
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiowaste-derived 3D honeycomb-like N and S dual-doped hierarchically porous carbons for high-efficient CO2 capture†
Weiwei Shia,
Rongzhen Wanga,
Huili Liua,
Binbin Chang *a,
Baocheng Yang*a and
Zuling Zhangb
*a,
Baocheng Yang*a and
Zuling Zhangb
aHenan Key Laboratory of Nanocomposites and Applications, Institute of Nanostructured Functional Materials, Huanghe Science and Technology College, Zhengzhou, Henan 450006, China. E-mail: binbinchang@infm.hhstu.edu.cn; baochengyang@infm.hhstu.edu.cn
bHenan Provincial Chemi-Industries Research Station Co., Ltd, Zhengzhou 450000, China
First published on 26th July 2019
Abstract
Considering the characteristics of abundant narrow micropores of <1 nm, appropriate proportion of mesopores/macropores and suitable surface functionalization for a highly-efficient carbon-based CO2 adsorbent, we proposed a facile and cost-effective strategy to prepare N and S dual-doped carbons with well-interconnected hierarchical pores. Benefiting from the unique structural features, the resultant optimal material showed a prominent CO2 uptake of up to 7.76 and 5.19 mmol g−1 at 273 and 298 K under 1 bar, and importantly, a superb CO2 uptake of 1.51 mmol g−1 at 298 K and 0.15 bar was achieved, which was greatly significant for CO2 capture from the post-combustion flue gases in practical application. A systematic study demonstrated that the synergetic effect of ultramicroporosity and surface functionalization determined the CO2 capture properties of porous carbons, and the synergistic influence mechanism of nitrogen/sulfur dual-doping on CO2 capture performance was also investigated in detail. Importantly, such as-prepared carbon-based CO2 adsorbents also showed an outstanding recyclability and CO2/N2 selectivity. In view of cost-effective fabrication, the excellent adsorption capacity, high selectivity and simple regeneration, our developed strategy was valid and convenient to design a novel and highly-efficient carbonaceous adsorbent for large-scale CO2 capture and separation from post-combustion flue gases.
Introduction
Currently, considering the continuous combustion of fossil fuels to meet the growing energy demand of the world, the ever-increasing CO2 emission has resulted in the quick rise of the atmospheric CO2 concentration. Abnormal environmental problems have occurred in response to the massive CO2 emission, such as global warming and its environmental effects, which include continuous rising sea levels, ocean acidification, increasing numbers of ocean storms and floods.1–3 However, CO2 is known as an important carbon source with wide applications in industry and daily life. In order to mitigate CO2 emission, CO2 capture and sequestration technology is regard as one of the promising strategies for the reduction of CO2 concentration and effective recycling of CO2.4–6 According to the relevant statistical data, power plants are one of the largest contributors for increasing atmospheric CO2 concentration, which discharge about 60% of the global CO2 emissions.7 Thus, it becomes an urgent need to capture and separate CO2 from post-combustion flue gases. Although CO2 adsorption from flue gases by liquid amine and ammonia solvents is an efficient strategy with satisfactory selectivity, this adsorption process suffers several tricky problems, which are high energy consumption, equipment corrosion, amine degradation, toxicity, and more severely the need for solvent regeneration.8,9In contrast, adsorption using porous solid materials as adsorbents for CO2 capture has been recognized as the most promising alternative technology owing to its low energy consumption, low cost, relatively high efficiency and easy handling.10–12 The key of this technology is to develop new porous solid sorbents with superior properties for CO2 capture. To this end, a number of porous solid materials, including zeolites, metal–organic frameworks, porous organic polymers, organic–inorganic hybrid adsorbents and porous carbon materials, have been extensively investigated.13–18 Among various types of adsorbent materials, porous carbons, as a special family of highly promising materials, have been extensively studied for high-efficiency CO2 capture by virtue of their unique structural advantages, including high accessible surface area, low-cost preparation, facile regeneration, easy-to-design surface functionality and porosity, inertness to both bases and acids and moderate heat of adsorption. Nevertheless, most traditional activated carbons exhibit a relatively low CO2 adsorption capacity of typically ca. 2–3 mmol g−1 under room temperature at 1 bar.19,20 In terms of CO2 capture by porous carbons, it has been widely reported that CO2 capture capacity at ambient pressure greatly depends on the proportion of narrow microporosity of smaller than 1 nm, and especially the ultramicroporosity of <0.7 nm, which plays a crucial role in determining CO2 uptakes of porous carbons.21–23 Thus, many efforts have been made to design microporous carbons with a large proportion of narrow micropores of <1 nm, especially to further tailor micropores to produce a more favorable ultramicroporous carbon for CO2 capture. For example, Sevilla et al. prepared microporous biomass-based carbon materials via KOH chemical activation of hydrothermal carbons derived from mixtures of algae and glucose, which possessed a large number of narrow micropores (<1 nm) and exhibited a superior CO2 adsorption capacity of 4.8 mmol g−1 at 298 K and 1 bar.24 Our group synthesized cross-linked microporous carbon beads by air-assisted activated method using glucose-derived carbon microspheres as precursor, which developed a large proportion of ultramicropores with primary pore size of 0.5–0.9 nm and showed a satisfactory CO2 uptake of 4.25 mmol g−1 at 298 K and 1 bar.25 Another valid strategy to further improve CO2 uptake of porous carbons is the incorporation of heteroatoms into carbon skeleton.26–28 Typically, nitrogen doping is the most attractive, which can provide more basic sites for enhanced interactions with acidic CO2 molecule. Specifically, it has been testified that the pyrrolic and amine nitrogen functionalities have the strongest interactions with CO2 molecules.29
In addition, it has been proposed that the enhancement of CO2 uptake on porous carbons can be related to the improvement in polar interaction and hydrogen bonding interaction.30,31 Since hydrogen bonding or polar interactions of CO2 within the carbon pore can more originate from other functional groups than nitrogen-based groups. Recently, CO2 capture over sulfur-doped porous carbons has attracted much attention. For sulfur-doped porous carbons, the lone pair of electrons in a sulfur atom induces polarizability and interactions with oxygen.32 Xia et al. reported an improved CO2 capture capacity of 2.4 mmol g−1 at 298 K and 1 bar on sulfur-doped porous carbon.33 Seema et al. obtained a reduced-graphene oxide/polythiophene complex via chemical activation route, which exhibited an excellent CO2 uptake of 4.5 mmol g−1 at 298 K and 1 bar.34 Bandosz et al. reported the chemical interactions between sulfur doped carbons and CO2.35 These investigations manifested sulfur-containing functional groups in carbon adsorbents could facilitate CO2 adsorption due to acid interactions of CO2 with neutral sulfur, polar interactions of CO2 with oxidized sulfur, and hydrogen bonding of CO2 with sulfonic acids. However, there are relatively few studies about the synergistic effect of nitrogen and sulfur dual-doping on hierarchically porous carbons for CO2 capture.36,37 Thus, it is significant for high-effective CO2 capture to design a novel porous carbon adsorbent comprising multiscale pores including abundant narrow micropores of <1 nm (which favor high CO2 uptake), appropriate proportion of mesopores/macropores (which facilitate efficient CO2 diffusion into and out of the adsorption sites) coupled with suitable surface nitrogen and sulfur functionalities (which further improve CO2 uptake).
Considering the characteristics described above, herein, nitrogen and sulfur dual-doped porous carbons with well-interconnected hierarchical pores were synthesized by a facile and cost-efficient strategy using bio-waste as carbon source and thiourea as nitriding and sulfurizing agent. Innovatively, the synergetic effect of ultramicropores (<0.7 nm) and nitrogen/sulfur dual-doping on CO2 capture was investigated in detail. Gratifyingly, the resultant optimal material exhibited a superior CO2 capture capacity of up to 7.76 and 5.19 mmol g−1 at 273 and 298 K under 1 bar, respectively. More importantly, such a CO2 adsorbent also showed an outstanding recyclability and CO2/N2 selectivity adsorption property. Hence, such results suggested that we proposed a valid strategy to exploit novel porous carbon sorbents for the removal of CO2 from post-combustion exhaust gases.
Experimental section
Material preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2:1 and 3:1, respectively). For comparison, 1 g of the puce product was directly carbonized at 800 °C for 1 h without chemical activating agent to obtain hierarchically porous carbon, which was defined as HPC.
