
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBranched EHNBR and its properties with enhanced low-temperature performance and oil resistance
Lu
Liang
 a,
Jianjun
Dong
ab and
Dongmei
Yue
*b
a,
Jianjun
Dong
ab and
Dongmei
Yue
*b
aAECC Beijing Institute of Aeronautical Material, 100095, Beijing, China
bBeijing University of Chemical Technology, 100029, Beijing, China. E-mail: yuedm@mail.buct.edu.cn
First published on 9th October 2019
Abstract
Epoxide nitrile butadiene rubber (ENBR) was prepared via in situ epoxidation from nitrile butadiene rubber (NBR) with acetic acid and hydrogen peroxide. ENBR had been selectively hydrogenated in the presence of a homogeneous Wilkinson catalyst. The hydrogenated epoxide nitrile butadiene rubber (EHNBR) and ENBR were characterized by infra-red and proton nuclear magnetic resonance. No change was noted in the epoxy content of the polymer after the reaction. The catalyst is highly selective in reducing carbon–carbon double bonds in the presence of epoxy groups. DSC analysis reveals the Tg of ENBR varied linearly with molar epoxide content and the Tg value increased by 0.82 °C per mol%. It also found that the introduction of epoxy groups can effectively reduce the extent of crystallization by impairing the regularity of the molecular chain, but crystalline structure was difficult to completely eliminate. Therefore, anhydrides were selected as ring-opening reagents to react with epoxy groups in EHNBR. The products, branched EHNBR, were characterized by infra-red and proton nuclear magnetic resonance. The conversion rate of the epoxide group was calculated by 1H NMR. The glass transition temperature of EHNBR-g-heptyl group was −34.1 °C, and its DSC curve demonstrated no crystal structure. The coefficient of cold resistance under compression of EHNBR grafted propyl ester was 0.36, which represented a superior low-temperature performance. Furthermore, residual epoxy groups and ester groups extremely enhanced the oil resistance of HNBR.
Introduction
Hydrogenated nitrile butadiene rubber (HNBR) is saturated by hydrogenation of acrylonitrile-butadiene rubber (NBR).1–3 Most butadiene chain segments (about 95% to 99%) are hydrogenated, save only a few (about 1% to 5%) for crosslinking.4 HNBR not only retains the advantages of original NBR, such as oil resistance and solvent resistance, but also exhibits additional characteristics such as aging resistance and high temperature resistance.5 Although HNBR has a very high application value, its vital disadvantage is the lack of low temperature performance after hydrogenation. After hydrogenation, the polybutadiene sequence in the molecule chain turns into a tetramethylene sequence, which is liable to crystallization. Therefore, the HNBR molecule chain can easily form crystalline structure in a low temperature environment, hardening the whole molecule chain and raising its glass transition temperature.6 These disadvantages limit the applications of HNBR in low temperature environments.7Crystalline rubber's applicable temperature shall not be lower than its crystallization temperature. When the ambient temperature goes lower than the crystallization temperature, the molecular chain facilitates crystallization, which leads to rigid materials. The key to improve the low temperature performance of HNBR is to reduce its glass transition temperature (Tg) and hinder its crystallization. Consequently, enhancing the flexibility of the molecular chain and decreasing the molecular regularity are general methods to obtain HNBR with low-temperature performance.8
So far, there are three main ways to improve the low temperature performance of HNBR in literatures: (a) adopting a third monomer in the molecular structure of HNBR through grafting reaction or copolymerization, as a flexible side group to improve rubber molecular flexibility and sabotage the molecular regularity;9,10 (b) adding a plasticizer and adjusting the curing agent, reinforcing the agent of rubber mixing system in the vulcanization of rubber modified process;11–14 (c) blending with other rubbers by physical or chemical blending methods, such as silicone rubber, fluorine rubber, and ethylene-propylene rubber, combining merits of these rubber to improve the low temperature performance of HNBR.15–17
Epoxidation is the common and effective methods to improve the properties of non-polar rubber,18–21 but few studies report epoxidation of polar rubber.22,23 Epoxide natural rubber (NR) has been widely studied24,25 and realized industrialization. Introducing an epoxy group endows ENR with air permeability and oil resistance.26,27 Besides that, the presence of epoxy group provides a potential gateway to abundant secondary modifications.28–31 A few articles studied the hydrogenation of epoxied rubber,32–34 such as different hydrogenated catalysts' effect on the epoxy group. Phan Trung Nghia et al.31 prepared noncrystallizable hydrogenated NR by the introduction of epoxy group to disturb the stereo-regular alternative ethylene–propylene units. Jia-rui Xu et al.34 revealed that oxirane unit's random distribution in rubber's molecular chain significantly impair its crystal structure. In this article, we prepared a series of epoxide hydrogenated nitrile butadiene rubber (EHNBR) in different level of epoxidation. Though crystal structure is impaired with the increase of epoxide content, continual increase of epoxy group enhances the chemical polarity of its molecular chain, which will decrease its Tg.35 Therefore, we adopted the way of anhydride reacting with epoxy group to introduce ester group in HNBR so that crystallization is further destroyed. In addition, ester group as a flexible side group can decrease the glass transition temperature. It's worth mentioning that residual epoxy group and ester group significantly enhance the oil resistance of HNBR.
