle's Medium (DMEM) (HyClone, LA, USA) supplemented with 10% fetal bovine serum (FBS) (BI, Israel) at 37 °C in an atmosphere of 5% CO2. The cell culture medium was replaced every 2 days until cells became confluent. HMCs were subcultured after partial digestion with 0.25% trypsin with 0.9 mM EDTA (HyClone). Homocysteine (Sigma-Aldrich, MO, USA) solution for cell treatments was prepared in a pre-warmed low-glucose DMEM cell culture medium without added FBS and passed through a 0.2 μm sterile filter.Cell viability assay
To evaluate cell viability, 5 × 103 HMCs per well were seeded into 96-well plates overnight before Hcy treatment. The cells were subsequently treated with different concentrations of Hcy (0–1000 μmol L−1) for 24 h and 250 μmol L−1 Hcy for different periods of time (0–48 h). Cell Counting Kit-8 (CCK8) Cell Proliferation and Cytotoxicity Test Kit (Beyotime, Shanghai, China) was used to assess the viability and proliferative capacity of cells in accordance with the manufacturer's protocol. Cell viability was measured by calculating the absorbance at 450 nm.Apoptosis assay by fluorescence-activated cell sorting
The number of cells induced to undergo apoptosis by Hcy was measured using the AnnexinV-FITC/PI Apoptosis Detection Kit (KeyGen Biotech, Nanjing, China). Cell samples were measured using the AnnexinV-APC/7-AAD Apoptosis Detection Kit (KeyGen Biotech). Cells were collected after different treatments and centrifuged at 1200 rpm for 3 min at 4 °C. After washing twice with PBS, cells were resuspended in 500 μL of 1× binding buffer containing 5 μL of PI and 5 μL of FITC-annexinV (or 5 μl of annexinV-APC) for 15 min without light at room temperature. Samples were analyzed by flow cytometry (CytoFLEX; Beckman). In a total of 1 × 104 cells per sample, both early (PI-negative and annexin V-positive cells) and late (PI-positive and annexin V-positive cells) apoptotic cells were analyzed.Protein extraction and western blotting
After the different treatments, the cells were washed twice with PBS and then lysed with RIPA buffer containing proteinase inhibitors [1% cocktail and 1 mmol L−1 phenylmethylsulfonyl fluoride (PMSF); Sigma-Aldrich] on ice for 30 min. The cell lysates were collected and centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min at 4 °C, after which the supernatants were collected. The total proteins were denatured by sodium dodecyl sulfate (SDS) sampling buffer and boiled for 5 min. SDS polyacrylamide gel electrophoresis (6–15%) was used to separate the total protein samples. The proteins were then transferred from gel to polyvinyl difluoride (PVDF) membrane (Millipore, MA, USA). After blocking with 5% nonfat milk for 1 h, the membrane was incubated with the primary antibody overnight at 4 °C (GAPDH, rabbit polyclonal antibody, 1:1000, Goodhere Biotechnology, Hangzhou, China; LC3, rabbit polyclonal antibody, 1:1000, affinity; cleaved caspase-3, ATF4, CHOP, phospho-eIF2α, rabbit monoclonal antibody, 1:1000, Cell Signaling Technology, MA, USA; Bax, rabbit monoclonal antibody, 1:3000; Bcl2, rabbit polyclonal antibody, 1:600; p62, rabbit monoclonal antibody, 1:1000; GRP78, rabbit polyclonal antibody, 1:800, Abcam, Cambridge, UK; Atg5, 1:500; beclin1, 1:1500; rabbit polyclonal antibody, Proteintech Group, Wuhan, China). The next day, the blots were incubated with peroxidase-conjugated secondary antibody (Boster, Wuhan, China) for 1 h at room temperature. Membranes were washed again three times and then developed with Enhanced Chemiluminescence (Thermo Scientific, MA, USA), followed by apposition of the membrane to autoradiographic films (Kodak, NY, USA). The immunoblots were quantified by densitometric analysis.
000 rpm for 20 min at 4 °C, after which the supernatants were collected. The total proteins were denatured by sodium dodecyl sulfate (SDS) sampling buffer and boiled for 5 min. SDS polyacrylamide gel electrophoresis (6–15%) was used to separate the total protein samples. The proteins were then transferred from gel to polyvinyl difluoride (PVDF) membrane (Millipore, MA, USA). After blocking with 5% nonfat milk for 1 h, the membrane was incubated with the primary antibody overnight at 4 °C (GAPDH, rabbit polyclonal antibody, 1:1000, Goodhere Biotechnology, Hangzhou, China; LC3, rabbit polyclonal antibody, 1:1000, affinity; cleaved caspase-3, ATF4, CHOP, phospho-eIF2α, rabbit monoclonal antibody, 1:1000, Cell Signaling Technology, MA, USA; Bax, rabbit monoclonal antibody, 1:3000; Bcl2, rabbit polyclonal antibody, 1:600; p62, rabbit monoclonal antibody, 1:1000; GRP78, rabbit polyclonal antibody, 1:800, Abcam, Cambridge, UK; Atg5, 1:500; beclin1, 1:1500; rabbit polyclonal antibody, Proteintech Group, Wuhan, China). The next day, the blots were incubated with peroxidase-conjugated secondary antibody (Boster, Wuhan, China) for 1 h at room temperature. Membranes were washed again three times and then developed with Enhanced Chemiluminescence (Thermo Scientific, MA, USA), followed by apposition of the membrane to autoradiographic films (Kodak, NY, USA). The immunoblots were quantified by densitometric analysis.
