
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a photocurable acrylated poly(ethylene glycol)-co-poly(xylitol sebacate) copolymers hydrogel 3D printing ink for tissue engineering†
Yicai Wanga,
Yuan Li *a,
Xiaoling Yua,
Qizhi Longa and
Tian Zhang*ab
*a,
Xiaoling Yua,
Qizhi Longa and
Tian Zhang*ab
aSchool of Chemistry, Chemical Engineering and Life Science, Wuhan University of Technology, Wuhan, 430070, China. E-mail: liyuan2015@whut.edu.cn; tzhang@whut.edu.cn
bState Key Laboratory of Silicate Materials for Architectures, Wuhan University of Technology, Wuhan 430070, PR China
First published on 11th June 2019
Abstract
Photocurable hydrogel scaffolds for tissue engineering must have excellent biocompatibility, tunable mechanical characteristics, and be biodegradable at a controllable rate. Hydrogels developed as ink for 3D printing require several other properties such as optimal viscosity and shorter photocrosslinking time to ensure continuous extrusion and to avoid untimely collapse of the printed structure. Here, a novel photocurable hydrogel made of acrylated poly(ethylene glycol)-co-poly(xylitol sebacate) (PEXS-A) is developed for tissue engineering and 3D printing applications. Synthesis of PEXS-A hydrogel with equilibrated water content above 90% is achieved via a quick and facile photopolymerization process. Changing the acrylation ratio of the PEXS-A hydrogel has an impact on its crosslinking density, mechanical properties and degradation rate, thus highlighting PEXS-A tunability. PEXS-A could be employed as ink as demonstrated by the 3D printing of a 30-layers cubic grid with high structural integrity. Furthermore, 3T3 fibroblast cells encapsulated into PEXS-A during photocrosslinking maintain a viability of 93.76% after seven days, which showed the good biocompatibility of this novel hydrogel. These results indicate that PEXS-A hydrogel could have multiple applications including as 3D printing ink and as tissue engineering scaffold.
1. Introduction
Hydrogels are polymeric materials forming hydrophilic three-dimensional structures capable of storing large quantity of water.1,2 They have been developed for multiple applications including medical diagnostic, drug delivery and screening, tissue engineering, disease modeling as well as localized cancer treatment.3–6 In the case of tissue engineering, hydrogels serve as scaffold mirroring natural extracellular matrix and harboring living cells.7,8 Hydrogel scaffolds populated with cells can be injected or transplanted in situ to activate the reparation of damaged tissue.9,10For tissue engineering, hydrogel materials encapsulating cells should ideally have high biocompatibility, be easy to shape, have good mechanical tunability and be biodegradable at a controllable rate.11–13 Additionally, the hydrogel polymerization process must be gentle under physiological conditions with no impact on the viability of encased cells.14–16 One of the main strategies to prepare cell-containing hydrogels is to ensure crosslinking via photopolymerization.17 Recently, a photocurable hydrogel made of methacrylated poly(ethylene glycol)-co-poly(glycerol sebacate) (PEGS-M) copolymer was developed for tissue engineering.16 PEGS-M has many of the desired characteristics for applications related to tissue engineering. For instance, PEGS-M has a good biocompatibility for long-term cell cultivation and its mechanical properties as well as its in vitro biodegradation rate can be controlled by adjusting the degree of methacrylation.
One major approach for the fabrication of transplantable cell-containing hydrogel networks with high-resolution and refined structure is 3D printing.18 Examples of complex biomedical structures fabricated by 3D-printing with hydrogel ink include ear, articular cartilage, bone, microporous scaffold and vascular network.19–21 3D printing methods with high-printing resolution can also relies on photopolymerization for the synthesis of complex hydrogel structures developed for tissue engineering or other applications.22–25 Beside all the characteristics required for tissue engineering, hydrogels employed as ink for 3D printing must also have optimal viscosity to enable uninterrupted extrusion from the 3D printer nozzle.26 Furthermore, photocurable hydrogel inks should have shorter photocrosslinking time to ensure that the integrity of the printed structure is maintained.27
Here, we describe the synthesis of the novel copolymer acrylated poly(ethylene glycol)-co-poly(xylitol sebacate) (PEXS-A) and demonstrate its potential as photocurable hydrogel for tissue engineering or as ink for 3D printing. In PEXS-A, the glycerol monomer found in PEGS-M16 was replaced by xylitol to increase the number of available hydroxyl groups28 and the acylating agent employed was acryloyl chloride (AC), which has a greater nucleophilicity than 2-(methacryloyloxy)ethyl isocyanate.29 Both changes aimed at accelerating the photocrosslinking reaction to make PEXS-A suitable for 3D-printing. PEXS-A hydrogels with different acrylation ratio were fabricated and the physicochemical and mechanical properties of the different PEXS-A were characterized. PEXS-A was also tested as ink for photopolymerization-based 3D printing. Finally, the biocompatibility of PEXS-A was studied by assessing the short- and long-term viability of encapsulated 3T3 fibroblast cells.
2. Materials and methods
2.1. Materials
Poly(ethylene glycol) (PEG) (Mw = 6000), sebacic acid (SAA) (analytical grade, 99% pure), xylitol (analytical grade, 99% pure), acryloyl chloride (AC) (analytical grade, 99% pure), 4-(dimethyamino)pyridine (DMAP) and 2-hydroxy-4-(2-hydroxy-ethoxy)-2-methylpropiophenone (Irgacure 2959) were all purchased from Sigma-Aldrich. CCK-8 kit was purchased from Dojindo.2.2. Synthesis of acrylated poly(ethylene glycol)-co-poly(xylitol sebacate) copolymers (PEXS-A)
The copolymer PEXS was synthesized in two steps. The first step involved the polycondensation of sebacic acid (SAA) and PEG with a strict molar ratio (Table 1) at 130 °C under a flow of nitrogen gas for 2 hours before the reaction continued for another 24 h in vacuum oven (133 Pa) (Fig. 1 and Table S1†). In the second step, an appropriate amount of xylitol (Table S1†) was added into the reactor containing the SAA-PEG prepolymer and the reaction was further carried out at 130 °C under a nitrogen atmosphere for 4 hours before the reaction continued for another 48 h in vacuum oven (133 Pa) (Fig. 1). To synthesize high-purity PEXS, the product from the reaction between SAA-PEG and xylitol was then precipitated from DMF in diethyl ether, and was dried at 50 °C for 48 hours in vacuum oven (133 Pa).| Samples | Molar ratio PEG![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SAA:xylitol :SAA:xylitol |
Molar ratio xylitol:AC |
Actual molar ratio calculated from 1H-NMR (xylitol:AC) |
AC incorporationa |
|---|---|---|---|---|
| a AC incorporation is the percentage of added AC to the reaction that were comprised in the copolymer. | ||||
| PEXS-2A | 0.9:10:10 |
1:2 |
1:1.63 |
81.63% |
| PEXS-3A | 0.9:10:10 |
1:3 |
1:2.75 |
91.61% |
| PEXS-4A | 0.9:10:10 |
1:4 |
1:3.92 |
97.96% |
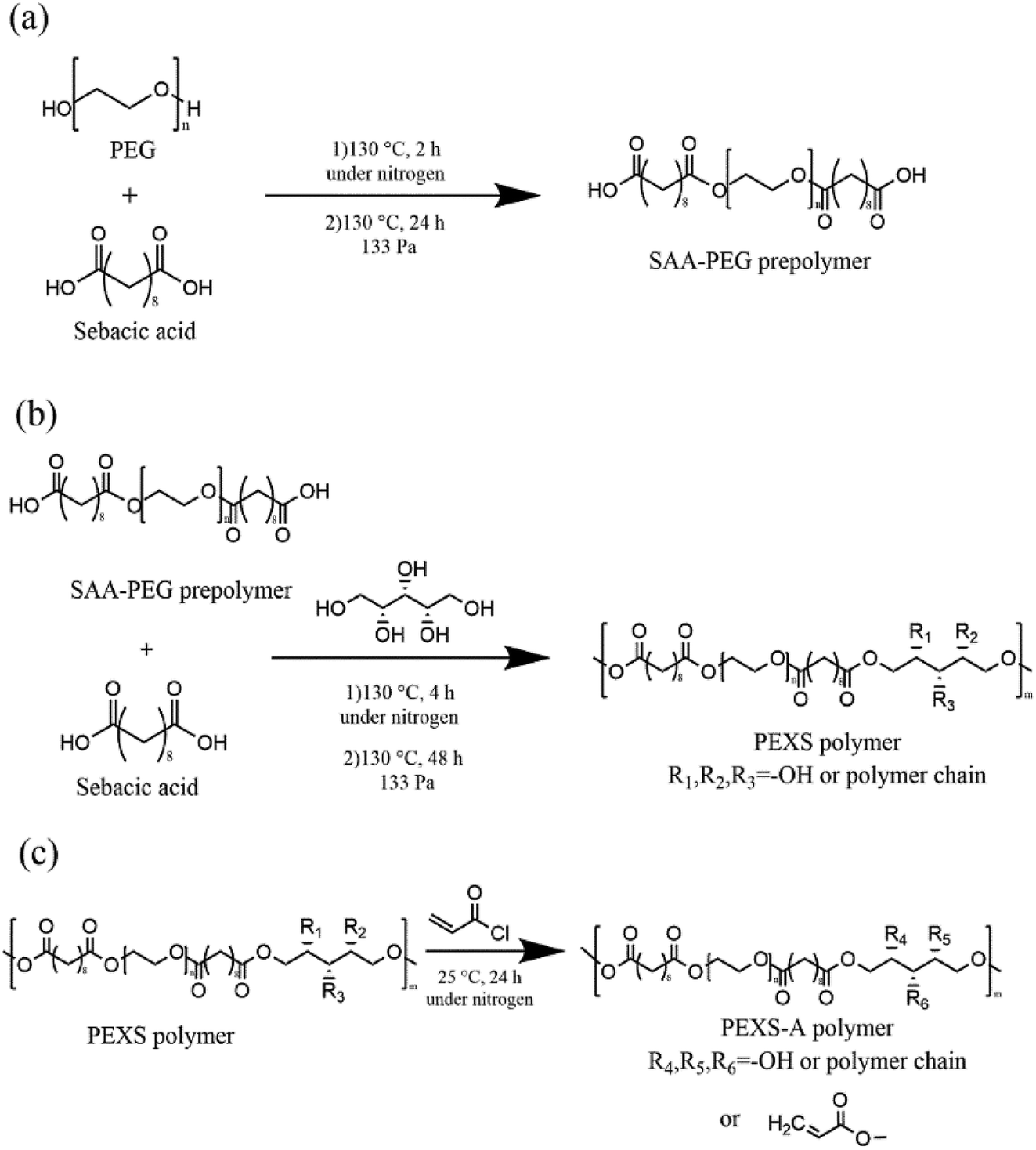 | ||
| Fig. 1 Synthesis of the PEXS-A copolymer. (a) Formation of the SAA-PEG prepolymer. (b) Synthesis of the PEXS copolymer following the addition of xylitol. R1, R2 and R3 represent –OH or polymer chain. (c) Acrylation of the PEXS copolymer into PEXS-A via a reaction between free hydroxyl groups in the PEXS copolymer and acyl groups from AC. R4, R5, R6 represent –OH, AC or polymer chain. | ||
For acrylation, the PEXS copolymer was dissolved in anhydrous dichloromethane (10% w/v) and then different molar ratios of AC were added (Table 1). After addition of the catalyst DMAP (0.1% w/v), the solution was stirred at 25 °C under nitrogen atmosphere for 24 hours (Fig. 1). Samples were named PEXS-2A, PEXS-3A and PEXS-4A, which means that the molar ratios of xylitol and AC was set as 1:2, 1:3 and 1:4, respectively. The products were then precipitated from DMF in diethyl ether before being freeze-dried.
