
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermal activation of charge carriers in ionic and electronic semiconductor β-AgIVVO3 and β-AgIVVO3@VV1.6VIV0.4O4.8 composite xerogels†
Roberto Fernández de Luis *a,
Edurne S. Larreab,
Joseba Orivec,
Luis Lezamaad,
C. M. Costae,
S. Lanceros-Méndezaf and
María I. Arriortuaab
*a,
Edurne S. Larreab,
Joseba Orivec,
Luis Lezamaad,
C. M. Costae,
S. Lanceros-Méndezaf and
María I. Arriortuaab
aBCMaterials (Basque Centre for Materials, Applications & Nanostructures), Bld. Martina Casiano, 3rd. Floor UPV/EHU Science Park Barrio Sarriena s/n, 48940 Leioa, Spain. E-mail: roberto.fernandez@bcmaterials.net; maribel@arriortua.ehu.es; Fax: +34 946013500; Tel: +34 946015984
bDepartamento de Mineralogía y Petrología, Facultad de Ciencia y Tecnología, Universidad del País Vasco, UPV/EHU, Apdo. 644, E-48080 Bilbao, Spain
cDepartamento de Ciencia de Materiales, Facultad de Ciencias Físicas y Matemáticas (FCFM), Universidad de Chile, Av. Beauchef 851, Santiago, Chile
dDepartamento de Química Inorgánica, Facultad de Ciencia y Tecnología, Universidad del País Vasco, UPV/EHU, Apdo. 644, E-48080 Bilbao, Spain
eCentre of Physics, University of Minho, 4710-057 Braga, Portugal
fIKERBASQUE, Basque Foundation for Science, 48013 Bilbao, Spain
First published on 20th December 2019
Abstract
Silver vanadium oxide (SVO) and Silver Vanadium Oxide/Vanadium Oxide (SVO@VO) composite hydrogels are formed from the self-entanglement of β-AgVO3 nanoribbons and slightly reduced vanadium oxide (VO) (VV1.6VIV0.4O4.8) nanoribbons; respectively. Starting from randomly distributed nanoribbons within hydrogels, and after a controlled drying process, a homogeneous xerogel system containing tuneable SVO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VO ratios from 1:0 to 1:1 can be obtained. The precise nanoribbons compositional control of these composite system can serve as a tool to tune the electrical properties of the xerogels, as it has been demonstrated in this work by impedance spectroscopy (IS) experiments. Indeed, depending on the composition and temperature conditions, composite xerogels can behave as electronic, protonic or high temperature ionic conductors. In addition, the electric and protonic conductivity of the composite xerogels can be enhanced (until a critical irreversible point), through the temperature triggered charge carrier creation. As concluded from thermogravimetry, IR, UV-Vis and EPR spectroscopy studies, besides the SVO:VO ratio, the thermal induced oxidation/reduction of V5+ to V4+, and thermally triggered release of strongly bonded water molecules at the nanoribbon surface are the two key variables that control the electric and ionic conduction processes within the SVO and composite SVO/VO xerogels.
:VO ratios from 1:0 to 1:1 can be obtained. The precise nanoribbons compositional control of these composite system can serve as a tool to tune the electrical properties of the xerogels, as it has been demonstrated in this work by impedance spectroscopy (IS) experiments. Indeed, depending on the composition and temperature conditions, composite xerogels can behave as electronic, protonic or high temperature ionic conductors. In addition, the electric and protonic conductivity of the composite xerogels can be enhanced (until a critical irreversible point), through the temperature triggered charge carrier creation. As concluded from thermogravimetry, IR, UV-Vis and EPR spectroscopy studies, besides the SVO:VO ratio, the thermal induced oxidation/reduction of V5+ to V4+, and thermally triggered release of strongly bonded water molecules at the nanoribbon surface are the two key variables that control the electric and ionic conduction processes within the SVO and composite SVO/VO xerogels.
1. Introduction
The rich crystal chemistry of vanadium oxides (VO) and silver vanadium oxides (SVO) is one of the key factors that govern their physical and chemical properties. Due to their labile nature, these complex oxides have been applied with outstanding performances as cathode materials for batteries,2–4 photo-catalysts,1,5–12 antibacterial agents,13–17 photochromic devices18–20 or as active components of sensing devices, just to mention some examples.21–23 In addition, the size-reduction of VO and SVO down to the nano-scale increases the surface area and modifies their surface chemistry, improving even more their performance in the above mentioned applications.24More concretely, from their electronic structure point of view, SVO are p-type semiconductors, and their electric properties has been studied both in bulk and single nanoparticles. The electric response of a single SVO nanoribbon shows ohmic electric transport, Schottky emission, and Pool–Frenkel mechanisms at low, medium and high electric fields, respectively. In view of their electric properties, the β-AgVO3 nanoribbons have been assembled in nanostructured electronic devices such as nano-Schottky barrier diodes and nano-field effect transistors.25 In bulk samples, the electric response changes, because the disorientation of the β-AgVO3 nanoribbons induces an ohmic electrical transport in all the electric field range.26
Ionic and electronic conduction processes in nano vanadium oxides have been also deeply studied. V2O5 undergoes V4+/V5+ oxidation/reduction reactions coupled to the loss or uptake of oxygen atoms.27–29 VO nanotubes are able to adapt the vanadium interlayer space and vanadium oxidation state to the amine and ionic interlayer species, and these parameter have a drastic influence on their electric properties.30–34 These layered structural architectures are repeated in other mixed valence vanadium oxides such as V2O5·H2O xerogels,27 BaV3O7, TMAV3O7, just to mention some of the multiple examples found in the literature. In general, the electron conduction of mixed valence vanadium oxides is governed by the electron hopping mechanism. Therefore, V4+/V5+ distribution within the structure has a great impact on the activation energy and electron migration on these layered materials. Last but not least, oxygen vacancies in V2O5, especially in anisotropic nano-particles, also play a crucial role on the electric and ionic conduction processes within these layered structures.35
Despite the interesting electron/ionic conduction properties reported for VO and SVO,26 impedance spectroscopy (IS) studies are scarcely reported for these types of materials.
Recently, our research team reported a room temperature synthesis methodology to generate inorganic composite hydrogels formed from the self-entanglement of β-AgVO3, and AgIVVO3@VV1.6VIV0.4O4.8 nanoribbons.24 This easy and highly reproducible synthesis strategy allows a high degree of compositional control of the SVO:VO nanoribbon ratio within the hydrogels. Therefore, after a controlled drying process, a homogenous distribution of both types of nanoribbon is present the final xerogel materials; offering a potential tool to tune the electric properties of the composite SVO@VO xerogels through the combination of semiconductor (AgIVVO3) and electric conductor (VV1.6VIV0.4O4.8) anisotropic nanoribbons in the same system.
Herein, the goal of this work is to confirm if the electric properties of composite xerogels can be tuned through the variation of SVO:VO ratio within the final xerogels. To this end temperature and moisture dependence of the electric properties of the bulk β-AgVO3 (SVO) and AgIVVO3@VV1.6VIV0.4O4.8 (SVO@VO) composite xerogels have been studied by impedance spectroscopy. In addition, in order to gain a further insight into the the underlying mechanisms governing the temperature dependent electrical properties, the samples have been characterized by powder X-ray diffraction (XRD), thermal analyses, Electron Paramagnetic Resonance (EPR), Fourier-transform infrared spectroscopy (FTIR) and Ultraviolet-Visible (UV-Vis) spectroscopies before and after different stages of the heating and cooling cycles.