Experiment of 4-PBA and Atg5 siRNA interference
HMCs were treated with Hcy (250 μmol L−1, 24 h) without or with pretreatment with 1 mmol L−1 4-phenylbutyric acid (4-PBA) (Sigma-Aldrich) for 2 h. Small interfering RNAs (siRNAs) against autophagy-related gene 5 (Atg5-homo-695) and negative control siRNA were designed and synthesized by GenePharma (Shanghai, China). A total of 5 × 105 per well HMCs were seeded in six-well plates 1 day before the experiments and cells were transiently transfected with siRNA using Lipofectamine™ 2000 siRNA Transfection Reagent (Invitrogen), in accordance with the manufacturer's protocol. Real-time PCR was used to analyze the success of gene silencing. Twenty-four hours after transfection, cells were treated in accordance with the experimental design and harvested for further assays.Statistical analysis
All statistical analyses were carried out using SPSS 18.0 software package (SPSS Inc., Chicago, IL, USA). Quantitative data are reported as the mean ± SEM. Differences between experimental and control groups were determined by Student's t-test, while comparisons among multiple groups were performed using one-way ANOVA. Differences were considered statistically significant at p < 0.05.Results
Effect of Hcy on the viability and apoptosis of HMCs
HMCs were treated with various concentrations of Hcy (25, 50, 100, 250, 500, 1000 μmol L−1) for 24 h and with 250 μmol L−1 Hcy for different time periods (0, 6, 12, 24, or 48 h) to evaluate cell viability. The results showed that cell viability significantly decreased in a dose-dependent manner (Fig. 1A) as well as in a time-dependent manner (Fig. 1B) after Hcy treatment, when compared with the control group (p < 0.05). To clarify whether apoptosis was involved in the Hcy-induced decrease in cell viability, annexin V/PI staining with flow cytometry was used. The results showed that Hcy also significantly induced apoptosis in a time-dependent manner (Fig. 1C and E, p < 0.05) and a dose-dependent manner (Fig. 1D and F, p < 0.05). Moreover, the levels of the apoptosis-related proteins Bax and cleaved caspase-3 were significantly increased, while the level of Bcl-2 protein was markedly decreased after treatment with Hcy at a concentration of 250 μmol L−1 for 24 h (Fig. 2A, p < 0.05). These results together showed that Hcy could induce HMC apoptosis.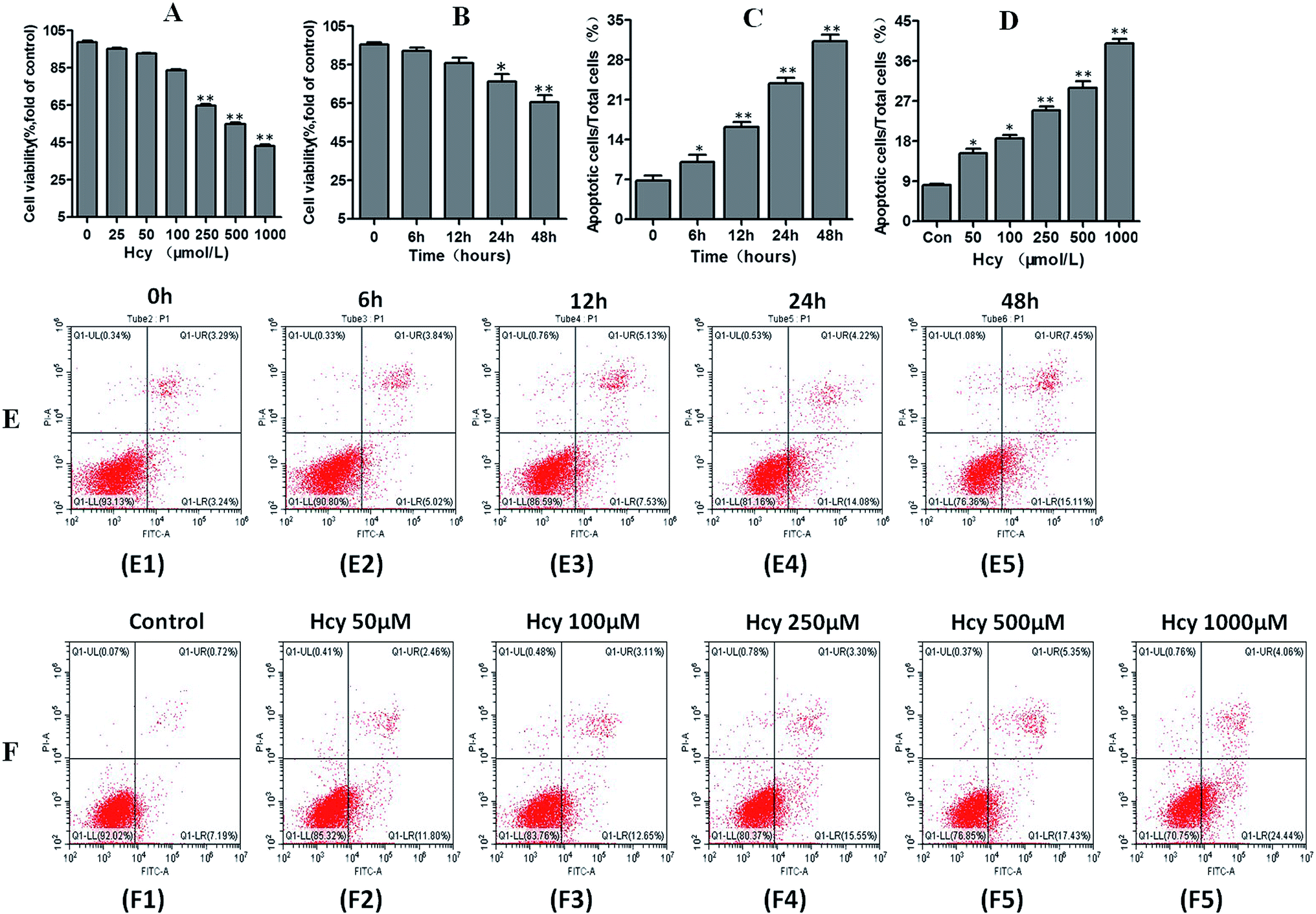 | ||
| Fig. 1 Effects of Hcy on mesangial cell viability and apoptosis proportion. HMCs were treated with various concentrations of Hcy (25, 50, 100, 250, 500 and 1000 μ mol L−1) for 24 h and different hours (0, 6, 12, 24, 48 h) of Hcy 250 μ mol L−1 to evaluate the cell viability by CCK-8 assay. With the increase of Hcy concentration and the prolongation of Hcy incubation time, the cell viability significantly decreased in a dose-dependent manner (A) as well as in a time-dependent manner (B) as compared with normal control group. Proportions of apoptotic cells after treated with various concentrations of Hcy(C and E) and different incubation hours (D and F) were determined by flow cytometry. Data are presented as mean ± SEM of three independent experiments performed in triplicates. *P < 0.05, **P < 0.01, compared with the control group. | ||
 | ||