2.3. Preparation of PEXS-A hydrogels via photopolymerization
The PEXS-A copolymers were first dissolved in 0.5% (w/v) of the photoinitiator Irgacure 2959 to reach a final concentration of 25% (w/v). 50 μl PEXS-A solution was placed in a mold and photopolymerized under 365 nm UV light at 12 mW cm−2 for 1 or 5 minutes.2.4. Characterization of PEXS-A copolymers and hydrogels
1H-NMR, 13C-NMR and FT-IR were used to characterize PEXS and PEXS-A. For 1H-NMR and 13C-NMR, PEXS and PEXS-A samples were dissolved in D2O and the 13C-NMR and 1H-NMR spectra were respectively recorded at 25 °C using AVANCE III HD (Bruker) and Ascend™ (Bruker) at 500 MHz. FT-IR spectra of PEG, PEXS and PEXS-3A copolymer and hydrogel were obtained with Bruker Vertex 80 V in the range of 400–4000 cm−1 over 65 scans. To maintain standard conditions, same mass of PEG, PEXS and PEXS-3A were employed for the preparation of the sample/KBr blends. Peak area of –OH in PEG, PEXS and PEXS-3A was calculated with Origin. For the photopolymerization kinetic of PEXS-3A, PEXS-3A copolymer and freeze-dried PEXS-3A hydrogels with different UV photocrosslinking time were analysed quantitatively by FT-IR. Peak area of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C at 1650 cm−1 was measured and normalized to the CO stretching peak at 1733 cm−1 as an internal standard to obtain CC conversion percentage. The molecular weight of PEXS and PEXS-3A copolymers were measured by gel permeation chromatography (GPC) (Polytech PL-GPC50) with tetrahydrofuran (THF) as the solvent. To observe the internal morphology of the photopolymerized hydrogel, PEXS-3A hydrogel was freeze-dried and broken in liquid nitrogen and section image were taken with a scanning electron microscope (SEM) (Hitachi S4800). The sample was platinum-coated and observed at an accelerating voltage of 10 kV.
C at 1650 cm−1 was measured and normalized to the CO stretching peak at 1733 cm−1 as an internal standard to obtain CC conversion percentage. The molecular weight of PEXS and PEXS-3A copolymers were measured by gel permeation chromatography (GPC) (Polytech PL-GPC50) with tetrahydrofuran (THF) as the solvent. To observe the internal morphology of the photopolymerized hydrogel, PEXS-3A hydrogel was freeze-dried and broken in liquid nitrogen and section image were taken with a scanning electron microscope (SEM) (Hitachi S4800). The sample was platinum-coated and observed at an accelerating voltage of 10 kV.
2.5. Swelling studies
First, PEXS-A hydrogels were soaked into phosphate-buffered saline (PBS) until water equilibrium was reached, and then rinsed with PBS for three times to remove uncrosslinked copolymer. Subsequently, PEXS-A hydrogels were freeze-dried and the dry weight (W1) was measured. Samples were then soaked for 24 hours into PBS at 37 °C until water equilibrium was reached. The surface of hydrogels was gently wiped to remove excessed PBS and the wet weight (W2) was measured. The equilibrium water content and swelling ratio of the hydrogels was calculated by using eqn (1) and (2):| equilibrium water content (%) = (W2 − W1)/W2 × 100 | (1) |
| equilibrium swelling ratio = W2/W1 × 100% | (2) |
2.6. Crosslink density (γ) and number average molecular weight between crosslinks (Mc)
The crosslink density (γ) and number average molecular weight between crosslinks (Mc) were determined from the volume fraction of the polymer in a water-swollen hydrogel (V) using eqn (5) and (6). They were calculated according to the modified Flory–Rehner's equation.30| V = 1/(1 + Δ) | (3) |
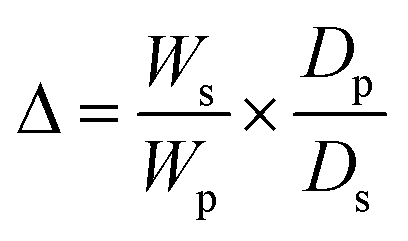 | (4) |
Crosslink density,
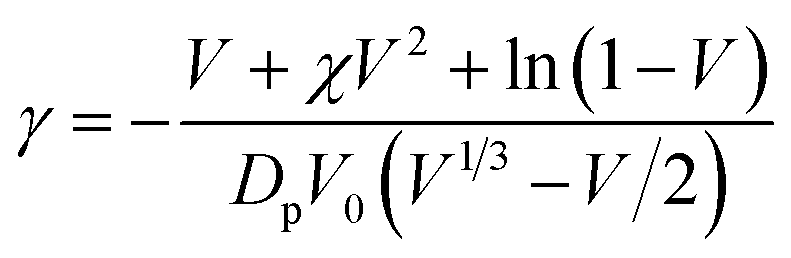 | (5) |
Molecular weight between crosslinks,
| Mc = 1/γ | (6) |
2.7. Compression testing
Compression testing was performed by the instrument of MTS (China) at room temperature. The samples were made into cylindrical shape with 4.64 × 4 mm (diameter × thickness) for testing and 500 N sensor of the MTS was used. The crosshead speed was 1 mm min−1 during the testing process. Hydrogels were compressed until broken and the strain of the hydrogels at this time was the max strain. Compressive stress, max strain and Young's modulus could obtain from this testing.2.8. Degradation of PEXS-A hydrogels
The physical stability of PEXS-A hydrogels was determined by conducting an in vitro degradation test for 28 days in PBS. Where indicated, 0.09 U esterase from pig liver (Yuanye Biotechnology, China) was added to the degradation reaction for each mg of hydrogel. At different time points, hydrogels were sampled from the degradation reaction and freeze-dried. Their weight (Wt) was measured and compared to the weight of freeze-dried hydrogels at day 0 (W0) according to eqn (7).| Remainning weight (%) = Wt/W0 × 100 | (7) |
Throughout the degradation test, the pH of the PBS solution was measured.
