
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recyclable iron(II) caffeine-derived ionic salt catalyst in the Diels–Alder reaction of cyclopentadiene and α,β-unsaturated N-acyl-oxazolidinones in dimethyl carbonate†
Di Meng ,
Dazhi Li and
Thierry Ollevier*
,
Dazhi Li and
Thierry Ollevier*
Département de chimie, Université Laval, 1045 Avenue de la Médecine, Québec, QC, Canada G1V 0A6. E-mail: thierry.ollevier@chm.ulaval.ca
First published on 15th July 2019
Abstract
Iron(II) triflate was used in combination with caffeine-derived salts as recyclable catalysts for the Diels–Alder reaction run in dimethyl carbonate (DMC) as a green solvent. The catalyst was prepared as an ionic salt from a xanthinium salt and Fe(OTf)2. Various substrates including α,β-unsaturated carbonyl and N-acyloxazolidinone derivatives were reacted with cyclopentadiene using this recyclable catalyst. The use of a low catalyst loading (1 mol%) afforded high yields (up to 99%) of the corresponding cycloadducts. The recycling and the efficiency of the catalyst were demonstrated for several runs.
Introduction
The Diels–Alder reaction is among the most powerful C–C bond forming transformations in synthetic chemistry.1 α,β-Dicarbonyl derivatives have been used as dienophiles in the Diels–Alder reaction2 ever since the introduction of N-acyloxazolinones as dienophiles 30 years ago by Evans.3 The reaction between α,β-unsaturated oxazolidinones and cyclopentadiene became a benchmark reaction4 to evaluate the catalytic activity of various metal Lewis acid catalysts, such as Mg,5 Cu,6 Sc,7 Ti,8 Ln,2 Ni,9 Pd,10 Fe,11 and Cr.12 Much progress was made through the development of more efficient Lewis acids and ligands.1–12 However, the use of large quantities of some of these catalysts to mediate this transformation, in addition to their limiting prices, render them difficult to use, thus creating a need for additional recyclable catalysts.13 Various approaches include heterogeneous catalysis,14 or replacing the reaction solvent by ionic liquids (ILs), which were originally used toward improved stereoselectivity.14,15 Ionic liquids have received considerable attention as new powerful reaction media and emerged as a potential alternative to conventional organic solvents.16 Furthermore, the combination of ionic liquids or ionic salts with transition metals has been reported as a promising area, and their scope of applicability is extending.17 An immobilized catalytic system of [bmim][FeCl4] was developed for aryl Grignard cross-coupling via a liquid–liquid biphasic process, in which FeCl3 was trapped in the ionic liquid.18 A similar system involving a Bi(OTf)3-trapped caffeine-derived salt (xanthinium salt) in a Diels–Alder reaction, in which the combined xanthinium–BiIII mixture of salts was recycled without loss of efficiency over several runs.19 However, one drawback of these methods was the use of dichloromethane as a solvent. In fact, the most commonly used solvents in Diels–Alder reactions are, among others, dichloromethane, chloroform, toluene, diethyl ether, and water. Hence, in order to develop a Diels–Alder reaction run in greener conditions, we were interested in seeking appropriate environmentally-benign solvents. Although ILs have many advantages, their high prices and waste disposal during large scale applications limit their widespread use, not to mention their adverse environmental impact in their life cycle assessment (LCA).20 Beside ILs, other ecofriendly solvents have also been studied.21 Dimethyl carbonate (DMC), which has been assessed as a green alternative to replace easily-flammable organic compounds (VOC) as solvents,22 appeared promising to use. It is environmentally benign and scores low on LCA scales.23 In the context of our research on iron catalysis and ionic liquids,24 a combination of a caffeine derivative, an iron salt, and dimethyl carbonate is disclosed herein.Iron has attracted considerable attention on its eco-friendliness, natural abundance, inexpensive price, and its promising applications in organic synthesis.25 Fe(OTf)2 was already used with an ionic liquid ethylmethylimidazolium bis-triflimide in an aziridination reaction.26 Also, FeII/FeIII-derived catalysts have been used in the Diels–Alder reaction, including asymmetric versions.4a,27 Moreover, many other examples of FeII/FeIII-catalyzed Diels–Alder reactions were reported in homogeneous and heterogeneous catalysis.28
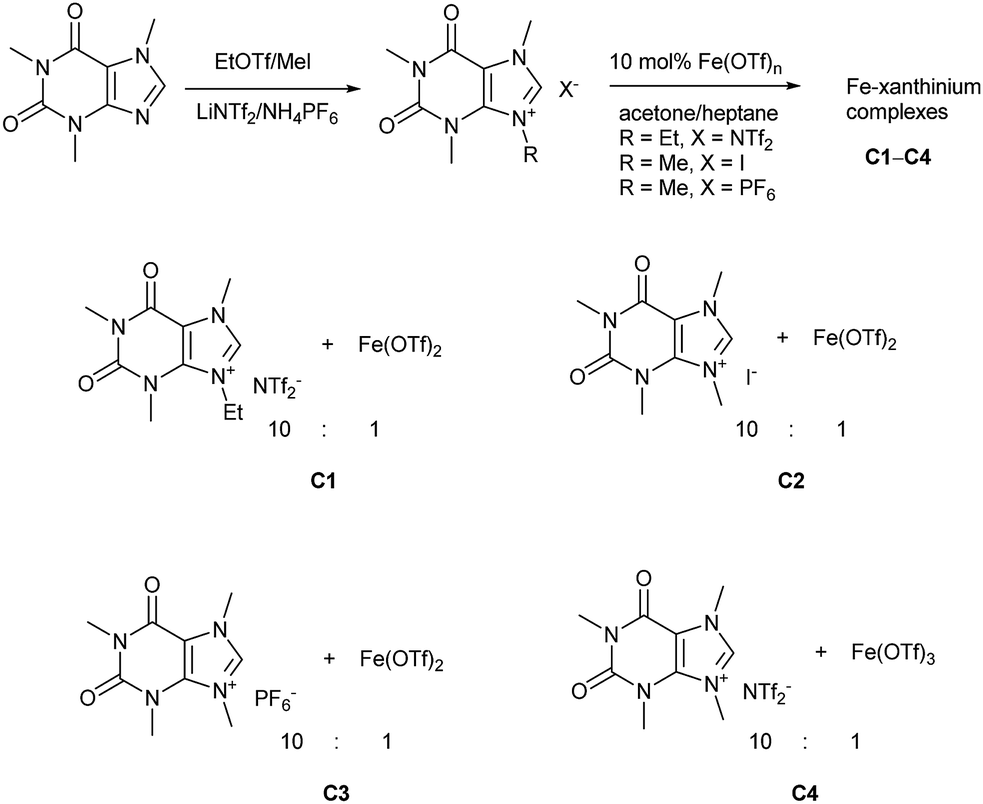
Caffeine, as one of the methylxanthines, is a green, natural, abundant, and biodegradable compound that can be further alkylated into xanthinium salts.29 These xanthinium salts have been used for the preparation of NHC-metal complexes in medicinal and organometallic chemistry.30 As already mentioned, a caffeine-derived ethylxanthinium salt was already used as an ionic solid in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio to Bi(OTf)3·4H2O and recycled as a combined ionic salt.18,19 The recycling of this catalyst was easily performed after its precipitation from heptane; however, the reaction was run in CH2Cl2 and only a few dienophiles were disclosed.18,19 Alkylated caffeines, N-methyl- and N-ethyl-substituted xanthinium salts were obtained with NTf2−, I−, and PF6− anions (Scheme 1).18,19,30b–g Next, the xanthiniums were mixed with Fe(OTf)2 or Fe(OTf)3 in acetone (Scheme 1). The ratio of the xanthiniums to FeII/FeIII salts was examined in a range of 2:1 to 10:1. For ratios 7:1 to 10:1, the iron salt was completely solubilized. A 10:1 ratio was used to ensure that there was no loss of the catalyst during the recycling process. Acetone and heptane were used to prepare catalysts C1, C2, C3, and C4. These catalysts were tested conjointly with Fe(OTf)2 and Fe(NTf2)2 in the Diels–Alder reaction of cyclopentadiene and 3-acryloyl-1,4-oxazolidin-2-one, chosen as the model reaction.
:1 ratio to Bi(OTf)3·4H2O and recycled as a combined ionic salt.18,19 The recycling of this catalyst was easily performed after its precipitation from heptane; however, the reaction was run in CH2Cl2 and only a few dienophiles were disclosed.18,19 Alkylated caffeines, N-methyl- and N-ethyl-substituted xanthinium salts were obtained with NTf2−, I−, and PF6− anions (Scheme 1).18,19,30b–g Next, the xanthiniums were mixed with Fe(OTf)2 or Fe(OTf)3 in acetone (Scheme 1). The ratio of the xanthiniums to FeII/FeIII salts was examined in a range of 2:1 to 10:1. For ratios 7:1 to 10:1, the iron salt was completely solubilized. A 10:1 ratio was used to ensure that there was no loss of the catalyst during the recycling process. Acetone and heptane were used to prepare catalysts C1, C2, C3, and C4. These catalysts were tested conjointly with Fe(OTf)2 and Fe(NTf2)2 in the Diels–Alder reaction of cyclopentadiene and 3-acryloyl-1,4-oxazolidin-2-one, chosen as the model reaction.
 | ||
| Scheme 1 Synthetic routes of chiral dihydroquinoxalinones. | ||
Results and discussion
Initially, Fe(OTf)2 and catalyst C1 were studied in dichloromethane. The ratio of diene to dienophile was set to 7:1. The yields and stereoselectivities obtained with Fe(OTf)2 (Table 1, entry 1) and catalyst C1 in CH2Cl2 were similar (entry 2). The same yield and endo/exo selectivity were obtained in dimethyl carbonate at room temperature (entry 3) and even at 2 °C (melting point of DMC, entry 4). Then, an optimization study was performed by lowering the ratio of reactants from 7:1 to 5:1 (entry 5) until 2:1, and a quantitative yield together with a slightly higher endo/exo ratio was obtained (entry 6). This result was even better than our previous study using FeIII catalyst in DMC (76% yield).24e The other three catalysts C2, C3, and C4 were tested in the same conditions as in entry 6. Catalysts C2 and C3 led to low yields of 3a (entries 7 and 8). Using the Fe(OTf)3 derived catalyst C4, the yield reached 97%, whereas the endo/exo ratio decreased to 90:10 (entry 9). Three control experiments were consequently performed (entries 10, 11, and 12). The yield dropped to 34% without using a catalyst (entry 10). While Fe(OTf)2 led to a lower yield than when using catalyst C1 (entry 11). A quantitative yield was obtained when using Fe(NTf2)2 alone (entry 12). Compared to Fe(OTf)2, catalyst C1 was shown to be more efficient in terms of yield and stereoselectivity.
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1a/2a | Catalyst | T (°C) | t (h) | Endo/exo | Yield 3ad (%) |
| a Conditions: 1a (1 mmol), 2a (0.5 mmol), DMC (1 mL); 3aendo/3aexo ratio was calculated by 1H NMR.b Reaction run in CH2Cl2 (1 mL).c Reaction was run on 7.1 mmol of 2a (1 g of scale).d Isolated yields. | ||||||
| 1b | 7:1 |
Fe(OTf)2 | rt | 13 | 87:13 |
89 |
| 2b | 7:1 |
C1 | rt | 13 | 88:12 |
92 |
| 3 | 7:1 |
C1 | rt | 13 | 91:9 |
99 |
| 4 | 7:1 |
C1 | 2 | 3 | 91:9 |
99 |
| 5 | 5:1 |
C1 | 2 | 3 | 91:9 |
99 |
| 6 | 2:1 |
C1 | 2 | 3 | 94:6 |
99 |
| 7 | 2:1 |
C2 | 2 | 3 | 94:6 |
33 |
| 8 | 2:1 |
C3 | 2 | 3 | 90:10 |
55 |
| 9 | 2:1 |
C4 | 2 | 3 | 90:10 |
97 |
| 10 | 2:1 |
— | 2 | 3 | 90:10 |
34 |
| 11 | 2:1 |
Fe(OTf)2 | 2 | 3 | 90:10 |
81 |
| 12 | 2:1 |
Fe(NTf2)2 | 2 | 3 | 93:7 |
100 |
| 13c | 2:1 |
C1 | 2 | 3 | 94:6 |
100 |
Although Fe(NTf2)2 alone was as efficient as catalyst C1, it was not recyclable from the reaction media. Similarly, Fe(OTf)2 was also lost during work-up (entries 1 and 11). On the contrary, catalyst C1 could be recycled after the addition of heptane and filtration. Since no detectable difference was observed among the results obtained with C1 and Fe(NTf2)2, it was hypothesized that OTf− and NTf2− can readily exchange within the catalytic system (entries 2 and 12). The reaction using gram scale of 2a was tested as well, and a quantitative yield was obtained (entry 13).
To explore the scope of the reaction solvent, a few green solvents were selected and compared with CH2Cl2 and THF (Table 2). CH2Cl2 afforded the lowest endo/exo selectivity, while THF led to a moderate yield and good endo/exo selectivity (entries 1 and 2). Me-THF resulted in an even lower yield than THF, but with a high stereoselectivity (entry 3). N-Methyl pyrrolidinone (NMP) was found less suitable due to its high boiling point, resulting in a difficult separation from the reaction product (entry 4). Cyclopentyl methyl ether (CPME), ethyl acetate, and methyl tert-butyl ether (MTBE) led to good results (entries 5, 6, and 7). Of all the solvents tested, DMC was selected for its polarity close to CH2Cl2.31 It provided a high endo/exo ratio combined with an excellent yield (entry 8) and was thus chosen for the next part of the study.
| Entry | Solvent (polarity)b | Endo/exo | Yield 3ac (%) |
|---|---|---|---|
| a Conditions: 1a (1.0 mmol), 2a (0.50 mmol), C1 (1 mol%), DMC (1 mL), 2 °C, 3 h; endo/exo ratio was determined by 1H NMR.b Relative polarity using water as reference (polarity = 1).31c Isolated yields. | |||
| 1 | CH2Cl2 (0.309) | 76:24 |
92 |
| 2 | THF (0.207) | 80:20 |
65 |
| 3 | Me-THF (0.179) | 90:10 |
50 |
| 4 | NMP (0.355) | 88:12 |
85 |
| 5 | CPME (—) | 77:23 |
100 |
| 6 | EtOAc (0.228) | 80:20 |
98 |
| 7 | MTBE (0.124) | 79:21 |
97 |
| 8 | DMC (0.232) | 94:6 |
99 |
To investigate the recoverability of the catalyst, recycling tests were performed using C1. Table 3 highlights the reaction yields and the mass of recycled catalyst after each of the 5 runs of the reaction. Firstly, 1 mol% catalyst was used for five runs, and after each run, heptane was added to the reaction mixture to precipitate the catalyst. Then, after filtration through a cotton-plugged pipet, the catalyst was recollected and washed with acetone, and was finally recycled after evaporation. Using 1 mol% of C1, the yields of both the product and the recycled catalyst slightly decreased over the 5 runs.32 However, when the catalyst loading was increased to 2 mol%, all of the catalyst was recycled after each run, and the reaction yield was maintained at the same level. According to 1H and 19F NMR analysis, the chemical composition of the catalyst resulted unchanged (or mostly unchanged) after recovery (see ESI†).
|
||||||
|---|---|---|---|---|---|---|
| x mol% | Run 1 | Run 2 | Run 3 | Run 4 | Run 5 | |
| a Conditions: 1a (1 mmol), 2a (0.5 mmol), C1 (1 mol%), DMC (1 mL); isolated yields. | ||||||
| 1 | Yield (%) | 99 | 98 | 96 | 95 | 95 |
| Cat. recycl. (%) | 100 | 99 | 97 | 95 | 94 | |
| 2 | Yield (%) | 99 | 99 | 99 | 99 | 99 |
| Cat. recycl. (%) | 100 | 100 | 100 | 100 | 100 | |
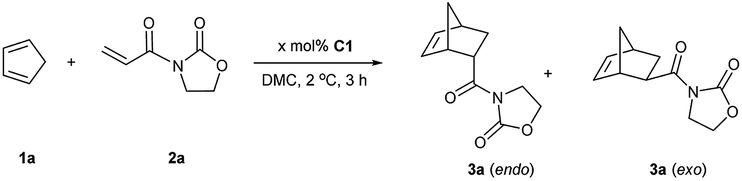
Finally, given the results above, the optimized conditions were applied to various dienophiles in the Diels–Alder reaction (Scheme 2). The general procedure was run as follows: cyclopentadiene was added into a solution of the catalyst and the dienophile in dimethylcarbonate. After the given time, heptane was added into the solution to precipitate the catalyst for recycling. 3-Crotonyl-oxazolidinone (2b) required a longer time than substrate acryloyl-oxazolidin-2-one (2a) but afforded a slightly higher stereoselectivity of 3b. Instead of a CH3 group, a β-CF3 to the carbonyl group led to higher endo/exo selectivity and high yield (3c). However, low solubility of cinnamoyl oxazolidinone resulted in a very low yield (3d).33 2-Alkenoyl pyridines were explored as bidentate dienophiles that can chelate to the metal center through both the pyridine and the carbonyl lone pairs.6a Here, the objective was to use them as substrates in the developed conditions (3e–h). High yields and high endo/exo ratios were obtained. The stereoselectivity decreased when using cinnamoyl pyridine N-oxide (2h), probably due to the weaker coordination of the oxide with iron than that with pyridine. Being similar on the cinnamoyl part, benzylidene acetone (2i) and acryloyl chloride (2j) were chosen to get a comparison with the bidentate dienophiles 2d and 2e. Benzylidene acetone showed a good reactivity with a higher endo/exo ratio (3i). Using acryloyl chloride, a moderate yield (60%, 3j) was obtained. Neat conditions were efficiently used with methyl acrylate at very low catalyst loading (0.1 mol%), and high yield (90%), and selectivity were obtained (77:23 endo/exo, 3k), which was even better than some obtained results using ionic liquids, but low yield was obtained for methyl propiolate.34 While keeping the dicarbonyl group, acryloyl 2-pyrrolidinone led to a 95% yield and a 83:17 endo/exo selectivity (3l). Without the second carbonyl group, the yield dropped to 25% when using acryloyl amide (2m), which revealed the importance of the oxazolidin-2-one or pyridine group to obtain a good conversion. Finally, cyclohexa-1,3-diene was selected to react with 3-acryloyl-1,4-oxazolidin-2-one, but this diene led to a slower reaction rate. A 20% yield and 77:23 endo/exo ratio were obtained for 4a.
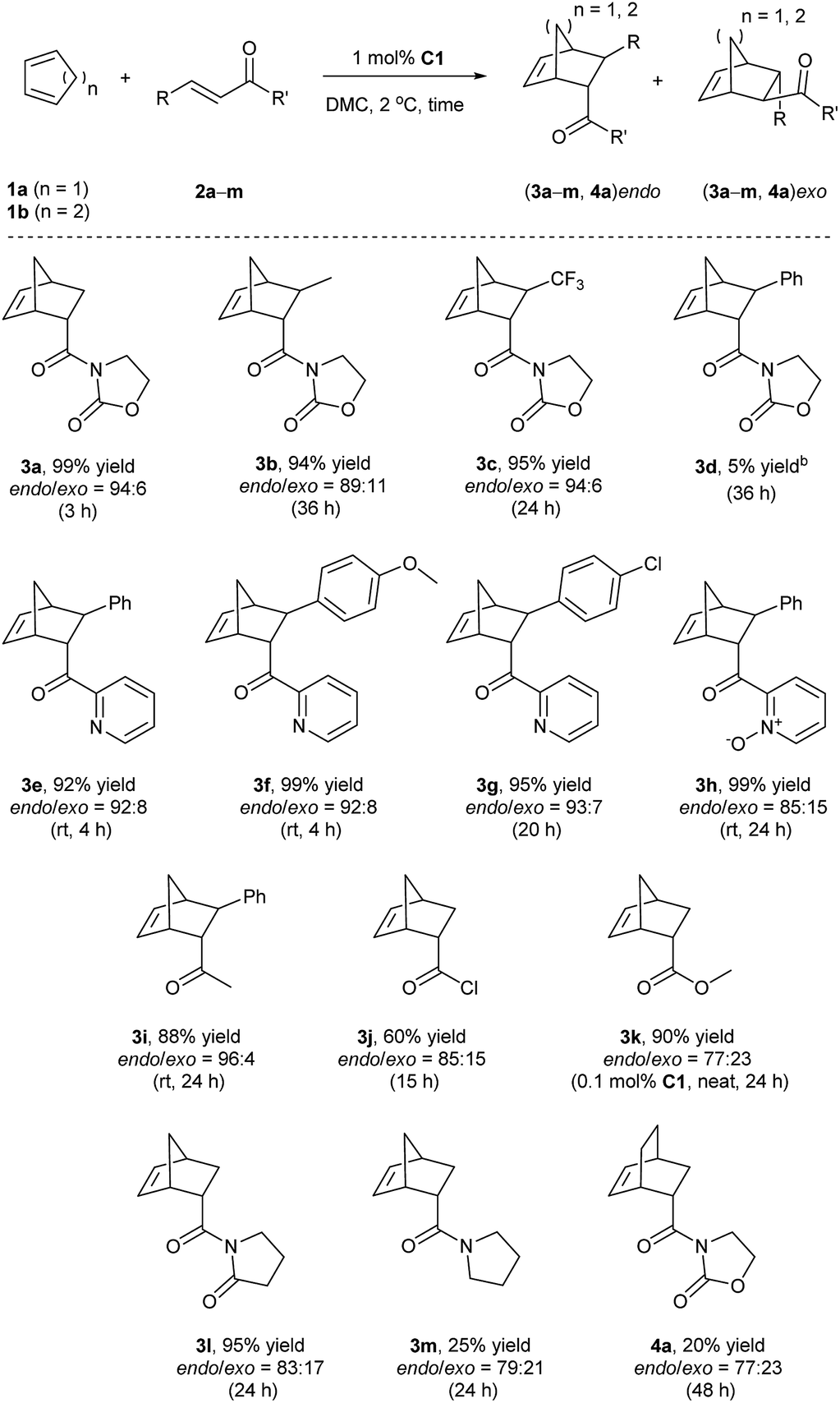 | ||
| Scheme 2 Reaction scope between cyclic dienes and various dienophiles.a a Conditions: 1a or 1b (1 mmol), 2a–2m (0.5 mmol), C1 (1 mol%), DMC (1 mL); isolated yields unless stated otherwise; endo/exo ratio was determined by 1H NMR. b Yield was calculated by 1H NMR. | ||
In order to shed light on the nature of the catalyst, the interaction between xanthinium and Fe(OTf)2 in the catalyst was studied by UV-vis, FTIR, 1H, 13C, and 19F NMR, and HRMS techniques (see ESI† for more details). As shown in Fig. 1, after mixing xanthinium and Fe(OTf)2 together, the 19F NMR signal of OTf− shifted from −56.9 ppm to −73.4 ppm, which is comparable to the chemical shift of unbound OTf− in Fe2+ complexes (−69.7 ppm,35 −78.96 ppm,36 and −79.59 ppm37). Since the OTf− signal shifted to high field (i.e. became more electron-enriched), the counterpart (Fe2+) was considered more electron-deficient, thus more Lewis acidic for the activation of dienophiles. The increase of Lewis acidity of Fe(II) in C1 thus led to higher yield (Table 1, entry 6 vs. entry 11). On the other hand, the NTf2− is a non-coordinating anion, so the 19F signal of NTf2− (−80.0 ppm) did not considerably change. According to the changes observed by FTIR, 19F NMR, and HRMS, the interaction between xanthinium and Fe(OTf)2 has been highlighted and provides evidence for anion metathesis.37 DFT calculations (B3LYP/6-31G/LANL2DZ level in gas phase) were run to shed light on the changes of Gibbs free energy and Mulliken charges of Fe(II) after the ion exchanges. According to the calculations, the anion exchange in C1 is favored (ΔG = −22.2 kcal mol−1), which is supported by the 19F NMR change and HRMS, allowing a qualitative evaluation of C1–C4 (see ESI† for more details).
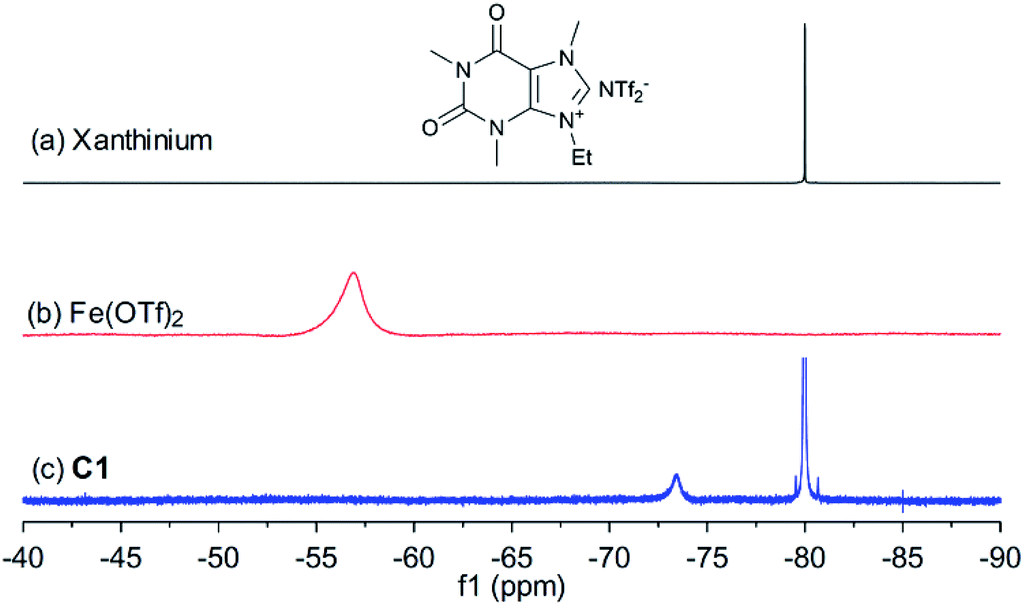 | ||
| Fig. 1 19F NMR spectrum of (a) xanthinium, (b) Fe(OTf)2 and (c) C1 (376 MHz, (CD3)2CO). | ||
Conclusions
In conclusion, a recyclable ionic salt/iron triflate catalyst was prepared from a xanthinium salt and iron(II) triflate. This green catalytic system using a caffeine-derived xanthinium–Fe(OTf)2 complex for the Diels–Alder reaction run in DMC was developed for a large scope of substrates. The xanthinium salt as a solid provides a new way of catalyst immobilization in comparison to ionic liquids. Several green solvents were examined and the recycling of the catalyst was demonstrated for several runs. The use of a caffeine derivative, an iron salt, and dimethyl carbonate represents a major advancement from a green chemistry point of view. Work is in progress on further applications of ionic salt catalysts and will be reported in due course.Experimental
General information
All materials are commercially available and were used as received without further purification. Thin-layer chromatography (TLC) was performed on commercial silica gel plates (250 μm, Silicycle F254) and compounds were visualized using UV light or KMnO4. The compounds were purified on silica gel column (200–300 mesh) unless stated otherwise. IR spectra were measured on a Bomem Michelson 100 Series FTIR spectrometer. 1H, 13C, and 19F NMR spectra were recorded on a Bruker AC 300 MHz, Varian Inova 400 MHz or Agilent Technologies DD2 500 MHz spectrometers. Chemical shifts are given in ppm and residual solvent peaks were used as reference. The coupling constants were reported in hertz. High-resolution mass spectra (HRMS) were recorded on a LC/MS-TOF Agilent 6210 mass spectrometer (electrospray ionization). Xanthiniums were synthesized according to published procedures.19,30a,38Preparation of catalysts C1–C4
The 1,3,7-trimethyl-9-ethylxanthinium bis(trifluoromethanesulfonyl)amide (1.26 g, 2.5 mmol) and anhydrous Fe(OTf)2 (89 mg, 0.25 mmol) were added to a 25 mL oven-dried flask. Acetone (7 mL) was then added into the flask to dissolve the salts, then heptane (15 mL) was added. A brown solution formed at the bottom of the flask. Then, the solvent was slowly evaporated at reduced pressure, and the flask was put under high vacuum overnight. Catalyst C1 was obtained in quantitative yield. Catalysts C2–C4 were prepared according to the same procedure. Characterization data for each catalyst is shown below.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 4.87 (q, J = 7.2 Hz, 2H, –CH2CH3), 4.26 (s, 3H, –CH3), 3.91 (s, 3H, –CH3), 3.35 (s, 3H, –CH3), 1.72 (t, J = 7.2 Hz, 3H, –CH2CH3); 1H NMR (400 MHz, (CD3)2SO) δ 9.33 (s, 1H), 4.53 (q, J = 7.2 Hz, 2H), 4.03 (s, 3H), 3.69 (s, 3H), 3.25 (s, 3H), 1.48 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, (CD3)2SO) δ 153.75 (C–CO–N), 150.77 (N–CO–N), 139.28 (Ar), 139.14 (N–CHN), 119.87 (q, JCF = 321.7 Hz, NTf2−), 108.30 (Ar), 45.43 (–CH2CH3), 36.10 (–CH3), 32.02 (–CH3), 28.78 (–CH3), 15.54 (–CH2CH3) (signal of OTf− was not observed). 19F NMR (282 MHz, (CD3)2CO) δ −73.40 (s, OTf−), −79.97 (s, NTf2−); 19F NMR (376 MHz, (CD3)2SO) δ −77.84 (s, OTf−), −78.78 (s, NTf2−). IR (ZnSe): 3429, 3164, 3100, 2960, 1721, 1670, 1580, 1545, 1450, 1347, 1321, 1176, 1048, 853, 739 cm−1.O–N), 150.59 (N–CO–N), 140.01 (Ar), 139.68 (N–CHN), 108.16 (Ar), 37.28 (–CH3), 36.04 (–CH3), 31.77 (–CH3), 28.82 (–CH3) (signal of OTf− was not observed). 19F NMR (376 MHz, (CD3)2CO) δ −79.06 (s, OTf−). IR (ZnSe): 3489, 3422, 3095, 3047, 1716, 1670, 1581, 1543, 1459, 1031, 1283, 1258, 1167, 1029, 1001, 876, 790, 776, 736 cm−1.
N), 4.87 (q, J = 7.2 Hz, 2H, –CH2CH3), 4.26 (s, 3H, –CH3), 3.91 (s, 3H, –CH3), 3.35 (s, 3H, –CH3), 1.72 (t, J = 7.2 Hz, 3H, –CH2CH3); 1H NMR (400 MHz, (CD3)2SO) δ 9.33 (s, 1H), 4.53 (q, J = 7.2 Hz, 2H), 4.03 (s, 3H), 3.69 (s, 3H), 3.25 (s, 3H), 1.48 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, (CD3)2SO) δ 153.75 (C–CO–N), 150.77 (N–CO–N), 139.28 (Ar), 139.14 (N–CHN), 119.87 (q, JCF = 321.7 Hz, NTf2−), 108.30 (Ar), 45.43 (–CH2CH3), 36.10 (–CH3), 32.02 (–CH3), 28.78 (–CH3), 15.54 (–CH2CH3) (signal of OTf− was not observed). 19F NMR (282 MHz, (CD3)2CO) δ −73.40 (s, OTf−), −79.97 (s, NTf2−); 19F NMR (376 MHz, (CD3)2SO) δ −77.84 (s, OTf−), −78.78 (s, NTf2−). IR (ZnSe): 3429, 3164, 3100, 2960, 1721, 1670, 1580, 1545, 1450, 1347, 1321, 1176, 1048, 853, 739 cm−1.O–N), 150.59 (N–CO–N), 140.01 (Ar), 139.68 (N–CHN), 108.16 (Ar), 37.28 (–CH3), 36.04 (–CH3), 31.77 (–CH3), 28.82 (–CH3) (signal of OTf− was not observed). 19F NMR (376 MHz, (CD3)2CO) δ −79.06 (s, OTf−). IR (ZnSe): 3489, 3422, 3095, 3047, 1716, 1670, 1581, 1543, 1459, 1031, 1283, 1258, 1167, 1029, 1001, 876, 790, 776, 736 cm−1.General procedure for Diels–Alder reaction in DMC
To an oven-dried vial, were added catalyst C1 (27.5 mg, 0.005 mmol) and dienophile 3-acryloyl-1,3-oxazolin-2-one (70.5 mg, 0.5 mmol), then DMC (1 mL) was injected. The solution was stirred for 10 minutes. After that, the vial was placed in an ice water bath at 2 °C. Then the pre-distilled cyclopentadiene (1 mmol, 66 mg) was injected into the vial. The solution was stirred for 3 hours. After the reaction was complete, heptane (5 mL) was added into the solution to precipitate the catalyst (other solvents, such as hexane, petroleum ether and Et2O, could also be used for recycling of catalyst). The precipitate of catalyst was recycled (27.5 mg) by filtration. The filtrate was concentrated, and the crude product was purified by silica gel column chromatography using hexanes/EtOAc as the eluent to obtain the product (103 mg, 99% yield). For other substrates, 0.25 mmol of dienophiles and 0.5 mmol of diene were used with 0.0025 mmol (14 mg) of the catalyst, and the crude products were purified by silica gel column chromatography using hexanes/EtOAc as the eluent unless stated otherwise.General procedure for Diels–Alder reaction under neat conditions
For liquid dienophiles, the reactions were carried out in neat conditions. Typically, to an oven-dried vial, was added catalyst C1 (27.5 mg, 0.005 mmol). The reaction vial was then cooled at 2–3 °C using a cryostat. Methyl acrylate (430 mg, 5 mmol) and distilled cyclopentadiene (396 mg, 6 mmol, 1.2 equiv.) were then injected into the vial. The reaction mixture was stirred for 1 day. Then, heptane (5 mL) was added into the vial to precipitate the catalyst, and the crude product was obtained through filtration of cotton-plugged pipet. The filtrate was evaporated and the product was dried under high vacuum for 5–10 minutes. Pure product was obtained without further purification in 90% yield (677 mg). The precipitated catalyst was dissolved by acetone, and filtered through cotton-plugged pipet to be recycled after evaporation of acetone. All the mass of the catalyst (27.5 mg) was recycled. Characterization data for each product is shown below.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant RGPIN-2017-04272, the FRQNT Centre in Green Chemistry and Catalysis (CGCC) Strategic Cluster FRQNT-2020-RS4-265155-CCVC, and Université Laval. D. M. and D. L. thank China Scholarship Council (CSC) for doctoral scholarships. The authors thank Mao Li (Département de chimie, Université Laval) for the synthesis of the dienophiles and Nour Tanbouza (Département de chimie, Université Laval) for proofreading of the manuscript.Notes and references
- K. Furuta, S. Shimizu, Y. Miwa and H. Yamamoto, J. Org. Chem., 1989, 54, 1481–1483 CrossRef CAS.
- G. Desimoni, G. Faita, M. Mella, F. Piccinini and M. Toscanini, Eur. J. Org. Chem., 2007, 1529–1534 CrossRef CAS.
- D. A. Evans, K. T. Chapman and J. Bisaha, J. Am. Chem. Soc., 1988, 110, 1238–1256 CrossRef CAS.
- (a) E. J. Corey, N. Imai and H. Y. Zhang, J. Am. Chem. Soc., 1991, 113, 728–729 CrossRef CAS; (b) K. Narasaka, N. Iwasawa, M. Inoue, T. Yamada, M. Nakashima and J. Sugimori, J. Am. Chem. Soc., 1989, 111, 5340–5345 CrossRef CAS.
- T. Ichiyanagi, M. Shimizu and T. Fujisawa, J. Org. Chem., 1997, 62, 7937–7941 CrossRef CAS.
- (a) S. Barroso, G. Blay and J. R. Pedro, Org. Lett., 2007, 9, 1983–1986 CrossRef CAS PubMed; (b) D. Rechavi and M. Lemaire, J. Mol. Catal. A: Chem., 2002, 182–183, 239–247 CrossRef CAS.
- D. Sarma and A. Kumar, Appl. Catal., A, 2008, 335, 1–6 CrossRef CAS.
- S. D. Bull, M. G. Davidson, A. L. Johnson, M. F. Mahon and D. E. J. E. Robinson, Chem.–Asian J., 2010, 5, 612–620 CrossRef CAS.
- H. Suga, A. Kakehi and M. Mitsuda, Bull. Chem. Soc. Jpn., 2004, 77, 561–568 CrossRef CAS.
- K. Hiroi and K. Watanabe, Tetrahedron: Asymmetry, 2002, 13, 1841–1843 CrossRef CAS.
- M. P. Sibi, S. Manyem and H. Palencia, J. Am. Chem. Soc., 2006, 128, 13660–13661 CrossRef CAS.
- K. Aikawa, R. Irie and T. Katsuki, Tetrahedron, 2001, 57, 845–851 CrossRef CAS.
- (a) G. Chollet, F. Rodriguez and E. Schulz, Org. Lett., 2006, 8, 539–542 CrossRef CAS PubMed; (b) G. Chollet, M.-G. Guillerez and E. Schulz, Chem.–Eur. J., 2007, 13, 992–1000 CrossRef CAS; (c) G. Chollet, D. Didier and E. Schulz, Catal. Commun., 2010, 11, 351–355 CrossRef CAS.
- (a) S. Doherty, P. Goodrich, C. Hardacre, J. G. Knight, M. T. Nguyen, V. I. Pârvulescu and C. Paun, Adv. Synth. Catal., 2007, 349, 951–963 CrossRef CAS; (b) C.-E. Yeom, H. W. Kim, Y. J. Shin and B. M. Kim, Tetrahedron Lett., 2007, 48, 9035–9039 CrossRef CAS.
- I. Meracz and T. Oh, Tetrahedron Lett., 2003, 44, 6465–6468 CrossRef CAS.
- C. Baudequin, J. Baudoux, J. Levillain, D. Cahard, A.-C. Gaumont and J.-C. Plaquevent, Tetrahedron: Asymmetry, 2003, 14, 3081–3093 CrossRef CAS.
- (a) C. Chiappe and M. Malvaldi, Phys. Chem. Chem. Phys., 2010, 12, 11191–11196 RSC; (b) A. G. Zazybin, K. Rafikova, V. Yu, D. Zolotareva, V. M. Dembitsky and T. Sasaki, Russ. Chem. Rev., 2017, 86, 1254–1270 CrossRef CAS.
- K. Bica and P. Gaertner, Org. Lett., 2006, 8, 733–735 CrossRef CAS.
- R. M. A. Pinto, J. A. R. Salvador and C. Le Roux, Catal. Commun., 2008, 9, 465–469 CrossRef CAS.
- (a) N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC; (b) G. W. Meindersma, S. A. F. Onink and B. A. de Haan, Green Separation Processes with Ionic Liquids, in Handbook of Green Chemistry, ed. P. T. Anastas, Wiley-VCH, Weinheim, 2010, vol. 6, pp. 137–190 Search PubMed; (c) H. Li, P. S. Bhadury, B. Song and S. Yang, RSC Adv., 2012, 2, 12525–12551 RSC; (d) Y. Zhang, B. R. Bakshi and E. S. Demessie, Environ. Sci. Technol., 2008, 42, 1724–1730 CrossRef CAS; (e) M. Cvjetko Bubalo, K. Radošević, I. Radojčić Redovniković, J. Halambek and V. Gaurina Srček, Ecotoxicol. Environ. Saf., 2014, 99, 1–12 CrossRef CAS; (f) P. L. Amado Alviz and A. J. Alvarez, J. Cleaner Prod., 2017, 168, 1614–1624 CrossRef CAS.
- C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC.
- (a) F. Aricò and P. Tundo, Russ. Chem. Rev., 2010, 79, 479–489 CrossRef; (b) H.-Z. Tan, Z.-Q. Wang, Z.-N. Xu, J. Sun, Y.-P. Xu, Q.-S. Chen, Y. Chen and G.-C. Guo, Catal. Today, 2018, 316, 2–12 CrossRef CAS; (c) G. Fiorani, A. Perosa and M. Selva, Green Chem., 2018, 20, 288–322 RSC.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef.
- (a) T. Ollevier, V. Desyroy, B. Debailleul and S. Vaur, Eur. J. Org. Chem., 2005, 4971–4973 CrossRef CAS; (b) T. Ollevier, Catal. Sci. Technol., 2016, 6, 41–48 RSC; (c) J. Kraïem and T. Ollevier, Green Chem., 2017, 19, 1263–1267 RSC; (d) H. Keipour and T. Ollevier, Org. Lett., 2017, 19, 5736–5739 CrossRef CAS; (e) M. Li, V. Carreras, A. Jalba and T. Ollevier, Org. Lett., 2018, 20, 995–998 CrossRef CAS.
- (a) A. Fürstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef; (b) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS.
- A. C. Mayer, A.-F. Salit and C. Bolm, Chem. Commun., 2008, 5975–5977 RSC.
- (a) S. Kanemasa, Y. Oderaotoshi, H. Yamamoto, J. Tanaka, E. Wada and D. P. Curran, J. Org. Chem., 1997, 62, 6454–6455 CrossRef CAS; (b) S. Kanemasa, Y. Oderaotoshi, S.-i. Sakaguchi, H. Yamamoto, J. Tanaka, E. Wada and D. P. Curran, J. Am. Chem. Soc., 1998, 120, 3074–3088 CrossRef CAS; (c) E. P. Kündig, B. Bourdin and G. Bernardinelli, Angew. Chem., Int. Ed. Engl., 1994, 33, 1856–1858 CrossRef; (d) M. E. Bruin and E. Peter Kündig, Chem. Commun., 1998, 2635–2636 RSC.
- (a) J. A. K. Howard, G. Ilyashenko, H. A. Sparkes and A. Whiting, Dalton Trans., 2007, 2108–2111, 10.1039/B704728B; (b) K. Fujiwara, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2012, 134, 5512–5515 CrossRef CAS; (c) J. W. Herndon, J. Org. Chem., 1986, 51, 2853–2855 CrossRef CAS; (d) S. Nakanishi, K. Kumeta, Y. Sawai and T. Takata, J. Organomet. Chem., 1996, 515, 99–101 CrossRef CAS; (e) B. Wang, B. Mu, D. Chen, S. Xu and X. Zhou, Organometallics, 2004, 23, 6225–6230 CrossRef CAS; (f) Z. Gültekin, Clay Miner., 2004, 39, 345–348 CrossRef; (g) S. P. Chavan and A. K. Sharma, Synlett, 2001, 667–669 CrossRef CAS; (h) K. Wei, S. Wang, Z. Liu, Y. Du, X. Shi, T. Qi and S. Ji, Tetrahedron Lett., 2013, 54, 2264–2266 CrossRef CAS; (i) M. C. Donatoni, G. A. B. Junior, K. T. de Oliveira, R. A. Ando, T. J. Brocksom and A. A. Dos Santos, Tetrahedron, 2014, 70, 3231–3238 CrossRef CAS; (j) N. Basavegowda, K. Mishra, Y. R. Lee and Y.-G. Joh, Bull. Korean Chem. Soc., 2016, 37, 142–147 CrossRef CAS.
- (a) J. W. Daly, J. Med. Chem., 1982, 25, 197–207 CrossRef CAS PubMed; (b) G. A. Spiller, in Basic Metabolism and Physiological Effects of the Methylxanthines in Caffeine, ed. G. A. Spiller, CRC press LLC, Boca Raton, Florida, 1st edn, 1997, ch. 10, pp. 199–255 Search PubMed; (c) B. F. Harland, Nutrition, 2000, 16, 522–526 CrossRef CAS; (d) M. A. Heckman, J. Weil and E. G. De Mejia, J. Food Sci., 2010, 75, 77–87 CrossRef; (e) Z. Baalbaki, E. Torfs, V. Yargeau and P. A. Vanrolleghem, Sci. Total Environ., 2017, 601–602, 874–885 CrossRef CAS PubMed; (f) E. Topp, J. G. Hendel, Z. Lu and R. Chapman, Can. J. Soil Sci., 2006, 86, 533–544 CrossRef CAS.
- (a) F.-T. Luo and H.-K. Lo, J. Organomet. Chem., 2011, 696, 1262–1265 CrossRef CAS; (b) B. Bertrand, L. Stefan, M. Pirrotta, D. Monchaud, E. Bodio, P. Richard, P. Le Gendre, E. Warmerdam, M. H. de Jager, G. M. M. Groothuis, M. Picquet and A. Casini, Inorg. Chem., 2014, 53, 2296–2303 CrossRef CAS; (c) A. Kascatan-Nebioglu, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Coord. Chem. Rev., 2007, 251, 884–895 CrossRef CAS; (d) A. Kascatan-Nebioglu, A. Melaiye, K. Hindi, S. Durmus, M. J. Panzner, L. A. Hogue, R. J. Mallett, C. E. Hovis, M. Coughenour, S. D. Crosby, A. Milsted, D. L. Ely, C. A. Tessier, C. L. Cannon and W. J. Youngs, J. Med. Chem., 2006, 49, 6811–6818 CrossRef CAS; (e) H. A. Mohamed, B. R. M. Lake, T. Laing, R. M. Phillips and C. E. Willans, Dalton Trans., 2015, 44, 7563–7569 RSC; (f) J. J. Hu, S.-Q. Bai, H. H. Yeh, D. J. Young, Y. Chi and T. S. A. Hor, Dalton Trans., 2011, 40, 4402–4406 RSC; (g) J.-J. Zhang, C.-M. Che and I. Ott, J. Organomet. Chem., 2015, 782, 37–41 CrossRef CAS.
- (a) C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS; (b) D. S. MacMillan, J. Murray, H. F. Sneddon, C. Jamieson and A. J. B. Watson, Green Chem., 2013, 15, 596–600 RSC.
- The loss of catalyst during its recovery process could have been the cause..
- Other previously listed solvents, e.g. NMP, EtOAc, and acetone, did not lead to improved yields..
- K. Erfurt, I. Wandzik, K. Walczak, K. Matuszek and A. Chrobok, Green Chem., 2014, 16, 3508–3514 RSC.
- D. W. Blakesley, S. C. Payne and K. S. Hagen, Inorg. Chem., 2000, 39, 1979–1989 CrossRef CAS.
- A. B. Grommet, J. L. Bolliger, C. Browne and J. R. Nitschke, Angew. Chem., Int. Ed., 2015, 54, 15100–15104 CrossRef CAS PubMed.
- E. M. Matson, J. A. Bertke and A. R. Fout, Inorg. Chem., 2014, 53, 4450–4458 CrossRef CAS.
- A. Kascatan-Nebioglu, M. J. Panzner, J. C. Garrison, C. A. Tessier and W. J. Youngs, Organometallics, 2004, 23, 1928–1931 CrossRef CAS.
- Y. Li, C. Wang, J. Hao, M. Cheng, G. Jia and C. Li, Chem. Commun., 2015, 51, 13174–13177 RSC.
- R. S. Singh, S. Adachi, F. Tanaka, T. Yamauchi, C. Inui and T. Harada, J. Org. Chem., 2008, 73, 212–218 CrossRef CAS.
- (a) O. A. Mukhina, N. N. Bhuvan Kumar, T. M. Arisco, R. A. Valiulin, G. A. Metzel and A. G. Kutateladze, Angew. Chem., Int. Ed., 2011, 50, 9423–9428 CrossRef CAS PubMed; (b) J.-L. Ripoll and M.-C. Lasne, Tetrahedron Lett., 1978, 19, 5201–5202 CrossRef.
- K. Matuszek, S. Coffie, A. Chrobok and M. Swadźba-Kwaśny, Catal. Sci. Technol., 2017, 7, 1045–1049 RSC.
- M. P. Sibi, J. Chen and L. Stanley, Synlett, 2007, 298–302 CrossRef CAS.
- D. A. Evans, D. M. Barnes, J. S. Johnson, T. Lectka, P. von Matt, S. J. Miller, J. A. Murry, R. D. Norcross, E. A. Shaughnessy and K. R. Campos, J. Am. Chem. Soc., 1999, 121, 7582–7594 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra. See DOI: 10.1039/c9ra04098f |
| This journal is © The Royal Society of Chemistry 2019 |