
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile and rapid synthesis of crystalline quadruply bonded Cr(II) acetate coordinated with axial ligands†
Intek Song a,
Jinyoung Koob and
Seok Min Yoon*c
a,
Jinyoung Koob and
Seok Min Yoon*c
aDepartment of Applied Chemistry, Andong National University, 1375 Gyeongdong-ro, Andong, Gyeongbuk 36729, Republic of Korea
bDepartment of Chemistry, Pohang University of Science and Technology, 77 Cheongam-ro, Pohang, Gyeongbuk 37673, Republic of Korea
cDepartment of Bio-Nano Chemistry, Wonkwang University, 460 Iksandae-ro, Iksan, Jeonbuk 54538, Republic of Korea. E-mail: smyoon1@wku.ac.kr
First published on 6th August 2019
Abstract
A quadruple bond formed between d-block or f-block atoms is an interesting research topic due to its unique nature including a supershort bonding distance and narrow energy gap between δ and δ*. Among various multiply bonded complexes, quadruply bonded Cr(II) acetates are considered useful to control the δ–δ* energy gap by the Lewis basicity of additional ligands. However, the synthesis and preparation of the high-quality, large-sized crystals of Cr(II) acetates coordinated with axial ligands (Cr2(OAc)4L2) have been difficult due to their vulnerability to O2, a representative oxidizing agent under aerobic conditions. In this study, we report a facile synthesis of sub-millimeter-scale crystals of Cr2(OAc)4L2 by simple dissolution of Cr2(OAc)4 in ligand solvents L. To obtain stably ligated Cr2(OAc)4L2, anhydrous Cr(II) acetates (Cr2(OAc)4) were dissolved in the ligand solvents L, which was degassed of dissolved O2. Also, sub-millimeter-scale single crystals of Cr2(OAc)4L2 were produced rapidly for less than an hour by the drop-drying process. The single-crystalline phase of the synthesized Cr(II) complexes was measured by X-ray diffraction techniques, confirming the dependency of Lewis basicity of the additional axial ligands on the Cr–Cr quadruple bond distance. Further, the Raman peaks of the quadruple bonds in Cr2(OAc)4L2 were observed to be red-shifted with the increased basicity of the axial ligands.
Introduction
There has been quite some progress in the research field on multiple bonds between transition metals since the Cotton group synthesized the quadruple bonded Cr(II) acetate (Cr2(OAc)4) in the early 1970s.1,2 More recently the Power group reported Cr2 quintuple bonds in addition to the quadruple bond,3 stimulating the in-depth research of the metal-to-metal (M–M) bonds.4–6 The multiple M–M bond, particularly the quadruple bond exists because the metals' d- or f-orbitals give rise to a delta bond (δ) as well as a sigma (σ) and two pi (π) bonds (Fig. 1a and b).1,2 Since the formation of quadruple bonds requires d-orbitals, they provide ultrashort (∼2 Å) metal-to-metal bonds1,2 and a narrow energy gap.7 Also, various ligands can coordinate to the axial position of a M–M multiple bonded complex. Thus, the axial coordination to the M–M bond has been an important research topic to control the M–M bond length and their reactivity depending on the types of ligand.3 Therefore, quadruple bonds have been used to construct a one-dimensional chain of transition metal atoms, to enable selective and reversible adsorption of O2, and a promising efficient catalyst.8,9 Besides, Cotton et al.7 reported that their magnetism can be changed at room temperature by the δ-to-δ* transition between the diamagnetic singlet state (σ2π2δ2) and the paramagnetic triplet state (σ2π2δδ*).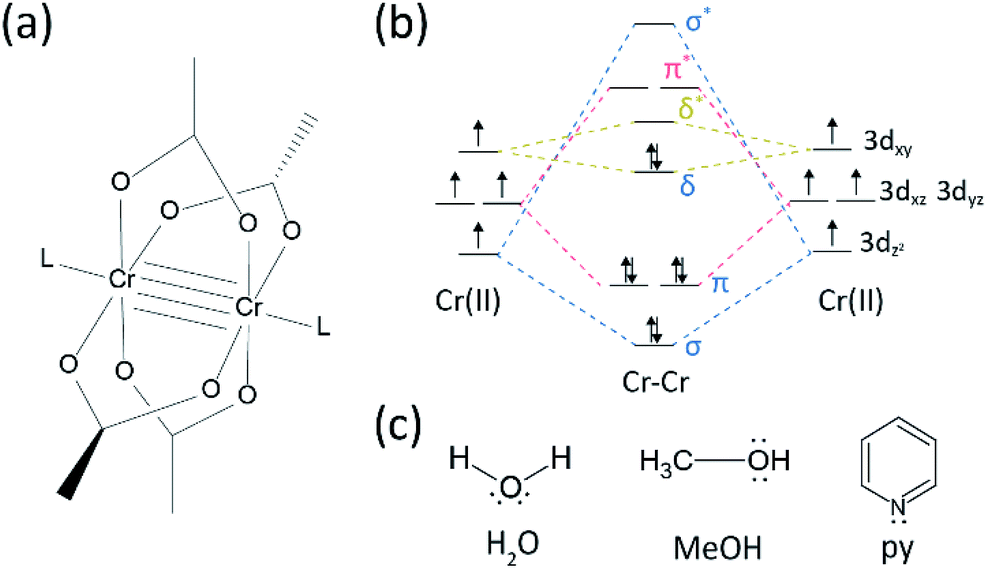 | ||
