
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiocatalytically active microgels by precipitation polymerization of N-isopropyl acrylamide in the presence of an enzyme†
Stefan Reinicke *ad,
Thilo Fischerad,
Julia Bramskib,
Jörg Pietruszkabc and
Alexander Bökerad
*ad,
Thilo Fischerad,
Julia Bramskib,
Jörg Pietruszkabc and
Alexander Bökerad
aFraunhofer Institute for Applied Polymer Research (IAP), Geiselbergstraße 69, Potsdam-Golm, 14476, Germany. E-mail: Stefan.Reinicke@iap.fraunhofer.de
bInstitut of Bioorganic Chemistry, Heinrich Heine University of Düsseldorf at Forschungszentrum Jülich, Stetternicher Forst, D-52426 Jülich, Germany
cIBG-1: Biotechnology, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany
dChair of Polymer Materials and Polymer Technologies, University of Potsdam, Potsdam-Golm, 14476, Germany
First published on 9th September 2019
Abstract
We present a novel protocol for the synthesis of enzymatically active microgels. The protocol is based on the precipitation polymerization of N-isopropylacrylamide (NIPAm) in the presence of an enzyme and a protein binding comonomer. A basic investigation on the influence of different reaction parameters such as monomer concentration and reaction temperature on the microgel size and size distribution is performed and immobilization yields are determined. Microgels exhibiting hydrodynamic diameters between 100 nm and 1 μm and narrow size distribution could be synthesized while about 31–44% of the enzyme present in the initial reaction mixture can be immobilized. Successful immobilization including a verification of enzymatic activity of the microgels is achieved for glucose oxidase (GOx) and 2-deoxy-D-ribose-5-phosphate aldolase (DERA). The thermoresponsive properties of the microgels are assessed and discussed in the light of activity evolution with temperature. The positive correlation of enzymatic activity with temperature for the GOx containing microgel originates from a direct interaction of the enzyme with the PNIPAm based polymer matrix whose magnitude is highly influenced by temperature.
Introduction
In many cases, in which enzymes are industrially used, it is necessary to immobilize the enzyme in order to provide a sufficient stability or to establish the opportunity to remove the biocatalyst from the reaction medium after the process without a lot of effort. The latter aspect is especially important as purification steps cause up to 80% of the overall costs of a biotechnological process.1 Often, nano scale materials with high specific surface area are used in order to provide a high enzymatic activity as well as a good dispersability while suppressing unwanted side effects like diffusion limitation.A vast number of respective immobilization protocols already exist,2,3 mostly relying on inorganic particles based on silica or magnetic material or on organic material like polymer lattices and –hydrogels. Normally, a two-step procedure is followed where the carrier material is produced first with the actual immobilization step taking place only afterwards. It is also possible to embed an enzyme in situ during the synthesis of the carrier material,3,4 yet this requires the compatibility of the enzyme with the reaction conditions and/or the introduction of reactive groups to the enzymes or the use of suitable stabilizing compounds. Coupling agents might be needed in order to ensure a stable binding of the enzyme to the carrier thereby avoiding enzyme leakage. On top of that, the synthesis of nano scale carrier systems typically requires the use of certain additives such as surfactants for stabilization and/or lipophilic substances to establish a two-phase system. If additional features such as temperature responsivity for activity control5,6 are wanted, the choice of involved materials is limited implementing an additional degree of complexity.
The precipitation polymerization of N-isopropylacrylamide (NIPAm) in water at temperatures above the lower critical solution temperature (LCST) of the resulting polymer promises a way to directly embed an enzyme into a microgel during the generation of the latter, while keeping the overall synthetic effort low. The incorporation of bioactive molecules in a polymeric microgel by precipitation polymerization of NIPAm or other suitable monomers has been shown already,7 however, either only non-enzymatic material has been immobilized or certain disadvantages had to be faced such as a long reaction time or the necessity to once again establish a two-phasic reaction medium. No broader applicability was demonstrated in any of the reported cases and often remaining side products had to be removed.
In this contribution we use a new synthetic procedure based on a modified protocol of the precipitation polymerization of NIPAm to demonstrate a fast and easy to perform immobilization of enzymes in well-defined microgel structures with high immobilization yield (Scheme 1). The procedure involves a number of advantages including a simple mixing of the involved components without complex preconditioning steps, an ultrashort reaction time, as well as the avoidance of expensive co-components and unwanted side products. By immobilizing enzymes of two different classes, 2-deoxy-D-ribose 5-phosphate aldolase (DERA) and glucose oxidase (GOx), we furthermore demonstrate the broad applicability of the protocol and finally we assess the temperature responsivity of the resulting microgels.
 | ||
| Scheme 1 Basic synthetic principle for enzyme containing microgels based on the precipitation polymerization of NIPAm (left) and idealized sketch of the internal microgel structure (right). | ||
Experimental
Materials
N-Isopropylacrylamide (NIPAm) (Sigma-Aldrich, 99%) was recrystallized from a mixture of n-hexane/toluene (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v). N-2-thiolactoneacrylamide (TlaAm) was synthesized according to a procedure described in a previous publication.8 N,N′-methylenebisacrylamide (Bis) (Sigma-Aldrich, 99%), N,N,N′,N′-tetramethylethylenediamine (TEMED) (Carl Roth, ≥99%), potassium persulfate (Sigma-Aldrich, ≥99.0%), nicotinamide adenine dinucleotide disodium salt (NADH) (Roche, ∼100%, grade I), 2-deoxy-D-ribose-5-phosphate sodium salt (DRP) (Sigma, ≥95%), glucose (Sigma-Aldrich, ≥99.5%), dimethyl sulfoxide (DMSO) (Carl Roth, ≥99.8%, <100 ppm H2O), deuterated chloroform (Carl Roth, 99.8% D), Atto-665-maleimide (Atto-Tec), bovine serum albumin (BSA) (Sigma-Aldrich, >98%, lyophilized powder, heat shock fraction), peroxidase from horseradish (HRP) (Sigma-Aldrich), (lyophilized powder, 150 U mg−1), glucose oxidase from Aspergillus niger (Carl Roth, lyophilized powder, 280 U mg−1) and α-glycerophosphate dehydrogenase-triosephosphate isomerase from rabbit muscle (Sigma-Aldrich), Type III, ammonium sulfate suspension, TPI 750–2000 units per mg protein, GDH 75–200 units per mg protein (biuret) were used as received.