:1, 2:1 and 3:1, respectively). For comparison, 1 g of the puce product was directly carbonized at 800 °C for 1 h without chemical activating agent to obtain hierarchically porous carbon, which was defined as HPC.Characterizations
X-ray diffraction (XRD) patterns were taken on a Bruker D8 diffractometer with Cu Kα radiation (λ = 0.15418 nm). Nitrogen adsorption–desorption isotherms were acquired at 77 K using a Micromeritics ASAP 2020HD88 system. Before adsorption, the samples were out-gassed at 200 °C for 10 h. The specific surface area (SBET) was evaluated according to the Brunauer–Emmett–Teller (BET) method at relative pressure of 0.05–0.25, and the pore size distribution was calculated according to the Density-Functional-Theory (DFT) method, and the micropores were analyzed using t-plot method, and the ultramicropores were analyzed by CO2 adsorption at 273 K using DFT model. The morphology was imaged on a Quanta 250 FEG scanning electron microscope (SEM). Fourier transform infrared spectroscopy (FTIR) was performed on a Nicolet Avatar 370 spectrometer. The samples for the FTIR measurements were mixed together with KBr, following the standard method. X-ray photoelectron spectra (XPS) were obtained on a VG ESCALAB MK II spectrometer with an exciting source of Mg Kα at 1253.6 eV.Results and discussion
Materials characterization
Scheme 1 illustrates the schematic diagram for the fabrication of waste paper-derived N and S dual-doped hierarchically porous carbons via a convenient strategy combining chemical activation and post-modification methods. The waste paper towels were collected from a cardboard box in large quantity, and then thoroughly washed, cleaned and dried at 60 °C. These dried waste paper towels were cut into pieces to be used as carbon source, and then were hydrothermal treatment at 200 °C for 4 h to obtain puce powders as precursor. These hydrothermally synthesized precursors were impregnated with activating agent solution for 4 h and then evaporated the solvent. Importantly, the resultant precursors not only contained abundant organic components, but also possessed an excellent adsorptive capacity to accommodate a large amount of activating agent, which favored generating well-developed porous structure, especially micropores. Followed, the dried activating-agent-impregnated precursors were carbonized and activated at 800 °C for 1 h in a nitrogen atmosphere, and washed by hydrochloric acid and water and dried to obtain hierarchically porous carbons. Subsequently, the resultant hierarchically porous carbons were further functionalized at 800 °C in a nitrogen atmosphere to prepare N and S dual-doped hierarchically porous carbons using thiourea as nitriding and sulfurizing agent. | ||
| Scheme 1 Schematic illustrating the preparation of the waste paper-derived N and S dual-doped hierarchically porous carbons. | ||
To reveal the effect of carbonization and activation processes on the microcrystalline texture, the XRD patterns of all the resultant materials were shown in Fig. 1a. Two weak and broad peaks at approximately 22.1° and 43.6° can be observed in all samples, which are attributed to the (002) and (100) reflections of turbostratic carbon structure, suggesting an amorphous carbon texture with a low crystallinity. The diffraction intensity of the (002) and (100) peak in activated materials is lower than those in HPC under the same conditions of detection, resulting from the chemical activation to break down the hexagonal symmetry of the graphite lattice and lead to lattice defects in HPCK-1 and HPCZn-1 samples.38 In addition, the high intensity in the low angle region indicates the existence of abundant micropores in all samples.39 The evolution of the chemical compositions of the waste paper precursor, activated samples and N and S dual-doped samples were characterized by FTIR (Fig. 1b). For hydrothermally synthesized precursor, it was similar to the chemical composition of the other plant fibers,40 the main component of waste paper towel was lignocelluloses. The broad beak at about 3380 cm−1 was assigned to O–H stretching vibration. The band at 2900 cm−1 was attributed to the C–H stretching vibrations in methyl and methylene groups, and the bands at 1425 and 1370 cm−1 were ascribed to aromatic skeletal vibrations combined the C–H deformation vibrations, while the band at 1320 cm−1 was due to the CH2 rocking vibration. The bands at 1660 and 1635 cm−1 were ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration and CC stretching vibration, respectively. The band at 1160 cm−1 was related to C–O–C asymmetric valence vibration, and the band at 1113 cm−1 was due to the C–C stretching or asymmetric in-phase ring stretching. The bands at 1060 and 1032 cm−1 were assigned to C–O valence vibration and C–O ether vibration, respectively. While, after carbonization and chemical activation, it was obvious that the bands in the resultant materials became broader and weaker, this could be related to the strong absorption of the carbon skeleton. Only the CC stretching vibration could be found in other activated samples, and the other characteristic absorption bands became weaker and even disappeared. Such results should be resulted from the carbonization and chemical activation, resulting in the elimination of surface functional groups.
O stretching vibration and CC stretching vibration, respectively. The band at 1160 cm−1 was related to C–O–C asymmetric valence vibration, and the band at 1113 cm−1 was due to the C–C stretching or asymmetric in-phase ring stretching. The bands at 1060 and 1032 cm−1 were assigned to C–O valence vibration and C–O ether vibration, respectively. While, after carbonization and chemical activation, it was obvious that the bands in the resultant materials became broader and weaker, this could be related to the strong absorption of the carbon skeleton. Only the CC stretching vibration could be found in other activated samples, and the other characteristic absorption bands became weaker and even disappeared. Such results should be resulted from the carbonization and chemical activation, resulting in the elimination of surface functional groups.
 | ||
| Fig. 1 The XRD patterns (a) and FTIR spectra (b) of the resultant materials. | ||
Scanning electron microscopy was conducted to reveal the evaluation of microstructure and morphology from paper towel to activated carbons and N, S dual-doped carbons material. Paper towel showed a long fiber structure with a diameter of range from ∼5 to ∼20 μm (Fig. S1a†), and the hydrothermally synthesized precursors completely inherited the original fiber morphology of paper towel (Fig. S1b†). As displayed in Fig. 2a, the fibrous texture could be obviously observed in HPC sample, which indicated the original structure could be retained during the high-temperature carbonization treatment. After KOH activation, HPCK-1and HPCK-2 presented a three-dimensional interconnected honeycomb-like microstructure with numerous irregular holes (Fig. 2b and c), which was a unique structure that could effectively prevent the small and thin blocks from agglomerating on a large scale. Such prominent honeycomb-like porous structure was attributed to the KOH chemical activation and the followed CO/CO2 physical activation in situ induced from the reactions of KOH and C.41 With an increase in the addition amount of KOH, the density of pores presented a developing trend. Whereas, as the mass ratio of KOH/precursor increased to 3, a harsh activation reaction would occur in carbon skeleton, which brought the pore widening and the degradation of 3D honeycomb-like structure. As a result, carbon sheets was too thin to support the honeycomb-like structure, resulting in the collapse and restacking of partial sections to form the block with thick wall (Fig. 2d). Different from the morphology of HPCK-x samples, after ZnCl2 activation, all the resultant HPCZn-x samples exhibited a porous structure with some macropores (Fig. 2e–g). Such results could be ascribed to the activation mechanism of ZnCl2 chemical activation, and ZnCl2 often serves as a dehydrating agent to chemically activate carbonaceous precursors. Similarly, with the increasing of ZnCl2 dosage, the density of generated pores was decreased, which could be related to pore coalescence. After further nitrogen and sulfur dual-doped, the N,S-HPCK-1 retained the original honeycomb-like porous structure of HPCK-1 sample (Fig. 2h), and N,S-HPCZn-1 also kept the initial porous structure of HPCZn-1 (Fig. 2i), which testified that the further functionalized process at high temperature could not destroy the intrinsic morphology.