2. Experimental
2.1. Materials and methods
Commercially available reagent grade chemicals AgNO3 (99.99%), NaVO3 (99.9%), HNO3 (70% v/v), ethanol (99.8% v/v), gallium (99%) and indium (99%) metals, were purchased from Sigma-Aldrich.2.2. Synthesis procedure of SVO hydrogels and compositional characterization
Synthesis and compositional characterization of the hydrogel and xerogel samples were performed based on ref. 24. Briefly described, SVO hydrogels are formed at room temperature after mixing silver nitrate and sodium vanadate solutions of specific concentrations. Immediately after the mixture of both solution an orange suspension is generated. Afterwards, the system is maintained undisturbed until the gel is stabilized approximately one hour later. The synthesis of composite hydrogels formed from the entanglement of SVO and VO nanoribbons is similar; but a sodium vanadate solution acidified with diluted HCl solution is used. Compositional ratio of β-AgVO3 and V(V)1.6V(IV)0.4O4.8 nanoribbons within the hydrogel depends on the acidification degree of sodium vanadate solution. Based on this methodology, seven xerogels with increasing amounts of V(V)1.6V(IV)0.4O4.8 nanoribbons were synthesized (Table 1). The final composition of the xerogels was determined combining chemical analysis, and vanadium double titration methodology, as described in our previous work.24
|
|
|---|---|
| Sample code | Chemical formula |
| H0A10 | AgIVVO3 |
| H1A9 | 0.90(AgIVVO3)0.10(VV1.6VIV0.4O4.8) |
| H1A10 | 0.92(AgIVVO3)0.08(VV1.6VIV0.4O4.8) |
| H2A8 | 0.82(AgIVVO3)0.18(VV1.6VIV0.4O4.8) |
| H3A7 | 0.75(AgIVVO3)0.25(VV1.6VIV0.4O4.8) |
| H4A4 | 0.70(AgIVVO3)0.30(VV1.6VIV0.4O4.8) |
| H4A6 | 0.65(AgIVVO3)0.35(VV1.6VIV0.4O4.8) |

In order to identify the xerogels studied in this work, simplified codes will be used to during the following sections. According to the synthesis conditions a HnAm code has been assigned to the xerogels, where n= milliliters of HNO3 added ×10 and m = grams of AgNO3 added ×10. Table 1 summarizes the code of each xerogel and its composition, after the washing, drying and grinding the hydrogels. H0A0 and H4A4 samples, studied by thermal dependence impedance spectroscopy, were characterized also after heating them to 100 °C, 200 °C, 300 °C and 400 °C.
2.3. Structural and thermal characterization of the xerogels
The samples were characterized using powder X-ray diffraction. The patterns were recorded on a Philips X'Pert PRO diffractometer (CuKα radiation) (2θ range = 5–60°, step size = 0.02°, exposure time = 1 s per step). A full pattern matching analyses of the patterns were carried out with the cell parameters and symmetry previously reported for β-AgVO3 (Fig. S1†). Thermal analyses were performed in air and nitrogen atmospheres, up to 150 and 400 °C, with a heating rate of 5 °C min−1 on a NETZSCH STA Simultaneous DSC-TGA. Average formulas of the composite xerogels were obtained by a combinatorial analysis approach, by ICP, EDX and XPS (Ag:V molar ratio at the materials), and vanadium oxidation quantification through double titration and EPR spectroscopy, as described in our previous work.
2.4. Spectroscopic characterization of the xerogels
Infrared spectra were obtained on a Jasco FT/IR-6100 spectrometer with pressed KBr pellets. UV-Vis diffuse reflectance spectra were obtained at room temperature on a Varian Cary 5000 spectrophotometer in the 50000–4000 cm−1 range. A Bruker ESP 300 spectrometer was used to obtain the EPR polycrystalline spectra. The magnetic field was measured using a Bruker BNM 200 gaussmeter and the frequency inside the cavity was determined using a Hewlett-Packard 5352B microwave frequency counter. The measurements were carried out at room temperature. To qualitatively compare the V4+ concentration, the samples were carefully prepared with the same mass and similar packing index of the powders within the quartz sample holders.
2.5. Morphological characterization of the xerogels
Transmission electron microscopy studies were conducted using a Philips Supertwin CM200 transmission microscope operated at 200 kV and equipped with a LaB6 filament and EDAX-DX-4 micro-analysis system. To avoid sample damage, the micrographs were acquired in the TEM low dose mode using a 100 nm C2 condenser aperture and a probe size of 25 nm. All samples were prepared by adding to the grids with a few drops of manually dispersed hydrogels, followed by drying at room temperature and plasma cleaning. It is very important to disperse the hydrogels/xerogels manually and not by ultrasonication, to avoid Ag(0) nanoparticle formation.2.6. Impedance spectroscopy measurements
Electrodes were fabricated on opposite pellet faces from Au and InGa alloy. Samples were ground in an agate mortar and pressed under 2 tons for 5 minutes to obtain 8 mm radius pellets. Impedance spectroscopy (IS) spectra of all the synthesized composites were acquired at RT. Temperature and moisture dependent IS measurements were carried out with selected xerogels (H0A10 and H4A4). For successive heating–cooling cycles, the InGa electrode was removed from the pellets, and a fresh one added to perform the new heating–cooling cycle. Samples with electrodes attached were placed in a conductivity jig and measured using LCR Agilent (10 Hz to 106 Hz), Solartron SI 1260 and Solartron Modulab Xm (10−2 Hz to 106 Hz) analyzers. Impedance data were corrected for overall pellet geometry and are reported in units of specific resistance (Ω cm) and capacitance (F cm−1). The data were not corrected for the geometry of the regions such as grain boundaries and sample porosity. Table 2 summarizes the measurements carried out in each equipment.| Analyzers | Sample | 1st cycle (°C) | 2nd cycle (°C) | 3rd cycle (°C) |
|---|---|---|---|---|
| a LCR Agilent.b Solartron Modulab.c Solartron SI. | ||||
| aLCR | H0A10@InGa | 22–150–22 | 22–154–22 | — |
| H1A10@Au | 22–306 | — | — | |
| H2A8@InGa | 22–138–22 | 22–146–22 | — | |
| H4A4@InGa | 22–145–22 | 22–157–22 | — | |
| H4A4@Au | 22–96–22 | 22–125–22 | 22–123–22 | |
| bS. Mod | H0A10@InGa | 22–137 | — | — |
| H1A10@InGa | 22–144 | — | — | |
| cS. SI | H0A10@InGa | Dry → wet | 22-65 | — |
3. Results and discussion
Prior to the temperature dependent electric behavior description, a full structural, thermal and spectroscopic characterization of samples heated at different temperatures was carried out. The impact of the temperature treatments on the (i) crystal domain sizes and nanoribbon morphology, (ii) loss of water species (weakly or strongly) adsorbed at the surface of the nanoparticles, or (iii) temperature dependent vanadium oxidation/reduction processes is described. These variables are considered in the dielectric properties section of the paper, since they help to understand the underlying process behind the electric responses of the composite xerogels.3.1. Structural and thermal properties
In agreement with our previous results, both for H0A10 and H4A4, the samples show the typical X-ray diffraction pattern of the monoclinic C2/m β-AgVO3 phase. (JCPDS card 29-1154) (Fig. 1).36 Furthermore, for both samples, no diffraction peaks related to impurity phases were observed. Due to the synthesis conditions, a more pronounced peak broadening is observed for H4A4, in comparison with H010, indicative of the existence of smaller crystalline domains within the nanoribbons (Fig. 1). | ||
| Fig. 1 XRD pattern of the H0A10 (a) and H4A4 (b) samples. | ||
It is difficult to acquire any quantitative information from the low crystalline diffraction patterns exhibiting broad peaks derived from nano-sized materials, but no additional phase formation, neither peak intensity loss is observed during heating. On the contrary, a crystallinity increase is detected in the xerogels cured at 300 and 400 °C (Fig. 1). In order to quantify the crystalline domains growth during the temperature curing of the samples, full pattern matching analyses of the patterns obtained at different temperatures were carried out (Fig. S1†). As concluded from the data obtained after the analysis, temperature has a negligible effect on the cell parameters of the compounds, but has a strong impact on the half-width of the diffraction maxima, a parameter that is directly related with the crystalline domain size of the materials. From RT to 300 °C, a slight decrease of the diffraction maxima width is observed, indicative of a slight increase of the crystal domain size of the SVO phase, a tendency that is highly enhanced from 300 °C to 400 °C (Fig. S2†)
Despite X-ray diffraction studies point towards the presence of β-AgVO3 anhydrous phase as the unique component of the composite hydrogels, the crystallization of the gel components as high surface/bulk ratio nanoribbons could favor the precedence of adsorbed or chemisorbed species, such as water, at the surface. In addition, the presence of VO type layered compounds in the composite samples, could contain also interlayer water species. In order to quantify and identify these surface chemistry features of the xerogels, thermogravimetric and IR spectroscopy techniques were applied.