:1 v/v). N-2-thiolactoneacrylamide (TlaAm) was synthesized according to a procedure described in a previous publication.8 N,N′-methylenebisacrylamide (Bis) (Sigma-Aldrich, 99%), N,N,N′,N′-tetramethylethylenediamine (TEMED) (Carl Roth, ≥99%), potassium persulfate (Sigma-Aldrich, ≥99.0%), nicotinamide adenine dinucleotide disodium salt (NADH) (Roche, ∼100%, grade I), 2-deoxy-D-ribose-5-phosphate sodium salt (DRP) (Sigma, ≥95%), glucose (Sigma-Aldrich, ≥99.5%), dimethyl sulfoxide (DMSO) (Carl Roth, ≥99.8%, <100 ppm H2O), deuterated chloroform (Carl Roth, 99.8% D), Atto-665-maleimide (Atto-Tec), bovine serum albumin (BSA) (Sigma-Aldrich, >98%, lyophilized powder, heat shock fraction), peroxidase from horseradish (HRP) (Sigma-Aldrich), (lyophilized powder, 150 U mg−1), glucose oxidase from Aspergillus niger (Carl Roth, lyophilized powder, 280 U mg−1) and α-glycerophosphate dehydrogenase-triosephosphate isomerase from rabbit muscle (Sigma-Aldrich), Type III, ammonium sulfate suspension, TPI 750–2000 units per mg protein, GDH 75–200 units per mg protein (biuret) were used as received.
Production of 2-deoxy-D-ribose-5-phosphate aldolase from Escherichia coli (DERAEC)
For preparation of DERA the protein was expressed in E. coli BL21(DE3) strain in TB-medium and purified by NiNTA as well as PD10 columns according to Dick et al.9 Before lyophilization the protein was dissolved in KPi-buffer at 20 mM and pH 7 and set to a concentration of ∼5 mg mL−1.Microgel synthesis
In a standard recipe the enzyme of choice (1/10 of the amount of BSA, that is added later on) was dissolved in a sodium borate buffer (4.5 mL, pH 9) followed by addition of TlaAm (30 mg) and stirring of the mixture for 24 h at room temperature. After rebuffering to Milli-Q grade water, NIPAm (360 mg), Bis (15 mg), BSA (20 mg), TEMED (20 μL) and again TlaAm (30 mg) were dissolved in the mixture which was then transferred to a round bottom flask equipped with a rubber septum. After degassing for 15 min by bubbling N2 through the mixture, the polymerization was initiated by adding 0.5 mL of degassed K2S2O8 solution (5 mg mL−1) to the reaction mixture. After stirring for 10 min at 50 °C the mixture was diluted to twice the original volume with preheated, non-degassed Milli-Q and pressurized air bubbled through the solution for 5 min. Finally the mixture was further diluted to the 5-fold volume using non-heated Milli-Q water and rebuffered to PBS (20 mmol, pH 7.4) by repeated centrifugation/redispersion. Since DERA is a bit more delicate than the other enzymes tested (see ESI†), the procedure was slightly adjusted. The reaction was carried out at only 35 °C, the amount of TEMED was halved to 10 μL and the reaction volume was doubled.GOx activity assay
For the GOx activity assay 50 μL HRP (15 U mL−1 in PBS), 50 μL ABTS (0.25 mg mL−1 in PBS), 50 μL microgel dispersion (5-fold diluted) or GOx (0.14 U mL−1 in PBS) and at last, to initiate the assay, 50 μL glucose (0.9 mg mL−1 in PBS) were added to a 96 well transparent microplate. The hydrogen peroxide formed by the enzymatic reaction of glucose oxidase and glucose oxidizes the dye ABTS catalyzed by HRP (Scheme S1, ESI†) resulting in a color change. The absorbance was measured at 405 nm by photometric spectroscopy using a microplate reader (Infinite M200 Pro, Tecan). For the measurements at 40 °C, the samples were incubated first at this temperature within the microplate before the glucose was added and the measurement started. The raw data was corrected for the contribution of the turbidity of the respective assay solution making use of the reference measurements in the presence and absence of microgel. In both cases, regions in the absorption/time plots were identified in which the contribution from the scattering appeared more or less constant. For these regions, an average absorption over time was calculated, respectively. The difference between both values was then subtracted from each measurement curve involving a microgel, assuming the apparent absorption being constructed additively from actual absorption and the turbidity.DERA activity assay
The DERA activity assay is based on the retro aldol type cleavage of DRP, which is followed by the oxidation of NADH using a coupled assay that involves the isomerization and subsequent reduction of the retro aldol product by the auxiliary enzymes TPI and GDH9,10 (Scheme S1, ESI†). In the standard setup, the reaction mixture was composed of PBS buffer, 1.5 mM DRP, 0.15 mM NADH, 1 U of TPI, 10 U of GDH, and 10 μL of a 0.2 mg mL−1 DERA solution or 20 μL of the non-diluted microgel dispersion. The volume was set to 1 mL in each case. The change in absorbance of NADH was assayed in 24-well plates at 340 nm using a microplate reader (Infinite M200 Pro, Tecan). The raw data was corrected for the contribution of the turbidity of the respective assay solution originating from the presence of microgel. First, regions of constant NADH concentration were identified for each measurement followed by calculating the average absorption value for each of these regions respectively. Assuming the detected apparent absorptions to be constructed additively by the actual absorption and the turbidity, the averaged values were subtracted from the measurement data in a way, that only the contribution from the actual absorption remained at the beginning of the measurement. Occasionally the measurement signal appeared to be unstable at the beginning of the measurement independent on the NADH concentration. In such a case, the recorded data points were not considered for evaluation.Protein labelling and quantification
4.9 mg of protein were dissolved in 2 to 2.5 mL PBS buffer. 11 μL of a solution of Atto-665 maleimide in dry DMSO (10 mg mL−1) were added and the reaction mixture was left for 3 hours being protected from light. After that, the labelled enzyme was purified by running the reaction mixture over a gel filtration column (Ø 2 cm, length 30 cm) packed with Sephadex G-25 using a Knauer Smartline system equipped with a S1050 pump, a S5050 manager unit and a 2550 UV detector (210 and 280 nm). At a flow rate of 5 mL min−1 4 increments à 500 μL could be passed through the column before the unbound dye was eluted.After synthesis of the microgel using the respective labeled protein, all microgel material was removed from the dispersion via centrifugation. The fluorescence intensity of the supernatant was recorded at 665 nm using a microplate reader (Infinite M200 Pro, Tecan). For calibration, solutions of labeled protein were exposed to the polymerization conditions except that the actual polymerization was not initiated.