| Fig. 1 (a) Molecular structure of ligated Cr–Cr acetates. The L stands for the axial ligand. The anhydrous Cr(II) acetate has the identical structure except that it lacks L. (b) Molecular orbital diagram of Cr–Cr quadruple bond. Note that non-bonding d orbitals are omitted for simplicity. (c) Molecular structure of the axial ligands used in this study. All of them have lone pair electrons. | ||
There are various organometallic complexes having delta bonds, such as Re2Cl82−,10 Mo2(mhp)4,11 Cr2(OAc)4,12 etc.1–6 Among them, significant examples are paddle-wheel-structured anhydrous Cr(II) acetate (Cr2(OAc)4), which lacks any axial ligands, and their derivatives that have axial ligands (Cr2(OAc)4L2, L = ligand) (Fig. 1a).1,13 Since Cr(II) has d4 electronic configuration, two Cr(II) ions in the acetates completely fill the four bonding orbitals of a quadruple bond (bond order = 4) (Fig. 1b). Cr(II) acetates are thus moderately air-stable unlike other Cr(II) compounds such as CrCl2; as dictated by the Pourbaix diagram,14 most Cr(II) compounds are very hygroscopic and quite easily oxidized to Cr(III). In addition, Cr(II) acetates are good Lewis acids that accept electron pairs at their axial positions. These electron donors contribute to the empty δ* antibonding orbitals (LUMO), so the Lewis basicity of the ligands affects the physicochemical properties by modulating the narrow energy gap between δ and δ* orbitals.1,12,13,15 Therefore, the synthesis of Cr(II) acetates with various axial ligands L (Cr2(OAc)4L2) would facilitate fundamental study on the nature of the unique delta bonds.
Traditionally Cr2(OAc)4L2 have been prepared by ligand exchange of the hydrate; for example, layering pyridine (py) on top of an aqueous solution of the hydrate (L = H2O) precipitates small crystals of Cr2(OAc)4(py)2.16 Such interfacial diffusion, however, may yield crystals of Cr2(OAc)4(H2O)2 as by-products (Fig. S1†). A rather straight forward way to coordinate the desired ligands is to dissolve Cr2(OAc)4 in the target ligand solvent, as pointed out by Cotton et al.1 However, this method has been restricted to few kinds of ligands, such as CH3OH.7
The Cr(II) acetates should be handled with care to prevent the oxidation of Cr(II) to Cr(III), since even the quadruple bonds are not strong enough to altogether avoid the oxidation;17 they instead expedite the oxidation process to some extent. This is because O2 in the air is such a good oxidizing agent that it effectively breaks the quadruple bond. Therefore, dissolved oxygen in the solvent should be removed, or degassed, before dissolving Cr(II) acetate. Nevertheless, the degassing process has not been explicitly mentioned or reported, to the best of our knowledge.1,7,12,16
Herein, we unveil a simple solvent degassing process for the various ligation of the anhydrous Cr(II) acetate at the axial position, to prepare crystalline Cr2(OAc)4L2 (L = H2O, CH3OH (MeOH), and pyridine (py)) (Fig. 1c). Then, the target crystals are obtained by lowering the temperature of the solution or drop-drying process (Fig. 3a). The whole process takes less than a day to yield sub-millimeters scale crystals, while the conventional synthetic methods took several days according to previous literatures.15,18 Our findings would contribute to promoting the exploration of quadruple bonds between metal atoms, the extension of metal-to-metal (M–M) multiple bonds-based networks to form coordination polymers19,20 or metal–organic frameworks8,21 as well as the study of their unique physical and chemical properties of the quadruple bonds.