| Fig. 2 Effects of Hcy on apoptosis, ERS, and autophagy-related protein expression. HMCs were treated with 250 μmol L−1 Hcy for 24 h and then subjected to analyses of apoptosis, ERS and autophagy-related protein expression by western blotting. The levels of apoptosis-related proteins Bax and cleaved caspase-3 were significantly increased along with a marked decrease of the level of Bcl-2 protein (A). The ratio of the autophagy-associated proteins LC3-II/LC3-I, and the expression levels of beclin1 and Atg5 proteins in HMCs were significantly increased after Hcy treatment, while the p62 protein level was significantly decreased after Hcy treatments (B). The levels of ERS-related proteins ATF4, CHOP, GRP78, and phospho-eIF2α were significantly upregulated (C). Data are presented as mean ± SEM of three independent experiments performed in triplicate. *P < 0.05, compared with the control group. | ||
Hcy induced ERS and autophagy in HMCs
To understand whether ERS is involved in the Hcy-induced apoptosis of MCs, ERS-related proteins were analyzed. HMCs treated with Hcy (250 μmol L−1) for 24 h also showed that Hcy can significantly upregulate the expression of ATF4, CHOP, GRP78, and phospho-eIF2α proteins (Fig. 2C, p < 0.05). To further explore whether autophagy was also involved in Hcy-induced apoptosis of MCs, the expression levels of proteins related to autophagy were also determined by western blotting. HMCs were treated with 250 μmol L−1 Hcy for 24 h. The levels of the autophagy-associated proteins cleaved LC3 (indicated by the LC3-II/LC3-I ratio), beclin1, and Atg5 in HMCs were significantly increased after Hcy treatment. p62 protein, as one of the key downstream signals of autophagy, was significantly decreased after Hcy intervention (Fig. 2B, p < 0.05). These results together indicate that Hcy induced ERS as well as increased autophagy activity in MCs, which may participate in Hcy-induced apoptosis of HMCs.4-PBA reversed ERS and apoptosis in HMCs induced by Hcy
4-PBA is an effective inhibitor of ERS. In this study, 4-PBA was used to verify the crucial role of ERS in Hcy-induced MC apoptosis. The results of annexin V-APC/7-AAD staining showed that HMCs pretreatment with 4-PBA (1 mmol L−1) before culture with Hcy (250 μmol L−1) significantly reduced the proportion of HMCs with Hcy-induced apoptosis compared with the Hcy only group (Fig. 3A, p < 0.05). Co-treatment with 4-PBA also dramatically reduced the levels of CHOP, phospho-eIF2α, and cleaved caspase-3 proteins in HMCs compared with the levels in the Hcy only group (p < 0.05). In addition, 4-PBA significantly reversed the increase of the LC3-II/LC3-I ratio and p62 protein degradation induced by Hcy (Fig. 3B, p < 0.05). These findings suggest that Hcy may induce apoptosis and autophagy through p-eIF2α/CHOP ERS pathway.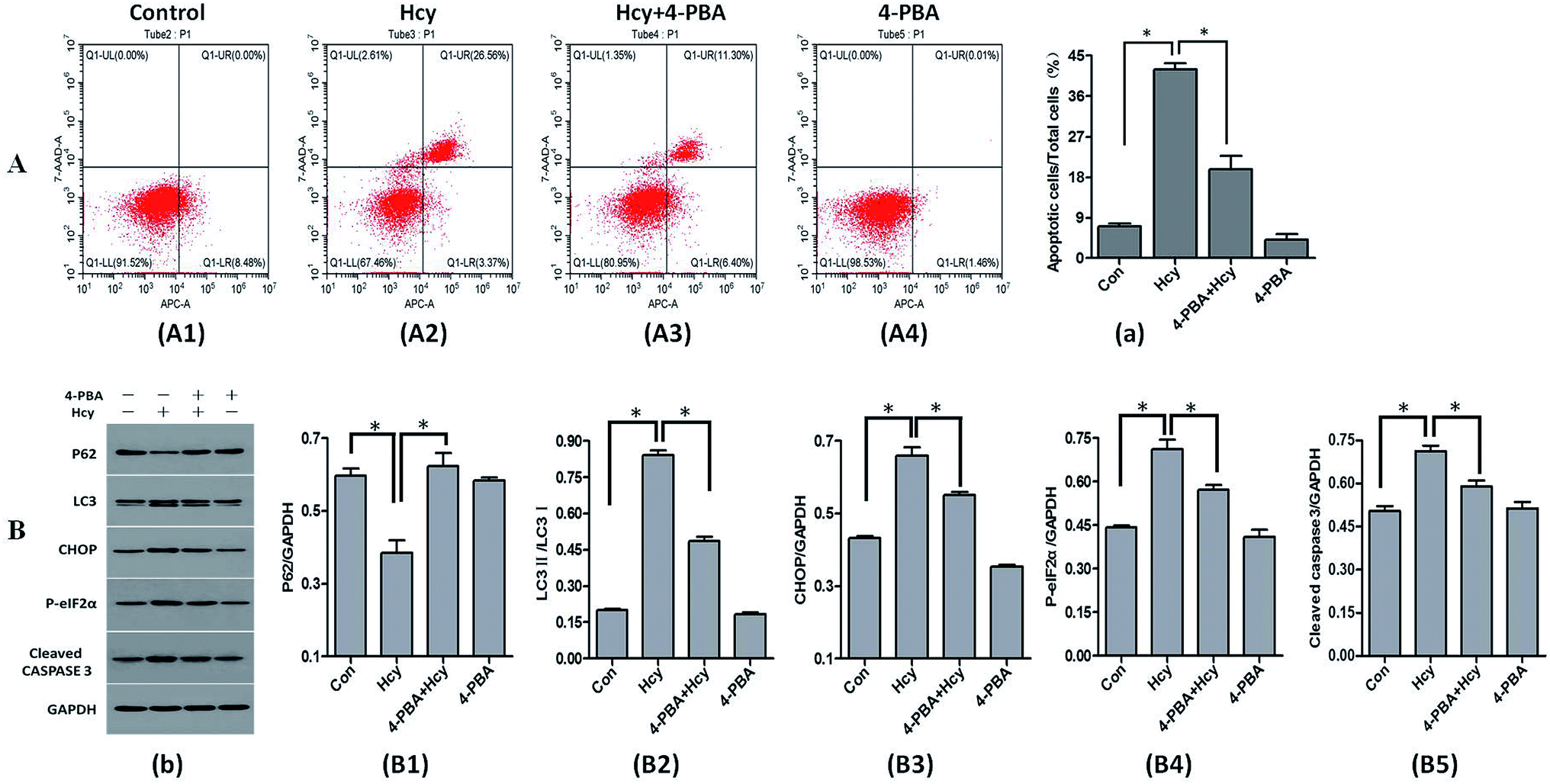 | ||
| Fig. 3 Effects of 4-PBA intervention on Hcy-induced apoptosis, ERS, and autophagy. The proportion of apoptotic HMCs was determined by flow cytometry and protein levels were determined by western blotting. Pretreatment with 4-PBA significantly reduced the proportion of Hcy-induced apoptotic mesangial cells compared with that in the Hcy group (A). The levels of ERS-related proteins CHOP and p-eIF2α were reduced. The ratio of the autophagy-associated proteins LC3-II/LC3-I was significantly decreased and the p62 protein level was significantly increased. The level of the apoptosis-related protein cleaved caspase-3 was also decreased (B). *P < 0.05, Hcy group vs. control group, and 4-PBA + Hcy group vs. Hcy group. All experiments were performed in at least triplicate. | ||