Experimental
Materials
Nitrile rubber (NANCAR1965) was obtained from Taiwan NANTEX with nitrile content in 19%; anhydrous ethanol and xylene were analytically pure; acetic acid (99%) and hydrogen peroxide (30% w/w) used were commercial grade, purchased from Beijing Chemical Plant; propionic anhydride, valeric anhydride and pentanoic anhydride were purchased from TGI; triphenylphosphine and deuterated chloroform were obtained from National Chemical Reagent Co.; Wilkinson catalyst was made in our laboratory.Epoxidation of NBR
24 g NBR was dissolved in 321 mL xylene in a 1 L three-necked glass flask equipped with a mechanical stirrer. Upon increasing the temperature to 65 °C, 21.6 g acetic acid were added to the polymer solution, and the hydrogen peroxide was then added drop by drop. After completion of the reaction, the reaction mixture was neutralized with 5% sodium carbonate solution. The polymer was coagulated in ethanol and washed three times. The samples were dried at 60 °C to a constant weight. Fig. 1 illustrates the process.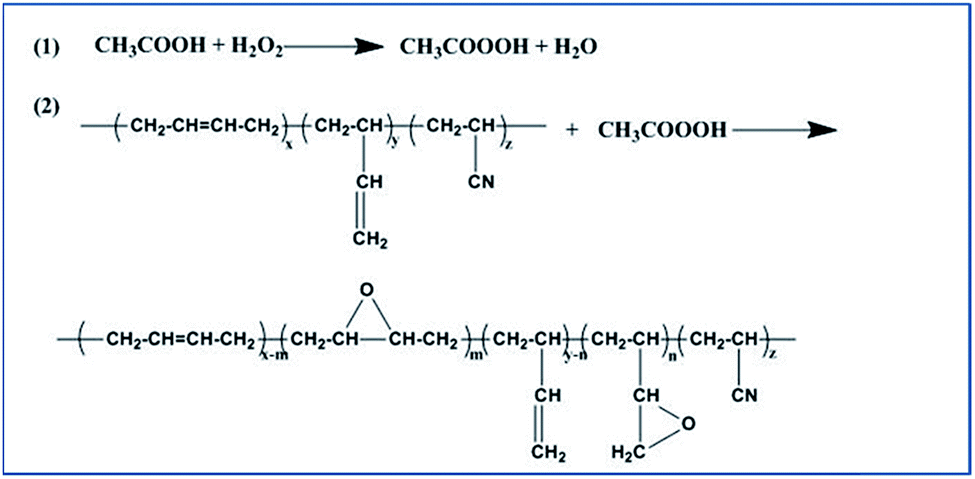 | ||
| Fig. 1 Schematic illustration of the epoxidation of NBR. | ||
Hydrogenation of ENBR
All the hydrogenation experiments were carried out in 500 mL autoclave reactor. Epoxide nitrile butadiene rubber (ENBR) (10 g) in xylene (232 mL) was added to autoclave, and stirred vigorously under hydrogen for a couple of minutes, followed by venting. This process was repeated five times to ensure that oxygen was completely removed. Then the required catalyst was transferred into the reactor. The autoclave was slowly heated to 110 °C and maintained at this temperature for 8 h. After reaction the autoclave was allowed to cool to room temperature. Fig. 2 illustrates the process.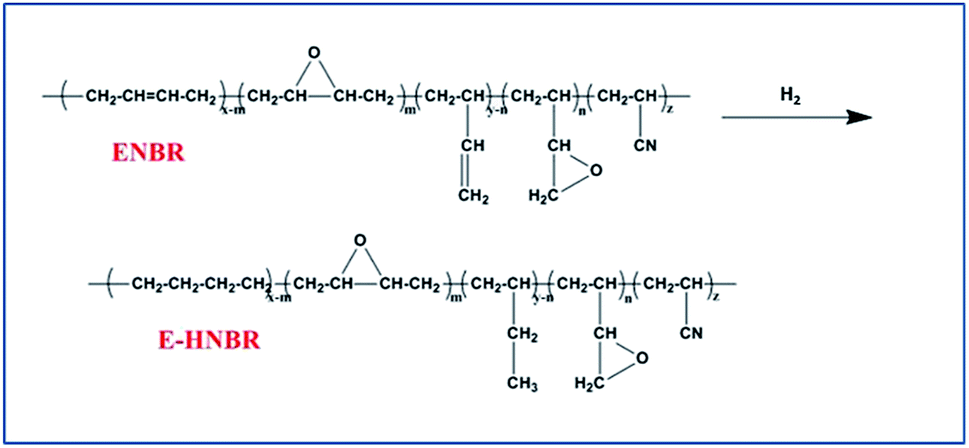 | ||
| Fig. 2 Schematic illustration of the hydrogenation of ENBR. | ||
Anhydride reacted with epoxy group in EHNBR
Ring-opening reaction was carried out in nitrogen atmosphere. Anhydride and tetrabutylammonium chloride were added in EHNBR solution (8% w/w in xylene). The three-necked glass flask was slowly heated to 110 °C and reacted in different times.Characterization
The changes in the structure of polymer were determined by Fourier transform infrared spectroscopy (FTIR) and proton nuclear magnetic resonance (NMR). FTIR spectra were obtained by a Bruker Tensor 27 spectrophotometer in the wave-number range of 400 to 4000 cm−1. NMR measurements were obtained from an AC600 spectrophotometer (Bruker Optik GmbH, Germany), with CDCl3 solution as solvent. Transition temperature of ENR samples was determined by differential scanning calorimetry (DSC). The sample was heated from 25 to 100 °C at a rate of 10 °C min−1 and cooled from 100 °C to −100 °C at a rate of 10 °C min−1 under a nitrogen purge. The glass transition temperature was determined from the heating curve from −100 °C to 100 °C at a rate of 10 °C min−1.Results and discussion
FTIR and 1H NMR characterization of epoxied nitrile butadiene rubber (ENBR)
Fig. 3 contains the FTIR spectra of the ENBR with different degree of epoxidation. The broad absorptions at 2920 and 2850 cm−1 are attributed to the stretching vibration of –CH2 in the nitrile butadiene rubber, and the absorption at 1450 cm−1 corresponds to the bending vibration of –CH2. The absorption at 2237 cm−1 corresponds to the stretching vibration of –CN in the acrylonitrile units. The absorption at 967 cm−1 and 915 cm−1 correspond to the bending vibration of![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H in the butadiene units. An absorbance band at 887 cm−1 associated with weak peak at 1239 cm−1 appears from the C–O–C ring vibration of epoxide groups, which means NBR successfully epoxied.22
C–H in the butadiene units. An absorbance band at 887 cm−1 associated with weak peak at 1239 cm−1 appears from the C–O–C ring vibration of epoxide groups, which means NBR successfully epoxied.22
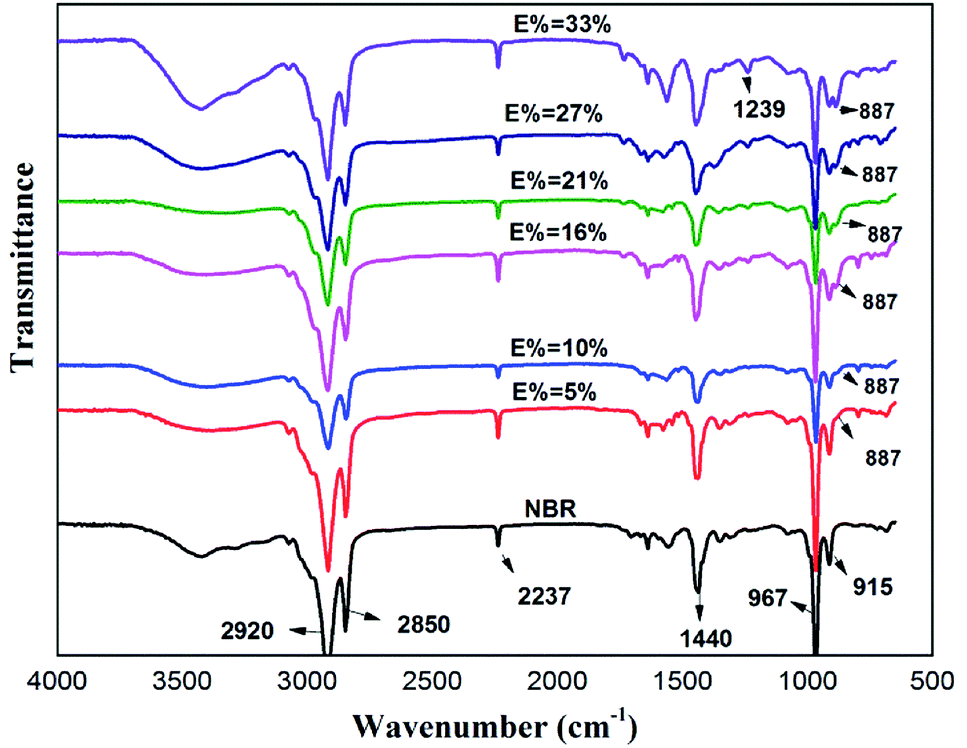 | ||
| Fig. 3 FTIR absorption spectrum of NBR and ENBR. | ||
In Fig. 4, to the NBR as an example, the peaks above 4.5 ppm indicate olefinic protons of 3,4-addition butadiene (4.8 to 5.2 ppm) and 1,4-addition butadiene (5.2 to 5.8 ppm). The peaks on 2.52 ppm indicate the protons of –CN.