Fig. 2 shows the TGA and DSC curves for the H4A4 (with 30% of VV1.6VIV0.4O4.8) and H0A10 (pristine of AgIVVO3) samples (Table 1). The thermal behaviour of samples with intermediate compositions is intermedaite between this shown in the Fig. 2 for H0A10 and H4A4 samples.
 | ||
| Fig. 2 Thermogravimetric and DSC curves for H0A10 and H4A4 xerogels. | ||
Thermogravimetric measurements show two weight losses associated to the release of: (i) weakly bonded water molecules between room temperature and 100 °C, and (ii) strongly interacting water or hydroxyl groups occurring between 175 °C and 250 °C. Hydrogels containing slightly reduced vanadium oxide nanoribbons (H4A4) exhibit a more pronounced weight loss ascribed to the weakly bonded water molecules (2.5%) in comparison with the pure β-AgVO3 hydrogels (H0A10 (0.75%)). The weight loss associated to the release of strongly bonded species is similar for both samples. It is worth mentioning that no weight gain ascribed to oxidation of V4+ species is observed in the TGA curves (Fig. 1).
Infrared spectra recorded after heating H0A10 and H4A4 to 80 °C, 150 °C and 200 °C evidenced the presence of O–H stretching vibrational mode associated to water molecules or hydroxyl groups. The O–H stretching vibrational modes are still observed even when the xerogels are heated up to 250 °C (Fig. S3†). This evidence further confirms that the second weight loss observed by TGA (175–250 °C) is associated to the loss of strongly interacting surface H2O or OH species.
During the first heating treatment up to 80 °C there is an evident position shift and absorbance reduction of IR band related with the V–O–V vibrational modes (900–800 cm−1) and V–O bonds (500 cm−1). This evidence suggests possible V–H2O and V–OH binding motifs at the surface of β-AgVO3 and vanadium oxide nanoribbons. Once the water molecules were released from the nanoribbons surface, the IR fingerprint became more similar to that of bulk β-AgVO3 and vanadium oxides.
Thus, from the IR, TGA and XRD results, we can assume that the water molecules or hydroxyl groups are generated at the surface of the nanoribbons during the synthesis, and their release does not affect the inner crystal structure or crystal ordering of the silver vanadium oxide phase.
3.2. Temperature induced silver migration
In order to disclose the thermal effect on the nanoribbons morphology, transmission electron microscopy (TEM) images of H0A10 (Fig. 1) and H4A4, previously annealed at different thermal conditions, were acquired. For sake of simplicity, the results for H0A10 sample are described, since the thermal effect on the morphology was similar for both compounds.In a first stage between RT and 300 °C, the migration of Ag+ ions from the AgVO3 nanoribbons inner to the surface occurs, crystallizing as Ag2O or Ag(0) nanoparticles (Fig. 3 H0A10-RT, H0A10-160, H0A10-200 micrographs). From RT to 300 °C SVO nanoribbon are poorly crystalline, since any attempt to obtain the electron diffraction pattern was unsuccessful, giving rise to the degradation of the nanoribbons as silver particles after their exposure to the beam. The electron diffraction patterns of thermally generated silver nanoparticles confirm their Ag(0) nature, but we cannot asses whether a Ag2O to Ag(0) transformation occurs during the electron beam exposure. Even more interesting is that Ag2O/Ag(0) nanoparticles are re-absorbed by the SVO nanoribbons when heated up to 400 °C. A side effect observed at high temperatures is that adjacent nanoribbons are fused forming platelets at 400 °C (Fig. 3 H0A10-300 and H0A10-400 micrographs). Crystallinity gain during temperature curing is also confirmed, since β-AgVO3 nanoplatelets formed at 400 °C present a well-defined diffraction pattern coincident with the theoretical one calculated for the [102] zone axis. The result obtained from TEM images agrees with the XRD data, since the main peak width sharpening is observed between 300 °C to 400 °C, the same temperature range as for the nanoribbon fusing forming nanoplatelet.
 | ||
| Fig. 3 TEM micrographs of H0A10 heated at 160, 300 and 400 °C. H0A10-400 inset: electron microscopy diffraction pattern of single β-AgVO3 nanoribbon. | ||
Temperature driven silver migration, and nanoplatelet formation can be described based on the reaction (1) and the process summarized in Scheme 1.
| (a) β-AgVO3 → (b) and (c) ½ Ag2O + ½ V2O5 → Ag(0) + ¼ O2 + ½ V2O5 → (c) and (d) β-AgVO3 |
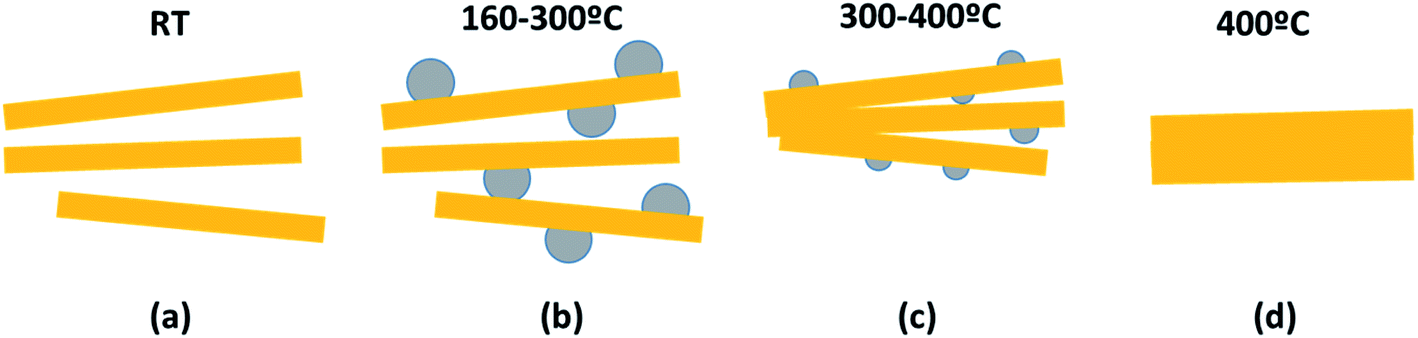 | ||
| Scheme 1 Thermally-induced silver migration in β-AgVO3 nanoribbons. | ||
3.3. Thermally-induced vanadium oxidation/reduction
The complexity of the SVO and composite SVO@VO systems goes beyond the temperature induced chemical and morphological changes, since vanadium is also likely to exhibit oxidation/reduction processes. Therefore, vanadium speciation within the compounds was studied by EPR and UV-Vis spectroscopies.Fig. 4 shows the EPR spectra for the as-synthesized H0A10 (Fig. 4(a)) and H4A4 (Fig. 3(b)) xerogels and after heating them at 120, 200, 300 and 400 °C. V4+ species formation can be followed by EPR spectroscopy, because the intensity of the axial signals centered at 3400 G in the EPR spectra can be correlated semi-quantitatively with the V4+ content.24
 | ||
| Fig. 4 EPR spectra for H0A10 (a) and H4A4 (b) xerogels treated at different temperatures. | ||
As expected, the initial EPR signal for H0A10 is less intense than this for H4A4, due to the higher content of V4+ arising from slightly reduced VO in H4O4 xerogels. Even without the addition of nitric acid, during the gelification of H0A10 xerogels some proportion of V4+ is detected.
The thermal dependence of the EPR signal clearly shows that a V5+ to V4+ reduction occurs when H0A10 and H4A4 are heated at 120 °C and 200 °C, respectively. Above these temperatures, an EPR intensity loss is observed, indicative of an oxidation of the V4+ species to V5+. The oxidation of V4+ species occurs within the same temperature range that the covalently attached water molecules are released from the nanoribbons' surface and the silver segregation is observed by TEM.
3.4. Thermal dependence of UV-Vis absorbance
The generation of mixed valence vanadium species within the H0A10 and H4A4 hydrogels and xerogels can be followed by UV-Vis spectroscopy, since it is well known that mixed valence vanadium oxides have darker colors, due to the charge transfer between different vanadium species.37,38 Thus, an absorbance increase due to the generation of V4+ species within the xerogels is expected.A red shift of the absorption edge and an increase of the absorption in all the spectral range are induced by the increasing incorporation of slightly reduced vanadium oxide nanoribbons into the β-AgVO3 hydrogels and xerogels (Fig. S4 and S5†).
Vanadium oxidation and reduction due to thermal annealing affects the optical properties of SVO and composite SVO/VO xerogels. For both samples, the vanadium reduction stage between RT and 120–200 °C induces an increase in the background absorbance, whilst the opposite effect occurs during oxidation between 200 and 400 °C. In addition, for β-AgVO3 a subtle slight red shift of the band gap is observed during vanadium reduction and a blue shift during oxidation. This trend is not so clear for the composite samples, and only a red shift of the absorbance edge is appreciated after the thermal curing. The close correlation between the optical properties and electronic structure of the materials suggest that the thermal induced variation of the vanadium average oxidation state will affect the electronic structure, and hence the electric properties of the studied xerogels.
As main conclusions of the above results, temperature induce a couple dehydration, reduction/oxidation, optical and morphological changes on H0A10 and H4A4 xerogels, that need to taken into consideration when describing the electric properties of these complex materials in the next section.
3.5. Electrical properties
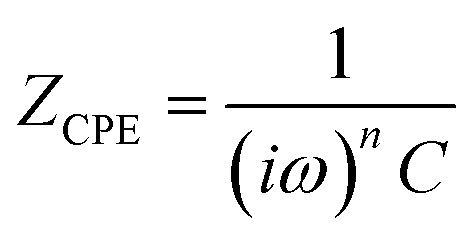 | (1) |
 | ||
| Fig. 5 (a) Impedance complex plots for β-AgVO3 and composite xerogels. In order to visualize more easily the main characteristics of the complex impedance spectra we have opted to use different scales at the x Z′ and Z′′ axis. Take into consideration that the impedance arcs are highly depressed, as described in the main text. (b) Resistivity values vs. slightly reduced vanadium oxide molar content of the xerogels. Resistivity values were taken from Table 3. | ||
Impedance complex planes for all the samples (Fig. 5(a)) exhibit a highly depressed arc at high frequencies, followed by an inclined spike with some curvature at low frequencies. Capacitance values of high frequency region varies depending on the sample, being within 10−10 F cm−1 order for silver vanadim oxide samples, and from 10−10 to 10−9 F cm−1 for composite samples (∼10−11 F cm−1). These values are between bulk to grain boundary responses; and they have been ascribed to the electric response of the bulk materials; but futher IS measurements at low temperature would be needed to fully confirm this hyphothesis; since they could be associated to electronic or ionic transport occuring at the surface of the nanoribbons too. With decreasing frequency, C′ rapidly increase to reach ∼10−5 F cm−1 at 0.01 Hz; a value within the typical ranges found for sample/electrode interphase phenomena.42,43 Table 3 shows the parameters obtained by the best fittings of the impedance spectra with the equivalent circuit for β-AgVO3 and composite xerogels, showing a good agreement between the experimental data and fittings (Fig. 5(a)).
| Parameters | Samples | ||||||
|---|---|---|---|---|---|---|---|
| H0A10 | H1A9 | H1A10 | H2A8 | H3A7 | H4A4 | H4A6 | |
| R1/kΩ | 19.2 | 281.9 | 2.0 | 4.0 | 18.2 | 5.0 | 5.1 |
| R2/kΩ | 83.4 | 247.6 | 10.0 | 13.3 | 24.5 | 8.1 | 8.2 |
| C1/F | 4.7 × 10−10 | 1.6 × 10−10 | 3.3 × 10−10 | 2.0 × 10−9 | 2.0 × 10−9 | 6.2 × 10−9 | 5.0 × 10−9 |
| n1 | 0.71 | 0.79 | 0.79 | 0.70 | 0.70 | 0.66 | 0.67 |
| C2/F | 6.1 × 10−7 | 2.0 × 10−8 | 8.6 × 10−6 | 4.4 × 10−6 | 8.6 × 10−7 | 8.9 × 10−6 | 1.2 × 10−5 |
| n2 | 0.45 | 0.58 | 0.33 | 0.38 | 0.44 | 0.35 | 0.32 |
Table 3 shows an initial high frequency capacitive response of ∼10−10 to 10−9 F dependent of the amount of VO nanoribbons. The capacitive element related to the response of the interfaces shows values in the range 10−8–10−5 F, associated to a double layer at the electrode-material interface where the electrons are trapped in grain boundaries for all samples.44 Further, the resistance value decreases for the composites except for H1A9 in comparison to pristine β-AgVO3 xerogels due to the amount of the transported electrons at the system through the addition of different amount of VO nanoribbons. This fact is also demonstrated by the decrease of C2 in composite xerogels.