Results and discussion
1. Synthesis of Anhydrous Cr2(OAc)4
Anhydrous Cr2(OAc)4, the precursor of the Cr2(OAc)4L2 has been prepared by annealing the hydrate or ethanolate (L = EtOH) in a vacuum. However, this process yields amorphous powder, which is easily oxidized.12,17 An altered approach to preparing Cr2(OAc)4 by a direct chemical reaction between acetic acid and chromocene (Cr(C2H5)2), but the chromocene is air-sensitive and flammable complex.18,22 Therefore, both methods are not suitable for preparing Cr2(OAc)4 that is stable in air.Recently, Levy et al.22 reported another synthetic method to prepare the crystalline Cr2(OAc)4, using such air-stable precursors as Cr(0) powder, acetic acid, acetic anhydride, and HBr. In addition, the product is stable in the air up to several hours, unlike that of the predecessors. Therefore, we adapted Levy et al.'s manner for the preparation of the high-purity crystals of Cr2(OAc)4 with slight modification (refer to Experimental section for details in ESI†).
In our synthetic process, concentrated (37%) HCl (aq) was used instead of HBr or KBr, and the Cr powder was halved in the amount of the reference. The total reaction time was less than an hour to prevent oxidation to Cr(III). Such modifications are to guarantee the exclusion of by-products or unreacted reactants (e.g. Cr(III) acetate and Cr(0) powder, respectively) in the final product. Therefore, red crystalline powder was prepared after simply filtering the product and washing it with excess acetone to remove Cr(III) acetates (Fig. 2a).
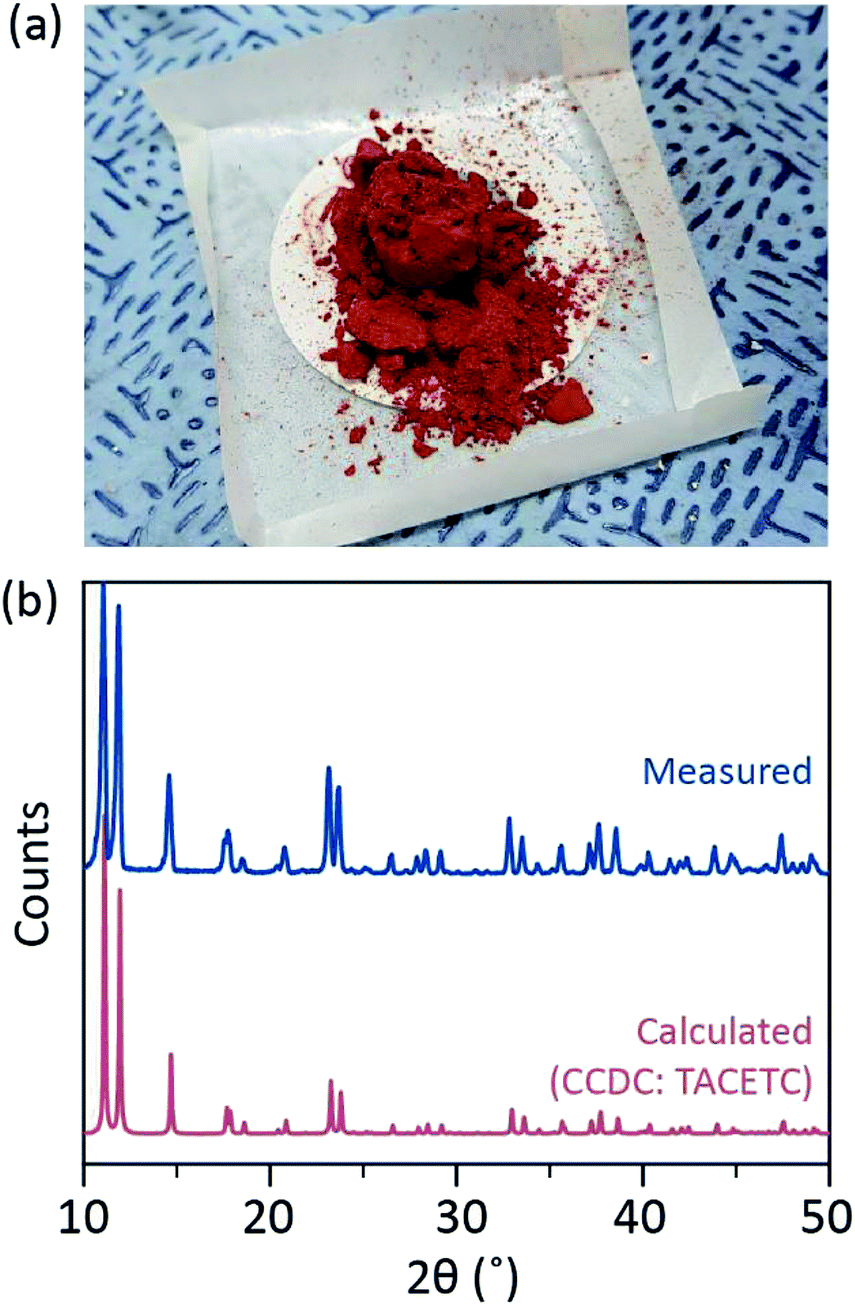 | ||
| Fig. 2 (a) Photograph of the as-prepared Cr2(OAc)4 powder. The image was taken in air. (b) X-ray diffraction of the powder (blue). The red graph is an estimated diffraction pattern based on the crystallographic information of Cr2(OAc)4 registered in the CCDC database. | ||
The powder was then dried in a vacuum at room temperature for a few hours. As shown in Fig. 2b, powder X-ray diffraction (PXRD) revealed that the red powder is Cr2(OAc)4 having a triclinic unit cell (P-1, a = 7.583 Å, b = 8.688 Å, c = 5.178 Å, α = 111.16°, β = 95.77°, γ = 98.16°, V = 310.626 Å3); the pattern is identical to a pattern calculated from previously reported crystallographic information of Cr2(OAc)4 (CCDC#: TACETC).