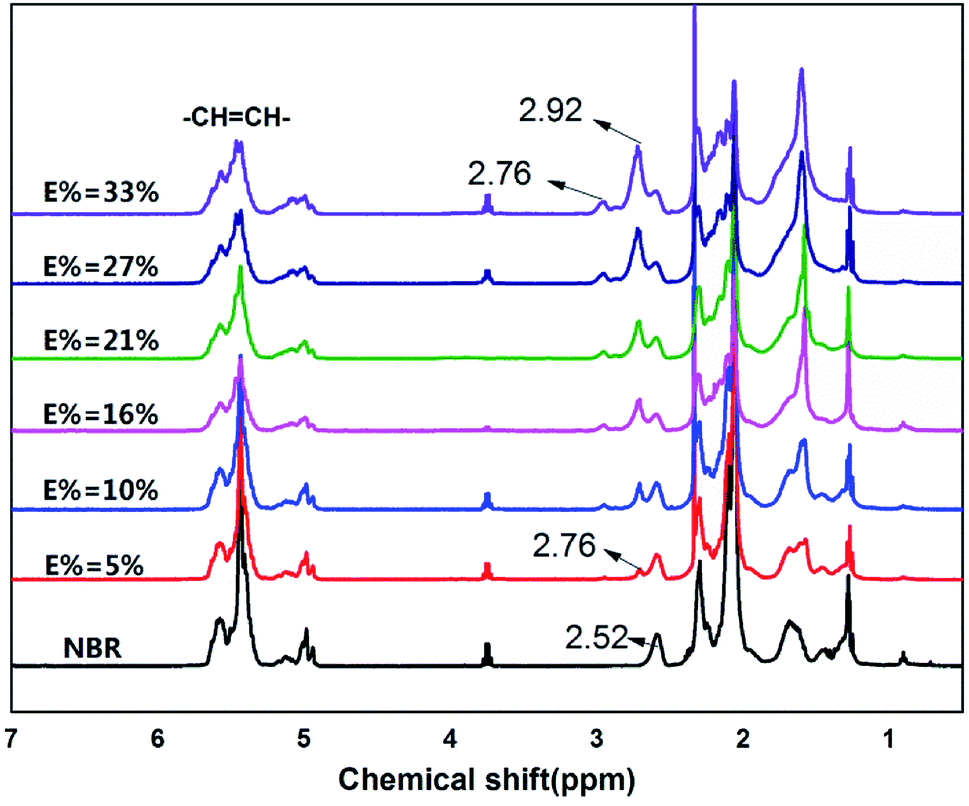 | ||
| Fig. 4 1H NMR spectrum of NBR and ENBR. | ||
In the 1H-NMR spectrum of ENBR, two new peaks appear at δ = 2.76 ppm and δ = 2.92 ppm. The peaks at δ = 2.76 ppm and δ = 2.92 ppm correspond to the methane resonance of the epoxy groups, in trans and cis position respectively.36 As the reaction progresses, there is an increase in the epoxidation content, resulting in an increase in the signals at 2.76 ppm and 2.92 ppm (trans and cis epoxy) and a decrease in the peak at 5.2 ppm (unsaturated 1,4-polybutadiene protons). And the peak at 4.9 ppm (vinyl group) remains practically constant for low and medium degrees of epoxidation. This peak decreased at 4.9 ppm only for high degrees of epoxidation. For all the rubbers studied, the behaviour was similar. These findings indicated that the reactivity of trans and cis 1,4 units is higher than that of vinyl 1,2 units. The peak at approximately 2.52 ppm in the NBR corresponds to the protons of –CN that remains constant during the epoxidation. Its area can be calculated from the area of the nitrile content. Taking this into account, the degree of epoxidation, E%, for all the ENBR rubbers has been calculated using eqn (1):
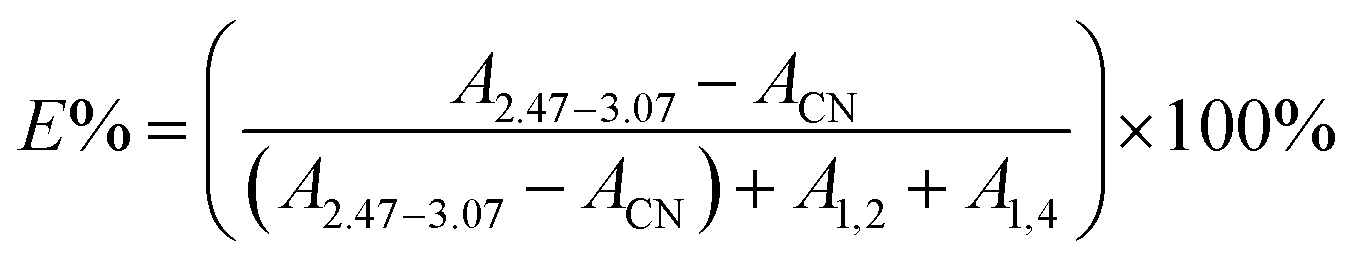 | (1) |
According to 1H-NMR and FTIR results, NBR rubber had been successfully epoxied. We prepared a series of ENBR in different level of epoxidation by controlling of react time.
NMR characterization of hydrogenated epoxied nitrile rubber (EHNBR)
As shown in Fig. 5, the degree of hydrogenation was determined by 1H-NMR. In comparison with NBR, the peaks at 4.8 ppm to 5.8 ppm in HNBR and HENBR indicate olefinic protons of 3,4-addition butadiene (4.8–5.2 ppm) and 1,4-addition butadiene (5.2–5.8 ppm) almost completely disappeared.37 However, no change of signal intensity is detectable at 2.76 ppm and 2.92 ppm, due to the epoxy methane proton for EHNBR. It turned out that Wilkinson catalyst owned highly selective hydrogenation of –CC– and had no reactive activity with epoxy group.