Fluorescence microscopy
Fluorescence and light microscope pictures were taken with a DMi8 (Leica) at appropriate magnification (dry for lower values and oil objective for 100×). For image processing the LAS X software (Version 2.0.0.14332) from Leica was used. A 7.5 μL drop of liquid sample was placed in between two microscope cover slides and to prevent solvent evaporation, at least 100 μL were used inside an eight-well chamber system.Dynamic light scattering (DLS)
Temperature-dependent dynamic light scattering measurements were done on a Malvern MAL1083122 Zetasizer using disposal cuvettes (ZEN 0040). The instrument uses a 633 nm “red” laser, and the detector position is 173°. For the measurement, the microgel dispersions, as obtained from the synthesis, were diluted by a factor of 44 using PBS buffer. At each temperature, three measurements were performed and averaged, respectively, with an automatic adjustment of measurement duration. Temperature was changed in steps of 1 °C with an equilibration time of 120 s before measurement.Results and discussion
Microgel synthesis
The basic principle of the microgel synthesis is based on the well established precipitation polymerization of N-isopropylacrylamide (NIPAm) in water.11,12 Here, the reaction mixture, containing monomer, cross-linker and the initiator, mostly K2S2O8, is heated to a temperature above the cloud point of the resulting polymer. The growing chains immediately separate from solution and aggregate into colloidal particles that form the base of the final microgel. Colloidal stabilization of the particles is provided by the charged fragments of the initiator that cause electrostatic repulsion between the gel particles.11,13 If the temperature is high enough (>70 °C), internal transfer reactions may also cause crosslinking, meaning that no additional cross-linker is necessary then.14 Normally, the polymerization time is settled in the range of minutes to hours, as the radical chain ends are not accessible so well anymore as soon as the growing chains are collapsed into the colloidal particles. In aqueous solution, NIPAm normally polymerizes within seconds.As a starting point for the synthesis, we used the recipe described by Gao et al.14 who polymerized NIPAm at 70 °C in order to have the cross-linking effect without using additional cross-linker. However, 70 °C is too high for our purposes given the fact that we want to incorporate proteins in our microgels. Consequently we set the temperature never higher than 50 °C. Unfortunately, at this temperature, efficient internal cross-linking is not taking place anymore, meaning that we need to use N,N′-methylenebisacrylamide (Bis) as an additional cross-linker. On top of that, the set temperature is insufficient for the decomposition of the initiator. Thus, an additional catalyst, triggering this decomposition at low temperatures, is necessary. Here, we make use of TEMED, which is applied in biotechnology for the room temperature curing of polyacrylamide gels used for gel electrophoresis.
With this basic recipe at hand, a first polymerization was attempted, which, however, only led to ill-defined macro-sized aggregates. This failure was attributed to the fact, that, in contrast to the work by Gao et al., a high amount of radicals was generated in a very short time due to catalytic action of TEMED, which is also manifested in a very quick reaction time. The polymerization took place within seconds and not minutes, as described by Gao. As mentioned by Pich et al., the stabilization by ionic initiator residues has its limits.13 It cannot stabilize extremely large surface areas that occur when a high amount of very small particles is present. In our case, the high radical generation rate is causing the quick generation of a large number of growing chains and, upon the collapse of these chains, a large number of very small primary particles. Consequently, colloidal stabilization fails and the particles agglomerate into large macroparticles. Although Pich and Richtering have also used the combination K2S2O8/TEMED as initiating system, they still achieved sufficient colloidal stabilization relying solely on the initiator fragments.15 A closer look, however, reveals that they used TEMED with a concentration an order of magnitude lower compared to our recipe while the reaction took somewhat longer (5 to 10 min) to complete. Thus, number of particles and thus particle surface area apparently was still in a range in which stabilization by the initiator fragments was still working, which is not anymore the case for our recipe. The picture, however, changes completely when a protein is supposed to be incorporated into the microgels. In this case, the reaction mixture additionally contains a protein and a certain amount of N-2-thiolactone acrylamide (TlaAm) as protein binding comonomer. For the start, we used bovine serum albumin (BSA) as a model protein. With this recipe, we obtained well defined microgels (Fig. 1) without presence of any macroparticles, although for both BSA and TlaAm certain threshold amounts were necessary in order to exclusively obtain micrometer sized objects. Reaction still takes place within seconds, as evidenced by turbidity that sets in almost instantly after addition of K2S2O8. After the removal of the microgels from the dispersion by centrifugation after their synthesis, the supernatant has been examined for residual soluble material. It turns out, that roughly 30% of all solid material from the initial reaction mixture remains in the supernatant. NMR reveals that this material is mainly composed of PNIPAm and less than 10% of the respective monomer (see Fig. S1, ESI†). Aromatic signals also indicate the presence of proteinic material.