Inhibition of Atg5 expression attenuated Hcy-induced apoptosis in HMCs
To further explore the role of autophagy in Hcy-induced apoptosis of HMCs, HMCs were transfected with Atg5 siRNA for 6 h before Hcy treatment. Notably, the results showed that Atg5 siRNA intervention significantly decreased the proportion of MCs exhibiting Hcy-induced apoptosis compared with the level in the Hcy only group (Fig. 4A, p < 0.05). The results also showed that transfection with Atg5 siRNA markedly reduced Atg5 protein expression as well as reversed the increase of in the LC3-II/LC3-I ratio and p62 protein degradation, which indicated that Atg5 siRNA transfection may inhibit Hcy-induced autophagy (p < 0.05). In addition, the increased levels of cleaved caspase-3 and CHOP proteins induced by Hcy were also significantly attenuated to varying degrees after transfection with Atg5 siRNA (Fig. 4B, p < 0.05). The above results indicated that Atg5-dependent autophagy played a role in exacerbating the apoptosis of MCs induced by Hcy.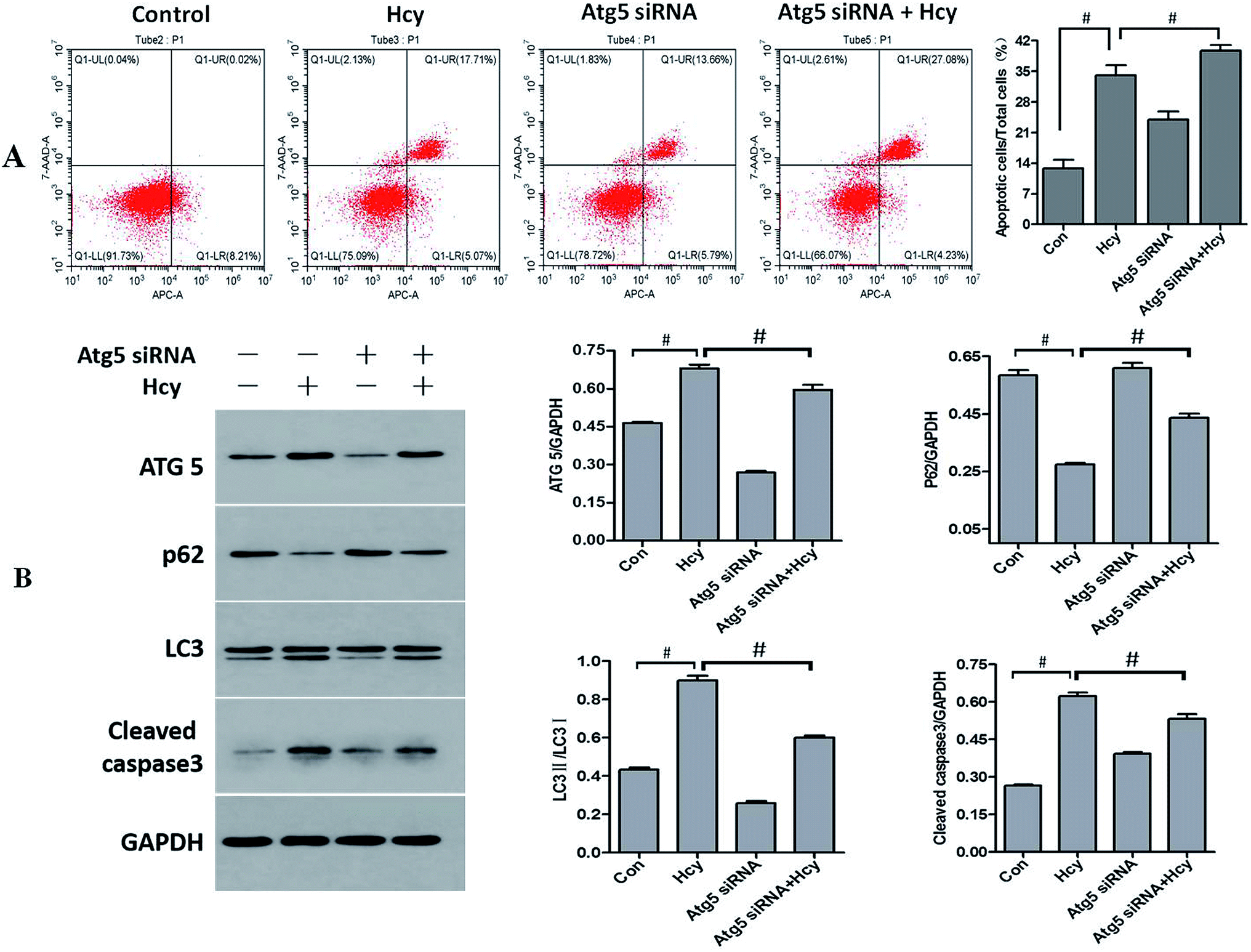 | ||
| Fig. 4 Effect of Atg5 siRNA intervention on Hcy-induced autophagy, ERS, and apoptosis. The proportion of apoptotic HMCs was determined by flow cytometry and related protein levels were determined by western blotting. Atg5 siRNA intervention decreased the proportion of Hcy-induced apoptotic HMCs compared with that in the Hcy group (A). The level of the autophagy-associated protein Atg5 and the LC3-II/LC3-I ratio were decreased, while the p62 protein level was significantly increased. The level of the apoptosis-related protein cleaved caspase-3 was also decreased (B). #P < 0.05, Hcy group vs. control group, and Atg5 siRNA + Hcy group vs. Hcy group. All experiments were performed in at least triplicate. | ||
Discussion
Hcy level characterizes a progressive increase in CKD. HHcy (plasma Hcy concentration >15 μmol L−1) is considered to be a serious risk factor for CKD that ultimately ends in renal failure. Recently, accumulating evidence has indicated that HHcy could directly act on glomerular cells to induce glomerular dysfunction and consequent glomerulosclerosis.4,26,27 In this study, we verified that Hcy induced glomerular mesangial cell apoptosis and reduced cell viability. We further found that both ERS and autophagy were involved in Hcy-induced MC apoptosis. The inhibition of ERS by 4-PBA could reverse the activity of Hcy-induced ERS, apoptosis, and autophagy in MCs. The inhibition of autophagy by Atg5 siRNA decreased the Atg5 protein level, LC3-II/LC3-I ratio, and p62 degradation, and attenuated apoptosis in Hcy-treated MCs. These results suggest that autophagy activated apoptosis under Hcy-induced ERS in MCs. Regarding the key role of MCs in glomerular function, the Hcy-induced apoptosis of MCs may be an important factor in the development and progression of CKD.Hcy has been reported to affect cell viability and induce apoptosis in various cell types, such as neuronal cells,8,9 vascular endothelial cells,12,13 SMCs,14 and podocytes.28 In the present study, we found that