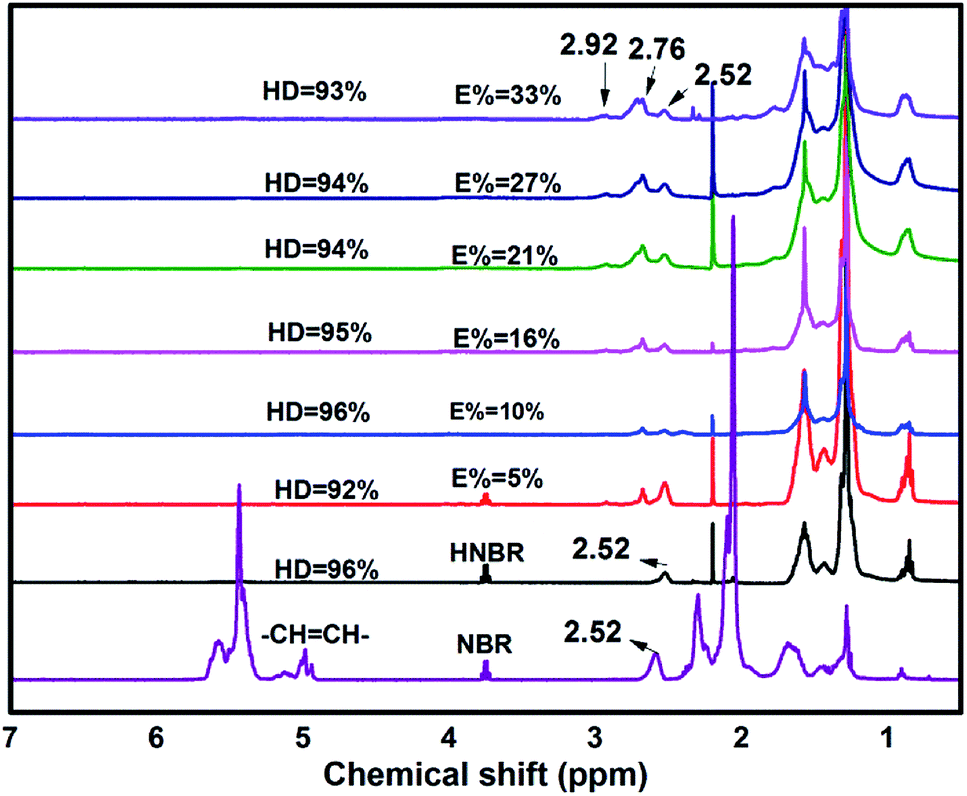 | ||
| Fig. 5 1H NMR spectra of NBR, HNBR and HENBR with different degree of epoxidation. | ||
FTIR and NMR characterization of branched EHNBR
Fig. 6 contains the FTIR spectra of branched EHNBR with different ester group. In contrast with original EHNBR, the peak at 889 cm−1 in the EHNBR grafted with ester group has sharply weakened. Also, two new peaks appear in the branched EHNBR. The intense CO vibration peak at 1735 cm−1 is associated with the formation of ester bonds, and the peak at 1179 cm−1 representing the asymmetric vibration of C–O–C indicate that anhydride has been successfully reacted with epoxy group. The results from FTIR spectra are qualitatively analyzed, so the detailed structure of the branched EHNBR, in particular the grafting ratio, should be quantified by 1H-NMR.
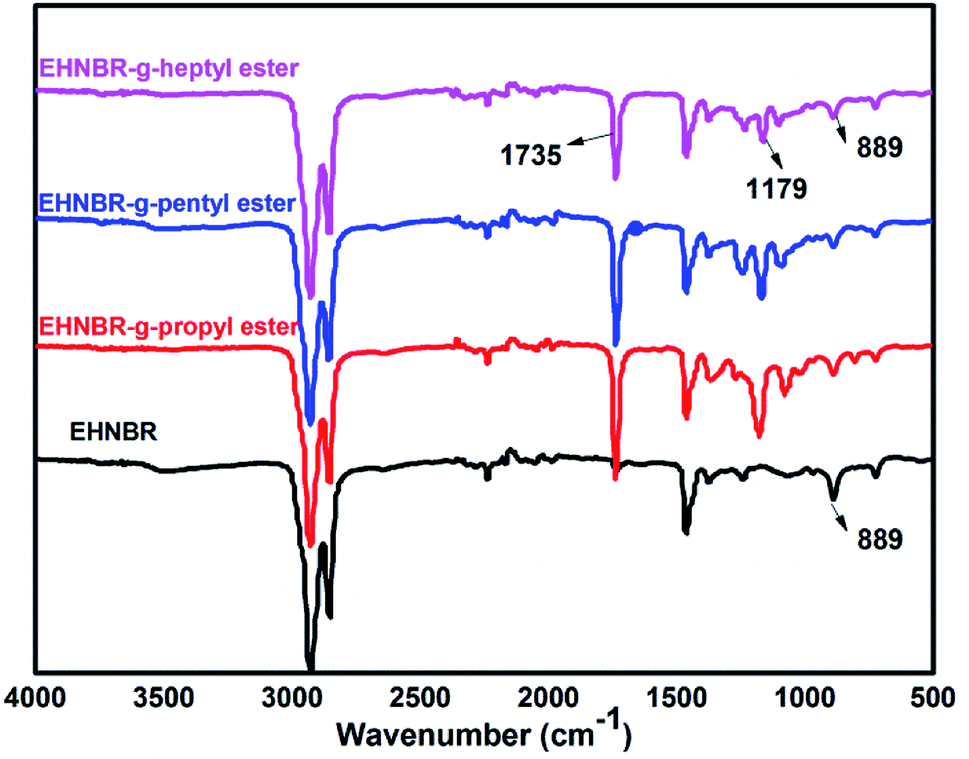 | ||
| Fig. 6 FTIR absorption spectrum of EHNBR and branched EHNBR. | ||
As shown in Fig. 7, the grafting rate of ester group was determined by 1H-NMR. In the 1H-NMR spectrum of ENBR, the peaks at 2.35 ppm in branched EHNBR indicate protons of –COOCH2–, whose appearance represents the introduction of ester group in EHNBR. Accompanied with appearance of peak at 2.35 ppm, the peaks at δ = 2.76 and δ = 2.92 ppm correspond to the methane resonance of the epoxy groups are obviously decreased. Then, the grafting rate, G%, can be calculated by eqn (2):