2.9. 3D printing of PEXS-A hydrogels
PEXS-3A with a 25% (w/v) concentration was used as ink for 3D printing. The viscosity of PEXS-3A was measured with a HBDV2TCP viscometer (Brookfield) at 5 rad min−1 and a shear rate (s−1) of 230 at 10, 20, 25 and 37 °C. A REGENOVO Bio-Architect WS extrusion-based 3D printer was used to print a cubic grid structure at room temperature, at a speed of 20 mm s−1 and an air pressure of 0.15 MPa. The printed scaffold had 30 layers with a height per layer of 0.11 mm. During 3D-printing, the UV light of the REGENOVO Bio-Architect was turned on for photopolymerization at an intensity of 85 mW cm−2. The rheological properties of the PEXS-3A copolymer ink and photocrosslinked PEXS-3A hydrogel were measured with a MCR 302 modular compact rheometer (Anton Paar) with 25 mm-diameter parallel plate. Dynamic frequency sweep test was conducted to monitor the storage (G′) and loss (G′′) moduli under shear amplitude of 0.1% with a frequency range of 1 to 100 rad s−1.2.10. NIH-3T3 cell culture
NIH 3T3 fibroblast cells (China Center for Type Culture Collection, Wuhan University, China) were cultured in DMEM medium supplemented with 10% fetal bovine serum, 1% penicillin and 1% streptomycin in T75 tissue culture flasks at 37 °C in a humidified 5% (v/v) CO2 incubator (Thermo Fisher Scientific). The cells were passaged when reaching 80–90% cell fusion. This study used cells from the third passage.2.11. Cytotoxicity of PEXS-A hydrogels
Two standard methods were employed to evaluate the cytotoxicity of PEXS-A hydrogels. In the first method, 3T3 cells were cultured in hydrogels DMEM extractant and cell viability was assessed with the CCK-8 kit (Dojindo) as described previously.33 Briefly, extractants were prepared by immerging 1 g biocompatible poly(D,L-lactic acid) (PDLLA), water-equilibrated PEXS-2A, PEXS-3A or PEXS-4A hydrogel in 10 ml DMEM supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin. The extraction reactions were then incubated at 37 °C for 24 hours before being filtered with 0.22 μm filter. Subsequently, DMEM extractants were used for 3T3 cells culture. 100 μl DMEM cell suspension containing 2000 cells were added to 96-well tissue culture plate and placed in the CO2 incubator. At this initial cell inoculation amount, the cells theoretically can expand to 80% fusion which is the optimal cell growth density in 48 hours. After six hours of incubation and establishment of cell adherence to the well, original DMEM medium was replaced by 100 μl of the different extractants. In the positive control, pristine DMEM medium was used instead of the extractants for medium substitution. After 2 days of incubation at 37 °C, culture medium was removed again and replaced by 100 μl pristine DMEM medium. 10 μl CCK-8 solution was then added to each well and the plate was further incubated for one hour. Finally, the medium absorbance was measured at 450 nm with a Multiskan FC microplate reader (Thermo Fisher Scientific).In the second method, 3T3 cells were cultured in DMEM medium in direct contact with the tested materials and the alamarBlue assay kit (Life Technology) was used to assess cell proliferation after 2, 4 and 6 days. AlamarBlue™ is non-toxic to cells and does not interrupt cell culture growth, which allows for a continuous measurement of cell proliferation kinetic. The reduction rate of alamarBlue is proportional to the number of metabolically active cells. Hence, the alamarBlue assay is appropriate for evaluating the long-term cytotoxicity of biomaterials that undergo biodegradation under physiological conditions.34 Briefly, materials were sterilized via UV exposure for two hours prior to hydrogel photopolymerization and cell cultivation. Before cytotoxicity test, hydrogels were washed with sterile DMEM medium to remove uncrosslinked copolymer. 500 μl cell suspension containing 2000 cells were added to 48-well tissue culture plate and placed in the CO2 incubator. After six hours of incubation and establishment of cell adherence to the well, 50 mg PDLLA, PEXS-2A, PEXS-3A or PEXS-4A hydrogel were added to cell culture plate. Material-free cell culture and sterile DMEM medium were also included as controls on the same plate. After 0, 2, 4 and 6 days of incubation, 50 μl of alamarBlue reagent was added to each well excluding background control and incubated under standard culture conditions for five hours more. Then, the medium was completely removed and transferred to a new 96-well plate for measuring alamarBlue reduction rate. Immediately, 500 μl fresh DMEM medium was added to each well of the 48-well tissue culture plate containing the active control cell cultures and the cell cultures with tested materials. AlamarBlue reduction rate was tested by measuring absorbance at 570 and 600 nm and calculated following the manufacturer's instructions.
2.12. Encapsulated cell viability of PEXS-A hydrogel
PEXS-3A was dissolved at a concentration of 25% (w/v) in sterile 0.5% (w/v) Irgacure 2959 solution prepared in PBS. In the next step, 3T3 cells were suspended at a concentration of 5 million per ml in the prepared PEXS-3A solution. 50 μl cell suspension was added to a sterile cylindrical mold and then hydrogel formation was activated via UV-photocrosslinking for one minute.The cell-laden PEXS-3A hydrogels were cultured in DMEM for 2 days and 7 days at 37 °C in a humidified 5% (v/v) CO2 incubator before cell viability testing. Live cells and dead cells were respectively stained by green fluorescent calcein AM and red fluorescent B using Calcein-AM/PI Double Stain Kit (Yeasen). The Live/Dead fluorescent staining of cells were observed under a confocal laser microscope (A1R HD25, Nikon). The relative cell viability and cell number were calculated by ImageJ.
2.13. Statistical analysis
All data are showed as mean and standard deviation (SD). GraphPad Prism 6.0 software was used to perform statistical analysis. Statistical significance was determined by one-way analysis of variance (ANOVA) and results were considered to be significant when p-value was below 0.05.3. Results and discussion
3.1. Synthesis of PEXS-A copolymer
The synthesis of the novel copolymer PEXS-A was done in three steps (Fig. 1). First, sebacic acid (SAA) and PEG were condensed to produce linear SAA-PEG pre-polymer. PEG with Mn of 6000 g mol−1 was chosen for the first step because it was shown to not cause significant adverse effects in animal models.35–37 In the second step, xylitol, which has five hydroxyl groups, was added to the SAA-PEG pre-polymer enabling the formation of the PEXS copolymer. In the last step, PEXS was acrylated via a reaction between AC and the available hydroxyl groups of PEXS to form PEXS-A.In steps 1 and 2 of the fabrication process (Fig. 1(a and b)), different PEXS copolymers with 50%, 60% or 70% weight percentage of PEG were synthesized with one to one molar ratio for SAA and xylitol (Table S1†). PEXS-70% and PEXS-60% were completely water soluble while PEXS-50% was only partially water soluble. Increasing PEG weight percentage in PEXS reduces the relative number of available hydroxyl group from xylitol in the copolymer, thus decreasing the quantity of reaction sites for acrylation by AC and possibly slowing down the photopolymerization process. PEXS-60% was used subsequently for PEXS-A synthesis to obtain a copolymer with good water solubility and lower PEG weight percentage. In step 3 (Fig. 1(c)), PEXS was acrylated with molar ratio of xylitol and AC of 1:2, 1:3 and 1:4 (Table 1). Later, PEXS-2A, PEXS-3A and PEXS-4A were the three novel copolymer materials tested for photopolymerization into hydrogel.
3.2. Characterization of PEXS
The structure of PEXS and PEXS-A copolymers was studied by 1H-NMR and FTIR spectroscopy (Fig. 2 and S1†). In the 1H-NMR spectrum of PEXS (Fig. 2(a)), peaks at 1.30 ppm (peak a), 1.60 ppm (peak b) and 2.37 ppm (peak c) corresponded to methylene protons of sebacic acid.33 Peak d at 3.70 ppm was identified as methylene protons from the PEG segment38 while peak e at 4.30 ppm (peak e) corresponded to methylene protons from xylitol. 1H-NMR results show that the PEXS copolymer comprising PEG, sebacic acid and xylitol was successfully synthesized.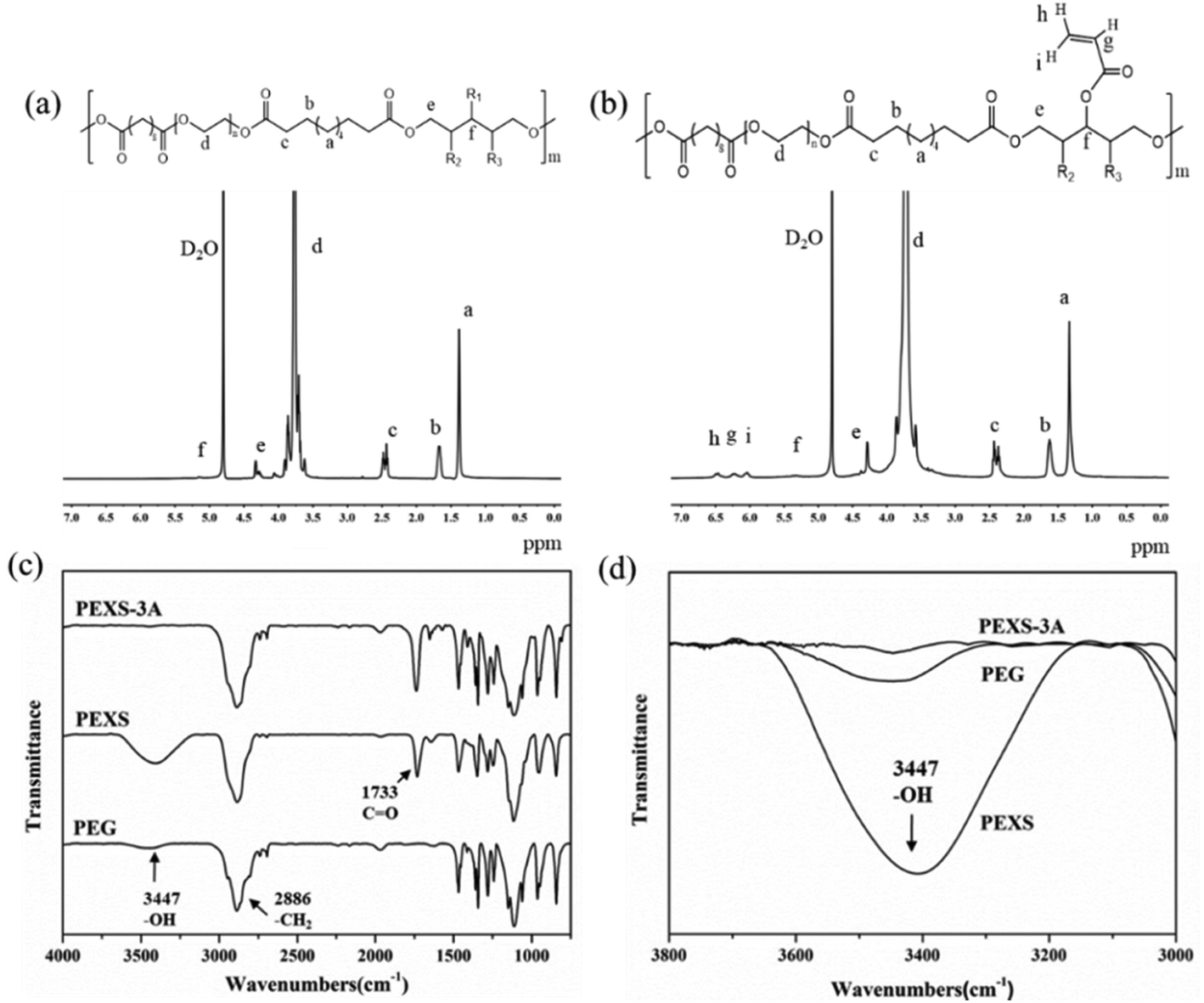 | ||
| Fig. 2 Characterization of PEXS and PEXS-A. 1H-NMR spectra of (a) PEXS and (b) PEXS-3A copolymers. (c) FT-IR spectra of PEG6000, PEXS and PEXS-A copolymer. (d) Decrease of the absorption peak for hydroxyl group (–OH) at 3447 cm−1 in PEXS-3A compared to PEXS. | ||
The incorporation of acrylate group was confirmed by the presence of new peaks at 6.04 ppm (peak i), 6.50 ppm (peak h) and 6.24 ppm (peak g) in the spectra of PEXS-A39 (Fig. 2(b) and S1†). To evaluate the acrylation degree of the PEXS-A copolymers, the signal intensity of protons at 6.04 ppm, which corresponds to vinyl group in the AC segment, was compared with the signal intensity of protons at 2.37 ppm from methylene groups in sebacic acid (Fig. 2(b) and S1†). For PEXS-2A, 81.63% of AC added in the acrylation reaction, was incorporated into the copolymer (Table 1). In the case of PEXS-3A and PEXS-4A, 91.61% and 97.96% of AC added to the reaction were comprised in the copolymer, respectively.
13C-NMR of PEXS-3A also demonstrated the successful synthesis of PEXS-A copolymers (Fig. S2†). The 13C-NMR spectrum included three peaks at 24.34 ppm, 28.14 ppm and 33.85 ppm corresponding to carbon atoms of sebacic acid. 60–70 ppm peaks corresponded to carbon atoms of PEG while 60–80 ppm peaks were related to carbon atoms of xylitol. Peaks at 127.37 ppm and 132.71 ppm corresponded to carbon atoms of acryloyl chloride.
FT-IR spectroscopy confirmed the formation of ester bonds in both PEXS and PEXS-3A with the presence of a significant absorption band at 1733 cm−1, which corresponds to carbonyl groups in ester linkages (Fig. 2(c)). A band at 3447 cm−1 and at 2886 cm−1 observed in the spectra for PEG, PEXS and PEXS-3A were respectively identified as free hydroxyl groups and methylene groups. In PEXS-3A, the absorption band of hydroxyl groups at 3447 cm−1 was significantly decrease, which may be due to substitution with AC (Fig. 2(d)).
The GPC result showed that PEXS has a Mw of 10521 Da and a polydispersity index of 1.18, while PEXS-3A has a Mw of 12389 Da and a polydispersity index of 1.07.
3.3. Formation of PEXS-A hydrogels
PEXS-A transited from liquid state to transparent gel state after photopolymerization by UV irradiation at 365 nm (Fig. 3). To induce photocrosslinking, the PEXS-A aqueous solution contained 0.5% (w/v) photoinitiator Irgacure 2959, which has a low cytotoxicity.40,41 PEXS-4A and PEXS-3A formed hydrogels after exposure to UV irradiation during short periods of time of one minute (Table 2). Hydrogel formation with PEXS-2A was slower and required five minutes of exposure to UV. Higher acrylation degree of PEXS-A reduced the period of time necessary for hydrogel formation by photopolymerization. For all three PEXS-A tested here, UV photocrosslinking time was well below ten minutes, which is the threshold where negative impact on cell viability starts being observed.42 Formation of PEXS-A hydrogels was confirmed by FT-IR spectroscopy (Fig. 3(c)). The intensity of the absorption band at 1650 cm−1, which corresponds to vinyl groups in the acrylate groups, was reduced in the PEXS-3A hydrogel compared to the PEXS-3A liquid copolymer. This observation can be explained by the consumption of vinyl groups by the photopolymerization process to form crosslink between the PEXS-A molecules. To establish precisely the minimum required time for optimal UV photocrosslinking of PEXS-3A hydrogel, the reduction of the vinyl groups absorption band at 1650 cm−1 was monitored after UV exposure for 15 seconds to 2 minutes (Fig. S3†). Photopolymerization was fast with most of the vinyl groups consumption occurring in the first 15 seconds before becoming constant after 60 seconds at 83.90 ± 0.89%. A SEM image of lyophilized PEXS-3A showed and interconnected porous structure (Fig. 3(d)). The average pore diameter was 31.11 ± 5.06 μm (n = 30). This microstructure could be promising for tissue regeneration applications. For instance, it has been shown that the optimum pore sizes for polymer scaffolds developed for adult mammalian skin morphogenesis was between 20 to 125 μm.43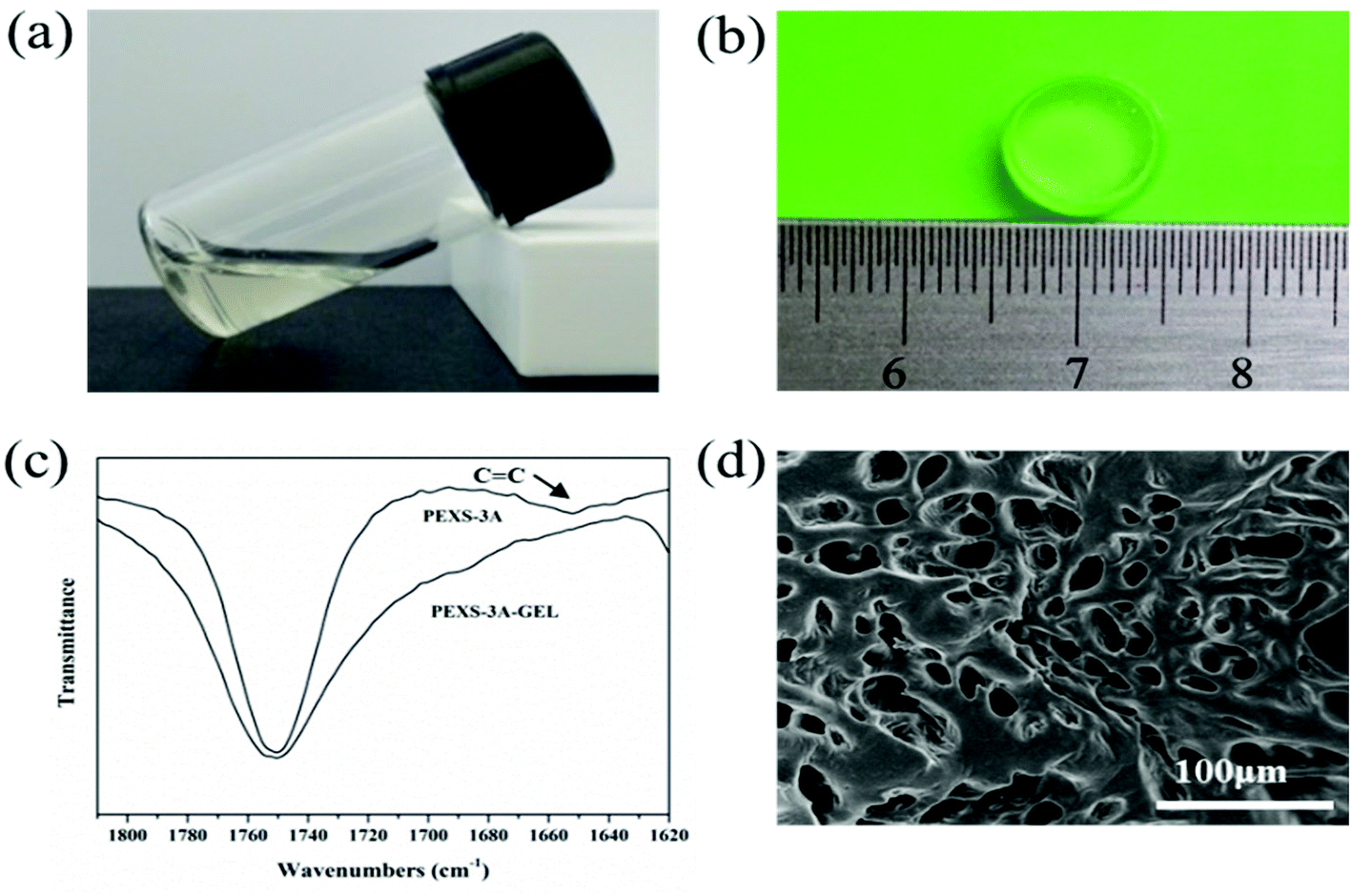 | ||
| Fig. 3 PEXS-A hydrogel formation via UV-activated photopolymerization. (a) PEXS-A copolymer solution. (b) The hydrogel image. (c) The decrease of the FT-IR absorption peak of the alkenyl group (CC) at 1650 cm−1 demonstrates the photocrosslinking of the PEXS-A hydrogel. (d) The SEM image of PEXS-3A hydrogel. | ||
| Hydrogels | Photo-polymerization time (min) | Crosslink density (γ)a (×10−4 mol cm−3) | Mca | Max. straina (%) | Max. compressive stressa (kPa) | Young's modulusa (kPa) |
|---|---|---|---|---|---|---|
| a The results are the mean and standard deviation of four replicates. | ||||||
| PEXS-2A | 5 | 1.65 ± 0.21 | 5942.05 ± 566.05 | 51.13 ± 0.30 | 74.72 ± 1.23 | 14.87 ± 4.40 |
| PEXS-3A | 1 | 4.12 ± 0.05 | 2428.17 ± 29.13 | 66.06 ± 2.84 | 449.97 ± 24.32 | 57.56 ± 0.72 |
| PEXS-4A | 1 | 8.13 ± 0.24 | 1230.31 ± 35.78 | 49.06 ± 3.97 | 178.57 ± 1.93 | 163.51 ± 5.39 |
3.4. Swelling and mechanical properties of PEXS-A hydrogels
PEXS-2A, PEXS-3A and PEXS-4A hydrogels were immersed in PBS for 24 hours at 37 °C to determine their equilibrated water contents as well as their swelling ratios (Fig. 4). All PEXS-A hydrogels reached above 88% equilibrium water content. PEXS-4A hydrogel had a lower equilibrated water content compared with PEXS-2A and PEXS-3A (Fig. 4(a)). Still, the three tested PEXS-A hydrogels had a water content suitable for encapsulated cells metabolism.44,45 PEXS-4A hydrogel also had a lower swelling ratio of 7.46 ± 0.19 compared to PEXS-2A and PEXS-3A, which had increases of 18.53 ± 1.29 and 10.91 ± 0.07 times their initial dry weight, respectively (Fig. 4(b)). Besides the swelling ratio, the crosslink density was modified by changing the acrylation degree of PEXS-A (Table 2). Crosslinking densities calculated with eqn (2) ranged from (1.65 ± 0.21) x 10−4 mol cm−3 for PEXS-2A to (8.13 ± 0.24) × 10−4 mol cm−3 for PEXS-4A. Higher acrylation degree of PEXS-4A resulted in higher crosslinking density and lower Mc, which explains why PEXS-4 could not absorb as much aqueous solution as PEXS-2A and PEXS-3A.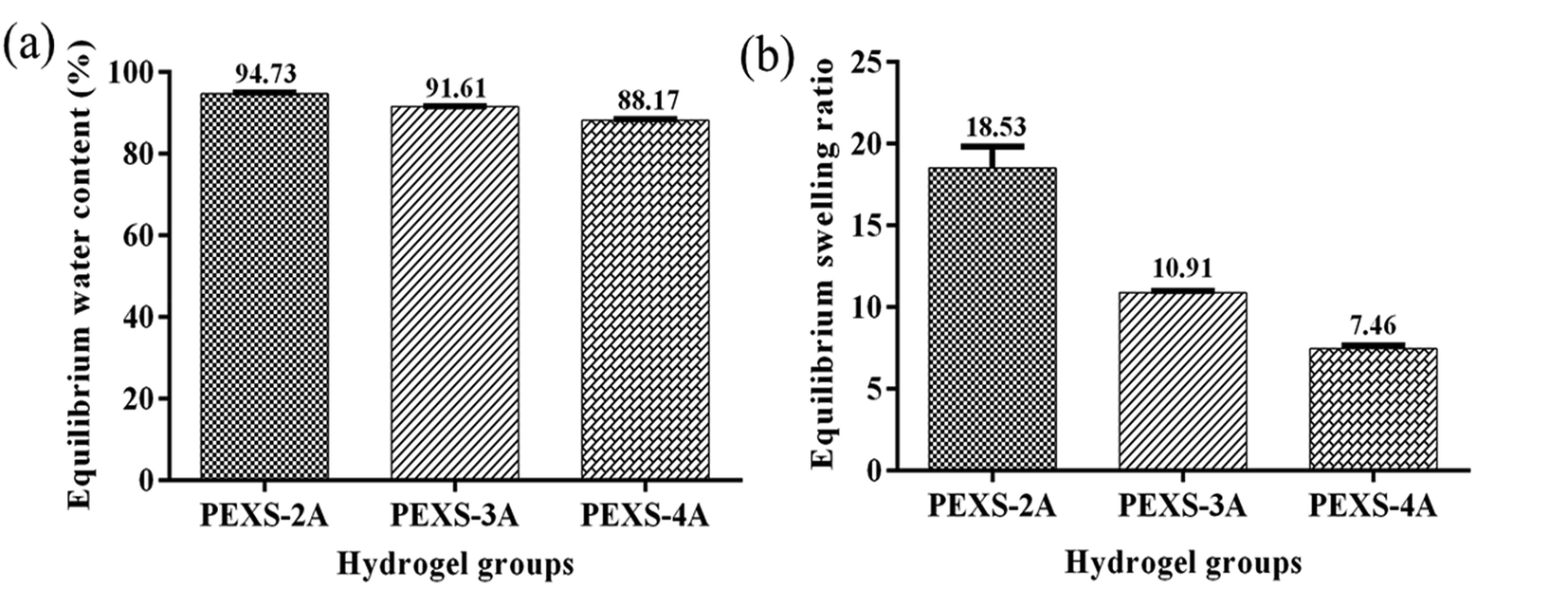 | ||
| Fig. 4 Swelling property of PEXS-A hydrogels immersed in PBS for 24 h at 37 °C. (a) Equilibrium water content and (b) swelling ratio of PEXS-2A, PEXS-3A and PEXS-4A hydrogels after 2 days of immersion in PBS at 37 °C (n = 4). | ||
Acrylation ratio also had an important impact on the mechanical properties of PEXS-A hydrogels (Table 2). PEXS-3A had the highest maximum strain of 66.06 ± 2.84% and maximum compressive stress of 449.97 ± 24.32 kPa. PEXS-4A hydrogel exhibited the highest Young's modulus of 163.51 ± 5.39 kPa compared to 14.87 ± 4.40 kPa for PEXS-2A and 57.56 ± 0.72 kPa for PEXS-3A. As expected, the observed Young's modulus variations were correlated with changes in swelling ratio and crosslinking density for hydrogels since higher crosslinking density lessens solvent content and increases interaction between polymer chains in the hydrogel network. Young's modulus of PEXS-A hydrogels matched the natural mechanical environment of various biological tissues such as the myocardium (10–50 kPa), collagenous bone (100 kPa), skin (∼85 kPa) and artery and vein (∼125 kPa).46,47
3.5. In vitro degradation of PEXS-A hydrogels
For tissue engineering applications, hydrogel scaffolds should be degraded at a suitable rate. An optimal scaffold should be durable long enough to ensure sufficient cell ingrowth. Afterward, it is expected that cells will generate their own extracellular matrix, which should gradually replace the biodegradable hydrogel scaffold.48 In vitro degradation rates of PEXS-2A, PEXS-3A and PEXS-4A hydrogels were assessed at 37 °C in PBS (Fig. 5). All PEXS-A had faster degradation rate in the first 7 days, which could be related to the leaching out of uncrosslinked copolymers from PEXS-A hydrogel. Subsequently, degradation rate significantly slowed down for the three tested hydrogels, which all lost less than 10% of their initial weight from day 7 to day 28 (Fig. 5(a)). The pH of the PBS buffer where PEXS-A hydrogels were submerged started at 7.4 ± 0.01 at day 0 before becoming acid at 6.24 ± 0.02, 6.50 ± 0.01, 6.58 ± 0.01 after two days for PEXS-2A, PEXS-3A and PEXS-4A, respectively (Fig. 5(b)). These pH drops were probably caused by the hydrolysis of the PXS segment and the release of SAA into PBS in the first two days.49 After that, the pH was relatively stable between 6.68 ± 0.02 to 7.17 ± 0.03 from day 4 to day 28 for all three hydrogels. During the four-week time period of the degradation test, the PEXS-4A hydrogel with the highest acrylation degree had the slowest degradation rate and lost only 21.26 ± 0.43% of its initial mass (Fig. 5(a)). PEXS-2A and PEXS-3A were degraded faster and ended up losing 54.60 ± 0.24 and 31.80 ± 0.95% of their initial weight, respectively. The higher degree of acrylation and the correlated higher crosslinking density of the PEXS-4A hydrogel may render the network formed by this copolymer more resistant to degradation. These results demonstrated that PEXS-A hydrogels are slowly degraded and that their degradation rates can be controlled by adjusting the degree of acrylation. These two properties indicate that PEXS-A hydrogels could be promising materials for tissue engineering applications where the scaffold harboring cells must be degraded at a tunable rate.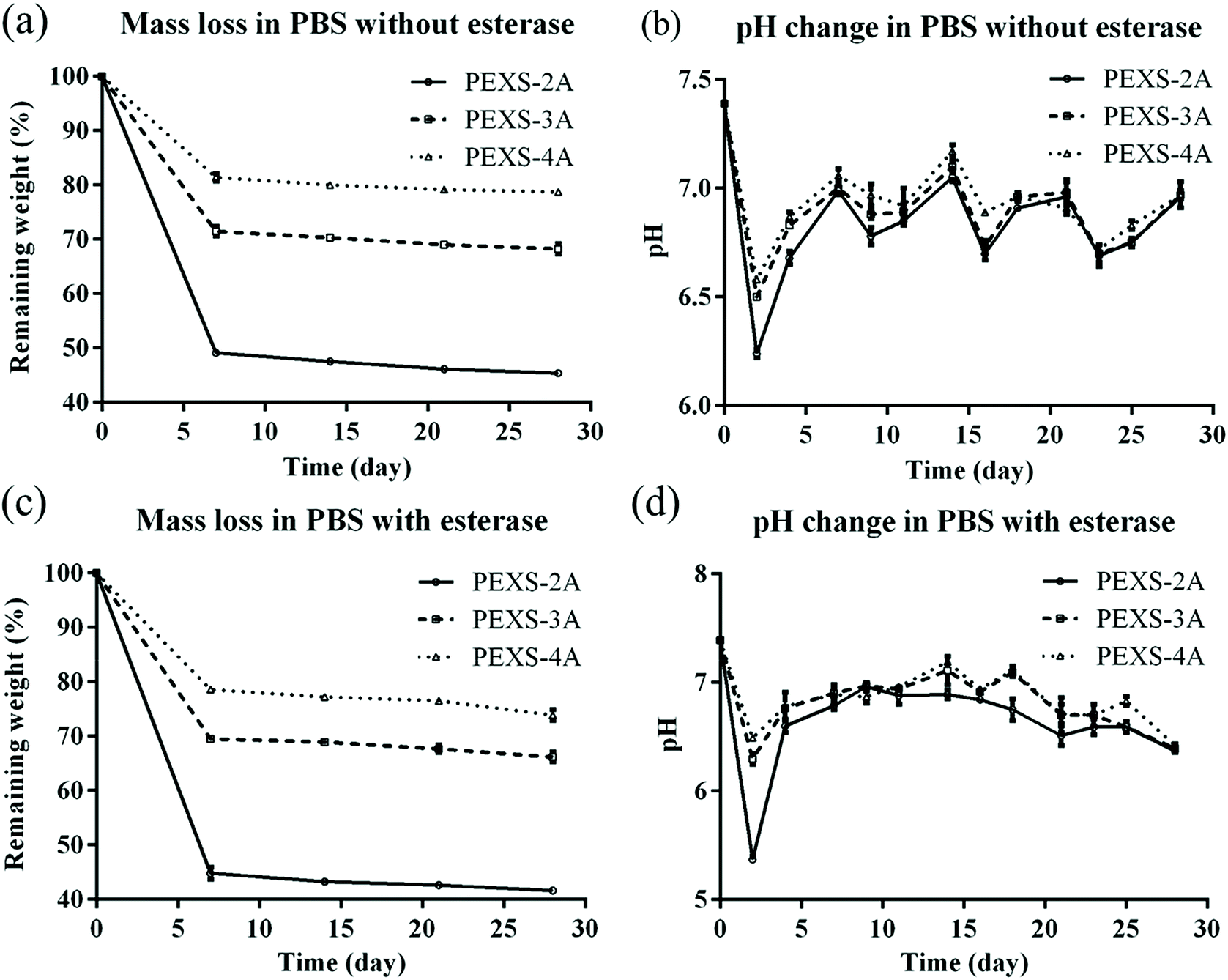 | ||
| Fig. 5 Degradation of PEXS-A hydrogels. (a) Weight loss of PEXS-A hydrogels and (b) pH change in PBS at 37 °C. (c) Weight loss of PEXS-A hydrogels and (d) pH change in PBS at 37 °C with added esterase (n = 4). | ||
A possible degradation mechanism of PEXS-A hydrogels would be via the breaking of ester bonds in the copolymer chain followed by the leaching out of monomers or small segments from the copolymer network. In a biological microenvironment, this could be accelerated by the presence of esterase enzyme. Here, we demonstrated that the degradation rate of PEXS-A hydrogels in PBS is accelerated by the presence of esterase (Fig. 5(c and d)). After 28 days, esterase-mediated degradation increased the mass loss of PEXS-2A, PEXS-3A and PEXS-4A hydrogels by 3.73 ± 0.16, 3.87 ± 0.40 and 4.22 ± 0.80%, respectively. These results provide initial evidences that PEXS-A hydrogels are biodegradable.
3.6. 3D printing with PEXS-A hydrogels
PEXS-A was tested as a potential ink for UV-curing based 3D printing with a REGENOVO Bio-Architect WS bioprinter (Fig. 6(a)). The viscosity of the extruded ink is a key parameter for 3D printing application. The viscosity of the PEXS-3A copolymer was measured at a concentration of 25% (w/v) at different temperature (Fig. S4†). From 10 to 37 °C, PEXS-3A had a viscosity varying between 5848 and 1029 mPa s, respectively. 3D printing test were done at a temperature of 20 °C at which PEXS-3A liquid copolymer had a viscosity of 4881 mPa s. Many hydrogel precursors previously employed as 3D printing ink have a viscosity in the same order of magnitude as PEXS-3A.26 The other important characteristic of PEXS-3A for printing is that it could form hydrogel by photopolymerization upon UV exposure quickly within one minute. The rheology of PEXS-3A prepolymer and hydrogel also demonstrate the rapid polymerization of PEXS-3A shown in Fig. S5,† where the storage and the loss moduli of PEXS-3A hydrogel photopolymerized in one minute were significant increased compared with that of PEXS-3A prepolymer. | ||
| Fig. 6 3D printing with PEXS-3A as ink. (a) 3D-printing procedure with a REGENOVO Bio-Architect WS bioprinter. (b) A top view and (c) a side view of a cubic grid printed with PEXS-3A hydrogel. (d) SEM image of the microstructure of a PEXS-3A printed cubic grid. | ||
A cubic grid pattern with a height of 30 layers was printed with PEXS-3A as ink (Fig. 6(b and c)). During the 3D printing process, the extrusion of PEXS-A from the printer nozzle was continuous and the UV-activated photopolymerization of the 3D scaffold was rapid. The printed PEXS-3A filament had a diameter of 478.57 ± 15.74 μm and the pore size of the cubic grid was 480 ± 14.14 μm (n = 30) (Fig. 6(d)). No collapse or deformation of the 3D scaffold structure and no pore occlusion was observed, which demonstrates that PEXS-A can be used as ink for 3D printing.
3.7. Cytotoxicity of PEXS-A hydrogels
To evaluate whether PEXS-A releases cytotoxic compounds, hydrogels were immerged in DMEM medium for 24 hour and then filtered out. DMEM medium possibly containing chemicals extracted from PEXS-A hydrogels was then used to cultivate NIH-3T3 fibroblast cells for two days before counting viable cells. The same process was done with PDLLA, and the resulting extractant was considered as a positive control for cell cultivation since PDLLA has been shown to be biocompatible.50 Cells cultivated in extractants from PDLLA, PEXS-3A and PEXS-4A hydrogels had a normal morphology and cell viability assessed with CCK-8 was comparable with cells cultivated in pristine DMEM medium that was not exposed beforehand to polymers (Fig. 7 and S6†). In the case of PEXS-2A hydrogel, exposing cells to the extractant resulted in a significant loss of viability to only 22.10 ± 0.19% (Fig. 7). This may be caused by the fact that there is less crosslinking in PEXS-2A and that larger quantity of cytotoxic compounds may be released from a more poorly organized structure.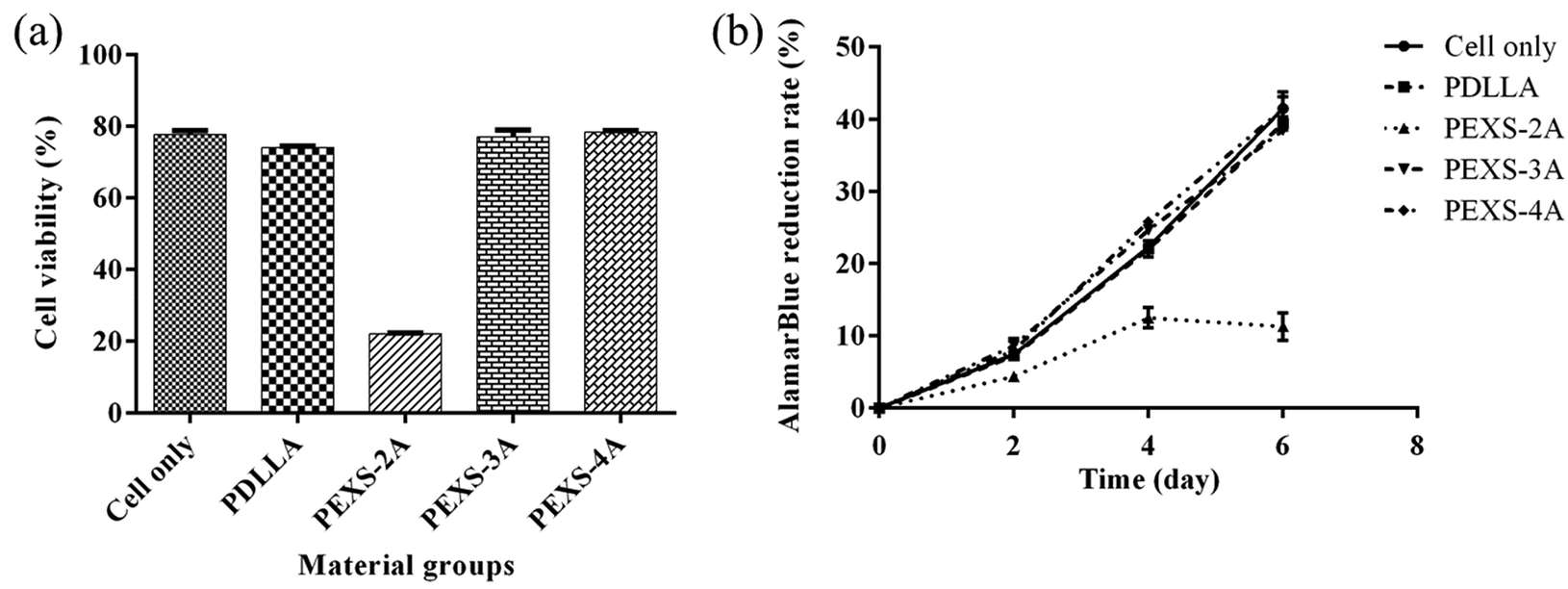 | ||
| Fig. 7 Cytotoxicity of PEXS-A hydrogels. (a) Cell viability after two-day exposure to DMEM extractant from PDLLA or PEXS-A hydrogels tested with CCK-8. (b) Cell proliferation measured with alamarBlue. AlamarBlue reduction rate is proportional to number of metabolically-active cell (n = 4). | ||
In a second method to evaluate the cytotoxicity of PEXS-A, 3T3 cells were incubated during six days in direct contact with PEXS-A hydrogels or PDLLA polymer. 3T3 cell proliferation was then assessed with the alamarBlue assay (Fig. 7(b)). Cell proliferation rate over six days in the presence of PEXS-3A and PEXS-4A hydrogels was comparable to the rates observed with cells cultivated alone or with PDLLA. This further confirmed that PEXS-3A and PEXS-4A are not cytotoxic, which is promising for biomedical applications. This assay also confirmed that PEXS-2A hydrogel is cytotoxic with a cell proliferation 3.67 ± 0.23 times slower than the cell-only control.
3.8. Cell encapsulation in PEXS-A hydrogels
To determine if PEXS-A copolymer can be used for tissue engineering, biocompatible PEXS-3A hydrogel was tested for 3T3 fibroblast cell encapsulation. Viability of cells incorporated before photocuration in PEXS-3A hydrogel was then tested with live/dead fluorescent staining after two and seven days of incubation in DMEM medium of (Fig. 8). CLSM images showed that a vast majority of encapsulated 3T3 cells stained green, which means that they were viable (Fig. 8(a and b)). A significantly lower number of cells stained red, which indicates cell death. 97.91 ± 0.13% and 93.76 ± 0.51% of cells were considered to be viable at day two and day seven, respectively (Fig. 8(c)). Furthermore, the number of cells encased in PEXS-3A hydrogel increased by 1.66 ± 0.04 times from day two to seven (Fig. 8(d)). These results demonstrate that PEXS-3A hydrogel is a suitable microenvironment for cell proliferation, which is a prerequisite for potential tissue engineering applications.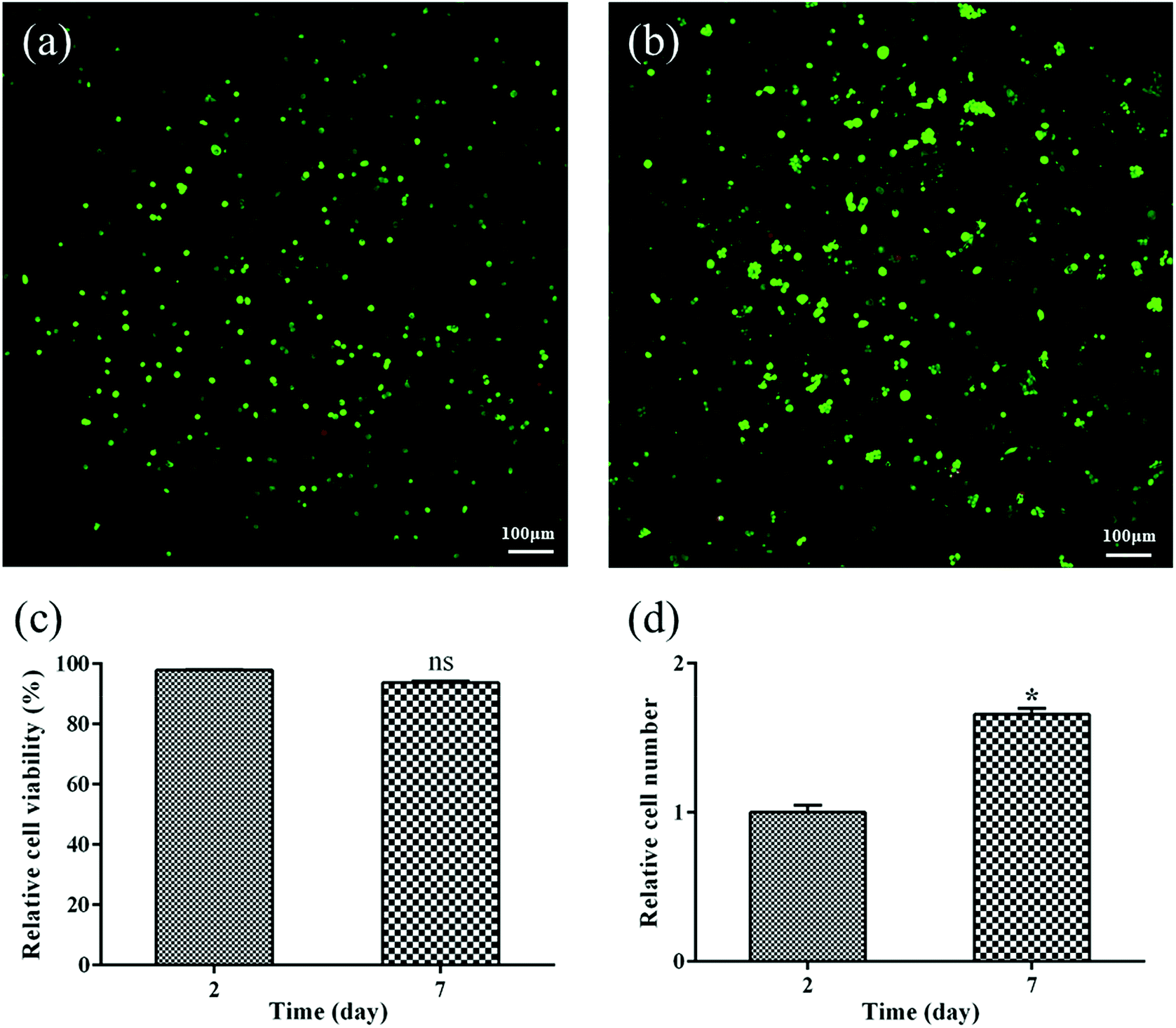 | ||
| Fig. 8 Cell encapsulation and proliferation in PEXS-3A hydrogel. CLSM images of encapsulated 3T3 cells after (a) two days or (b) seven days of incubation at 37 °C. Live/Dead fluorescent viability assay stained viable cells in green and dead cells in red. (c) Percentage of viable cells after two and seven days. (d) Increase of cell number from day 2 to day 7. nsDifference in relative cell viability between day 7 and day 2 is not statistically significant. *Difference in relative cell number between day 7 and day 2 is statistically significant (n = 4). | ||
4. Conclusions
PEXS-A hydrogels with different degree of acrylation were successfully synthesized via a quick photocrosslinking method under physiological conditions. The physicochemical and mechanical properties of PEXS-A hydrogels suggested that they are suitable microenvironments for cell proliferation. Cytotoxicity and viability assays as well as the proliferation of 3T3 cells after encapsulation in PEXS-3A hydrogel demonstrated the potential of these materials for tissue engineering applications. Interestingly, the characteristics of PEXS-A hydrogels such as the degradation rate or the Young's modulus could be modified by changing the acrylation ratio. The tunability of PEXS-A hydrogels is a major advantage for a wide range of biomedical purposes requiring adjustable scaffolds. Furthermore, the PEXS-3A copolymer has suitable viscosity and photopolymerization to serve as ink as demonstrated by the 3D printing of a stable complex structure of 30 layers. These results show that PEXS-A hydrogels could find applications either as tissue engineering platforms or for the fabrication of more elaborated scaffolds via 3D printing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. L. acknowledges the financial support of the Hubei Provincial Natural Science Foundation of China (2016CFB338) and Wuhan University of Technology, the Fundamental Research Funds for the Central Universities (WUT: 163120001), (WUT: 2016IVA084), (WUT: 2018IB023). T. Z. acknowledges the financial support of the Chinese Thousand Talents Plan Program and of Wuhan University of Technology.Notes and references
- R. Gauvin, R. Parenteau-Bareil, M. R. Dokmeci, W. D. Merryman and A. Khademhosseini, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 235–246 CAS.
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 CrossRef CAS PubMed.
- K. L. Fujimoto, Z. Ma, D. M. Nelson, R. Hashizume, J. Guan, K. Tobita and W. R. Wagner, Biomaterials, 2009, 30, 4357–4368 CrossRef CAS PubMed.
- X. Ma, X. Qu, W. Zhu, Y. S. Li, S. Yuan, H. Zhang, J. Liu, P. Wang, C. S. Lai and F. Zanella, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2206–2211 CrossRef CAS PubMed.
- J. Yang, J. Lee, J. Kang, S. J. Oh, H. J. Ko, J. H. Son, K. Lee, J. S. Suh, Y. M. Huh and S. Haam, Adv. Mater., 2010, 21, 4339–4342 CrossRef PubMed.
- W. Hong, X. Zhao and Z. Suo, J. Mech. Phys. Solids, 2010, 58, 558–577 CrossRef CAS.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- B. Guo, A. Finnewistrand and A. C. Albertsson, Biomacromolecules, 2011, 12, 2601–2609 CrossRef CAS PubMed.
- F. Byette, F. Bouchard, C. Pellerin, J. Paquin, I. Marcotte and M. A. Mateescu, Polym. Bull., 2011, 67, 159–175 CrossRef CAS.
- C. K. Kuo and P. X. Ma, J. Biomed. Mater. Res., Part A, 2010, 84, 899–907 Search PubMed.
- L. Saludas, S. Pascual-Gil, F. Prósper, E. Garbayo and M. Blanco-Prieto, Int. J. Pharm., 2017, 523, 454–475 CrossRef CAS PubMed.
- H. Tan and K. G. Marra, Materials, 2010, 3, 1746–1767 CrossRef CAS.
- Y. Li, J. Rodrigues and H. Tomás, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- L. Fertier, H. Koleilat, M. Stemmelen, O. Giani, C. Joly-Duhamel, V. Lapinte and J. J. Robin, Prog. Polym. Sci., 2013, 38, 932–962 CrossRef CAS.
- J. L. Ifkovits and J. A. Burdick, Tissue Eng., 2007, 13, 2369–2385 CrossRef CAS PubMed.
- Y. Wu, W. Ling, B. Guo and P. X. Ma, J. Mater. Chem. B, 2014, 2, 3674–3685 RSC.
- S. V. Vinogradov, T. K. Bronich and A. V. Kabanov, Adv. Drug Delivery Rev., 2002, 54, 135–147 CrossRef CAS PubMed.
- J. Hou, R. Chen, J. Liu, H. Wang, S. Qiang, Z. Xin, S. C. Wong and J. Yin, J. Mater. Chem. B, 2018, 6, 4792–4798 RSC.
- D. B. Kolesky, R. L. Truby, A. S. Gladman, T. A. Busbee, K. A. Homan and J. A. Lewis, Adv. Mater., 2014, 26, 2966 CrossRef.
- J. S. Miller, K. R. Stevens, M. T. Yang, B. M. Baker, D. H. T. Nguyen, D. M. Cohen, E. Toro, A. A. Chen, P. A. Galie and Y. Xiang, Nat. Mater., 2012, 11, 768–774 CrossRef CAS PubMed.
- T. Billiet, E. Gevaert, S. T. De, M. Cornelissen and P. Dubruel, Biomaterials, 2014, 35, 49–62 CrossRef CAS PubMed.
- A. P. Zhang, X. Qu, P. Soman, K. C. Hribar, J. W. Lee, S. Chen and S. He, Adv. Mater., 2012, 24, 4266–4270 CrossRef CAS PubMed.
- M. Gou, X. Qu, W. Zhu, M. Xiang, J. Yang, K. Zhang, Y. Wei and S. Chen, Nat. Commun., 2014, 5, 3774 CrossRef CAS PubMed.
- Z. Wei, J. Li, Y. J. Leong, I. Rozen, Q. Xin, R. Dong, Z. Wu, G. Wei, P. H. Chung and J. Wang, Adv. Mater., 2015, 27, 4411–4417 CrossRef PubMed.
- A. Ovsianikov, A. Deiwick, V. S. Van, P. Dubruel, L. Moller, G. Dräger and B. Chichkov, Biomacromolecules, 2011, 12, 851–858 CrossRef CAS PubMed.
- J. Malda, J. Visser, F. P. Melchels, T. Jüngst, W. E. Hennink, W. J. A. Dhert, J. Groll and D. W. Hutmacher, Adv. Mater., 2013, 25, 5011–5028 CrossRef CAS PubMed.
- F. P. W. Melchels, M. A. N. Domingos, T. J. Klein, J. Malda, P. J. Bartolo and D. W. Hutmacher, Prog. Polym. Sci., 2012, 37, 1079–1104 CrossRef CAS.
- Y. Li, W. Huang, W. D. Cook and Q. Chen, Biomed. Mater., 2013, 8, 035006 CrossRef PubMed.
- Q. Feng, F. Chen and X. Zhou, J. Biobased Mater. Bioenergy, 2012, 6, 336–342 CrossRef CAS.
- S. L. Liang, W. D. Cook, G. A. Thouas and Q. Z. Chen, Biomaterials, 2010, 31, 8516–8529 CrossRef CAS PubMed.
- M. Jayabalan, P. P. Lizymol and V. Thomas, Polym. Int., 2015, 49, 88–92 CrossRef.
- L. Krishna and M. Jayabalan, J. Mater. Sci.: Mater. Med., 2009, 20, 115–122 CrossRef PubMed.
- Y. Li, W. D. Cook, C. Moorhoff, W. C. Huang and Q. Z. Chen, Polym. Int., 2013, 62, 534–547 CrossRef CAS.
- Q. Z. Chen, Y. Li, L. Y. Jin, J. M. Quinn and P. A. Komesaroff, Acta Biomater., 2010, 6, 4143–4153 CrossRef CAS PubMed.
- G. Niemeyer, J. Am. Pharm. Assoc., 1947, 8, 194 CAS.
- M. J. Strauss, Arch. Dermatol. Syphilol., 1950, 61, 420–425 CrossRef CAS.
- H. F. Smyth, C. P. Carpenter and C. S. Weil, J. Am. Pharm. Assoc., 1955, 44, 27–30 CrossRef CAS.
- J. M. Dust, H. F. Zhi and J. M. Harris, Macromolecules, 1990, 22, 2077–2080 Search PubMed.
- C. L. E. Nijst, J. P. Bruggeman, J. M. Karp, F. Lino, Z. Andreas, C. J. Bettinger and L. Robert, Biomacromolecules, 2007, 8, 3067–3073 CrossRef CAS PubMed.
- C. G. Williams, A. N. Malik, K. Tae Kyun, P. N. Manson and J. H. Elisseeff, Biomaterials, 2005, 26, 1211–1218 CrossRef CAS PubMed.
- S. J. Bryant, C. R. Nuttelman and K. S. Anseth, J. Biomater. Sci., Polym. Ed., 2000, 11, 439–457 CrossRef CAS PubMed.
- I. Mironi-Harpaz, D. Y. Wang, S. Venkatraman and D. Seliktar, Acta Biomater., 2012, 8, 1838–1848 CrossRef CAS PubMed.
- I. V. Yannas, E. Lee, D. P. Orgill, E. M. Skrabut and G. F. Murphy, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 933–937 CrossRef CAS PubMed.
- A. K. Shung, E. S. Behravesh and A. G. Mikos, Tissue Eng., 2003, 9, 243–254 CrossRef CAS PubMed.
- M. A. Daniele, A. A. Adams, J. Naciri, S. H. North and F. S. Ligler, Biomaterials, 2014, 35, 1845–1856 CrossRef CAS PubMed.
- Q. Z. Chen, E. S. Harding, N. N. Ali, R. A. Lyon and R. A. Boccaccini, Mater. Sci. Eng., R, 2008, 59, 1–37 CrossRef.
- J. C. Bodle, A. D. Hanson and E. G. Loboa, Tissue Eng., Part B, 2011, 17, 195–211 CrossRef PubMed.
- F. J. O'Brien, Mater. Today, 2011, 14, 88–95 CrossRef.
- Y. Li, G. A. Thouas, H. Shi and Q. Chen, J. Biomater. Appl., 2014, 28, 1138–1150 CrossRef CAS PubMed.
- Y. M. Lin, A. R. Boccaccini, J. M. Polak, A. E. Bishop and V. Maquet, J. Biomater. Appl., 2006, 21, 109–118 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03637g |
| This journal is © The Royal Society of Chemistry 2019 |