the viability of MCs was clearly decreased with the increase of Hcy concentration from 50 to 1000 μmol L−1. As shown in previous studies,29–31 the concentrations of Hcy used here were also observed in the clinical context and readily induced MC injury. We also verified that Hcy led to significant increase in the proportion of MCs undergoing apoptosis with the prolongation of incubation time. Besides, the increased levels of apoptosis-related proteins Bax and cleaved caspase-3 and the markedly decreased level of Bcl-2 protein after Hcy treatment also indicated that Hcy could induce the apoptosis of MCs. These results reveal that Hcy induced apoptosis in both time- and dose-dependent manners, which is consistent with the findings from studies on other cells, such as vascular endothelial cells and podocytes.21,32 It's worth mentioning that the apoptosis of MCs may be influenced by many other factors in vitro experiments. The disruption of mitochondrial bioenergetic function caused by energy depletion33 and dynamic fatigue caused by shear stress34 could also lead to the apoptosis of MCs. Therefore, the strict experimental quality control is needed to exclude above-mentioned factors. The follow tips were all important for our study to confirm the effect of Hcy on MCs' apoptosis: guaranteeing the same survival activity and the same cell density for cells in each group before treatment, ensuring that cell densities were never too dense or too sparse in each group at the end of the experiment to avoid different mechanical forces due to cell–cell interaction, minimizing the difference in shear stress caused by lifting and unnecessary fluid exchange, and supplying adequate energy for all cells to avoid energy depletion. Notably, since MC apoptosis plays a crucial role in both clinical and experimental kidney diseases, the much higher proportion of apoptotic cells in Hcy-treated cells than those in normal MCs and in rat glomeruli as reported previously may contribute significantly to disease progression.35
With respect to the pathogenicity of Hcy in cell injury, several important cellular and molecular mechanisms have been particularly focused on, such as oxidative stress,36 ERS,10 homocysteinylation, and hypomethylation.37 Recently, the Hcy-induced ERS response has received considerable attention, and evidence has increasingly shown that this ERS may represent an important fundamental mechanism mediating the Hcy-induced cell injury.38–40 It has been speculated that the basis for the impairment in ER function by Hcy involves disrupting disulfide bond formation to perturb protein folding. In addition, Hcy was found to activate the expression of genes known to be under the control of signaling pathways that respond to loading on the ER. On the other hand, the pathophysiological role of ERS in glomerular injury in vitro and in vivo has also been reported. Miyata et al. showed that ERS was detected in isolated glomeruli in IgA nephropathy.41 Inagi et al. reported that complement attack against podocytes induced ERS by the upregulation of ERS proteins such as GRP78 and GRP94.42 Moreover, Park et al. indicated that treatment with palmitate, which mimics cellular lipotoxicity, could exacerbate diabetic nephropathy by inducing the apoptosis of MCs via protein kinase RNA-like endoplasmic