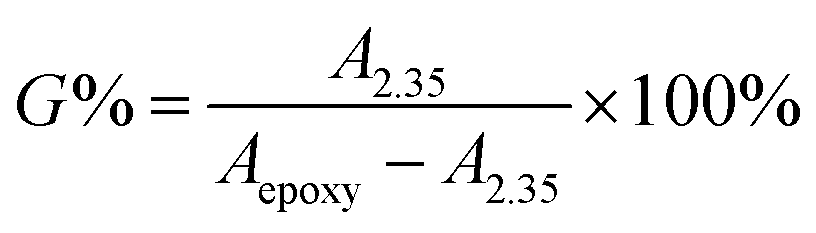 | (2) |
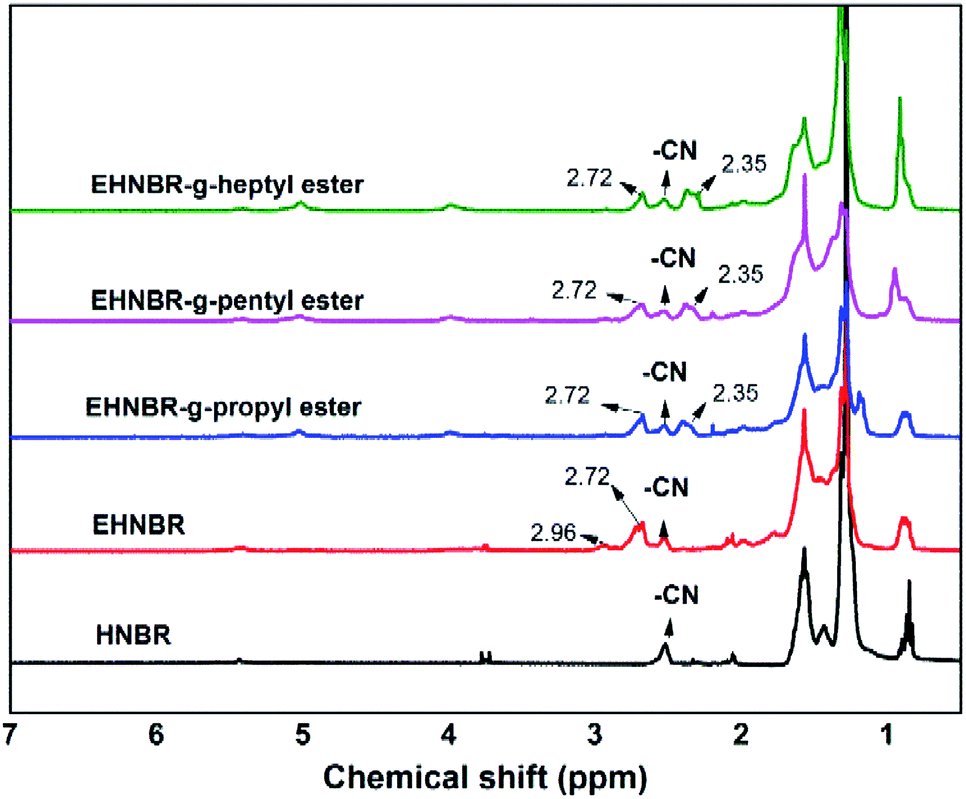 | ||
| Fig. 7 1H NMR spectra of HNBR, EHNBR and branched HENBR. | ||
DSC analysis of ENBR, EHNBR and branched EHNBR
Glass-transition temperatures of ENBR samples obtained from DSC are shown in Fig. 8 and are fitting curve with degree of epoxidation in Fig. 9. It is similar to ENR that Tg of ENBR varies linearly with molar epoxide content and the Tg value increases by 0.82 °C per mol%. The difference of the trend of ENBR and ENR lies in the increase of Tg with molar epoxide content. The nonpolar molecular structure of NR maybe responsible for the more sensitive Tg with epoxide content.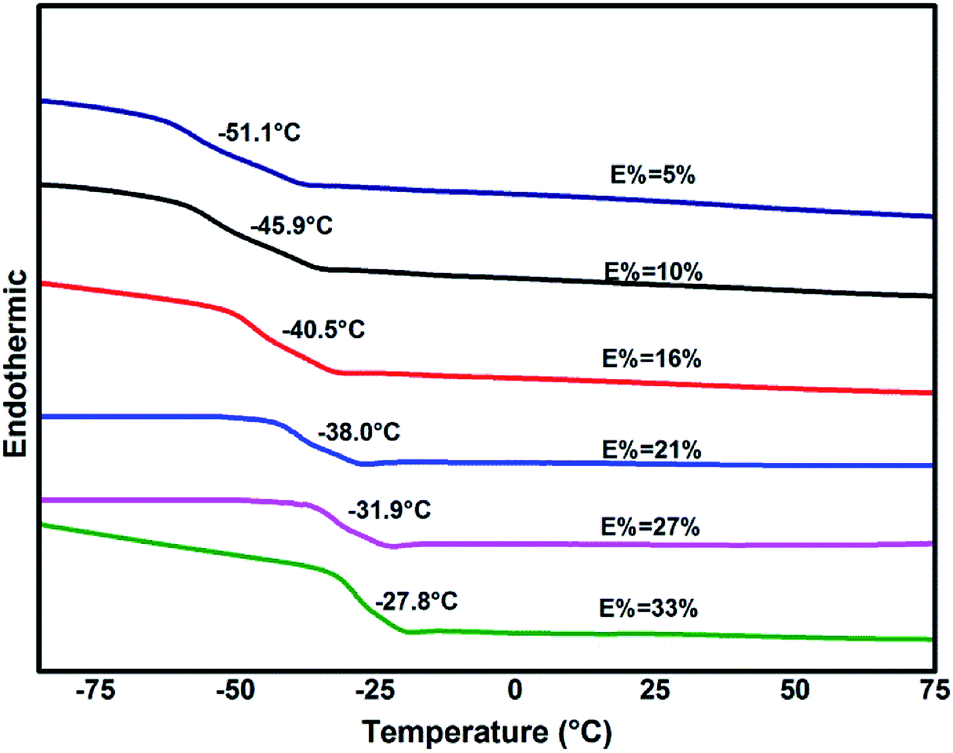 | ||
| Fig. 8 DSC thermograms of ENBR with different epoxide content. | ||
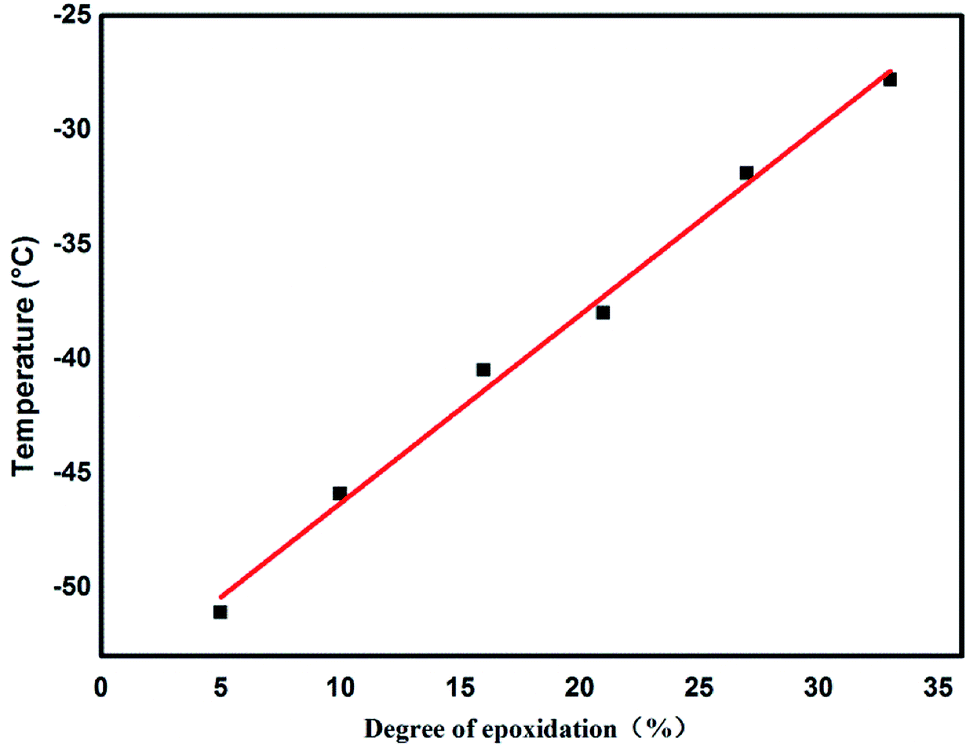 | ||
| Fig. 9 The relation of the degree of epoxidation and Tg of ENBR. | ||
DSC curves (Fig. 10) show the different condition of crystallization in EHNBRs with different epoxide content. The curve of HNBR presents a huge defect from −30 °C to 70 °C, rather than platform of glass transition, which serves as crystal-melting areas could impact the low-temperature properties of HNBR. HNBR forms the crystalline structure caused by highly regular structure of methylene, particularly by tetramethylene sequences. Takush Kobatake found that the length of tetramethylene sequences in a