
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTime-controlled synthesis of the 3D coordination polymer U(1,2,3-Hbtc)2 followed by the formation of molecular poly-oxo cluster {U14} containing hemimellitate uranium(IV)†
Maxime Dufaye a,
Nicolas P. Martina,
Sylvain Duvala,
Christophe Volkringerab,
Atsushi Ikeda-Ohnoc and
Thierry Loiseau*a
a,
Nicolas P. Martina,
Sylvain Duvala,
Christophe Volkringerab,
Atsushi Ikeda-Ohnoc and
Thierry Loiseau*a
aUniversité de Lille, CNRS, Centrale Lille, ENSCL, Univ. Artois, UMR 8181 - UCCS - Unité de Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: thierry.loiseau@univ-lille.fr; Fax: +33 320 434 895; Tel: +33 3 74 95 13 58
bInstitut Universitaire de France (IUF), 1, rue Descartes, 75231 Paris Cedex 05, France
cCollaborative Laboratories for Advanced Decommissioning Science (CLADS), Japan Atomic Energy Agency (JAEA), 2-4 Shirakata, Tokai-mura, Naka-gun, Ibaraki 319-1195, Japan
First published on 23rd July 2019
Abstract
Two coordination compounds bearing tetravalent uranium were synthesized in the presence of tritopic hemimellitic acid in acetonitrile with a controlled amount of water (H2O/U ≈ 8) and structurally characterized. Compound 1, [U(1,2,3-Hbtc)2]·0.5CH3CN is constructed around an eight-fold coordinated uranium cationic unit [UO8] linked by the poly-carboxylate ligands to form dimeric subunits, which are further connected to form infinite corrugated ribbons and a three-dimensional framework. Compound 2, [U14O8(OH)4Cl8(H2O)16(1,2,3-Hbtc)8(ox)4(ac)4] ({U14}) exhibits an unprecedented polynuclear {U14} poly-oxo uranium cluster surrounded by O-donor and chloride ligands. It is based on a central core of [U6O8] type surrounded by four dinuclear uranium-subunits {U2}. Compound 1 was synthesized by a direct reaction of hemimellitic acid with uranium tetrachloride in acetonitrile (+H2O), while the molecular species ({U14} (2)) crystallized from the supernatant solution after one month. The slow hydrolysis reaction together with the partial decomposition of the starting organic reactants into oxalate and acetate molecules induces the generation of such a large poly-oxo cluster with fourteen uranium centers. Structural comparisons with other closely related uranium-containing clusters, such as the {U12} cluster based on the association of inner core [U6O8] with three dinuclear sub-units {U2}, were performed.
Introduction
For the last decade, interest in the synthesis of coordination polymers containing actinides has grown considerably.1,2 Indeed, many literatures reported a wide range of studies related to the formation of such polymers for the natural 5f elements, thorium or uranium, involving hybrid organic–inorganic networks with the association of O- and/or N-donor organic linkers, such as carboxylate ligands. Most of the reported studies have utilized the most stable oxidation states of these 5f elements, such as Th(IV) for thorium or U(VI) (as uranyl(VI):UO22+) for uranium, under ambient conditions. Among these precedent works on coordination polymer compounds, significant efforts have been made on Metal–Organic Frameworks (MOF) materials, which exhibit remarkable open-framework architecture with a large porous cavity or channel systems. Since the first report of the thorium-based MOF-like solid,3,4 stable porous thorium-based coordination networks have been discovered,5 offering new applications to the fields of gas sorption (Kr/Xe) and separation,6 trapping anionic persistent organic pollutants7 or selective photoexcitation of mustard gas simulant.8 Some of these MOF compounds are related to the well-known topologies, belonging to the UiO-66/67/68 series,9 which exist for other tetravalent metals such as Zr(IV),10 Hf(IV)11 or Ce(IV).12,13 The latter is based on a hexanuclear core [M6O8] unit (M = Th(IV),14,15 U(IV)16–18 or Np(IV)19) connected by ditopic carboxylate ligands derived from the terephthalate molecule. The crystal chemistry of uranyl–organic framework is investigated and documented much more in detail, while there are fewer examples of three-dimensional architectures, limiting the application of their porous cavities to sorption, etc. One of the most outstanding structural organization is the uranyl carboxylate (NU-1301) recently described by the Farha's group,20 who has shown the complex assembly of discrete uranyl nodes via a tritopic ligand derived from a trimethyl-terphenyl-phenyl-tricarboxylate in order to generate a framework with five levels of cages of internal diameter up to 5.0 and 6.2 nanometers.20 The resulting anionic framework possesses interesting porous properties with the capacity to adsorb small proteins such as cytochrome c or α-lactalbumin. Although some uranyl-based compounds with smaller pores have been recently described for their molecular sorption capacities,21–24 other uranyl 3D networks are quite complicated and very often involved complex arrangements with interwoven structures in which no space is available for real sorption properties.25,26In contrast to a large number of studies devoted to Th(IV) and U(VI), the use of uranium with the tetravalent oxidation state (U(IV)) has been less investigated in terms of coordination polymers. Although, as mentioned above, the elaboration of UiO-66/67/68 series materials has been previously reported for U(IV),16,17 the combination of various polytopic ligands still has huge potential to lead to the formation of diverse coordination networks. For instance, the use of the trimesate ligand resulted in the formation of channel-like structure with a honeycomb lattice based on a trinuclear U(IV)-centered building unit,27 whereas the use of terephthalate, isophtalate, pyromellitate, mellitate linkers generated rather dense 3D frameworks with either tetranuclear,18 dinuclear28 or discrete units.29 With the phthalate molecule, a layered structure has been obtained with ribbons involving U–O–U linkage connected to each other via the organic ditopic ligand.28
Moreover, it was also demonstrated that the use of monotopic ligands, such as formate, triflate, glycine or benzoate, favors the formation of discrete molecular poly-oxo clusters30 with a wide range of nuclearities from a classical motif containing six U(IV) centers31–35 up to a giant unit containing thirty eight U(IV) centers.36 Between these two end members of this series, different middle-size clusters such as {U10},37,38 {U12},33,39 {U13},37 {U16}38 and {U24},37 have been identified, mostly by the use of benzoate ligands with controlling the water content and/or reaction time in organic solvents. Other U(IV)-based poly-oxo moieties have been stabilized by the DOTA ligand or isopropanol molecules.40,41 Such clusters were also characterized in the solid-state for the heavier 5f elements, such as Np(IV)40,42 or Pu(IV).40,43–46 It has been also reported that monotopic ligands may also act as a bidentate linker between isolated [UO8] unit (with acetate)47 or hexanuclear [U6O8] unit with formate48 to generate one-dimensional chain structure.
Based on these precedent studies, this study aims at further exploring the chemical system of U(IV) combined with other aromatic polydentate carboxylate ligands. Herein, we investigated the reactivity of the hemimellitic acid (1,2,3-benzenetricarboxylic acid) with uranium tetrachloride (UCl4) in acetonitrile and a controlled amount of water under solvothermal conditions. A novel coordination polymer [U(1,2,3-Hbtc)2]·0.5CH3CN (1), was thus isolated and structurally characterized. After filtration of crystalline product of 1, the supernatant solution was left at room temperature, and a new type of crystals appeared. This second phase, was then further analyzed and is related to a new molecular poly-oxo cluster containing fourteen uranium centers {U14} (compound 2). The crystal structures of the two compounds 1–2 have been determined by single-crystal X-ray diffraction. The thermal behavior of compound 1 has been analyzed (TG, X-ray thermodiffraction).
Experimental section
Synthesis
Caution! Natural U is a radioactive and chemically toxic material. Therefore, precautions with suitable equipments and facility for radiation protection are required for handling these elements.The compounds have been solvothermally synthesized under autogenous pressure using a 2 mL glass vial with a Teflon cap. Uranium tetrachloride (UCl4, obtained according to the protocol27 using the reaction of hexachloropropene with uranium oxide UO3), hemimellitic acid (C9H6O6, 1,2,3-H3btc, Sigma Aldrich 99%), and acetonitrile (Sigma Aldrich 99.8%) were mixed in the glass vial. The mixtures were then sealed with the cap, removed from an inert glove box and then heated in an oven under ambient atmosphere. The chemicals used in this study (except UCl4) are commercially available and were used without any further purification.
