
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, structural studies and biological properties of some phosphono-perfluorophenylalanine derivatives formed by SNAr reactions†‡
Joanna Kwiczak-Yiğitbaşıab,
Jean-Luc Piratb,
David Virieux b,
Jean-Noël Volle*b,
Agnieszka Janiaka,
Marcin Hoffmanna,
Jakub Mrzygłóda,
Dariusz Wawrzyniakc,
Jan Barciszewskicd and
Donata Pluskota-Karwatka*a
b,
Jean-Noël Volle*b,
Agnieszka Janiaka,
Marcin Hoffmanna,
Jakub Mrzygłóda,
Dariusz Wawrzyniakc,
Jan Barciszewskicd and
Donata Pluskota-Karwatka*a
aAdam Mickiewicz University in Poznań, Faculty of Chemistry, Umultowska 89b, 61-614 Poznań, Poland. E-mail: donatap@amu.edu.pl
bAM2N, UMR 5253, ICGM, ENSCM, 8 Rue de L'Ecole Normale, 34296 Montpellier Cedex 5, France. E-mail: jean-noel.volle@enscm.fr
cInstitute of Bioorganic Chemistry, Polish Academy of Sciences, Noskowskiego 12/14, 61-704 Poznań, Poland
dNanoBioMedical Center of Adam Mickiewicz University, Umultowska 85, 61-614, Poznań, Poland
First published on 5th August 2019
Abstract
Several novel phosphono-perfluorophenylalanine derivatives, as mimetics of phenylalanine, were synthesized by subjecting diethyl (2-(perfluorophenyl)-1-(phenylamino)ethyl)-phosphonate to SNAr reactions with different types of nucleophiles such as thiols, amines and phenols. The structure of the products was confirmed using spectroscopic and spectrometric techniques. For two compounds X-ray single crystal diffraction analysis and DFT investigations were performed providing information in regard to the preferable conformation, hydrogen bonds and other interactions. The antiproliferative potency of some of the new phosphono-perfluorophenylalanine derivatives obtained as well as representatives of previously synthesized perfluorophenyl phosphonate analogues of phenylalanine was studied on selected glioma cell lines. Preliminary evaluation of the compounds drug likeness was examined with respect to Lipinski's and Veber's rules, and showed that they meet the criteria perfectly. MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) assay results demonstrated that the compounds exhibit moderate activity against the glioblastoma multiforme cell lines (T98G and U-118 MG). Moreover most of the studied SNAr reaction products displayed significantly higher inhibitory activity against both cancer cell lines than the parent diethyl (2-(perfluorophenyl)-1-(phenylamino)ethyl)phosphonate.
1. Introduction
Chemistry of α-aminophosphonates has played a significant role in the development of organophosphorus compounds, and still remains a field of great interest.1–3 As structural analogues of α-amino acids, α-aminophosphonates have found application in organic and medicinal chemistry,4 mainly due to their anticancer,5–9 antiviral10 and antibacterial11,12 activities. Moreover, phosphonates represent classical phosphate bioisosters, in which the labile O–P bond is replaced by an enzymatically and chemically stable C–P bond. This change makes α-aminophosphonates structurally similar to phosphate esters or anhydrides, yet increases their stability under physiological and chemical conditions.13,14 Also, the tetrahedral geometry on the phosphorus atom mimics the transition state of peptide hydrolysis thus, α-aminophosphonates can act as enzyme inhibitors.15–17Among α-aminophosphonates, the fluorinated ones constitute a particularly important group of compounds. The incorporation of fluorine atoms in the structure of α-aminophosphonates provides access to more lipophilic molecules, which often exhibit better biological activities than the parent compounds.18,19 What is more, the presence of fluorine and phosphorus atoms in the α-aminophosphonates structure gives us a chance to use 19F and 31P NMR spectroscopy to follow the location and to study the molecular interactions in biological systems.20,21
Introduction of fluorine has also become an important strategy in protein biochemistry. Fluorinated amino acids serve as powerful tools for exploring polar π-interactions in proteins,22,23 and enabling novel recognition mechanisms for protein design.24–26 Heavily fluorinated aromatic residues are particularly desirable because multifluorination gives maximum electronic perturbation of aromatic rings.
Fluorinated aromatic rings may undergo nucleophilic aromatic substitution reactions (SNAr). Due to higher electronegativity of carbon comparing to hydrogen, benzene exhibits a negative potential on the π face and a positive one around the periphery.27 Fluorine as strongly electronegative element, reverses this distribution to give a negative potential on the periphery and a positive on the aromatic ring,27 or, as theoretical studies suggest, localized more to carbon skeleton than the π-electrons cloud.28,29 Thus fluorination clearly enhances the rate of nucleophilic attack. In general, the reaction of pentafluorophenyl moiety is highly regioselective; nucleophile replaces almost exclusively the fluorine atom in the para position.27 The SNAr reactions found application for fluorinated amino acids syntheses. Regioselective nucleophilic addition–elimination reaction of pentafluorobenzyl moiety was the key step on the synthetic route to tetrafluorotyrosine.30 This strategy was then extended to preparation of a series of para-substituted tetrafluorophenylalanines.31 Also cysteine arylation was achieved via SNAr reactions.32 Reaction between cysteine thiolate and perfluoroaromatic molecules afforded exclusively 1,4-disubstituted products. Such approach enabled selective modification of cysteine residues in unprotected peptides.32
As part of our investigations, we have recently reported a convenient synthetic method for the preparation of a series of phosphonate analogues of phenylglycine, homophenylalanine, and phenylalanine that differ in the number and position of fluorine atoms in the phenyl ring.33,34 These studies were supported by single-crystal X-ray diffraction analysis and quantum chemical calculations that provided information concerning the conformational preferences both in the solid and isolated states. Indeed, some of the obtained aminophosphonates underwent intramolecular SNAr reactions yielding indolinylphosphonates as minor products.33 Since SNAr reaction is an attractive and effective way for modifying structure of fluorinated aromatic compounds, we decided to study the ability of the synthesized α-aminophosphonates to undergo such a transformation. It gave us a chance to obtain a library of diversely substituted α-aminophosphonates and opened a new perspectives to seek for original bioactive molecules. Therefore in this paper we describe the synthesis of para substituted derivatives of diethyl (2-(perfluorophenyl)-1-(phenylamino)ethyl)phosphonate 1a (Scheme 1) as well as results of their X-ray and DFT studies. Structural variations on products were achieved by subjecting the phenylalanine analogue 1a to reactions with various nucleophiles, i.e. thiols, amines and phenols (Scheme 1).
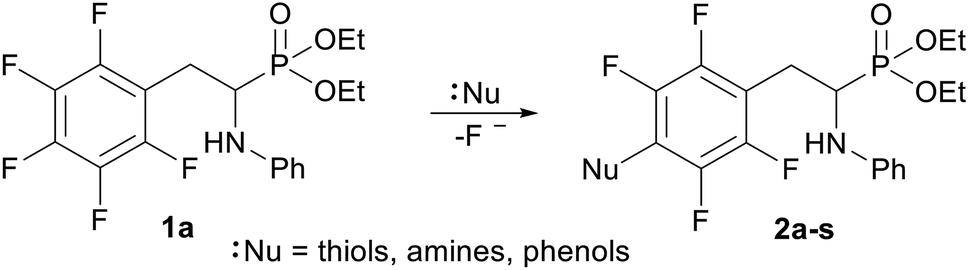 | ||
| Scheme 1 Synthesis of 2a–s. | ||
Some of the phosphono-perfluorophenylalanine derivatives obtained together with representatives of previously synthesized33 perfluorophenyl phosphonate analogues of phenylalanine were subjected to studies aimed at evaluation of the compounds antiproliferative potency on chosen glioblastoma multiforme (GBM) cell lines.
GBM is highly infiltrative tumour which displays extreme resistance to conventional radiotherapy and chemotherapy.35 It contains self-renewing, tumorigenic cancer stem cells (CSCs) that contribute to tumour initiation and therapeutic resistance. These cells in malignant gliomas were called glioblastoma stem cells (GSCs).36 Recent studies uncovered increased expression, aberrant localization and disturbed functions of certain cysteine cathepsins in GSCs.37 Cathepsins B and L are overexpressed in glioma cells and responsible for glioblastoma cell invasion.38,39 The scientific literature shows ability of aminophosphonic acids and aminophosphonates to act as inhibitors of esterases,17,40 protease inhibitors41 and cathepsins. Therefore, targeting cathepsin activity by specific protease inhibitors in GBM is a logical consequence and might interfere with proliferation, and migration to avoid recurrence of glioblastoma.
2. Results and discussion
2.1 Synthesis
Diethyl (2-(perfluorophenyl)-1-(phenylamino)ethyl)phosphonate (1a, Scheme 1) was subjected to SNAr reactions with thiols, amines and phenols resulted in the formation of para-substituted products (2a–s, Scheme 1, Table 1) with full regioselectivity.| Entry | Compound | Nucleophile | Conditions | Yield [%] |
|---|---|---|---|---|
| 1 | 2a | C6H5SH | TRIS, DMF, rt, 2 h | 87 |
| 2 | 2b | 4-CH3C6H4SH | 89 | |
| 3 | 2c | 3,5-(CH3)2C6H3SH | 81 | |
| 4 | 2d | 4-CH3OC6H4SH | 95 | |
| 5 | 2e | 3-CH3OC6H4SH | 84 | |
| 6 | 2f | 3-NH2C6H4SH | 76 | |
| 7 | 2g | 4-BrC6H4SH | 86 | |
| 8 | 2h | C6H5CH2SH | 65 | |
| 9 | 2i | CH3(CH2)8CH2SH | 37 | |
| 10 | 2j | N-acetyl-L-cysteine methyl ester | 71 | |
| 11 | 2k | CH3NH2 | DMSO, 80 °C, 3 h | 71 |
| 12 | 2l | CH3(CH2)2NH2 | 74 | |
| 13 | 2m | CH3(CH2)3NH2 | 60 | |
| 14 | 2n | CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2NH2 CHCH2NH2 |
65 | |
| 15 | 2o | C6H5CH2NH2 | 63 | |
| 16 | 2p | C6H5OH | K2CO3, DMF, 80 °C, 24 h | 68 |
| 17 | 2q | 4-CH3OC6H4OH | 48 | |
| 18 | 2r | 4-ClC6H4OH | 56 | |
| 19 | 2s | 3-NO2C6H4OH | 48 |
Several conditions already described in the literature for these SNAr reactions32,42–44 were applied to the reaction of 1a with both hard and soft nucleophiles. Hard nucleophiles, such as hydroxide or alcoxides, had a clear tendency to attack the phosphonate ester while soft nucleophiles cleanly afforded the SNAr reactions (Table 1).
Following the procedure developed by Spokoyny et al.,32 thiols reacted smoothly in DMF, in the presence of tris(hydroxymethyl)aminomethane (TRIS) at room temperature within short reaction time (2 h). In order to study the scope of the reaction, syntheses with different thiols were performed (Table 1, entries 1–10). Nucleophilic aromatic substitution of 1 with thiophenols (Table 1, entries 1–7) occurred in good to excellent yields (76–95%). Both EDG (Table 1, entries 2–6) and EWG (Table 1, entry 7) substituents did not affect the reaction yields in comparison to the reaction performed with unsubstituted thiophenol. Decylthiol was chosen to introduce long aliphatic chain into the structure (Table 1, entry 9). However, compound 2i was obtained in a modest 37% yield. Introduction of other aliphatic thiols, such as benzyl mercaptan (Table 1, entry 8) and N-acetyl-L-cysteine methyl ester (Table 1, entry 10), resulted in better yields. Nevertheless, reactions performed with the aliphatic thiols occurred with lower yields than those with the aromatic ones. Formation of 2h–i was monitored by 19F NMR analysis. 31P NMR spectra of crude material confirmed the full conversion of 1a. However, lower yields which the reactions occurred with, were caused by formation of several minor by-products. This can be rationalized by the fact that aliphatic thiols are better nucleophiles in comparison to thiophenols thus, their reactivity is increased.
Reaction of 1a with a large excess of alkylamine in DMSO after 3 hours of heating at 80 °C gave aryl aminophosphonates 2k–o in yields ranging from 60 to 74% (Table 1, entries 11–15). Progress of the reactions was monitored by 19F NMR, and after 3 hours a total conversion of the starting α-aminophosphonate was generally observed.
Different phenols were consecutively used as nucleophiles. The expected products were obtained in moderate yields (Table 1, entry 16–19). Following known procedure for this kind of reactions,42,43 the syntheses were performed in DMF in the presence of K2CO3, at 80 °C for 24 hours. In comparison to alcoxides in which the negative charge is located only on the oxygen atom, phenoxides are much softer nucleophiles therefore, the reactions proceeded efficiently.
2.2 X-ray and DFT studies
In order to comprehensively understand the structural features of compounds investigated we made attempts to grow their crystals by slow evaporation of toluene solutions under ambient conditions. This process resulted in obtaining crystals suitable for X-ray single crystal diffraction analysis only for 2k and 2l. In terms of chemistry, compounds 2k and 2l differ in the chain length of substituent in para position of the fluorinated ring; 2k contains the methylamine group, while propylamine group is present in 2l. Molecules of 2k and 2l are chiral, however, they are not enantiomerically pure since the racemic mixture of substrate (1a) was used for their synthesis. Thus, crystallization yielded single crystals containing both enantiomers. Since the nitrogen atoms included in the molecular structure of 2k and 2l are pyramidal in shape, i.e. are bonded to three different groups and possess lone pair of electrons, they constitute stereogenic centres. This, in turn, may affect the occurrence of diastereomers in the crystals.Both compounds examined crystallize in the triclinic space group P-1. 2k crystallizes with two molecules (mol 1 and mol 2) in the asymmetric unit that adopt a similar molecular conformation described by two torsion angles Csp2–N–Csp3–Csp3 and Csp2–Csp3–Csp3–N. The torsion angle values are −131.3(3)° and −83.9(3)° for mol 1, and 147.3(2)° and −68.9(3)° for mol 2, while the dihedral angle of two aromatic rings are 68.6(3)° and 78.0(3)° in mol 1 and 2, respectively. Adoption of very similar conformation (marked as a conformation B), by some phosphonates analogues of fluorophenylalanine was previously observed in our research group.33 The main structural differences are displayed within the aliphatic diethyl phosphonate chains (Csp3–Csp3–O–P) where the chains adopt antiperiplanar–antiperiplanar conformation with the corresponding torsion angle values of −171(3) and 165.7(2)° for mol 1, while the synclinal–antiperiplanar conformation is characterized by the values of 65.5(4) and 176.1(2)° for mol 2. Interestingly, the configuration of the aniline nitrogen atom in both symmetrically independent molecules shows the opposite stereochemistry i.e. S in mol 1 and R in mol 2; however, the methylamine moieties have the same R configuration. As a consequence these two molecules possess R(C5), S(N1), R(N2) and R(C24), R(N3), R(N4) configurations, respectively. Therefore they are diastereomers. Since the crystals of 2k are centrosymmetric, two pairs of diastereomers coexist in the crystal structure. The pyramidal environment of N atom in methylamine group also affects significant deviation of this group from the tetrafluorophenyl plane; the Csp2–Csp2–N–Csp3 torsion angle is 18.6(5)° in mol 1 and 21.2(4)° in mol 2. In contrast, 2l crystallizes with one molecule in the asymmetric unit and adopts previously described conformation C (extended)33 with the corresponding torsion angles of 80.3(2)° and −172.4(2)°. The anticlinal–antiperiplanar combination of the aliphatic diethyl phosphonate chains have torsion angles of 107.5(2) and 178.8(2)°. The propylamine group adopts a folded conformation with an N–Csp3–Csp3–Csp3 torsion angle of 62.5(3)°. The stereochemistry of nitrogen atoms in this group is S while in the aniline moiety is R, hence the molecule in the asymmetric unit has a R(C5), R(N1), S(N2) configuration. The crystals investigated were chosen such that the R stereogenic centre at the carbon atom constitutes the asymmetric unit. The molecular structures of 2k and 2l are shown in Fig. 1 and 2.