polymer chain is the key factor for improving the low-temperature flexibility. As a consequence, the introduction of epoxy group can effectively reduce extent of crystallization by shortening the length of tetramethylene sequences. As shown in Fig. 10, the defects represented crystal-melting areas narrow down with the increase of epoxide content. ENBR with epoxide content up to 32% largely impairs the trend of crystallization, however, it does not completely eliminate crystallization yet.
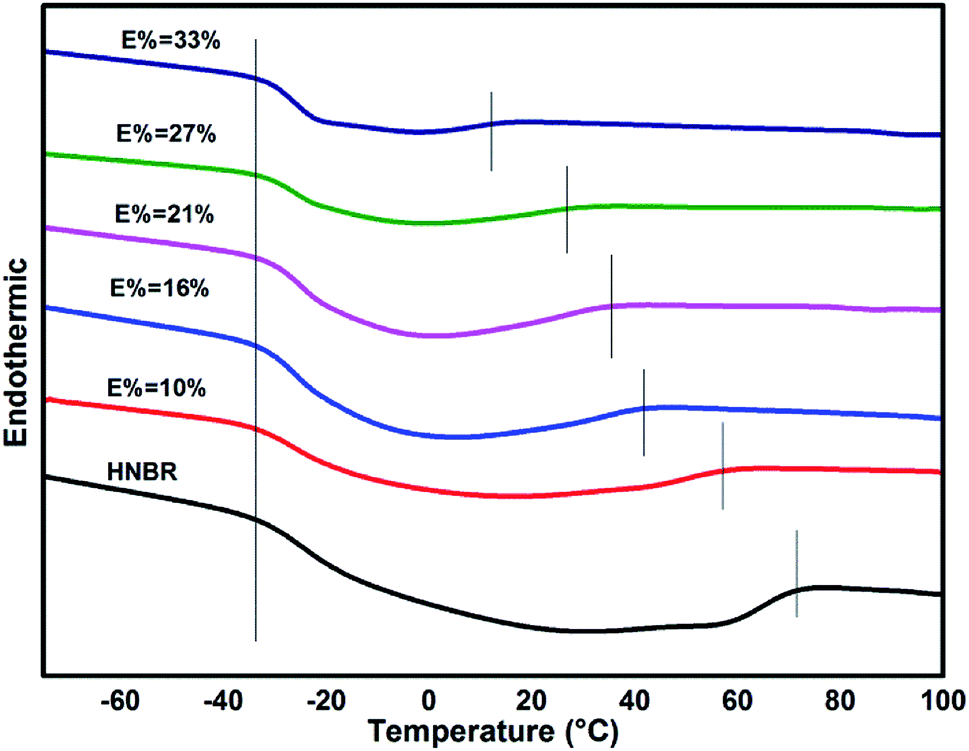 | ||
| Fig. 10 DSC thermograms of EHNBR with different epoxide content. | ||
Although EHNBR with epoxide content in 32% does not successfully collapse the crystal structure, its ring-opened products represent typical platform of glass transition, as shown in Fig. 11. The grafting rates of EHNBR grafted propyl ester, pentyl ester and propyl ester are 47%, 48% and 46% respectively. One epoxy transforms into two ester group after EHNBR reacted with anhydride, which further hinders the crystallization of methylene sequences along the polymer backbone. Furthermore, flexibility of ester group is excellent, and the flexibility increases with the length of molecular chain.9 The glass transition temperature of EHNBR-g-heptyl group is −34.1 °C, and its DSC curve demonstrates typical platform of glass transition and has no endothermic melting peak, which indicate rubber in amorphous state.