Note that the obtained powder was used without further purification, since it was very difficult to purify. When vapor-phase recrystallization of Cr2(OAc)4 was attempted for the purification, nothing was evaporated below 270 °C under Ar flow. Only a small portion of the red powder evaporated at temperature above 270 °C and below 300 °C, and the remnant was decomposed into black powder (Fig. S2†). The evaporated powder was recrystallized into reddish thin film, but it was so quickly decomposed into green-implying oxidation to Cr(III)-that it could not be retrieved for further reactions. At temperature above 300 °C the powder was fully decomposed without evaporation. Such difficulty in vapor-phase recrystallization is consistent with previous reports.1,13,15
2. Synthesis of Cr2(OAc)4L2
In order to synthesize the Cr(II) acetates coordinated with axial ligands (Cr2(OAc)4L2), we first selected target ligand solvents as follows: MeOH, H2O, and py (Fig. 1c). These ligands have electron lone pairs to form a stable coordination bond to Cr2(OAc)4. They have different Lewis basicity (MeOH < H2O < py), and accordingly the internuclear distance of the quadruple bond in the ligated complex is known to span from 2.288 Å (anhydrous), 2.329 Å (MeOH), 2.362 Å (H2O), and to 2.369 Å (py), according to the Lewis basicity of the axial ligands.7,12,16,23The ligand solvents were degassed by heating it just below its boiling point with an injection of Ar gas (Fig. 3a). This degassing process is prerequisite for the dissolution of Cr2(OAc)4 in the target solvent for preventing degradation of the quadruple bonds, as obstructing vigorous oxidation by the dissolved O2. When Cr2(OAc)4 was dissolved into a degassed solvent, the color of the solution instantly changed to red like Cr2(OAc)4 (Fig. 3b). Note that dissolution in degassed H2O is not instantaneous; when a small amount of Cr2(OAc)4 was put in degassed H2O, it was blue at first, and then adding an extra amount of the solute eventually turned it reddish brown. Such two-step dissolution may be attributed to the equilibrium between Cr2(OAc)4 (aq) and Cr2+ (aq), as follows:1,24 (i) Cr2(OAc)4 ⇄ 2Cr(OAc)2, (ii) Cr(OAc)2 ⇄ Cr(OAc)+ + OAc−, (iii) Cr(OAc)+ ⇄ Cr2+ + OAc−.
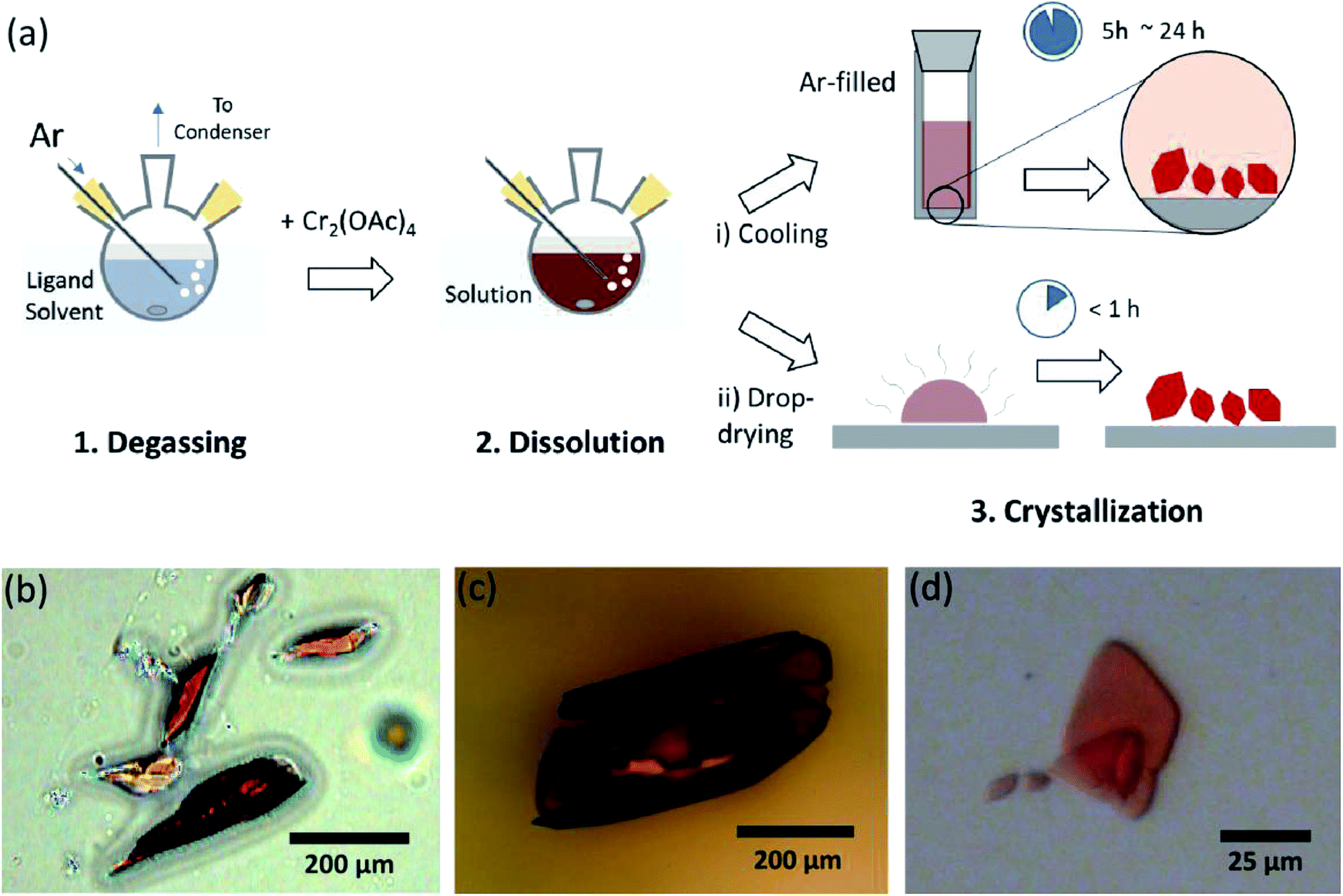 | ||
| Fig. 3 (a) Scheme of the ligand coordination process. (b–d) Microscope images of obtained crystals of Cr2(OAc)4L2, where L = H2O (b), MeOH (c), py (d). | ||