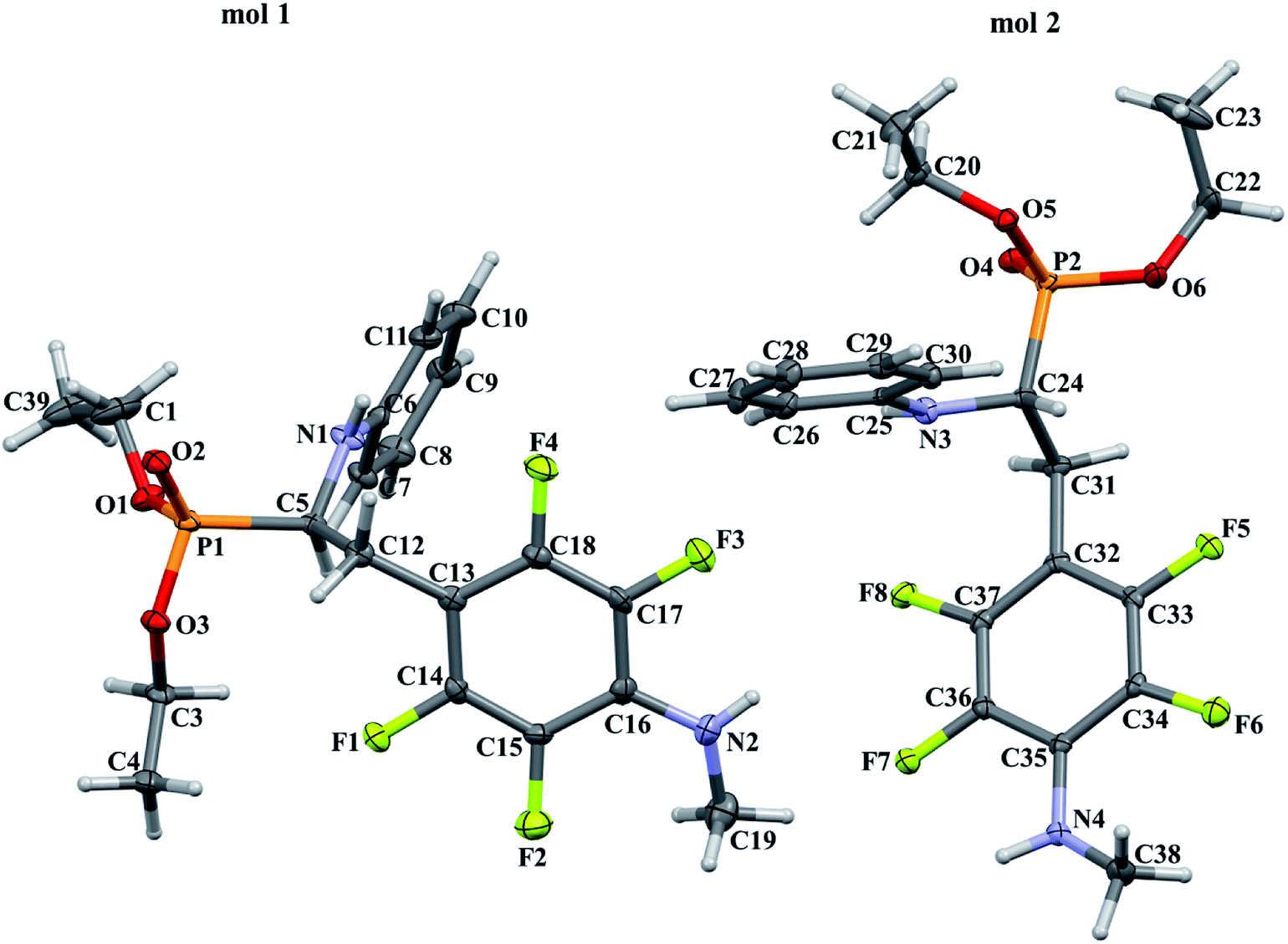 | ||
| Fig. 1 A perspective view showing the asymmetric part of unit cell of 2k together with the labelling atoms scheme. Ellipsoids are drawn at the 30% probability level, hydrogen atoms are represented by spheres of arbitrary radii. | ||
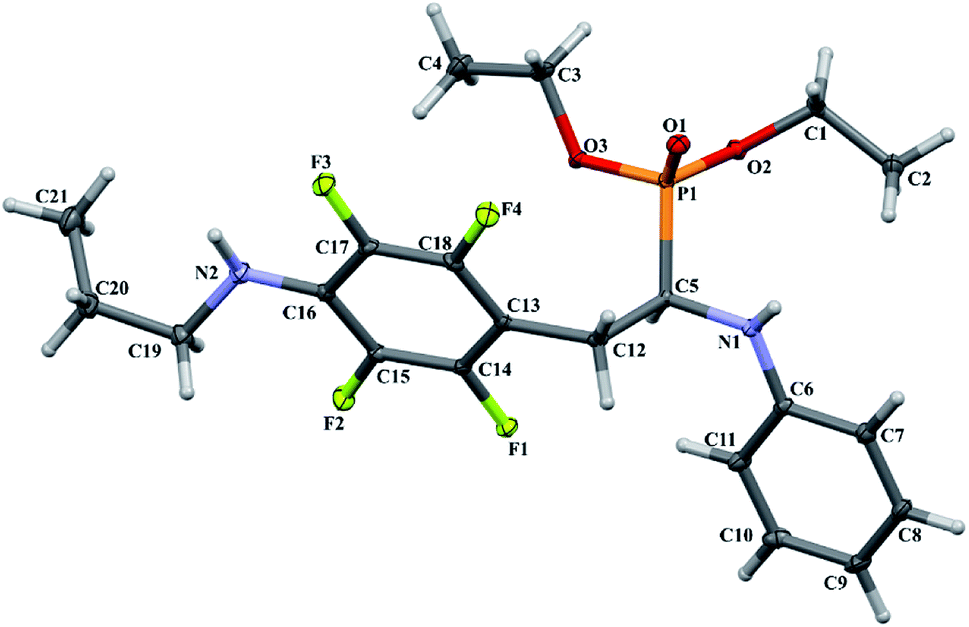 | ||
| Fig. 2 A perspective view showing the asymmetric part of unit cell of 2l together with the labelling atoms scheme. Ellipsoids are drawn at the 30% probability level, hydrogen atoms are represented by spheres of arbitrary radii. | ||
The different configuration exhibited by the aniline nitrogen atoms in 2k, prompted us to use quantum-chemical methods to study energy differences between the diastereomers. For this purpose all possible stereomers (R,R,R; R,S,S; R,S,R; R,R,R) were built based on the geometry of mol 1 derived from the crystal. The calculations were performed in vacuo, as well as using the polarisable continuum model (PCM) to take into account the effect of chloroform solution as NMR spectra of 2k were recorded for the compound sample dissolved in CDCl3.
The results obtained support structural diversity of the possible stereomers. The results show that the R,R,R stereomer is energetically favourable at WB97XD/6-31+G(d) level of theory including PCM, while in vacuo the lowest relative energy is assigned to the R,S,R stereomer (Table 2).
| Stereomer | ΔE in vacuo | ΔE in chloroform | ZPE | TCG | Pop in vacuo | Pop in chloroform |
|---|---|---|---|---|---|---|
| a Relative energies calculated with respect to the lowest energy structure at WB97XD/6-31+G(d) level is equal to −1813.40587669 hartree.b Zero-point energy; the lowest value was equal to 0.412559 hartree.c Thermal correction to Gibbs free energy at 298 K; the lowest value was equal to 0.348198 hartree. The relative energy in chloroform was equal to −1813.41658767 hartree. | ||||||
| R,S,R | 0.00 | 5.12 | 0.00 | 0.00 | 98.9 | <1.0 |
| R,S,S | 2.35 | 5.25 | 0.15 | 0.50 | 1.0 | <1.0 |
| R,R,S | 5.77 | 1.88 | 0.39 | 0.97 | <1.0 | 20.2 |
| R,R,R | 5.90 | 0.00 | 0.78 | 2.00 | <1.0 | 79.1 |
These findings suggest a strong influence of surrounding medium on the energy preferences of stereomers (diastereomers). In vacuo the R,S,R stereomer constitutes over 98% in the population of the calculated diastereomers (Table 2). Its population drops below 1% when chloroform solvent is taken into consideration via PCM approximation. In chloroform R,R,R stereomer seems to be predominant as it constitutes almost 80% in the population of diastereomers – a striking difference with its contribution (below 1%) in vacuo.
Analysis of the nitrogen atoms pyramidal environment in both the optimized and crystal structures of 2k shows strong deviation from the ideal tetrahedral geometry, described by set of valence angles equal to 109.5°, towards the planar one. As seen in Table 3, the valence angles values in both structures are similar. For example, in the crystal structure the values of C6–N1–H1, C5–N1–H1 and C6–N1–C5 valence angles are equal to 116.7°, 115.8°, and 125.4°, in mol 1, and 115.2°, 117.1°, and 124.9° for C25–N3–H3, C24–N3–H3 and C24–N3–C25 in mol 2, while in the calculated R,R,R stereomer the corresponding valence angles adopt values of 113.6°, 112.3° and 123.1° (Table 3). The sum of the valence angles values in the crystal structure is equal to 357.9° and 357.2°, in mol 1 and mol 2, respectively, and 349° in the R,R,R stereomer calculated. All these values correspond to forms between the ideally planar (360°) and ideally tetrahedral (328.5°) geometry but closer to the planar one.
| The valence angle [°] | Stereomer | |||||
|---|---|---|---|---|---|---|
| R,S,R DFT | X-ray | R,R,R DFT | X-ray | R,R,S DFT | R,S,S DFT | |
| C6–N1–H1 or C25–N3–H3 | 112.3 | 116.7 | 113.6 | 115.2 | 114.2 | 113.1 |
| C5–N1–H1 or C24–N3–H3 | 113.2 | 115.8 | 112.3 | 117.1 | 113.1 | 113.3 |
| C6–N1–C5 or C25–N3–C24 | 126.1 | 125.4 | 123.1 | 124.9 | 123.5 | 126.2 |
| Sum of the valence angles | 351.6 | 357.9 | 349.0 | 357.2 | 350.8 | 352.6 |
| C16–N2–H2 or C35–N4–H4 | 111.6 | 115.9 | 111.1 | 113.9 | 111.5 | 111.5 |
| C19–N2–H2 or C38–N4–H4 | 113.4 | 116.9 | 112.8 | 113.2 | 113.2 | 113.3 |
| C16–N2–C19 or C35–N4–C38 | 121.8 | 125.2 | 120.7 | 123.3 | 121.6 | 121.5 |
| Sum of the valence angles | 346.8 | 358.0 | 344.9 | 350.4 | 346.3 | 346.3 |
The bond order analysis showed that bond order for the bond between C6–N1 (nitrogen atom attached to the carbon atom in the phenyl ring) is ca 1.10. For the bond between C16–N2 (nitrogen atom attached to the carbon atom in the perfluorinated phenyl ring) the bond order is ca 1.12, further explaining partially planar arrangement at the nitrogen atoms. Indeed the pyramidal shape at the N atom can invert its configuration in the structures investigated. The energy barrier of transition between the diastereomers (invertomers) was estimated in DFT calculations to ca 2.7 kcal mol−1 in vacuo. The temperature of coalescence (Tc) was approximated to ca 53 K in accordance to eqn (2) (Experimental section). The transition energy in chloroform differed from the value calculated in vacuo conditions, and was estimated to ca 2.5 kcal mol−1. The Tc temperature was approximated to ca 50 K.
Our quantum chemical computational results refer to isolated molecule or a molecule in a polarisable continuum that model chloroform solution. On the other hand in the solid state the molecules can interact with the actual molecules in the crystal environment. Unfortunately, the attempt to perform calculations with PBC (Periodic Boundary Conditions) for such a large system resulted in failure due to restrictions on supercomputer's resources.
In the crystal structures of 2k and 2l the principal interactions are N–H⋯O(P) hydrogen bonds that hold two enantiomers of opposite stereochemistry together around a centre of symmetry, as shown in Fig. 3. Geometrical parameters describing hydrogen bonds are listed in Table 4. It is worth noting that in 2k this motif occurs between enantiomers but not between diastereomers.