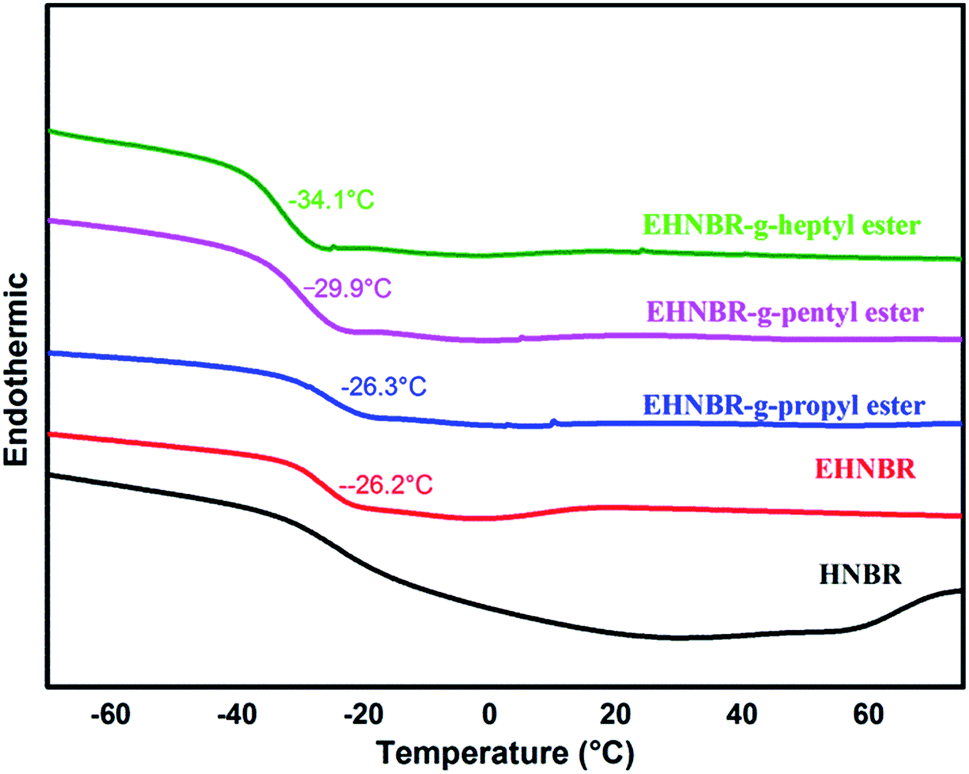 | ||
| Fig. 11 DSC thermograms of EHNBR with different ester side group. | ||
We also determine the coefficient of cold resistance under compression of HNBR, EHNBR grafted propyl ester, pentyl ester and propyl ester, which is 0.21, 0.22, 0.29 and 0.36 respectively. It's well-known that the coefficient of cold resistance under compression is key indicator of mechanical properties of materials at low service temperatures. Therefore, the introduction of ester side groups improves the low temperature performance of HNBR.
Oil resistance of vulcanized branched EHNBR
Vulcanized rubbers were weighed and immersed in ASTM no. 3 oil (IRM903) and aviation hydraulic oil no. 15 at 150 °C for 48 h. These samples were removed from the oils and wiped out excess oil from surface. Changes in the weight of test specimens are shown in Fig. 11. The oil resistance was determined as a change in weight calculated as follows:where Wt is the weight after immersion in oils and W0 is the original weight.
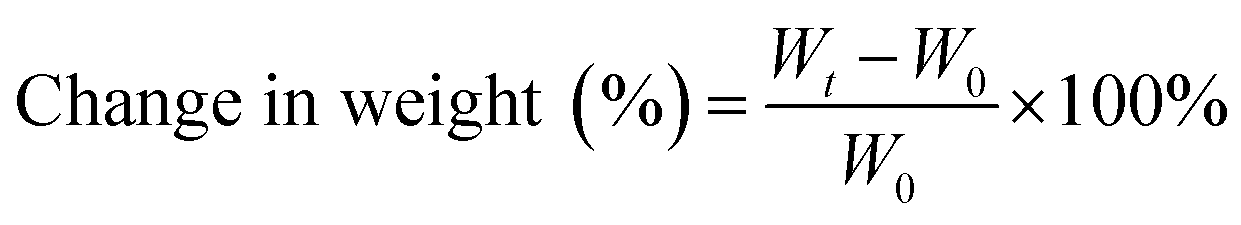
Column diagram of Fig. 12 in black is representative for samples in no. 3 oil, column diagram in red for samples in no. 15 oil. Weights change rate in no. 3 oil are all larger than no. 15 oil, which is attributable to difference of polar. A large number of polar groups, epoxy group and ester group, was introduced in HNBR, resulting in improvement of oil resistance of HNBR, especially for HNBR with low content of nitrile group. Take EHNBR-g-heptyl ester as example, its oil resistance in no. 3 oil increase three times than unmodified HNBR, eight times in no. 15 oil. It means that the method of introduction of epoxy group, and thus reacted with anhydride, is not only effectively improvement of low-temperature performance of HNBR but also enhancement of its oil resistance.
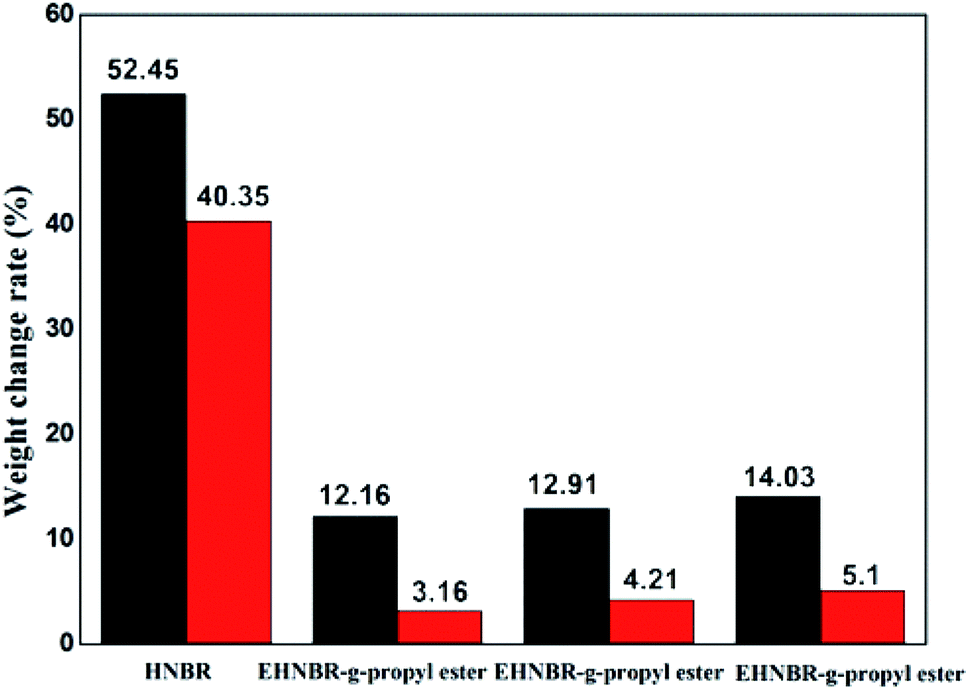 | ||
| Fig. 12 Column chart of oil resistance of branched EHNBR in no. 3 and no. 15 oil. | ||
Conclusions
In method of in situ epoxidation, we prepared ENBR with controllable epoxide content by control the reacted time. The DSC analysis reveals that Tg of ENBR varies linearly with molar epoxide content and the Tg value increases by 0.82 °C per mol%.ENBR has been selectively hydrogenated in the presence of Wilkinson catalyst. FTIR and 1H NMR characterization of HNBR reveals that the epoxy content has no change after hydrogenated reaction. The presence of epoxy group can effectively impair crystal structure, proved by DSC characterization of EHNBR.