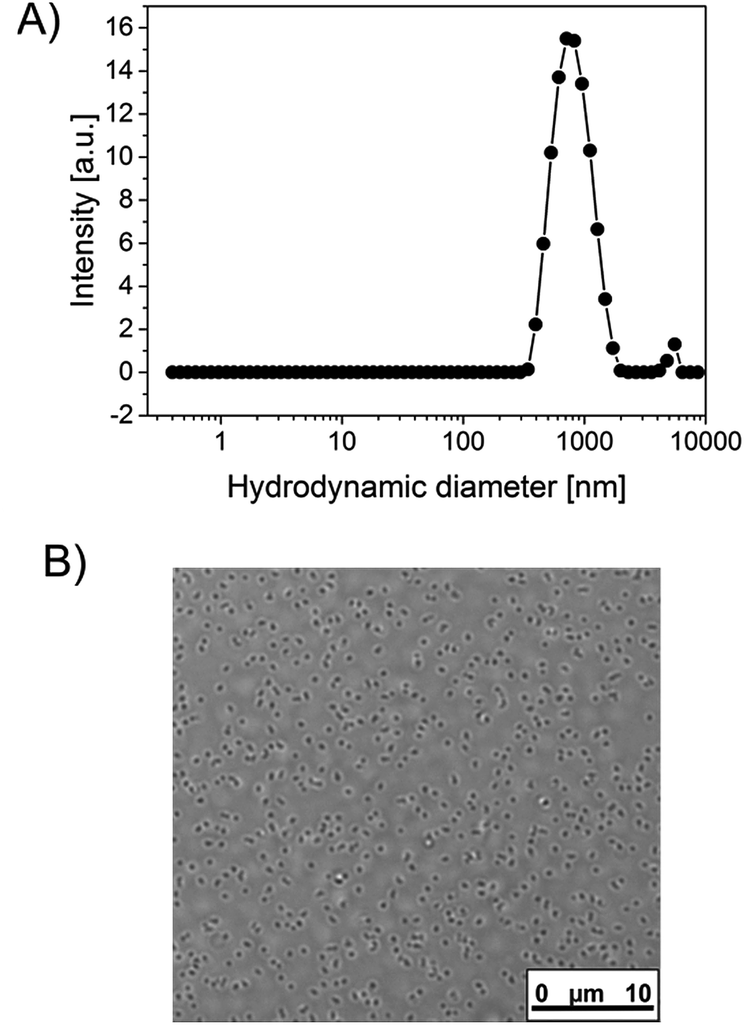 | ||
| Fig. 1 Size specifications of a BSA containing microgel synthesized according to the standard synthesis protocol (no extra enzyme embedded): (A) CONTIN plot derived from DLS, (B) optical microscope image. | ||
Applying labeled BSA for the synthesis and using photometric quantification (see Exp. part) (Fig. S3, ESI†), the amount of protein that remains in the soluble fraction is detected with 29% for the standard recipe, which coincides very well with the fraction of material that remained soluble. Additionally, SDS-PAGE reveals, that large portions of the BSA in the soluble fraction are polymer bound (Fig. S2, ESI†). Fluorescence microscopy of the microgels also clearly indicates that a substantial amount of BSA has been incorporated in the gel particles (Fig. 2). As most proteins, and especially BSA, show surface activity,16 we believe that it is mainly the BSA that provides the colloidal stabilization in our case. Interestingly, the TlaAm comonomer also has a positive impact on the stabilization of the microgels (Table 1). Lowering its concentration will also lead to the formation of macroparticles, regardless of the presence of BSA. In addition, the TlaAm content has an influence on the microgel size. The correlation between TlaAm content in the reaction mixture and fraction of BSA getting incorporated into the microgel (Fig. 2B) may give a satisfying explanation here. Thus it is not sufficient to simply provide enough stabilizing protein for the reaction, we also need to make sure, that enough protein-reactive comonomer is present that firmly binds the BSA to the newly formed microgel structure. It is noted that the thiolactone group of TlaAm can bind the protein but may also serve as chain transfer agent. In fact, the binding of the protein to the thiolactone as well as the hydrolysis of the latter in the basic aqueous reaction environment releases thiol groups that are well known for their chain transfer ability. Normally, such a chain transfer would suppress branching of a growing chain and lower the average molar mass of the polymer.17 It would thus counteract the cross-linker actions. Yet, the thiol is linked to a polymerizable C–C double bond in our case, meaning that chain transfer via the hydrolyzed thiolactone unit would just lead to another branching point and not to the generation of a new growing chain. The precise role, TlaAm plays in the synthesis and how it affects the internal structure of the microgel will be subject of further investigations. The microgel size can be controlled further by the extent of dilution of the reaction mixture and to some extent by the initiator concentration. With the current toolbox we can set the hydrodynamic diameter of the microgels between 150 nm and ∼1 μm (Table 1).
 | ||
| Fig. 2 (A) Fluorescence microscopy image of a representative batch of a microgel synthesized by the standard protocol (see Exp. part) using labeled BSA. (B) Apparent fluorescence intensities of microgel dispersions as a function of the relative TlaAm content in the polymerization mixture. The data was recorded after removal of the soluble fractions in each dispersion by repeated centrifugation/redispersion. | ||
| Parameter | Parameter valuesa | DLS results Z-Avg. [nm] (Std Dev.)/PDI |
|---|---|---|
| a Given concentrations/values are relative values with respect to the standard recipe: cNIPAm = 65.2 mg mL−1; cTlaAm = 5.3 mg mL−1; cBis = 2.7 mg mL−1; cinitiator = 0.45 mg mL−1; cBSA = 3.5 mg mL−1; Vreaction = 5.52 mL – for the standard recipe the resulting microgel had an z-average size of 650 nm with a PDI of 0.1.b The standard recipe includes a polymerization temperature of 50 °C.c Apparent values; additional macroscopic particles are present that settle in the measuring cell with time and are thus not captured by the measurement. | ||
| BSA concentration | 0 | 1900 (277)/0.04c |
| 0.25 | 920 (22)/0.23 | |
| 0.5 | 780 (107)/0.07 | |
| Bis concentration | 0.33 | 450 (3.5)/0.04 |
| 0.5 | 450 (10)/0.02 | |
| TlaAm concentration | 0 | 1900 (285)/0.7c |
| 0.33 | 1230 (35)/0.18c | |
| 0.5 | 720 (6)/0.09c | |
| 2 | 440 (10)/0.04 | |
| 4 | 180 (1)/0.05 | |
| Initiator concentration | 0.1 | 1060 (23)/0.16c |
| 0.2 | 900 (23)/0.15 | |
| Reaction volume | 10 | 210 (2)/0.07 |
| Temperatureb | 30 °C | Macro scale precipitate |
| 35 °C | 920 (22)/0.23 | |
| 40 °C | 850 (20)/0.08 | |
Not surprisingly, the synthesized microgels exhibit thermoresponsive behavior. Fig. 3A shows the scattering intensity as well as the hydrodynamic diameters of representative microgels, recorded via DLS, as a function of temperature. The volume phase transitions appear relatively broad, an observation that is not uncommon for cross-linked, thermoresponsive polymers.18 In the case of microgels this effect is mostly owing to an inhomogeneous crosslinker distribution.19 With increasing initial size of the microgels, the volume phase transition occurs sharper and also at a slightly lower temperature. Generally the increase in scattering intensity at the transition point comes along with a decrease in microgel size. As smaller colloidal objects should scatter light to a lesser extent, the rise in scattering intensity must therefore be attributed to a respective change in dn/dc, which makes sense considering the fact that the microgel matrix expels most of the water in which it is swollen below the transition temperature. At temperatures above 35 °C agglomeration and settling occurs, as evidenced by a strongly increasing size and the onset of a drop in scattering intensity. The tendency for agglomeration of the collapsed microgels above the cloud point is apparently increasing with microgel size. The microgels that contain enzyme (DERA and GOx respectively, see next section), behave similarly (Fig. 3B), which is not surprising given the fact that only 10% of the BSA within the standard recipe has been replaced by the respective enzyme in the cases. The onset of the transition is detected at 25.6 °C for GOx and 26.7 °C for DERA.
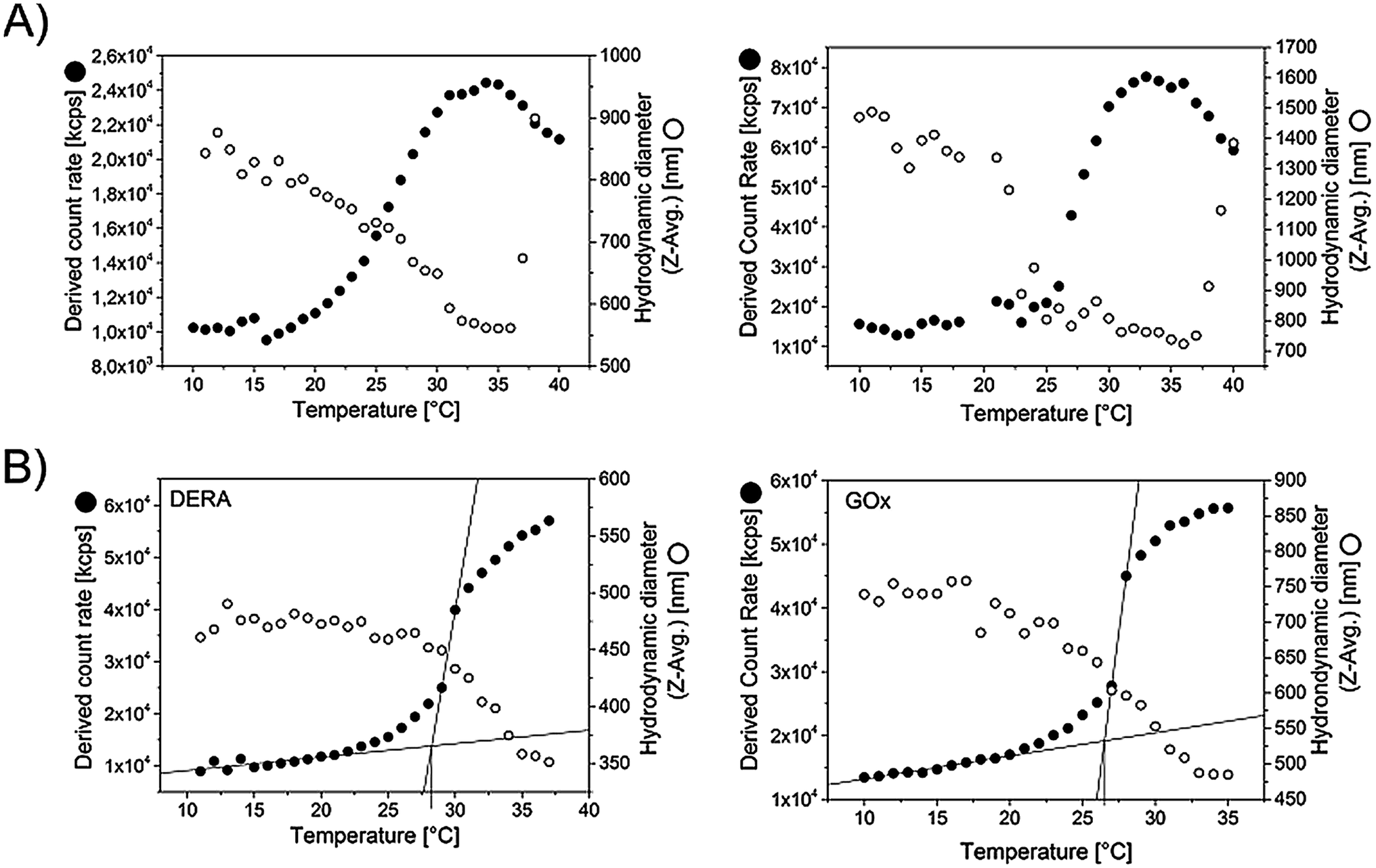 | ||
| Fig. 3 Evaluation of microgel size and scattering intensity of the microgel dispersions as a function of temperature. The data was recorded via DLS, taking data points in steps of 1 °C with an equilibration time of 120 s in each case. For the measurement the microgel dispersions were diluted by a factor of 44 using PBS buffer. (A) Microgels with different size (no extra enzyme embedded) (B) DERA and GOx containing microgels (specifications see Table 2). | ||
Enzyme containing microgels
In order to incorporate an enzyme of interest, the BSA in the basic polymerization recipe may be simply replaced by that enzyme. However, depending on the specific enzyme in use and its production costs, it may be worthwhile to only replace a part of the BSA, also considering the fact that the surface activity of certain enzymes is inferior to that of BSA, meaning that a poorer stabilization of the microgels can be expected. Consequently, the protein mixture we use always contains 90% BSA and 10% of the enzyme of choice. In order to show a broad applicability, we tested several different enzymes for our immobilization protocol, including glucose oxidase (GOx), 2-deoxy-D-ribose-5-phosphate aldolase (DERA) and horseradish peroxidase (HRP). GOx and HRP represent typical enzymes that are used in biosensing applications.20 DERA on the other hand is an enzyme useful for biocatalytic synthesis purposes.21 In general, these enzymes represent typical cases in which immobilization can have a benefit.22 For sensing applications, thin layers of enzymatically active microgels could be deposited on electrode surfaces while DERA containing microgels could be used to pack columns for continuously operated biocatalysis. As the polymerization conditions are potentially harmful to the enzymes, we exposed each of them to these conditions and assessed the course of the respective specific activity (Fig. S4, ESI†). GOx is mostly affected by the polymerization temperature, but still keeps a certain activity in the end. DERA on the other hand loses a lot of activity upon exposure to TEMED and gets completely inactivated upon the temperature rise. However, activity loss remains limited when the TEMED concentration is lowered to half of the original value and the temperature set to only 35 °C. The latter is the minimum for a successful microgel synthesis, at least when NIPAm is used as main monomer. As mentioned, horseradish peroxidase (HRP) has also been considered for immobilization. A test for its resilience against the polymerization conditions revealed, that HRP also survives the polymerization procedure, just as DERA and GOx. However, the respective microgel later on showed no enzymatic activity. The reason for that is either a disturbance of the enzymes confirmation upon the actual incorporation into the microgel structure, or an incompatibility of the substrate in use with the microgel matrix. The size and size distribution of the two synthesized microgels were again determined by DLS. Specifications are given in Table 2. The respective CONTIN plots are shown in Fig. 6.| Sample code | Immobilized enzyme | DLS resultsa Z-Avg. [nm] (Std Dev.)/PDI | Immobilization yieldb [%] (Std Dev.) | Transition temperaturec [°C] (Std Dev.) |
|---|---|---|---|---|
| a For DERA, polymerization was done at 35 °C and the TEMED concentration lowered to 1/2 compared to the standard recipe with a doubled reaction volume.b Fraction of enzyme that can be removed from the solution after the polymerization via centrifugation.c Defined as the intersection of the two tangents applied to the two linear regimes of the scattering intensity curve (Fig. 3). | ||||
| GOx | Glucose oxidase | 651 (10)/0.14 | 31 (8.5) | 25.6 (1.2) |
| DERA | 2-Deoxy-D-ribose-5-phosphate aldolase | 430 (24)/0.2 | 44 (3.8) | 26.7 (2.1) |
Quantification of the amount of respective enzyme that is incorporated is done by using fluorescently labeled versions of the enzymes analogue to the protocol used for quantification of BSA as described in the previous subsection. Measuring the residual fluorescence of the supernatants after removal of solid microgel material reveals the respective amount of enzyme that has been incorporated. For the two enzymes, different values are obtained while the lower quantity was achieved for GOx (31%) and the higher for DERA (44%). The remaining parts are bound to the soluble fraction of the polymeric material or have remained unaffected by the thiolactone groups of the TlaAm comonomer, as stated above. Nevertheless, the amounts of incorporated enzymes are fairly substantial.
Enzymatic activity of the microgels
After incorporation of the respective enzymes into the microgels, their activity was assessed in comparison to their counterparts in solution. Prior to the measurements the microgel dispersions have been purified by repeated centrifugation/redispersion in order to get rid of all soluble material that is left after the synthesis. The dispersions should then only contain microgel bound enzyme. This is verified by testing for the enzymatic activity of the supernatants, as will be discussed later. For the measurement of the activities, the overall concentrations of enzyme have been kept within the same order of magnitude within one measurement series if not otherwise stated. Yet, a direct comparison of the initial slopes in the recorded curves, which are direct measures for the respective activity in all cases, has to be done with caution, as the presence of turbidity that is brought in by the microgels means that the Lambert–Beer-Law does not apply anymore. Consequently, we only show the raw absorption data and draw conclusions only where differences in the detected slopes are substantial. Although temperature control was used, there was a risk that turbidity changes may still occur when performing measurements as the microgel particles may settle at the bottom of the well at a slow pace. As the detection device cannot distinguish between actual absorption and turbidity (detected also as apparent absorption due to the fact that part of the incident light is scattered away) we thus needed to additionally follow the course of the measurement signal in the presence of microgel but absence of substrate. By that we could judge whether the obtained data indeed indicate enzymatic activity. End point detection would have opened the opportunity to first get rid of microgel via centrifugation before recording the actual data point. However, we tried and reproducibility was very poor. Thus we did not follow that particular approach.Both GOx as well as the DERA microgel show enzymatic activity, as shown in Fig. 4 and 5. In both cases the activity of the microgel bound enzyme is somewhat decreased compared to the one of the respective enzyme in solution. For the GOx-microgel at 25 °C the detected activity equates the activity measured for 1/25 of the amount of non-immobilized biocatalyst. A clear distinction can be made between the measurements for the GOx-microgel in the presence and absence of substrate, indicating that we are indeed dealing with actual enzymatic activity and not just a change in solution turbidity. This is also supported by the observation that the assay solution exhibits green color after the measurement is finished, as seen by the naked eye. The absence of activity in the supernatant shows that the detected activity stems exclusively from biocatalyst that is bound to the microgel, indicating the absence of enzyme leakage during storage of the microgel dispersion.
 | ||
| Fig. 4 Activity assay for the GOx containing microgel (specifications see Table 2) at 25 and 40 °C. The slopes of the linear regimes indicate the extent of enzymatic activity. Note, that the enzyme in solution was much more active than the immobilized enzyme so that the action of the HRP in the coupled assay (see Exp. part) would have become the rate limiting step when sticking to the same amount of GOx. Consequently, the enzyme concentration had to be lowered to 1/25 of the initial value in these cases. The slope for the GOx-microgel at 25 °C was analyzed for the time period between 500 and 1000 s as slight turbidity changes contributed to the measurement signal at the initial measurement period <500 s. The underlying reaction mechanism for the assay is shown in Scheme S1 (ESI†). | ||
 | ||
| Fig. 5 Activity assay for the DERA containing microgel at 25 °C. The concentrations of enzyme are in the same order of magnitude in order to be able to directly compare the detected slopes in the linear regime of the curves. The underlying reaction mechanism is shown in Scheme S1 (ESI†). | ||
DERA immobilization within the microgel structure also comes along with a significant decline in apparent enzymatic activity. While the maximum signal drop occurs within minutes for DERA in solution, the microgel has converted only a fraction of the substrate even after 2 hours. Although the apparent absorption is also affected by a slight, counteracting increase of turbidity as indicated by the measurement in the absence of substrate, one can conclude that the activity of DERA embedded in the microgel is significantly lower than the activity of DERA in solution. The significantly decreased activity for DERA must not necessarily be the result of a deactivation of the enzyme upon immobilization but may also stem from other factors such as diffusion limitation due to a low compatibility of the microgel matrix with the substrates in use. For DERA the substrate in use is 2-deoxy-D-ribose-5-phosphate (DRP), which is an anion. At the same time the microgel matrix carries plenty of deprotonated and thus also negatively charged carboxyl groups. Consequently, charge repulsion might hamper the substrate to diffuse to the enzyme. The assumption of diffusion limitations playing a strong role would be supported by the fact that DERA experiences only a moderate loss of activity when being exposed to the polymerization conditions without being actually incorporated into the microgel (Fig. S4, ESI†). Establishing an alternative quantitative assay for the detection of the enzymatic activity of DERA using a more suitable substrate, however, is out of scope of the current contribution, yet will be pursued in the nearer future. In case of GOx, the lower activity of the immobilized enzyme is rather the result of the exposure to the polymerization conditions, as the detected activity loss upon this exposure is comparable to the loss in activity that occurs upon the actual immobilization (Fig. 4 and S4, ESI†).
As the PNIPAm based microgel matrix shows a distinct response on temperature increase, originating from the thermosensitivity of the PNIPAm, it was further investigated, how the picture on the enzymatic activity changes upon temperature increase from room temperature to 40 °C. Unfortunately, measurements at such an elevated temperature gave conclusive results only in case of GOx. For DERA, substrate conversion is so slow for the microgel bound enzyme that the respective signal change observed in the 2 hour measurement time is significantly smaller than the signal rise and drop stemming from the turbidity change as a result of the aggregation and settling of the microgels above the cloud point. Thus, signal change originating from enzymatic activity cannot really be distinguished from the change that is the result of the turbidity change. With respect to the temperature effect on the enzymatic activity of the GOx microgel meaningful data could be generated both at 25 and at 40 °C (Fig. 4). Assessing the slopes of the linear regimes of the curves, one observes a meaningful increase both for the microgel bound enzyme and the enzyme in solution upon the temperature change. However, the relative change of the slope is significantly more pronounced for the microgel bound enzyme than it is for the enzyme in solution. In the first case the slope at 40 °C is increased by about 40% compared to the one at 25 °C, in the latter case it is only ∼15%. Thus, the transgression of the microgel through the volume phase transition at the cloud point has indeed an effect on the enzymatic activity. It is not expected for an enzyme embedded within a thermoresponsive microgel with LCST characteristics, that an increased activity is occurring at elevated temperatures, as the microgel is only swollen in the aqueous dispersion medium below the cloud point.6,23 The microgel is in the collapsed state above the transition temperature, therefore, substrate diffusion through the gel network should be hampered. Consequently, activities should be higher at lower temperatures relative to the enzymes in solution. However, the incorporated proteins (BSA and the enzyme) as non-responsive, water soluble macromolecules may form channels that allow for substrate diffusion even when the gel is in the collapsed state. Besides, a large portion of protein can be expected to be located in the outer parts of the microgel as the presence of protein appeared to be beneficial for the colloidal stability of the microgel, thus acting as a surfactant as discussed earlier. This fraction of protein thus cannot be shielded by the collapsed gel matrix. These considerations explain the absence of activity decline upon temperature increase, yet, they still do not explain the actually observed contrary effect. One might argue that the increased microgel size below the volume phase transition (Fig. 3) does prolong the diffusion pathways. Yet, other effects may be considered as well. Depending on the state of the microgel matrix (swollen or collapsed), we expect the enzymes conformation to be disturbed to different extents. There are two possible explanations for that effect, the first one including different magnitudes of interaction between the PNIPAm gel matrix and the enzyme at temperatures above and below the cloud point. The other explanation considers the observed effect as a direct consequence of the fact that the enzyme gets incorporated into the gel while the latter is in the collapsed state (remember that the polymerization temperature is set to a value above the cloud point of the polymer). Assuming a multi-point attachment of the protein to the gel matrix via the thiolactone based comonomer, a swelling of the gel upon cooling of the reaction mixture after the polymerization would then also lead to a disturbance in the conformation of the enzyme. In order to get an idea, which case is true in case of GOx, the activity of the enzyme was assessed in solution in the presence of an enzyme free microgel. Indeed, the presence of microgel material does not only cause a certain general decrease of the enzymatic activity, the temperature change has the same effect as in the case of the microgel bound enzyme. Thus, the temperature effect on the enzymatic activity is the result of a direct interaction of the enzyme with the microgel matrix. Another observed feature worth to be mentioned is the fact that for GOx in solution, the slope of the signal starts to decrease quite soon after the start of the measurement at 40 °C (at 600 s for the pure enzyme and at 400 s for the enzyme in presence of microgel). Even though the curve for the microgel bound enzyme shows more or less the slope as the curve for the enzyme in solution in presence of the microgel, this effect is only observable in the latter case, pointing to a respective stabilization effect of the immobilization.
As a final remark, it is mentioned that the synthesized microgels appear pretty robust. It is possible to cross the transition temperature multiple times without leaking any enzyme out of the microgel or cause a decline of activity (results not shown). Also, freeze drying for prolonged storage is possible. Respective samples can be freeze dried without any special measure (such as the use of lyophilization additives) and easily redispersed (Fig. 6) and still show decent enzymatic activity. DERA does not show any activity loss both in solution (not shown) and in immobilized form (Fig. 6B, left graph). The activity of microgel bound GOx, on the other hand, is significantly decreased upon lyophilization. However, the respective enzyme in solution is almost completely deactivated (Fig. 6B, inset in the right graph), which leads to the conclusion that the immobilization of GOx within the microgel has a beneficial effect on the enzymes stability. For both, the DERA and the GOx-containing microgel, lyophilization goes along with an increased hydrodynamic ratio and a slightly higher dispersity. One should consider, however, that no extra treatment for redispersion, such as sonication was needed and the microgels still appear to be rather defined in size. Finally, it is noted, that the dispersed samples do not deteriorate in terms of colloidal stability and enzymatic activity for at least 3 months.
 | ||
| Fig. 6 Assessment of enzymatic and colloidal stability upon freeze drying of the microgels: (A) CONTIN plots before and after lyophilization derived from DLS measurements; (B) activity assays conducted before and after lyophilization (conditions see Exp. part). For the DERA-microgel, the starting values for the absorption were normalized for a better comparison. | ||
Conclusions
We have shown how enzymes can be easily incorporated within thermo-responsive microgel structures using a synthetic protocol based on a modification of the well-known precipitation polymerization of NIPAm. The protocol appears to be simple, in principle scalable and may be applicable for many different enzymes without the necessity of a major adjustment of the reaction conditions. The protocol therefore has the potential to significantly reduce costs that normally come along with an immobilization procedure and thus bears relevance for industrial biocatalysis and specific sensing applications. So far, we have shown the proof-of-principle. Next steps will include investigations on the internal structure of the microgels including the determination of the spatial distribution of the incorporated enzyme. We will also have a deeper look into what role each polymerization parameter plays on the molecular level. Although researchers like Richtering have already given a lot of insight into the polymerization kinetics and their correlation to the hydrogel structure,15,24 the presence of protein as well as a chain transferring comonomer adds additional degrees of complexity to the system. Hence, more investigations are necessary.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Marc Zimmermann, Dr Ruben Rosencrantz and Dr Sophia Rosencrantz of the Fraunhofer Institute for Applied Polymer Research for the assistance in carrying out experiments and for giving helpful input during manuscript writing.References
- W. Storhas, Bioverfahrensentwicklung, Wiley-VCH Verlag GmbH, 2003 Search PubMed.
- (a) A. Petri, P. Marconcini and P. Salvadori, J. Mol. Catal. B: Enzym., 2005, 32, 219–224 CrossRef CAS , http://www.sciencedirect.com/science/article/pii/S1381117704002917; (b) K. Min and Y. J. Yoo, Biotechnol. Bioprocess Eng., 2014, 19, 553–567 CrossRef CAS; (c) G. S. Chauhan, Polym. Int., 2014, 63, 1889–1894 CrossRef CAS.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- M. Yan, J. Ge, Z. Liu and P. Ouyang, J. Am. Chem. Soc., 2006, 128, 11008–11009 CrossRef CAS PubMed.
- N. Welsch, M. Ballauff and Y. Lu, in Chemical Design of Responsive Microgels, ed. A. Pich and W. Richtering, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 129–163 Search PubMed.
- X. Yang, X. Zhang, W. Shang and S. Zhang, Clean: Soil, Air, Water, 2016, 44, 189–194 CAS.
- (a) E. Gau, F. Flecken, A. N. Ksiazkiewicz and A. Pich, Green Chem., 2018, 20, 431–439, 10.1039/c7gc03111d; (b) F. Li, C. Wang and W. Guo, Adv. Funct. Mater., 2018, 28, 1705876 CrossRef; (c) S. Schachschal, H.-J. Adler, A. Pich, S. Wetzel, A. Matura and K.-H. van Pee, Colloid Polym. Sci., 2011, 289, 693–698, DOI:10.1007/s00396-011-2392-1.
- S. Reinicke, P. Espeel, M. M. Stamenović and F. E. Du Prez, ACS Macro Lett., 2013, 2, 539–543 CrossRef CAS.
- M. Dick, O. H. Weiergräber, T. Classen, C. Bisterfeld, J. Bramski, H. Gohlke and J. Pietruszka, Sci. Rep., 2016, 6, 17908 CrossRef CAS PubMed.
- (a) H. Fei, G. Xu, J.-P. Wu and L.-R. Yang, J. Mol. Catal. B: Enzym., 2014, 101, 87–91 CrossRef CAS; (b) M. Dick, R. Hartmann, O. H. Weiergräber, C. Bisterfeld, T. Classen, M. Schwarten, P. Neudecker, D. Willbold and J. Pietruszka, Chem. Sci., 2016, 7, 4492–4502 RSC.
- R. H. Pelton and P. Chibante, Colloids Surf., 1986, 20, 247–256 CrossRef CAS.
- M. Stieger, W. Richtering, J. S. Pedersen and P. Lindner, J. Chem. Phys., 2004, 120, 6197–6206 CrossRef CAS PubMed.
- A. Pich and W. Richtering, Adv. Polym. Sci., 2010, 234, 1–37 CrossRef CAS.
- J. Gao and B. J. Frisken, Langmuir, 2003, 19, 5212–5216 CrossRef CAS.
- O. L. J. Virtanen, M. Kather, J. Meyer-Kirschner, A. Melle, A. Radulescu, J. Viell, A. Mitsos, A. Pich and W. Richtering, ACS Omega, 2019, 4, 3690–3699 CrossRef CAS PubMed.
- (a) H. Neurath and H. B. Bull, Chem. Rev., 1938, 23, 391–435 CrossRef CAS; (b) F. Heitz and N. van Mau, Biochim. Biophys. Acta, 2002, 1597, 1–11 CrossRef CAS.
- C. Henríquez, C. Bueno, E. A. Lissi and M. V. Encinas, Polymer, 2003, 44, 5559–5561 CrossRef.
- (a) S. Reinicke, P. Espeel, M. M. Stamenovic and F. E. Du Prez, Polym. Chem., 2014, 5, 5461–5470 RSC; (b) T. Tanaka and D. J. Fillmore, J. Chem. Phys., 1979, 70, 1214–1218 CrossRef CAS.
- (a) X. Wu, R. H. Pelton, A. E. Hamielec, D. R. Woods and W. McPhee, Colloid Polym. Sci., 1994, 272, 467–477, DOI:10.1007/bf00659460; (b) L. C. Kröger, W. A. Kopp and K. Leonhard, J. Phys. Chem. B, 2017, 121, 2887–2895 CrossRef; (c) O. L. J. Virtanen, A. Mourran, P. T. Pinard and W. Richtering, Soft Matter, 2016, 12, 3919–3928 RSC.
- (a) P. N. Bartlett and P. R. Birkin, Anal. Chem., 1994, 66, 1552–1559 CrossRef CAS; (b) T. Ruzgas, E. Csöregi, J. Emnéus, L. Gorton and G. Marko-Varga, Anal. Chim. Acta, 1996, 330, 123–138 CrossRef CAS.
- (a) J. M. Patel, J. Mol. Catal. B: Enzym., 2009, 61, 123–128 CrossRef CAS; (b) C. F. Barbas, Y. F. Wang and C. H. Wong, J. Am. Chem. Soc., 1990, 112, 2013–2014 CrossRef CAS; (c) S. Jennewein, M. Schürmann, M. Wolberg, I. Hilker, R. Luiten, M. Wubbolts and D. Mink, Biotechnol. J., 2006, 1, 537–548 CrossRef CAS PubMed; (d) W. A. Greenberg, A. Varvak, S. R. Hanson, K. Wong, H. Huang, P. Chen and M. J. Burk, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5788–5793 CrossRef CAS PubMed.
- (a) S. Reinicke, H. C. Rees, P. Espeel, N. Vanparijs, C. Bisterfeld, M. Dick, R. R. Rosencrantz, G. Brezesinski, B. G. de Geest, F. E. Du Prez, J. Pietruszka and A. Böker, ACS Appl. Mater. Interfaces, 2017, 9, 8317–8326 CrossRef CAS PubMed; (b) S. Zhang, C. Bisterfeld, J. Bramski, N. Vanparijs, B. G. de Geest, J. Pietruszka, A. Böker and S. Reinicke, Bioconjugate Chem., 2018, 29, 104–116 CrossRef CAS.
- Chemical Design of Responsive Microgels, ed. A. Pich and W. Richtering, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011 Search PubMed.
- O. L. J. Virtanen and W. Richtering, Colloid Polym. Sci., 2014, 292, 1743–1756, DOI:10.1007/s00396-014-3208-x.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and SDS-PAGE data recorded from the extractable soluble fraction of the synthesized microgels, data from fluorescence recordings performed for the sake of enzyme quantification, information on the reaction mechanisms that apply for the activity assays in use and finally supplemental data on enzyme activity. See DOI: 10.1039/c9ra04000e |
| This journal is © The Royal Society of Chemistry 2019 |