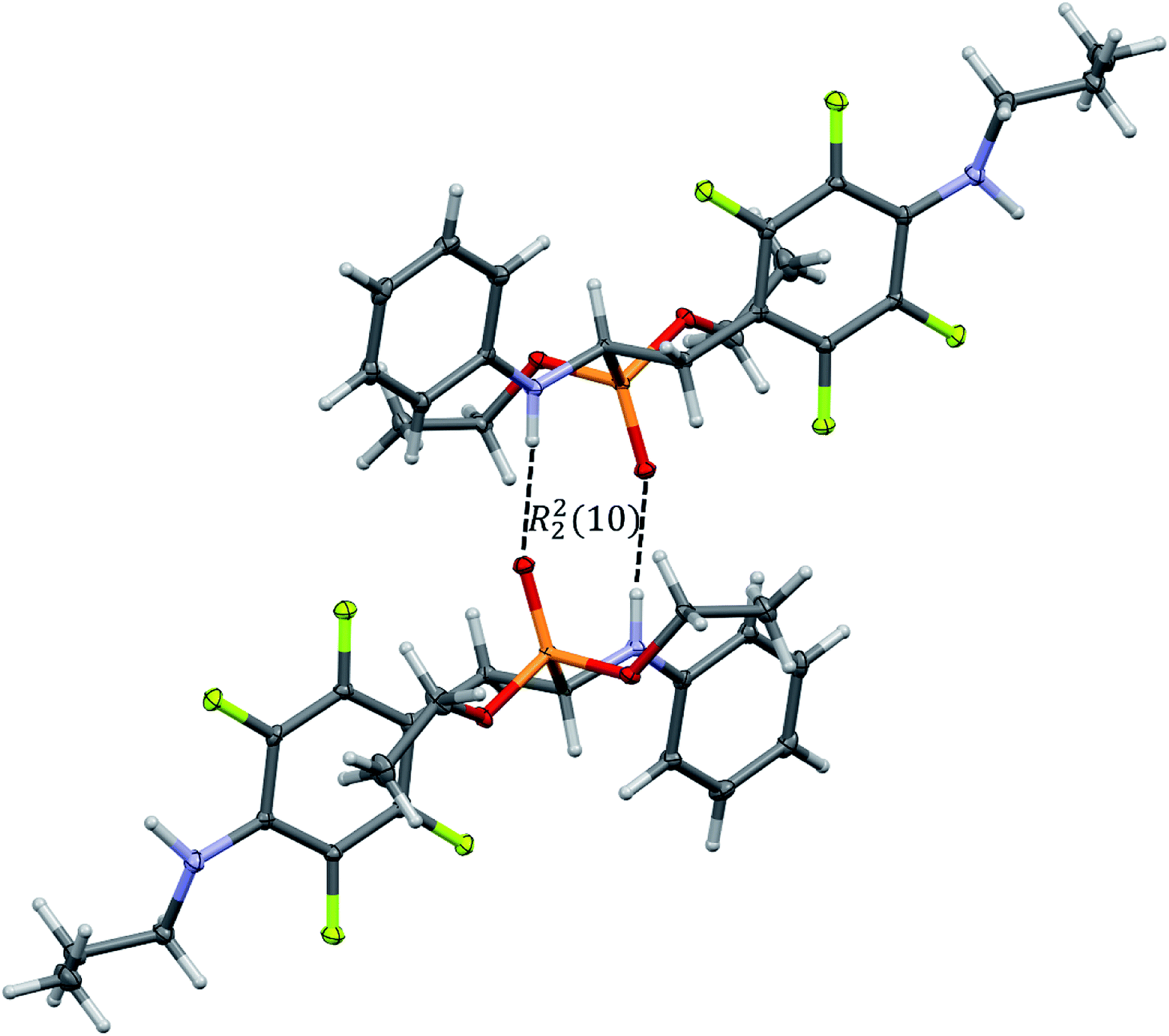 | ||
| Fig. 3 Dimeric motif of N–H⋯O(P) hydrogen bonds between two enantiomers of opposite stereochemistry in the crystals of 2l. This motif is also observed in the crystals of 2k. | ||
| D–H [Å] | H⋯A [Å] | D⋯A [Å] | D–H⋯A [°] | SYMM | |
|---|---|---|---|---|---|
| 2k | |||||
| N1–H1⋯O1 | 0.81(2) | 2.16(2) | 2.975(3) | 177(2) | [−x + 2, −y + 1, −z + 1] |
| N3–H3⋯O4 | 0.86(2) | 2.12(3) | 2.964(2) | 168(3) | [−x + 1, −y + 1, −z] |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| 2l | |||||
| N1–H1⋯O1 | 0.85(3) | 2.03(3) | 2.871(3) | 173(2) | [−x + 1, −y + 2, −z + 1] |
The self-sorting phenomenon of diastereomers at supramolecular level is observed in the crystals of 2k. Since there are two diastereomers in the asymmetric unit, each of them forms separate layer perpendicular to c lattice direction containing pairs of enantiomers. The layers are stabilized by the previously mentioned N–H⋯O(P) hydrogen bonds, which are supported by interactions involving π-electrons. The fluorinated rings of two neighbouring mol 1 oriented parallel to each other favours the formation of π⋯π interactions between these moieties, while a tilted arrangement of fluorinated and non-fluorinated aromatic rings of two mol 2 leads to formation of H⋯π and H⋯F contacts (2.77(3) and 2.67(3) Å, respectively) that are equal or shorter than the sum of van der Waals radii of hydrogen and carbon atoms (HvdW = 1.2 Å, CvdW = 1.7 Å, the sum = 2.9 Å),45 and hydrogen and fluorine atoms (FvdWr = 1.47 Å, the sum is 2.67 Å).45 The geometrical parameters describing π⋯π interactions are 3.319(3) and 0.88 Å for distance between the planes of aromatic rings along with the offset, respectively. The two layer types are alternately arranged in the crystal as shown in Fig. 4, and are related to each other through N–H⋯O(P) hydrogen bonds in which the methylamine group acts as a donor of hydrogen bonds. The geometrical parameters of the hydrogen bond listed in Table 5 suggest that the interactions between diastereomers are slightly weaker than those between enantiomers in the layer. Moreover, the proximity of the aromatic rings and ester group belonging to diastereomers in two neighbouring layers promotes the formation of multiple H⋯F and H⋯F interactions and thus substantially affects the crystal structure stabilization.
 | ||
| Fig. 4 Self-sorting of diastereomers on supramolecular level in the crystals of 2k. Two symmetry independent molecules that represent two diastereomers (mol 1 and mol 2) were distinguished by green and blue colors. | ||
| D–H [Å] | H⋯A [Å] | D⋯A [Å] | D–H⋯A [°] | SYMM | |
|---|---|---|---|---|---|
| 2k | |||||
| C11–H11⋯F7 | 0.95 | 2.55(3) | 3.333(4) | 140(3) | [x + 1, y, z] |
| C22–H22A⋯F3 | 0.99 | 2.55(3) | 3.387(4) | 143(3) | [−x + 1, −y + 1, −z] |
| C26–H26⋯F3 | 0.95 | 2.42(2) | 3.153(3) | 134(3) | |
| N4–H4⋯O1 | 0.85(3) | 2.29(4) | 3.074(3) | 154(3) | [−x + 1, −y + 1, −z + 1] |
| N2–H2⋯F7 | 0.79(3) | 2.53(3) | 3.017(3) | 121(3) | |
| C22–H22A⋯π | 0.99 | 2.78(4) | 3.486(5) | 129(4) | [−x + 1, −y + 1, −z] |
| C22–H22B⋯π | 0.99 | 2.67(5) | 3.529(6) | 145(5) | [−x + 1, −y + 1, −z] |
| C28–H28⋯F5 | 0.95 | 2.67(3) | 3.299(3) | 125(3) | [−x, −y, −z] |
|
|||||
| 2l | |||||
| C1–H1B⋯F1 | 0.99 | 2.59(3) | 3.339(2) | 133(2) | [−x + 2, −y + 2, −z + 1] |
| C5–H5⋯O2 | 1.00 | 2.62(2) | 3.526(2) | 152(2) | [−x + 2, −y + 2, −z + 1] |
| N2–H2⋯F3 | 0.98(3) | 2.19(3) | 3.159(2) | 171(2) | [−x + 1, −y + 1, −z] |
The extended C molecular conformation of 2l favours self-association forming dimers in which the two molecules interact via N–H⋯F hydrogen bonding and strong affects the formation of isolated π⋯π intermolecular interactions between fluorinated and non-fluorinated aromatic rings as shown in Fig. 5. The interacting rings are tilted toward each other by 16° while the interplanar distance between the centres of gravity is 3.796 Å. The intermolecular interactions present in the crystals are reported in Table 5.
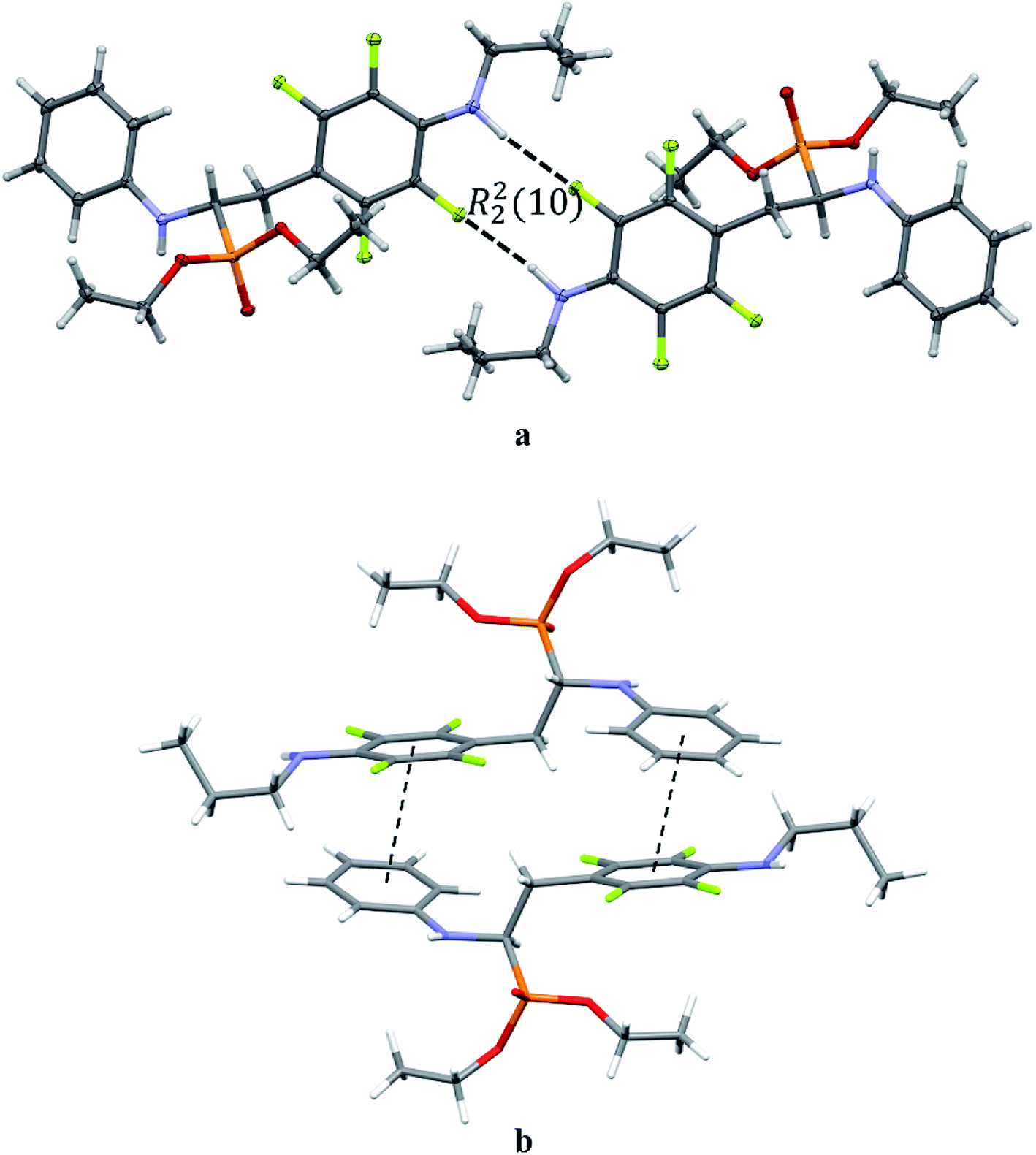 | ||
| Fig. 5 Dimeric motif of N–H⋯F interactions (a) and π⋯π interactions between two enantiomers of opposite stereochemistry in the crystals of 2l (b). | ||
2.3 Biological evaluation
Six randomly chosen SNAr reactions products, and also six of the fluorinated phosphonate analogues of phenylalanine, including 1a, previously synthesized in our research group,33 were subjected to biological studies. The first group of compounds contained mainly different thiophenols derivatives, while various α-aminophosphonates differing in number and position of fluorine substituents in one of the phenyl rings constituted the second group.In the compounds molecules, apart from amino group, two additional pharmacophores are present; fluorine substituents and phosphonate group that replaced an amino acid carbonyl one. A considerable number of α-aminophosphonates is known to exhibit various biological activities including anticancer one. On the other hand, a lot of existing drugs, for example synthetic statins and 5-fluorouracil, contain fluorine. This shows a remarkable potential of fluorine in pharmaceutical chemistry and provide a source for drug discovery.18 Therefore combination of fluorine pharmacophore with amino and phosphonate group in molecules of target compounds was expected to have an impact on their bioactivity.
The most common criteria used for preliminary evaluation of drug likeness of a compound encompass the Lipinski's “rule of 5”.46,47 In this respect, the physicochemical parameters of the examined compounds generally match the rule (Table 6).
| Cpd | MW | aPSA [Å2] | PSA [Å2] | logP |
HBD | HBA | RB | Caco-2 [nm s−1] | BB | MDCK [nm s−1] |
|---|---|---|---|---|---|---|---|---|---|---|
| a MW – molecular weight, aPSA – apolar surface area, PSA – polar surface area, logP – an octanol–water partition coefficient calculated with ALOGPS 2.1, HBD – number of hydrogen bond donors, HBA – number of hydrogen bond acceptors, RB – rotatable bonds number. |
||||||||||
| 1a | 423.31 | 303.9 | 8.7 | 4.22 | 1 | 4 | 9 | 21.72 | 1.38 | 86.84 |
| 1b | 385.80 | 294.6 | 13.5 | 4.27 | 1 | 4 | 9 | 21.79 | 1.30 | 102.26 |
| 1c | 385.80 | 312.7 | 8.7 | 4.25 | 1 | 4 | 9 | 21.74 | 1.27 | 104.23 |
| 1d | 383.37 | 295.7 | 13.5 | 4.18 | 1 | 4 | 9 | 21.72 | 1.26 | 116.53 |
| 1e | 351.35 | 297.4 | 8.7 | 3.64 | 1 | 4 | 9 | 21.72 | 0.98 | 168.46 |
| 1f | 387.34 | 283.1 | 13.5 | 3.95 | 1 | 4 | 9 | 21.72 | 1.00 | 109.96 |
| 2b | 527.52 | 364.9 | 27 | 6.25 | 1 | 4 | 11 | 21.72 | 1.06 | 42.40 |
| 2d | 543.52 | 363.8 | 35.3 | 5.75 | 1 | 5 | 12 | 21.72 | 0.18 | 12.35 |
| 2e | 543.52 | 367.5 | 36 | 5.75 | 1 | 5 | 12 | 21.72 | 0.18 | 14.77 |
| 2f | 528.51 | 325.4 | 52 | 5.18 | 3 | 5 | 11 | 21.70 | 0.19 | 3.94 |
| 2j | 580.15 | 329.0 | 81.8 | 3.75 | 2 | 8 | 15 | 21.69 | 0.01 | 0.06 |
| 2r | 531.87 | 361.8 | 13.4 | 5.92 | 1 | 5 | 11 | 21.73 | 3.06 | 44.20 |
According to Veber's rule, reduced molecular flexibility, as measured by the number of rotatable bonds, and low polar surface area or total hydrogen bond count (sum of donors and acceptors) are found to be important predictors of good oral bioavailability, independent of molecular weight.48 Veber's observations suggest that compounds which meet only the two criteria: 10 or fewer rotatable bonds and polar surface area equal to or less than 140 Å2 (or 12 or fewer H-bond donors and acceptors) will have a high probability of good oral bioavailability. All of the examined compounds meet these criteria perfectly (Table 6).