The ring-opened product has been characterized by infra-red and nuclear magnetic resonance spectroscopies, which indicates ester group grafted EHNBR successfully in presence of Bu4NCl catalyst. The results of DSC testify that ester side group completely hindered the crystallization of methylene sequences along the polymer backbone. Moreover, Tg of branched HNBR decreased with the length of carbon chain of ester group. EHNBR-g-heptyl ester group has superior low-temperature performance, because its Tg is up to −34.1 °C and in amorphous state. Low temperature test measured the coefficient of cold resistance under compression of EHNBR-g-heptyl ester group is 3.6.
The branched EHNBR has well-performance in no. 3 oil and no. 15 oil, whose oil resistance increases with the decrease of carbon chain of ester group.
Therefore, considering other reports on HNBR, the present study offers a simple and cost-effective preparation technique to obtain branched EHNBR with superior low-temperature performance and excellent oil resistance.
Conflicts of interest
There are no conflicts to declare.References
- N. K. Singha, S. Bhattacharjee and S. Sivaram, Rubber Chem. Technol., 1997, 70(3), 309–367 CrossRef CAS.
- T. Yoshimura and H. Magamori, Nippon Gomu Kyokaishi, 2005, 78(2), 51–55 CrossRef CAS.
- N. T. McManus and G. L. Rempel, J. Macromol. Sci., Rev. Macromol. Chem. Phys., 1995, 35(2), 239–285 CrossRef.
- G. L. Rempel, Q. Pan and J. Wu, Handbook of Homogeneous Hydrogenation, 2007, pp. 547–583 Search PubMed.
- H. Wang, L. Yang and G. L. Rempel, Polym. Rev., 2013, 53(2), 192–239 CrossRef CAS.
- S. Hayashi, H. Sakakida, M. Oyama and T. Nakagawa, Rubber Chem. Technol., 1991, 64(4), 534–544 CrossRef CAS.
- L. Liu, T. Ma, S. Li, F. Liu and R. Li, Elastomer, 2008, 18(02), 69–74 Search PubMed.
- G. Severe and J. L. White, Kautsch. Gummi Kunstst., 2002, 55(4), 144 CAS.
- T. Kobatake, K. Kodama, S. Hayashi and A. Yoshioka, Rubber Chem. Technol., 1997, 70(5), 839–854 CrossRef CAS.
- H. Benner, P. Ngu and J. Gan Lin, China Pat., CN1471540, 2001.
- S. Ito and M. Oyama M, Gummi, Fasern, Kunstst., 2006, 59(1), 25–28 CAS.
- N. Sandland, S. Ito, M. Oyama and E. Files, presented in part at the American Chemical Society Spring Technical Meeting – Rubber Division, San Antonio, TX, United States, May, 2005 Search PubMed.
- S. Kotova, N. Buhtiyarova and S. Mikhailov, Rubber World, 2013, 248(5), 18–20 CAS.
- M. A. Kader and A. K. Bhowmick, Polym. Degrad. Stab., 2003, 79(2), 283–295 CrossRef CAS.
- K. Hirano, K. Suzuki, K. Nakano and M. Tosaka, J. Appl. Polym. Sci., 2005, 95(1), 149–156 CrossRef CAS.
- Y. Yeo, H. Park and C. Lee, J. Ind. Eng. Chem., 2013, 19(5), 1540–1548 CrossRef CAS.
- Y. Zhao, B. Huang, W. Yao, H. Cong, H. Shao and A. Du, J. Appl. Polym. Sci., 2008, 107(5), 2986–2993 CrossRef CAS.
- M. A. Rahman, M. Penco, I. Peroni, G. Ramorino, G. Janszen and L. Di Landro, Smart Mater. Struct., 2012, 21(3), 35014–35020 CrossRef.
- G. Hsiue and J. Yang, J. Polym. Sci., Part A: Polym. Chem., 1990, 28(13), 3761–3773 CrossRef CAS.
- M. M. A. Nikje and Z. Mozaffari, Des. Monomers Polym., 2007, 10(1), 67–77 CrossRef.
- K. Zhao, X. Song, C. Liang, J. Wang and S. Xu, Iran. Polym. J., 2015, 24(5), 425–435 CrossRef CAS.
- R. A. Al-Mansob, A. Ismaila, A. N. Alduri, C. H. Azhari, M. R. Karim and N. I. M. Yusoffa, Constr. Build. Mater., 2014, 63, 242–248 CrossRef.
- S. Roy, B. R. Gupta and B. R. Maiti, Ind. Eng. Chem. Res., 1991, 30(12), 2573–2576 CrossRef CAS.
- N. Z. Noriman and H. Ismail, J. Appl. Polym. Sci., 2012, 123(2), 779–787 CrossRef CAS.
- S. Chuayjuljit, C. Yaowsang, N. Na-Ranong and P. Potiyaraj, J. Appl. Polym. Sci., 2006, 100(5), 3948–3955 CrossRef CAS.
- V. Tanrattanakul, B. Wattanathai, A. Tiangjunya and P. Muhamud, J. Appl. Polym. Sci., 2003, 90(1), 261–269 CrossRef CAS.
- T. Lin and B. Guo, Ind. Eng. Chem. Res., 2013, 52(51), 18123–18130 CrossRef CAS.
- M. Pire, S. Norvez, I. Iliopoulos, B. Le Rossignol and L. Leibler, Polymer, 2011, 52(23), 5243–5249 CrossRef CAS.
- M. Pire, S. Norvez, I. Iliopoulos, B. Le Rossignol and L. Leibler, Polymer, 2010, 51(25), 5903–5909 CrossRef CAS.
- L. Mascia, J. Clarke, K. S. Ng, K. S. Chua and P. Russo, J. Appl. Polym. Sci., 2015, 132(6), 24–39 CrossRef.
- P. T. Nghia, H. Onoe, Y. Yamamoto and S. Kawahara, Colloid Polym. Sci., 2008, 286(8), 993–998 CrossRef CAS.
- J. Samran, P. Phinyocheep, P. Daniel and S. Kittipoom, J. Appl. Polym. Sci., 2005, 95(1), 16–27 CrossRef CAS.
- S. Bhattacharjee, A. K. Bhowmick and B. N. Avasthi, Polymer, 1993, 34(24), 5168–5173 CrossRef CAS.
- J. R. Xu and A. E. Woodward, Macromolecules, 1988, 21(10), 2994–2997 CrossRef CAS.
- G. H. Hsiue and J. M. Yang, J. Polym. Sci., Part A: Polym. Chem., 1990, 28(13), 3761–3773 CrossRef CAS.
- X. Wei, L. Zhang, W. Wang and D. Yue, J. Appl. Polym. Sci., 2012, 124(2), 1716–1722 CrossRef CAS.
- M. J. R. Loadman and B. K. Tidd, in Natural Rubber Science and Technology, ed. A. D. Robberts, Oxford University Press, Oxford, UK, 1990, ch. 22 Search PubMed.
| This journal is © The Royal Society of Chemistry 2019 |