[U(1,2,3-Hbtc)2]·0.5CH3CN (1): a mixture of 25 mg (0.0625 mmol) of UCl4, 28 mg (0.17 mmol) of hemimellitic acid, 1 mL (55.6 mmol) of acetonitrile and 25 μL (1.39 mmol) of H2O were placed in a closed glass vial and then heated statically at 120 °C for 24 hours. The resulting product of 1 (green blocks crystallites – Fig. S1†) was then filtered off, and washed with ethanol to eliminate unreacted species (mainly unreacted ligand). Scanning electron microscopy showed the formation of block-shape crystallites (Fig. S1†). Compound 1 was obtained as a pure phase, which was confirmed by powder X-ray diffraction measurements (Fig. S2†).
[U14O8(OH)4Cl8(H2O)16(1,2,3-Hbtc)8(ox)4(ac)4] (2, noted {U14}): a mixture of 25 mg (0.0625 mmol) of UCl4, 28 mg (0.17 mmol) of hemimellitic acid, 1 mL (55.6 mmol) of acetonitrile and 25 μL (1.39 mmol) of H2O were placed in a closed glass vial and then heated statically at 120 °C for 24 hours. Green blocks crystallites (compound 1) appeared instantly and the solution was left to crystallize for a longer period. After one month, compound 2 appeared as brown squared crystals (Fig. S1†). These crystals were manually selected for solid state analyses. Since the amount of crystals of 2 is very limited, only the single crystal X-ray diffraction and scanning electron microscopy characterizations have been performed.
Single-crystal X-ray diffraction
Crystals of the uranium compounds (1 and 2) were analyzed on a Bruker DUO-APEX2 CCD area-detector diffractometer at 300 K, using microfocused Mo-Kα radiation (λ = 0.71073 Å) with an optical fiber as collimator. Crystals of 1–2 were selected under a polarizing optical microscope and glued on a glass fiber for a single-crystal X-ray diffraction measurements. Several sets of narrow data frames (20 s per frame) were collected at different values of θ for two initial values of ϕ and ω, respectively, with 0.3° increments. Data reduction was accomplished using SAINT V7.53a.49 The substantial redundancy in the collected data allowed a semi-empirical absorption correction (SADABS V2.10) to be applied, on the basis of multiple measurements of equivalent reflections. The structure was solved by direct methods, developed by successive difference Fourier syntheses, and refined by full-matrix least-squares on all F2 data using SHELX50 program suite and OLEX2 (ref. 51) software. Hydrogen atoms of the benzene ring were included in the calculated positions and allowed to ride on their parent atoms. The final refinements include anisotropic thermal parameters of all non-hydrogen atoms, except the oxygen atoms of the water molecules. The CheckCif of compound 2 reveals several A and B rank alerts. These alerts are primarily due to the low quality of the crystal. Further details are given in the ESI (Fig. S3†). The crystal data are given in Table 1. CCDC file numbers: 1916502 and 1916503.| 1 | 2 | |
|---|---|---|
| Formula | C19H8NO12U | C8H44Cl8NO124U14 |
| Formula weight | 673.29 | 6701.25 |
| Temperature/K | 300 | 100 |
| Crystal type | Green block | Brown plate |
| Crystal size/mm | 0.09 × 0.08 × 0.04 | 0.04 × 0.03 × 0.01 |
| Crystal system | Tetragonal | Tetragonal |
| Space group | I41/a | I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) |
| a/Å | 11.7099(16) | 18.247(2) |
| b/Å | 11.7099(16) | 18.247(2) |
| c/Å | 27.227(4) | 29.034(3) |
| α/° | 90 | 90 |
| β/° | 90 | 90 |
| γ° | 90 | 90 |
| Volume/Å3 | 3733.4(12) | 9667(2) |
| Z, ρcalculated/g cm−3 | 8, 2.396 | 2, 2.302 |
| μ/mm−1 | 8.769 | 11.890 |
| θ range/° | 1.89–28.36 | 1.32–16.59 |
| Limiting indices | −15 ≤ h ≤ 15 | −14 ≤ h ≤ 14 |
| −15 ≤ k ≤ 15 | −13 ≤ k ≤ 13 | |
| −36 ≤ l ≤ 36 | −23 ≤ l ≤ 23 | |
| Collected reflections | 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 345 345 |
25791 |
| Unique reflections | 2335 [R(int) = 0.0630] | 2633 [R(int) = 0.1273] |
| Parameters | 147 | 263 |
| Goodness-of-fit on F2 | 1.024 | 1.051 |
| Final R indices [I > 2σ(I)] | R1 = 0.0208 | R1 = 0.0542 |
| wR2 = 0.0422 | wR2 = 0.1339 | |
| R indices (all data) | R1 = 0.0408 | R1 = 0.0835 |
| wR2 = 0.0471 | wR2 = 0.1563 | |
| Largest diff. peak and hole/e Å−3 | 0.507 and −0.580 | 0.894 and −0.899 |
Powder X-ray diffraction
The powder X-ray diffraction pattern of compound 1 was collected at room temperature with a D8 advance A25 Bruker apparatus with Bragg–Brentano geometry (θ–2θ mode).X-ray thermodiffraction
X-ray thermodiffractometry was performed for compound 1 under a 5 L h−1 nitrogen flow in an Anton Paar HTK1200N of a D8 Advance Bruker diffractometer (θ–θ mode, CuKα radiation) equipped with a Vantec1 linear position sensitive detector (PSD). Each powder pattern was recorded in the range 5–50° (2θ) (at intervals of 20 °C between RT and 800 °C) with a 1 s per step scan, corresponding to an approximate duration of 37 min. The temperature ramps between two patterns were 5 °C min−1.Thermogravimetric analysis
Thermogravimetric experiment was carried out for compound 1, on a thermo-analyzer TGA 92 SETARAM under air atmosphere with a heating rate of 5 °C min−1 from room temperature up to 800 °C.Infrared spectroscopy
Infrared spectrum of compound 1 (see ESI†) was measured between 4000 and 400 cm−1 on PerkinElmer Spectrum Two™ spectrometer equipped with a diamond Attenuated Total Reflectance (ATR) accessory.UV/visible spectroscopy
UV/Vis spectrum of compound 1 was collected on a PerkinElmer Lambda 650 spectrophotometer equipped with a powder sample holder set.Gas sorption
Gas sorption experiments were carried out for compound 1 using a Micromeritics ASAP 2020 apparatus. Prior to measurements, the solid was degassed at 120 °C for approximately 6 h until the outgas rate reached below 4 μmHg. The BET surface was estimated by gas sorption isotherm experiments in liquid nitrogen (77 K) from the p/p0 range: 0.02–0.3. CO2 adsorption was performed from 3 to 760 mmHg at 273.15 K.Results
Structure description
The crystal structure of U(1,2,3-Hbtc)2·0.5CH3CN (compound 1) is built up from the three-dimensional assembly of one distinct crystallographically independent uranium center (U1) linked to each other by the hemimellitate ligands. The uranium atom is located on the special position 8e (along the 2 axis) and is eight-fold coordinated in a square antiprismatic geometry with the oxo groups of carboxylate arms of the hemimellitate linker (Fig. 1). Typical U–O bonds distances are ranging from 2.282(3) up 2.442(3) Å and agree well with those for U(IV) based on the bond valence considerations52 (calculated to be of 4.23 for 4+). This square antiprismatic coordination environment for the discrete tetravalent uranium center, was previously reported in some networks involving carboxylate linkers. The first example is the chain-like coordination polymer with acetate ligands.47 Other compounds also exhibit isolated anti-prismatic [UO8] units linked to one another via oxalate ligands in a layered network53 or engaged in 3D frameworks for the desolvated uranium isophthalate (U(1,3-bdc)2).29 The unique crystallographically independent hemimellitate ligand acts as a tridentate linker with uranium; two of the carboxylate arms are linked to one uranium atom through a monodentate fashion whereas one carboxylate group is linked to two adjacent uranium centers in a bidentate bridging fashion (Fig. 1). The remaining non-bonded carboxyl group is either in terminal position (with C6 = O6 length of 1.234(5) Å) or attaches a hydrogen atom related to C–OH, or the protonated state of the tricarboxylate molecule (C1–O5 = 1.270(5) Å). This configuration is hardly observable by infrared spectroscopy, as it is hidden under a very broad band centered at 1610 cm−1, which is typical for the C–O bonds connected to uranium (Fig. S5†). | ||
| Fig. 1 (Top) Coordination environment around the uranium (U1) atom in a square antiprismatic geometry (green UO8 polyhedra) in the compound 1, U(1,2,3-Hbtc)2·0.5CH3CN, (Bottom) coordination mode of the tritopic hemimellitate ligand with the three uranium centers U1. | ||
The structure is constructed from a dimeric sub-unit of uranium atoms bridged to each other via four carboxylate groups from the four distinct organic molecules. This uranium-centered dimer is linked to each other via the monodentate carboxylate arms from two other hemimellitate ligands to generate infinite corrugated chains along the a and b axes (Fig. 2). The ribbons are further connected to each other through the bidentate carboxylate arms of the organic components along the perpendicular directions (b or a axis) to eventually form a three-dimensional framework (Fig. 3). Narrow channels are then generated along the a and b directions, but are obstructed by the aromatic rings of the tritopic ligands pointing toward the centers, limiting the pore accessibility (aperture diameter ≈ 2–3 Å). However, acetonitrile species are found to be trapped within these small pores and are statistically located on two equivalent positions (50% occupancy) around the −4 axis of the structure. These solvent molecules are removed after the washing step using ethanol. This was confirmed by the fact that the infrared spectrum of the activated compound does not reveal vibrations associated to acetonitrile (Fig. S5†). The resulting framework is neutral, with the U/[1,2,3-btc] stoichiometry of 1/2, involving the monoprotonated organic ligand that is negatively charged with −2.