 | ||
| Fig. 2 SEM images. (a) HPC; (b) HPCK-1; (c) HPCK-2; (d) HPCK-3; (e) HPCZn-1; (f) HPCZn-2; (g) HPCZn-3; (h) N,S-HPCK-1; (i) N,S-HPCZn-1. | ||
The porous structure features of the resultant carbonaceous sorbents were analyzed by N2 adsorption–desorption at 77 K. In Fig. S2,† it can be clearly found that the hydrothermally synthesized precursors possess an only 1.79 m2 g−1 of BET total surface area and a 0.006 cm3 g−1 of pore volume, which indicated that such carbonaceous precursors obtained by sole hydrothermal treatment exhibited extremely poor porosity. After directly carbonized precursors at 800 °C, the HPC material depicted a typical type I curve (Fig. 3a), meaning a microporous characteristic pore structure, and these micropores could mainly originate from the decomposition of volatile matter and the elimination of surface O- and H-groups during the carbonization process. But, the relatively small N2 adsorbed amount manifested the low total surface area and pore volume (Table 1). With the KOH chemical activation, the HPCK-x still presented a type I isotherm of microporous structure, and the sharply increased and highly adsorbed quantity at a low relative pressure (P/P0 < 0.01) owed to the capillary filling of micropores, which testified the pore structure of HPCK-x mainly consisted of micropores. Comparatively, an abrupt increase of the isotherm occurred at the higher relative pressure (P/P0 > 0.9) for HPCK-2 and HPCK-3 samples, which was ascribed to the capillary condensation, implying that a larger pore size was generated when more KOH was employed as activating agent. Fig. 3b showed the pore size distribution of HPC and HPCK-x samples. HPC owned a micropore size distribution of 1.17/1.86 nm, and the proportion of 1.17 nm micropores was larger. Apparently, with the use of different dosage of KOH activating agent, the pore size gradually extended from bi-modal distribution to multi-modal distribution, and larger pore size and proportion stepwise enlarged with the enhancement of KOH dosage. The pore size distribution of HPCK-1 was 0.47–0.86 and 1.27 nm, and the overall pore sizes were still in the micropore range. The majority of the pore sizes centered at 0.47–0.86 nm, while the minority of the pore sizes was 1.27 nm. With the increase of KOH activating agent concentration, some new and larger pore size distributions of 1.61/2.02 nm and 2.10 nm were produced in HPCK-2 and HPCK-3 sample, respectively. More noteworthy, the majority of pore sizes distributed in 1.61 and 2.10 nm, and the minority of pore sizes was in micropores of 0.47–1.27 nm. When used ZnCl2 as activating agent, the HPCZn-1 sample exhibited a similar isotherm with HPCK-x, meaning a characteristic micropore structure of HPCZn-1. Whereas, HPCZn-2 and HPCZn-3 presented a transitional isotherm from type I to type IV (Fig. 3c), and even a marked H3 type hysteresis loop in the relative pressure region between 0.4 and 0.7 could be clearly observed in HPCZn-3. Such results manifested the coexistence of micropores and slit-shaped mesopores. The pore size distributions of HPCZn-x samples were shown in Fig. 3d. The pore size of HPCZn-1 centered at 0.68 and 1.27 nm, and overall pore size were in micropore, and the pore proportion was almost isometric. With the increase of ZnCl2 dosage, some new mesopores of 2.34 and 3.07 nm were generated in HPCZn-2 and HPCZn-3 samples, respectively, and these mesopores were dominant in porosity. By comparing the pore size distributions of HPCK-x and HPCZn-x samples, we could speculate such conclusions: (i) the hierarchical pore size distribution significantly depend on the activation of activating agents; (ii) KOH activating agent exhibits a more effective impact on tailoring multi-modal pore size distribution, especially in micropores; (iii) ZnCl2 activating agent plays a high-effective role in developing mesopores. In addition, the BET surface area and other textural properties of all the resultant materials are summarized in Table 1. It can be clearly found that the activating agents have a vital influence on developing porosity of materials. With the increase of activating agent dosage, both surface area and pore volume were greatly improved owing to the further etch of activating agent on carbonaceous skeleton. However, as the activating agent dosage further increase to 3, because of the excessive activation, numerous micropores coalesced to form a mesopore, which resulted in the continuous enhancement of mesoporous surface area and mesopore volume and decrease of total surface area and micropore volume. Therefore, varying the dosages of the activating agent is an effective strategy to engineer the proportion of mesopore/micropore in porosity. Moreover, after N and S dual-doping, the specific surface area and pore volume changed slightly, which testified that surface functionalization process could not destroy the original porosity of materials.
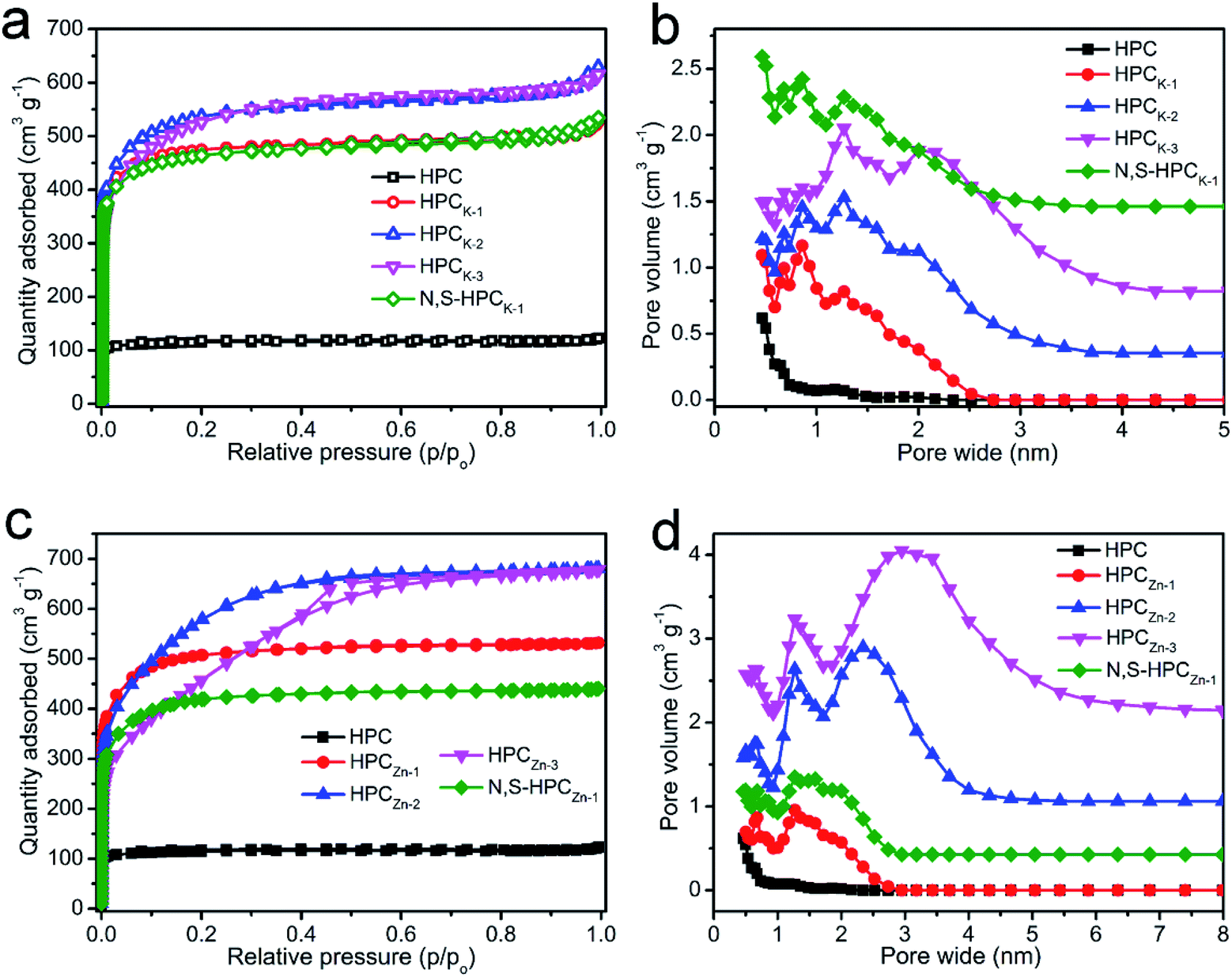 | ||
| Fig. 3 N2 adsorption–desorption isotherms (a and b) and pore size distributions (c and d) of all the resultant materials. | ||
| Sample | SBETa (m2 g−1) | Smicrob (m2 g−1) | Smesoc (m2 g−1) | Sultramicrod (m2 g−1) | Vtotale (cm3 g−1) | Vmicrof (cm3 g−1) | Vultramicrog (cm3 g−1) |
|---|---|---|---|---|---|---|---|
| a BET surface area.b Micropore surface area calculated using the V–t plot method.c Smeso = SBET − Smicro.d The cumulative ultramicropore <0.7 nm surface area measured by CO2 adsorption at 273 K using DFT model.e The total pore volume calculated by single point adsorption at P/P0 = 0.9945.f The micropore volume calculated using the V–t plot method.g The cumulative ultramicropore <0.7 nm volume measured by CO2 adsorption at 273 K using DFT model. | |||||||
| HPC | 453.4 | 431.9 | 21.5 | 294.9 | 0.19 | 0.17 | 0.078 |
| HPCK-1 | 1825.1 | 1698.7 | 126.4 | 499.9 | 0.81 | 0.68 | 0.122 |
| HPCK-2 | 2016.2 | 1871.5 | 144.7 | 337.8 | 0.97 | 0.78 | 0.081 |
| HPCK-3 | 1916.2 | 1676.7 | 239.5 | 237.7 | 0.95 | 0.73 | 0.071 |
| HPCZn-1 | 1564.8 | 1488.4 | 76.4 | 202.1 | 0.68 | 0.62 | 0.058 |
| HPCZn-2 | 2040.4 | 1572.8 | 467.6 | 156.7 | 1.05 | 0.73 | 0.050 |
