
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne pot conversion of glucose to ethyl levulinate over a porous hydrothermal acid catalyst in green solvents†
Monika Bosilj ab,
Johannes Schmidte,
Anna Fischerbcd and
Robin J. White*af
ab,
Johannes Schmidte,
Anna Fischerbcd and
Robin J. White*af
aFraunhofer Institute for Solar Energy Systems ISE, Heidenhofstraße 2, 79110 Freiburg im Breisgau, Germany. E-mail: robin.white666@googlemail.com
bDepartment of Inorganic and Analytical Chemistry, University of Freiburg, Albertstrasse 21, 79104 Freiburg, Germany
cFreiburg Materials Research Center (FMF), University of Freiburg, Stefan-Meier-Straße 21, 79104 Freiburg im Breisgau, Germany
dFIT – Freiburg Center for Interactive Materials and Bioinspired Technologies, University of Freiburg, Georges-Köhler-Allee 105, 79110 Freiburg im Breisgau, Germany
eTechnische Universität Berlin, Fakultät II Institut für Chemie, Sekretariat BA 2, Hardenbergstr. 40, 10623 Berlin, Germany
fThe Netherlands Organisation for Applied Scientific Research (TNO), High Tech Campus 25, 5656AE Eindhoven, The Netherlands
First published on 1st July 2019
Abstract
The one-pot conversion of glucose to ethyl levulinate over an acid-functionalised hydrothermal catalyst (derived from glucose) provides high initial yields up to 37 mol%, comparable to the homogeneous H2SO4 catalyst, whilst catalyst performance is strongly influenced by green solvent choice.
With regard to further elaboration of biorefinery concepts,1 glucose is a key platform compound for the production of a range of fuels and chemicals, including levulinic acid2 and its esters.3–5 Obtaining these products in high yield from glucose is a challenging task in comparison to fructose conversion due to hindering isomerisation of glucose to fructose. Nevertheless, using fructose as a starting compound is more costly than glucose. Levulinic ester ethyl levulinate (EL) is of interest, as it is a potential bio-derived solvent, flavouring agent or plasticizer,6 as well as an octane fuel additive.7 Mechanistically, a one-pot synthesis of EL from C6-sugars (at 200 °C) could proceed via two routes. The first one starts with glucose ethanolysis followed by ethyl D-glucoside isomerisation to ethyl fructoside. This rapidly dehydrates to 5-ethoxymethylfurfural (Scheme 1a). 5-Ethoxymethylfurfural is further converted to EL.4,8 The second route is possible with glucose isomerisation to fructose, its dehydration to 5-HMF and final esterification (Scheme 1b).6,9 High reaction temperatures and greater equivalents of alcohol could favour intermolecular alcohol dehydration.6 It was reported that at lower temperatures (140 °C) glucose reacts preferentially with ethanol (EtOH) to form ethyl D-glucoside, preventing further glucose isomerisation and dehydration.10
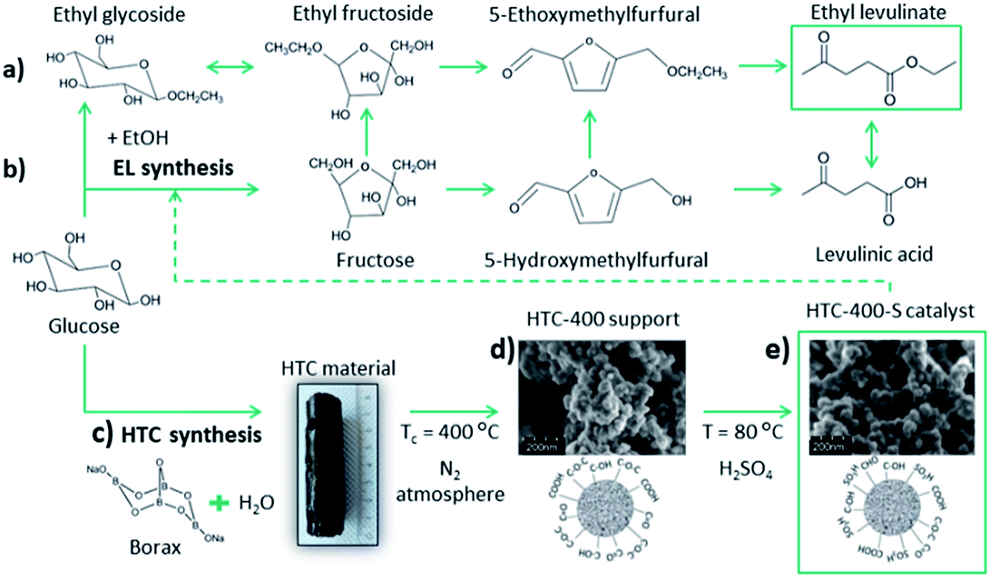 | ||