reticulum kinase (PERK) and ATF6-mediated ERS signaling.43 In this study, our results showed that Hcy markedly upregulated the ERS-related proteins GRP78, phospho-eIF2α, and ATF4 as well as apoptosis in MCs. Phospho-eIF2α is one of the major signal transducers in ERS; it can induce the expression of CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP), which plays an important role in ERS-induced apoptosis. In the present study, the upregulation of CHOP and the apoptosis-related protein levels of Bax and cleaved caspase-3 was also observed in Hcy-treated MCs. Notably, we also found that the ERS inhibitor 4-PBA significantly reversed the Hcy-induced upregulation of CHOP and the apoptosis-related proteins, then significantly inhibited the apoptosis of MCs induced by Hcy. These results together indicate that Hcy induce the apoptosis of MCs through the ERS-related p-eIF2α/CHOP signaling pathway.
The present study also showed that Hcy induced the expression of autophagy-relevant proteins, such as cleaved LC3 (reflected in the LC3-II/LC3-I ratio), beclin1, and Atg5, and decreased the expression of p62 in MCs. Furthermore, all of the above changes were reversed by the ERS inhibitor 4-PBA. These findings indicated that autophagy was not only involved in Hcy-induced MC injury, but was also activated by ERS. Autophagy is considered to be a pivotal and evolutionarily conserved mechanism for maintaining cellular homeostasis and is closely related to the apoptosis induced by ERS.24 As one of the essential components in the initial stage of autophagy, beclin-1 interacts with Bcl-2 (or its related family member, Bcl-xL) to form beclin-1–Bcl-2/BclxL complex.44 The complex inhibits the proautophagic function of beclin-1 to constrain autophagy level during normal conditions, and the disruption of the complex leads to the upregulation autophagy.45 As shown in previous study,46 CHOP could decrease the expression of Bcl-2 and disrupt the beclin-1-Bcl-2 complex, therefore increase autophagy effectively. Combining with our findings in present study, we deduced that upregulation of CHOP may be the key mechanism underlying ERS-induced autophagy in Hcy-treated HMCs. Interestingly, whether autophagy promotes or inhibits apoptosis depends on the particular situation. In a previous study, autophagy was demonstrated to play a protective role in podocytes under hyperglycemic conditions.47 However, in our study, autophagy appeared to be an activator of apoptosis because the inhibition of autophagy by Atg5 siRNA not only markedly inhibited autophagy but also decreased the apoptotic effect in Hcy-treated MCs. Atg5 is indispensable for autophagosome formation. Knocking down or knocking out Atg5 results in the downregulation or complete inhibition of autophagy. In addition to its role in the formation of autophagosomes, recent study showed that an Atg5 fragment produced by calpain cleavage has pro-apoptotic properties, which could switch autophagy to apoptosis.48 The present study showed that the knockdown of Atg5 significantly increased the LC3-II/LC3-I ratio and p62 protein degradation, and decreased the expression of the apoptosis-related protein cleaved caspase-3 under Hcy-induced ERS. This indicates that Atg5-dependent autophagy may play a relatively harmful role in Hcy-induced MC injury.