In contrast, when Cr2(OAc)4 was dissolved in non-degassed solvents, the solution was blue for H2O and MeOH and orange for pyridine, which are attributed to the decomposition of Cr2(OAc)4 (Fig. S3†).1 Note that there are also certain solvents that do not dissolve Cr2(OAc)4 even after the degassing process. When Cr2(OAc)4 was put in hexane or toluene, which does not have any electron lone pairs to donate, neither dissolution nor coordination was observed; all the powder sank to the bottom of the reaction vessel (Fig. S4†). Such selective solubility shows that the coordination of the ligands accompanies the dissolution of Cr2(OAc)4, and vice versa.1
3. Crystallization of Cr2(OAc)4L2
When the ligation in anoxic condition was finished, stirring was stopped for precipitation of remaining reactants like Cr powder or undissolved Cr2(OAc)4 powder. The temperature was kept identical to the degassing and dissolution process (i.e., just below the boiling point of the ligand solvent). To obtain the crystals of the ligated complexes (Cr2(OAc)4L2), clear supernatant was carefully put in an Ar-filled vial for cooling under anoxic condition at room temperature, since the solution is very air-sensitive; only a brief exposure changes the color from red to blue or green, implying the oxidation of the complex. Small red crystals in the Ar-filled vial had been grown at the bottom of the vial less than 24 hours (crystallization process (i) in Fig. 3a). The crystals can also be obtained very rapidly by even drop-drying the supernatant onto a substrate, and it takes less than an hour under the ambient condition (crystallization process (ii) in Fig. 3a). The crystalline products by both methods are identical; they are sub-millimeter-scale, reddish crystals, of which colors are subtly different depending on the ligands (Fig. 3b–d).4. Characterization of Cr2(OAC)4L2
Further, single-crystal X-ray diffraction (SCXRD) studies confirmed that the crystallographic structure of the obtained Cr2(OAc)4L2 crystals are matched with those of previously reported crystals (Table 1).7,12,16 Note that SCXRD was attempted for structural confirmation of the large sized single crystal instead of PXRD, since the yield of the crystals were not high (7.94% for L = H2O, 11.26% for L = MeOH, and 3.70% for L = py). Such low yield is attributed to that the solubility of Cr2(OAc)4 in the ligand solvents is poor,1 and also that most of the ligated complex molecules remain as the solute even after the crystallization. Moreover, through the SCXRD studies, it is confirmed that the internuclear distance between M–M is elongated depending on the Lewis basicity of the additional axial ligands (Lewis basicity: MeOH < H2O < py). Thus, the order of M–M distances of the Cr2(OAc)4L2 are disclosed by the SCXRD as follows: Cr2(OAc)4 < Cr2(OAc)4(MeOH)2 < Cr2(OAc)4(H2O)2 < Cr2(OAc)4(py)2. Besides, the rapidly synthesized ligated crystals were also stable in the air for, but not limited by, 12 hours just as the original Cr2(OAc)4 crystals. A probable