| Scheme 1 (a) and (b) Proposed reaction pathway for one pot synthesis of EL from glucose with EtOH, (c) HTC synthesis, (d) post-thermal carbonisation and (e) introduction of S-containing functional groups into HTC-400 support. | ||
Along similar lines to levulinic acid production, the treatment of sugar-based biomass (e.g. glucose), under acidic conditions generates carbonaceous humin by-products.11 In the context of chemicals and fuel production, this solid formation is undesired as it lowers product yield and often deactivates the catalyst via pore and active site blocking.12 Whilst the carbonaceous by-product can be of value, efforts have been made to suppress the formation of these polymeric carbonaceous products. It has been reported that humin formation can be inhibited to some extent via non-aqueous solvent use such as methanol,3 EtOH,4 γ-valerolactone (GVL)13 or ethylene glycol.14
Different types of solid acid catalysts have been proposed for one-pot conversion of glucose to EL, from sulphonated ionic liquids,15 zeolites,16 sulphated metal oxides4 to sulphonated carbons.17 Carbon-based materials containing sulphonic acid groups are especially attractive catalysts in biorefinery processes due to expected high stability and resistance to acidic and chelating media. The advantage of carbon material is to allow catalyst hydrophilicity and acid–base character to be readily tuned and usually have lower production costs than that of other conventional supports, such as alumina or silica. Regarding the biomass derived catalysts, the sulphonation of carbons synthesized by incomplete carbonisation of biomass precursors has been an attractive way due to its simple synthesis. However, these materials exhibit very low specific surface area (<10 m2 g−1) and no porosity, which hinder the diffusion of bulky, viscous reactants to active sites.18
With regard to biomass derived catalysts, synthesised by hydrothermal carbonisation (HTC) of sugar-based biomass (e.g. glucose) with addition of sodium borate (borax) generates structurally similar to sol–gel monoliths mesoporous xerogels with high specific surface area (Scheme 1c).19 The synthesis, characterisation and application of (meso)porous high specific surface area sulphonated HTC catalysts have thus not been explored. Typically, large micron sized, low surface, low porosity HTC spheres have been sulphonated and investigated as potential solid acids.20–22 From a catalytic point of view (e.g. site loading per catalyst bed volume), the introduction of porosity/high surface area is clearly beneficial. In this regard, the use of borax in the glucose (aq) solution is partly catalytic, partly surface stabilising agent. It binds with glucose via the inherent diol structure allowing access to higher yielding pathway to HMF and the forming HTC material structure (e.g. via inhibition of acetalisation of glucose with 5-HMF), whilst also acting to direct primary carbon nanoparticle size (via control of the borax/glucose ratio).23 After the HTC was completed, borax was washed out and the carbonaceous solid product was thermally carbonised at 400 °C under N2 for 5 h (Scheme 1d). To introduce Brønsted acid functionality (i.e. –SO3H), the carbonised xerogel was dispersed in 98% w/w H2SO4 at 80 °C for 4 h under N2 (Scheme 1e, catalyst denoted as HTC-400-S). Further synthesis details are reported in the ESI.† Porous HTC-400-S catalyst with high specific surface area significantly differs from the previously reported sulphonated hydrothermal carbons in the literature20–22 due to the presence of the structure directive agent borax in the HTC reaction, which tailors the material porosity based on the HTC reaction time and its concentration. Further post-thermal treatment at 400 °C opens up interstitial porosity between the primary particles due to the condensation process. Consequently, high pore volume (Vpore = 0.42 cm3 g−1) and specific surface area (SBET = 506 m2 g−1) determined by N2 sorption of HTC support were obtained (Table S1 and Fig. S1†), which improve mass transport and diffusion through the catalyst. The amount of acid groups on the HTC-400-S was initially determined through back titration. A loading of 2.6 mmol g−1 acidic groups was recorded for HTC-400-S. CHNS elemental analysis (Table S1†) confirmed the presence of sulphur (1.1 mmol g−1/3.6 wt%). As might be anticipated for a carbonaceous material of this nature, a higher number of Brønsted acid sites were determined through titration, attributed to other surface acidic groups (e.g. –OH, –COOH) in addition to the expected sulphonic (–SO3H) acidity. Complementary, an analysis of the X-ray photoelectron spectroscopy (XPS) survey for HTC-400-S indicated a loading of 1.94 at%/4.73 wt% for “S” containing surface functionality (Fig. S2a†). The high resolution S2p core state for HTC-400-S revealed beside the S2p3/2–S2p1/2 spin orbital splitting, the existence of two doublets (Fig. 1A) confirming the evidence of two chemically different sulphur species: a lower energy doublet S2p3/2 at 167.4 eV and 168.3 eV attributed to sulphonic (–SO3H) groups24 and a higher energy doublet S2p1/2 at 168.5 eV and 169.5 eV assignment to sulphate functionalities (–SO42−).25 In comparison to other glucose-derived sulphonated materials,21 S-containing functionalities at lower binding energies such as thiol (C–SH)26 are not present in our catalyst.
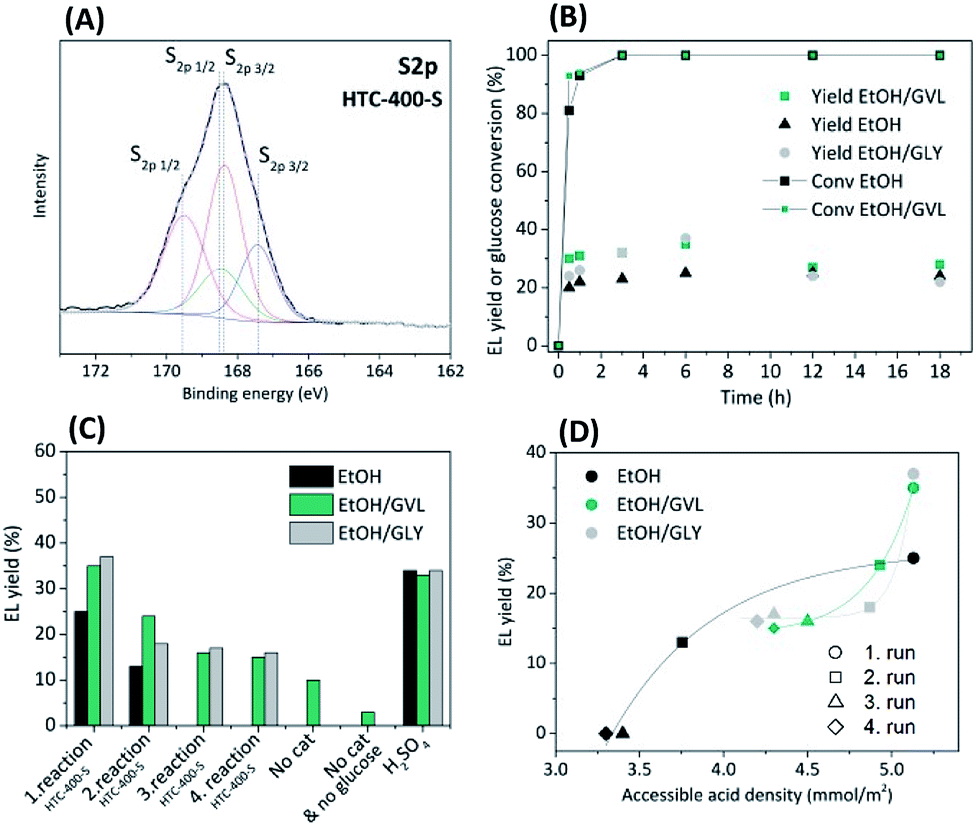 | ||
| Fig. 1 (A) X-ray photoelectron spectra of S2p core level for HTC-400-S, (B) effect of different solvents on EL yield (0.5 wt% HTC-400-S), (C) EL yields obtained over 0.5 wt% HTC-400-S and 0.05 wt% H2SO4 in comparison to non-catalysed reaction and (D) accessible acid sites vs. EL yield in different solvents. | ||
In this communication, an acid-functionalised “carbonaceous” xerogel has been synthesised and applied as a catalyst in the one pot synthesis of EL from glucose. The inhibition of humin formation via the use of non-aqueous “green” solvents such as EtOH, GVL and glycerol (GLY) in tandem with this functional solid catalyst is also discussed. To our knowledge GLY has so far not been investigated as a humin formation inhibiting solvent. Catalytic reactions were performed with 2.4 wt% glucose dissolved in 20 g of solvent (i.e. EtOH, EtOH/GVL or EtOH/GLY; mass ratio of mEtOH/mGVL = 1.86/1 or mEtOH/mGLY = 1.86/1). 0.5 wt% of HTC-400-S was loaded with the desired reaction solution into a stainless-steel high pressure autoclave (v = 50 mL) and reactions were performed at 200 °C. The data in Fig. 1B show the EL yields and glucose conversions in different solvents achieved over time starting from 0.5 wt% of HTC-400-S. It was observed that already after t = 30 min, 81% conversion of glucose was reached in the reaction with EtOH as a solvent. Using EtOH/GVL mixture increases the rate of glucose conversion to 91% in comparison to the reaction in EtOH. Glucose was completely converted t > 3 h. Maximum EL yields (in mol%) over HTC-400-S were achieved after t = 6 h. Using GVL or GLY as a co-solvent with EtOH in the reaction obtained higher EL yields than EtOH alone. The highest EL yield of 37 mol% was achieved in EtOH/GLY (t = 6 h), a slightly lower of 35 mol% by using mixture of EtOH/GVL and the lowest EL yield of 25 mol% was measured by using EtOH as a solvent. This suggests that GVL and GLY promote the conversion of glucose to EL. To investigate if they initiate the conversion of glucose, the blank reactions without the catalyst were performed. EL yield of 10 mol% was reached in EtOH/GVL mixture (Fig. 1C), whereas in EtOH or EtOH/GLY solvent mixture no EL was observed. A control reaction with EtOH/GVL solvent mixture without the catalyst and glucose was also performed to explain if EL could be formed only from GVL and EtOH. 3 mol% yield of EL was obtained (Fig. S7†).
Most probably GVL undergoes dehydration to α-angelica lactone and hydration to levulinic acid, which could further form EL with EtOH. Another pathway could be through the hydration of GVL to γ-hydroxyvaleric acid and its dehydrogenation to levulinic acid.27 This observation warrants more study. Regarding the enhanced EL yield in EtOH/GLY solvent mixture with HTC-400-S catalyst, GLY could potentially affect the reactivity of the aldehyde group in glucose or in 5-HMF, as He et al. proved that glycerol as a solvent can promote activity of aldehydes.28
One pot conversion of glucose performed at longer reaction times (t > 6 h) revealed lower EL yields due to the side reactions (Fig. 1B). GC-MS analysis proves different side products depending on the used solvent (Fig. S4–S6†). This indicates solvent rearrangement/etherification reactions at longer reaction times at 200 °C.
For comparison and to further ascertain solvent influence on the EL yield over HTC-400-S, control experiments were performed using the homogeneous equivalent H2SO4 (0.05 wt%) (Fig. 1C). As might be expected, H2SO4 performed well with EL yield of 34 mol% but no significant differences in EL yields were observed when using different solvent mixtures. Higher EL yields (ca. 48 mol%) obtained by H2SO4 have been reported by Xu et al. after shorter reaction times (<1 h) however, using higher H2SO4 concentrations (1 wt%).29
To demonstrate the advantage aspect of HTC-400-S catalyst in the one pot conversion of glucose to EL in green solvents, our catalyst was compared to other carbonaceous acid catalysts as well as to commonly applied sulphated metal oxides (Table S2†). It is observed that among carbonaceous catalysts, HTC-400-S has an outstanding performance in acid catalysed conversion of glucose as well as of fructose to EL probably due to its high specific surface area, mesoporosity and high acid density. In regard to the sulphate metal oxides they perform well and offer better recyclability due to the possible calcination step, which removes the humin deposits from the catalyst surface. EL yields of mesoporous HTC-400-S catalyst were compared to the conventional non-porous HTC catalyst (denoted as HTC-S), which was synthesised without the addition of borax and post-thermal treatment. Despite its high acidity (2.9 mmol g−1), HTC-S obtained lower EL yields probably as a result of its low specific surface area (43.9 m2 g−1) and non-porous structure.
Regarding HTC-400-S catalyst re-use, complete deactivation was observed after the 2nd recycle in EtOH (Fig. 1C). The reusability of HTC-400-S was found to be better in EtOH/GVL and EtOH/GLY solvent mixtures. The measured yield decreased from 37 mol% to 16 mol% for EtOH/GLY mixture and from 35 mol% to 15 mol% for EtOH/GVL mixture after four cycles. In EtOH/GVL mixture, GVL may participate in the reaction and in turn improving EL yields. Complete loss of catalytic activity in EtOH presumably results from humin formation and possible sulphur leaching. The EtOH/GLY mixture prevented visible humin formation (e.g. on the reactor wall), whereas EtOH/GVL only partially inhibited a visible deposition of humins. It has been reported that GVL is capable of solubilising humin30 but there are currently no reports on humin solubility in GLY. Despite decreased humin formation in GLY/EtOH and GVL/EtOH solvents, spent HTC-400-S catalyst after 4th catalytic cycles presented a lower specific surface area, reduced pore volume and S content (Table S1†). SEM images (Fig. S8†) do not show morphological changes at the micron level in the structure of HTC-400-S after usage in different solvents, but at higher resolution analysis indicates “smoothing” of surface features. The mass increase of the catalyst was observed after 4 catalytic cycles as well as the increased amount of carbon presumed to be from humin deposition on the HTC-400-S surface. The lowest content of S on HTC-400-S catalyst after 4 catalytic cycles was observed when only EtOH was used as a solvent and higher S contents were detected by using GVL/EtOH and GLY/EtOH solvent mixtures (Table S1†). Fig. 1D shows the EL yields in quantitative terms with respect to μmol of accessible acidic sides per m2 determined by back titration of the fresh HTC-400-S catalyst and spent catalysts after 2nd, 3rd and 4th cycle. The number of accessible acid sites per m2 of HTC-400-S significantly decreases after 2nd catalyst reuse in EtOH and therefore no EL in 3rd and 4th cycle was obtained. Higher catalyst activity after the 2nd cycle in GVL/EtOH and GLY/EtOH mixtures is due to the higher number of accessible acid sites which catalyse the conversion of glucose to EL. CHNS elemental analysis and ICP-OES of liquid product solutions after 1st catalytic run confirmed the leached sulphur in the product solution (Table S1†). As expected, S loss from HTC-400-S in EtOH was the highest, suggesting GLY and GVL partially inhibit S leaching. This confirms our findings in regard to complete deactivation of HTC-400-S after the 2nd recycle in EtOH due to the combination of S-leaching and humin deposition on the catalyst. Leached S in the product mixture could be removed by desulphurisation using ion exchange resins or precipitation methods with salts.31
In conclusion, the use of EtOH/GVL and EtOH/GLY solvent mixtures in one pot synthesis of EL from glucose was found to improve EL yield as well the reusability of HTC-400-S catalyst partly by preventing the leaching of S-containing functional groups and visible humin formation. Interestingly, EtOH/GVL solvent mixture was found to initiate the conversion of glucose to EL and also yielded very small amounts of EL. EtOH/GLY mixture completely inhibited humin deposition on the reactor wall – an advantage with regard to performing the same reaction under continuous flow operation (e.g. avoidance of blockages). Further work should focus on the optimisation of reaction conditions (e.g. lower reaction temperature) to further inhibit humin formation and consequently improve catalyst recyclability. Likewise, the scope for further modification of the carbonaceous support chemistry (e.g. via higher carbonisation temperature) opens opportunities to further manipulate surface–reagent interactions and optimisation of catalyst performance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by an “Attract” award for R. J. White (as financed by the Fraunhofer Society and Fraunhofer Institute for Solar Energy Systems ISE). A. Fischer thanks the University of Freiburg and the BMBF for core funding (01FP13033F) as well as the BMBF for generous funding of the HR-SEM via the project EDELKAT (FKZ 03X5524). The authors would like to thank A. Siegel for CHNS elemental analysis, A. Becherer for SEM support and P. Hügenell for N2 sorption analysis.Notes and references
- T. M. C. Hoang, E. R. H. van Eck, W. P. Bula, J. G. E. Gardeniers, L. Lefferts and K. Seshan, Green Chem., 2015, 17, 959–972 RSC.
- (a) P. P. Upare, J.-W. Yoon, M. Y. Kim, H.-Y. Kang, D. W. Hwang, Y. K. Hwang, H. H. Kung and J.-S. Chang, Green Chem., 2013, 15, 2935 RSC; (b) S. Kang, J. Fu and G. Zhang, Renewable Sustainable Energy Rev., 2018, 94, 340–362 CrossRef CAS.
- X. Hu and C.-Z. Li, Green Chem., 2011, 13, 1676–1679 RSC.
- L. Peng, L. Lin, J. Zhang, J. Shi and S. Liu, Appl. Catal., A, 2011, 397, 259–265 CrossRef CAS.
- F. D. Pileidis and M.-M. Titirici, ChemSusChem, 2016, 9, 562–582 CrossRef CAS PubMed.
- A. Démolis, N. Essayem and F. Rataboul, ACS Sustainable Chem. Eng., 2014, 2, 1338–1352 CrossRef.
- (a) H. Joshi, B. R. Moser, J. Toler, W. F. Smith and T. Walker, Biomass Bioenergy, 2011, 35, 3262–3266 CrossRef CAS; (b) B. C. Windom, T. M. Lovestead, M. Mascal, E. B. Nikitin and T. J. Bruno, Energy Fuels, 2011, 25, 1878–1890 CrossRef CAS.
- G. Morales, A. Osatiashtiani, B. Hernández, J. Iglesias, J. A. Melero, M. Paniagua, D. R. Brown, M. Granollers, A. F. Lee and K. Wilson, Chem. Commun., 2014, 50, 11742–11745 RSC.
- E. Ahmad, M. I. Alam, K. K. Pant and M. A. Haider, Green Chem., 2016, 18, 4804–4823 RSC.
- S. Saravanamurugan and A. Riisager, Catal. Commun., 2012, 17, 71–75 CrossRef CAS.
- I. van Zandvoort, Y. Wang, C. B. Rasrendra, E. R. H. van Eck, P. C. A. Bruijnincx, H. J. Heeres and B. M. Weckhuysen, ChemSusChem, 2013, 6, 1745–1758 CrossRef CAS PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- (a) J. Heltzel and C. R. F. Lund, Catal. Today, 2016, 269, 88–92 CrossRef CAS; (b) E. I. Gürbüz, J. M. R. Gallo, D. M. Alonso, S. G. Wettstein, W. Y. Lim and J. A. Dumesic, Angew. Chem., Int. Ed., 2013, 125, 1308–1312 CrossRef.
- W. Hao, X. Tang, X. Zeng, Y. Sun, S. Liu and L. Lin, BioResources, 2015, 10, 4191–4203 CAS.
- S. Saravanamurugan, O. N. van Buu and A. Riisager, ChemSusChem, 2011, 4, 723–726 CrossRef CAS PubMed.
- S. Saravanamurugan and A. Riisager, ChemCatChem, 2013, 5, 1754–1757 CrossRef CAS.
- Z. Yuan, Z. Zhang, J. Zheng and J. Lin, Fuel, 2015, 150, 236–242 CrossRef CAS.
- (a) M. Hara, T. Yoshida, A. Takagaki, T. Takata, J. N. Kondo, S. Hayashi and K. Domen, Angew. Chem., Int. Ed. Engl., 2004, 43, 2955–2958 CrossRef CAS PubMed; (b) M. Toda, A. Takagaki, M. Okamura, J. N. Kondo, S. Hayashi, K. Domen and M. Hara, Nature, 2005, 438, 177–178 CrossRef PubMed.
- T.-P. Fellinger, R. J. White, M.-M. Titirici and M. Antonietti, Adv. Funct. Mater., 2012, 22, 3254–3260 CrossRef CAS.
- J. M. Fraile, E. García-Bordejé, E. Pires and L. Roldán, J. Catal., 2015, 324, 107–118 CrossRef CAS.
- J. A. Maciá-Agulló, M. Sevilla, M. A. Diez and A. B. Fuertes, ChemSusChem, 2010, 3, 1352–1354 CrossRef PubMed.
- F. D. Pileidis, M. Tabassum, S. Coutts and M.-M. Titirici, Chin. J. Catal., 2014, 35, 929–936 CrossRef CAS.
- T. S. Hansen, J. Mielby and A. Riisager, Green Chem., 2011, 13, 109–114 RSC.
- (a) Q. Guan, Y. Li, Y. Chen, Y. Shi, J. Gu, B. Li, R. Miao, Q. Chen and P. Ning, RSC Adv., 2017, 7, 7250–7258 RSC; (b) A. P. Terzyk, Colloids Surf., A, 2001, 177, 23–45 CrossRef CAS.
- G. S. Duesberg, R. Graupner, P. Downes, A. Minett, L. Ley, S. Roth and N. Nicoloso, Synth. Met., 2004, 142, 263–266 CrossRef CAS.
- E. Cano-Serrano, G. Blanco-Brieva, J. M. Campos-Martin and J. L. G. Fierro, Langmuir, 2003, 19, 7621–7627 CrossRef CAS.
- W. R. H. Wright and R. Palkovits, ChemSusChem, 2012, 5, 1657–1667 CrossRef CAS PubMed.
- F. He, P. Li, Y. Gu and G. Li, Green Chem., 2009, 11, 1767 RSC.
- G.-Z. Xu, C. Chang, W.-N. Zhu, B. Li, X.-J. Ma and F.-G. Du, Chem. Pap., 2013, 67, 57 Search PubMed.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC.
- Handbook of Refinery Desulfurization, N. S. El-Gendy and J. G. Speight, CRC Press, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC, and GC-MS of liquid products, N2 sorption analysis, XPS and SEM analysis of HTC-400-S. See DOI: 10.1039/c9ra03902c |
| This journal is © The Royal Society of Chemistry 2019 |