| HPCZn-3 | 1664.5 | 494.2 | 1170.3 | 143.9 | 1.08 | 0.23 | 0.027 |
| N,S-HPCK-1 | 1770.7 | 1668.5 | 102.2 | 431.2 | 0.83 | 0.67 | 0.098 |
| N,S-HPCZn-1 | 1262.1 | 1189.5 | 72.6 | 189.3 | 0.63 | 0.57 | 0.052 |
CO2 capture property
The CO2 adsorption performance of the resultant HPC, HPCK-x and HPCZn-x samples were investigated at two representative temperatures of 273 and 298 K under the ambient pressure, and the CO2 adsorption isotherms are depicted in Fig. 4a and b. At the atmospheric pressure (1 bar), all the samples exhibit a higher CO2 uptake at 273 K than that of at 298 K, which should be related to the exothermic feature of CO2 adsorption.42 HPC sample exhibits a CO2 uptake of 3.12 mmol g−1 at 273 K and 2.35 mmol g−1 at 298 K under 1 bar. After activated by KOH and ZnCl2, all HPCK-x and HPCZn-x (except HPCZn-3) samples present the improved CO2 uptakes in the ranges of 3.82–7.26 mmol g−1 and 2.69–4.68 mmol g−1 at 273 K and 298 K under 1 bar, respectively. Obviously, HPCK-1 exhibits the highest CO2 capture capacity of 7.26 and 4.68 mmol g−1 at 273 and 298 K in KOH-activated system, respectively. However, the further increase in activating agent dosage of KOH brings a negative effect on CO2 adsorption capacity, and the declined CO2 uptake values of 6.84 (6.25) and 4.25 (3.76) mmol g−1 are obtained over HPCK-2 (HPCK-3) at 273 and 298 K, respectively. The similar variation tendency of CO2 uptake can be found over HPCZn-x samples in ZnCl2-activated system. Noticeably, HPCK-x samples exhibit the better CO2 capture performance than HPCZn-x samples, which should be benefited from the more prominent porosity of HPCK-x, especially the higher microporosity and ultra-microporosity. In addition, although HPC sample presents a low CO2 uptake at the atmospheric pressure, and it shows a more superior CO2 capture property than those of the other resultant samples below 0.15 bar at 273 K but 0.3 bar at 298 K (Fig. S3†), which suggest that CO2 capture capacity is also related to the adsorption conditions. Under high adsorption temperature and low pressure, small micropores contribute more to CO2 capture. But, with declining adsorption temperature and increasing adsorption pressure, CO2 capture is more dependent on large micropores and even mesopores. Such result is ascribed to the developed multiscale micropore of <1 nm size distribution, especially its more prominently hierarchical ultramicropore size distribution (Fig. S4,† CO2 micropore size of <1 nm distribution), and such behavior is closely related to the pore filling mechanism of CO2 adsorption.21 Furthermore, though HPCK-2 possesses the higher total surface area, micropore surface area and micropore volume than those of HPCK-1, HPCK-2 sample presents a lower CO2 uptake of 6.84 and 4.25 mmol g−1 at 273 and 298 K under 1 bar than that of HPCK-1, and the similar result can be found over HPCZn-2 and HPCZn-1 samples. Such behavior should be attributed to their declined proportion of ultramicropores, giving rise to the decreased ultramicropore surface area and ultramicropore volume. This result testifies that the efficient CO2 capture of porous carbons does not only depend on the micropore surface area/pore volume, but also more depend on ultramicropore surface area/volume, as ultramicropores are more beneficial to the CO2 molecule filling.21,43 Thus, the inferior CO2 capture property of HPCZn-2 and HPCZn-3 should also be due to the over-activated in micropore structure, which results in forming a larger proportion of mesopores but a poorer ultramicroporosity. After nitrogen and sulfur dual-doped, the obtained N,S-HPCK-1 and N,S-HPCZn-1 samples both exhibit an enhanced CO2 capture performance at 273 K and 298 K (Fig. 4c and d), manifesting that the nitrogen and sulfur functionalization of carbon skeleton brings an effective improvement for the CO2 capture, which should be benefit from the increasing number of CO2 adsorption sites on the pore surface. Especially, N,S-HPCK-1 sample displays an excellent CO2 uptake of 7.76 and 5.19 mmol g−1 at 273 and 298 K under 1 bar, respectively, which is much higher than other carbon-based sorbents and even some of the reported nitrogen-doped porous carbons and hierarchical porous carbons (Table S1†). Of particular importance is that the hierarchically porous carbons prepared in this work using cost-free biomass waste as carbon precursor, exhibits a comparable CO2 uptake to those of N-doped carbonaceous adsorbents that are considered as good CO2 adsorbents.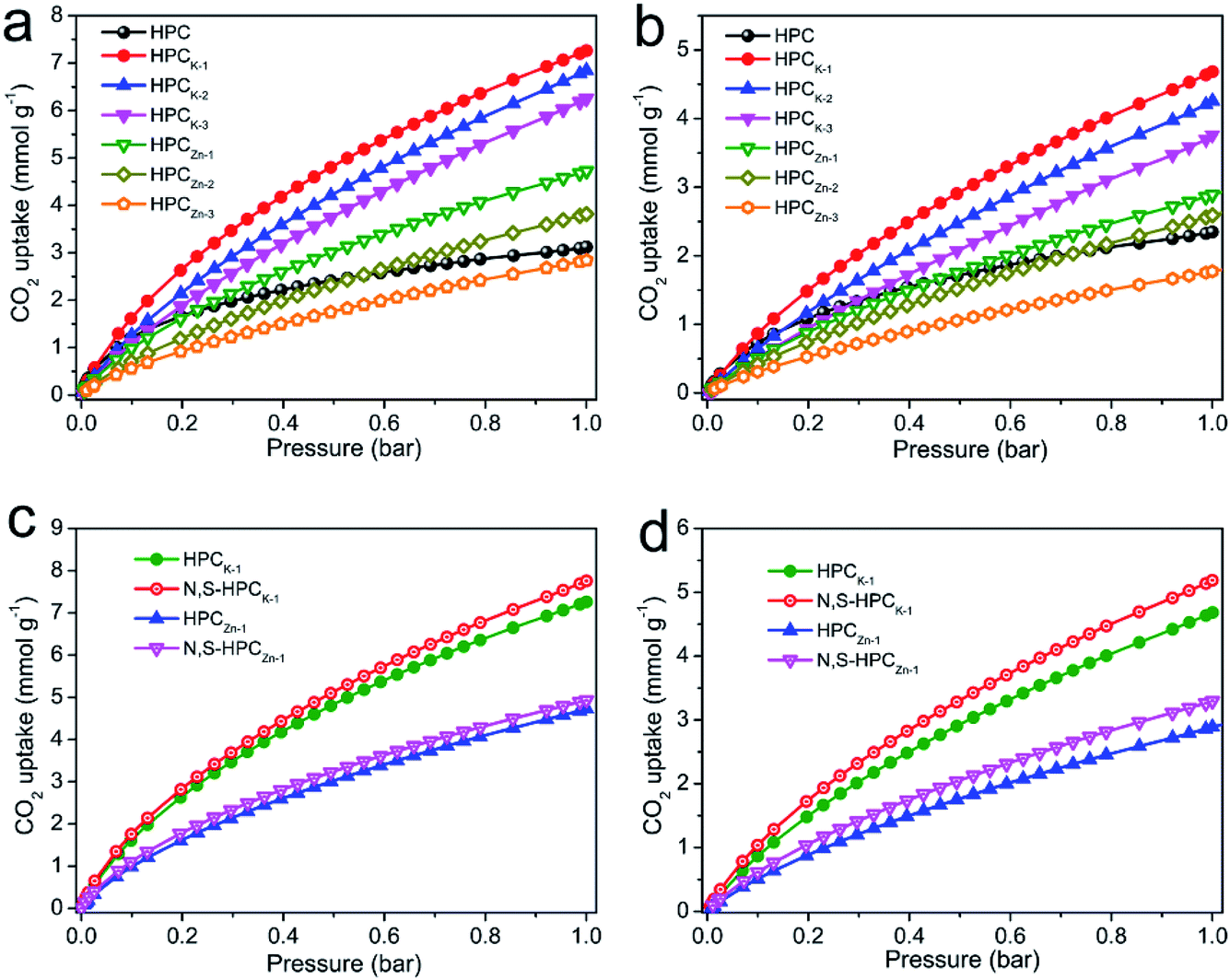 | ||
| Fig. 4 CO2 adsorption isotherms of all the prepared materials. (a) at 273 K; (b) 298 K; (c) the comparison of HPCK-1, HPCZn-1, N,S-HPCK-1 and N,S-HPCZn-1 at 273 K; (d) the comparison of HPCK-1, HPCZn-1, N,S-HPCK-1 and N,S-HPCZn-1 at 298 K. | ||
As is well known, the post-combustion flue gas streams produced in industrial processes (e.g., fossil fuel-fired power stations) contain a large proportion of CO2. The partial pressure of CO2 in the flue gas streams is typically around 0.15 bar, and it is still a challenge to capture CO2 from post-combustion flue gases. In order to examine the possibility of the resultant samples to act as the CO2 adsorbents for the flue gas streams, we also list the adsorbed CO2 amounts at 298 K under 0.15 bar in Fig. 5. The CO2 uptakes at 0.15 bar vary with the amount of the activating agent. The variation trend at 0.15 bar is slightly different from that at 1 bar. It can draw a conclusion that the CO2 capture amount more rely on the proportion of ultramicropores and pore size distribution of these ultramicropores, and thus the high CO2 uptake amount of 1.29 at 298 K at 0.15 bar for HPCK-1 arises from its prominent ultramicroporosity. More importantly, after modification with nitrogen and sulfur, N,S-HPCK-1 sample exhibits a more superior CO2 capture capacity of up to 1.51 mmol g−1 at 0.15 bar at 298 K, greatly surpassing the uptakes of previously reported carbon-based CO2 adsorbents (Table S1†). Consequently, the currently designed nitrogen and sulfur dual-doped hierarchically porous carbons have excellent potential for capturing CO2 from the post-combustion flue gases.
 | ||
| Fig. 5 The CO2 uptakes of all materials at 298 K under 0.15 bar and the correlation with ultramicropore volume. | ||
Effect of hierarchical porosity on CO2 capture capacity
To better understand the CO2 uptake behaviors of the resultant samples, we further analyzed the effect of the hierarchical porosity and structure on the CO2 adsorption capacity. A comparison of the porosity characteristics and the CO2 uptake amounts shows that the CO2 uptake amount of the resultant samples is not directly determined by the BET total surface area and total pore volume (shown in Fig. 6a and b). | ||
| Fig. 6 (a) Dependence of the CO2 uptake amount at 273 K and 1 bar on the total and microporous surface areas. (b) Dependence of the CO2 uptake amount at 273 K and 1 bar on the total pore and microporous volumes. (c) The correlation of CO2 uptake at 273 and 298 K under 0.15 bar with ultramicropore volume. (d) The cumulative ultramicropore volume measured by CO2 adsorption at 273 K using DFT model. | ||
But, the CO2 capture capacities of the resultant materials are strongly correlated with their microporosity of pore size <1 nm, especially the ultramicropore volume between 0.3 and 0.7 nm. Especially, it can be clearly found that the CO2 uptakes at 0.15 bar greatly depend on the ultramicropore volume, and the CO2 capture capacity enhances with the increase of ultramicropore volume (Fig. 6c). Such result suggests that the micro and narrow mesopores were mainly responsible for the high CO2 sorption performance of the materials at low pressure, which is in agreement with previous findings.3,44 By analyzed the data in Fig. 6d, it can be concluded that the best CO2 capture capacity of HPCK-1 should be owed to the largest ultramicropore volume. In addition, HPCZn-x samples exhibit the lower CO2 uptakes compared to HPCK-x samples although they possess the comparative BET surface area, which should be correlated with the lower ultramicropore volumes of HPCZn-x. Thus, to achieve an outstanding CO2 capture capability, it is more important to tailor the ultramicropore size than to have a high surface area for porous carbon-based CO2 adsorbents. But, the CO2 uptakes at 1 bar are also still affected by the BET total surface area, as revealed by Fig. 6a, which should be related to the important impact large micropores and even mesopores on CO2 capture capacity at high pressure region. Therefore, the large proportion of microporosity and suitable ultramicroporosity combined with the largest surface area might be an essential factor for the outstanding CO2 uptake at 273 and 298 K under ambient pressure.
Effect of surface functionalization on CO2 capture capacity
In general, surface chemical modification, especially nitrogen or sulfur doping in carbon skeleton, is another non-negligible factor for determining the CO2 capture capacity of porous carbon-based sorbents. In this work, the resultant CO2 adsorbents should consist mainly of nitrogen, sulfur and oxygen atoms on the surface of carbon, and therefore we investigated the effects of N, S and O on the CO2 capture capacity. Fig. S5† depicts the elemental mapping of C, N, S and O collected by scanning N,S-HPCK-1 porous microstructure. The homogeneous distribution of N, S and O element in N,S-HPCK-1 carbon framework indicates that the successful modification of N and S on the surface of carbon skeleton in N,S-HPCK-1 sample. To better understand the contribution of the chemical interactions towards the high CO2 sorption, X-ray photoelectron spectroscopy (XPS) analysis was conducted on the optimally performing materials to study the nature of the surface groups. Fig. 7a presents the full XPS spectrum of the N,S-HPCK-1 sample with four main peaks at binding energies of 163.6, 284.1, 398.2 and 532.5 eV, which corresponds to S, C, N and O element, respectively. The C, N, O and S atomic percentages of N,S-HPCK-1 determined by XPS are 89.95%, 4.32%, 4.57% and 1.16%, respectively.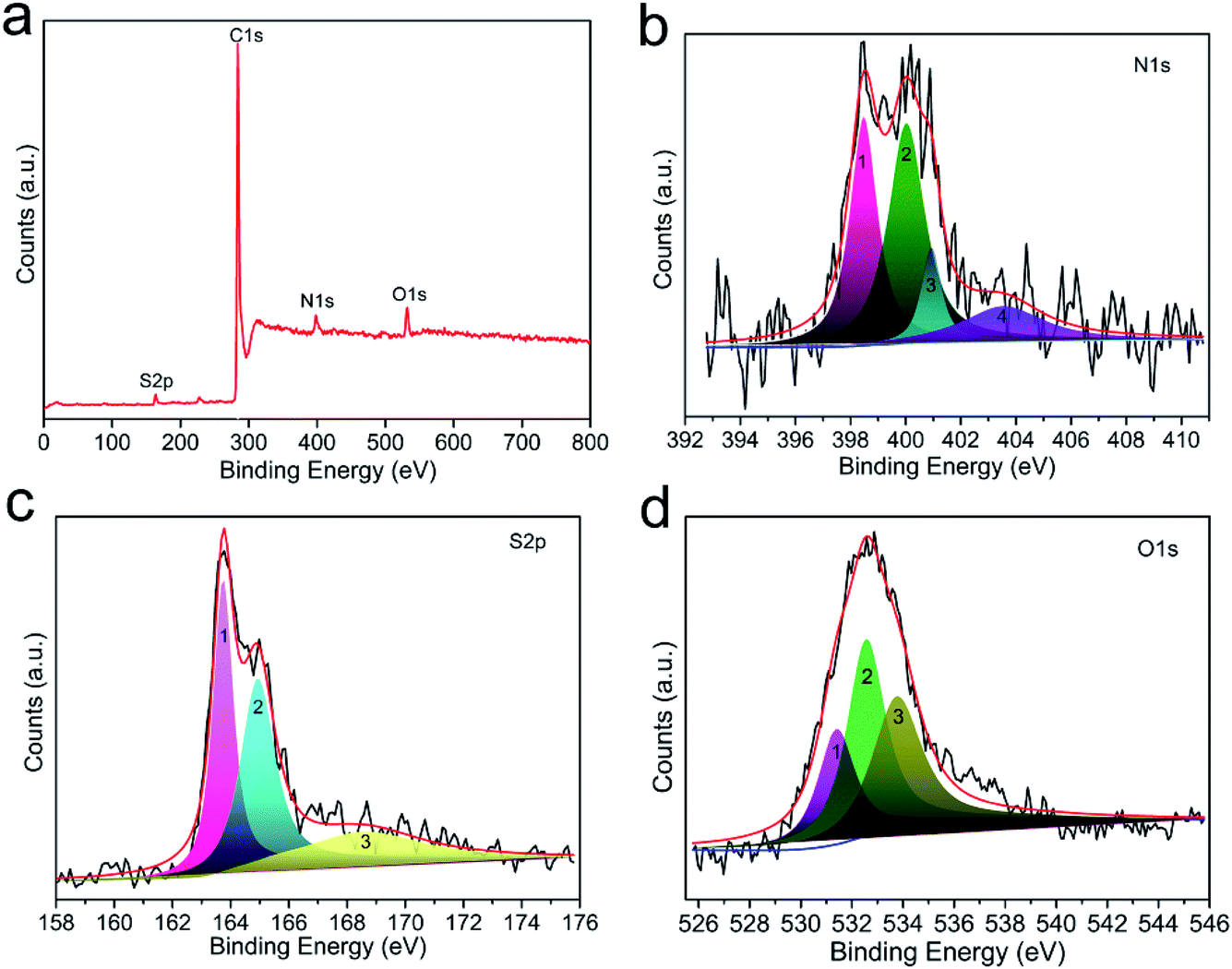 | ||
| Fig. 7 The XPS spectrum of N,S-HPCK-1. (a) The survey; (b) N 1s; (c) S 2p; (d) O 1s. | ||
N 1s core spectrum contributes to a further understanding of the types and local environment of the nitrogen atoms modified on the surface of carbon matrix. The deconvolution of N 1s spectrum reveals the bonding of N with C (Fig. 7b), and the N 1s signal can be resolved into four individual peaks centered at binding energies of 398.5, 400.1, 400.9, 403.6 eV, which are related to four different types of nitrogen functional groups, corresponding to pyridinic-N (N–H), amine (–NH2), pyrrolic/pyridone-N (–N–H) and quaternary-N (N+–H), respectively. Apparently, the pyridinic-N and amine nitrogen groups are the most stable and abundant, which has been identified the pyridinic-N or amine nitrogen groups to have stronger interactions with CO2 molecules,29,45 resulting in bringing the more contribution for CO2 capture. In addition, it was also revealed that the introduction of nitrogen into carbon matrix facilitated hydrogen-bonding interactions between the carbon surface and CO2 molecules,30 favoring the improvement of CO2 uptake.
The deconvolution of S 2p core level peak of N,S-HPCK-1 sample displays the peaks located at 163.8 and 164.9 eV corresponding to neutral S, and the peak centered at 168.5 eV assigns to oxidized S (Fig. 7c). Sulfur-containing functional groups in carbon skeleton could facilitate CO2 adsorption owing to the acid interactions of CO2 with neutral sulfur (163.8 and 164.9 eV) and polar interactions of CO2 with oxidized sulfur (168.5 eV).37 As a result, N,S-HPCK-1 exhibits an enhanced CO2 capture capacity than that of HPCK-1. Additionally, to examine the synergistic effect of N and S dual-doping on CO2 capture capacity, a conjecture is made to ascertain whether there is an optimum N/S ratio, which furthest promoting the CO2 capture capacity. As presented in Fig. S6,† unexpectedly, there is no obvious correlation between N/S ratio and CO2 uptake, which could be related with the heterogeneous distribution of nitrogen and sulfur in the surface of carbon matrix and the limited sites for the doping of nitrogen and sulfur groups.