Caco-2 cell permeability model classifies compounds into 3 classes of permeability (high, medium and low). Compounds with tPapp below 4 nm s−1 are classified as low permeable; compounds with tPapp above 70 nm s−1 are classified as high permeable and compounds with permeability values between 4–70 nm s−1 are classified as medium.49 For blood–brain barrier penetration we calculated the BB value which is defined as the ratio of the concentration of a drug in the brain and in the blood, measured at equilibrium (BB = CBrain/CBlood). Compounds with BB > 2.0 cross the blood–brain barrier readily (high absorption to CNS (central nervous system)) while molecules with BB < 0.1 are poorly distributed to the brain (low absorption to CNS).50 Results show that all compounds are characterized by a good intestinal absorption (20–80%) and relatively high brain penetration, therefore making these compounds more druggable (Table 6).
| Compound | Compound structure | T98G IC50 [μM] | U-118 MG IC50 [μM] | HaCaT IC50 [μM] |
|---|---|---|---|---|
| 1a | 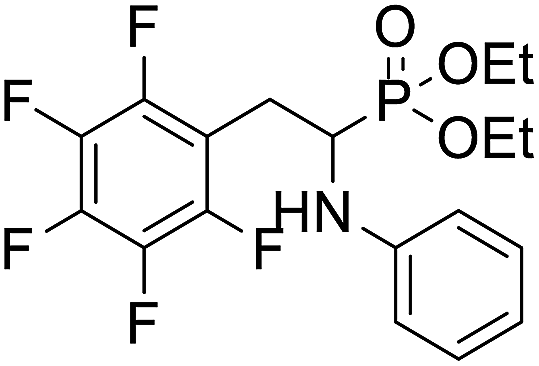 |
40.4 ± 6.2 | 60.67 ± 8.9 | 56.8 ± 7.4 |
| 1b | 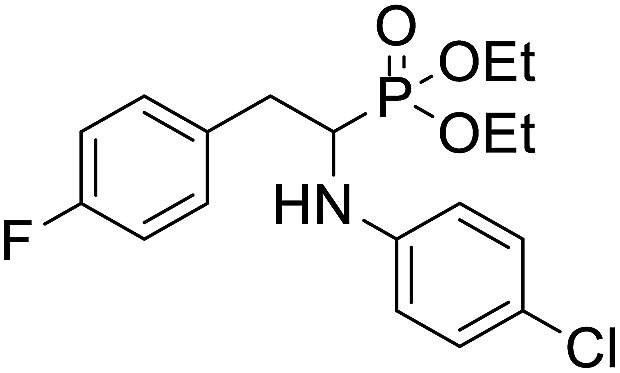 |
73.5 ± 9.2 | 88.2 ± 7.5 | >100 |
| 1c | 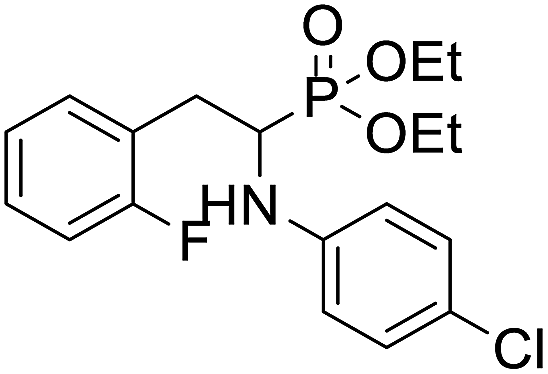 |
67.9 ± 11.2 | 91.2 ± 15.5 | >100 |
| 1d | 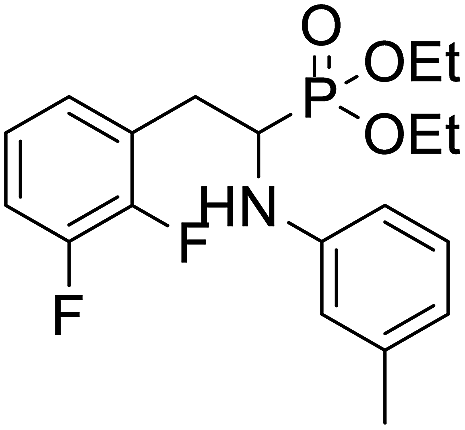 |
71.3 ± 8.7 | 48.6 ± 10.4 | 67.9 ± 11.8 |
| 1e | 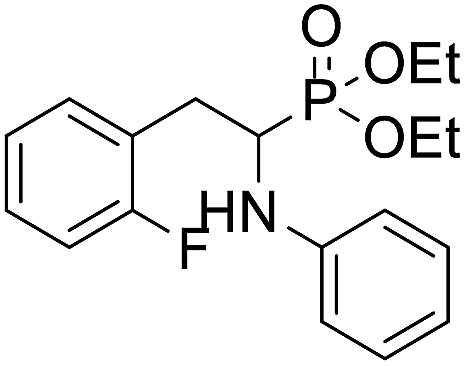 |
106.4 ± 14.7 | 86.8 ± 7.2 | 61.8 ± 9.1 |
| 1f | 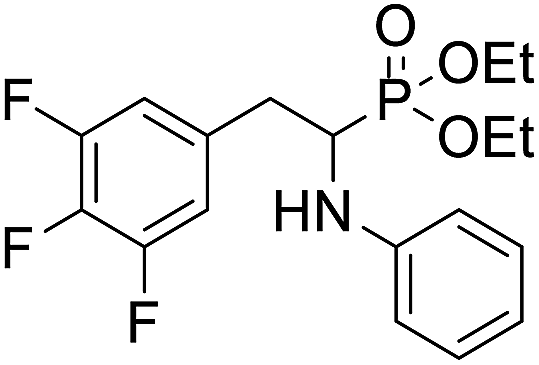 |
25.1 ± 4.5 | 40.8 ± 8.4 | 33.2 ± 5.1 |
| 2b | 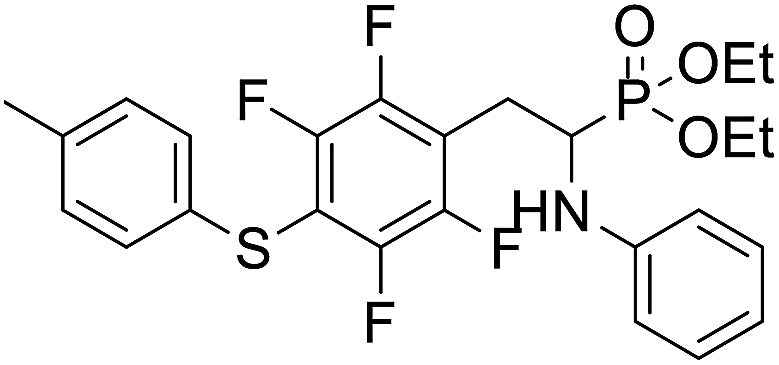 |
14.5 ± 3.3 | 37.9 ± 2.5 | 26.5 ± 4.7 |
| 2d | 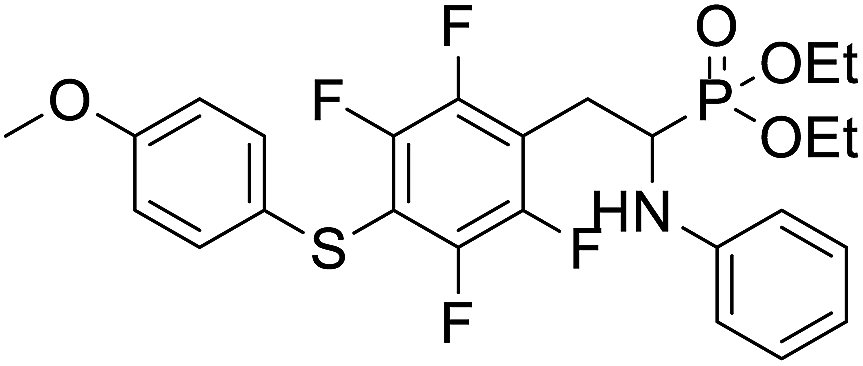 |
36.8 ± 5.7 | 33.2 ± 7.3 | 19.8 ± 3.9 |
| 2e | 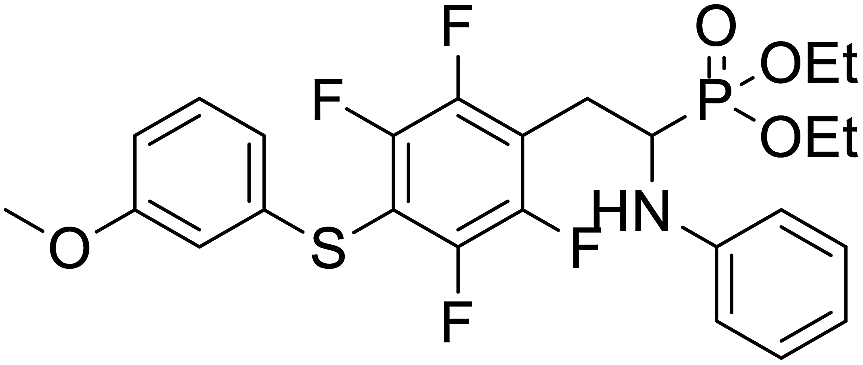 |
20.4 ± 3.1 | 21.4 ± 2.7 | 10.1 ± 2.2 |
| 2f | 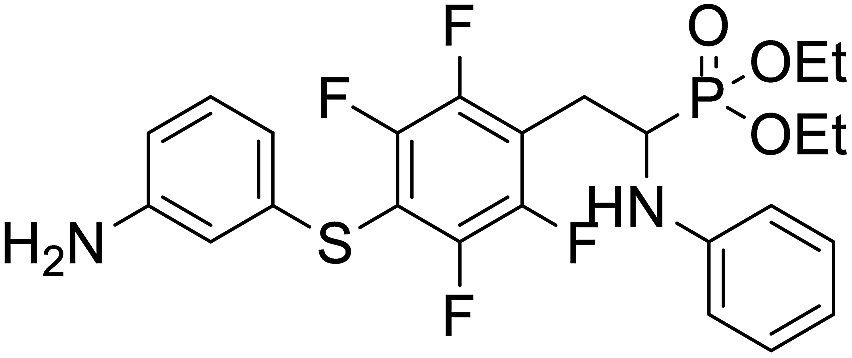 |
25.5 ± 1.9 | 28.1 ± 3.6 | 18.2 ± 3.4 |
| 2j | 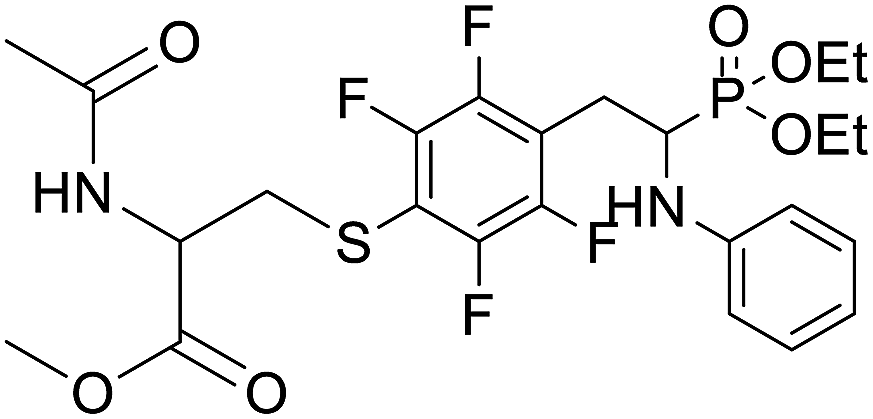 |
82.7 ± 15.7 | >100 | >100 |
| 2r | 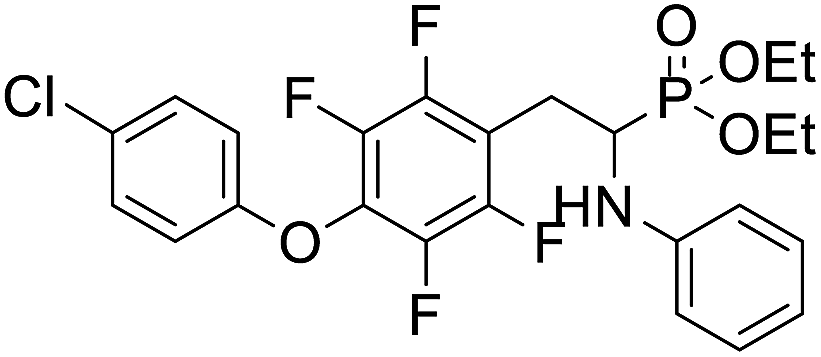 |
70.3 ± 8.3 | 79.2 ± 6.4 | 48.4 ± 7.4 |
| 5-FdU (5-fluoro-2′-deoxyuridine) | 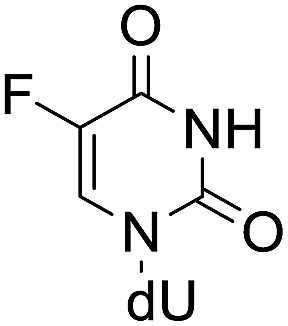 |
5.57 ± 0.9 | 23.40 ± 1.6 | 4.42 ± 1.1 |
As shown in this table, all compounds exhibited moderate cytostatic activity in the both glioblastoma cell lines. Most of the compounds showed higher activity against T98G cell line. Only 1d, 1e and to a small degree also 2d occurred to be better inhibitors of U-118MG cell line. Most of the studied SNAr reactions products displayed significantly higher inhibitory activity against both glioblastoma multiforme cell lines than the parent α-aminophosphonate 1a. This indicates that the introduction of thiophenols, but not aliphatic thiols nor phenols, to the para position in the fluorinated phenyl ring of 1a molecule improve the antiglioma activity. IC50 values also demonstrate that for compounds 2b, 2d, 2e and 2f, the substituents in the thiophenol moiety had an influence on the cytotoxic activity (Table 7). All these substituents belong to EDG and it can be concluded that less electrodonating character of CH3 group than of OCH3 and NH2 groups, results in higher activity of 2b in comparison with 2d–2f. Among the SNAr reactions products studied, 2b exhibiting the highest cytotoxic activity against T98G cell line and good selectivity can be considered as the best candidate for antiglioma drug.
Analysis of IC50 values for α-aminophosphonates 1a–1f, indicates that number of fluorine substituents in one aromatic ring as well as presence and character of substituents in the second ring contribute to the anticancer activity displayed by these compounds. Compounds 1b and 1c containing one fluorine and one chlorine atoms, exhibit very similar inhibitory activity against both cancer cell lines indicating that position of this fluorine atom does not influence much the activity. Comparing these compounds with the 1e demonstrates that chlorine substituent may constitute an important factor contributing to anticancer activity. Number of fluorine substituents also seems to have impact on the cytotoxic inhibition, though the influence is irregular.
The inhibitory effects of α-aminophosphonates in T98G cells were further assessed by performing clonogenic assays (Fig. 6). Compounds 2d–f were also significantly more potent than other compounds in this assay. Treatment with 20 μM 2d–f resulted in clonogenic survival of 85%, 30%, and 10%, respectively, compared to control cells. When we tested the effect of each compound at 50 μM, we found that 2d reduces cancer cell clonogenic potential by 25%, 2e by 85%, and 2f by 98%.
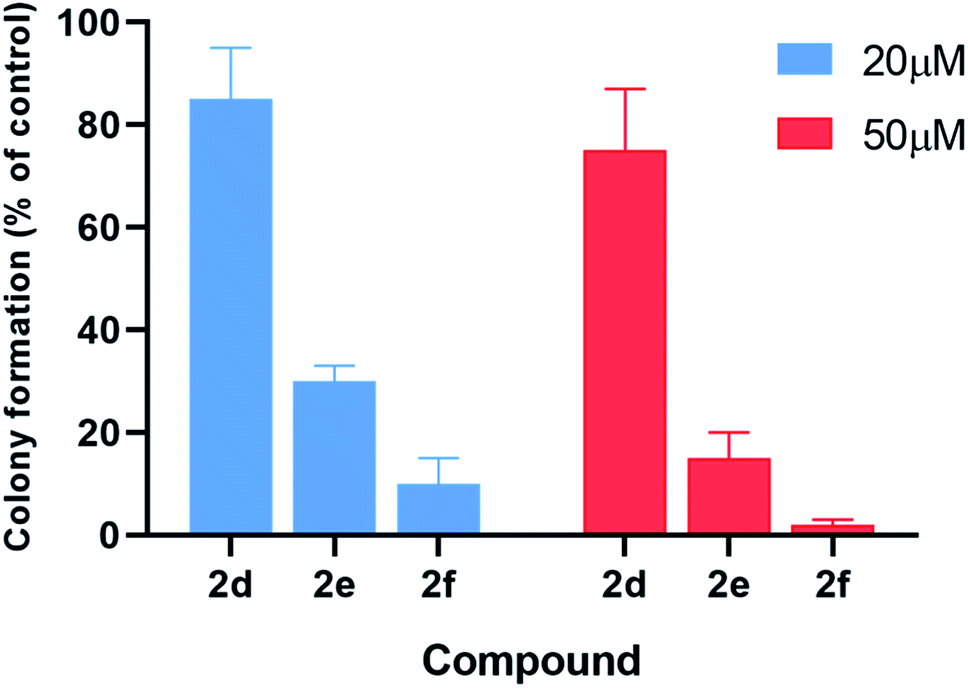 | ||
| Fig. 6 Treatment with compounds 2d–f significantly reduces the colony-forming ability of T98G cells as compared to untreated cells. Error bars display the standard deviation from at least three independent measurements. | ||
3. Conclusions
Several diethyl (2-(perfluorophenyl)-1-(phenylamino)ethyl)phosphonate derivatives were synthesised by SNAr reactions, and subjected to structural and biological studies. X-ray single crystal diffraction analysis performed for two of the obtained compounds (2k and 2l) revealed their conformational preferences, self-sorting at supramolecular level and different conformation at the aniline nitrogen atoms. It was found that in the solid state, 2k molecules adopt conformation with almost perpendicular arrangement of both aromatic rings, while molecule of 2l prefers extended conformation with the aromatic rings arranged nearly parallel. Interestingly, in the crystals of parent 1a its molecules also adopt conformation with parallel oriented rings. This indicates that introduction of substituent into para position in the fluorinated ring may or may not change the conformation of the resulted molecule comparing to the starting material.Due to the different configuration at the aniline nitrogen atoms, two molecules in 2k crystal structure constitute a pair of diastereomers. This phenomenon was studied with the use of quantum-chemical methods. Results obtained at WB97XD/6-31+G(d) level of theory for isolated molecule show energetic preference of R,S,R stereomer, while for a molecule in a polarisable continuum mimicking chloroform solution indicate the R,R,R stereomer as an energetically favourable. These results generally support the stereomers structural diversity. Coexistence of both stereomers observed in the crystal structure is most likely due to intermolecular interactions.
Structurally various α-aminophosphonates are known to exhibit activity against different cancer cell lines. However, to the best of our knowledge there is no studies referring to α-aminophosphonates as potential antiglioma agents. Malignant gliomas remain almost always fatal, and none of the current state of the art treatments can be regarded as effective.51,52 Synthesised by SNAr reactions phosphono-perfluorophenylalanine derivatives perfectly meet the criteria of Lipinski's and Veber's rules. MTT assay results demonstrated that the compounds, particularly these formed by introduction of thiophenol moiety to para position in the fluorinated ring, exhibited significantly higher inhibitory activity against T98G and U118 MG cell lines than the parent 1a. This enables to conclude that phosphono-perfluorophenylalanine core constitute promising scaffold for further studies focused on developing of new agents active against glioblastoma multiforme.
4. Experimental Section
4.1 Synthesis
:1, v/v).
4.1.2.1 Diethyl (1-(phenylamino)-2-(2,3,5,6-tetrafluoro-4-(phenylthio)phenyl)ethyl)phosphonate (2a). Pale yellow oil (48 mg, 87%). 1H NMR (400 MHz, acetone-d6): δ = 7.33–7.28 (m, 3H, 3 CHar), 7.18–7.16 (m, 2H, 2CHar), 7.06–7.02 (m, 2H, 2CHar), 6.70 (d, J = 7.9 Hz, 2H, 2CHar), 6.60 (t, J = 7.3 Hz, 1H, CHar), 4.86–4.83 (m, 1H, NH) 4.16–4.05 (m, 5H, 2OCH2, CH), 3.36–3.30 (m, 1H, ArCH2), 3.24–3.14 (m, 1H, ArCH2), 1.25 (t, J = 7.1 Hz, 3H, CH3), 1.20 (t, J = 7.1 Hz, 3H, CH3). 19F NMR (377 MHz, acetone-d6) δ = −135.96 to −136.06 (m, 2F), −142.90 to −142.99 (m, 2F). 31P NMR (162 MHz, acetone-d6) δ = 22.95 (s). 13C NMR (101 MHz, CDCl3) δ = 148.05–145.45 (m, 2Car) 146.31 (d, J = 7.3 Hz, Car), 146.79–144.11 (m, 2Car) 133.39 (s, Car), 129.83 (s, 2CHar), 129.32–129.26 (m, 4 CHar) 127.50 (s, CHar), 118.77 (s, CHar), 118.25–117.89 (m, Car), 113.28 (s, 2CHar), 112.14–111.74 (m, Car), 63.73 (d, J = 7.0 Hz, OCH2), 62.46 (d, J = 7.4 Hz, OCH2), 50.46 (d, J = 158.3 Hz, CH), 25.16 (d, J = 7.4 Hz, ArCH2), 16.56, 16.51, 16.45, and 16.39 (2CH3). HRMS (ESI+) calcd for C24H25F4NO3PS (M + H)+: 514.1229, found: 514.1230.
4.1.2.2 Diethyl (1-(phenylamino)-2-(2,3,5,6-tetrafluoro-4-(m-tolylthio)phenyl)ethyl)phosphonate (2b). Pale yellow solid (55 mg, 89%), mp = 103–105 °C. 1H NMR (400 MHz, acetone-d6) δ = 7.20–7.10 (m, 4H, 4 CHar), 7.05–7.01 (m, 2H, 2CHar), 6.70 (d, J = 7.9 Hz, 1H, 2CHar), 6.60 (t, J = 7.3 Hz, 1H, CHar), 4.87–4.83 (m, 1H, NH), 4.13–4.04 (m, 5H, CH, 2OCH2), 3.33–3.28 (m, 1H, ArCH2), 3.22–3.13 (m, 1H, ArCH2), 2.30 (s, 3H, ArCH3), 1.24 (t, J = 7.1 Hz, 3H, CH3), 1.19 (t, J = 7.1 Hz, 3H, CH3). 19F NMR (377 MHz, CDCl3) δ = −134.05 (dd, J = 23.6, 11.5 Hz, 2F), −142.52 to −142.62 (m, 2F). 31P NMR (162 MHz, acetone-d6) δ = 23.01 (s). 13C NMR (101 MHz, CDCl3) δ = 147.96–146.36 (m, 2Car), 146.77–144.05 (m, 2Car), 146.32 (d, J = 7.2 Hz, Car), 137.99 (s, Car), 130.83 (s, 2CHar), 130.11 (s, 2CHar), 129.62 (s, Car), 129.31 (s, 2CHar), 118.80 (s, CHar), 117.83–117.47 (m, Car), 113.36 (s, 2CHar), 112.87 (s, Car), 63.75 (d, J = 7.0 Hz, OCH2), 62.48 (d, J = 7.4 Hz, OCH2), 50.51 (d, J = 158.2 Hz, CH), 25.13 (d, J = 7.4 Hz, ArCH2), 21.23 (s, ArCH3), 16.59, 16.54, 16.48, and 16.42 (2CH3). HRMS (ESI+) calcd for C25H27F4NO3PS (M + H)+: 528.1385, found: 528.1387.
4.1.2.3 Diethyl (2-(4-((3,5-dimethylphenyl)thio)-2,3,5,6-tetrafluorophenyl)-1-(phenylamino)ethyl)phosphonate (2c). Pale yellow solid (52 mg, 81%). 1H NMR (400 MHz, acetone-d6) δ = 7.05–7.01 (m, 2H, 2CHar), 6.91–6.89 (m, 3H, 3 CHar), 6.72 (d, J = 7.8 Hz, 2H, 2CHar), 6.57 (t, J = 7.3 Hz, 1H, CHar), 4.89–4.86 (m, 1H, NH), 4.13–4.02 (m, 5H, CH, 2OCH2), 3.33–3.30 (m, 1H, ArCH2), 3.23–3.13 (m, 1H, ArCH2), 2.83 (s, 6H, 2 ArCH3), 1.23 (t, J = 7.1 Hz, 3H, CH3), 1.17 (t, J = 7.1 Hz, 3H, CH3). 19F NMR (377 MHz, CDCl3) δ = −133.72 to −133.81 (m, 2F), −142.38 (dd, J = 24.4, 11.9 Hz, 2F). 31P NMR (162 MHz, acetone-d6) δ = 23.10 (s). 13C NMR (101 MHz, CDCl3) δ = 148.13–145.45 (m, 2Car), 146.78–144.11 (m, 2Car) 146.30 (d, J = 6.5 Hz, Car), 139.06 (s, Car), 132.63 (s, 2Car), 129.69 (s, CHar) 129.31 (s, 2CHar), 128.05 (s, 2CHar), 118.87 (s, CHar), 118.13–117.64 (m, Car), 113.49 (s, 2CHar), 112.67–112.26 (m, Car), 63.69 (d, J = 7.0 Hz, OCH2), 62.44 (d, J = 7.4 Hz, OCH2), 50.60 (d, J = 157.4 Hz, CH), 25.09 (d, J = 7.3 Hz, ArCH2), 21.28 (s, 2 ArCH3), 16.56, 16.51, 16.44, and 16.38 (2CH3). HRMS (ESI+) calcd for C26H29F4NO3PS (M + H)+: 542.1542, found: 542.1542.
4.1.2.4 Diethyl (1-(phenylamino)-2-(2,3,5,6-tetrafluoro-4-((-methoxyphenyl)thio)phenyl)ethyl)phosphonate (2d). Pale yellow solid (61 mg, 95%), mp = 93–95 °C. 1H NMR (400 MHz, acetone-d6) δ = 7.30–7.28 (m, 2H, 2CHar), 7.05–6.98 (m, 2H, 2CHar), 6.92–6.90 (m, 2H, 2CHar), 6.68 (d, J = 7.8 Hz, 2H, 2CHar), 6.58 (t, J = 7.2 Hz, 1H, CHar), 4.85–4.82 (m, 1H, NH), 4.11–4.06 (m, 5H, CH, 2OCH2), 3.80 (s, 3H, ArCH3), 3.31–3.27 (m, 1H, ArCH2), 3.19–3.10 (m, 1H, ArCH2), 1.23 (t, J = 7.1 Hz, 3H, CH3), 1.17 (t, J = 7.1 Hz, 3H, CH3). 19F NMR (377 MHz, CDCl3) δ = −134.67 (dd, J = 24.0, 11.8 Hz, 2F), −142.70 (dd, J = 24.3, 12.2 Hz, 2F). 31P NMR (162 MHz, acetone-d6) δ = 23.04 (s). 13C NMR (101 MHz, CDCl3): δ = 159.95 (s, Car), 147.79–145.13 (m, 2Car), 146.70–144.00 (m, 2Car), 146.30 (d, J = 7.1 Hz, Car), 134.07 (s, 2CHar), 129.25 (s, 2CHar), 123.25 (s, Car), 118.74 (s, CHar), 117.61–117.11 (m, Car), 114.88 (s, 2CHar), 114.14–113.69 (m, Car), 113.36 (s, 2CHar), 63.69 (d, J = 7.0 Hz, OCH2), 62.43 (d, J = 7.4 Hz, OCH2), 55.44 (s, OCH3), 50.47 (d, J = 158.1 Hz, CH), 25.00 (s, ArCH2), 16.54, 16.49, 16.42, 16.36 (2CH3). HRMS (ESI+) calcd for C25H27F4NO4PS (M + H)+: 544.1335, found: 544.1339.
4.1.2.5 Diethyl (1-(phenylamino)-2-(2,3,5,6-tetrafluoro-4-((3-methoxyphenyl)thio)phenyl)ethyl)phosphonate (2e). Yellow oil (61 mg, 95%). 1H NMR (400 MHz, acetone-d6) δ = 7.23 (t, J = 8.0 Hz, 1H, CHar), 7.06–7.02 (m, 2H, 2CHar), 6.85–6.80 (m, 2H, 2CHar), 6.71 (d, J = 7.9 Hz, 1H, 2CHar), 6.66 (d, J = 7.9 Hz, 1H, CHar), 6.59 (t, J = 7.3 Hz, 1H, CHar), 4.88–4.86 (m, NH), 4.15–4.04 (m, 5H, 2OCH2, CH), 3.75 (s, 3H, OCH3), 3.36–3.30 (m, 1H, ArCH2), 3.24–3.14 (m, 1H, ArCH2), 1.25 (t, J = 7.1 Hz, 3H, CH3), 1.19 (t, J = 7.0 Hz, 3H, CH3). 19F NMR (377 MHz, CDCl3): δ = −133.45 to −133.55 (m, 2F), −142.22 (dd, J = 24.4, 12.4 Hz, 2F). 31P NMR (162 MHz, acetone-d6): δ = 23.04 (s). 13C NMR (101 MHz, CDCl3): δ = 159.99 (s, Car), 148.10–145.44 (m, 2Car), 147.90–144.05 (m, 2Car), 146.28 (d, J = 6.9 Hz, Car), 134.52 (s, Car), 130.15 (s, CHar), 129.28–129.24 (s, 2CHar), 121.84 (s, CHar), 118.81 (s, CHar), 118.40–118.04 (m, Car), 115.25 (s, CHar), 113.34 (s, 2CHar), 113.12 (s, CHar), 111.88–111.47 (m, Car), 63.68 (d, J = 7.0 Hz, OCH2), 62.45 (d, J = 7.4 Hz, OCH2), 55.34 (s, OCH3), 50.48 (d, J = 157.9 Hz, CH), 25.12 (d, J = 7.3 Hz, ArCH2), 16.53, 16.47, 16.42, and 16.36 (2CH3). HRMS (ESI+) calcd for C25H27F4NO4PS (M + H)+: 544.1335, found: 544.1328.
4.1.2.6 Diethyl (2-(4-((3-aminophenyl)thio)-2,3,5,6-tetrafluorophenyl)-1-(phenylamino)ethyl)phosphonate (2f). Pale yellow oil (47 mg, 76%). 1H NMR (400 MHz, acetone-d6) δ = 7.06–6.96 (m, 3H, 3 CHar), 6.71 (d, J = 8.3 Hz, 2H, 2CHar), 6.57 (m, 3H, 3 CHar), 6.35 (d, J = 8.2 Hz, 1H, CHar), 4.86–4.83 (m, 1H, NH2), 4.76–4.74 (m, 1H, NH2), 4.13–4.06 (m, 6H, CH, NH2OCH2), 3.35–3.29 (m, 1H, ArCH2), 3.23–3.14 (m, 1H, ArCH2), 1.24 (t, J = 7.1 Hz, 3H, CH3), 1.19 (t, J = 7.1 Hz, 3H, CH3). 19F NMR (377 MHz, CDCl3) δ = −133.46 (dd, J = 23.5, 11.4 Hz, 2F), −142.32 to −142.42 (m, 2F). 31P NMR (162 MHz, acetone-d6) δ = 23.04 (s). 13C NMR (101 MHz, CDCl3) δ = 148.19–145.53 (m, 2Car), 147.21 (s, Car), 146.65–144.08 (m, 2Car), 146.35 (d, J = 6.6 Hz, Car), 134.31 (s, Car), 130.12 (s, CHar), 129.35 (s, 2CHar), 119.61 (s, CHar), 118.80 (s, CHar), 118.37–117.88 (m, Car), 115.64 (s, CHar), 114.24 (s, CHar), 113.41 (s, 2CHar), 112.18–111.78 (m, Car), 63.68 (d, J = 7.0 Hz, OCH2), 62.51 (d, J = 7.4 Hz, OCH2), 50.56 (d, J = 157.9 Hz, CH), 25.09 (s, ArCH2), 16.58, 16.52, 16.47, and 16.42 (2CH3). HRMS (ESI+) calcd for C24H26F4N2O3PS (M + H)+: 529.1338, found: 529.1335.
4.1.2.7 Diethyl (2-(4-((4-bromophenyl)thio)-2,3,5,6-tetrafluorophenyl)-1-(phenylamino)ethyl)phosphonate (2g). Pale yellow solid (60 mg, 86%), mp = 77–79 °C. 1H NMR (400 MHz, acetone-d6) δ = 7.52–7.49 (m, 2H, 2CHar), 7.14–7.04 (m, 2H, 2CHar), 7.05–7.04 (m, 2H, 2CHar), 6.70 (d, J = 8.6 Hz, 2H, 2CHar), 6.61 (t, J = 7.4 Hz, 1H, CHar), 4.87–4.83 (m, 1H, NH), 4.16–4.05 (m, 5H, CH, 2OCH2), 3.36–3.29 (m, 1H, ArCH2), 3.24–3.18 (m, 1H, ArCH2), 1.26 (t, J = 7.1 Hz, 3H, CH3), 1.20 (t, J = 7.0 Hz, 3H, CH3). 19F NMR (377 MHz, acetone-d6) δ = −135.90 to −136.00 (m, 2F), −142.59 to −142.69 (m, 2F). 31P NMR (162 MHz, acetone-d6) δ = 22.89 (s). 13C NMR (101 MHz, CDCl3) δ = 148.05–145.38 (m, 2Car), 146.87–144.21 (m, 2Car) 146.33 (d, J = 7.2 Hz, Car), 132.59 (s, Car), 132.46 (s, 2CHar), 131.50 (s, 2CHar), 129.34 (s, 2CHar), 121.74 (s, Car), 118.79 (s, CHar), 118.49 (s, Car), 113.27 (s, 2CHar), 111.45 (s, Car), 63.78 (d, J = 7.0 Hz, OCH2), 62.54 (d, J = 7.4 Hz, OCH2), 50.43 (d, J = 158.6 Hz, CH), 25.23 (d, J = 7.5 Hz, ArCH2), 16.61, 16.56, 16.51, and 16.45 (2CH3). HRMS (ESI+) calcd for C24H24BrF4NO3PS (M + H)+: 592.0334, found: 592.0336.
4.1.2.8 Diethyl (2-(4-(benzylthio)-2,3,5,6-tetrafluorophenyl)-1-(phenylamino)ethyl)phosphonate (2h). Yellow oil (40 mg, 65%). 1H NMR (400 MHz, acetone-d6) δ = 7.33–7.20 (m, 1H, CHar), 7.17 (s, 4H, 4 CHar), 7.08–7.04 (m, 2H, 2CHar), 6.70 (d, J = 7.9 Hz, 2H, 2CHar), 6.59 (t, J = 7.3 Hz, 1H, CHar), 4.82–4.79 (m, 1H, NH), 4.13–4.02 (m, 7H, SCH2, CH, 2OCH2), 3.30–3.24 (m, 1H, ArCH2), 3.17–3.08 (m, 1H, ArCH2), 1.24 (t, J = 7.1 Hz, 3H, CH3), 1.17 (t, J = 7.1 Hz, 3H, CH3). 19F NMR (377 MHz, acetone-d6) δ = −136.30 to −136.40 (m, 2F), −143.81 to −143.90 (m, 2F). 31P NMR (162 MHz, acetone-d6) δ = 23.13 (s). 13C NMR (101 MHz, CDCl3) δ = 148.18–145.53 (m, 2Car), 146.48–143.90 (m, 2Car) 146.45 (d, J = 5.7 Hz, Car), 136.55 (s, Car), 129.32 (s, 2CHar), 128.85 (s, CHar), 128.66 (s, 2CHar), 127.75 (s, 2CHar), 118.81 (s, CHar), 117.27–116.91 (m, Car), 113.51 (s, 2CHar), 112.49–112.08 (m, Car), 63.59 (d, J = 7.0 Hz, OCH2), 62.45 (d, J = 7.4 Hz, OCH2), 50.62 (d, J = 157.1 Hz, CH), 39.14 (s, ArCH2S), 24.93 (d, J = 6.9 Hz, ArCH2), 16.56, 16.51, 16.48, and 16.42 (2CH3). HRMS (ESI+) calcd for C25H27F4NO3PS (M + H)+: 528.1385, found: 528.1387.
4.1.2.9 Diethyl (2-(4-(decylthio)-2,3,5,6-tetrafluorophenyl)-1-(phenylamino)ethyl)phosphonate (2i). Yellow oil (25 mg, 36%). 1H NMR (400 MHz, CDCl3) δ = 7.07–7.03 (m, 2H, 2CHar), 6.64 (t, J = 7.3 Hz, 1H, CHar), 6.53 (d, J = 7.9 Hz, 2H, 2CHar), 4.18–4.01 (m, 5H, CHP, 2OCH2), 3.82–3.79 (m, 1H, NH), 3.28–3.24 (m, 1H, ArCH2), 3.17–3.14 (m, 1H, ArCH2), 2.80 (t, J = 7.3 Hz, 2H, CH2S), 1.48–1.19 (m, 22H, 2CH3, CH3(CH2)8), 0.88 (t, J = 6.9 Hz, 3H, CH3(CH2)9). 19F NMR (377 MHz, CDCl3) δ = −134.89 (dd, J = 24.3, 12.0 Hz, 2F), −143.26 (dd, J = 24.3, 12.1 Hz, 2F). 31P NMR (162 MHz, CDCl3) δ = 23.52 (s). 13C NMR (101 MHz, CDCl3) δ = 148.15–145.53 (m, 2Car), 146.68–143.97 (m, 2Car) 146.41 (d, J = 6.9 Hz, Car), 129.27 (s, 2CHar), 118.76 (s, CHar), 116.81–116.44 (m, Car), 113.44 (s, 2CHar), 113.15 (s, Car), 63.67 (d, J = 7.0 Hz, OCH2), 62.46 (d, J = 7.4 Hz, OCH2), 50.61 (d, J = 157.7 Hz, CH), 34.84 (s, SCH2), 32.01 (s, CH3(CH2)8), 29.83 (s, CH3(CH2)8), 29.63 (s, CH3(CH2)8), 29.59 (s, CH3(CH2)8), 29.41 (s, CH3(CH2)8), 29.16 (s, CH3(CH2)8), 28.48 (s, CH3(CH2)8), 25.03 (d, J = 7.3 Hz, ArCH2), 22.80 (s, CH3(CH2)8), 16.59, 16.53, 16.49, and 16.43 (2CH3), 14.23 (s, CH3(CH2)9) ppm. HRMS (ESI+) calcd for C28H41F4NO3SP (M + H)+: 578.2481, found: 578.2477.
4.1.2.10 Methyl 2-acetamido-3-((4-(2-(diethoxyphosphoryl)-2-(phenylamino)ethyl)-2,3,5,6-tetrafluorophenyl)thio)propanoate (2j). Yellow oil (49 mg, 71%). 1H NMR (400 MHz, CDCl3) δ = 7.06 (t, J = 7.7 Hz, 2H, 2CHar), 6.65 (t, J = 7.2 Hz, 1H, CHar), 6.54–6.51 (m, 2H, 2CHar), 6.38–6.34 (m, 1H, NHCO), 4.76–4.72 (m, 1H, CHN), 4.14–4.08 (m, 5H, CHP, 2OCH2), 3.86, 3.85 (2d, J = 3.6, 3.6 Hz, 1H, ArNH), 3.44, 3.41 (2s, 3H, CH3O), 3.35–3.14 (m, 4H, ArCH2, SCH2), 1.92 (s, 3H, CH3CO), 1.29 (t, J = 7.1 Hz, 3H, CH3), 1.17 (td, J = 7.1, 3.4 Hz, 3H, CH3). 19F NMR (377 MHz, CDCl3) δ = −133.64 to −133.74 (m, 2F), −142.36 to −142.49 (m, 2F) ppm. 31P NMR (162 MHz, CDCl3): δ = 23.45 and 23.41 (2s). 13C NMR (101 MHz, CDCl3) δ = 170.28 (s, CO), 169.92, 169.90 (2s, CO), 148.16–145.56 (m, 2Car), 146.31, 146.29 (2d, J = 5.0, 4.8 Hz, Car), 145.71–144.24 (m, 2Car), 129.34 (s, 2CHar), 118.86, 118.83 (2s, CHar), 118.10–117.59 (m, Car), 113.51, 113.44 (2s, 2CHar), 111.81–111.40 (m, Car), 63.6 (d, J = 7.0 Hz, POCH2), 62.56, 62.48 (2d, J = 7.5, 7.4 Hz, POCH2), 52.56, 52.55 (2s, CH3O), 52.08, 52.02 (2s, CHCH2S), 50.5, 50.4 (2d, J = 156.1, 156.2 PCH), 36.31, 36.24 (2t, J = 2.2, 2.1 Hz, SCH2), 24.89, 24.82 (2d, J = 5.1, 6.2 Hz, ArCH2), 22.95 (s, COCH3), 16.5, 16.4 (2d, J = 5.3, 5.7 Hz, CH3). HRMS (ESI+) calcd for C24H30F4N2O6PS (M + H)+: 581.1498, found: 581.1503.
:1, v/v).
4.1.3.1 Diethyl (1-(phenylamino)-2-(2,3,5,6-tetrafluoro-4-(methylamino)phenyl)ethyl)phosphonate (2k). Pale yellow solid (31 mg, 60%), mp = 74–76 °C. 1H NMR (400 MHz, CDCl3) δ = 7.10–7.06 (m, 2H, 2CHar), 6.65 (t, J = 7.4 Hz, 1H, CHar), 6.55 (d, J = 7.9 Hz, 2H, 2CHar), 4.17–3.97 (m, 5H, CH, 2OCH2), 3.77 (dd, J = 10.6, 3.4 Hz, 1H, NH), 3.66 (bs, 1H, NH), 3.19–3.13 (m, 1H, ArCH2), 3.06–2.99 (m, 4H, NCH3, ArCH2), 1.30 (t, J = 7.0 Hz, 3H, CH3), 1.20 (t, J = 7.1 Hz, 3H, CH3). 19F NMR (377 MHz, CDCl3) δ = −146.46 to −146.55 (m, 2F), −161.22 to −161.30 (m, 2F). 31P NMR (162 MHz, CDCl3) δ = 24.13 (s). 13C NMR (101 MHz, CDCl3) δ = 147.18–144.46 (m, 2Car), 146.75 (d, J = 5.9 Hz, Car), 138.76–136.11 (m, 2Car), 129.22 (s, 2CHar), 124.77–124.68 (m, Car), 118.42 (s, CHar), 113.48 (s, 2CHar), 103.17–102.67 (m, Car), 63.44 (d, J = 7.0 Hz, OCH2), 62.34 (d, J = 7.5 Hz, OCH2), 51.06 (d, J = 156.3 Hz, CH), 33.18 (s, NCH3), 24.03 (d, J = 6.6 Hz, ArCH2), 16.60, 16.55, 16.52, 16.46 (2CH3). HRMS (ESI+) calcd for C19H24F4N2O3P (M + H)+: 435.1461, found: 435.1465.
4.1.3.2 Diethyl (1-(phenylamino)-2-(2,3,5,6-tetrafluoro-4-(propylamino)phenyl)ethyl)phosphonate (2l). Pale yellow solid (40 mg, 74%), mp = 78–80 °C. 1H NMR (400 MHz, CDCl3) δ = 7.08–7.04 (m, 1H, 2CHar), 6.64 (t, J = 7.3 Hz, 1H, CHar), 6.54 (d, J = 7.9 Hz, 2H, 2CHar), 4.17–3.98 (m, 5H, CH, 2OCH2), 3.80–3.65 (m, 1H, NH), 3.65 (s, 1H, CH2NH), 3.27–3.22 (m, 2H, CH3CH2CH2N), 3.18–3.13 (m, 1H, ArCH2), 3.06–3.00 (m, 1H, ArCH2), 1.52 (dd, J = 14.4, 7.2 Hz, 2H, CH3CH2CH2N), 1.30 (t, J = 7.1 Hz, 3H, CH3), 1.20 (t, J = 7.1 Hz, 3H, CH3), 0.92 (t, J = 7.4 Hz, 3H, CH3CH2CH2N). 19F NMR (377 MHz, CDCl3) δ = −146.46 to −146.53 (m, 2F), −160.68 to −160.72 (m, 2F). 31P NMR (162 MHz, CDCl3) δ = 24.13 (s). 13C NMR (101 MHz, CDCl3) δ = 147.16–144.51 (m, 2Car), 146.72 (d, J = 6.3 Hz, Car), 138.79–136.16 (m, 2Car), 129.17 (s, 2CHar) 127.17–126.90 (m, Car), 118.37 (s, CHar), 113.42 (s, 2CHar), 103.16–102.64 (m, Car), 63.45 (d, J = 7.0 Hz, OCH2), 62.31 (d, J = 7.5 Hz, OCH2), 51.03 (d, J = 156.8 Hz, CH), 47.82 (t, J = 4.0 Hz, CH3CH2CH2N), 24.04 (d, J = 7.0 Hz, ArCH2), 23.93 (s, CH3CH2CH2N), 16.57, 16.52, 16.49, and 16.43 (2CH3), 11.17 (s, CH3CH2CH2N). HRMS (ESI+) calcd for C21H28F4N2O3P (M + H)+: 463.1774, found: 463.1773.
4.1.3.3 Diethyl (2-(4-(butylamino)-2,3,5,6-tetrafluorophenyl)-1-(phenylamino)ethyl)phosphonate (2m). Yellow oil (40 mg, 71%). 1H NMR (400 MHz, CDCl3) δ = 7.06 (t, J = 7.9 Hz, 2H, 2CHar), 6.64 (t, J = 7.3 Hz, 1H, CHar), 6.54 (d, J = 7.9 Hz, 2H, 2CHar), 4.17–3.97 (m, 5H, CH, 2OCH2), 3.78 (dd, J = 10.8, 3.1 Hz, 1H, NH), 3.60 (bs, 1H, NH), 3.31–3.25 (m, 2H, NCH2), 3.18–3.13 (m, 1H, ArCH2), 3.06–3.00 (m, 1H, ArCH2), 1.49–1.28 (m, 7H, CH3, CH2CH2), 1.20 (t, J = 7.1 Hz, 3H, CH3), 0.92 (t, J = 7.3 Hz, 3H, CH3(CH2)3). 19F NMR (377 MHz, CDCl3) δ = −146.45 to −146.52 (m, 2F), −160.68 to −160.72 (m, 2F). 31P NMR (162 MHz, CDCl3) δ = 24.13 (s) ppm. 13C NMR (101 MHz, CDCl3) δ = 147.16–146.70 (m, 2Car), 146.73 (d, J = 6.2 Hz, Car), 138.77–136.15 (m, 2Car), 129.18 (s, 2CHar) 127.20–126.97 (m, Car) 118.38 (s, CHar) 113.43 (s, 2CHar) 103.13–102.61 (m, Car) 63.45 (d, J = 7.0 Hz, OCH2), 62.31 (d, J = 7.5 Hz, OCH2), 51.05 (d, J = 156.8 Hz, CH), 45.82 (t, J = 4.0 Hz, NCH2), 32.88 (s, CH2CH2CH2), 24.04 (d, J = 6.7 Hz, ArCH2), 19.88 (s, CH3CH2CH2), 16.58, 16.52, 16.49, and 16.43 (2CH3), 13.87 (s, CH3(CH2)3). HRMS (ESI+) calcd for C22H30F4N2O3P (M + H)+: 477.1930, found: 477.1932.
4.1.3.4 Diethyl (2-(4-(allylamino)-2,3,5,6-tetrafluorophenyl)-1-(phenylamino)ethyl)phosphonate (2n). Pale yellow oil (35 mg, 65%). 1H NMR (400 MHz, CDCl3) δ = 7.09–7.05 (m, 2H, 2CHar), 6.64 (t, J = 7.3 Hz, 1H, CHar), 6.54 (d, J = 7.9 Hz, 2H, 2CHar), 5.88–5.81 (m, 1H, CH2
CH), 5.15 (ddd, J = 13.7, 11.5, 1.3 Hz, 2H, CH2CH), 4.17–4.02 (m, 5H, CHP, 2OCH2), 3.89–3.88 (m, 2H, NCH2), 3.77–3.76 (m, 2H, 2 NH), 3.20–3.14 (m, 1H, ArCH2), 3.07–2.97 (m, 1H, ArCH2), 1.30 (t, J = 7.1 Hz, 3H, CH3), 1.20 (t, J = 7.1 Hz, 3H, CH3). 19F NMR (377 MHz, CDCl3) δ = −146.13 to −146.22 (m, 2F), −159.83 to −159.92 (m, 2F). 31P NMR (162 MHz, CDCl3) δ = 24.08 (s). 13C NMR (101 MHz, CDCl3) δ = 147.14–146.66 (m, 2Car), 146.70 (d, J = 6.3 Hz, Car), 138.93–136.29 (m, 2Car), 135.27 (s, CH2CH), 129.20 (s, 2CHar), 126.66–126.39 (m, Car), 118.42 (s, CHar), 116.86 (s, CH2CH), 113.43 (s, 2CHar), 103.89–103.38 (m, Car), 63.50 (t, J = 6.8 Hz, OCH2), 62.37 (t, J = 8.9 Hz, OCH2), 51.01 (d, J = 156.8 Hz, CHP), 48.35 (t, J = 4.3 Hz, NCH2), 24.09 (d, J = 7.0 Hz, ArCH2), 16.58, 16.52, 16.49, 16.43 (2CH3). HRMS (ESI+) calcd for C21H26F4N2O3P (M + H)+: 461.1617, found: 461.1616.
4.1.3.5 Diethyl (1-(phenylamino)-2-(2,3,5,6-tetrafluoro-4-(methylamino)phenyl)ethyl)phosphonate (2o). Pale yellow oil (38 mg, 63%). 1H NMR (400 MHz, acetone-d6) δ = 7.33–7.27 (m, 5H, 5 CHar), 7.04–7.00 (m, 2H, 2CHar), 6.67–6.65 (m, 2H, 2CHar), 6.57 (t, J = 7.3 Hz, 1H, CHar), 5.60 (bs, 1H, NHCH2), 4.71–4.67 (m, 1H, NH), 4.52 (d, J = 7.0 Hz, 2H, NCH2), 4.08–4.01 (m, 5H, CH, 2OCH2), 3.16–3.10 (m, 1H, ArCH2), 3.00–2.90 (m, 1H, ArCH2), 1.21 (t, J = 7.1 Hz, 3H, CH3), 1.15 (t, J = 7.0 Hz, 3H, CH3). 19F NMR (377 MHz, acetone-d6) δ = −147.27 to −147.39 (m, 2F), −161.21 to −161.32 (m, 2F). 31P NMR (162 MHz, acetone-d6) δ = 23.69 (s). 13C NMR (101 MHz, CDCl3): δ = 146.91–144.51 (m, 2Car), 146.69 (d, J = 6.0 Hz, Car), 139.10 (s, Car), 138.95–136.33 (m, 2Car), 129.23 (s, 2CHar), 128.89 (s, 2CHar), 127.81 (s, CHar), 127.67 (s, 2CHar), 126.72–126.57 (m, Car), 118.45 (s, CHar), 113.47 (s, 2CHar), 104.00–103.63 (m, Car), 63.47 (d, J = 7.0 Hz, OCH2), 62.33 (d, J = 7.5 Hz, OCH2), 51.01 (d, J = 156.0 Hz, CH), 50.18 (s, ArCH2N), 24.09 (d, J = 7.1 Hz, ArCH2), 16.58, 16.53, 16.49, 16.43 (2 × CH3). HRMS (ESI+) calcd for C25H28F4N2O3P (M + H)+: 511.1774, found: 511.1774.
:1, v/v).
4.1.4.1 Diethyl (1-(phenylamino)-2-(2,3,5,6-tetrafluoro-4-phenoxyphenyl)ethyl)phosphonate (2p). Yellow oil (40 mg, 68%). 1H NMR (400 MHz, CD3C(O)CD3) δ = 7.39–7.35 (m, 2H, 2CHar), 7.17–7.10 (m, 1H, CHar), 7.07–7.05 (m, 2H, 2CHar), 6.81 (d, J = 8.2 Hz, 2H, 2CHar), 6.72 (d, J = 7.9 Hz, 2H, 2CHar), 6.65 (t, J = 7.3 Hz, 1H, CHar), 4.84–4.82 (m, 1H, NH), 4.19–4.09 (m, 5H, CH, 2OCH2), 3.33–3.30 (m, 1H, ArCH2), 3.21–3.12 (m, 1H, ArCH2), 1.29 (t, J = 7.1 Hz, 3H, CH3), 1.22 (t, J = 7.2 Hz, 3H, CH3). 19F NMR (377 MHz, CDCl3) δ = −143.74 to −143.83 (m, 2F), −154.95 to −155.03 (m, 2F). 31P NMR (162 MHz, CD3C(O)CD3) δ = 23.02 (s). 13C NMR (101 MHz, CDCl3) δ = 157.27 (s, Car), 147.14–144.45 (m, 2Car), 146.50 (d, J = 8.0 Hz, Car), 142.77–140.05 (m, 2Car) 132.48–132.17 (m, Car), 129.83 (s, 2CHar), 129.37 (s, 2CHar), 123.61 (s, CHar), 118.66 (s, CHar), 115.38 (s, 2CHar), 113.17 (s, 2CHar), 112.74–112.23 (m, Car), 63.82 (d, J = 7.0 Hz, OCH2), 62.55 (d, J = 7.4 Hz, OCH2), 50.53 (d, J = 159.1 Hz, CH), 24.75 (d, J = 7.8 Hz, ArCH2), 16.63, 16.57, 16.55 and 16.49 (2CH3). HRMS (ESI+) calcd for C24H25F4NO4P (M + H)+: 498.1457, found: 498.1459.
4.1.4.2 Diethyl (1-(phenylamino)-2-(2,3,5,6-tetrafluoro-4-(4-methoxyphenoxy)phenyl)ethyl)phosphonate (2q). Yellow solid (30 mg, 48%), mp = 97–99 °C. 1H NMR (400 MHz, CD3C(O)CD3) δ = 7.08–7.01 (m, 2H, 2CHar), 6.92–6.88 (m, 2H, 2CHar), 6.78–6.76 (m, 2H, 2CHar), 6.72–6.69 (m, 2H, 2CHar), 6.65 (t, J = 7.4 Hz, 1H, CHar), 4.83–4.80 (m, 1H, NH), 4.16–4.09 (m, 5H, CH, 2OCH2), 3.78 (s, 3H, OCH3), 3.33–3.27 (m, 1H, ArCH2), 3.19–3.10 (m, 1H, ArCH2), 1.30–1.26 (m, 3H, CH3), 1.24–1.15 (m, 3H, CH3). 19F NMR (377 MHz, CDCl3) δ = −143.91 to −144.00 (m, 2F), −155.33 to −155.42 (m, 2F). 31P NMR (162 MHz, CDCl3) δ = 23.42 (s). 13C NMR (101 MHz, CDCl3) δ = 155.85 (s, Car), 151.42 (s, Car), 147.13–144.49 (m, 2Car) 146.51 (d, J = 7.9 Hz, Car), 142.78–140.10 (m, 2Car), 133.40–133.04 (m, Car) 129.36 (s, 2CHar), 118.64 (s, CHar), 116.70 (s, 2CHar), 114.79 (s, 2CHar), 113.21 (s, 2CHar), 112.34–111.83 (m, Car), 63.80 (d, J = 7.0 Hz, OCH2), 62.55 (d, J = 7.4 Hz, OCH2), 55.82 (s, OCH3), 50.57 (d, J = 158.7 Hz, CH), 24.70 (d, J = 7.6 Hz, ArCH2), 16.63, 16.58, 16.55, and 16.50 (2CH3). HRMS (ESI+) calcd for C25H27F4NO5P (M + H)+: 528.1563, found: 528.1567.
4.1.4.3 Diethyl (2-(4-(4-chlorophenoxy)-2,3,5,6-tetrafluorophenyl)-1-(phenylamino)ethyl)phosphonate (2r). Yellow solid (35 mg, 56%), mp = 94–99 °C. 1H NMR (400 MHz, CD3C(O)CD3) δ = 7.40–7.38 (m, 2H, 2CHar), 7.08–7.02 (m, 2H, 2CHar), 6.85–6.83 (m, 2H, 2CHar), 6.72 (d, J = 7.9 Hz, 2H, 2CHar), 6.66 (t, J = 7.3 Hz, 1H, CHar), 4.87–4.83 (m, 1H, NH), 4.17–4.10 (m, 5H, CH, 2OCH2), 3.34–3.28 (m, 1H, ArCH2), 3.21–3.14 (m, 1H, ArCH2), 1.29 (t, J = 7.1 Hz, 3H, CH3), 1.22 (t, J = 7.1 Hz, 3H, CH3). 19F NMR (377 MHz, CDCl3) δ = −143.37 to −143.45 (m, 2F), −154.94 to −155.02 (m, 2F). 31P NMR (162 MHz, CDCl3) δ = 23.24 (s). 13C NMR (101 MHz, CDCl3) δ = 155.79 (s, Car), 147.12–142.57 (m, 2Car), 146.47 (d, J = 8.1 Hz, Car), 142.57–139.85 (m, 2Car) 132.10–131.84 (m, Car), 129.77 (s, 2CHar), 129.36 (s, 2CHar), 128.73 (s, Car), 118.59 (s, CHar), 116.75 (s, 2CHar), 113.07 (s, 2CHar), 112.95–112.80 (m, Car), 63.87 (d, J = 7.0 Hz, OCH2), 62.59 (d, J = 7.5 Hz, OCH2), 50.38 (d, J = 159.4 Hz, CH), 24.74 (d, J = 7.9 Hz, ArCH2), 16.62, 16.57, 16.55, and 16.49 (2CH3) ppm. HRMS (ESI+) calcd for C24H24ClF4NO4P (M + H)+: 532.1068, found: 532.1066.
4.1.4.4 Diethyl (1-(phenylamino)-2-(2,3,5,6-tetrafluoro-4-(3-nitrophenoxy)phenyl)ethyl)phosphonate (2s). Yellow solid (31 mg, 48%), mp = 77–79 °C. 1H NMR (400 MHz, CD3C(O)CD3) δ = 8.05 (ddd, J = 8.2, 2.1, 0.8 Hz, 1H, CHar), 7.84 (t, J = 2.3 Hz, 1H, CHar), 7.71 (t, J = 8.3 Hz, 1H, CHar), 7.24 (dd, J = 8.3, 2.6 Hz, 1H, CHar), 7.10–7.06 (m, 2H, 2CHar), 6.74 (d, J = 7.9 Hz, 2H, 2CHar), 6.63 (t, J = 7.3 Hz, 1H, CHar), 4.86 (d, J = 13.7 Hz, 1H, NH), 4.17–4.10 (m, 5H, CH, 2OCH2), 3.35–3.30 (m, 1H, ArCH2), 3.23–3.14 (m, 1H, ArCH2), 1.28 (t, J = 7.1 Hz, 3H, CH3), 1.22 (t, J = 7.1 Hz, 3H, CH3). 19F NMR (377 MHz, CDCl3) δ = −142.44 (dd, J = 22.6, 9.4 Hz, 2F), −154.52 to −154.61 (dd, J = 22.0, 9.3 Hz, 2F). 31P NMR (162 MHz, CDCl3) δ = 23.25 (s). 13C NMR (101 MHz, CDCl3) δ = 157.42 (s, Car), 149.36 (s, Car), 147.24–144.60 (m, 2Car), 146.42 (d, J = 7.7 Hz, Car), 142.42–139.69 (m, 2Car), 131.44–130.97 (m, Car), 130.67 (s, CHar), 129.41 (s, 2CHar), 121.39 (s, CHar), 118.79 (s, CHar), 118.74 (s, CHar), 113.97–113.75 (m, Car), 113.28 (s, 2CHar), 111.18 (s, CHar), 63.83 (d, J = 7.0 Hz, OCH2), 62.60 (d, J = 7.4 Hz, OCH2), 50.47 (d, J = 158.6 Hz, CH), 24.85 (d, J = 7.8 Hz, ArCH2), 16.63, 16.58, 16.55, and 16.50 (2CH3). HRMS (ESI+) calcd for C24H24F4N2O6P (M + H)+: 543.1308, found: 543.1312.
4.2 X-ray and DFT studies
4.2.1.1 Crystals of 2k. C19H23F4N2O3P, Mr = 434.36, 0.35 × 0.21 × 0.18 mm3, triclinic, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 11.7691(9) Å, b = 12.2935(5) Å, c = 16.8735(9) Å, α = 101.120(4)°, β = 99.040(5)°, γ = 118.147(6)°, V = 2023.5(2) Å3, Z = 4, Dx = 1.426 g cm−3, F000 = 904, MoKα radiation, λ = 0.71073 Å, T = 100(2) K, 2θmax = 57.01°, 15277 reflections collected, 8589 unique (Rint = 0.027), Final GooF = 1.028, R1 = 0.0527, wR2 = 0.1144, R indices based on 6237 with I > 2σ(I) (refinement on F2), 545 parameters, μ = 0.195 mm−1.
(no. 2), a = 11.7691(9) Å, b = 12.2935(5) Å, c = 16.8735(9) Å, α = 101.120(4)°, β = 99.040(5)°, γ = 118.147(6)°, V = 2023.5(2) Å3, Z = 4, Dx = 1.426 g cm−3, F000 = 904, MoKα radiation, λ = 0.71073 Å, T = 100(2) K, 2θmax = 57.01°, 15277 reflections collected, 8589 unique (Rint = 0.027), Final GooF = 1.028, R1 = 0.0527, wR2 = 0.1144, R indices based on 6237 with I > 2σ(I) (refinement on F2), 545 parameters, μ = 0.195 mm−1.
4.2.1.2 Crystals of 2l. C21H27F4N2O3P, Mr = 462.41, 0.56 × 0.46 × 0.18 mm3, triclinic, space group P
(no. 2), a = 8.8604(4) Å, b = 9.8978(8) Å, c = 13.1171(7) Å, α = 91.889(5)°, β = 91.350(4)°, γ = 109.119(6)°, V = 1085.59(12) Å3, Z = 2, Dx = 1.415 g cm−3, F000 = 484, MoKα radiation, λ = 0.71073 Å, T = 100(2) K, 2θmax = 56.88°, 8189 reflections collected, 4658 unique (Rint = 0.0249), Final GooF = 1.040, R1 = 0.0447, wR2 = 0.0951, R indices based on 3666 with I > 2σ(I) (refinement on F2), 291 parameters, μ = 0.186 mm−1, CCDC 1912530 and 1912531 contain the supplementary crystallographic data for this paper.
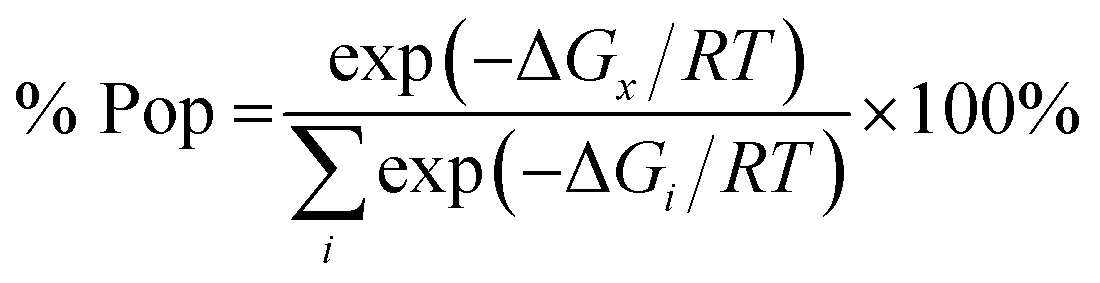 | (1) |
The temperature for interconversion between possible stereomers was calculated on the basis of NMR studies using Earing's equation modified by Shanan-Atidi and Bar-Eli61
| ΔG = 4.57Tc{10.62 + log[X/2π(1 − ΔP)] + log(Tc/Δv)} | (2) |
The relative energies of stereomers calculated at WB97XD/6-31+G(d) level of theory are summarized in Table 2.
4.3 Biological evaluation
P values (octanol/water partition coefficient), PSA (topological polar surface area) and aPSA (apolar surface area). The logP values were calculated by ALOGPS 2.1 software (http://www.vcclab.org/lab/alogps).65 PSA and aPSA descriptors were calculated using the VEGA ZZ program (http://www.vegazz.net).66 The pharmacokinetic profiles were also evaluated according to Lipinski's “rule of five”67 by using Molinspiration application (http://www.molinspiration.com), which analyses molecular weight (MW), number of hydrogen-bond acceptors (HBA), and number of hydrogen-bond donors (HBD). The Caco-2 prediction model based on descriptors generated by preADMET (http://preadmet.bmdrc.org) was used to compute Caco-2 apparent permeability (tPapp), for the tested compounds. In this model a number of hydrogen bond donors and three molecular surface area properties determine membrane permeability of compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported in part by PL-Grid Infrastructure (http://www.plgrid.pl/en).References
- Z.-Q. Zhu, L.-J. Xiao, D. Guo, X. Chen, J.-J. Ji, X. Zhu, Z.-B. Xi and Z.-G. Le, J. Org. Chem., 2019, 84, 435–442 CrossRef CAS PubMed
.
- Z. Chen, P. Marce, R. Resende, P. M. Alzari, A. Carlos Frasch, J. M. H. van den Elsen, S. J. Crennell and A. G. Watts, Eur. J. Med. Chem., 2018, 158, 25–33 CrossRef CAS PubMed
- V. M. S. Isca and A. C. Fernandes, Green Chem., 2018, 20, 3242–3245 RSC
- A. Mucha, P. Kafarski and Ł. Berlicki, J. Med. Chem., 2011, 54, 5955–5980 CrossRef CAS PubMed
- X.-C. Huang, M. Wang, Y.-M. Pan, G.-Y. Yao, H.-S. Wang, X.-Y. Tian, J.-K. Qin and Y. Zhang, Eur. J. Med. Chem., 2013, 69, 508–520 CrossRef CAS PubMed
- Q. Wang, L. Yang, H. Ding, X. Chen, H. Wang and X. Tang, Bioorg. Chem., 2016, 69, 132–139 CrossRef CAS PubMed
- Z. Rezaei, H. Firouzabadi, N. Iranpoor, A. Ghaderi, M. R. Jafari, A. A. Jafari and H. R. Zare, Eur. J. Med. Chem., 2009, 44, 4266–4275 CrossRef CAS PubMed
- I. Kraicheva, A. Bogomilova, I. Tsacheva, G. Momekov and K. Troev, Eur. J. Med. Chem., 2009, 44, 3363–3367 CrossRef CAS PubMed
- Y.-C. Yu, W.-B. Kuang, R.-Z. Huang, Y.-L. Fang, Y. Zhang, Z.-F. Chen and X.-L. Ma, MedChemComm, 2017, 8, 1158–1172 RSC
- B.-A. Song, Y.-L. Wu, S. Yang, D.-Y. Hu, X.-Q. He and L.-H. Jin, Molecules, 2003, 8, 186–192 CrossRef CAS
- F. R. Atherton, C. H. Hassall and R. W. Lambert, J. Med. Chem., 1986, 29, 29–40 CrossRef CAS PubMed
- P. Kafarski and B. Lejczak, Phosphorus, Sulfur Silicon Relat. Elem., 1991, 63, 193–215 CrossRef CAS
- F. Westheimer, Science, 1987, 235, 1173–1178 CrossRef CAS PubMed
- V. D. Romanenko and V. P. Kukhar, Chem. Rev., 2006, 106, 3868–3935 CrossRef CAS PubMed
- S. A. Bernhard and L. E. Orgel, Science, 1959, 130, 625–626 CrossRef CAS PubMed
- K. V. Turcheniuk, V. P. Kukhar, G.-V. Röschenthaler, J. L. Aceña, V. A. Soloshonok and A. E. Sorochinsky, RSC Adv., 2013, 3, 6693–6716 RSC
- G. F. Makhaeva, A. Y. Aksinenko, V. B. Sokolov, I. I. Baskin, V. A. Palyulin, N. S. Zefirov, N. D. Hein, J. W. Kampf, S. J. Wijeyesakere and R. J. Richardson, Chem.-Biol. Interact., 2010, 187, 177–184 CrossRef CAS PubMed
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed
- B. E. Smart, J. Fluorine Chem., 2001, 109, 3–11 CrossRef CAS
- S. L. Cobb and C. D. Murphy, J. Fluorine Chem., 2009, 130, 132–143 CrossRef CAS
- L. D. Quin and G. S. Quin, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2001, 128, 173–185 CrossRef CAS
- M. Salwiczek, E. K. Nyakatura, U. I. M. Gerling, S. Ye and B. Koksch, Chem. Soc. Rev., 2012, 41, 2135–2171 RSC
- D. M. Ryan, S. B. Anderson, F. T. Senguen, R. E. Youngman and B. L. Nilsson, Soft Matter, 2010, 6, 475–479 RSC
- H. Zheng, K. Comeforo and J. Gao, J. Am. Chem. Soc., 2009, 131, 18–19 CrossRef CAS PubMed
- C. J. Pace, H. Zheng, R. Mylvaganam, D. Kim and J. Gao, Angew. Chem., Int. Ed., 2012, 51, 103–107 CrossRef CAS PubMed
- G. Akçay and K. Kumar, J. Fluorine Chem., 2009, 130, 1178–1182 CrossRef PubMed
- C. J. Pace and J. Gao, Acc. Chem. Res., 2013, 46, 907–915 CrossRef CAS PubMed
- S. E. Wheeler and K. N. Houk, J. Chem. Theory Comput., 2009, 5, 2301–2312 CrossRef CAS PubMed
- T. Siodla, W. P. Oziminski, M. Hoffmann, H. Koroniak and T. M. Krygowski, J. Org. Chem., 2014, 79, 7321–7331 CrossRef CAS PubMed
- F. Wang, L. Qin, P. Wong and J. Gao, Org. Lett., 2011, 13, 236–239 CrossRef CAS PubMed
- L. Qin, C. Sheridan and J. Gao, Org. Lett., 2012, 14, 528–531 CrossRef CAS PubMed
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y.-S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed
- J. Kwiczak-Yiğitbaşı, J.-L. Pirat, D. Virieux, J.-N. Volle, A. Janiak, M. Hoffmann and D. Pluskota-Karwatka, Arabian J. Chem., 2018 DOI:10.1016/j.arabjc.2018.05.002
- A. Pawlowska, J.-N. Volle, D. Virieux, J.-L. Pirat, A. Janiak, M. Nowicki, M. Hoffmann and D. Pluskota-Karwatka, Tetrahedron, 2018, 74, 975–986 CrossRef CAS
- S. A. Azizi and C. Miyamoto, J. NeuroVirol., 1998, 4, 204–216 CrossRef CAS PubMed
- J. D. Lathia, S. C. Mack, E. E. Mulkearns-Hubert, C. L. L. Valentim and J. N. Rich, Genes Dev., 2015, 29, 1203–1217 CrossRef CAS PubMed
- P. Anja, J. Anahid and K. Janko, Semin. Cancer Biol., 2018, 53, 168–177 CrossRef CAS PubMed
- M. Sivaparvathi, R. Sawaya, S. W. Wang, A. Rayford, M. Yamamoto, L. A. Liotta, G. L. Nicolson and J. S. Rao, Clin. Exp. Metastasis, 1995, 13, 49–56 CrossRef CAS PubMed
- M. Sivaparvathi, M. Yamamoto, G. L. Nicolson, Z. L. Gokaslan, G. N. Fuller, L. A. Liotta, R. Sawaya and J. S. Rao, Clin. Exp. Metastasis, 1996, 14, 27–34 CrossRef CAS PubMed
- G. F. Makhaeva, S. V. Lushchekina, O. G. Serebryakova, A. Y. Aksinenko, T. V. Goreva, R. J. Richardson and I. V. Martynov, Dokl. Biochem. Biophys., 2013, 451, 203–206 CrossRef CAS PubMed
- M. Sieńczyk and J. Oleksyszyn, Curr. Med. Chem., 2009, 16, 1673–1687 CrossRef
- J. Ajenjo, M. Greenhall, C. Zarantonello and P. Beier, Beilstein J. Org. Chem., 2016, 12, 192–197 CrossRef CAS PubMed
- I. A. Khalfina and V. M. Vlasov, Russ. J. Org. Chem., 2005, 41, 978–983 CrossRef CAS
- C. S. Gutsche, M. Ortwerth, S. Gräfe, K. J. Flanagan, M. O. Senge, H.-U. Reissig, N. Kulak and A. Wiehe, Chem.–Eur. J., 2016, 22, 13953–13964 CrossRef CAS PubMed
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS
- C. A. Lipinski, Adv. Drug Delivery Rev., 2016, 101, 34–41 CrossRef CAS PubMed
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed
- M. Yazdanian, S. L. Glynn, J. L. Wright and A. Hawi, Pharm. Res., 1998, 15, 1490–1494 CrossRef CAS
- X. Ma, C. Chen and J. Yang, Acta Pharmacol. Sin., 2005, 26, 500–512 CrossRef CAS PubMed
- S. Lapointe, A. Perry and N. A. Butowski, Lancet, 2018, 392, 432–446 CrossRef
- Q. T. Ostrom, L. Bauchet, F. G. Davis, I. Deltour, J. L. Fisher, C. E. Langer, M. Pekmezci, J. A. Schwartzbaum, M. C. Turner, K. M. Walsh, M. R. Wrensch and J. S. Barnholtz-Sloan, Neuro-Oncology, 2014, 16, 896–913 CrossRef CAS PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC
- J. J. P. Stewart, J. Comput. Chem., 1989, 10, 209–220 CrossRef CAS
- C. C. J. Roothaan, Rev. Mod. Phys., 1951, 23, 69–89 CrossRef CAS
- J. B. Collins, P. von R. Schleyer, J. S. Binkley and J. A. Pople, J. Chem. Phys., 1976, 64, 5142–5151 CrossRef CAS
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- P. Atkins and J. de Paula, Physical Chemistry, W. H. Freeman, New York, 9th edn, 2009 Search PubMed
- H. Shanan-Atidi and K. H. Bar-Eli, J. Phys. Chem., 1970, 74, 961–963 CrossRef CAS
- K. Salus, M. Hoffmann, T. Siodła, B. Wyrzykiewicz and D. Pluskota-Karwatka, New J. Chem., 2017, 41, 2409–2424 RSC
- C. P. R. Xavier, C. F. Lima, M. Rohde and C. Pereira-Wilson, Cancer Chemother. Pharmacol., 2011, 68, 1449–1457 CrossRef CAS PubMed
- L. C. Crowley, M. E. Christensen and N. J. Waterhouse, Cold Spring Harb. Protoc., 2016 DOI:10.1101/pdb.prot087171
- I. V. Tetko, J. Gasteiger, R. Todeschini, A. Mauri, D. Livingstone, P. Ertl, V. A. Palyulin, E. V. Radchenko, N. S. Zefirov, A. S. Makarenko, V. Yu. Tanchuk and V. V. Prokopenko, J. Comput.-Aided Mol. Des., 2005, 19, 453–463 CrossRef CAS PubMed
- A. Pedretti, L. Villa and G. Vistoli, J. Mol. Graphics Modell., 2002, 21, 47–49 CrossRef CAS PubMed
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed
Footnotes |
| † Dedication to Professor Henryk Koroniak in honour of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1912530 and 1912531. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra03982a |
| This journal is © The Royal Society of Chemistry 2019 |