reason would be the packing of the molecules in the crystals, which can hinder the attack by certain oxidizing agents like O2. These shreds of evidence tell us that our simple crystallization method makes the diverse ligands readily attach to the axial position of the Cr–Cr quadruple bond with increased stability.Raman spectroscopy has been used as a powerful tool for studying vibration modes of multiply-bonded binuclear complexes containing Cr, Mo, W, or Re and a correlation between the M–M bond strength and distance according to various ligands.1,25–27 As shown in Fig. 4, confocal Raman spectroscopy (Alpha 300R, WITec) revealed vibration modes of the crystals of Cr(II) acetates. The Raman spectral data of Cr(II) acetates were collected less than a few minutes, to prevent degradation of the complexes while the crystals were irradiated by laser excitation source (532 nm, 0.5 mW, Nd:YAG) under the ambient condition.
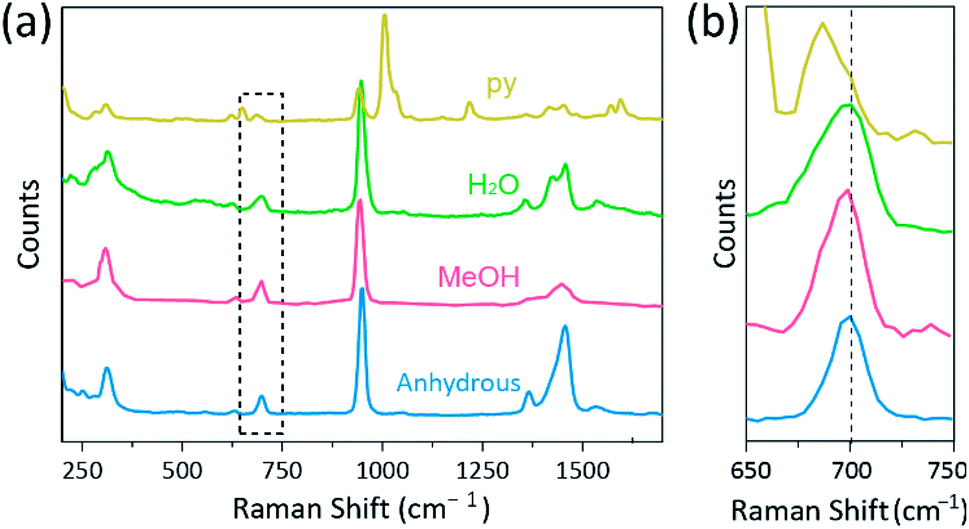 | ||
| Fig. 4 (a) Raman spectra of Cr2(OAc)4 (blue) and Cr2(OAc)4L2 (L = MeOH (red), H2O (green), and py (yellow)). (b) Detailed Raman spectra inside the dashed box in (a). The dashed line represents the center of the overtone mode of Cr2(OAc)4. The spectra were obtained by confocal Raman spectroscope with 532 nm excitation wavelength. | ||
In our experiment, the measured Raman spectrum of Cr2(OAc)4(H2O)2 is identical to a previous report,25 but the Raman spectra of the other synthesized complexes including anhydrous Cr2(OAc)4 are not reported, to the best our knowledge. The Cr–Cr quadruply bonded complexes are shown in Fig. 4 have a similar pattern of the Raman spectrum of the Cr2(OAc)4(H2O)2, possibly due to the similarity of the molecular structures. In the Raman spectra, bands around 550 cm−1 and 350 cm−1 have been assigned to Cr–Cr stretching and twisting/bending modes of Cr(II) acetates, respectively.25,28,29 Nonetheless, the exact band positions are barely distinguished from the measured Raman spectra of all Cr(II) acetates due to their low intensity18 or broad width (Fig. S5†). As an alternative, the overtone envelopes of the Cr–Cr quadruple bonds' vibration are obviously displayed around 700 cm−1 (2 × ∼350 cm−1) (Fig. 4b).29 Indeed, as the basicity of the axial ligand increases, the overtone band is red-shifted accordingly.30 This clue reflects that the bond strength of the quadruple bonds being weaker, as increasing the Lewis basicity of the axial ligands.29 Also, the degree of red-shifts of the Cr(II) acetates in Raman spectra is correlated to its Cr–Cr distances. Thus, the overtone band of Cr2(OAc)4 shows the highest wavenumber, but Cr2(OAc)4(py)2 have the lowest wavenumber among the Cr–Cr quadruply bonded complexes.
Conclusion
We developed a feasible and rapid way to synthesize sub-millimeter-scale single crystals of Cr2(OAc)4L2, where L = H2O, MeOH, and py. The key to success is the removal of oxidizing agents dissolved in the ligand solvents before dissolving crystalline powder of Cr2(OAc)4. Also, dissolution of Cr2(OAc)4 in certain ligand solvents occurs only when the coordination bonds are formed between anhydrous Cr(II) acetate (Lewis acid) and the ligand molecules (Lewis base). Our result demonstrates that various molecules can coordinate as axial ligands to multiply bonded organometallic complexes by a facile process. Such versatility and simplicity can further be expanded to replace the paddle-wheel ligands and metal atoms, achieving an even more diverse combination to develop the large extent of M–M based network system such as M–M chained coordination polymers and metal–organic frameworks. Design and preparation of such novel complexes would contribute to the fundamental understanding of delta orbitals and multiple bonds and their applications to various fields of physics, chemistry, and related engineering.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate Prof. Dr Minjoong Yoon at Chungnam National University and Prof. Dr Hee Cheul Choi at Pohang University of Science and Technology for valuable comments. This work was supported by Wonkwang University in 2018.Notes and references
- F. A. Cotton, C. A. Murillo and R. A. Walton, Multiple Bonds Between Metal Atoms, Springer-Verlag, New York, 2005 Search PubMed.
- F. A. Cotton, B. G. DeBoer, M. D. LaPrade, J. R. Pipal and D. A. Ucko, J. Am. Chem. Soc., 1970, 92, 2926–2927 CrossRef CAS.
- T. Nguyen, A. D. Sutton, M. Brynda, J. C. Fettinger, G. J. Long and P. P. Power, Science, 2005, 310, 844–847 CrossRef CAS PubMed.
- F. R. Wagner, A. Noor and R. Kempe, Nat. Chem., 2009, 1, 529–536 CrossRef CAS PubMed.
- K. A. Kreisel, G. P. A. Yap, O. Dmitrenko, C. R. Landis and K. H. Theopold, J. Am. Chem. Soc., 2007, 129, 14162–14163 CrossRef CAS PubMed.
- A. Falceto, K. H. Theopold and S. Alvarez, Inorg. Chem., 2015, 54, 10966–10977 CrossRef CAS PubMed.
- F. A. Cotton, H. Chen, L. M. Daniels and X. Feng, J. Am. Chem. Soc., 1992, 114, 8980–8983 CrossRef CAS.
- E. D. Bloch, W. L. Queen, M. R. Hudson, J. A. Mason, D. J. Xiao, L. J. Murray, R. Flacau, C. M. Brown and J. R. Long, Angew. Chem., Int. Ed., 2016, 55, 8605–8609 CrossRef CAS PubMed.
- L. J. Murray, M. Dinca, J. Yano, S. Chavan, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2010, 132, 7856–7857 CrossRef CAS PubMed.
- F. A. Cotton and C. B. Harris, Inorg. Chem., 1965, 4, 330–333 CrossRef CAS.
- W. Clegg, C. D. Garner, L. Akhter and M. H. Al-Samman, Inorg. Chem., 1983, 22, 2466–2468 CrossRef CAS.
- F. A. Cotton, C. E. Rice and G. W. Rice, J. Am. Chem. Soc., 1977, 99, 4704–4707 CrossRef CAS.
- S. N. Ketkar and M. Fink, J. Am. Chem. Soc., 1985, 107, 338–340 CrossRef CAS.
- M. Pourbaix, Atlas of electrochemical equilibria in aqueous solutions, National Association of Corrosion Engineers, 1974 Search PubMed.
- F. A. Cotton, E. A. Hillard, C. A. Murillo and H. C. Zhou, J. Am. Chem. Soc., 2000, 122, 416–417 CrossRef CAS.
- F. A. Cotton and T. R. Felthouse, Inorg. Chem., 1980, 19, 328–331 CrossRef CAS.
- J. C. Reeve, J. Chem. Educ., 1985, 62, 444 CrossRef CAS.
- F. A. Cotton, M. W. Extine and G. W. Rice, Inorg. Chem., 1978, 17, 176–186 CrossRef CAS.
- H.-C. Chang, J.-T. Li, C.-C. Wang, T.-W. Lin, H.-C. Lee, G.-H. Lee and S.-M. Peng, Eur. J. Inorg. Chem., 1999, 1999, 1243–1251 CrossRef.
- M. Yang, T.-W. Lin, C.-C. Chou, H.-C. Lee, H.-C. Chang, G.-H. Lee, M. Leung and S.-M. Peng, Chem. Commun., 1997, 2279–2280 RSC.
- L. J. Murray, M. Dinca, J. Yano, S. Chavan, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2010, 132, 7856–7857 CrossRef CAS PubMed.
- O. Levy, B. Bogoslavsky and A. Bino, Inorg. Chim. Acta, 2012, 391, 179–181 CrossRef CAS.
- F. A. Cotton, B. G. DeBoer, M. D. LaPrade, J. R. Pipal and D. A. Ucko, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 1664–1671 CrossRef CAS.
- R. D. Cannon and J. S. Stillman, Inorg. Chem., 1975, 14, 2207–2214 CrossRef CAS.
- Y. M. Huang, H. R. Tsai, S. H. Lai, S. J. Lee, I. C. Chen, C. L. Huang, S. M. Peng and W. Z. Wang, J. Phys. Chem. C, 2011, 115, 13919–13926 CrossRef CAS.
- R. E. Da Re, J. L. Eglin, C. N. Carlson, K. D. John, D. E. Morris, W. H. Woodruff, J. A. Bailey, E. Batista, R. L. Martin, F. A. Cotton, E. A. Hillard, C. A. Murillo, A. P. Sattelberger and R. J. Donohoe, J. Am. Chem. Soc., 2010, 132, 1839–1847 CrossRef CAS PubMed.
- W. C. Trogler and H. B. Gray, Acc. Chem. Res., 1978, 11, 232–239 CrossRef CAS.
- F. A. Cotton, P. E. Fanwick, R. H. Niswander and J. C. Sekutowski, J. Am. Chem. Soc., 1978, 100, 4725–4732 CrossRef CAS.
- R. E. Da Re, J. L. Eglin, C. N. Carlson, K. D. John, D. E. Morris, W. H. Woodruff, J. A. Bailey, E. Batista, R. L. Martin, F. A. Cotton, E. A. Hillard, C. A. Murillo, A. P. Sattelberger and R. J. Donohoe, J. Am. Chem. Soc., 2010, 132, 1839–1847 CrossRef CAS PubMed.
- Note that in case of Cr2(OAc)4(py)2 a weak peak near 680 cm−1 was assigned as the overtone mode instead of the strong peak near 650 cm−1, because it too much deviates from 700 cm−1. This assignment is nonetheless acceptable, because the Raman peaks of Cr2(OAc)(py)2 that are common to other complexes are substantially weaker than the uncommon peaks..
- Cr2(OAc)4(H2O)2: C8H12O10Cr2, Mr = 372.18, crystal dimensions 0.10 × 0.09 × 0.02 mm3, monoclinic, C2/c, a = 13.105(3) Å, b = 8.5680(17) Å, c = 13.838(3) Å, β = 116.71(3)°, V = 1388.0(6) Å3, T = −173 °C, Z = 4, rcal= 1.781 g cm−3, m = 0.15 cm−1, 1968 unique reflections out of 2076 with I > 2s(I), 94 parameters, 2.925° < q < 29.848°, R1 = 0.0425, wR2 = 0.1183, GOF = 0.927. CCDC deposit number 1131602..
- Cr2(OAc)4(MeOH)2: C10H18O10Cr2, Mr = 402.24, crystal dimensions 0.05 × 0.04 × 0.05 mm3, monoclinic, P21/n, a = 8.0650(16) Å, b = 7.4060(15) Å, c = 13.296(3) Å, β = 92.84(3)°, V = 793.2(3) Å3, T = −173 °C, Z = 2, rcal = 1.684 g cm−3, m = 0.135 cm−1, 2197 unique reflections out of 2277 with I > 2s(I), 105 parameters, 3.102° < q < 29.841°, R1 = 0.0439, wR2 = 0.1198, GOF = 0.862. Refer to ref. 7..
- Cr2(OAc)4(py)2: C18H22O8N2Cr2, Mr = 496.36, crystal dimensions 0.09 × 0.08 × 0.09 mm3, orthorhombic, Pbca, a = 12.875(3) Å, b = 8.5890(17) Å, c = 19.524(4) Å, V = 2159.0(7) Å3, T = −173 °C, Z = 4, rcal= 1.533 g cm−3, m = 0.10 cm−1, 2797 unique reflections out of 3243 with I > 2s(I), 138 parameters, 2.579° < q < 29.860°, R1 = 0.0452, wR2 = 0.1206, GOF = 1.043. CCDC deposit number 1100915..
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04189c |
| This journal is © The Royal Society of Chemistry 2019 |