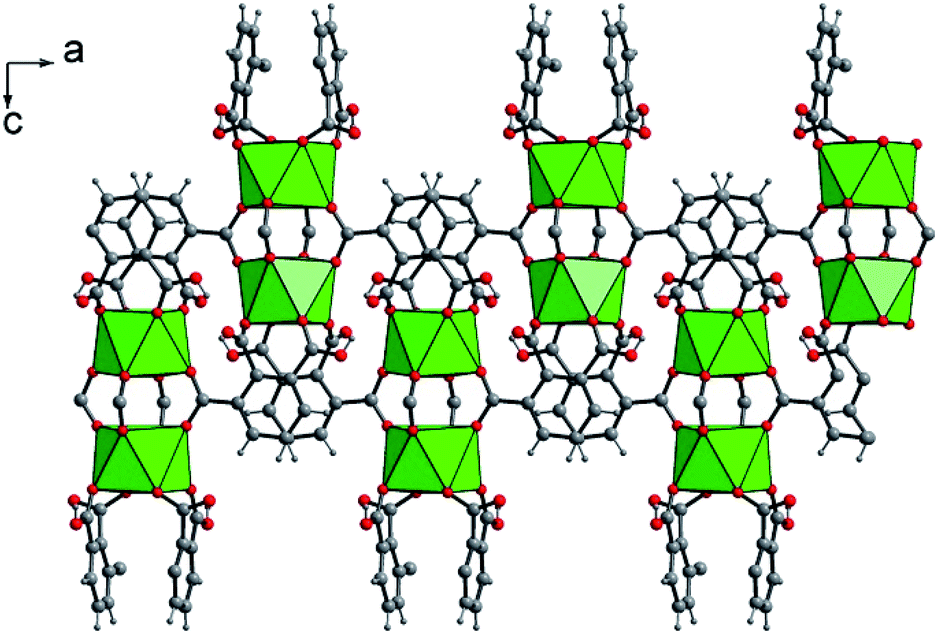 | ||
| Fig. 2 Connection mode of the dinuclear uranium-centered sub-unit through the protonated hemimellitate ligands along the a axis in U(1,2,3-Hbtc)2·0.5CH3CN (1). Green polyhedra: square antiprisms UO8. | ||
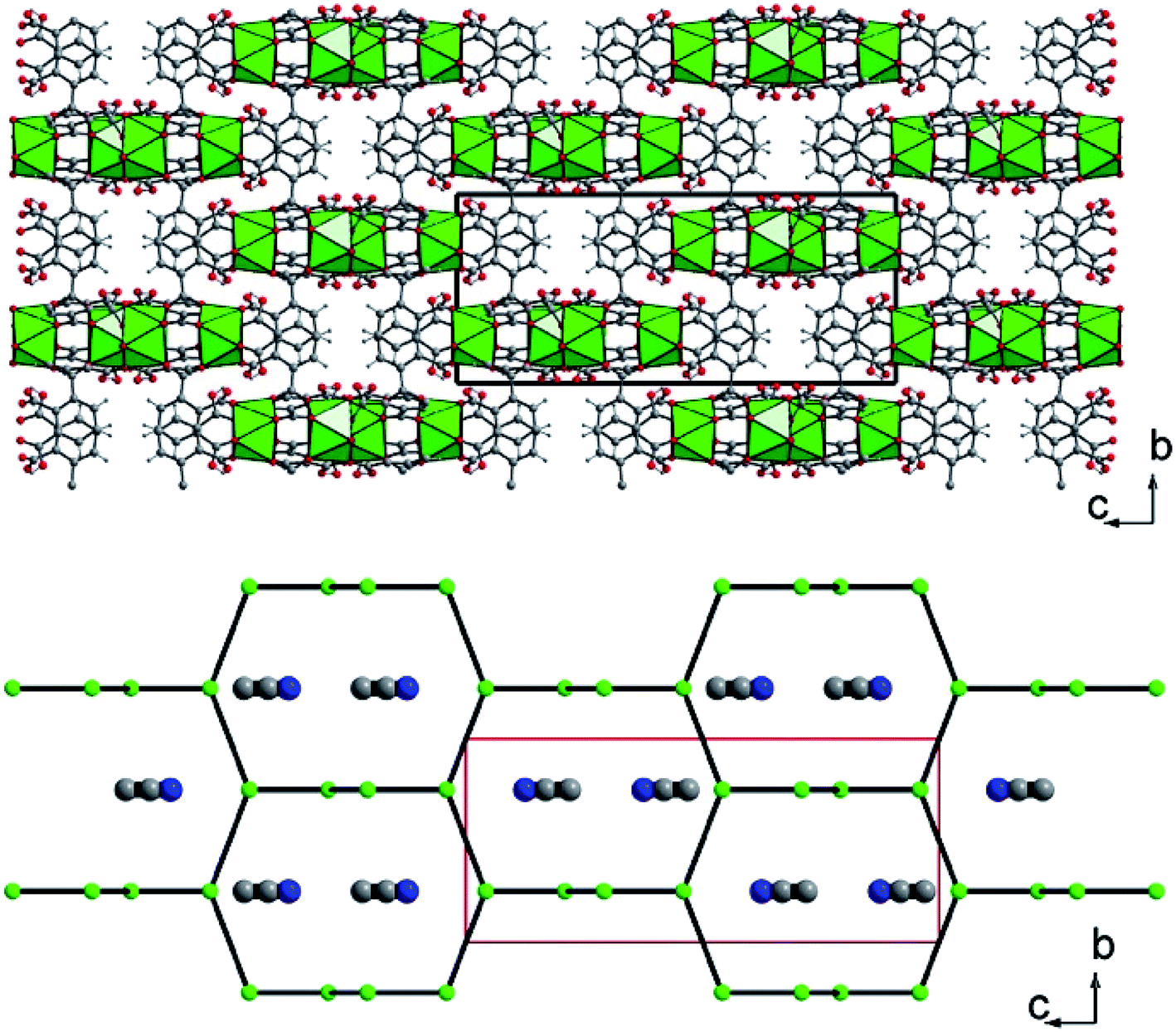 | ||
| Fig. 3 (Top) Three-dimensional framework of U(1,2,3-Hbtc)2·0.5CH3CN (1). The blocks related to the four discrete UO8 units correspond to the infinite corrugated ribbons shown in Fig. 2. (Bottom) Schematic representation of the three-dimensional network showing the channels developed along the a (or b) axis, in which disordered acetonitrile species are encapsulated. The acetonitrile molecules are shown randomly due to their statistically disordered positions within the channels. Green polyhedral and circles; uranium, grey circles; carbon, blue circles; nitrogen (from acetonitrile). | ||
Whereas N2 sorption at 77 K did not indicate any porosity associated with the 3D framework (BET surface value close to zero), the capture of CO2 at 273.15 K revealed a small gas uptake (12 cm3 g−1) at 760 mmHg (Fig. S9†). The better affinity for CO2 could attribute to the smaller kinetic diameter of CO2 (∅ = 3.3 Å) compared to that of N2 (∅ = 3.6–3.8 Å),54 as well as the existence of structural defects allowing the access to microporous cages.
The solid-state UV-Vis spectrum of compound 1 (Fig. S6†) exhibits a typical signature for the U(IV). The different adsorption bands corresponds to the f–f transitions from the 3H4 ground state to various excited states. Between 350 and 575 nm, a series of signals with moderate intensity are observed and assigned to the different transitions 3H4 → P2 (433 nm); 3H4 → 1I6 (455–520 nm) and 3H4 → 3P1 (548 nm). The more intense band is located between 619 and 667 nm, which is due to the transitions from 3H4 → 3P0/1D2/1G4. The last transition 3H4 → 3H6 is characterized by a quite broad signal, centered around 820 nm. These values were previously reported in diverse uranium(IV)-containing solid compounds (phosphate,55 phosphonate,56 fluoride,57 chloride,58 carboxylate18,29,48).
The X-ray thermodiffractogram of the washed compound 1 (Fig. S7†) revealed that the solid of compound 1 is stable up to 280 °C. Above this temperature, the intensity of Bragg peaks decreased and eventually resulted in the formation of an amorphous product at 300 °C. The increase of the temperature induces the formation of an uranium oxide over 420 °C, which was identified as U3O8 (pdf file: 00-020-1345). From 500 °C, new intense Bragg peaks was growing at 21.4, 26 and 34° (in 2θ), corresponding to the formation of UO3 (pdf file: 00-018-1429).
These observed structural transformations were also well supported by thermogravimetric analysis (Fig. S8†). The decomposition of the organic part was ranging from 200 to 470 °C, and was composed of two steps (exp: 55.5%, calc.: 57.1%). The first step ends at 410 °C and could be assigned to a partial decomposition of the carboxylate/carboxylic functions into CO2. The subsequent step can be assigned to the degradation of the remaining part of the ligand. U3O8 was identified as the final residue and lead to a final plateau at 44.5%.
Another type of crystalline product was observed in the supernatant that was obtained 24 hours after the solvothermal reaction of UCl4 with 1,2,3-benzene tricarboxylic acid in acetonitrile. After leaving this solution for one month in closed cell under argon atmosphere, brownish plate-like crystals appeared. Its single-crystal X-ray diffraction analysis revealed a new crystal structure of U(IV) engaged in the molecular poly-oxo cluster system containing fourteen uranium centers (denoted as {U14} or compound 2 hereafter). The structure consists of an inner core with six uranium atoms that are linked to each other through μ3-oxo (O1) or μ3-hydroxo (O2) groups (Fig. 4). This is a typical hexanuclear motif, reported for many coordination complexes bearing tetravalent actinides (Th,59–62 U,32,35 Np,19 Pu46) surrounded by bidentate ligands (mainly carboxylate groups). In the hexanuclear motif of compound 2, there are two distinct crystallographically independent uranium centers (U1 and U2), which are either eight-fold coordinated in a square antiprismatic geometry (U1) or nine-fold coordinated in a mono-capped square antiprismatic geometry (U2). They are bridged to each other either via two μ3-oxo groups (U–O1 = 2.21(5)–2.28(5) Å) and two μ3-hydroxo groups (U–O2 = 2.41(5)–2.55(5) Å). Bond valence calculations52 give the values of 1.88 and 1.10 for the oxo and hydroxo species. In case of U1, additional four oxo groups from the carboxylate arms of the hemimellitate molecules coordinate to the metal center with U1–O distance of 2.41(5) Å. The same configuration occurs for U2 that is coordinated by two carboxyl oxygens of two distinct hemimellitate molecules with U2–O bond lengths of 2.46(5)–2.48(5) Å and are further coordinated by carboxyl oxygens of two distinct oxalate molecules with U2–O bond lengths of 2.58(5)–2.61(5) Å. A ninth oxygen atom (O11) capping one square face of the antiprismatic polyhedron is attached to the U2 atom with U2–O bond length of 2.45(4) Å, bridging two adjacent uranium centers (U3 and U4) in a μ3 configuration. Through this μ3-O11 oxo group, the hexanuclear unit is then further connected to four pairs of uranium centers located in a plane perpendicular to the −4 axis. These exterior uranium atoms (U3 and U4) are nine-fold coordinated in a distorted tri-capped trigonal prismatic geometry with several different ligands (Fig. 5). The U3 center is surrounded by five oxygen atoms from carboxyl functions, in which two from oxalate with U3–O distances of 2.52(4)–2.59(5) Å, two from hemimellitate with U3–O distance of 2.37(4) Å, and one from acetate with U3–O distance of 2.35(7) Å. The U3 center is further coordinated by one terminal aquo species (U3–O21 = 2.58(6) Å), one bridging μ3-O11 group between U3 and U4 (U3–O11 = 2.20(4) Å), one terminal chloro species (U3–Cl1 = 2.89(3) Å), and one bridging chloro group between U3 and U4 (U3–Cl2 = 2.63(4) Å). This results in the coordination type of [UO(Ocarboxyl)5(H2O)Cl2]. The adjacent U4 atom is linked to the U3 polyhedron via the μ3-O11 group (U4–O11 = 2.15(4) Å), four carboxylate oxygens (two from oxalate with U4–O distances of 2.49(4)–2.61(5) Å and two from hemimellitate U4–O distances of 2.36(6)–2.47(7) Å), one bridging chloro group (U4–Cl2 = 2.71(4) Å) and three terminal water molecules (U4–O = 2.44(8)–2.68(5) Å). The coordination environment of U4 center is therefore [UO(Ocarboxyl)4(H2O)3Cl]. The bond valence calculations52 of these different oxygen species adequately reflect the difference in charge between oxo-(μ3-O11: 2.09) and terminal aquo species (O9: 0.40; O10: 0.22, O12: 0.29 and O21: 0.28).
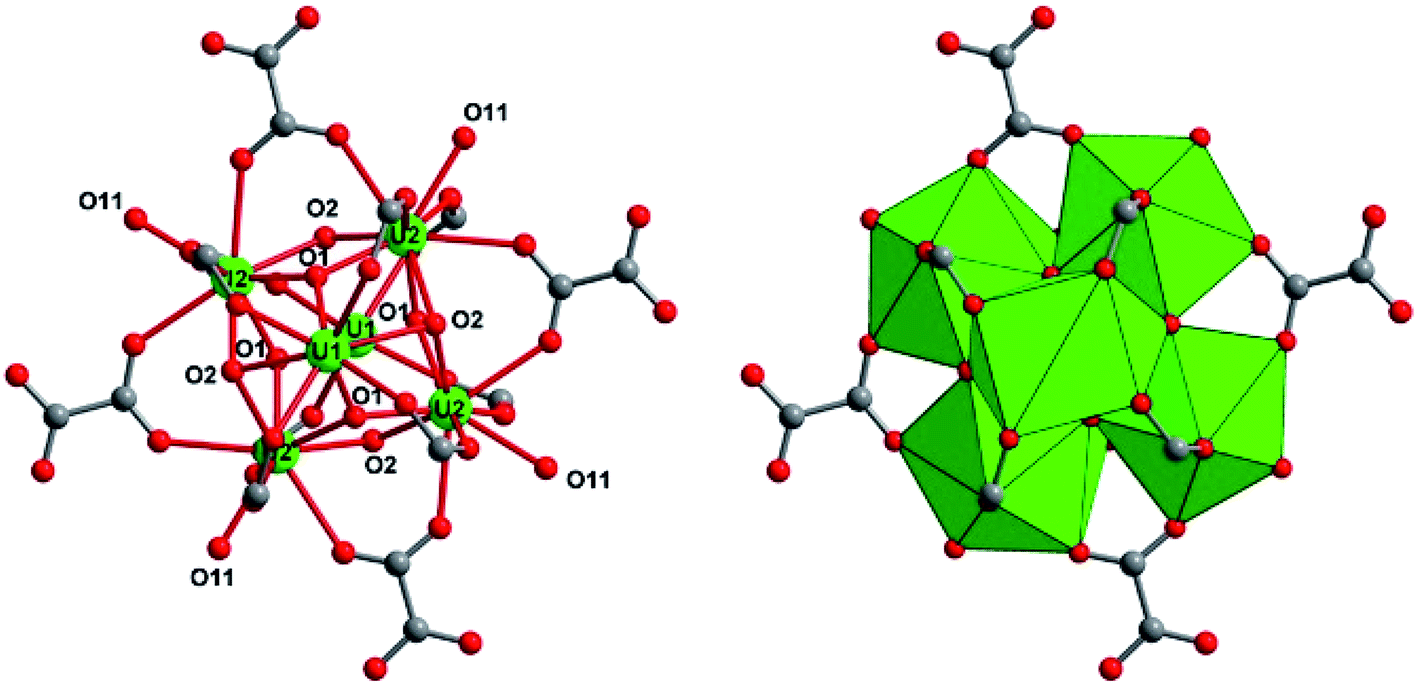 | ||
| Fig. 4 Structure arrangement of the internal hexanuclear core [U6O4(OH)4] in the poly-oxo cluster {U14} (2). The carboxylate groups of oxalate and hemimellitate ligands act bidentately to bridge two adjacent uranium centers (U1 and U2). Green circles and polyhedral; uranium U1 or U2, red circles; oxygen, grey circles; carbon. | ||
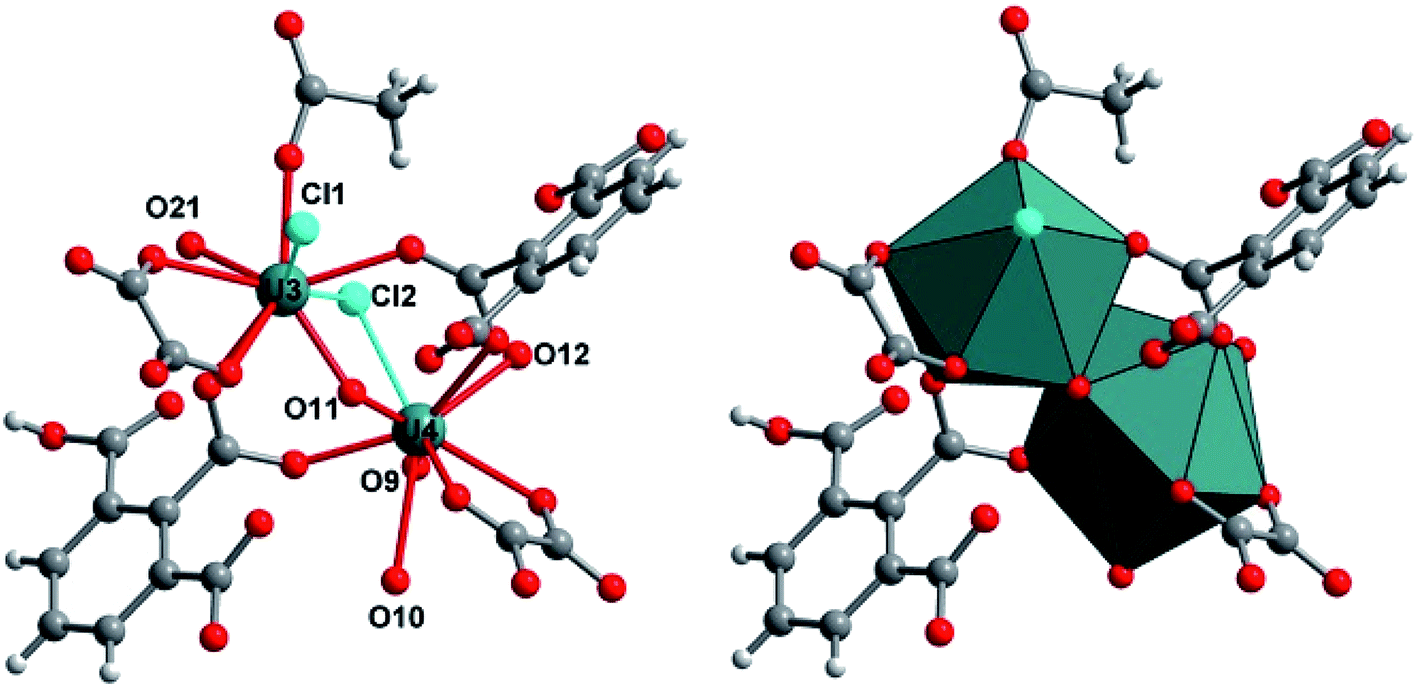 | ||
| Fig. 5 Structure arrangement of the exterior uranium centers (U3, U4) surrounding the central hexanuclear core [U6O4(OH)4] in the poly-oxo cluster {U14} (2). O9, O10, O12, and O21 are terminal aquo species. Slate circles and polyhedral; uranium U3 or U4, red circles; oxygen, grey circles; carbon, cyan circles; chlorine. | ||
The two exterior uranium atoms U3 and U4 are therefore linked through the common vertices formed by one μ3-oxo and μ2-chloro groups, forming a dinuclear sub-unit which are further linked via the oxalate species to form an eight-membered ring along a plane perpendicular to the −4 axis of the crystal structure (Fig. 6). This resulting crown-like [U8O4Cl4(ox)4] ring encapsulates the hexanuclear core [U6O4(OH)4] located at the center via μ3-oxo groups (O11) and two carboxyl oxygen atoms from the arm of oxalate molecule. This particular combination of internal hexanuclear core and external U8 ring eventually results in the formation of a new type of cluster involving the mixed-oxo/hydroxo-bridged fourteen uranium atoms with the chemical formula [U14O8(OH)4Cl8(H2O)16(1,2,3-Hbtc)8(ox)4(ac)4]. In this {U14} motif, the hemimellitate ligand exists as a monoprotonated molecule as it is observed in compound 1. One of the three carboxylate arms is non-bonded with a typical short C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond distance of 1.23(10) Å and a long C–OH bond length of 1.33(11) Å. The other two carboxylate arms adopt a bidentate bridging mode toward either U1 or U2 atoms of the hexanuclear core, or U3 or U4 atoms of the exterior dinuclear sub-units, resulting in a tetradentate connection fashion with the uranium centers. The oxalate linker also connects four uranium atoms: one carboxylate group links two uranium atoms (U3 and U4) in a bidentate bridging manner, whereas the other carboxylate arm is bis-bidentate and connect four uranium atoms. Another organic molecule found in this crystal structure is acetate molecules acting monodentately to link the U3 atoms. The presence of these oxalate and acetate ligands is unexpected since they were not present in the initial reaction medium. They were presumably produced by the slow hydrolysis process during a long reaction duration of one month. In fact, acetonitrile is known to undergo hydrolysis under acidic conditions to generate acetamide and subsequently acetic acid. The in situ formation of the oxalate from polycarboxylate molecules are also described in detail in literature, even in terms of the formation of coordination polymers of actinides.63–69 The generation of oxalate can be reasonably interpreted as a result of the oxidative benzene ring-opening followed by decarboxylation to generate the dicarboxylate moiety.70 The low synthetic yield of the {U14} cluster is therefore correlated to the slow in situ degradation of the initial 1,2,3-benzene tricarboxylic acid and acetonitrile into the oxalate and acetate species, respectively. The resulting chemical formula of {U14} ([U14O8(OH)4Cl8(H2O)16(1,2,3-Hbtc)8(ox)4(ac)4]) is neutral when taking into account all the different ligands (monoprotonated hemimellitate, oxalate, acetate, oxo, hydroxo, chloro, and aquo groups). In the crystal packing, free water molecule are intercalated among the {U14} units. However, due to the large uncertainty of the X-ray diffraction data, the hydrogen-bond network could not be adequately defined.
O bond distance of 1.23(10) Å and a long C–OH bond length of 1.33(11) Å. The other two carboxylate arms adopt a bidentate bridging mode toward either U1 or U2 atoms of the hexanuclear core, or U3 or U4 atoms of the exterior dinuclear sub-units, resulting in a tetradentate connection fashion with the uranium centers. The oxalate linker also connects four uranium atoms: one carboxylate group links two uranium atoms (U3 and U4) in a bidentate bridging manner, whereas the other carboxylate arm is bis-bidentate and connect four uranium atoms. Another organic molecule found in this crystal structure is acetate molecules acting monodentately to link the U3 atoms. The presence of these oxalate and acetate ligands is unexpected since they were not present in the initial reaction medium. They were presumably produced by the slow hydrolysis process during a long reaction duration of one month. In fact, acetonitrile is known to undergo hydrolysis under acidic conditions to generate acetamide and subsequently acetic acid. The in situ formation of the oxalate from polycarboxylate molecules are also described in detail in literature, even in terms of the formation of coordination polymers of actinides.63–69 The generation of oxalate can be reasonably interpreted as a result of the oxidative benzene ring-opening followed by decarboxylation to generate the dicarboxylate moiety.70 The low synthetic yield of the {U14} cluster is therefore correlated to the slow in situ degradation of the initial 1,2,3-benzene tricarboxylic acid and acetonitrile into the oxalate and acetate species, respectively. The resulting chemical formula of {U14} ([U14O8(OH)4Cl8(H2O)16(1,2,3-Hbtc)8(ox)4(ac)4]) is neutral when taking into account all the different ligands (monoprotonated hemimellitate, oxalate, acetate, oxo, hydroxo, chloro, and aquo groups). In the crystal packing, free water molecule are intercalated among the {U14} units. However, due to the large uncertainty of the X-ray diffraction data, the hydrogen-bond network could not be adequately defined.
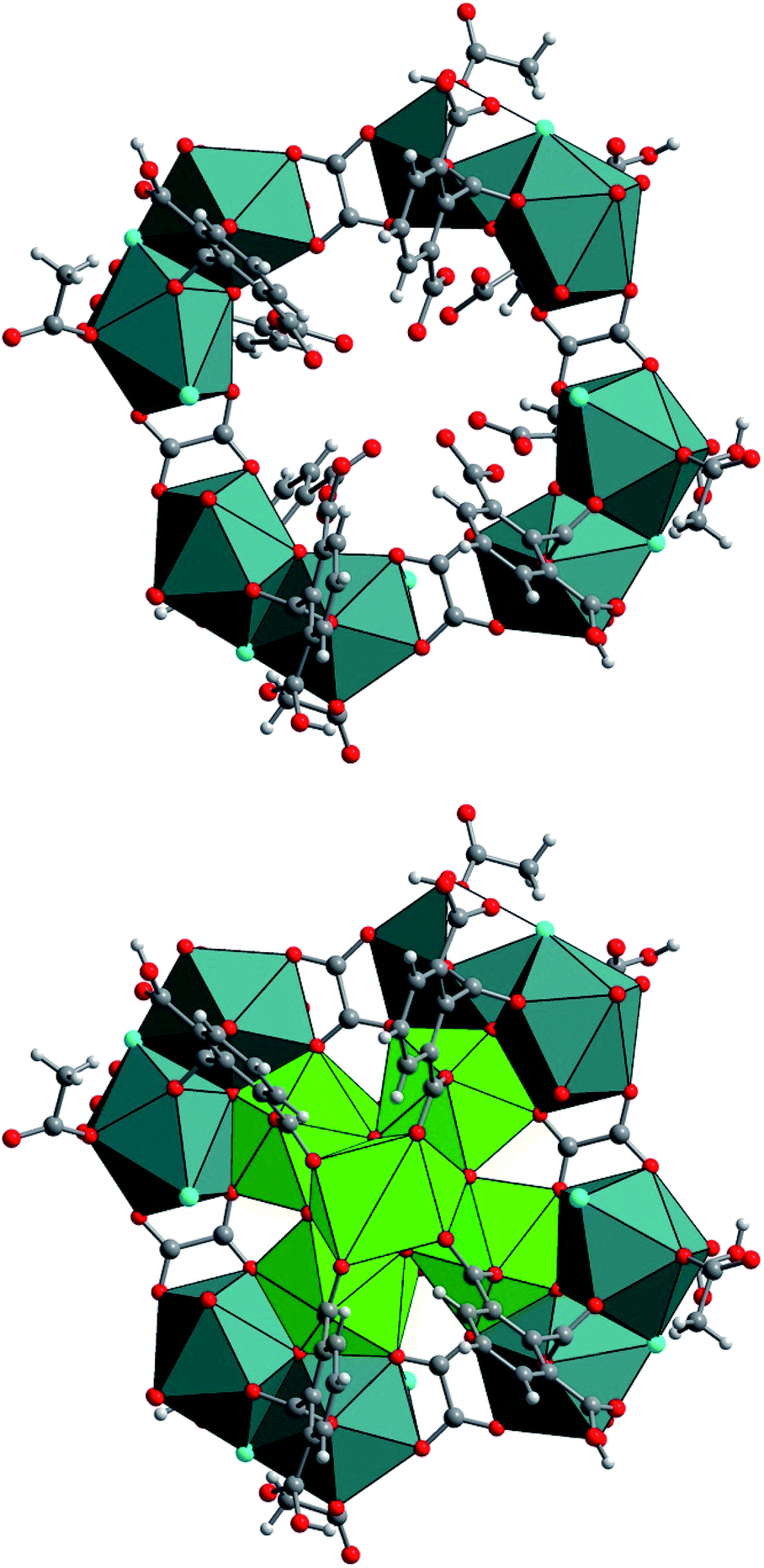 | ||
| Fig. 6 (Top) Structure arrangement of the external crown-like [U8O4Cl4(ox)4] ring composed of four dinuclear units (U3 and U4) that are further linked to each other via the oxalate molecules. (Bottom) Molecular structure of the poly-oxo cluster [U14O8(OH)4Cl8(H2O)16(1,2,3-Hbtc)8(ox)4(ac)4] (2). Green/slate circles and polyhedral; uranium, red circles; oxygen, grey circles; carbon, cyan circles; chlorine. | ||
The formation of such a poly-oxo cluster has been previously described for the formation of the {U38} cluster with a similar synthetic procedure.36 In the case of {U38}, a transient phase has been observed in the supernatant solution after 4 hours of reaction.39 The resulting product, which crystallized with a very low yield, consists of another dodecanuclear cluster {U12} with the comparable structure arrangement consisting of an inner hexanuclear core [U6O4(OH)4] surrounded by three dinuclear uranium sub-units. The {U12} motif also involves benzoate molecules that introduced in the initial reaction medium, together with oxalate groups (and glycolate) generated by the partial decomposition of the initial benzoic acid. In this context, the formation of the {U14} cluster can be considered as a derivative of the {U12} cluster. As illustrated in Fig. 7, the difference between {U12} and {U14} clusters is only the number of peripheral dinuclear uranium subunits surrounding the central hexanuclear core [U6O4(OH)4]. This suggests that other U(IV)-based poly-oxo clusters with the nuclearity of “6 + 2n” (i.e. n = 1–6, which correspond to {U8} up to {U18}) could be potentially feasible by adjusting the number of the exterior dinuclear subunits. This far, only n = 3 ({U12}) and 4 ({U14}) members were isolated. These two clusters differ significantly in terms of structure as compared with those reported by Mazzanti, in which the intermediates members ({Ux} where 6 ≤ x ≤ 38) are formed via the association of hexanuclear motifs.37 In this cluster series, the uranium centers in the inner core of the poly-oxo clusters adopt a cubic coordination UO8 environment of UO8 for the higher nuclearity (x > 12), instead of the square antiprism observed in our system.
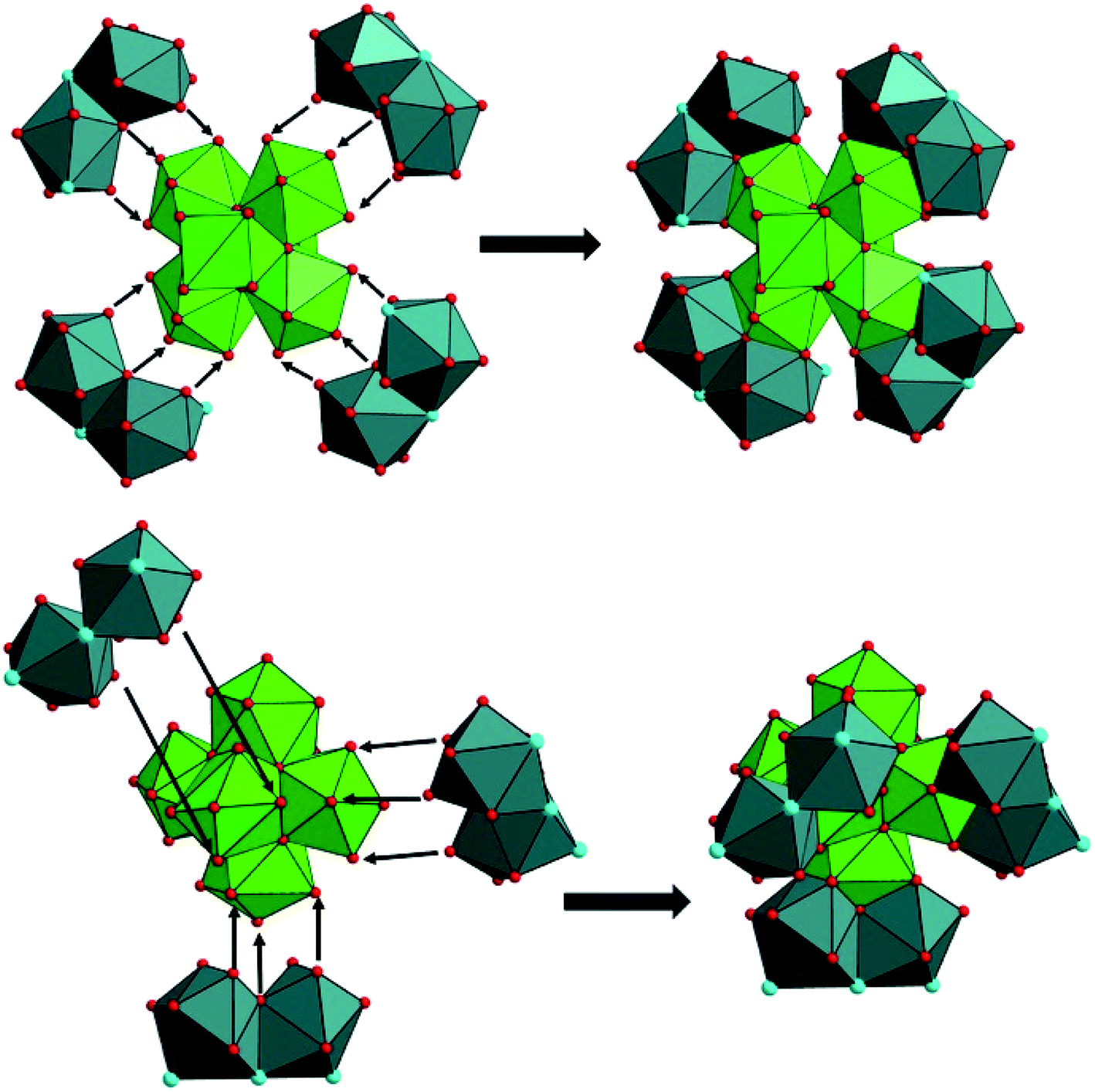 | ||
| Fig. 7 Different connection manners between the central hexanuclear core [U6O4(OH)4] and peripheral dinuclear uranium sub-units to form different uranium clusters (top; {U14}, bottom; {U12}39). | ||
Conclusion
The solvothermal reaction of tetravalent uranium (U(IV)) with hemimellitic acid (1,2,3-H3btc) in acetonitrile solvent with a controlled amount of water (molar ratio H2O/U ≈ 8) resulted in the crystallization of two distinct coordination cluster complexes of U(IV). The compound 1, [U(1,2,3-Hbtc)2]·0.5CH3CN, was obtained after a solvothermal treatment for 24 hours. Its crystal structure exhibits a three-dimensional open-framework consisting of inorganic [UO8] discrete units connected through the hemimelitate ligands, showing a thermal stability up to 280 °C. Although solvent acetonitrile molecules are encapsulated within the network, the porosity in 1 is very limited due to very small channel apertures. It however allows the small amount of CO2 sorption (12 cm3 g−1 at 760 mmHg). In the same chemical system, another cluster compound (2) appeared when leaving the supernatant solution for one month. This yielded a new molecular poly-oxo cluster involving fourteen uranium atoms ([U14O8(OH)4Cl8(H2O)16(1,2,3-Hbtc)8(ox)4(ac)4]). During the formation of 2, the partial decomposition of hemimellitate and acetonitrile molecules presumably occurs to generate oxalate and acetate that are incorporated in 2. In addition to the hemimellitate ligand, these supplementary organic anions promote the stabilization of the hexanuclear [U6O8] core surrounded by four dinuclear {U2} subunits. This resultant structural arrangement indicates the slow hydrolysis process during the synthesis to form oxo/hydroxo bridges between the uranium centers, eventually forming the particular {U14} motif supported and stabilized by different O-donor organic ligands (hemimellitate, oxalate, and acetate). The elucidation of this {U14} cluster together with the previously reported {U12} cluster potentially opens a new route for the synthesis of a series of other cluster systems deriving from the hexanuclear [U6O8] core surrounded by dinuclear uranium subunits with the uranium nuclearity of “6 + 2n”.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Mrs Nora Djelal, Mr Laurence Burylo, and Mr Philippe Devaux for their technical assistance to collect SEM images and to perform TG measurements and powder XRD at UCCS. The Chevreul Institute (FR 2638), “Fonds Européen de Développement Régional (FEDER)”, “CNRS”, “Région Hauts de France”, and “Ministère de l'Education Nationale de l'Enseignement Supérieur et de la Recherche” are acknowledged for financial support to acquire X-ray diffractometers. The authors also thank TALISMAN for financial support (Project No. TALI_C05-18).References
- T. Loiseau, I. Mihalcea, N. Henry and C. Volkringer, Coord. Chem. Rev., 2014, 266–267, 69 CrossRef CAS.
- E. A. Dolgopolova, A. M. Rice and N. B. Shustova, Chem. Commun., 2018, 54, 6472 RSC.
- K. M. Ok, J. Sung, G. Hu, R. M. J. Jacobs and D. O'Hare, J. Am. Chem. Soc., 2008, 130, 3762 CrossRef CAS PubMed.
- J.-Y. Kim, A. J. Norquist and D. O'Hare, J. Am. Chem. Soc., 2003, 125, 12688 CrossRef CAS PubMed.
- K. P. Carter, J. A. Ridenour, M. Kalaj and C. L. Cahill, Chem.–Eur. J., 2019, 25, 7114–7118 CrossRef CAS PubMed.
- Y. Wang, W. Liu, Z. Bai, T. zheng, M. A. Silver, Y. Li, Y. Wang, X. Wang, J. Diwu, Z. Chai and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 5783 CrossRef CAS PubMed.
- Y. Li, Z. Yang, Y. Wang, Z. Bai, T. Sheng, X. Dai, S. Liu, D. Gui, W. Liu, M. Chen, L. Chen, J. Diwu, L. SZhu, R. Zhou, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Nat. Commun., 2017, 8354, 1354 CrossRef.
- P. Li, S. Goswami, K.-I. Otake, X. Wang, Z.-R. Chen, S. L. Hanna and O. K. Farha, Inorg. Chem., 2019, 58, 3586 CrossRef CAS PubMed.
- T. Islamoglu, D. Ray, P. Li, M. B. Majewski, I. Akpinar, X. Zhang, C. J. Cramer, L. Gagliardi and O. K. Farha, Inorg. Chem., 2018, 57, 13246 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef PubMed.
- S. Jakobsen, D. Gianolio, S. Wragg, M. H. Nilsen, H. Emerich, S. Bordiga, C. Lamberti, U. Olsbye, M. Tilset and K. P. Lillerud, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 125429 CrossRef.
- M. Lammert, M. T. Wharmby, S. Smolders, B. Bueken, A. Lieb, K. A. Lomachenko, D. D. Vos and N. Stock, Chem. Commun., 2015, 51, 12578 RSC.
- M. Lammert, H. Reinsch, C. A. Murray, M. T. Wharmby, H. Terraschke and N. Stock, Dalton Trans., 2016, 45, 18822 RSC.
- C. Falaise, J.-C. Charles, C. Volkringer and T. Loiseau, Inorg. Chem., 2015, 54, 2235 CrossRef CAS PubMed.
- E. A. Dolgopolova, O. A. Ejegbavwo, C. R. Martin, M. D. Smith, W. Setyawan, S. G. Karakalos, C. H. Henager, H.-C. zur Loye and N. B. Shustova, J. Am. Chem. Soc., 2017, 139, 16852 CrossRef CAS PubMed.
- C. Falaise, C. Volkringer, J.-F. Vigier, N. Henry, A. Beaurain and T. Loiseau, Chem.–Eur. J., 2013, 19, 5324 CrossRef CAS PubMed.
- C. Falaise, A. Assen, I. Mihalcea, C. Volkringer, A. Mesbah, N. Dacheux and T. Loiseau, Dalton Trans., 2015, 44, 2639 RSC.
- C. Falaise, C. Volkringer and T. Loiseau, Cryst. Growth Des., 2013, 13, 3225 CrossRef CAS.
- N. P. Martin, J. März, H. Feuchter, S. Duval, P. Roussel, N. Henry, A. Ikeda-Ohno, T. Loiseau and C. Volkringer, Chem. Commun., 2018, 54, 6979 RSC.
- P. Li, N. A. Vermeulen, C. D. Maliakas, D. A. Gomez-Gualdron, A. J. Howarth, B. L. Mehdi, A. Dohnalkova, N. D. Browning, M. O'Keeffe and O. K. Farha, Science, 2017, 356, 624 CrossRef CAS PubMed.
- P. Li, N. A. Vermeulen, X. Gong, C. D. Maliakas, J. F. Stoddart, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2016, 55, 10358 CrossRef CAS.
- F. Hu, Z. Di, P. Lin, P. Huang, M. Wu, F. Jiang and M. Hong, Cryst. Growth Des., 2018, 18, 576 CrossRef CAS.
- J. Ai, F.-Y. Chen, C.-Y. Gao, H.-R. Tian, Q.-J. Pan and Z.-M. Sun, Inorg. Chem., 2018, 57, 4419 CrossRef CAS PubMed.
- K.-Q. Hu, Z.-W. Huang, Z.-H. Zhang, L. Mei, B.-B. Qian, J.-P. Yu, Z.-F. Chai and W.-Q. Shi, Chem.–Eur. J., 2018, 24, 16766 CrossRef CAS PubMed.
- Y.-C. Ge, L. Mei, F.-Z. Li, K.-Q. Hu, C.-Q. Xia, Z.-F. Chai and W.-Q. Shi, Cryst. Growth Des., 2018, 18, 3073 CrossRef CAS.
- K.-Q. Hu, Q.-Y. Wu, L. Mei, X.-L. Zhang, L. Ma, G. Song, D.-Y. Chen, Y.-T. Wang, Z.-F. Chai and W.-Q. Shi, Chem.–Eur. J., 2017, 23, 18074 CrossRef CAS.
- C. Volkringer, I. Mihalcea, J.-F. Vigier, M. Visseaux and T. Loiseau, Inorg. Chem., 2011, 50, 11865 CrossRef CAS PubMed.
- N. P. Martin, J. März, C. Volkringer, N. Henry, C. Hennig, A. Ikeda-Ohno and T. Loiseau, Inorg. Chem., 2017, 56, 2902 CrossRef CAS PubMed.
- C. Falaise, J. Delille, C. Volkringer and T. Loiseau, Eur. J. Inorg. Chem., 2015, 2813 CrossRef CAS.
- K. E. Knope and L. Soderholm, Chem. Rev., 2013, 113, 944 CrossRef CAS PubMed.
- J.-C. Berthet, P. Thuéry and M. Ephritikhine, Chem. Commun., 2005, 3415 RSC.
- S. Takao, K. Takao, W. Kraus, F. Emmerling, A. C. Scheinost, G. Bernhard and C. Hennig, Eur. J. Inorg. Chem., 2009, 4771 CrossRef CAS.
- G. Nocton, F. Burdet, J. Pécaut and M. Mazzanti, Angew. Chem., Int. Ed., 2007, 46, 7574 CrossRef CAS PubMed.
- V. Mougel, B. Biswas, J. Pécaut and M. Mazzanti, Chem. Commun., 2010, 8648 RSC.
- C. Falaise, H. A. Neal and M. Nyman, Inorg. Chem., 2017, 56, 6591 CrossRef CAS PubMed.
- C. Falaise, C. Volkringer, J.-F. Vigier, A. Beaurain, P. Roussel, P. Rabu and T. Loiseau, J. Am. Chem. Soc., 2013, 135, 15678 CrossRef CAS PubMed.
- L. Chatelain, R. Faizova, F. Fadaei-Tirani, J. Pécaut and M. Mazzanti, Angew. Chem., Int. Ed., 2019, 58, 3021 CrossRef CAS PubMed.
- B. Biswas, V. Mougel, J. Pécaut and M. Mazzanti, Angew. Chem., Int. Ed., 2011, 50, 5745 CrossRef CAS PubMed.
- C. Falaise, C. Volkringer, C. Hennig and T. Loiseau, Chem.–Eur. J., 2015, 21, 16654 CrossRef CAS PubMed.
- C. Tamain, T. Dumas, C. Hennig and P. Guilbaud, Chem.–Eur. J., 2017, 23, 6864 CrossRef CAS PubMed.
- N. P. Martin, C. Volkringer, N. Henry, X. Trivelli, G. Stoclet, A. Ikeda-Ohno and T. Loiseau, Chem. Sci., 2018, 9, 5021 RSC.
- N. P. Martin, C. Volkringer, P. Roussel, J. März, C. Hennig, T. Loiseau and A. Ikeda-Ohno, Chem. Commun., 2018, 54, 10060 RSC.
- L. Soderholm, P. M. Almond, S. Skanthakumar, R. E. Wilson and P. C. Burns, Angew. Chem., Int. Ed., 2008, 47, 298 CrossRef CAS PubMed.
- R. E. Wilson, S. Skanthakumar and L. Soderholm, Angew. Chem., Int. Ed., 2011, 50, 11234 CrossRef CAS PubMed.
- G. E. Sigmon and A. E. Hixon, Chem.–Eur. J., 2019, 25, 2463 CrossRef CAS PubMed.
- K. E. Knope and L. Soderholm, Inorg. Chem., 2013, 52, 6770 CrossRef CAS PubMed.
- I. Jelenic, D. Grdenic and A. Bezjak, Acta Crystallogr., 1964, 17, 758 CrossRef CAS.
- J. Ling, H. Lu, Y. Wang, K. Johnson and S. Wang, RSC Adv., 2018, 8, 34947 RSC.
- SAINT Plus Version 7.53a, Bruker Analytical X-ray Systems, Madison, WI, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192 CrossRef.
- L. Duvieubourg-Garela, N. Vigier, F. Abraham and S. Grandjean, J. Solid State Chem., 2008, 181, 1899 CrossRef CAS.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC.
- P. Benard, D. Louër, N. Dacheux, V. Brandel and M. Genet, Chem. Mater., 1994, 6, 1049 CrossRef CAS.
- J. Diwu and T. E. Albrecht-Schmitt, Inorg. Chem., 2012, 51, 4432 CrossRef CAS PubMed.
- A. T. Chemey, J. M. Sperling and T. E. Albrecht-Schmitt, RSC Adv., 2018, 8, 28642 RSC.
- J. N. Wacker, M. Vasiliu, K. Huang, R. E. Baumbach, J. A. Bertke, D. A. Dixon and K. E. Knope, Inorg. Chem., 2017, 56, 9772 CrossRef CAS PubMed.
- K. E. Knope, R. E. Wilson, M. Vasiliu, D. A. Dixon and L. Soderholm, Inorg. Chem., 2011, 50, 9696 CrossRef CAS PubMed.
- C. Hennig, S. Takao, K. Takao, S. Weiss, W. Kraus, F. Emmerling and A. C. Scheinost, Dalton Trans., 2012, 41, 12818 RSC.
- Y.-J. Hu, K. E. Knope, S. Skanthakumar and L. Soderholm, Eur. J. Inorg. Chem., 2013, 4159 CrossRef CAS.
- N. A. Vanagas, J. N. Wacker, C. L. Rom, E. N. Glass, I. Colliard, Y. Qiao, J. A. Bertke, E. Van Keuren, E. J. Schelter, M. Nyman and K. E. Knope, Inorg. Chem., 2018, 57, 7259 CrossRef CAS PubMed.
- K. E. Knope and C. L. Cahill, Inorg. Chem., 2007, 46, 6607 CrossRef CAS PubMed.
- P. Thuéry, CrystEngComm, 2008, 10, 808 RSC.
- K. L. Ziegelgruber, K. E. Knope, M. Frisch and C. L. Cahill, J. Solid State Chem., 2008, 181, 373 CrossRef CAS.
- C. E. Rowland and C. L. Cahill, Inorg. Chem., 2010, 49, 6716 CrossRef CAS PubMed.
- M. B. Andrews and C. L. Cahill, CrystEngComm, 2011, 13, 7068 RSC.
- H.-H. Li, X.-H. Zeng, H.-Y. Wu, X. Jie, S.-T. Zheng and Z.-R. Chen, Cryst. Growth Des., 2015, 15, 10 CrossRef CAS.
- N. P. Martin, C. Falaise, C. Volkringer, N. Henry, P. Farger, C. Falk, E. Delahaye, P. Rabu and T. Loiseau, Inorg. Chem., 2016, 55, 8697 CrossRef CAS PubMed.
- K. E. Knope, H. Kimura, Y. Yasaka, M. Nakahara, M. B. Andrews and C. L. Cahill, Inorg. Chem., 2012, 51, 3883 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optical and SEM photographs of 1 and 2, optical photographs of 1 and 2, powder X-ray diffraction pattern of 1, thermogravimetric curve of 1, X-ray thermodiffraction diagram of 1, infrared and UV-Vis spectra of 1. CCDC 1916502 and 1916503. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra03707a |
| This journal is © The Royal Society of Chemistry 2019 |