The n1 values are similar for all samples, being tentatively ascribed both to the resistive and capacitive behavior of the bulk region or nanoparticles surface.
The n2 values indicate that the dependence of the capacitance with frequency is different for the regions at the grain boundary, with a resistor-like behavior. These values obtained for pristine β-AgVO3 xerogels are similar to the ones reported in the literature for this material.45
Three are the consequences of incorporating slightly reduced vanadium oxide nanoribbons within β-AgVO3 xerogels.
• A further depression of the semi-arc.
• A global resistivity decreases.
• Length and inclination reduction of the Warburg spike.
The electrical inhomogeneity of the samples is the most plausible origin of the semi-arc depression in the impedance complex planes, being directly related with the existence of two nanoribbon types with different electric properties within the composite xerogels. Nevertheless the pristine β-AgVO3 xerogels (H0A10) also exhibit a depressed complex impedance semi-arc, revealing some electrical inhomogeneity inherent to AgVO3 nanoribbons.
For H0A10, formed from AgVO3 nanoribbons, the ion blocking at the sample electrode interface, arising from proton mobility within the xerogels is the most plausible origin for the Warburg spike, interpreted as a double capacitance from the electronic circuit point of view. In composite xerogels, the competition between the proton conduction at the AgVO3 nanoribbons, and the electric conduction ascribed to the VO nanoribbons, induces the progressive horizontalization of the Warburg spike as the content of VO component increase. In these SVO@VO composite electronic conductors, the resistance obtained from the semicircle and spike intercept with the Z′ axis is associated to the energy difference between the electronic conduction levels of composite xerogels and the electrodes.
The content of slightly reduced vanadium oxide nanoribbons of the xerogels, versus the resistivity plot (Fig. 5(b)), shows a jump in the conductivity of the samples containing more than 0.1 moles of slightly reduced vanadium oxide component. This conductivity jump from proton conductor to electric conductor xerogels can be explained based on the percolation threshold. When a critical ratio of slightly reduced V(V)1.6V(IV)0.4O4.8 nanoribbons are included in the composite xerogels, effective electron conduction pathways are stablished between them, and the migration of electron through the composite xerogels becomes possible (Scheme 2).
 | ||
| Scheme 2 Percolation threshold in composite xerogels. (a) Proton conductor β-AgVO3 xerogels, (b) mixed proton and electric conductor composite xerogels, and (c) electron conductor composite xerogels. | ||
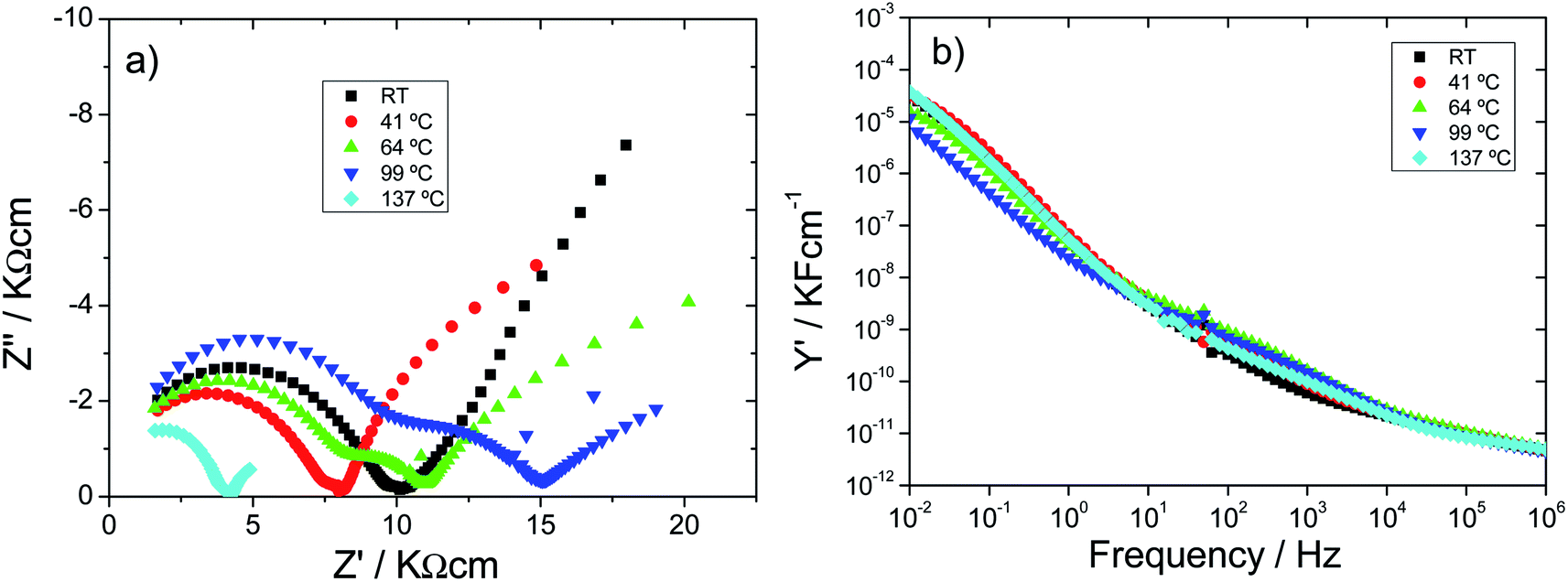 | ||
| Fig. 6 (a) Impedance complex (b) and Y′ spectroscopic plots for H0A10 xerogel during the 1st heating cycle. The readers need to take into consideration that the scale of real and imaginary components of Z is different with the end to highlight the changes occuring during the heating process. | ||
As it has been abovementioned, the inclined spike at low frequencies is attributed to the proton conduction towards and away from sample–electrode interface. Three stages can be distinguished during the heating process:
• At room temperature, protonic conduction of H0A10 xerogels was confirmed by moisture dependent IS measurements (Fig. S6(a)†) since a continuous increase on the conductivity, from 1.46 × 10−5 S cm−1 (100 Hz) to 2.69 10−5 S cm−1 after the exposure of H0A10 to a wet atmosphere, is observed (Fig. S6(b)†). After reaching the water uptake equilibrium, the temperature dependence of the electric properties follows the same tendency described before for H0A10 in the Fig. 6.
• A continuous resistivity increases between RT and 100 °C along with the appearance of a second depressed arc in the impedance complex plane associated to an inhomogeneous dehydration of the pellet during the water release process (Fig. 6(a)).
• A conductivity enhancement between 100 °C and 137 °C, once the sample is partially dehydrated (as derived from TGA results). After the loss of weakly bonded water molecules, the second arc in the impedance complex spectra disappears, and the Warburg spike at low frequencies gains again inclination, indicating that a new ion-blocking phenomenon is occurring at the electrode sample interphase at high temperature. Therefore, the high temperature impedance complex spectra suggest that H0A10 samples are high temperature ion (more probably silver ion) conductors.
In order to further understand the thermal dependence of the electric behaviour of H010 and H1A10 samples, total sample resistances were obtained from intercepts of either the arc or the Warburg spike on the Z′ axis and are shown as conductivity Arrhenius plots as a function of the reciprocal temperature in Fig. 7. Total resistance values for the samples were obtained from different impedance spectroscopy experiments specifically designed to unravel different points:
 | ||
| Fig. 7 (a) Arrhenius plot for H0A10 and H1A10 xerogels during the 1st heating treatment and (b) Arrhenius plot for H0A10 xerogel in two heating and cooling cycles. | ||
• A more complete set of data were acquired to define more precisely the conductivity dependence on the sample dehydration (H1A10@InGa) (Fig. 7(a)).
• Experiments with samples prepared from gold electrodes (H1A10@Au – Fig. 7) were developed to acquire a more accurate data at higher temperatures (up to 300 °C) than with InGa electrodes (150 °C) (Fig. 7(a)).
• In a third a heating–cooling cycle was performed to confirm the reversibility of the electric properties (H1A10@InGa) (Fig. 7(b)).
Both for H0A10 and H1A10, the above-described stages (proton conduction enhancement, dehydration and ionic conduction) are clearly distinguished in conductivity Arrhenius plots (Fig. 7(a)). There are slight differences in the conductivity values obtained for H1A10 samples prepared with InGa and Au electrodes, suggesting that both the dehydration of the sample and the proton conduction process is electrode dependent.
After the dehydration (above 100 °C), activation energies Ea close to 0.3 eV are obtained for the ionic conduction process occurring at the high temperature regime of the experiments (Table 3). This value was confirmed more accurately in the high temperature experiment developed with gold electrode until 300 °C. It is worthy to mention that above 300 °C the H0A10 sample becomes insulator, which confirms that after the loss of strongly bonded hydroxyl and water molecules anchored to the surface of the nanoribbons the high temperature ion migration is blocked.
In order to gain further insights about the possible nature of the high temperature ion conduction mechanisms, a comparison between previously reported activation energy values for proton conductors and silver conductors with the studied compounds have been stablished. It is well known that Ea strongly depends on the ion transport mechanisms. For pure protonic conductors, Ea values from 0.8 to 0.4 eV are usually ascribed to a Grotthuss mechanism46 in which the proton donors and acceptors are anchored to a surface, and hence, the proton exchange occurs between adjacent proton carriers. Below 0.4 eV Ea, a vehicle transport mechanism is assumed to govern the proton mobility, which involves the existence of mobile proton carriers. Considering that at high temperatures the potential proton carrier species in the studied xerogels are water or hydroxyl groups anchored to the nanoribbons surface, a Grotthuss type proton mobility would be expected, but the obtained experimental activation energies (Table 3) are below the expected ones for this type of proton transport mechanisms.
If the specific case of silver ion conduction scenario is considered, the activation energy would be determined by the energy needed for an ion hopping to an empty position within the nanoribbons. The silver ionic conduction Ea found in silver superconductors depends strongly on the considered material, ranging from 0.1 to 0.5 eV in different AgXM2O5 ((X = Cl−,Br−, I−; M = P, V, Mo) amorphous glasses, silver defective perovskites and silver phosphates, just to mention some examples. These values are closer to the 0.3 eV activation energy obtained for the studied xerogels. Therefore, obtained experimental Ea match better with a silver ionic conduction process, but without forgetting that the water or hydroxyl groups attached to the nanoribbons' surface also play a crucial role as charge carriers, since their release blocks the ionic conductivity at temperatures higher than 300 °C (Table 4).
| Sample | Ea (eV) |
|---|---|
| H1A10@InGa cycle | 0.279(14) |
| H1A10@Au | 0.315(13) |
| H1A10@InGa | 0.281(24) |
| H0A10@InGa | 0.272(—) |
| H4A4@InGa 1st cycle | 0.1225(37) |
| H4A4@InGa 2nd cycle | 0.1081(13) |
| H4A4@Au 1st cycle | 0.074(24) |
| H4A4@Au 3rd cycle | 0.0632(30) |
The reversibility of the electrical behavior was studied by Impedance measurements upon heating and cooling. The Arrhenius plots Fig. 7(b) confirm some degree irreversibility on the conductivity when samples are heated above 100 °C. Conductivity values for first and second heating/cooling cycles show a meaningful reduction ascribed to the non-complete rehydration of the sample, which reduces the number of proton carriers, and hence the conductivity.
Temperature dependence on electronic conductor composite xerogels was also studied. Arrhenius plots as a function of the reciprocal temperature for β-(AgIVVO3)0.7@(VV1.6VIV0.4O4.8)0.3 composite xerogel's pellets prepared with InGa and gold electrodes are shown in Fig. 8(a). For sake of comparison samples were studied with InGa and gold electrodes.
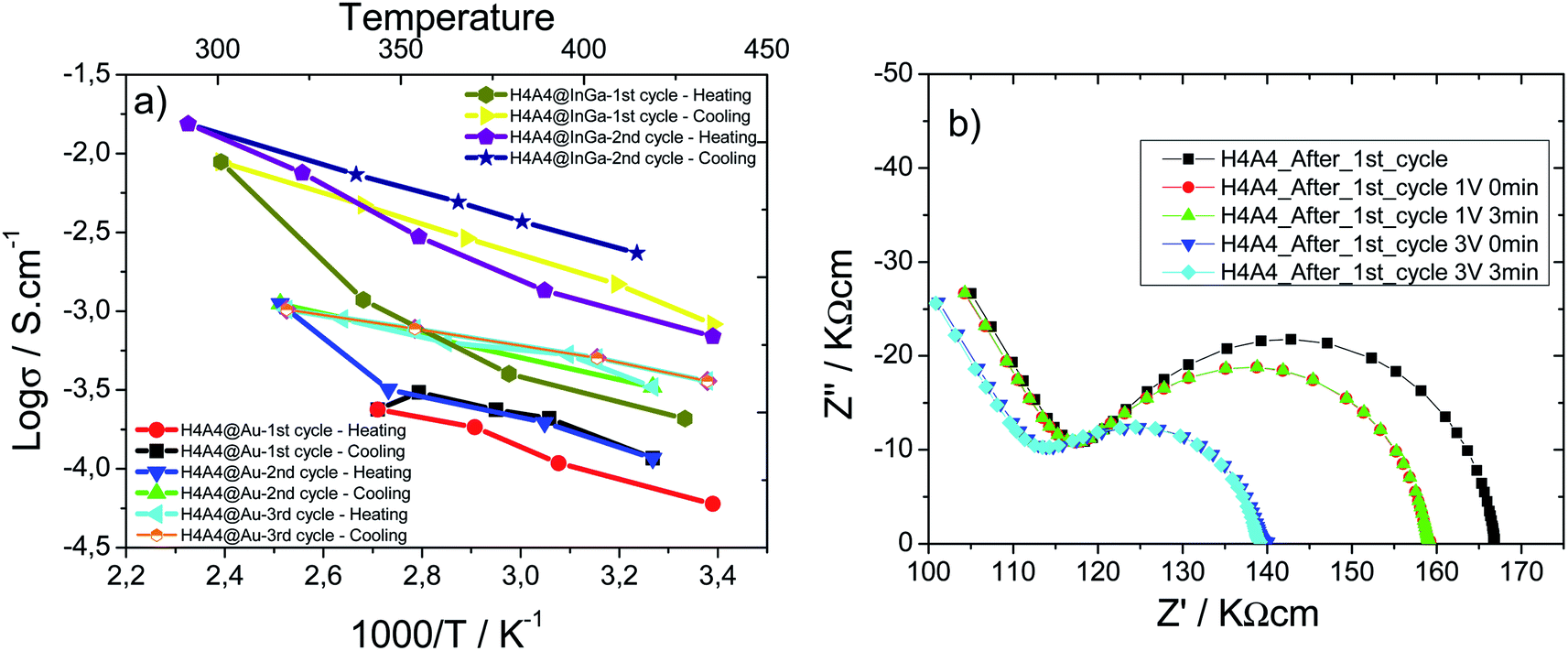 | ||
| Fig. 8 (a) Arrhenius plots for H4A4 samples prepared with InGa and Au electrodes. (b) Room temperature impedance complex plots applying 1 V and 3 V bias for H4A4 after the first heating/cooling cycle (sampled prepared with InGa electrode). | ||
A reduction of the resistivity of the sample and a shortening of the low frequency spike is induced by an increase of temperature, independent of the electrode type (Au or InGa) used in the measurements. Impedance complex data were recorded for successive heating/cooling cycles; which show some irreversibility during cooling. In fact, the thermal variation of the conductivity during heating does not follow a completely linear tendency, being higher the deviation from the linearity at higher temperatures. Once the maximum temperature is reached, during cooling the thermal response of the log(σ) follows a linear tendency. A qualitative fit of the data obtained during cooling indicates that the activation energy is similar for Au and InGa electrodes, and for the first, second and third cycles (Table 3). The generation of new electron carriers during the heating process could explain the positive deviation from the linear tendency, and more probably is closely related with the V5+ to V4+ reduction between RT and 200 °C. If fact, the higher the temperature is reached during the heating cycle, the more is the deviation from the linear tendency, and therefore, the more the V4+ charge carriers that are generated. This thermal activation of charge carriers has a critic temperature, since H4A4 samples heated up at 400 °C are insulators, because the complete oxidation of V4+ species suppresses the electron carrier species, disrupting the electric conduction.
After the first heating/cooling cycle, a Schottky barrier is stablished between the sample and the InGa electrodes. The vias increase induces an immediate resistivity decrease related with the reduction of the Schottky barrier height due to the approaching of vanadium oxide nanoribbons and InGa electrode Fermi levels (Fig. 8(b)).47
4. Conclusions
Control of the silver vanadium oxide (SVO) and vanadium oxide (VO) nanoribbons ratio content of the composite SVO@VO xerogels can be used as an effective tool to modify their electric properties from protonic/ionic conductor systems to electron conductor composite materials.Structural, textural and compositional complexity of these nanoribbon based materials induce complex thermal dependence changes at morphological, surface adsorbed species and vanadium oxidation average state levels. Considered jointly, these thermal induced changes can help us to understand the underlying processes giving rise to the thermal dependent electric response reported in this study.
SVO nanoribbons behave as a proton conductor at room temperature, being the proton conductivity dependent on the moisture degree. After the removal of weakly adsorbed water species (>100 °C), SVO xerogels begin to behave as a high ionic conductor (100–300 °C). Considering the activation energies of the process, this high temperature ionic process is ascribed to silver ion mobility. Nevertheless, the role of strongly bonded surface water species is undoubted; since after their loss, the ionic conduction is completely blocked.
For composite SVO@VO xerogels, their electronic conductivity is ascribed to the mixed vanadium V5+/V4+ oxidation, being the electronic conductivity enhanced during the thermal V5+ to V4+ reduction between RT and 250 °C; and completely blocked at higher temperatures, when the V4+ species are completely oxidized to V5+.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been financially supported by the “Ministerio de Economía y Competitividad” MAT2016-76739-R (AEI/FEDER, EU), and the “Gobierno Vasco” Basque University Research System Group, IT-630-13 which we gratefully acknowledge. Joseba Orive wishes to thank Millenium Nucleus Multimat for funding. The authors thank the technicians of SGIker (UPV/EHU). The authors thank the FCT (Fundação para a Ciência e Tecnologia) for financial support under the framework of Strategic Funding grants UID/FIS/04650/2018 and grant SFRH/BPD/112547/2015 (C.M.C.). Also, to the project PTDC/FIS-MAC/28157/2017 funded by national funds through FCT and by the ERDF through the COMPETE2020 - Programa Operacional Competitividade e Internacionalização (POCI). Financial support from the Basque Government under the ELKARTEK, HAZITEK and PIBA (PIBA-2018-06) programs is also acknowledged. Authors thank to the Department of Materials Science and Engineering of the University of Sheffield for supporting the work with the impedance spectroscopy facilities; and especially to Prof. A. R. West for his scientific guidance during the experimental data acquisition and initial results interpretation.References
- C. C. Chen, J. Shaya, H. J. Fan, Y. K. Chang, H. T. Chi and C. S. Lu, Sep. Purif. Technol., 2018, 206, 226–238 CrossRef CAS.
- H. Kim, D. Y. Kim, Y. Kim, S. S. Lee and K. Park, ACS Appl. Mater. Interfaces, 2015, 7, 1477–1485 CrossRef CAS PubMed.
- R.-H. Kim, J.-S. Kim, H.-J. Kim, W.-S. Chang, D.-W. Han, S.-S. Lee and S.-G. Doo, J. Mater. Chem. A, 2014, 2, 20636–20641 RSC.
- Y. Wu, G. Gao and G. Wu, J. Mater. Chem. A, 2015, 3, 1828–1832 RSC.
- Y. Sang, L. Kuai, C. Chen, Z. Fang and B. Geng, ACS Appl. Mater. Interfaces, 2014, 6, 5061–5068 CrossRef CAS PubMed.
- R. C. De Oliveira, M. Assis, M. M. Teixeira, M. D. P. Da Silva, M. S. Li, J. Andres, L. Gracia and E. Longo, J. Phys. Chem. C, 2016, 120, 12254–12264 CrossRef CAS.
- R. Konta, H. Kato, H. Kobayashi and A. Kudo, Phys. Chem. Chem. Phys., 2003, 5, 3061–3065 RSC.
- W. Zhao, Y. Guo, Y. Faiz, W. T. Yuan, C. Sun, S. M. Wang, Y. H. Deng, Y. Zhuang, Y. Li, X. M. Wang, H. He and S. G. Yang, Appl. Catal., B, 2015, 163, 288–297 CrossRef CAS.
- R. Wang and L. Cao, J. Alloys Compd., 2017, 722, 445–451 CrossRef CAS.
- M.-Y. Ye, Z.-H. Zhao, Z.-F. Hu, L.-Q. Liu, H.-M. Ji and Z.-R. Shen, Angew. Chem., Int. Ed., 2017, 56, 8407–8411 CrossRef CAS PubMed.
- X. Zhang, J. Zhang, J. Yu, Y. Zhang, Z. Cui, Y. Sun and B. Hou, Appl. Catal., B, 2018, 220, 57–66 CrossRef CAS.
- Z.-D. Lei, J.-J. Wang, L. Wang, X.-Y. Yang, G. Xu and L. Tang, J. Hazard. Mater., 2016, 312, 298–306 CrossRef CAS PubMed.
- R. D. Holtz, A. G. Souza Filho, M. Brocchi, D. Martins, N. Durán and O. L. Alves, Nanotechnology, 2010, 21, 185102 CrossRef CAS PubMed.
- M. C. Artal, R. D. Holtz, F. Kummrow, O. L. Alves and G. de A. Umbuzeiro, Environ. Toxicol. Chem., 2013, 32, 908–912 CrossRef CAS PubMed.
- A. P. de Melo Monteiro, R. Dias Holtz, L. Carneiro Fonseca, C. H. Zanini Martins, M. de Sousa, L. A. V. de Luna, D. L. de Sousa Maia and O. L. Alves, Chem. Rec., 2018, 18, 1–14 CrossRef PubMed.
- J. Ren, W. Wang, M. Shang, S. Sun, L. Zhang and J. Chang, J. Hazard. Mater., 2010, 183, 950–953 CrossRef CAS PubMed.
- R. C. De Oliveira, C. C. De Foggi, M. M. Teixeira, M. D. P. Da Silva, M. Assis, E. M. Francisco, B. N. A. D. S. Pimentel, P. F. D. S. Pereira, C. E. Vergani, A. L. Machado, J. Andres, L. Gracia and E. Longo, ACS Appl. Mater. Interfaces, 2017, 9, 11472–11481 CrossRef CAS PubMed.
- Y. Wang, L. Pan, Y. Li and A. I. Gavrilyuk, Appl. Surf. Sci., 2014, 314, 384–391 CrossRef CAS.
- R. J. Colton, A. M. Guzman and J. W. Rabalais, Acc. Chem. Res., 1978, 11, 170–176 CrossRef CAS.
- S. Nishio and M. Kakihana, Chem. Mater., 2002, 14, 3730–3733 CrossRef CAS.
- G. S. Zakharova, Y. Liu, A. N. Enyashin, X. Yang, J. Zhou, W. Jin and W. Chen, Sens. Actuators, B, 2018, 256, 1021–1029 CrossRef CAS.
- S. Azizi, M. Salah, H. Nefzi, C. Khaldi, F. Sediri, E. Dhahri and J. Lamloumi, J. Alloys Compd., 2015, 648, 244–252 CrossRef CAS.
- J. Huotari, J. Lappalainen, J. Eriksson, R. Bjorklund, E. Heinonen, I. Miinalainen, J. Puustinen and A. Lloyd Spetz, J. Alloys Compd., 2016, 675, 433–440 CrossRef CAS.
- R. Fernández de Luis, A. Martínez-Amesti, E. S. Larrea, L. Lezama, A. T. Aguayo and M. I. Arriortua, J. Mater. Chem. A, 2015, 3, 19996–20012 RSC.
- M. Feng, L. B. Luo, B. Nie and S. H. Yu, Adv. Funct. Mater., 2013, 23, 5116–5122 CrossRef CAS.
- S. J. Bao, Q. L. Bao, C. M. Li, T. P. Chen, C. Q. Sun, Z. L. Dong, Y. Gan and J. Zhang, Small, 2007, 1174–1177 CrossRef CAS PubMed.
- K. Bali, L. Kiss, T. Szörényi, M. Török and I. Hevesi, J. Phys., 1987, 48, 431–434 CrossRef CAS.
- Z. R. Xiao and G. Y. Guo, J. Chem. Phys., 2009, 130, 214704 CrossRef CAS PubMed.
- M. Harth, R. Mitdank, D. Habel, O. Görke, M. Tovar, H. Winter and H. Schubert, Int. J. Mater. Res., 2013, 104, 657–665 CrossRef CAS.
- V. V Porsev, A. V Bandura and R. A. Evarestov, ChemPhysChem, 2015, 16, 3007–3014 CrossRef PubMed.
- X. Liu, C. Täschner, A. Leonhardt, M. H. Rümmeli, T. Pichler, T. Gemming, B. Büchner and M. Knupfer, Phys. Rev. B, 2005, 72, 115407 CrossRef.
- K. Won Lee, H. Kweon and C. E. Lee, J. Appl. Phys., 2009, 106, 044313 CrossRef.
- B. Sipos, M. Duchamp, A. Magrez, L. Forró, N. Barišić, A. Kis, J. W. Seo, F. Bieri, F. Krumeich, R. Nesper and G. R. Patzke, J. Appl. Phys., 2009, 105, 074317 CrossRef.
- H. Nefzi and F. Sediri, J. Solid State Chem., 2013, 201, 237–243 CrossRef CAS.
- (a) J. M. Li and Y.-B. Hu, Appl. Phys. Lett., 2019, 114, 203101 CrossRef; (b) V. Katari, S. J. Patwe, S. N. Achary and A. K. Tyagi, J. Am. Ceram. Soc., 2013, 96, 166–173 CrossRef CAS.
- J. M. Song, Y. Z. Lin, H. B. Yao, F. J. Fan, X. G. Li and S. S. H. Yu, ACS Nano, 2009, 3, 653–660 CrossRef CAS PubMed.
- B. Jiang, X. Peng, Y. Qu, H. Wang, C. Tian, Q. Pan, M. Li, W. Zhou and H. Fu, ChemCatChem, 2014, 6, 2553–2559 CrossRef CAS.
- M. F. Goes, M. A. Sinhoreti, S. Consani and M. A. Silva, Braz. Dent. J., 1998, 9, 3–10 CAS.
- J. T. S. Irvine, D. C. Sinclair and A. R. West, Adv. Mater., 1990, 2, 132–138 CrossRef CAS.
- A. Kaushal, S. M. Olhero, B. Singh, D. P. Fagg, I. Bdikin and J. M. F. Ferreira, Ceram. Int., 2014, 40, 10593–10600 CrossRef CAS.
- A. Reyes-Montero, P. Ramos-Alvarez, A. M. González, R. López-Juárez and M. E. Villafuerte-Castrejón, Appl. Sci., 2017, 7, 214 CrossRef.
- I. A. Alagdal and A. R. West, J. Mater. Chem. C, 2016, 4, 4770–4777 RSC.
- M. Jovaní, H. Beltrán-Mir, E. Cordoncillo and A. R. West, Inorg. Chem., 2017, 56, 7081–7088 CrossRef PubMed.
- F. Greuter and G. Blatter, Semicond. Sci. Technol., 1990, 5, 111–137 CrossRef CAS.
- J. Zheng, L. Calvillo, C. Valero-Vidal, C. Marega, P. Sekar and S. Shuang, ACS Omega, 2017, 2, 2792–2802 CrossRef CAS PubMed.
- T. Miyake and M. Rolandi, J. Phys.: Condens. Matter, 2015, 28, 023001 CrossRef PubMed.
- L. Gil Escrig, M. Prades, H. Beltrán, E. Cordoncillo, N. Masõ and A. R. West, J. Am. Ceram. Soc., 2014, 97, 2815–2824 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04227j |
| This journal is © The Royal Society of Chemistry 2019 |