In conclusion, this study has demonstrated that Hcy can induce apoptosis in MCs and provided evidence that the activation of Atg5-dependent autophagy by ERS plays a crucial role in this process. These findings may offer a novel strategy against Hcy toxicity in kidney injury and should help us to further clarify the pathogenesis of CKDs. Future studies will be aimed at testing these hypotheses using in vivo models.
Conflicts of interest
The authors declare that they have no competing interests.Abbreviations
| 4-PBA | 4-phenylbutyric acid |
| CCK8 | cell counting kit-8 |
| C/EBP | CCAAT/enhancer-binding protein |
| CHOP | C/EBP homologous protein |
| CKD | chronic kidney disease |
| DMEM | Dulbecco's modified Eagle's medium |
| ER | endoplasmic reticulum |
| ERS | endoplasmic reticulum stress |
| FBS | fetal bovine serum |
| Hcy | homocysteine |
| HHcy | hyperhomocysteinemia |
| HMCs | human mesangial cells |
| MCs | mesangial cells |
| PERK | protein kinase RNA-like endoplasmic reticulum kinase |
| PMSF | phenylmethylsulfonyl fluoride |
| PVDF | polyvinyl difluoride |
| SDS | sodium dodecyl sulfate |
| SMCs | smooth muscle cells |
| siRNAs | small interfering RNAs |
| UPR | unfolded protein response |
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No.81570670), the Major Scientific and Technological Innovation Project of Shaanxi Province (Grant No. 2017ZDXM-SF-057), the Social Science and Technology Development Project of Shaanxi Province (Grant No. 2018SF-162) and the International Science and Technology Cooperation Project of Shaanxi Province (Grant No. 2019KW-32).References
- E. A. Ostrakhovitch and S. Tabibzadeh, Adv. Clin. Chem., 2015, 72, 77–106 CrossRef CAS.
- Y. Long and J. Nie, Kidney Dis., 2016, 2, 80–87 CrossRef.
- A. B. Lawrence de Koning, G. H. Werstuck, J. Zhou and R. C. Austin, Clin. Biochem., 2003, 36, 431–441 CrossRef CAS.
- Y. F. Chen, P. L. Li and A. P. Zou, Circulation, 2002, 106, 1275–1281 CrossRef CAS.
- H. E. Abboud, Exp. Cell Res., 2012, 318, 979–985 CrossRef CAS.
- L. Chen, Y. Lu, J. Wen, X. Wang, L. Wu, D. Wu, X. Sun, B. Fu, Z. Yin, H. Jiang and X. Chen, Cell. Physiol. Biochem., 2016, 39, 1761–1776 CrossRef CAS.
- G. A. Herrera, E. A. Turbat-Herrera and J. Teng, Contrib. Nephrol., 2011, 169, 6–22 CAS.
- J. W. Zhang, R. Yan, Y. S. Tang, Y. Z. Guo, Y. Chang, L. Jing, Y. L. Wang and J. Z. Zhang, Int. J. Immunopathol. Pharmacol., 2017, 30, 371–382 CrossRef CAS.
- H. J. Wei, J. H. Xu, M. H. Li, J. P. Tang, W. Zou, P. Zhang, L. Wang, C. Y. Wang and X. Q. Tang, Acta Pharmacol. Sin., 2014, 35, 707–715 CrossRef CAS.
- Z. Zhang, L. Zhao, Y. Zhou, X. Lu, Z. Wang, J. Wang and W. Li, Apoptosis, 2017, 22, 647–661 CrossRef CAS.
- T. P. Vacek, J. C. Vacek, N. Tyagi and S. C. Tyagi, Cell Biochem. Biophys., 2012, 62, 1–11 CrossRef CAS.
- Z. Zhang, C. Wei, Y. Zhou, T. Yan, Z. Wang, W. Li and L. Zhao, Oxid. Med. Cell. Longevity, 2017, 2017, 5736506 Search PubMed.
- Y. Zhang, Y. Zhang, J. Tang, S. Zhao, C. Li, Y. P. Huang and M. Yi, Cell. Physiol. Biochem., 2018, 45, 1893–1903 CrossRef CAS.
- Q. Yuan, D. J. Jiang, Q. Q. Chen, S. Wang, H. Y. Xin, H. W. Deng and Y. J. Li, Biochem. Biophys. Res. Commun., 2007, 356, 880–885 CrossRef CAS.
- M. E. Francis, P. W. Eggers, T. H. Hostetter and J. P. Briggs, Kidney Int., 2004, 66, 303–312 CrossRef CAS.
- W. Herrmann, H. Schorr, R. Obeid, J. Makowski, B. Fowler and M. K. Kuhlmann, Clin. Chem., 2005, 51, 891–897 CrossRef CAS.
- X. Li, L. Meng, F. Wang, X. Hu and Y. Yu, Mol. Cell. Biochem., 2019, 454, 77–85 CrossRef CAS.
- R. Inagi, Curr. Opin. Pharmacol., 2010, 10, 156–165 CrossRef CAS.
- J. G. Dickhout and J. C. Krepinsky, Antioxid. Redox Signaling, 2009, 11, 2341–2352 CrossRef CAS.