The surface oxygen functionalities on the carbon matrix was also been revealed to contribute to the enhancement of CO2 adsorption.46 The O 1s spectrum shown in Fig. 7d exposes the bonding of N with C, which can be fitted into three deconvoluted peaks located at ca. 531.4, 532.6 and 533.8 eV, respectively. The peak at about 531.4 eV ascribes to the contribution of oxygen in carboxyl groups and the peak at ca. 532.6 eV assigns to the –CO band, and the one centered at ca. 533.8 eV should be related to the band of –C–O–C. The oxygen content detected from XPS is correlated with the CO2 capture capacity of the as-obtained materials (Fig. S7†), and there is no obvious trend between the oxygen content and the CO2 uptake. Moreover, a guess is made to ascertain whether there is an optimum N/O ratio, which favors improving the CO2 uptake. As displayed in Fig. S8,† no correlation is observed with N/O ratio, which could be ascribed to the heterogeneous distribution of oxygen in the surface of carbon framework. Similarly, such no correlation relationship between CO2 capture capacity and the oxygen content was found in porous carbon sorbents.47
CO2 isosteric heat of adsorption (Qst)
In order to reveal the combination interaction strength between the CO2 molecule and adsorbents, and evaluate the energetic heterogeneity of surfaces of adsorbents, the isosteric heat of adsorption (Qst) was calculated from the CO2 adsorption isotherms measured at 273 and 298 K based on Clausius–Clapeyron equation. As depicted in Fig. 8, the Qst values of the resultant HPCK-x and HPCZn-x adsorbents at low surface coverage are in the range of 30–35 and 18–27 kJ mol−1, respectively. It can be clearly found that the Qst gradually declines at the low CO2 uptake, and then reaches a near plateau as the continuous occupation of adsorption active sites with the increasing CO2 uptake, manifesting the heterogeneity of interaction between CO2 molecules and the surface of adsorbents, which could be related to the heterogeneity of the surface chemistry and pore sizes. Obviously, the Qst values of HPCK-x are much higher than those of HPCZn-x samples, which should be benefit from their more prominent microporosity, especially the developed micropores of <1 nm. Moreover, it is worth of notice that the Qst values of HPCK-x samples gradually decline with the increase of activating agent dosage, suggesting the weaker and weaker interactions between adsorbent surfaces and CO2 molecule. Such result should be related to the poorer and poorer microporosity, especially the decreased proportion of ultramicropores with the rise of activating agent dosage, resulting in the weaker interaction. The similar trend can be observed in HPCZn-x samples, and such result further confirms the crucial role of micropore proportion and size in capturing CO2. Even though HPCK-3 has a higher Qst value at low CO2 coverage, its Qst value continuously declines with the enhanced CO2 uptake, indicative of the nonuniform and unstable CO2 adsorption on HPCK-3 adsorbent surface, resulting in the poor regeneration and reversibility, which seriously restricts it to be an efficient CO2 adsorbent. In addition, it is clearly observed that the samples functionalized by nitrogen and sulfur groups show an enhanced Qst values, meaning an improved interaction, which should be ascribed to the more adsorption sites contributed by nitrogen and sulfur doping. Importantly, the initial Qst of N,S-HPCK-1 is up to 39 kJ mol−1, is higher than those of porous carbons and is comparable to that of nitrogen doped carbons.48–52 For sulfur doped carbons, since the radius of sulfur is larger than that of carbon, sulfur atoms tend to protrude out of the carbon layer.53 The disruption of the carbon structure will induce strain and defects, and facilitate charge localization creating favorable conditions for the adsorption of CO2. Furthermore, due to the large and polarizable d-orbitals of sulfur, the lone pair of electrons in doped sulfur atom can interact easily with the oxygen.54 In the current work, the doped sulfur interacts with the oxygen of CO2 molecule and thus strengthens the adsorption of CO2, as reflected by the high initial Qst value. The high Qst at low CO2 loading is helpful for CO2 capture at a low pressure, and the moderate Qst is beneficial for easy desorption to regenerate the sorbents. | ||
| Fig. 8 Isosteric heat of CO2 adsorption on all the as-obtained samples calculated from the adsorption isotherms at 273 and 298 K. (a) HPC, HPCK-x and N,S-HPCK-1; (b) HPCZn-x and N,S-HPCZn-1. | ||
CO2/N2 selectivity and cycling stability
Highly efficient CO2 adsorbents need to meet the following characteristics: (i) large CO2 uptake; (ii) fast adsorption kinetics; (iii) moderate heat of adsorption; (iv) good selectivity against other gas molecules; (v) easy regeneration. To evaluate the CO2 separation performance of the selected sample of HPCK-1 and N,S-HPCK-1 samples, their CO2 and N2 isotherms at 273 and 298 K are presented in Fig. 9a and b. Obviously, the adsorption capacity of N2 is much lower than that of CO2 at the same condition for both HPCK-1 and N,S-HPCK-1 samples. At 298 K and 1 bar, N,S-HPCK-1 sample has a CO2 uptake of 5.19 mmol g−1 and a N2 adsorption capacity of 0.67 mmol g−1. The equilibrium CO2/N2 adsorption ratio of 8 is higher than the value of 7 for HPCK-1 sample, indicative of the better selectivity of N,S-HPCK-1 for CO2 from N2. | ||
| Fig. 9 Adsorption selectivities. (a) CO2 and N2 adsorption isotherms of HPCK-1 at 273 and 298 K. (b) CO2 and N2 adsorption isotherms of N,S-HPCK-1 at 273 and 298 K. (c) IAST-calculated selectivities of CO2/N2 on HPCK-1 and N,S-HPCK-1 at 273 K. (d) IAST-calculated selectivities of CO2/N2 on HPCK-1 and N,S-HPCK-1 at 298 K. | ||
The ideal adsorbed solution theory (IAST) model was employed to evaluate the selectivity for CO2 adsorption from simulated post-combustion flue gas, which has been widely used to predict the selectivity of adsorbents for any two gases in a binary gas mixture by using the isotherms of pure gas. The calculated CO2/N2 selectivity of HPCK-1 and N,S-HPCK-1 samples with IAST model are displayed in Fig. 9c and d. The CO2/N2 ratio is 15/85 in the calculation, representing the typical composition of flue gas. Apparently, the selectivity of HPCK-1 and N,S-HPCK-1 samples significantly decrease in the low pressure region, finally reaching a plateau despite the increase in pressure. The highest selectivity of N,S-HPCK-1 in the low pressure are 72 and 73 at 273 and 298 K, respectively, and the corresponding values are 70 and 69 for HPCK-1 sample. Evidently, the selectivity of N,S-HPCK-1 is superior than that of HPCK-1 under the same conditions, manifesting that the introduction of nitrogen and sulfur groups into the HPCK-1 framework brings an improvement in CO2 capture and CO2/N2 selectivity. Such results should be attributed to the strengthened affinity of CO2 molecule and N,S-HPCK-1 skeleton owing to the more adsorption sites derived from the doping of nitrogen and sulfur. The same result can be found in N,S-HPCZn-1 and HPCZn-1 samples (Fig. S9†), which further confirms the improved effect of nitrogen/sulfur doping in carbon matrix for enhancing CO2/N2 selectivity. Importantly, these values are even greatly higher than most of reported porous carbons and nitrogen-rich carbon materials.55–58 Moreover, it is noticeable that the selectivity has no decrease with the increase of adsorption temperature.
Besides prominent separation performance, the recyclability of an adsorbent is a critical property determining the potential of practical utilization. After the adsorbent was saturated with CO2 up to 1 bar, the adsorbent was recycled. Then, the recycled adsorbent was degassed at the room temperature for 10 min to apply for the next adsorption. The regeneration test of N,S-HPCK-1 sample was conducted for ten consecutive cycles at 273 and 298 K. As presented in Fig. 10a and b, the CO2 uptakes are almost similar without noticeable loss, indicative of the outstanding recyclability of N,S-HPCK-1 with relatively low energy requirement for regeneration. Moreover, such recyclability test suggests CO2 can be completely desorbed from the adsorbent by only changing the pressure. It offers a chance for such adsorbents to be applied in the pressure swing adsorption technology.
 | ||
| Fig. 10 Recyclability test on CO2 capture for N,S-HPCK-1. (a) at 273 K; (b) at 298 K. | ||
Conclusions
In this work, a series of hierarchically porous carbons with well-interconnected porosity were successfully prepared by one-step carbonization and activation route using no-cost biomass waste as carbon source. Followed by a facile nitrogen and sulfur binary doping using thiourea as nitriding and sulfurizing agent, N,S-co-doped porous carbons were obtained with more surface active sites for efficient CO2 capture. By further investigating activation mechanism of KOH/ZnCl2 activating agents, it can be drawn that KOH activating agent mainly produce micropores with different pore sizes and ZnCl2 activating agent tends to develop mesopores. As expected, the resultant N,S-HPCK-1 material exhibits an excellent CO2 capture capacity of up to 7.76 and 5.19 mmol g−1 at 273 and 298 K under 1 bar, respectively, which are much higher than those of reported carbon-based CO2 adsorbents. By analyzing the CO2 capture performance in detail, it can be found that CO2 capture at atmosphere pressure is a combined action of the ultramicropore structure and the surface chemistry, in which the ultramicropore plays a determined role. Further investigations shows that the synergetic effect of nitrogen and sulfur dual-doping has an improvement in CO2 capture, but there is no an obvious connection between nitrogen/sulfur doping content and CO2 uptake. More importantly, N,S-HPCK-1 material also exhibits a moderate isosteric heat of adsorption, a satisfactory recyclability and high selectivity for CO2 capture from the gas mixture of CO2 and N2. Considering the attractive features for CO2 capture, the low-cost preparation as well as the aspect of environmental friendliness, our prepared N/S dual-doped hierarchically porous carbons are believed to be a promising candidate for capturing CO2 from flue gas.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51702114), the National Natural Science Foundation of China (51872110), Natural Science Foundation of Henan Province (172102210381), Natural Science Foundation of Education Department of Henan Province (18A150011 and 19A150032).References
- S. Mane, Z. Y. Gao, Y. X. Li, D. M. Xue, X. Q. Liu and L. B. Sun, J. Mater. Chem. A, 2017, 5, 23310–23318 RSC.
- L. M. Yue, L. L. Rao, L. L. Wang, L. Y. An, C. Y. Hou, C. D. Ma, H. DaCosta and X. Hu, Energy Fuels, 2018, 32, 6955–6963 CrossRef CAS.
- B. B. Chang, W. W. Shi, H. Yin, S. R. Zhang and B. C. Yang, Chem. Eng. J., 2019, 358, 1507–1518 CrossRef CAS.
- M. Idrees, V. Rangari and S. Jeelani, J. CO2 Util., 2018, 26, 380–387 CrossRef CAS.
- W. J. Tian, H. Y. Zhang, H. Q. Sun, A. Suvorova, M. Saunders, M. Tade and S. B. Wang, Adv. Funct. Mater., 2016, 26, 8651–8661 CrossRef CAS.
- Y. Lin, C. Kong, Q. Zhang and L. Chen, Adv. Energy Mater., 2017, 7, 1601296 CrossRef.
- Y. W. Chen, Z. W. Qiao, J. L. Huang, H. X. Wu, J. Xiao, Q. B. Xia, H. X. Xi, J. Hu, J. Zhou and Z. Li, ACS Appl. Mater. Interfaces, 2018, 10, 38638–38647 CrossRef CAS PubMed.
- M. Sevilla, P. Valle-Vigón and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781–2787 CrossRef CAS.
- Y. H. Abdelmoaty, T. D. Tessema, N. Norouzi, O. M. El-Kadri, J. B. M. G. Turner and H. M. El-Kaderi, ACS Appl. Mater. Interfaces, 2017, 9, 35802–35810 CrossRef CAS PubMed.
- A. M. Fracaroli, H. Furukawa, M. Suzuki, M. Dodd, S. Okajima, F. Gandara, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 8863–8866 CrossRef CAS PubMed.
- G. K. Parshetti, S. Chowdhury and R. Balasubramanian, RSC Adv., 2014, 4, 44634–44643 RSC.
- J. Gong, M. Antonietti and J. Y. Yuan, Angew. Chem., Int. Ed., 2017, 56, 7557–7563 CrossRef CAS PubMed.
- H. Li, M. M. Sadiq, K. Suzuki, R. Ricco, C. Doblin, A. J. Hill, S. Lim, P. Falcaro and M. R. Hill, Adv. Mater., 2016, 28, 1839–1844 CrossRef CAS PubMed.
- Z. Xiang, R. Mercado, J. M. Huck, H. Wang, Z. Guo, W. Wang, D. Cao, M. Haranczyk and B. Smit, J. Am. Chem. Soc., 2015, 137, 13301–13307 CrossRef CAS PubMed.
- J. Chen, J. Yang, G. S. Hu, X. Hu, Z. M. Li, S. Wshen, M. Radosz and M. H. Fan, ACS Sustainable Chem. Eng., 2016, 4, 1439–1445 CrossRef CAS.
- L. H. Zhang, W. C. Li, L. Tang, Q. G. Wang, Q. T. Hu, Y. Zhang and A. H. Lu, J. Mater. Chem. A, 2018, 6, 24285–24290 RSC.
- L. W. Wang, L. L. Rao, B. B. Xia, L. L. Wang, L. M. Yue, Y. Q. Liang, H. Dacosta and X. Hu, Carbon, 2018, 130, 31–40 CrossRef CAS.
- J. Wang, P. Zhang, L. Liu, Y. Zhang, J. Yang, Z. Zeng and S. Deng, Chem. Eng. J., 2018, 348, 57–66 CrossRef CAS.
- D. Qian, C. Lei, E. M. Wang, W. C. Li and A. H. Lu, ChemSusChem, 2014, 7, 291–298 CrossRef CAS PubMed.
- D. Qian, C. Lei, G. P. Hao, W. C. Li and A. H. Lu, ACS Appl. Mater. Interfaces, 2012, 4, 6125–6132 CrossRef CAS PubMed.
- Z. Zhang, J. Zhou, W. Xing, Q. Xue, Z. Yan, S. Zhuo and S. Z. Qiao, Phys. Chem. Chem. Phys., 2013, 15, 2523–2529 RSC.
- V. Presser, J. Mcdonough, S. H. Yeon and Y. Gogotsi, Energy Environ. Sci., 2011, 4, 3059–3066 RSC.
- N. Wickramaratne and M. Jaroniec, J. Mater. Chem. A, 2013, 1, 112–116 RSC.
- M. Sevilla, C. Falco, M. Titirici and A. Fuertes, RSC Adv., 2012, 2, 12792–12797 RSC.
- B. B. Chang, L. Sun, W. W. Shi, S. R. Zhang and B. C. Yang, ACS Omega, 2018, 3, 5563–5573 CrossRef CAS.
- F. Q. Yang, J. Wang, L. Liu, P. X. Zhang, W. K. Yu, Q. Deng, Z. L. Zeng and S. G. Deng, ACS Sustainable Chem. Eng., 2018, 6, 15550–15559 CrossRef CAS.
- P. Zhang, Y. Zhong, J. Ding, J. Wang, M. Xu, Q. Deng, Z. Zeng and S. Deng, Chem. Eng. J., 2019, 355, 963–973 CrossRef CAS.
- Y. H. Abdelmoaty, T. D. Tessema, N. Norouzi, O. M. EI-Kadri, J. B. Mcgee Turner and H. M. EI-Kaderi, ACS Appl. Mater. Interfaces, 2017, 9, 35802–35810 CrossRef CAS PubMed.
- A. Sanchez-Sanchez, F. Suarez-Garcia, A. Martinez-Alonso and J. M. Tascon, ACS Appl. Mater. Interfaces, 2014, 6, 21237–21247 CrossRef CAS PubMed.
- W. Xing, C. Liu, Z. Zhou, L. Zhang, J. Zhou, S. Zhuo, Z. Yan, H. Gao, G. Wang and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 7323–7327 RSC.
- J. Wang, Y. Lin, Q. Yue, K. Tao, C. Kong and L. Chen, RSC Adv., 2016, 6, 53017–53024 RSC.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 RSC.
- Y. Xia, Y. Zhu and Y. Tang, Carbon, 2012, 50, 5543–5553 CrossRef CAS.
- H. Seema, K. C. Kemp, N. H. Le, S. W. Park, V. Chandra and J. W. Lee, et al., Carbon, 2014, 66, 320–326 CrossRef CAS.
- M. Seredych, J. Jagiello and T. J. Bandosz, Carbon, 2014, 74, 207–217 CrossRef CAS.
- T. J. Bandosz, M. Seredych, E. Rodríguez-Castellon, Y. Cheng, L. L. Daemen and A. J. Ramírez-Cuesta, Carbon, 2016, 96, 856–863 CrossRef CAS.
- Y. H. Sun, K. X. Li, J. H. Wang, N. Tang, D. Zhang, T. T. Guan and Z. Jin, J. Colloid Interface Sci., 2018, 526, 174–183 CrossRef CAS PubMed.
- G. Y. Xu, J. P. Han, B. Ding, P. Nie, J. Pan, H. Dou, H. S. Li and X. G. Zhang, Green Chem., 2015, 17, 1668–1674 RSC.
- L. J. Xie, G. H. Sun, F. Y. Su, X. Q. Guo, Q. Q. Kong and X. M. Li, et al., J. Mater. Chem. A, 2016, 4, 1637–1646 RSC.
- L. C. Zhang, X. Sun, Z. Hu, C. C. Yuan and C. H. Chen, J. Power Sources, 2012, 204, 149–154 CrossRef CAS.
- Y. X. Liu, Z. C. Xiao, Y. C. Liu and L. Z. Fan, J. Mater. Chem. A, 2018, 6, 160–166 RSC.
- C. Ge, J. Song, Z. F. Qin, J. G. Wang and W. B. Fan, ACS Appl. Mater. Interfaces, 2016, 8, 18849–18859 CrossRef CAS PubMed.
- H. Wei, S. Deng, B. Hu, Z. Chen, B. Wang, J. Huang and G. Yu, ChemSusChem, 2012, 5, 2354–2360 CrossRef CAS PubMed.
- S. Ello, L. K. C. de Souza, A. Trokourey and M. Jaroniec, Microporous Mesoporous Mater., 2013, 180, 280–283 CrossRef.
- E. M. Kutorglo, F. Hassouna, A. Beltzung, D. Kopecky, I. Sedlařova and M. Šooš, Chem. Eng. J., 2019, 360, 1199–1212 CrossRef CAS.
- W. Xing, C. Liu, Z. Zhou, J. Zhou, G. Wang, S. Zhuo, Q. Xue, L. Song and Z. Yan, Nanoscale Res. Lett., 2014, 9, 1–8 CrossRef CAS PubMed.
- J. He, J. W. F. To, P. C. Psarras, H. Yan, T. Atkinson, R. T. Holmes, D. Nordlund, Z. Bao and J. Wilcox, Adv. Energy Mater., 2016, 6, 1–11 Search PubMed.
- J. C. Wang and Q. Liu, Nanoscale, 2014, 6, 4148–4156 RSC.
- Z. Liu, Z. Zhang, Z. J. Jia, L. Zhao, T. T. Zhang, W. Xing, S. Komarneni, F. Subhan and Z. F. Yan, Chem. Eng. J., 2018, 337, 290–299 CrossRef CAS.
- S. Ghosh and S. Ramaprabhu, Carbon, 2019, 141, 692–703 CrossRef CAS.
- X. Li, Z. Y. Sui, Y. N. Sun, P. W. Xiao, X. Y. Wang and B. H. Han, Microporous Mesoporous Mater., 2018, 257, 85–91 CrossRef CAS.
- L. S. Shao, M. Q. Liu, J. H. Huang and Y. N. Liu, J. Colloid Interface Sci., 2018, 513, 304–313 CrossRef CAS PubMed.
- W. Kiciński, M. Szala and M. Bystrzejewski, Carbon, 2014, 68, 1–32 CrossRef.
- S. A. Wohlgemuth, R. J. White, M. G. Willinger, M. M. Titirici and M. Antonietti, Green Chem., 2012, 14, 1515–1523 RSC.
- X. M. Ren, H. Li, J. Chen, L. J. Wei, A. Modak, H. Q. Yang and Q. H. Yang, Carbon, 2017, 114, 473–481 CrossRef CAS.
- L. S. Shao, Y. Li, J. H. Huang and Y. N. Liu, Ind. Eng. Chem. Res., 2018, 57, 2856–2865 CrossRef CAS.
- J. C. Geng, D. M. Xue, X. Q. Liu, Y. Q. Shi and L. B. Sun, AIChE J., 2017, 63, 1648–1658 CrossRef CAS.
- M. Q. Liu, L. S. Shao, J. H. Huang and Y. N. Liu, Microporous Mesoporous Mater., 2018, 264, 104–111 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03659h |
| This journal is © The Royal Society of Chemistry 2019 |