
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoordinating gallium hexacyanocobaltate: Prussian blue-based nanomaterial for Li-ion storage
Kaiqiang Zhang ab,
Tae Hyung Leea,
Bailey Bubachc,
Mehdi Ostadhassan*c,
Ho Won Jang*a,
Ji-Won Choi*b and
Mohammadreza Shokouhimehr*ac
ab,
Tae Hyung Leea,
Bailey Bubachc,
Mehdi Ostadhassan*c,
Ho Won Jang*a,
Ji-Won Choi*b and
Mohammadreza Shokouhimehr*ac
aDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea. E-mail: hwjang@snu.ac.kr; mrsh2@snu.ac.kr
bElectronic Materials Center, Korea Institute of Science and Technology (KIST), Seoul 136-791, Republic of Korea. E-mail: jwchoi@kist.re.kr
cDepartment of Petroleum Engineering, University of North Dakota, Grand Forks, ND 58202, USA. E-mail: mehdi.ostadhassan@und.edu
First published on 27th August 2019
Abstract
Prussian blue analogs (PBAs) are a type of metal–organic framework and have drawn significant attention recently. To date, most are constructed with divalent transition metal ions coordinated to the N end of a cyanide bridge. In this report, we studied a trivalent gallium ion-based Ga hexacyanocobaltate (GaHCCo), which depicted a face-centered cubic crystal structure. In addition, the synthesized GaHCCo was demonstrated as a cathode material of lithium-ion batteries (LIBs) and was found to exhibit long-term stability, having a capacity retention of 75% after 3000 cycles of repeated charge–discharge cycling and an extremely high coulombic efficiency of 98%, which was achieved because of a solid-state diffusion controlled Li-ion storage process. After ex situ XRD analysis on the different charge stages, the Li-ion storage in the GaHCCo was attributed to the Co species via the formation of a Li/Co compound. This work will pave the way toward the study of PBAs constructed with trivalent metal ions and provide more insights into the development of high-performance LIBs in the future.
Introduction
Batteries are considered necessary devices for the integration of renewable and green natural energy resources into the electric grid.1–4 Lithium-ion batteries (LIBs) have seen great advances because of increases in the reliability and durability of the anodes and cathodes, and LIBs now have highly improved energy and power densities.5–7 However, improvements in anodic safety and in cost efficiency of electrode materials are still required. Typically, the further modification of anode materials to prevent or minimize the formation of dendrites and to form a stable solid electrolyte interface8,9 and the development of low-cost cathode materials synthesized by facile and environmentally friendly processes are required for sustainable developments. To date, several research groups have explored different types of high-performing cathode materials for LIBs,10,11 but further work is required to reduce the complexity of cathode material synthesis while maintaining excellent electrochemical properties.Metal–organic frameworks (MOFs) are multi-functional materials that have been studied in diverse fields including catalysts, gas sensors, and solid electrolytes and have shown great potential.12–16 With regard to the use of MOFs as cathode materials for LIBs, two different methods (direct use or as sacrificial precursors) are typically employed.17,18 In particular, Prussian blue analogs (PBAs), which are typical MOFs, are considered one of the most attractive candidates because of their easy of synthesis in aqueous solution at room temperature without the need of additives or post-processing.19 Recently, the study of PBAs as cathode materials for LIBs has mainly focused on functionalized PBAs (for example, those treated by etching, oxidation, or combination with other materials such as carbon nanotubes),20,21 which results in highly improved electrochemical properties. However, this extra processing increases the fabrication costs during a massive fabrication. Thus, the further study of naked PBAs is logical. With regard to the study of naked PBAs, research groups have mainly focused on A3[MIII(CN)6]2 and A2MII(CN)6 systems having A2+ ions and B4[MII(CN)6]3 and BMIII(CN)6 systems having B3+ ions (M: transition metal ions coordinated to C; A and B: transition metal ions coordinated to N). However, the latter case (i.e., those constructed with B3+ ions) is limited to Fe3+ such as Fe3+Fe(CN)6, Fe3+Co(CN)6, and Fe3+Cr(CN)6.22,23 Therefore, studies of PBAs constructed with other types of N-coordinated trivalent metal ions are necessary to provide an in-depth understanding for this overlooked issue. To obtain a trivalent metal ion that is soluble and stable in an aqueous solution, we systematically studied the elements in the periodic table. The elements of the boron group in the periodic table have typical trivalent characteristics. We, thus, fabricated the six-fold coordinated PBAs using the lighter Ga species. In particular, we synthesized the Ga hexacyanocobaltate (GaHCCo) and further studied its electrochemical properties as a cathode material of LIBs, revealing a solid-state diffusion-controlled Li-ion storage mechanism by utilizing the Co species.
Experimental
Materials
GaHCCo nanoparticles (NPs) were synthesized by a co-precipitation method involving the simultaneous dropwise addition of 100 mL Ga(NO3)3 (0.01 M) (Sigma-Aldrich) and 100 mL K3[Co(CN)6] (0.01 M) (Sigma-Aldrich) to 200 mL deionized H2O. The entire synthesis process was carried out at 80 °C with vigorous stirring. The formation of a precipitate was observed after a period of heating. After a cooling of the mixture to room temperature, the precipitate was separated and rinsed with large amounts of deionized water several times to remove the impurity ions (such as K+) and subsequently dried in a vacuum oven at 60 °C; it was then ready for subsequent use.Physical characterization
X-ray diffraction (XRD, D8 Advance, Bruker, Cu Kα radiation at a fixed incident angle of 2°), X-ray photoelectron spectroscopy (XPS, PHI 5000 VersaProbe with an Al Kα source (Sigma Probe, VG Scientific)), Raman spectroscopy (inVia Raman Microscope), and Fourier transform infrared (FTIR, Nicolet iS50, Thermo Fisher) spectroscopy measurements were carried out. The morphology and composition of the samples were investigated using field emission-scanning electron microscopy (FE-SEM, SUPRA 55VP, Carl Zeiss AG), transmission electron microscopy (TEM, JEOL JEM-2100F), energy-dispersive X-ray spectroscopy (EDX). The analysis for a composition of the as-prepared GaHCCo was also carried out using thermogravimetric analysis (TGA) which was performed under N2 flow from room temperature to 700 °C with a temperature ramp of 10 °C min−1, as well as X-ray fluorescence measurements (XRF, ZSX-PRIMUS, Rigaku). Quantitative measurements were achieved by using inductively coupled plasma (ICP) for Ga, Co, and K elements and EA(CHNS) analysis for C, H, and N elements.Electrode preparation
A slurry containing GaHCCo, carbon black (Super P Li), and poly(vinylidene)difluoride in a mass ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 was prepared by manually grinding the mixed powders and subsequently dispersing them in N-methyl-2-pyrolidinone (NMP) in a manner similar to the standard slurry preparation method widely employed in the LIB research communities. In addition, before the injection of NMP, the mixed powders were dried overnight in a vacuum oven at 60 °C. The mixed powder weight was measured before and after vacuum drying to ensure that water was eliminated as much as possible. A working electrode with a mass loading of approximately 3 mg cm−2 was prepared by spreading the slurry on a graphite paper current collector (Alfa Aesar), followed by drying overnight in a vacuum oven at 60 °C.
:2:1 was prepared by manually grinding the mixed powders and subsequently dispersing them in N-methyl-2-pyrolidinone (NMP) in a manner similar to the standard slurry preparation method widely employed in the LIB research communities. In addition, before the injection of NMP, the mixed powders were dried overnight in a vacuum oven at 60 °C. The mixed powder weight was measured before and after vacuum drying to ensure that water was eliminated as much as possible. A working electrode with a mass loading of approximately 3 mg cm−2 was prepared by spreading the slurry on a graphite paper current collector (Alfa Aesar), followed by drying overnight in a vacuum oven at 60 °C.
Electrochemical characterization
To perform electrochemical measurements, a two-electrode setup was used: the GaHCCo active material was used as the working electrode and sufficient lithium metal to ensure that the capacity was limited solely by the mass of active materials was used as the anode. The electrodes were placed in 1.0 M LiPF6 in a 1:1 mixture (by volume) of ethylene carbonate and diethylene carbonate in an argon-filled glove box.
The electrochemical impedance spectrum (EIS) of GaHCCo was measured using an Im6ex ZAHNER instrument in the assembled half-cell. The frequency range used was from 10 mHz to 1 MHz, at a voltage amplitude of 10 mV.
Cyclic voltammetry (CV) measurements were performed on an electrochemical workstation (WBCS3000, WonATech Co., Ltd., Korea) in the potential range of 2.2–4.5 V vs. Li+/Li at a scan rate of 0.5 mV s−1. Galvanostatic charge/discharge cycling measurements were performed between 2.2 and 4.5 V vs. Li+/Li at various current densities corresponding to 100, 200, 400, 600, 800, and 1000 mA g−1. Unless otherwise specified, all the current densities and specific capacities in the present study were calculated based on the weight of the active material, GaHCCo.
Results and discussion
The proposed crystal structure is shown in Fig. 1; the structure consists of a face-centered cubic crystal structure containing cyanide bridges alternately bonded with Co3+ and Ga3+ centers, having eight cavities (sub-units) within each unit with a size as small as ca. 1 nm. Morphologies of the GaHCCo particles are observed using FE-SEM, and the NPs are found with a size of ca. 500 nm (Fig. 1b). Furthermore, the comprising elements were uniformly distributed throughout the NPs (Fig. 1c). The further magnification images of the particles are obtained using TEM (Fig. 1d) displaying the particle with a diameter of ca. 500 nm and uniformly distributed elements as shown in the elemental mapping. To investigate this further, TGA experiments were conducted (Fig. 1e), and it revealed a constant decrease in a weight of the product upon heating, confirming the structural weakness of GaHCCo, which is unlike that of other PBAs being able to withstand a temperature up to 400 °C.24 The crystalized feature is depicted in the XRD result. Furthermore, the crystal structure is further confirmed by the Rietveld refinement showing well matched XRD curves for experimentally obtained result and calculated one. The refinement parameters are given in Table 1 demonstrating a space group 225 and lattice parameters of a = b = c = 10.0971 Å. The characteristic lattice diffraction peaks in the XRD patterns (Fig. 1f) indicate a typical face-centered cubic crystal structure for the GaHCCo. The characteristic Bragg diffraction peaks (PDF card no. 01-072-1431) are well indexed. Here, the standard diffraction peaks are referred to the PBA Co3[Co(CN)6]2 with a cubic crystal structure due to the absence of the corresponding standard card for GaHCCo. The presence of cyanide bridges was inferred from FTIR (Fig. 1g), which showed characteristic bands at ca. 2200 cm−1. Furthermore, the cyanide bridges were further detected using Raman spectroscopy (Fig. 1h), revealing characteristic bands at ca. 2210 and 2240 cm−1, consistent with reports concerning other PBAs.25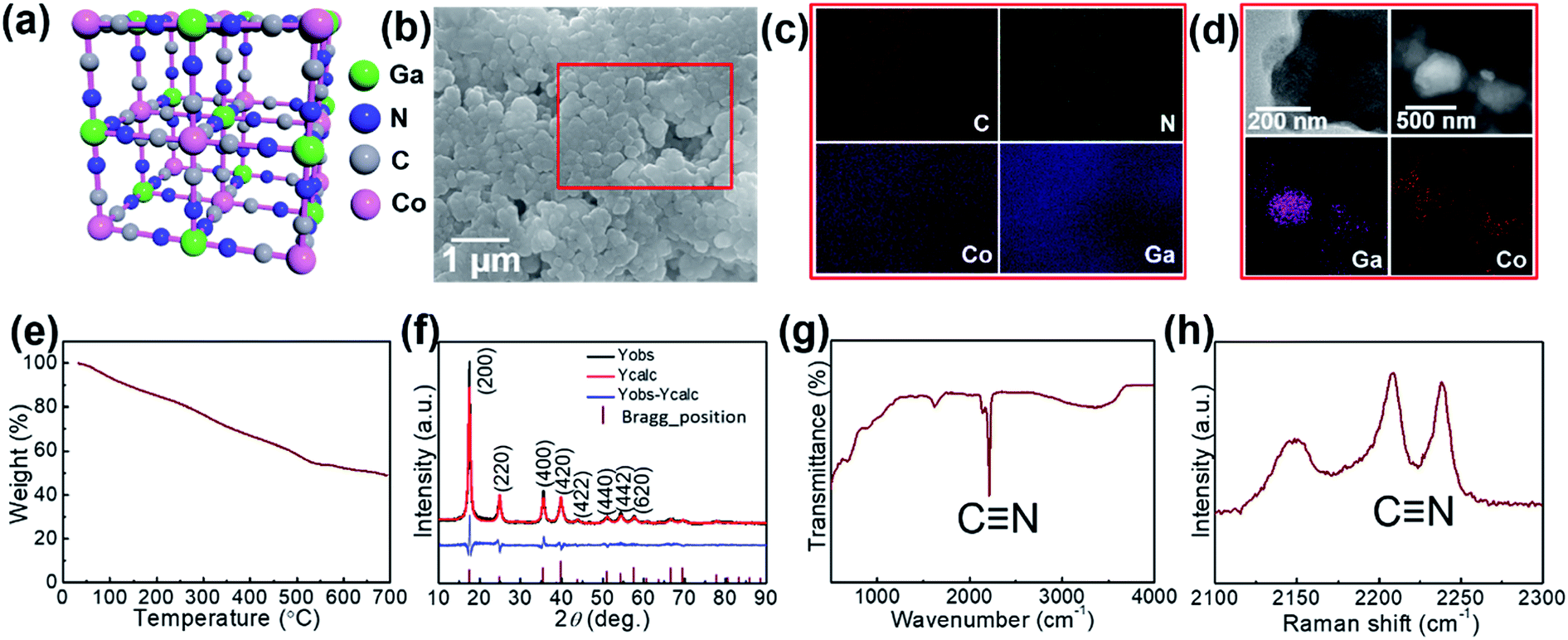 | ||
| Fig. 1 (a) Crystal structure (b) FE-SEM image, (c) EDX mapping, (d) TEM images, (e) TGA curve, (f) XRD and Rietveld refinement curves, (g) Raman spectrum, and (h) FT-IR spectrum of the GaHCCo. | ||
| Parameters | Values |
|---|---|
| Space group | 225 |
| a (Å) = b = c | 10.0971 |
| V (Å3) | 1029.4 |
| C (x, y, z) | (0.303, 0.000, 0.000) |
| N (x, y, z) | (0.1928, 0.000, 0.000) |
| Ga (x, y, z) | (0.500, 0.500, 0.500) |
| Co (x, y, z) | (0.000, 0.000, 0.000) |
| RBragg | 3.607 |
| Rwp | 16.42 |
| Rexp | 10.41 |
| Rp | 13.17 |
| GOF | 1.58 |
The comprising elements are also clearly confirmed in XRF spectra (Fig. 2). The composition elements are screened out at different energies as shown by peaks with high intensities for each element. A quantitative elemental analysis for the GaHCCo by ICP and EA(CHNS) (Table 2) demonstrates the molecular formula K0.017Ga0.99Co(CN)6·0.32H2O. Furthermore, the surface chemical properties of the GaHCCo product are studied using XPS. The wide-survey spectrum (Fig. 3a) indicates bonding nature in GaHCCo. The C 1s and N 1s peaks in the deconvoluted spectra are consistent with the presence of cyanide bridges, as indicated by the FTIR and Raman spectra (Fig. 1g and h). Concomitant Co3+ and Co2+ ions in the deconvoluted Co 2p spectra with different energies are consistent with the split Raman peaks. Unlike the Co 2p peak, the deconvoluted Ga 2p peak indicates pure Ga3+ after the aqueous synthesis process. Thus, the XPS results demonstrate the bonding state of the formed GaHCCo.
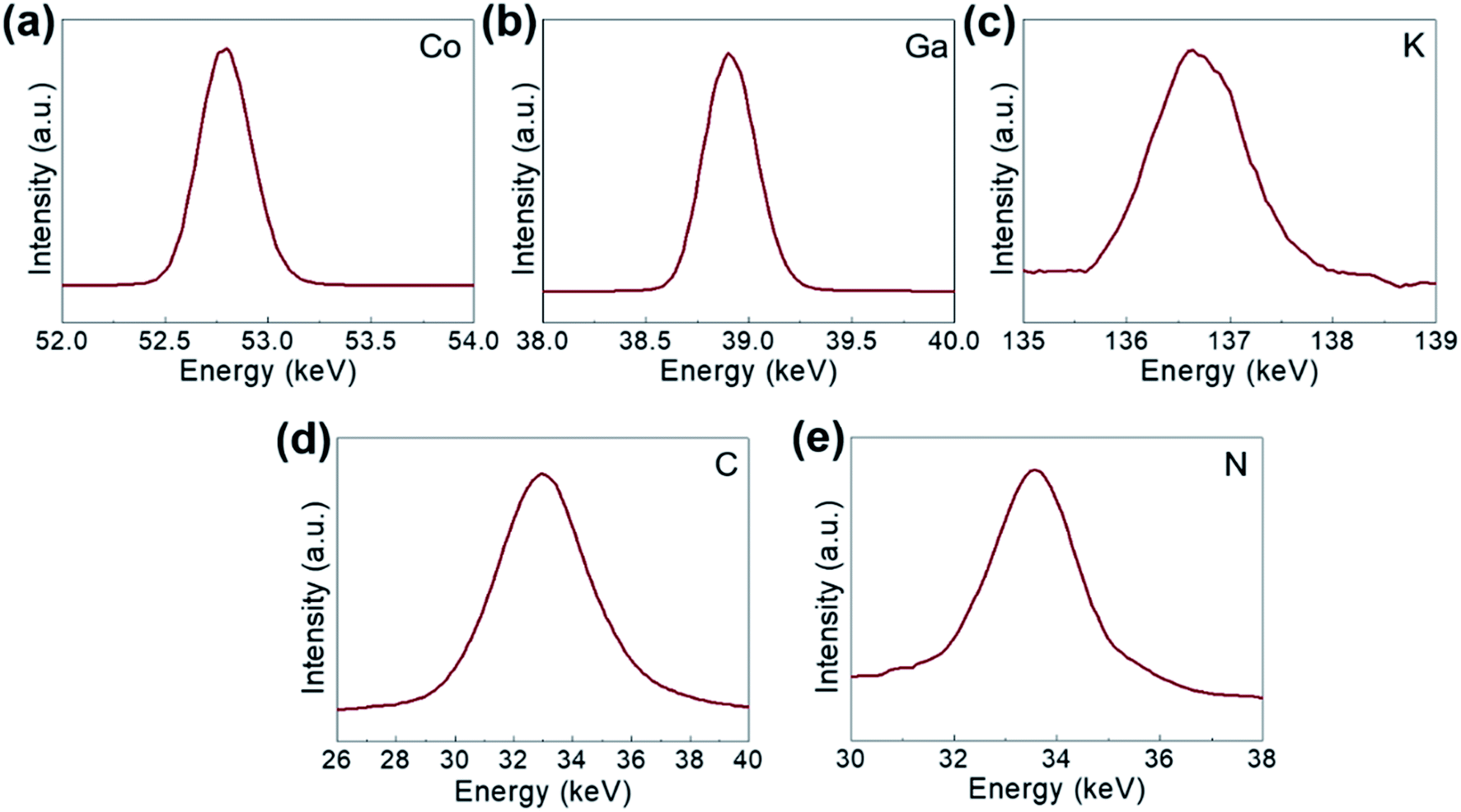 | ||
| Fig. 2 XPS spectra of the synthesized GaHCCo: (a) Co 2p, (b) Ga 2p, (c) K 2p, (d) C 1s, and (e) N 1s. | ||
| Element | C (wt%) | H (wt%) | N (wt%) | Co (wt%) | Ga (wt%) | K (wt%) |
| Amount | 22.51 | 1.29 | 24.19 | 13.30 | 15.60 | 0.07 |
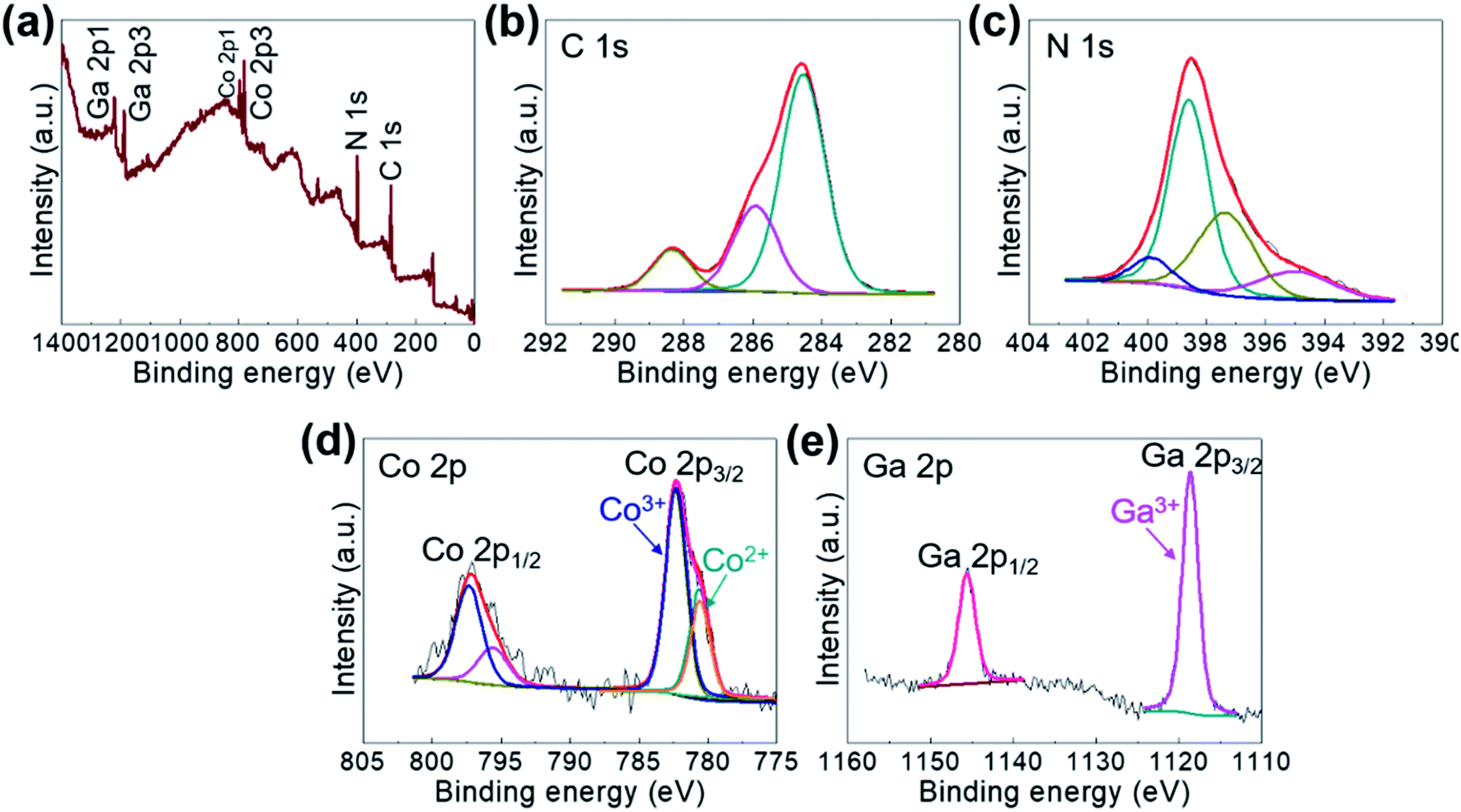 | ||
| Fig. 3 (a) Wide-survey and deconvoluted (b) C 1s, (c) N 1s, (d) Co 2p, and (e) Ga 2p XPS spectra of the GaHCCo. | ||
Electrochemical properties of GaHCCo as a cathode material are studied using CV measurements to determine the electrochemical activity toward Li-ion storage. A stable potential window of 2.2–4.5 V vs. Li+/Li without obvious distortion was observed in the CV profile (Fig. 4a). The quasi-rectangular CV curves indicate a predominant double-layer capacitive charge storage feature. The rate performance of GaHCCo is determined at current densities of 100, 200, 400, 600, 800, and 1000 mA g−1, with a capacity retention of 42.2% at 1000 mA g−1. The corresponding charge/discharge potential profiles exhibit quasi-plateaus at ca. 4.0 V vs. Li+/Li (Fig. 4c). A capacity differential (dQ/dV) (Fig. 4d) exhibits a pair of depressed humps as marked in the differential curves suggesting the charge/discharge plateaus in the voltage profiles (Fig. 4c). The long-term repeated charge/discharge process is shown in Fig. 4e, where an initial capacity of ca. 80 mA h g−1 after the activation phase is obtained. A high capacity retention of 75% is achieved after 3000 cycles of successive charge/discharge testing, corresponding to an average loss in capacity of 0.008% for each cycle, although some fluctuation in the electrochemical capacities is observed. Notably, a high coulombic efficiency of ca. 98% is achieved during the long-term cycling measurements (Fig. 4e). The voltage profiles during the long-term capacity test are shown in Fig. 4f, revealing the tedious charge/discharge process with quasi-plateaus at around 3.9 V vs. Li+/Li (charge) and 3.3 V vs. Li+/Li (discharge) for each cycle, which can be described as Li-ion redox reactions with local equilibrium.26,27
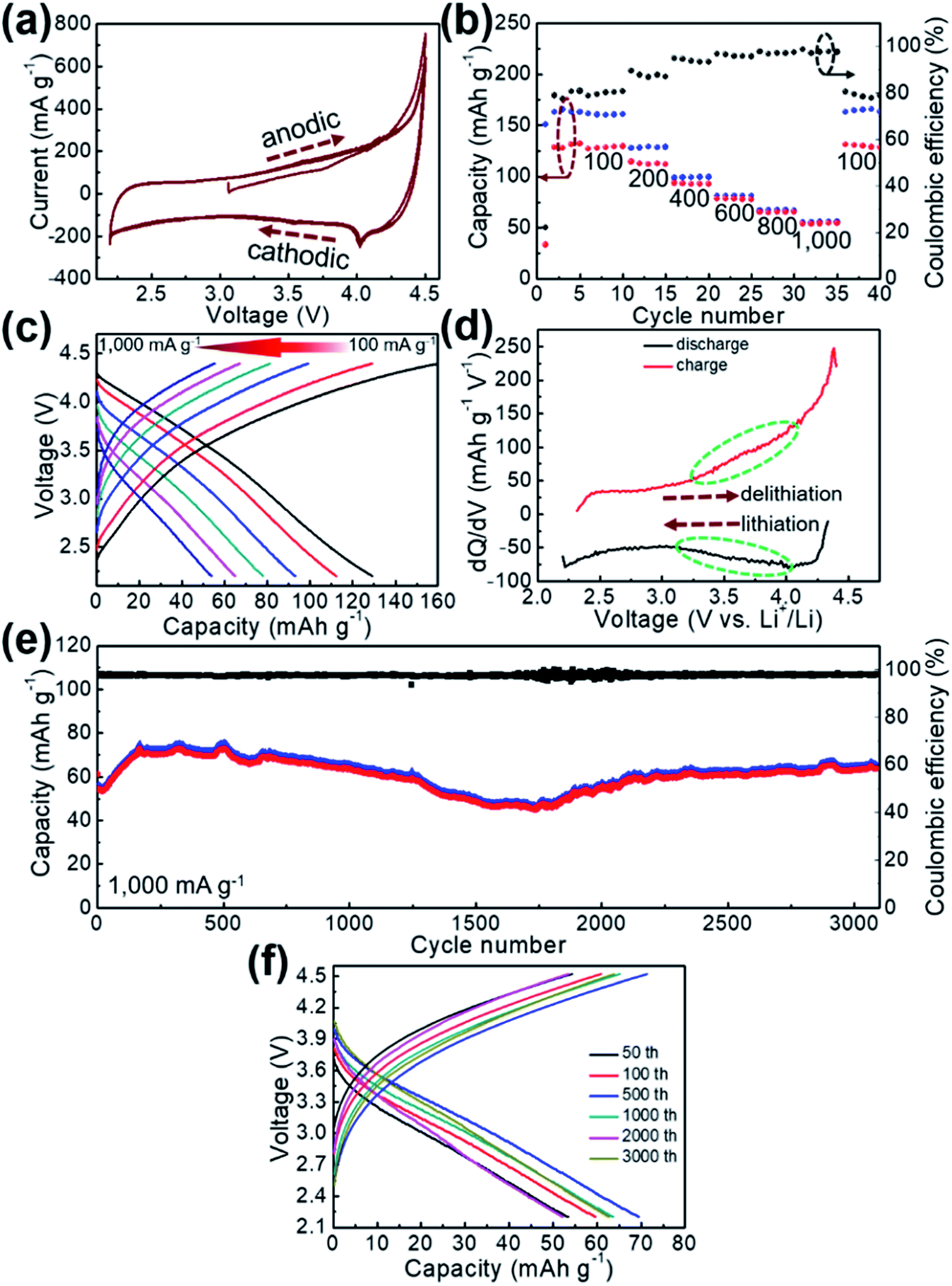 | ||
| Fig. 4 (a) CV curves, (b) rate performance, (c) corresponding voltage profiles, (d) dQ/dV curves, (e) long-term repeated cycling test, and (f) representative voltage profiles of the GaHCCo. | ||
The Li-ion storage process of GaHCCo is further studied using the ex situ XRD method (Fig. 5). The XRD spectra of the samples charged to 2.2, 3.0, and 3.6 V vs. Li+/Li reveal the collapse of the GaHCCo structure, which is similar with the previously reported Co3[Co(CN)6]2 as anode for potassium-ion batteries.28 This can be further observed in the sample after a repeated charge/discharge cycling test (Fig. 6b), where the characteristic peaks of GaHCCo are greatly eliminated. A newly formed peak (corresponding to Li0.73CoO2) at ca. 18° in the XRD pattern (Fig. 5d) indicates that the corresponding charge/discharge process is mainly achieved by bonding with Co species instead of Ga. On increasing the cutoff voltage, the intensity of this phase was reduced, illustrating the underlying delithiation process. This can be further demonstrated in the ex situ XPS results by the gradually increase of the deconvoluted Co2+ peak intensity with discharge proceeding (Fig. 7). Other deconvoluted XPS results (C 1s, N 1s, and Ga 2p) exhibit an absence of obvious changes (Fig. 7). This is the first example of Li-ion storage in a collapsed PBAs via bonding with one of the host species.
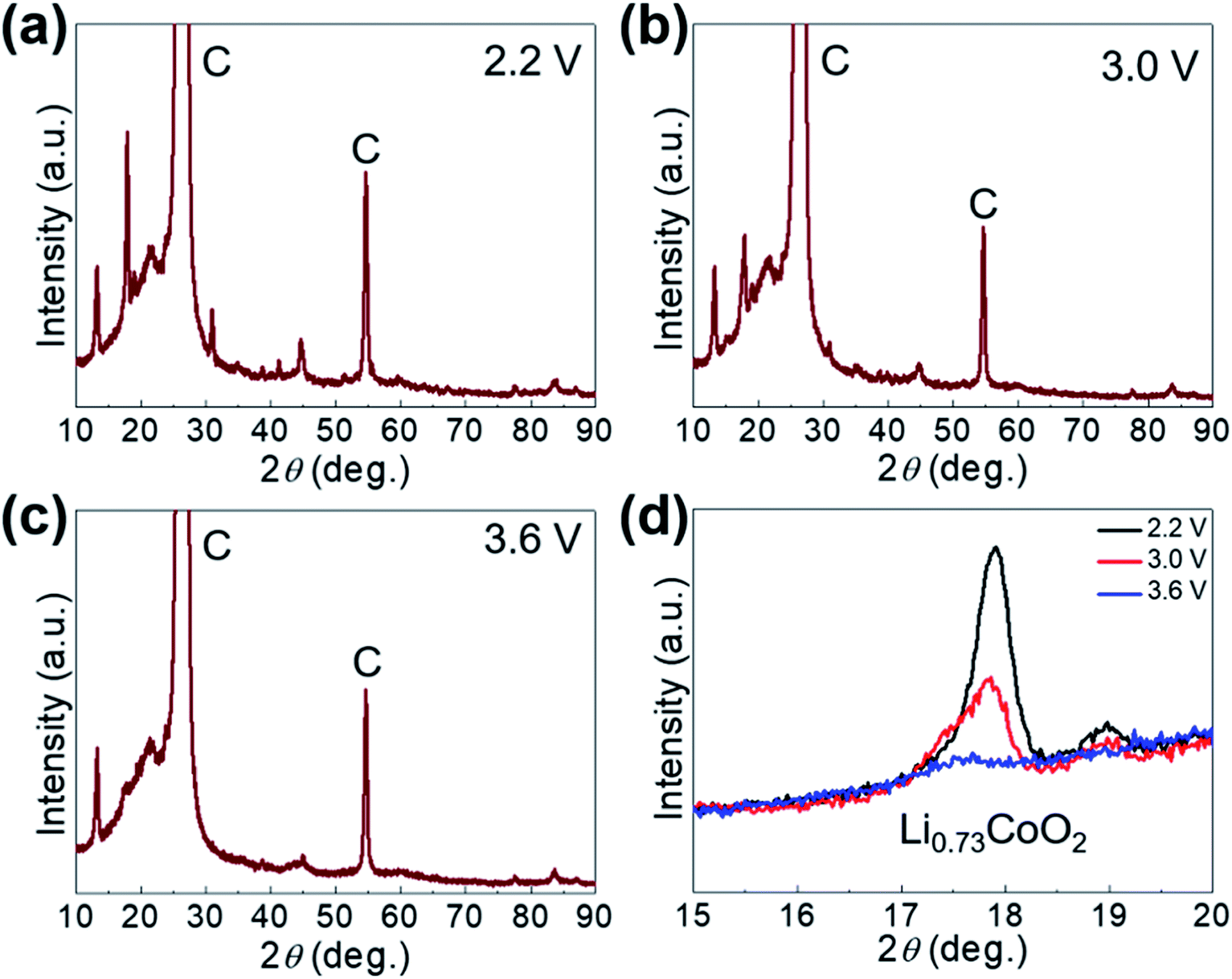 | ||
| Fig. 5 (a) Ex situ XRD patterns of GaHCCo at (a) 2.2 V, (b) 3.0 V, (c) 3.6 V, and (d) magnified XRD peaks. | ||
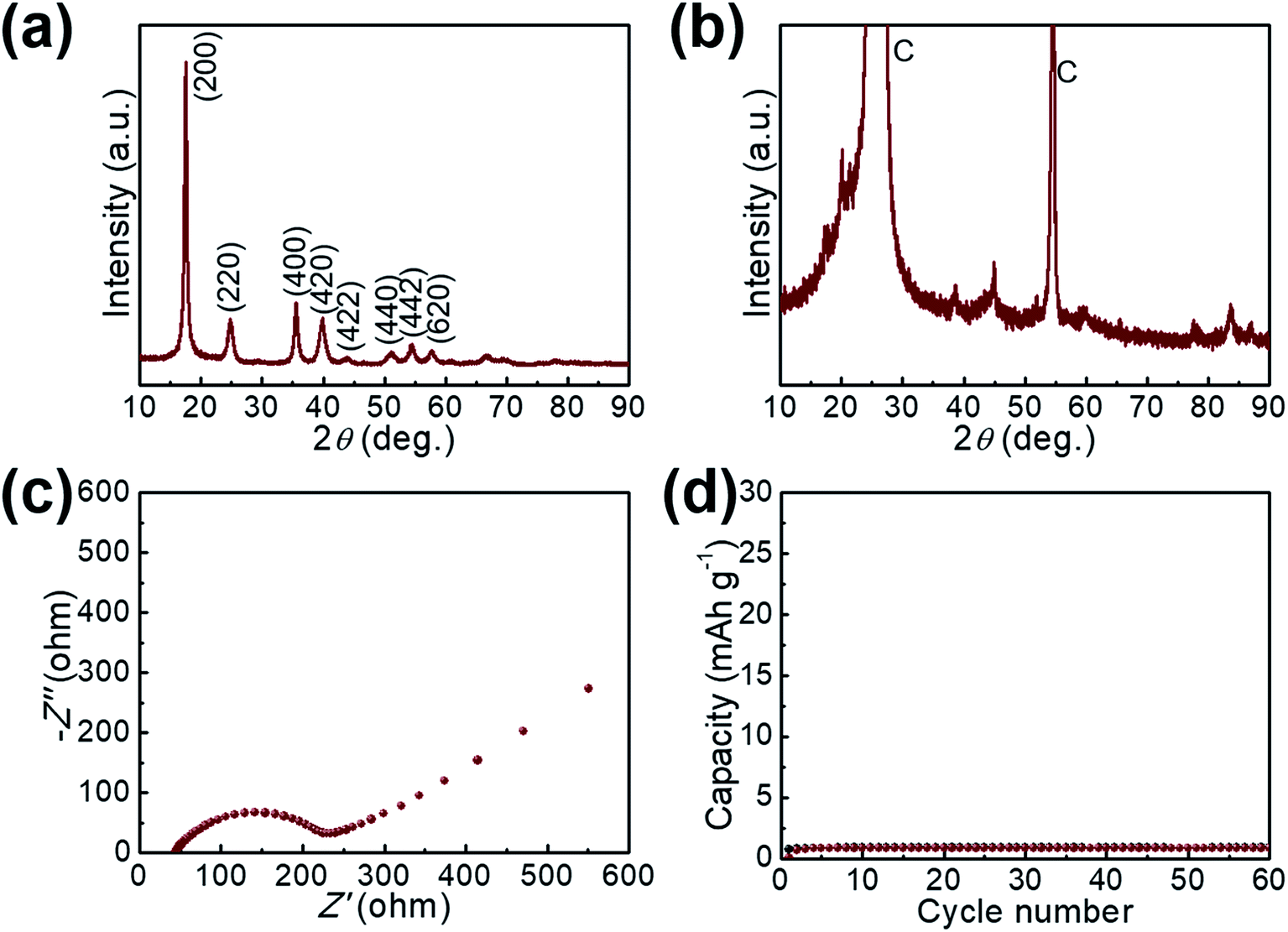 | ||
| Fig. 6 XRD pattern of the GaHCCo (a) before and (b) after cycling test. (c) EIS spectrum of the GaHCCo. (d) The charge/discharge cycling measurement of the bare graphite paper current collector. | ||
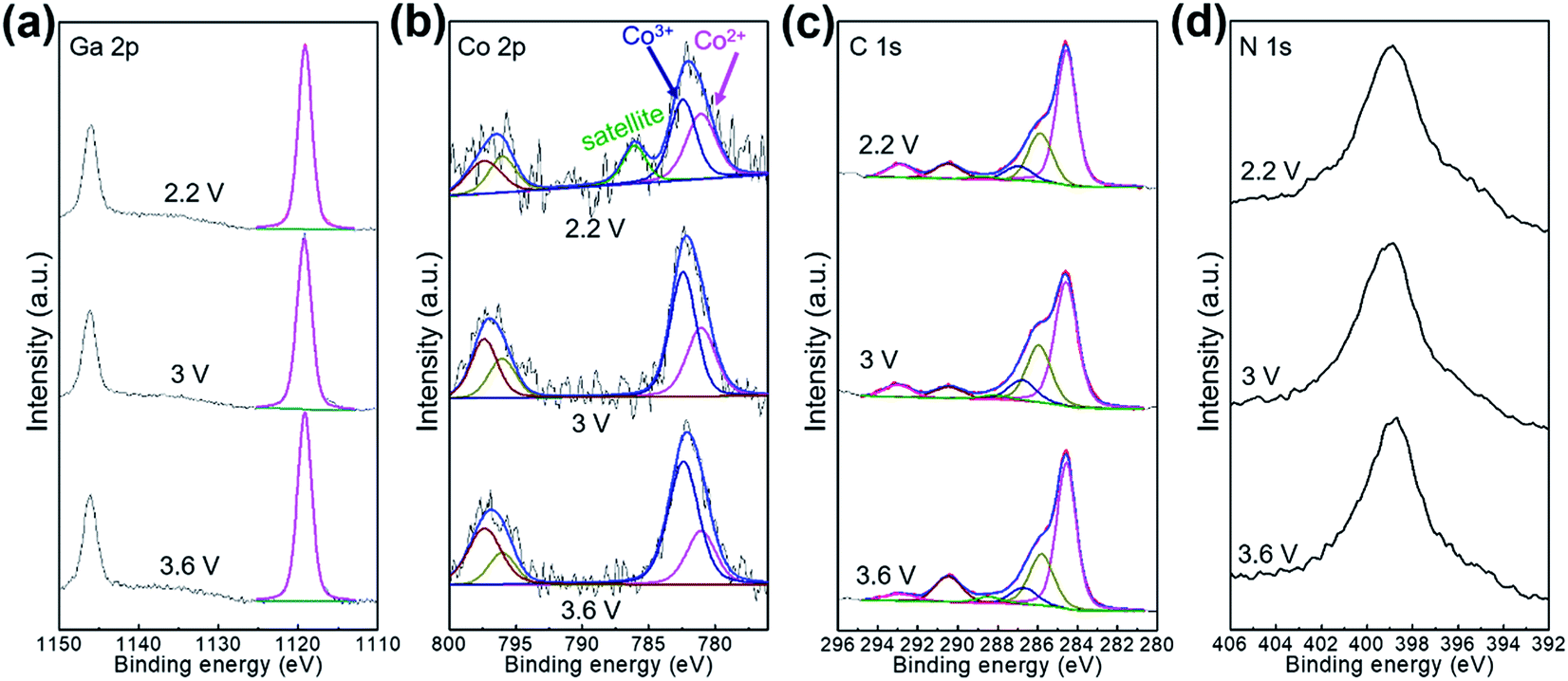 | ||
| Fig. 7 Ex situ XPS characterizations for the electrode charged at 2.2, 3.0, and 3.6 V. | ||
The electrochemical impedance spectra of GaHCCo is shown in Fig. 6c, with a depressed semicircle (charge transfer process) connected with an oblique line (mass transfer process). The material exhibits a charge transfer impedance of ca. 200 Ω. In a controlled experiment, we also measure the capacity of the bare graphite paper current collector, which shows a negligible capacity (Fig. 6d).
To analyze the contributions of diffusion-controlled and capacitive Li-ion storage contributions to the total Li-ion storage capacity of GaHCCo qualitatively and quantitatively, we carried out CV measurements for the GaHCCo cathode at different scan rates (0.5–2.5 mV s−1) within the same potential window of 2.2–4.5 V vs. Li+/Li (Fig. 8a). The curves obtained at all scan rates displayed a quasi-rectangular shape. To investigate this capacitive feature further, the dependence of response current (i) on scan rate (v) is modeled using the following power law: i = avb.
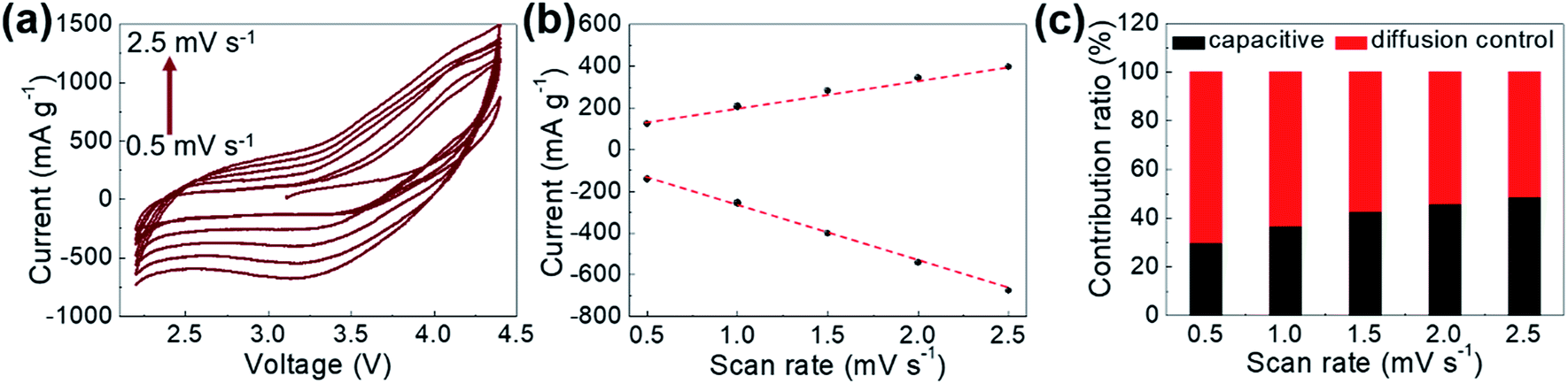 | ||
| Fig. 8 (a) CV curves obtained at various scan rates, (b) response currents at 3.1 V when scanned at different rates, and (c) contribution ratios of capacitive and diffusion-controlled Li-ion storage. | ||
The fitting of a log(i) vs. log(v) plot is expected to afford a straight line with slope b; b = 0.5 corresponds to diffusion-controlled Li-ion storage, indicating a faradaic electrochemical reaction, whereas b = 1.0 corresponds to capacitive Li-ion storage. Here, b-values between 0.5 and 1.0 are observed, which indicates the concomitant nature of diffusion-controlled and capacitive Li-ion storage (Fig. 8b). The relative contributions of these storage mechanisms are further calculated by re-expressing eqn (1) as:
| i = k1v + k2v1/2, | (1) |
Conclusions
In this study, we conduct the formation of GaHCCo with an N-coordinated trivalent metal ion (Ga3+) as a cathode material for LIBs. The PBA depicts a face-centered cubic structure with a unique thermally sensitive property. In addition, the synthesized GaHCCo exhibits long-term charge/discharge stability, having a capacity retention of 75% after 3000 cycles of repeated cycling test with an extremely high coulombic efficiency of 98%, benefiting from a solid-state diffusion-controlled Li-ion storage process. In particular, the Li-ion storage in the GaHCCo is mainly attributed to the Co species by forming a Li/Co compound. This work will pave the way toward the study of PBAs constructed with other trivalent metal ions coordinated to N end of cyanides and provides more insights into the development of high-performing LIBs in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Korea Institute of Science and Technology Future Resource Program (2E29400). Furthermore, the financial supports of the Future Material Discovery Program (2016M3D1A1027666), the Basic Science Research Program (2017R1A2B3009135) through the National Research Foundation of Korea are appreciated, and China Scholarship Council (201808260042).References
- K. Zhang, R. S. Varma, H. W. Jang, J.-W. Choi and M. Shokouhimehr, J. Alloys Compd., 2019, 791, 911–917 CrossRef CAS.
- W. Li, Y. Yang, G. Zhang and Y.-W. Zhang, Nano Lett., 2015, 15, 1691–1697 CrossRef CAS PubMed.
- S.-H. Yu, M. Shokouhimehr, T. Hyeon and Y.-E. Sung, ECS Electrochem. Lett., 2013, 2, A39–A41 CrossRef CAS.
- M. Shokouhimehr, S. H. Yu, D. C. Lee, D. Ling, T. Hyeon and Y. E. Sung, Nanosci. Nanotechnol. Lett., 2013, 5, 770–774 CrossRef CAS.
- Q. Xu, J.-Y. Li, J.-K. Sun, Y.-X. Yin, L.-J. Wan and Y.-G. Guo, Adv. Energy Mater., 2017, 7, 1601481 CrossRef.
- J. Zhou, J. Qin, X. Zhang, C. Shi, E. Liu, J. Li, N. Zhao and C. He, ACS Nano, 2015, 9, 3837–3848 CrossRef CAS PubMed.
- S.-H. Yu, X. Guo, D. Ling, D. Y. Chung, A. Jin, M. Shokouhimehr, T. Hyeon and Y.-E. Sung, RSC Adv., 2014, 4, 37365–37370 RSC.
- S.-H. Yu, M. Park, H. S. Kim, A. Jin, M. Shokouhimehr, T.-Y. Ahn, Y.-W. Kim, T. Hyeon and Y.-E. Sung, RSC Adv., 2014, 4, 12087–12093 RSC.
- Y. Sun, N. Liu and Y. Cui, Nat. Energy, 2016, 1, 16071 CrossRef CAS.
- K. Chen, Z. Sun, R. Fang, Y. Shi, H.-M. Cheng and F. Li, Adv. Funct. Mater., 2018, 28, 1707592 CrossRef.
- Q. Pang, X. Liang, C. Y. Kwok and L. F. Nazar, Nat. Energy, 2016, 1, 16132 CrossRef CAS.
- Y.-B. Huang, J. Liang, X.-S. Wang and R. Cao, Chem. Soc. Rev., 2017, 46, 126–157 RSC.
- W.-T. Koo, S.-J. Choi, S.-J. Kim, J.-S. Jang, H. L. Tuller and I.-D. Kim, J. Am. Chem. Soc., 2016, 138, 13431–13437 CrossRef CAS PubMed.
- Z. Wang, R. Tan, H. Wang, L. Yang, J. Hu, H. Chen and F. Pan, Adv. Mater., 2018, 30, 1704436 CrossRef PubMed.
- Y. You, X. Yu, Y. Yin, K.-W. Nam and Y.-G. Guo, Nano Res., 2015, 8, 117–128 CrossRef CAS.
- Y. You, X. L. Wu, Y. X. Yin and Y.-G. Guo, Energy Environ. Sci., 2014, 7, 1643–1647 RSC.
- T. Wei, M. Zhang, P. Wu, Y.-J. Tang, S.-L. Li, F.-C. Shen, X.-L. Wang, X.-P. Zhou and Y.-Q. Lan, Nano Energy, 2017, 34, 205–214 CrossRef CAS.
- C. Guan, X. Liu, W. Ren, X. Li, C. Cheng and J. Wang, Adv. Energy Mater., 2017, 7, 1602391 CrossRef.
- M. Shokouhimehr, E. S. Soehnlen, J. Hao, M. Griswold, C. Flask, X. Fan, J. P. Basilion, S. Basu and S. D. Huang, J. Mater. Chem., 2010, 20, 5251–5259 RSC.
- X. Hou, G. Zhu, X. Niu, Z. Dai, Z. Yin, Q. Dong, Y. Zhang and X. Dong, J. Alloys Compd., 2017, 729, 518–525 CrossRef CAS.
- J. Shao, J. Feng, H. Zhou and A. Yuan, Appl. Surf. Sci., 2019, 471, 745–752 CrossRef CAS.
- K. Zhang, T. H. Lee, H. W. Jang, M. Shokouhimehr and J.-W. Choi, Electron. Mater. Lett., 2019, 1–10 Search PubMed.
- F. J. Luque, I. A. Kowalik, J. P. P. Ruiz, M. Á. Niño, H. Prima-García, F. M. Romero, D. Arvanitis, E. Coronado, R. Miranda and J. J. Miguel, J. Mater. Chem. C, 2018, 6, 8171–8186 RSC.
- P. Bhatt, S. S. Meena, M. D. Mukadam, B. P. Mandal, A. K. Chauhan and S. M. Yusuf, New J. Chem., 2018, 42, 4567–4578 RSC.
- Q. Wang, S. He, N. Wang, J. Zhao, J. Fang and W. Shen, New J. Chem., 2016, 40, 3244–3251 RSC.
- B. Mazinani, M. Kazazi, G. Mobarhan and M. Shokouhimehr, JOM, 2019, 71, 1499–1506 CrossRef CAS.
- R. A. Huggins, J. Power Sources, 1999, 81, 13–19 CrossRef.
- L. Deng, Z. Yang, L. Tan, L. Zeng, Y. Zhu and L. Guo, Adv. Mater., 2018, 30, 1802510 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2019 |