- F. J. Guo, Y. Liu, J. Zhou, S. Luo, W. Zhao, X. Li and C. Liu, Histochem. Cell Biol., 2012, 138, 447–460 CrossRef CAS.
- Z. Zhang, C. Wei, Y. Zhou, T. Yan, Z. Wang, W. Li and L. Zhao, Oxid. Med. Cell. Longevity, 2017, 2017, 5736506 Search PubMed.
- A. V. Cybulsky, Kidney Int., 2013, 84, 25–33 CrossRef CAS.
- G. P. Kaushal, Kidney Int., 2012, 82, 1250–1253 CrossRef CAS.
- T. Yorimitsu, U. Nair, Z. Yang and D. J. Klionsky, J. Biol. Chem., 2006, 281, 30299–30304 CrossRef CAS.
- S. Song, J. Tan, Y. Miao, M. Li and Q. Zhang, J. Cell. Physiol., 2017, 232, 2977–2984 CrossRef CAS.
- E. A. Ostrakhovitch and S. Tabibzadeh, Adv. Clin. Chem., 2015, 72, 77–106 CAS.
- C. Zhang, K. M. Boini, M. Xia, J. M. Abais, X. Li, Q. Liu and P. L. Li, Hypertension, 2012, 60, 154–162 CrossRef CAS.
- C. Zhang, J. J. Hu, M. Xia, K. M. Boini, C. Brimson and P. L. Li, Biochim. Biophys. Acta, 2010, 1803, 482–491 CrossRef CAS.
- C. Zhang, J. J. Hu, M. Xia, K. M. Boini, C. Brimson and P. L. Li, Biochim. Biophys. Acta, 2010, 1803, 482–491 CrossRef CAS.
- S. Liang, S. Liu, H. Liu, X. He, L. Sun, L. Chen, M. Wei, F. Gao and H. Jiang, Kidney Blood Pressure Res., 2018, 43, 1516–1528 CrossRef CAS.
- S. Majumder, L. Ren, S. Pushpakumar and U. Sen, Cell. Signalling, 2019, 61, 66–77 CrossRef CAS.
- C. Zhang, K. M. Boini, M. Xia, J. M. Abais, X. Li, Q. Liu and P. L. Li, Hypertension, 2012, 60, 154–162 CrossRef CAS.
- Y. Kushnareva and D. D. Newmeyer, N. Y. Acad. Sci., Ann., 2010, 1201, 50–57 CrossRef CAS.
- Y. Qiang, J. Liu and E. Du, Acta Biomater., 2017, 57, 352–362 CrossRef CAS.
- S. Shastry, A. J. Ingram, J. W. Scholey and L. R. James, Kidney Int., 2007, 71, 304–311 CrossRef CAS.
- P. M. Kanani, C. A. Sinkey, R. L. Browning, M. Allaman, H. R. Knapp and W. G. Haynes, Circulation, 1999, 100, 1161–1168 CrossRef CAS.
- R. Esse, M. Barroso, I. Tavares de Almeida and R. Castro, Int. J. Mol. Sci., 2019, 20 Search PubMed.
- B. L. Chen, M. L. Sheu, K. S. Tsai, K. C. Lan, S. S. Guan, C. T. Wu, L. P. Chen, K. Y. Hung, J. W. Huang and C. K. Chiang, Antioxid. Redox Signaling, 2015, 23, 1233–1245 CrossRef CAS PubMed.
- H. M. Zeeshan, G. H. Lee, H. R. Kim and H. J. Chae, Int. J. Mol. Sci., 2016, 17, 327 CrossRef.
- F. Yi and P. L. Li, Am. J. Nephrol., 2008, 28, 254–264 CrossRef CAS.
- T. Miyata, M. Nangaku, D. Suzuki, R. Inagi, K. Uragami, H. Sakai, K. Okubo and K. Kurokawa, J. Clin. Invest., 1998, 102, 828–836 CrossRef CAS.
- R. Inagi, M. Nangaku, N. Usuda, A. Shimizu, H. Onogi, Y. Izuhara, K. Nakazato, Y. Ueda, H. Oishi, S. Takahashi, M. Yamamoto, D. Suzuki, K. Kurokawa, C. van Ypersele de Strihou and T. Miyata, J. Am. Soc. Nephrol., 2005, 16, 1339–1349 CrossRef CAS.
- M. J. Park, H. J. Han and D. I. Kim, Int. J. Mol. Sci., 2017, 18 Search PubMed.
- X. H. Liang, S. Jackson, M. Seaman, K. Brown, B. Kempkes, H. Hibshoosh and B. Levine, Nature, 1999, 402, 672–676 CrossRef CAS.
- K. Wang, Cell Cycle, 2015, 14, 1631–1642 CrossRef CAS PubMed.
- K. Liu, Y. Shi, X. Guo, S. Wang, Y. Ouyang, M. Hao, D. Liu, L. Qiao, N. Li, J. Zheng and D. Chen, Cell Death Dis., 2014, 5, e1323 CrossRef CAS.
- K. Yamahara, M. Yasuda, S. Kume, D. Koya, H. Maegawa and T. Uzu, J. Diabetes Res., 2013, 2013, 193757 Search PubMed.
- S. Yousefi, R. Perozzo, I. Schmid, A. Ziemiecki, T. Schaffner, L. Scapozza, T. Brunner and H. U. Simon, Nat. Cell Biol., 2006, 8, 1124–1132 CrossRef CAS PubMed.
Footnote |
| † Shanshan Liang, Hua Liu and Sixiu Liu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |