
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of amphiphilic branched silica derivatives associated with oligomeric medium
R. S. Davletbaevc,
I. I. Zaripov *c,
Z. Z. Faizulinab,
I. M. Davletbaevab,
D. S. Domrachovaa and
A. M. Gumerovb
*c,
Z. Z. Faizulinab,
I. M. Davletbaevab,
D. S. Domrachovaa and
A. M. Gumerovb
aNIOST LLC (SIBUR Corporate R&D Center), 2 Kuzovlevskiy tract, bld. 270, Tomsk, 634067, Russian Federation
bKazan National Research Technological University, 68 Karl Marx str, Kazan, Republic of Tatarstan 420015, Russian Federation
cKazan National Research Technical University after A.N. Tupolev-KAI, 10 Karl Marx str., Kazan, Republic of Tatarstan 420111, Russian Federation. E-mail: zaripovilnaz@gmail.com
First published on 9th July 2019
Abstract
Amphiphilic branched silica derivatives associated with oligomeric medium (ASiP) were obtained using tetraethoxysilane, polyoxyethylene glycol and low molecular weight polydimethylsiloxane. The creation of a silica core was based on tetraethoxysilane hydrolysis and condensation reactions by using water and a potassium diethylene glycolate as the catalyst. These reactions proceeded with the sequential participation of polyoxyethylene glycol and polydimethylsiloxane in parallel transetherification reactions. Microporous polymer film based on 2,4-toluene diisocyanate and block copolymers of propylene and ethylene oxides with terminal potassium-alcoholate groups were modified by ASiP. It has been shown that ASiP at the phase interface between thermodynamically incompatible macrochains performs the function of a link. It leads to a significant increase of intermolecular interactions and the supramolecular organization of the modified microporous polymers.
Introduction
Amphiphilic macromolecules with different affinities for polar and non-polar solvents have recently attracted the interest of researchers. Hydrophobic and hydrophilic branches in amphiphilic polymers can alternate in chains randomly or be arranged in blocks. Covalently bonded hydrophilic and hydrophobic structural units, which are part of the macrochains, are capable of self-organization in selective solvents.1,2 Such features of amphiphilic polymers as sensitivity to temperature, ionic strength, the nature of the solvent are important for their application in medicine, biochemistry and in other modern high-tech areas. Change of the system physical parameters can be the cause of both the formation of supramolecular formations and their destruction as a result of changes in the intermolecular interactions and macromolecules conformations. With the development of new polymerization methods, amphiphilic copolymers of various architectures were synthesized, including linear block copolymers, polymer brushes, star polymers, hyperbranched polymers.The big interest of these copolymers was attracted by amphiphilic macromolecular brushes due to their unique chemical and physical properties. Amphiphilic macromolecular brushes are copolymers with narrowly dispersed side chains grafted to the main chain.3–13 The properties of polymer brushes can be changed in a wide range compared with similar properties of linear polymers. The main design elements here are the length regulation of the main and side chains, the density of grafting, the chemical nature and architecture of the side chains. All these parameters are determined by the method of polymer brushes synthesis. Usually, there are two different approaches to the synthesis of macromolecular brushes. The first is a chemical method. In this case, a brush is grafted to a certain surface by terminal functional groups. Thus, some regularly branched polymers and flat brushes can be obtained, in which the chains are grafted to a flat surface. Spherically convex brushes can originate from star-shaped macromolecules that contain a large number of branches, and cylindrical convex brushes are obtained from comb-shaped polymers. The second way to form brushes is based on the ability of macromolecules to self-organize or self-assemble. A prerequisite for this is the presence of various groups in macromolecules. In simple cases, the formation of two-component copolymers is sufficient for these purposes, and the driving force of self-organization is intermolecular interaction. There are limitations of synthesis and certain advantages in molecular structures for each approach and method of obtaining macromolecular brushes. Important achieved parameters of synthesized macromolecular brushes are the grafting density, chemical composition, size, morphology, uniformity, length and distribution of the main and side chains. Asymmetric macromolecular brushes with polyethylene oxide and hydrocarbon side chains in polar solvents are assembled into micellar structure, whose crowns consist of polyoxyethylene side chains. The biggest part of researches in the field of amphiphilic multicomponent macromolecular brushes is devoted to the study of carbon-chain brushes. Self-organization processes of amphiphilic copolymers in selective solvents cause the formation of a large number of self-organized structures with different morphology. It's spheres, rods, cylinders, tubes, vesicles, disks.14–18 Amphiphilic copolymers are ionic and non-ionic, which is determined by the presence or absence of a charge in a hydrophilic block. In most cases, polyethylene oxide and its derivatives are non-ionic hydrophilic fragments.19–25 Long aliphatic or aromatic hydrocarbon radicals can be as a hydrophobic fragment in the structure of an amphiphilic copolymer. There are works on the synthesis of hyperbranched polymers and amphiphilic silica particles. Now, receipt mini-emulsions with side polystyrene branches are the most common works.26–29 Amphiphilic silica particles were obtained using tetraethoxysilane by high-temperature reaction gel–sol synthesis in ethylene glycol.30,31 Amphiphilic hyperbranched macromolecular compounds may influence polymerization processes32–35 and lead to improved homopolymer blends.36–40
In the work41 to control the interphase properties of glass fibre/PP composites, poly(ethylene glycol) (PEG) chains with different average molar mass were grafted onto the glass fibre surface by treating the fibres with aminopropyltriethoxysilane, 1,6-diisocyanatohexane and PEG stepwise.
Thus, the synthesis, research and use of amphiphilic branched macromolecular substances is the current direction of modern polymer science. The areas of their use are associated with the modification of polymer-forming systems and polymer compositions. Silica particles that contain grafted hydrophobic and hydrophilic branches on the surface are the most effective and universal compounds. Structural homogeneity and regular grafting of amphiphilic side branches can be achieved by using reactive oligomers in reactions with alkoxysilanes.
The aim of the work is the synthesis and characterization of the new amphiphilic branched silica derivatives associated with oligomeric medium. They are of particular interest as modifiers that have a significantly impact influence on the microphase separation processes of block copolymers.
Experimental
(A) Materials
Polyoxyethylene glycol (PEO, molecular weight is 400 g mol−1) was purchased from PJSC Nizhnekamskneftekhim (Nizhnekamsk, Russia), tetraethoxysilane (TEOS) was purchased from CJSC Vekton (St. Petersburg, Russia). Polydimethylsiloxane (PDMS, average molecular weight is 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), was purchased from JSC Kazan factory of Synthetic Rubber (Kazan, Russia). Potassium diethylene glycolate (DEG-K) with formula H(O–CH2–CH2)2–OK was obtained in laboratory conditions. The block copolymer of propylene and ethylene oxide (PPEG) with formula HO[CH2CH2O]n[CH2(CH3)CH2O]m[CH2CH2O]nK, where n ≈ 14 and m ≈ 48, molecular weight 4200 g mol−1, containing 30 wt% of peripheral polyoxyethylene blocks, where the content of potassium alcoholate groups is 10.9% from the total number of functional groups, was purchased from PJSC Nizhnekamskneftekhim (Nizhnekamsk, Russia). The 2,4-toluene diisocyanate (TDI, 98%) was purchased from Sigma-Aldrich (St. Louis, MO, USA). PEO, PPEG and PDMS were additionally dried at reduced pressure (approximately 0.1 kPa) and at elevated temperature of 95 °C down to 0.01% moisture concentration. All other reagents were used without further purification.
000 g mol−1), was purchased from JSC Kazan factory of Synthetic Rubber (Kazan, Russia). Potassium diethylene glycolate (DEG-K) with formula H(O–CH2–CH2)2–OK was obtained in laboratory conditions. The block copolymer of propylene and ethylene oxide (PPEG) with formula HO[CH2CH2O]n[CH2(CH3)CH2O]m[CH2CH2O]nK, where n ≈ 14 and m ≈ 48, molecular weight 4200 g mol−1, containing 30 wt% of peripheral polyoxyethylene blocks, where the content of potassium alcoholate groups is 10.9% from the total number of functional groups, was purchased from PJSC Nizhnekamskneftekhim (Nizhnekamsk, Russia). The 2,4-toluene diisocyanate (TDI, 98%) was purchased from Sigma-Aldrich (St. Louis, MO, USA). PEO, PPEG and PDMS were additionally dried at reduced pressure (approximately 0.1 kPa) and at elevated temperature of 95 °C down to 0.01% moisture concentration. All other reagents were used without further purification.
(B) Synthesis of amphiphilic branched silica derivatives associated with oligomeric medium (ASiP)
The synthesis of ASiP was multistage. At the first stage, PEO (9.64 ml), TEOS (20 wt% by weight of the total amount of oligomers and TEOS, 4.76 ml), water (0.04 wt% of the total weight of components PEG-TEOS, 20 μl) and DEG-K catalyst (0.1 wt% relative to the mass of the total reaction system, 10 μl) were added to the flask. The mixtures were stirred at temperature 90 °C for at least 6 h. Transetherification of ethoxysilane TEOS's groups, sequential reactions of their hydrolysis with condensation of the resulting Si–OH groups simultaneously proceeded in the DEG-K presence and water. Then, the second part of TEOS (10 wt% by weight of the total amount of TEOS, 2.38 ml) was introduced into the reaction system. The synthesis was carried out for 6 hours, after the final part of TEOS (5 wt% by weight of the total number of TEOS, 1.4 ml) was introduced in the mixture with stirring for 4 h. After each stage mixtures were put in vacuum for 10 min to remove alcohol and water and shift the balance towards the product.As a result of the described reaction processes, including the interaction of PEO, TEOS with a catalyst DEG-K and water, was obtained silica branched by polyoxyethylene glycol (PEOASiP).
Further, PDMS (5 ml) was added in PEOASiP with stirring at temperature 90 °C, after which the viscosity increased in reaction system and ASiP was obtained.
(C) Synthesis of modified by ASiP microporous polymers based on TDI and PPEG
The reaction was carried out in toluene at 20 °C in a flask equipped with a reflux condenser. The polymerization proceeded with the constant stirring, a magnetic stirrer being used. PPEG (1 g), toluene (7.9 g), bisphenol A (0.004 g), acetic acid (0.007 g) and calculated amount of ASiP were added to the flask. The reaction mass was mixed at a given temperature until the complete dissolution of PPEG took place, then TDI (0.62 g.) at a molar ratio [PPEG]:[TDI] = 1:15 was introduced. Five minutes after mixing with TDI, triethylamine (0.0072 g) and water (0.001 g) were loaded into the reaction system. After ten minutes the reaction mass was dispensed to Petri dish, and then it was cured at room temperature during 72 hours.
(D) Polymer characterization
Results and discussion
(A) ASiP characterization
The reagents were preliminarily examined to analyze the size distribution of ASiP particles. PEO in toluene exists in the associated state. This is the reason that the average size of PEO determined using dynamic light scattering was 450 nm (Fig. 1a). The average particle size of the PDMS is 10 nm (Fig. 1b). The average particle size of PEOASiP is 60–400 nm with a maximum at 150 nm. There are particles with a size distribution in the region of 10–30 nm, which exhibit relatively low intensity (Fig. 1c).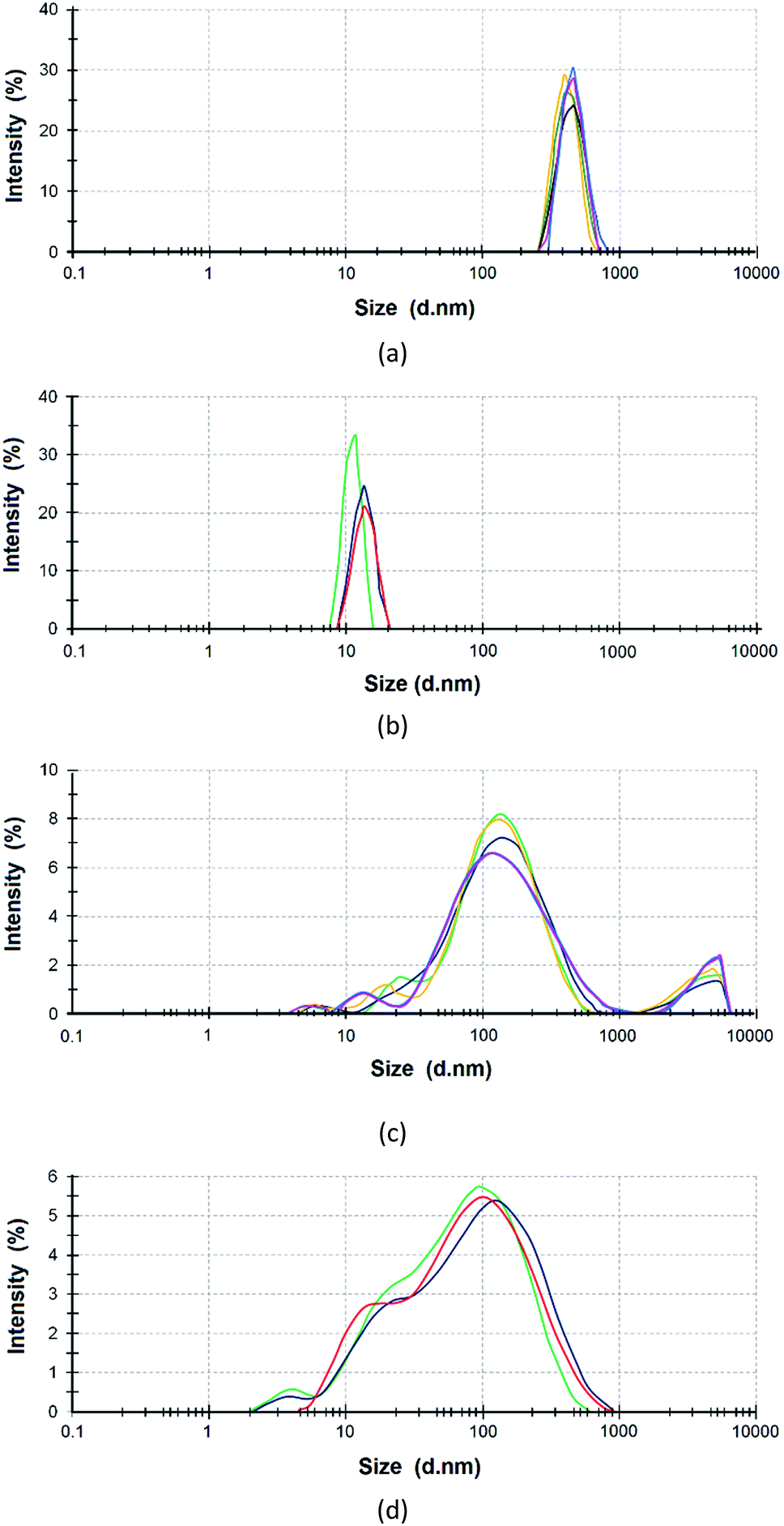 | ||
| Fig. 1 Particle size distribution of PEO (a), PDMS (b), PEOASiP (c), ASiP (d) in toluene. | ||
In the case of ASiP, there is an increase in the proportion of particles with a size distribution in the region of 10–30 nm. The intensity in the region of the size distribution of 60–400 nm falls from 8 to 5.5% compared with PEOASiP (Fig. 1d). There is no increase in particle size. This is because of PEOASiP is associated in toluene due to intermolecular interactions of PEO. As a result, the share of associates falls because of PDMS addition to PESOASiP and consequently ASiP formation.
From these results we expect that PEOASiP particles exist in the associated state. The sizes of associates are 60–400 nm. The redistribution of intensity (a decrease in the region of 60–400 nm and an increase in the region of 10–30 nm) is associated with the consumption of PEOASiP particles for the formation of ASiP particles.
29Si NMR and 1H NMR spectra were measured for TEOS, PDMS and ASiP. The 1H NMR spectra of the TEOS (Fig. 2) contains triplets at δ = 1.224 ppm and δ = 3.84 ppm assigned to the protons of the methyl and methylene groups, respectively. The 1H NMR spectra of PDMS (Fig. 3) exhibits a resonance signal at δ = 0.066 ppm, corresponding to the protons of the methyl groups in the CH3–Si–CH3. The 1H NMR spectrum of ASiP (Fig. 4), the signal at δ = 1.224 ppm almost disappears, corresponding to the protons of the methyl groups of TEOS. There is a signal at δ = 3.7–3.5 ppm assigned to the protons of methylene groups in the composition of PEO links. According to the 1H NMR spectra, low intensity peaks are present in the ASiP composition and corresponds to the presence of unreacted TEOS and the absence of ethanol (the product of TEOS hydrolysis). At the 29Si NMR spectra of the TEOS, a signal is present at δ = −81.963 ppm (Fig. 5).
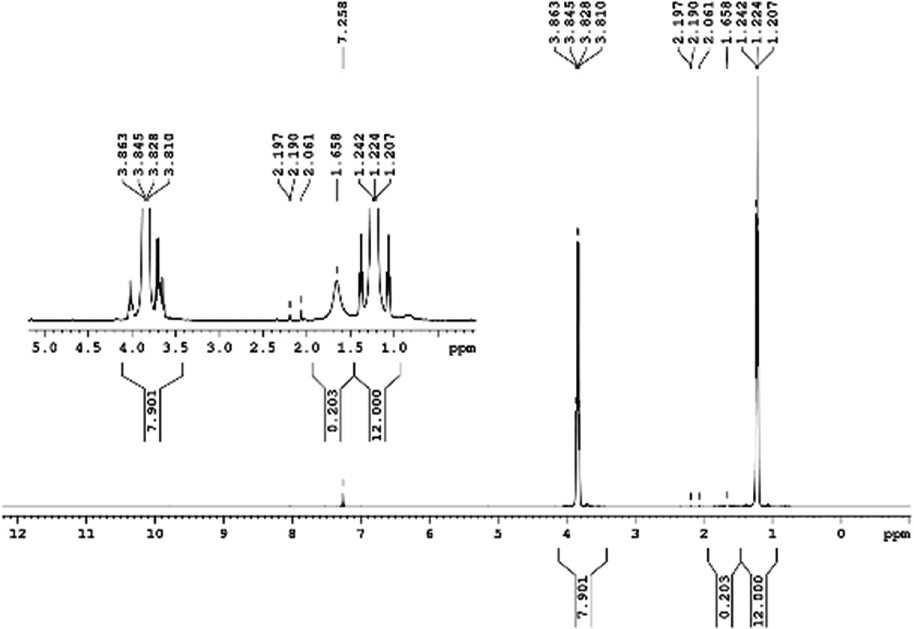 | ||
| Fig. 2 1H NMR spectra of TEOS. | ||
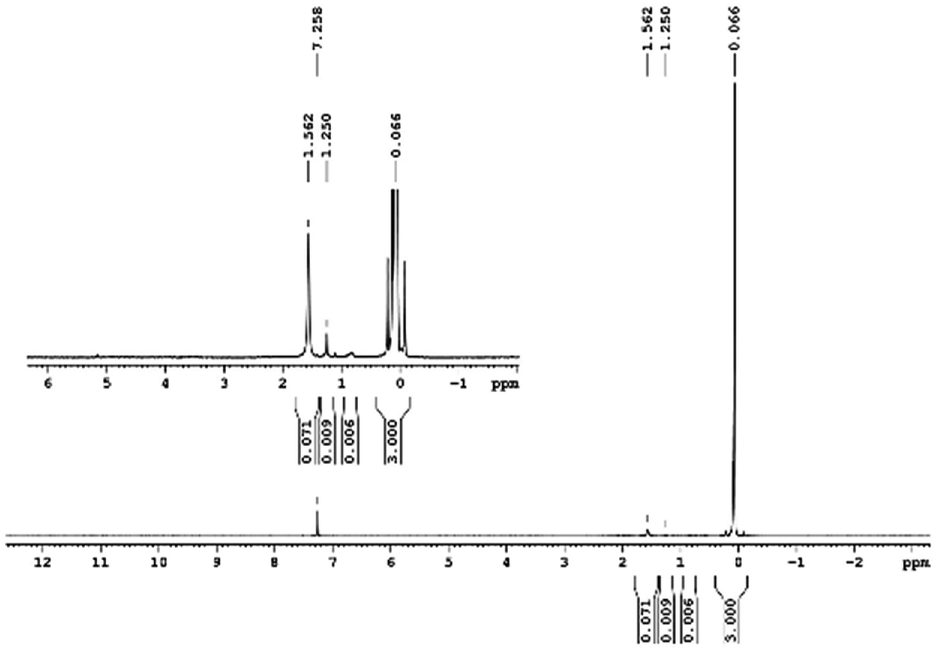 | ||
| Fig. 3 1H NMR spectra of PDMS. | ||
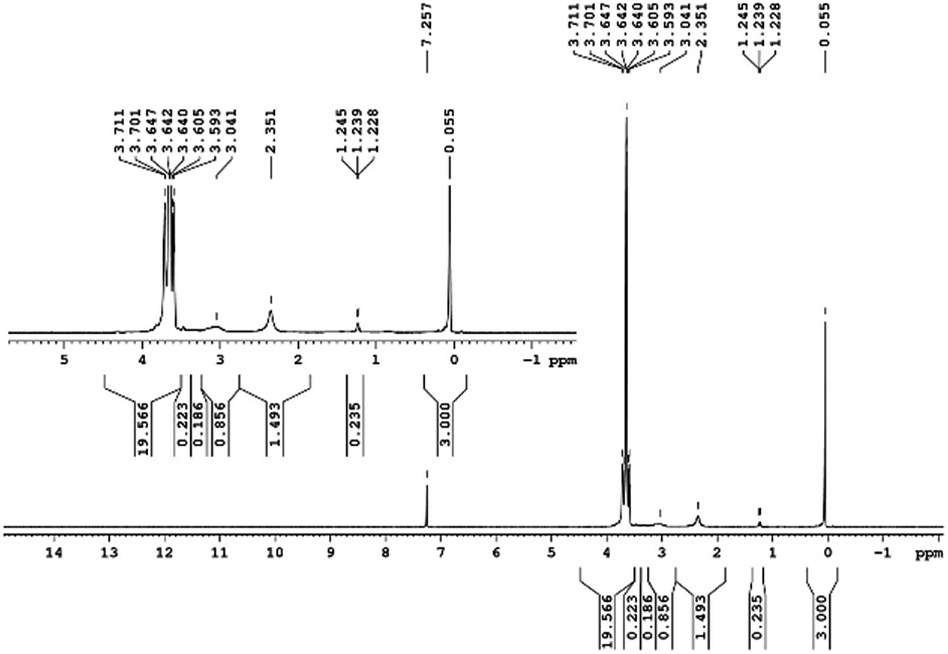 | ||
| Fig. 4 1H NMR spectra of ASiP. | ||
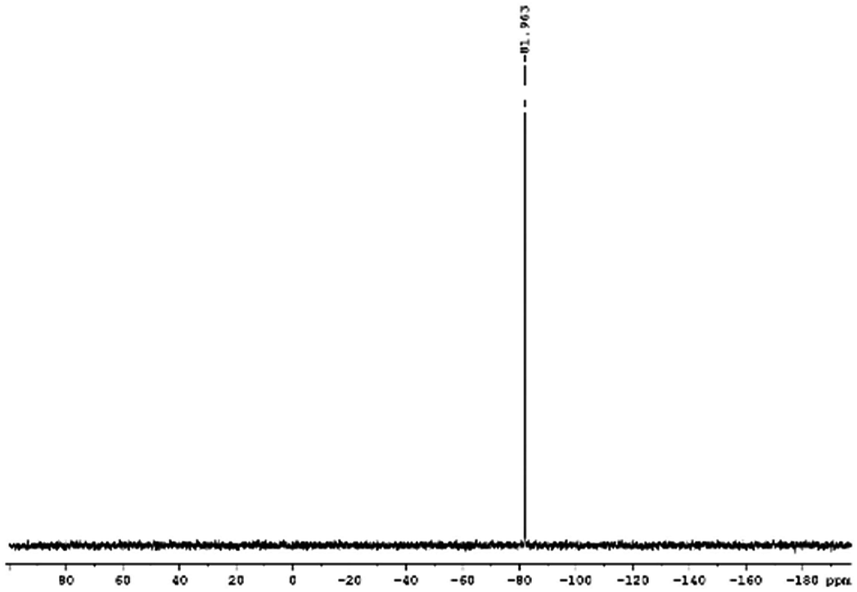 | ||
| Fig. 5 29Si NMR spectra of TEOS. | ||
Analysis of the 29Si NMR spectra of PEOASiP confirms the absence of TEOS in the ASiP (Fig. 6). The absence of signals at δ = −82 ppm enables to conclude that tetraalkoxide silane derivatives are practically absent in the composition of ASiP. The appearance of the δ = −108 ppm signal indicates the formation of the cube silica, substituted by alkoxide groups.42
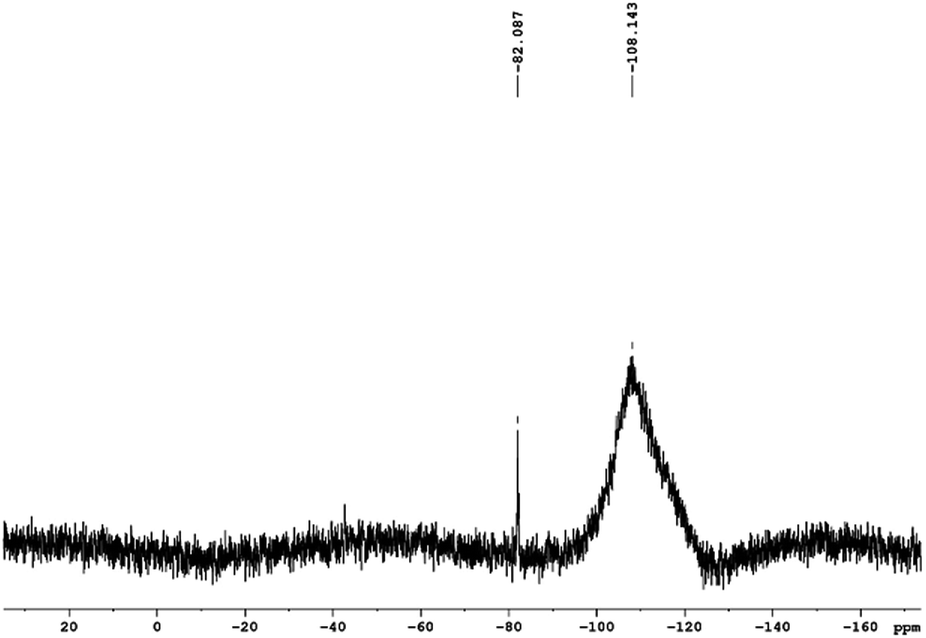 | ||
| Fig. 6 29Si NMR spectra of PEOASiP. | ||
The reactions occurring during the formation of PEOASiP and ASiP are shown at Scheme 1.
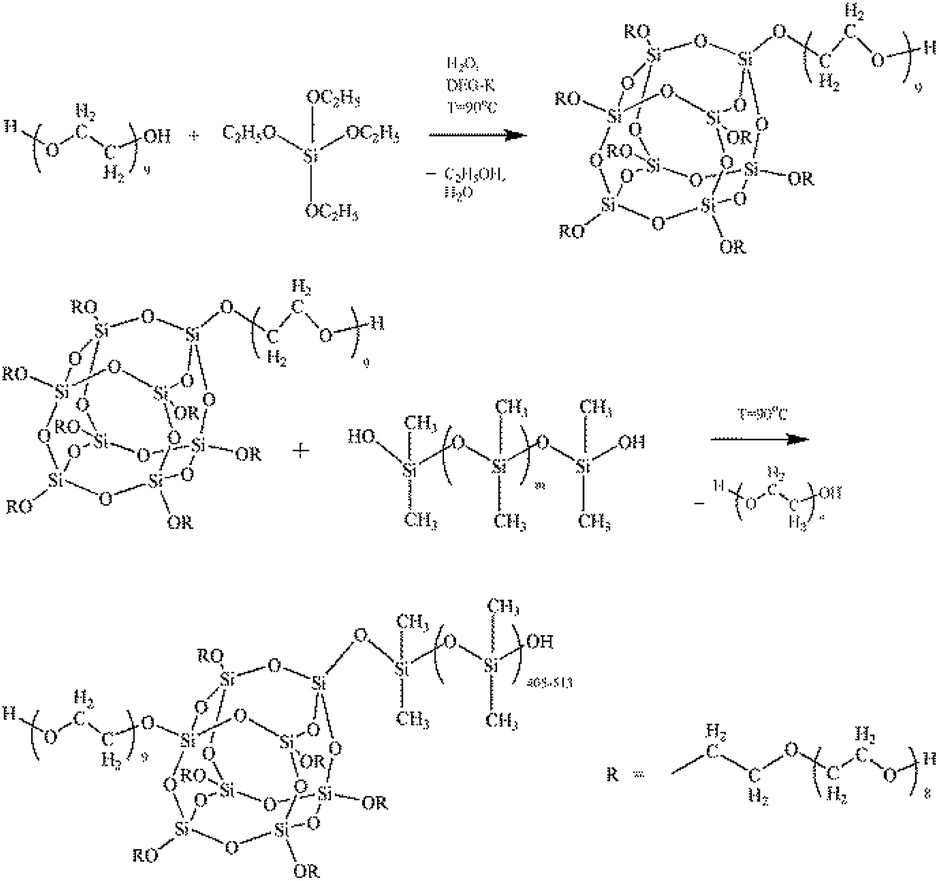 | ||
| Scheme 1 The scheme of experimental approaches to form ASiP. | ||
The bands at 965, 1075, and 1102.5 cm−1 are present on the TEOS FTIR spectra, the corresponding valence vibrations of the Si–O bonds in the Si–O–C group of tetraethoxysilan (Fig. 7). Absorption band at 1010 cm−1 corresponding to the stretching vibrations of the Si–C bond (as part of the Si–CH3 group) and band at 1070 cm−1 assigned to the stretching vibrations of the Si–O bond in the composition of the Si–O–Si present in the FTIR spectra of PDMS.
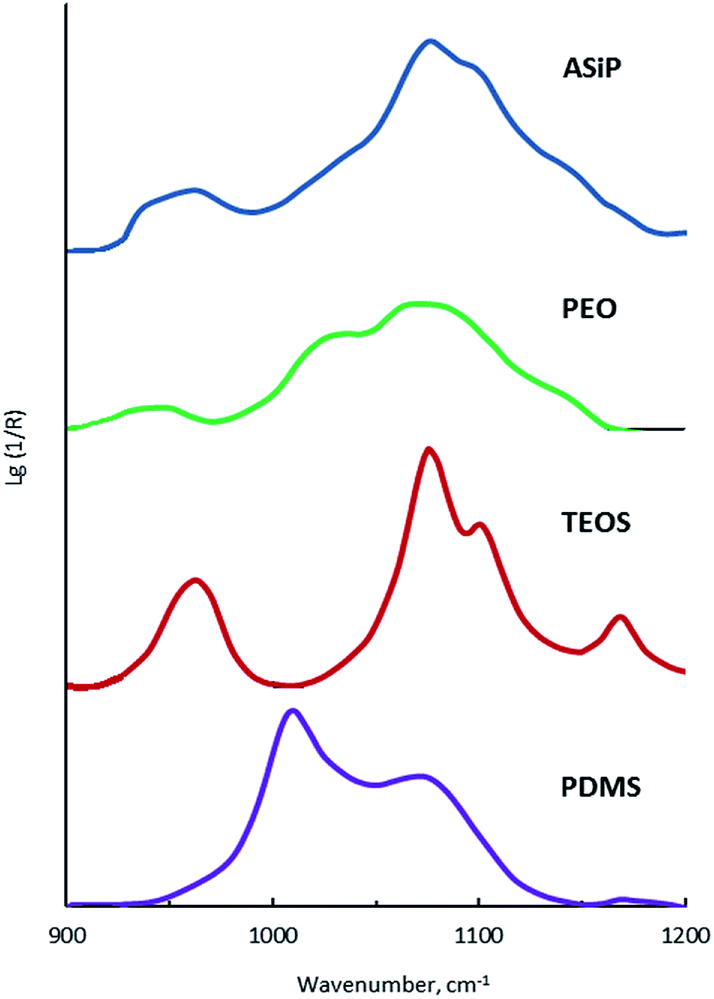 | ||
| Fig. 7 FTIR spectra of TEOS, PEO, PDMS and ASiP. | ||
On the FTIR spectra of ASiP, the band at 965 cm−1 corresponding to the stretching vibrations of the Si–O bond in the Si–O–C group of TEOS has completely changed shape and location. The spectral manifestation turned out to be more gentle and it's the superposition of the absorption bands at 950 and 965 cm−1. It can be due to the existence of the Si–O–C group, but not as part of TEOS. The bands at 1075 and 1100 cm−1 correspond to the stretching vibrations of the Si–O bond in the Si–O–C group. It indicates the existence of the group Si–O–C in the composition of polyoxyethylene silane fragments ASiP. It should be noted that the intensities of these bands differ slightly compared with TEOS, and the band at 1102.5 cm−1 has shifted to 1100 cm−1. PEO and PDMS are attached to a cubic silica derivative. It can be judged by the characteristic absorption regions for the Si–O–Si bonds in cubic structures42 that are present in the FTIR spectra of ASiP. These are pronounced shoulders in the region of 1140, 1179 cm−1 and a low-intensity band at 1200 cm−1.
(B) Thermal – gravimetric behavior of PEOASiP and ASiP
The beginning of the mass loss for PEOASiP and ASiP is in the region of 150 °C (Fig. 8). The maximum decomposition rate of PEOASiP starts at 390 °C and for ASiP is observed at a temperature that exceeds the maximum decomposition rate of PEOASiP by 20 °C. PEOASiP shows a lower heat resistance compared to ASiP due to the fact that in this case cubic silica has only polyoxyethylene environment. Partial substitution of PEO with PDMS naturally leads to an increase in the heat resistance of ASiP.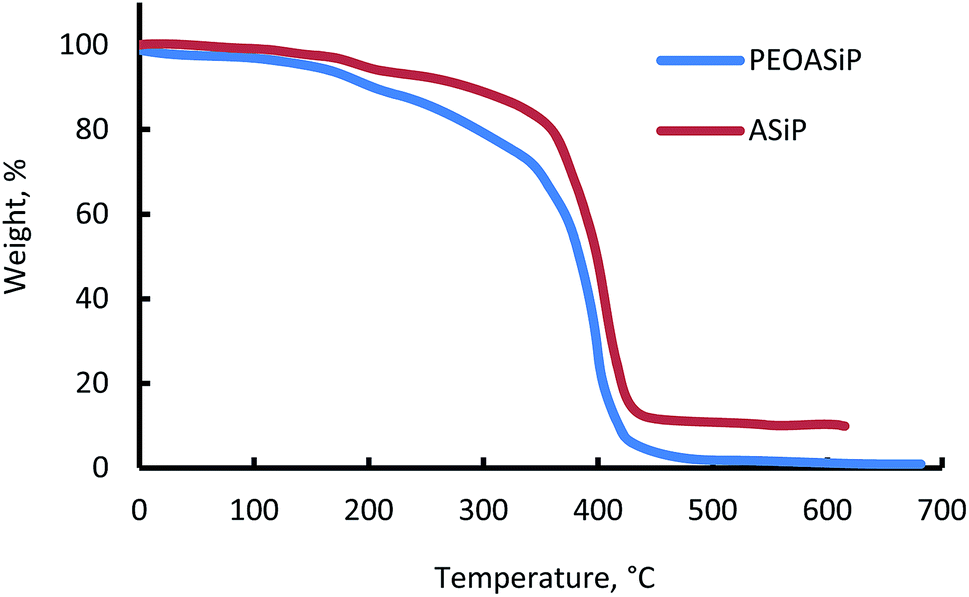 | ||
| Fig. 8 TGA of PEOASiP and ASiP. | ||
(C) Surface active properties
An important feature of amphiphilic compounds is the ability to self-organize processes, which are accompanied by the formation of micelles. The self-assembly of small-molecule amphiphiles has been studied for many decades, and various morphologies have been observed in bulk and in aqueous solutions. The morphologies include spherical and cylindrical micelles, lamellae and vesicles, among others.43–45Amphiphilic polymers form thermodynamically more stable structures in comparison with low-molecular surfactants. Values of CMC of polymer micelles are, as a rule, 10−6 to 10−7 M, while for low molecular weight surfactants this value is of the order 10−3 to 10−4 M.46 Structures of amphiphilic block copolymers are also kinetically more stable, since their dissociation into individual macromolecules is slowed down.47
According to measurements of surface tension, ASiP exhibit the ability to form micelles in water (Fig. 9). All particles are characterized the manifestation of two CMC because of their ability to form different micellar structure with a change of ASiP concentration.
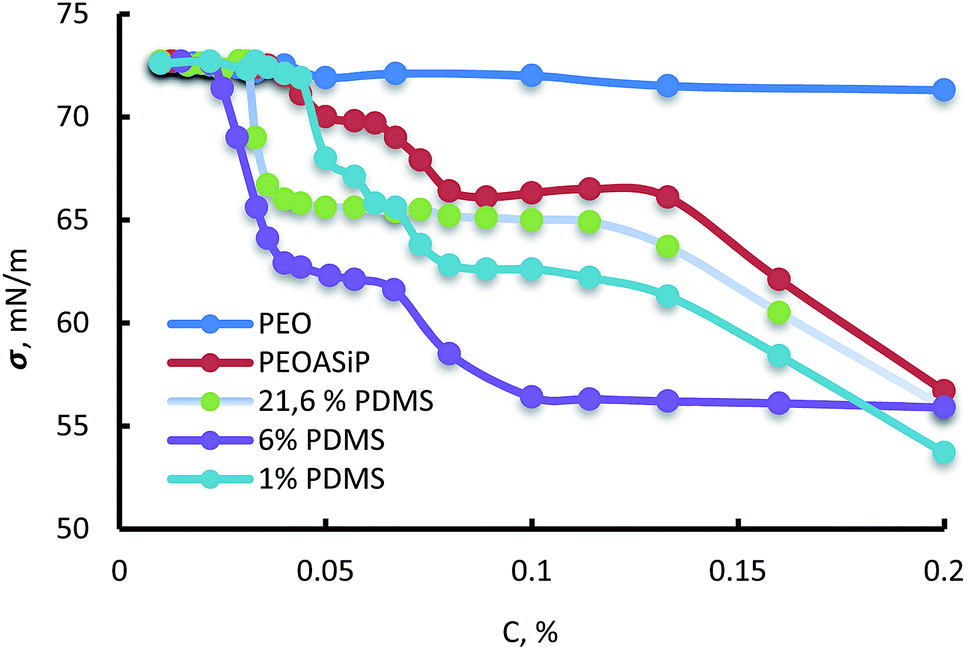 | ||
| Fig. 9 Dependencies of surface tension (σ, mN m−1) on concentration in water (C, %) for PEO, PEOASiP and ASiP, obtained using 1% PDMS, 6% PDMS, 21.6% PDMS. | ||
It turned out that the content of PDMS in the ASiP composition influences the values of CMC and surface tension. Values of CMC and surface tension fall to a certain value with increasing of PDMS content in the composition of ASiP. The critical micelle concentration for ASiP, obtained using 0%, 1%, 6% and 21.6% of PDMS are 1 o10−4, 9 910−5, 3 310−5 and 2.5 an10−5 M, respectively.
This fact confirms that PDMS is included in ASiP and enhances their amphiphilicity. The scheme of ASiP formation and the scheme of ASiP micelle formation in water are shown in Schemes 2 and 3, respectively.
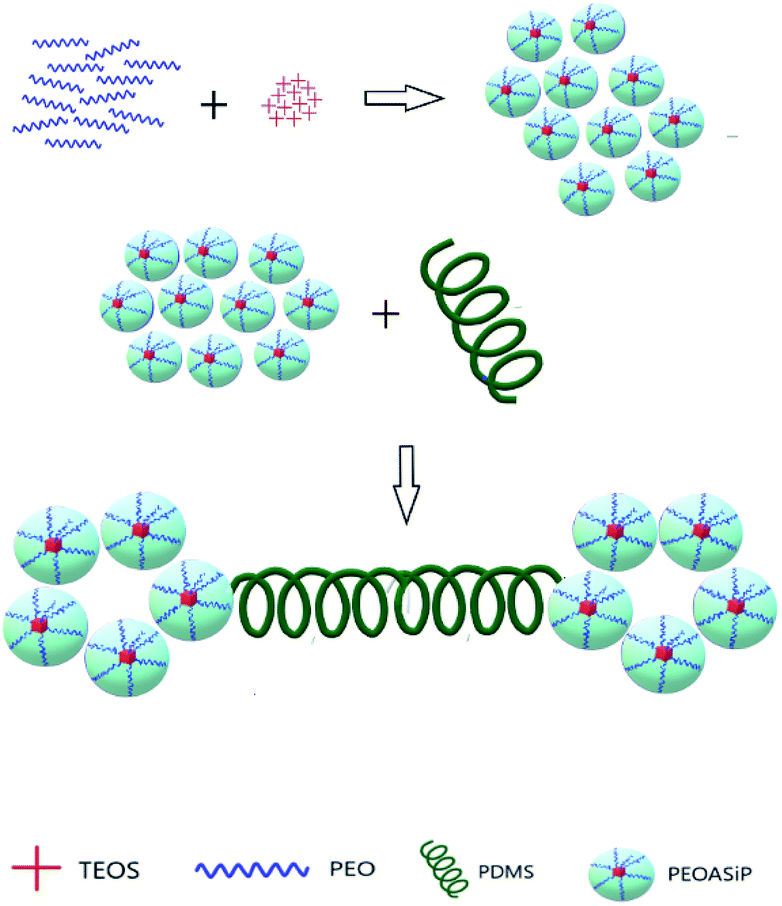 | ||
| Scheme 2 The scheme of ASiP formation. | ||
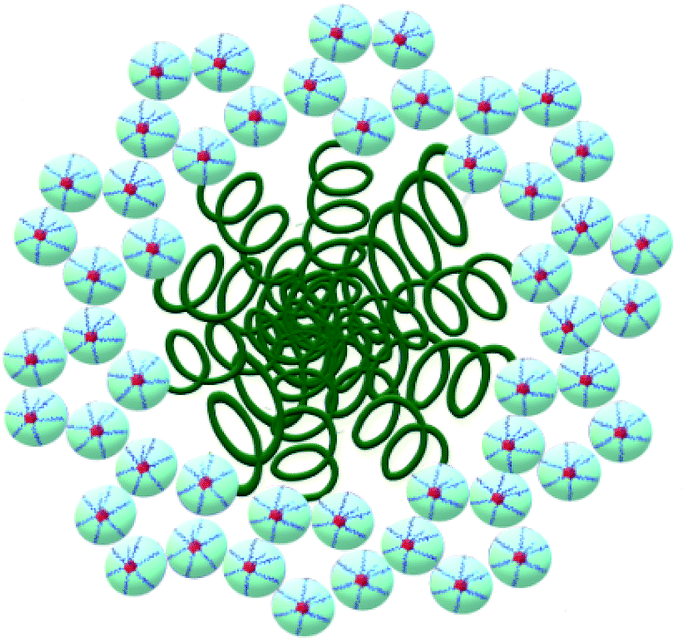 | ||
| Scheme 3 The structure of ASiP micelles. | ||
(D) SEM analyses
The ASiP aqueous emulsion was treated with ultrasound for SEM analysis, and then a drop of the emulsion was placed on the substrate. The measurements were carried out after preliminary evacuation of the sample (Fig. 10). | ||
| Fig. 10 SEM image of ASiP aqueous emulsion. | ||
The particle diameter is 20–70 nm. According to the SEM measurements, the particles of the prepared aqueous emulsion are uniform in size; the internal component of the particles (core) is denser than the shell. The size of the prepared aqueous emulsion determined using SEM correlates with the size of the ASiP in toluene determined using dynamic light scattering method. The particle size distribution of ASiP determined using DLS measurements are in the range of 10–30 nm (Fig. 1d).
(E) Modification of polymers based on TDI and PPEG with ASiP
ASiP was used to modify microporous polymers based on TDI and PPEG. These polymers are of interest due to their high gas transport characteristics.48–54 The specific characteristics of theirs supramolecular structure is polyisocyanate blocks of acetal nature (O-polyisocyanates) as a component with a rigid chain. A scheme of polymers synthesis by the reaction of polyaddition of 2,4-toluene diisocyanate (TDI) and a macroinitiator, which is a potassium-substituted block copolymer of propylene and ethylene (PPEG) is shown in Scheme 4.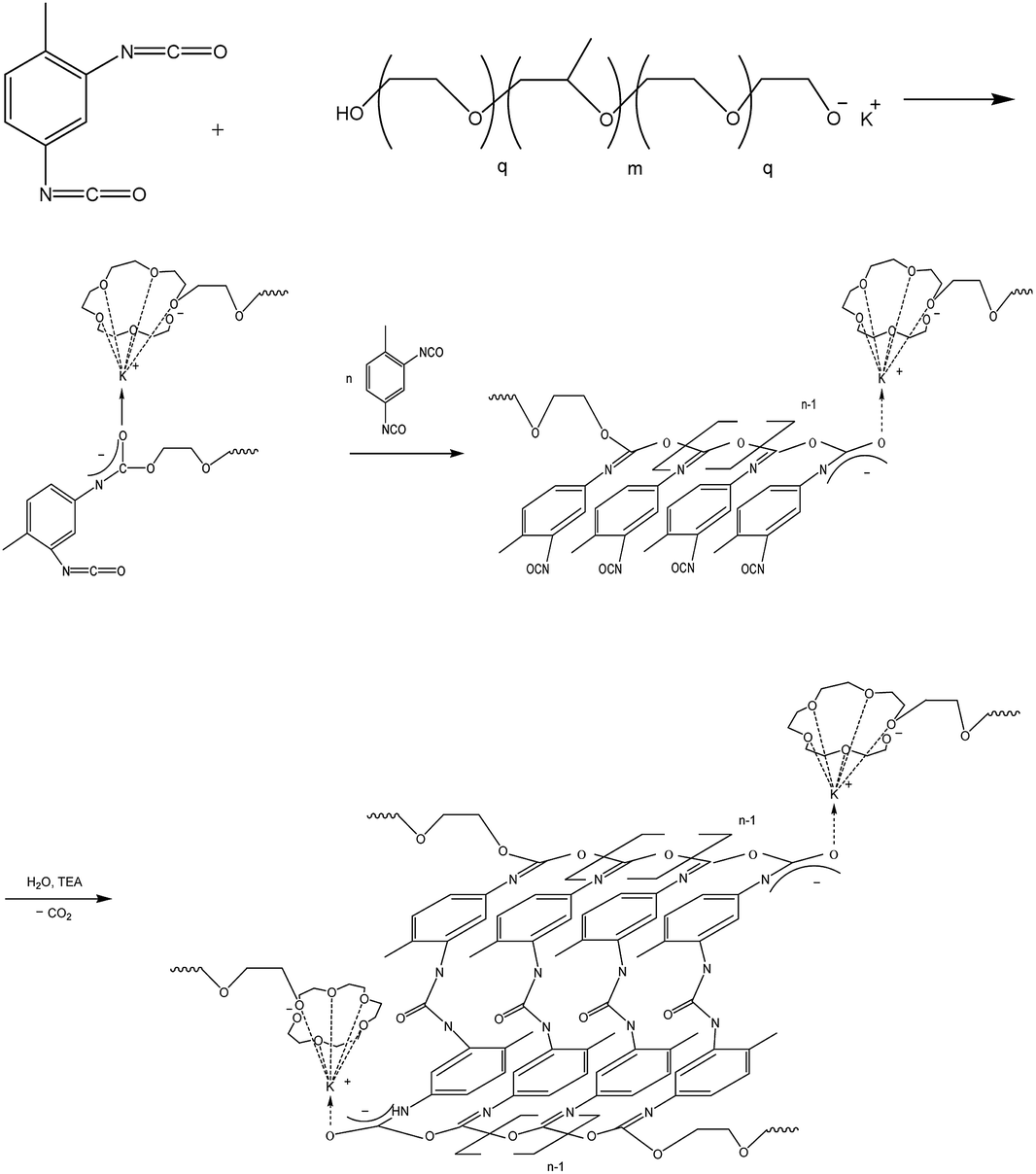 | ||
| Scheme 4 Synthesis of polymers based on TDI and PPEG. | ||
Microporous polymers modified with ASiP was investigated using FTIR spectroscopy. There aren't C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in the structure of O-polyisocyanates. Thus, the NC bond, which exists in the structure, is characterized by a band at 1670 cm−1. According to Fig. 11, changes in the area of 1670 cm−1 aren't observed and the intensity of the band in the area of 1710 cm−1 doesn't increase. It's characteristic of polyisocyanurates, which are formed during a possible competing reaction. It leads to the disclosure of the NCO group by the NC component and the use of ASiP doesn't interfere with the reaction by the mechanism, which is presented in Scheme 4.
O bonds in the structure of O-polyisocyanates. Thus, the NC bond, which exists in the structure, is characterized by a band at 1670 cm−1. According to Fig. 11, changes in the area of 1670 cm−1 aren't observed and the intensity of the band in the area of 1710 cm−1 doesn't increase. It's characteristic of polyisocyanurates, which are formed during a possible competing reaction. It leads to the disclosure of the NCO group by the NC component and the use of ASiP doesn't interfere with the reaction by the mechanism, which is presented in Scheme 4.
 | ||
| Fig. 11 FTIR spectra of polymers. | ||
(F) Thermomechanical and dynamic mechanical analysis of polymers
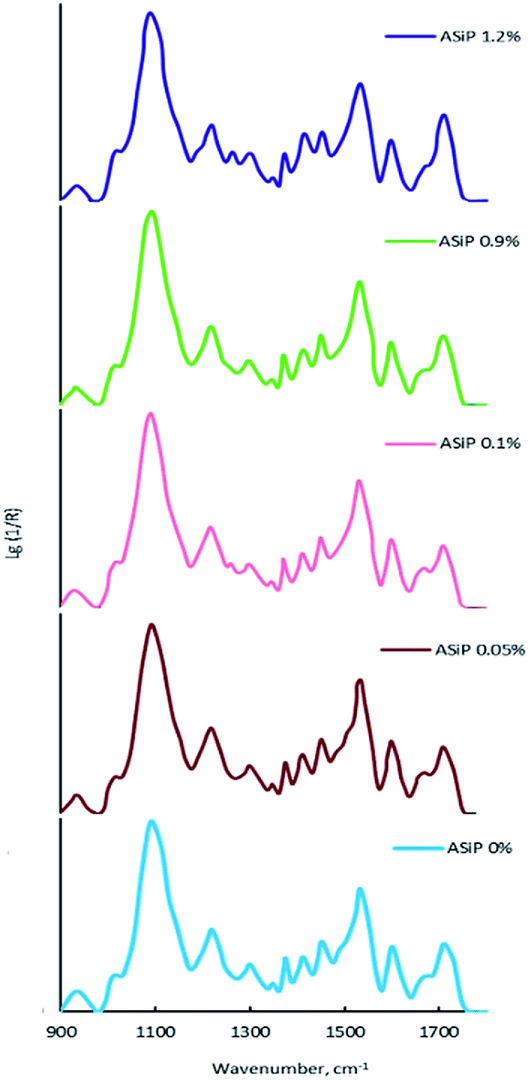
According to Fig. 12a, the use of ASiP has a significant impact on the thermomechanical behaviour of polymers.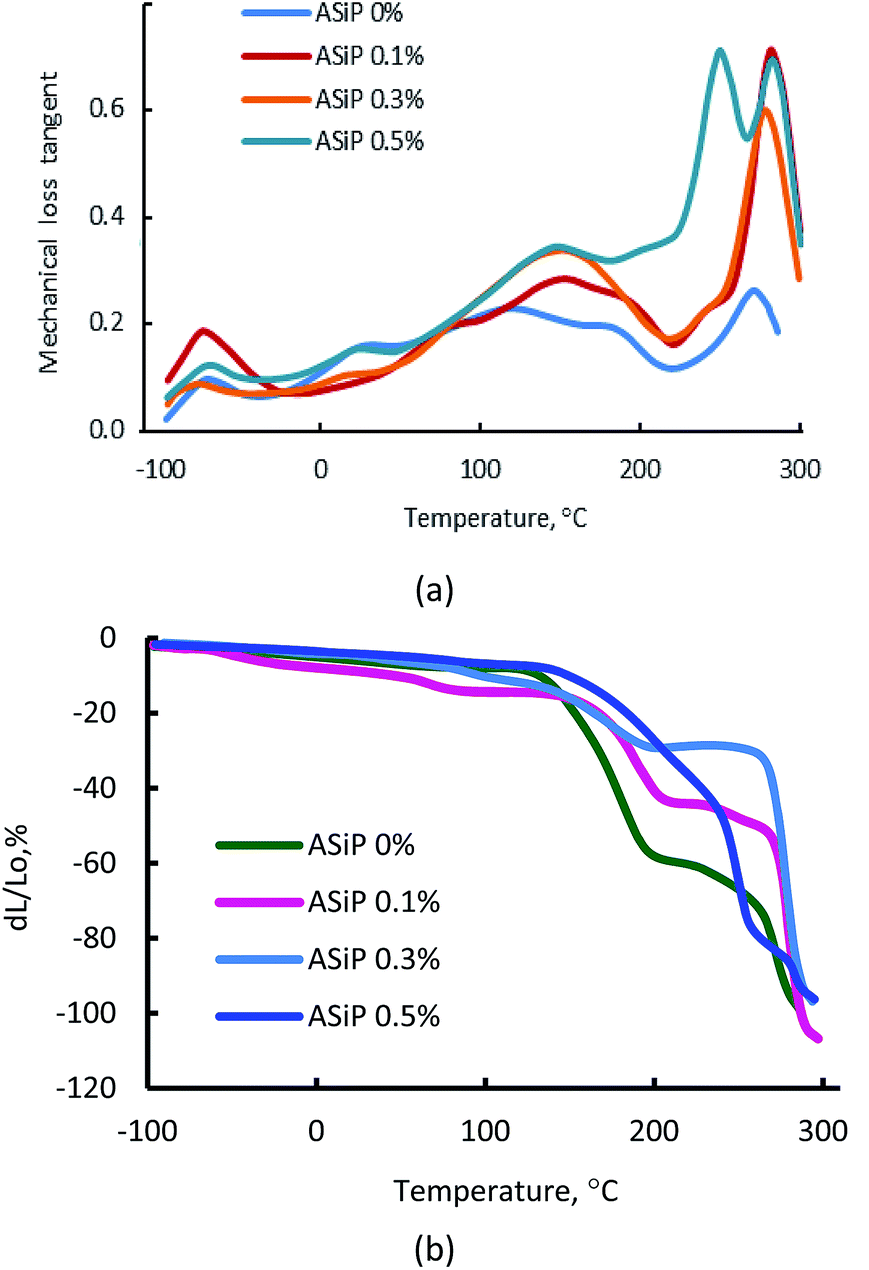 | ||
| Fig. 12 MLT (a) and TMA (b) curves of polymers. | ||
This figure depicts, that the region of the α-transition, which corresponds to the beginning of the segmental mobility of the flexible polyether component, practically unchanged, but significant changes are observed in the region of relatively high temperatures. This is an increase of the structural elements mobility of the polymer matrix in the region of 60–150 °C. The most significant deformation effect is observed in the temperature range above 200 °C, which corresponds to relaxation processes with the participation of polyisocyanate hard blocks. Modification of films by ASiP to its content of 0.3 wt% leads to a decrease in the strain from 60% to 20% (Fig. 12b). Such a significant effect occurs because of the enhancement of intermolecular interactions and the overall structural organization of the rigid-chain component, which are O-polyisocyanates.
(G) Mechanical behavior and density of polymers
Modification of polymers based on TDI and PPEG by ASiP leads to an increase in strength, relative elongation (Fig. 13), and decrease in density in the low concentrations of ASiP (Table 1). The decrease in the density of microporous polymers is a consequence of the active influence of ASiP on the processes of their supramolecular organization.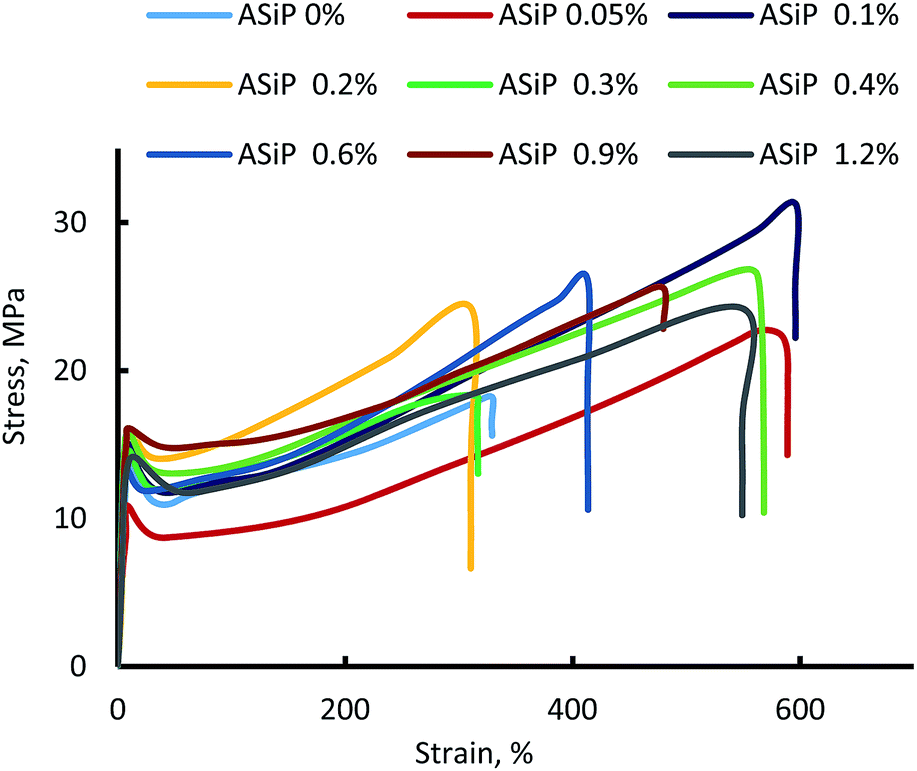 | ||
| Fig. 13 Tensile tests of polymers. | ||
| [ASiP], wt% | Density, g cm−3 | E, MPa |
|---|---|---|
| 0.00 | 1.156 | 198 |
| 0.03 | 1.112 | 198 |
| 0.05 | 1.096 | 217 |
| 0.08 | 1.129 | 349 |
| 0.10 | 1.165 | 469 |
| 0.20 | 1.167 | 121 |
| 0.30 | 1.168 | 413 |
| 0.40 | 1.163 | 219 |
| 0.60 | 1.158 | 313 |
Conclusions
Amphiphilic branched silica derivatives associated with oligomeric medium were obtained using tetraethoxysilane, polyoxyethylene and low molecular weight polydimethylsiloxane. The creation of a silica core was based on TEOS hydrolysis and condensation reactions using latent water and a DEG-K catalyst. These reactions proceeded with the sequential participation of PEO and PDMS in parallel transetherification reactions.Microporous polymers based on 2,4-toluene diisocyanate and block copolymers of propylene and ethylene oxides terminated by potassium-alcoholate groups were modified by ASiP. It has been shown that ASiP at the phase interface between thermodynamically incompatible macrochains performs the function of a link. It leads to a significant increase of intermolecular interactions, the structural organization of the rigid-chain component and the growth of the mechanical properties of the modified microporous polymers. The decrease in the density of microporous polymers is a consequence of the active influence of ASiP on the processes of their supramolecular organization.
Abbreviations
| PEO | Polyoxyethylene glycol |
| TEOS | Tetraethoxysilane |
| DEG-K | Potassium diethylene glycolate |
| PDMS | Polydimethylsiloxane |
| PEOASiP | Silica branched by polyoxyethylene glycol |
| ASiP | Amphiphilic branched silica derivatives associated with oligomeric medium |
| TDI | 2,4-Toluene diisocyanate |
| PPEG | Potassium-substituted block copolymer of propylene and ethylene oxide |
| FTIR | Fourier transform infrared spectroscopy analysis |
| TGA | Thermogravimetric analysis |
| NMR | Nuclear magnetic resonance |
| SEM | Scanning electron microscopy |
| TMA | Thermomechanical analysis |
| MLT | Mechanical loss tangent |
| CMC | Critical micelle concentration |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The reported study was funded by RFBR according to the research project no. 18-43-160002.References
- W. Zhao, P. Fonsny, P. Fitzgerald, G. G. Warr and S. Perrier, Polym. Chem., 2013, 4, 2140 RSC.
- J. Wang, K. Yao, C. Wang, C. Tang and X. Jiang, J. Mater. Chem. B, 2013, 1, 2324 RSC.
- S. Sheiko, B. S. Sumerlin and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 759 CrossRef CAS.
- S. S. Sheiko, S. A. Prokhorova, K. L. Beers, K. Matyjaszewski, I. I. Potemkin, A. R. Khokhlov and M. Möller, Macromolecules, 2001, 34(23), 8354 CrossRef CAS.
- A. V. Yakimansky, T. K. Meleshko, D. M. Ilgach, M. A. Bauman, T. D. Anan'eva, L. G. Klapshina, S. A. Lermontova, I. V. Balalaeva and W. E. Douglas, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4267 CrossRef CAS.
- A. Krasova, E. Belyaeva, E. Tarabukina, A. Filippov, T. Meleshko, D. Ilgach, N. Bogorad and A. Yakimansky, Macromol. Symp., 2012, 316(1), 32 CrossRef CAS.
- M. Zhang and A. H. E. Müller, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3461 CrossRef CAS.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129(20), 6633 CrossRef CAS.
- M. Schappacher and A. Deffieux, Science, 2008, 319(5869), 1512 CrossRef CAS.
- Y. Yan, Y. Shi, W. Zhu and Y. Chen, Polymer, 2013, 54(21), 5634 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136(18), 6513 CrossRef CAS.
- H.-Y. Chang, Y.-L. Lin and Y.-J. Sheng, Macromolecules, 2012, 45(11), 4778 CrossRef CAS.
- X. Lian, D. Wu, X. Song and H. Zhao, Macromolecules, 2010, 43(18), 7434 CrossRef CAS.
- J. Raez, I. Manners and M. A. Winnik, J. Am. Chem. Soc., 2002, 124, 10381 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- S. J. Holder and N. A. J. M. Sommerdijk, Polym. Chem., 2011, 2, 1018 RSC.
- M. Zhang and A. H. E. Müller, J. Polym. Sci., Part A: Polym. Chem., 2005, 43(16), 3461 CrossRef CAS.
- A. P. Martinez, Zh. Cui, Ch. Hire, T. A. P. Seery and D. H. Adamson, Macromolecules, 2015, 48(13), 4250 CrossRef CAS.
- M. Higa, K. Yaguchi and R. Kitani, Electrochim. Acta, 2010, 55(4), 1380 CrossRef CAS.
- A. H. E. Müller, Y. Cai, M. Hartenstein, M. Gradzielski, M. Zhang, H. Mori and D. V. Pergushov, Polym. Prepr., 2004, 45(2), 267 Search PubMed.
- D. Neugebauer, Polymer, 2007, 48(17), 4966 CrossRef CAS.
- G. Wu, S. C. Chen, Q. Zhan and Y. Z. Wang, Macromolecules, 2011, 44(4), 999 CrossRef CAS.
- F. P. Wang, X. Z. Du, H. P. Mu, D. X. Zhang and Y. J. Ma, Adv. Mater. Res., 2011, 233–235, 2138 CAS.
- Zh. Cheng, X. Zhu, E. T. Kang and K. G. Neoh, Langmuir, 2005, 21(16), 7180 CrossRef CAS.
- J. Zhao, G. Mountrichas, G. Zhang and S. Pispas, Macromolecules, 2010, 43(4), 1771 CrossRef CAS.
- W. Lu, M. Chen and L. M. Wu, J. Colloid Interface Sci., 2008, 328, 98 CrossRef CAS.
- J. N. Zhang, M. Y. Wu, Q. Y. Wu, J. J. Yang, N. N. Liu and Z. L. Jin, Chem. Lett., 2010, 39(3), 206 CrossRef CAS.
- C. L. Zhang, B. Liu, C. Tang, J. G. Liu, X. Z. Qu, J. L. Li and Z. Z. Yang, Chem. Commun., 2010, 46(1), 4610 RSC.
- M. Teo, S. K. Suh, T. A. Hatton, Ma. Ashokkumar and F. Grieser, Langmuir, 2011, 27(1), 30 CrossRef.
- L. Zhang, F. Zhang, W. F. Dong, J. F. Song, Q. S. Huob and H. B. Sun, Chem. Commun., 2011, 47(4), 1225 RSC.
- M. M. Rahman, F. Montagne, H. Fessi and A. Elaissari, Soft Matter, 2011, 7, 1483 RSC.
- N. B. Bowden, M. Dankova, W. Wiyatno, C. J. Hawker and R. M. Waymouth, Macromolecules, 2002, 35(25), 9246 CrossRef CAS.
- A. Walther, M. Hoffmann and A. H. E. Muller, Angew. Chem., Int. Ed., 2008, 47(4), 711 CrossRef CAS.
- J. Y. Yuan, Y. Y. Xu, A. Walther, S. Bolisetty, M. Schumacher, H. Schmalz, M. Ballauff and A. H. E. Muller, Nat. Mater., 2008, 7, 718 CrossRef CAS.
- R. Erhardt, A. Böker, H. Zettl, H. Kaya, W. Pyckhout-Hintzen, G. Krausch, V. Abetz and A. H. E. Muller, Macromolecules, 2001, 34(4), 1069 CrossRef CAS.
- A. Adedeji, S. Lyu and C. W. Macosko, Macromolecules, 2001, 34(25), 8663 CrossRef CAS.
- G. He, V. V. Ginzburg and A. C. Balazs, J. Polym. Sci., Part B: Polym. Phys., 2006, 44(17), 2389 CrossRef CAS.
- V. V. Ginzburg, Macromolecules, 2005, 38(6), 2362 CrossRef CAS.
- J. J. Chiu, B. J. Kim, G.-R. Yi, J. Bang, E. J. Kramer and D. J. Pine, Macromolecules, 2007, 40(9), 3361 CrossRef CAS.
- B. J. Kim, J. J. Chiu, G.-R. Yi, D. J. Pine and E. J. Kramer, Adv. Mater., 2005, 17(21), 2618 CrossRef CAS.
- L. Zeyu, B. Haoab and Y. Zhang, RSC Adv., 2015, 5, 40668 RSC.
- Y. Abe and G. Takahiro, Prog. Polym. Sci., 2004, 29(3), 149 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- J. N. Israelachvili, Intermolecular and Surface Forces, Elsevier, Amsterdam, 2011 Search PubMed.
- C. Fong, T. Le and C. J. Drummond, Chem. Soc. Rev., 2012, 41, 1297 RSC.
- M. L. Adams, A. Lavasanifar and G. S. Kwon, J. Pharm. Sci., 2003, 92, 1343 CrossRef CAS.
- A. Halperin and S. Alexander, Macromolecules, 1989, 22, 2403 CrossRef CAS.
- I. M. Davletbaeva, A. I. Akhmetshina, R. S. Davletbaev, I. I. Zaripov, A. M. Gumerov and R. R. Sharifullin, Polym. Sci., Ser. B, 2014, 56(6), 781 CrossRef.
- R. Davletbaev, A. Akhmetshina, A. Gumerov, I. Davletbaeva and V. Parfenov, Composite Interfaces Special Issue: The Fourth Asia Symposium on Advanced Materials (ASAM-4), 2014, vol. 7, p.611 Search PubMed.
- I. M. Davletbaeva, V. F. Shkodich, A. M. Gumerov, O. Yu. Emelina and A. V. Naumov, Polym. Sci., Ser. A, 2010, 52(4), 392 CrossRef.
- A. I. Akhmetshina, R. S. Davletbaev, I. M. Davletbaeva, A. V. Mikhailova, A. M. Gumerov and R. Ya. Deberdeev, Russ. J. Appl. Chem., 2015, 88(3), 494 CrossRef CAS.
- I. M. Davletbaeva, G. R. Nurgaliyeva, A. I. Akhmetshina, R. S. Davletbaev, A. A. Atlaskin, T. S. Sazanova, S. V. Efimov, V. V. Klochkov and I. V. Vorotyntsev, RSC Adv., 2016, 6, 111109 RSC.
- I. M. Davletbaeva, A. I. Mazil’nikov, I. I. Zaripov, R. S. Davletbaev, A. M. Gumerov and V. V. Parfenov, Polym. Sci., Ser. B, 2018, 60(1), 51 CrossRef CAS.
- I. M. Davletbaeva, I. I. Zaripov, A. I. Mazilnikov, R. S. Davletbaev, R. R. Sharifullin, A. A. Atlaskin, T. S. Sazanova and I. V. Vorotyntsev, Membranes, 2019, 9(3), 42 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |