
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular chains of coordinated dimolybdenum isonicotinate paddlewheel clusters†
Minyuan M. Li‡
 ab,
F. James Claire‡a,
Marina A. Solomos‡a,
Stephanie M. Tenney§
a,
Sergei A. Ivanovb,
Maxime A. Sieglera and
Thomas J. Kempa*ac
ab,
F. James Claire‡a,
Marina A. Solomos‡a,
Stephanie M. Tenney§
a,
Sergei A. Ivanovb,
Maxime A. Sieglera and
Thomas J. Kempa*ac
aDepartment of Chemistry, Johns Hopkins University, 3400 North Charles Street, Baltimore, MD 21218, USA. E-mail: tkempa@jhu.edu
bCenter for Integrated Nanotechnologies, Los Alamos National Laboratory, PO Box 5800, MS 1315, Albuquerque, NM 87185, USA
cDepartment of Materials Science & Engineering, Johns Hopkins University, 3400 North Charles Street, Baltimore, MD 21218, USA
First published on 28th May 2019
Abstract
A growing focus on the use of coordination polymers for active device applications motivates the search for candidate materials with integrated and optimized charge transport modes. We show herein the synthesis of a linear coordination polymer comprised of Mo2(INA)4 (INA = isonicotinate) metal–organic clusters. Single-crystal X-ray structure determination shows that this cluster crystallizes into one-dimensional molecular chains, whose INA-linked Mo2 cores engage in alternate axial and equatorial binding motifs along the chain axis. Electron paramagnetic resonance spectra, absorption spectra, and density functional theory calculations show that the aforementioned linear coordination environment significantly modifies the electronic structure of the clusters. This work expands the synthetic foundation for assembly of coordination polymers with tailorable dimensionalities and charge transport properties.
Over the past few decades, researchers have developed and investigated coordination polymers (CP) encompassing an extensive range of architectures.1–3 Interest in CPs most frequently focuses on their large accessible surface areas, especially in the case of porous metal–organic frameworks, and on their significantly tunable geometries. These features have led to the application of CPs in molecular sieving,4–6 contaminant encapsulation,7–9 catalysis,10–12 and sensing.13,14 With increased attention towards use of CPs as electrochemical and electronic materials,13,15,16 there is a need to incorporate and optimize charge transport pathways within the framework of these extended solids. The integration of coupled redox-active entities as part of the CP framework can facilitate charge delocalization and thereby engender long-range electronic transport pathways. This strategy has recently been employed to produce electrochemically- and electronically-active CP frameworks.16,17
We drew our attention to di-metallic tetracarboxylate clusters with metal–metal multiple bonds because of their tendency to form Class III Robin-Day mixed-valence complexes.18–20 These four-fold symmetric paddlewheel clusters exhibit redox behavior, and there is spectroscopic evidence for electronic communication within small supramolecular units of these clusters joined by organic or organometallic linkers.21–25 By comparison, the assembly of these clusters into extended solids and an interrogation of their properties is less developed. Obtaining well-defined and structurally robust CPs from these clusters can also be challenging.26–31
Motivated by these challenges and opportunities, we have recently focused on Mo2(INA)4 as a new building block for CPs. We previously reported the preparation of CP crystals comprised of commensurately stacked two-dimensional lattices of fully coordinated Mo2(INA)4 clusters.32 In this work, we were interested in accessing other crystal topologies containing the same molecular cluster. A red crystalline precipitate forms after refluxing a solution of Mo2(OAc)4, excess isonicotinic acid (H-INA), and trace acetic acid in dimethylformamide (DMF) for 2 days (Scheme 1). Electrospray ionization mass spectrometry of a solution of this red precipitate in dimethylsulfoxide (DMSO) and acetonitrile reveals peaks centered around m/z = 681 with isotopic patterning (Fig. S1†). This value matches the theoretical mass of Mo2(INA)4. The organic composition of the crystalline product is further verified by nuclear magnetic resonance (NMR) experiments following acid digestion of the precipitate (Fig. S2†). These data confirm that the crystalline product (1) of the reaction outlined in Scheme 1 is comprised of the Mo2(INA)4 cluster.
 | ||
| Scheme 1 Ligand exchange followed by polymerization yields product 1 after 2 days. | ||
Large single crystals of 1 were obtained by stirring the aforementioned reaction (Scheme 1) at 60 rpm (Fig. S3†). The structure of these single crystals was determined by X-ray crystallography (Fig. 1 and Table S1†). The crystal structure of 1 contains one-dimensional (1D) chains comprised of Mo2(INA)4 clusters that alternately bind through axial and equatorial binding sites. One DMF solvent molecule is coordinated to each of two Lewis acidic axial Mo sites on every other cluster. This coordination motif is supported by proton NMR data showing a DMF-to-INA ratio of approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.07 in acid-digested samples of 1 (Fig. S2†). The Mo–Mo bond lengths in the two clusters are 2.1219(5) Å and 2.1189(5) Å. These distances are within the anticipated range of lengths for Mo–Mo quadruple bonds (2.06–2.17 Å as described by Murillo et al.) and consistent with axial coordination of a strongly donating ligand such as pyridine.22,33,34 Furthermore, we observe a Raman band at 383 cm−1 (Fig. S4†), which is relatively weaker than that anticipated for axially under-coordinated Mo2 complexes, and treat this observation as further evidence for the axial donation effect.22,34 We note that the Mo2 centers within each molecular chain are coordinatively saturated and do not bond to pyridines from neighboring chains (Fig. S5†).
:4.07 in acid-digested samples of 1 (Fig. S2†). The Mo–Mo bond lengths in the two clusters are 2.1219(5) Å and 2.1189(5) Å. These distances are within the anticipated range of lengths for Mo–Mo quadruple bonds (2.06–2.17 Å as described by Murillo et al.) and consistent with axial coordination of a strongly donating ligand such as pyridine.22,33,34 Furthermore, we observe a Raman band at 383 cm−1 (Fig. S4†), which is relatively weaker than that anticipated for axially under-coordinated Mo2 complexes, and treat this observation as further evidence for the axial donation effect.22,34 We note that the Mo2 centers within each molecular chain are coordinatively saturated and do not bond to pyridines from neighboring chains (Fig. S5†).
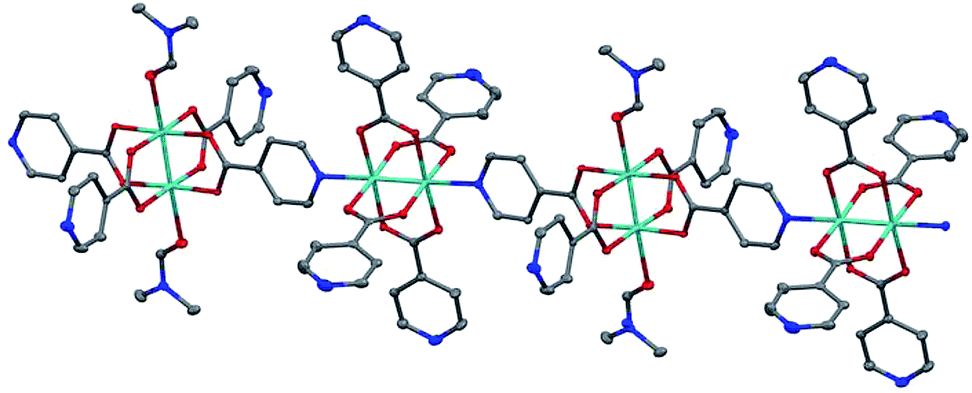 | ||
| Fig. 1 Structure of the 1D coordination polymer (1) showing the paddlewheel units that alternately bond through axial Mo (cyan) and equatorial N (blue) binding sites. Ellipsoids are shown at 50% probability level. Disorder and H atoms have been omitted for clarity. | ||
We next characterized the phase purity, stability, and porosity of bulk samples of 1. First, powder X-ray diffraction (pXRD) patterns collected from products precipitated across a range of stir rates (60–300 rpm) match well with the pXRD pattern simulated from the single-crystal XRD data discussed above (Fig. 2 and S3†). These data suggest that a single common crystal phase is produced regardless of stir rate, though stir rates <100 rpm are conducive to the formation of high-quality single crystals. Second, Fourier transform infrared (FT-IR) spectroscopy data show no evidence of H-INA or Mo2(OAc)4 in the bulk crystalline powder (Fig. S6†), suggesting good purity of our bulk samples. Third, thermogravimetric analysis shows a mass loss of 10.1% between 190 °C and 252 °C that can be attributed to the loss of coordinated DMF molecules, which account for 9.7% of the 1D CP (Fig. S7†). Upon further heating, the sample rapidly loses mass above 350 °C and likely degrades. Finally, nitrogen gas adsorption measurements on the crystalline powder yield a Brunauer–Emmett–Teller (BET) surface area of 325.8 m2 g−1 (Fig. S8†). We note that this modest BET surface area is reasonable given the tight packing of the 1D chains within 1 (Fig. S5†).
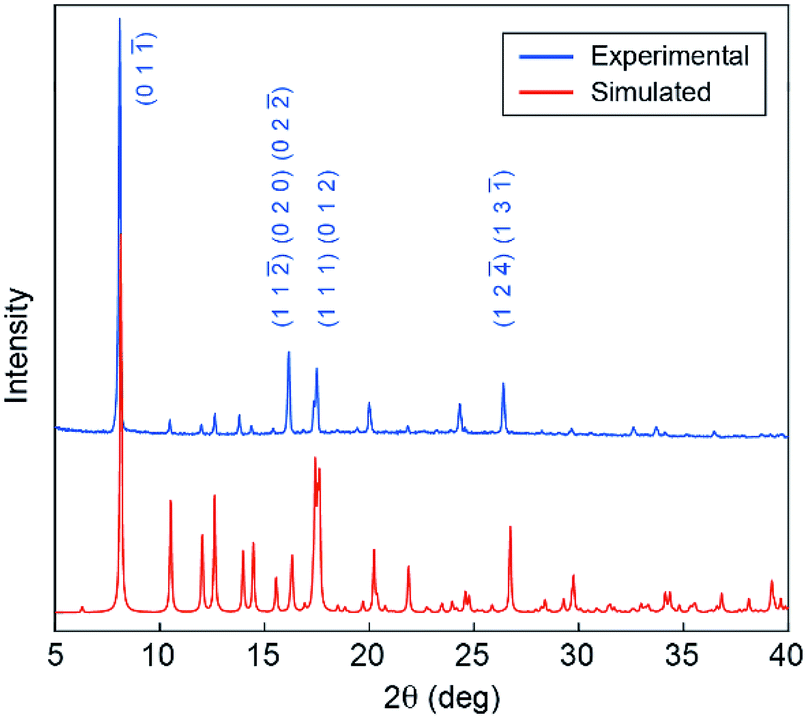 | ||
| Fig. 2 Experimental pXRD pattern for powder precipitated from a DMF solution stirred at 300 rpm (blue), and simulated pXRD pattern obtained from single-crystal XRD data of 1 (red). | ||
Finally, we sought to analyze the electronic structure of 1 with absorption spectroscopy, density-functional theory (DFT) calculations, and electron paramagnetic resonance (EPR) spectroscopy. A diffuse reflectance UV-vis spectrum collected from a powder sample of 1 exhibits a broad absorption feature between ∼400 and 600 nm (Fig. 3a). The UV-vis absorption spectrum of a solution sample of 1 dissolved in DMF exhibits a significant feature centered at ∼470 nm. We ascribe the modest absorption shoulder at ∼570 nm to absorption by remnant 1D CP chains. We used computational methods35,36 to examine orbital interactions and electronic transitions that may be responsible for the absorption features identified above. Single point energies and molecular orbitals were calculated from coordinates taken directly from the single-crystal structure of 1. The B3LYP functional was used with the 6-31g* basis set for H, C, N, O and the Stuttgart/Dresden effective core potential for Mo. In the ground state, the first four highest-energy occupied molecular orbitals are all metal-centered, with discrete energy levels, localized electron density, and little sharing between the two coordinatively distinct clusters (Fig. S9†). These computational results suggest that charge (e.g. introduced from an external circuit or generated chemically in situ) would likely remain localized on the individual clusters comprising 1.
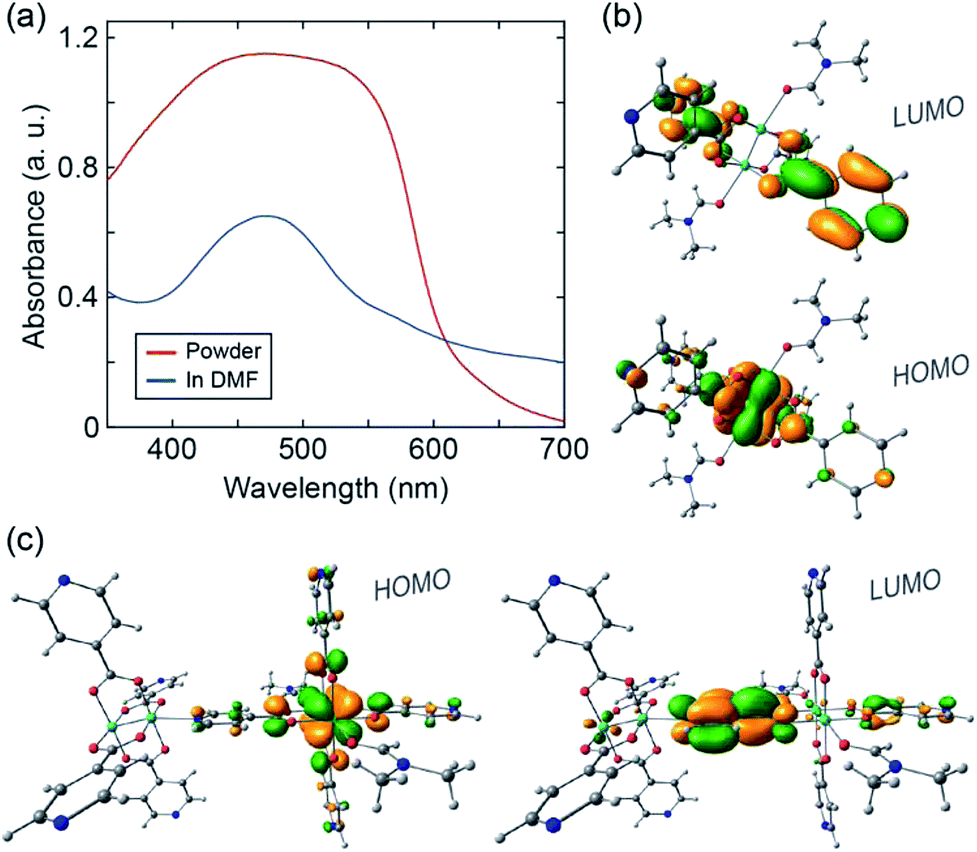 | ||
| Fig. 3 (a) UV-vis spectra of 1 in powder form (red) and dissolved in DMF (blue). (b) DFT calculated HOMO and LUMO orbitals of the monomer. (c) DFT calculated HOMO and LUMO orbitals of the dimer. | ||
We performed time-dependent DFT (TD-DFT) calculations to identify possible optical transitions in 1. We did so by considering the electronic structure of a single Mo2(INA)4 cluster coordinated by a DMF molecule at both axial sites, and of two Mo2(INA)4 clusters having the same coordination environment as observed in the crystal structure of 1. Henceforth we will refer to the former as the monomer and the latter as the dimer. For the monomer, calculations identify a 2.44 eV (508 nm) transition primarily between a HOMO of Mo2δ character and a ligand-based π* LUMO, which is nearly degenerate with several higher lying LUMO orbitals (Fig. 3b and S10†). The character of this transition is typical of the metal–ligand charge transfer (MLCT) transitions reported in analogous transition-metal complexes. We note that the calculated HOMO–LUMO transition energy for the monomer is in reasonable agreement with the 470 nm feature observed in the absorption spectrum of the solution sample of 1 dissolved in DMF. On the basis of these data we can assign the aforementioned 470 nm feature to absorption by the isolated monomer.
Turning our attention to the dimer, TD-DFT calculations identify a 2.19 eV (566 nm) transition primarily between a HOMO of Mo2δ character and a ligand-based π* LUMO orbital (Fig. 3c and S11†). We also note two higher energy MLCT transitions primarily from HOMO to LUMO+5 and from HOMO−1 to LUMO+1 with energies of 2.59 eV (479 nm) and 2.61 eV (475 nm), respectively (Fig. S11†). Notably, while the two higher energy MLCT transitions involve LUMO orbitals located on the INA ligands perpendicular to the dimer axis, the lower energy MLCT transition at 2.19 eV involves a LUMO orbital of the INA ligand along the dimer axis.
The calculated dimer transition energies cover much of the broad absorption envelope observed in the spectrum of the powder of 1. In particular, the low energy MLCT transition at 566 nm calculated for the dimer likely accounts for the significant absorption at wavelengths greater than 500 nm (Fig. 3a). These data suggest the dimer is a good simulant of the electronic properties of 1. This is a reasonable expectation given that 1 can be viewed, from a structural standpoint, as a linear polymer chain of the dimers. We can conclude that our 1D CP has an electronic structure comprised of dimer-like MLCT states arising from a lifting of the degeneracy of monomer orbitals due to their altered symmetry in the 1D chain.
Our results show that the alternating coordination environment along the backbone of our 1D CP gives rise to a unique chain topology with a distinct electronic structure and function. Further evidence for the functionally distinct nature of 1 over the monomer is provided by X-band electron paramagnetic resonance (EPR) spectroscopy. An EPR spectrum collected from a powder sample of 1 at 20 K reveals an EPR active system with weak hyperfine structure owing to 95Mo and 97Mo isotopes, which have I = 5/2 nuclear spin (Fig. 4). This spectrum is qualitatively similar to spectra reported for EPR active Mo2V species in transition metal alkoxide-bridged dimolybdenum clusters.23 This suggests the presence of MoII and MoIII centers in 1 that may have formed during partial oxidation of the powder sample. It is anticipated that the hole is equally present on both Mo atoms as expected from the Class III Robin-Day classification of such systems. We simulated the experimental EPR spectrum by modelling our 1D CP system as a single axial paramagnet and obtained g values of g‖ = 1.895 and g⊥ = 1.935 and a perpendicular hyperfine coupling constant A⊥ of 3.9 mT (Fig. 4 and S12†).37 The hyperfine coupling observed for our 1D CP is consistent with values reported for systems containing isolated Mo2V ions.23 Moreover, the fact that a single paramagnet model was sufficient to simulate the EPR spectrum suggests that the two distinct coordination environments are fairly similar electronically. Though our EPR data provide evidence for charge localization38 on Mo2 centers, we cannot exclude the possibility for photo-induced intra-chain (or indeed inter-chain) charge transport arising from population of the low energy LUMO orbital on each bridging INA ligand.
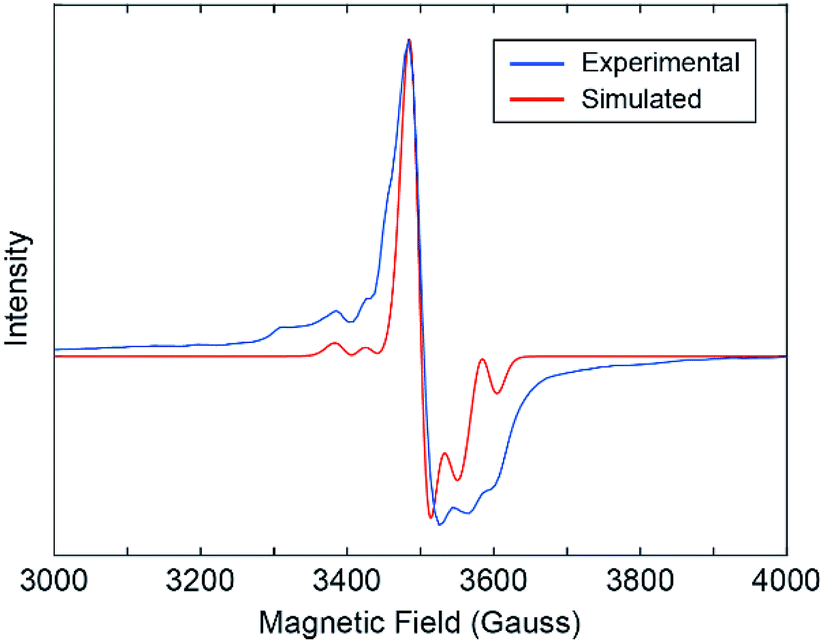 | ||
| Fig. 4 Experimental EPR spectrum of 1 collected at 20 K (blue) and 9.44 GHz. In EasySpin, a fifth-order background correction was applied and the system was modeled as an axial paramagnet with g‖ = 1.895 and g⊥ = 1.935 (red). | ||
In summary, we have synthesized through a one-step ligand substitution reaction a new inorganic cluster comprised of a quadruply bonded dimolybdenum core coordinated by 4 equatorial isonicotinate ligands: Mo2(INA)4. Single-crystal XRD data show that recrystallization of this cluster from DMF yields a 1D molecular chain wherein the clusters alternately coordinate to one another through axial and equatorial binding sites (1). pXRD, FT-IR, and NMR data confirm the phase and compositional purity of this 1D CP in the bulk. UV-vis spectroscopy and DFT calculations reveal an electronic structure for 1 that is distinct from an individual solvated Mo2(INA)4 monomer owing to the alternating coordination environment present along the chain. This work establishes the synthetic foundation for assembly of CPs from discrete molecular clusters that have the potential to support through-framework charge transport due to mixed-valence mechanisms. We also envision utilizing the molecular building block discussed herein for the assembly of CPs with tailorable dimensionalities or hierarchies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. J. K. acknowledges funding from the National Science Foundation (DMR-1848046) CAREER grant. T. J. K. and M. M. L. acknowledge funding from a Camille and Henry Dreyfus Foundation under a grant from the Postdoctoral Program in Environmental Chemistry (EP-15-046). We thank Austin Herzog (Dept. of Chemistry, Johns Hopkins University) for assistance with EPR, and the Center for Integrated Nanotechnologies, a user facility of the U.S. Department of Energy Office of Science.Notes and references
- J. J. Perry IV, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400–1417 RSC.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- M. J. Van Vleet, T. Weng, X. Li and J. R. Schmidt, Chem. Rev., 2018, 118, 3681–3721 CrossRef CAS PubMed.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- X. Li, Y. Liu, J. Wang, J. Gascon, J. Li and B. Van der Bruggen, Chem. Soc. Rev., 2017, 46, 7124–7144 RSC.
- S. Mukherjee, A. V. Desai and S. K. Ghosh, Coord. Chem. Rev., 2018, 367, 82–126 CrossRef CAS.
- L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- J. Li, X. Wang, G. Zhao, C. Chen, Z. Chai, A. Alsaedi, T. Hayat and X. Wang, Chem. Soc. Rev., 2018, 47, 2322–2356 RSC.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC.
- Q. Yang, Q. Xu and H.-L. Jiang, Chem. Soc. Rev., 2017, 4774, 4774–4808 RSC.
- K. Vellingiri, L. Philip and K.-H. Kim, Coord. Chem. Rev., 2017, 353, 159–179 CrossRef CAS.
- A. Chidambaram and K. C. Stylianou, Inorg. Chem. Front., 2018, 5, 979–998 RSC.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- J. Zhou and B. Wang, Chem. Soc. Rev., 2017, 46, 6927–6945 RSC.
- I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC.
- L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed.
- F. A. Cotton, J. P. Donahue and C. A. Murillo, Inorg. Chem. Commun., 2002, 5, 59–63 CrossRef CAS.
- N. J. Patmore, in Organometallic Chemistry, Royal Society of Chemistry, Cambridge, 2010, pp. 77–92 Search PubMed.
- F. A. Cotton, A. Chun Lin and C. A. Murillo, Acc. Chem. Res., 2001, 34, 759–771 CrossRef CAS PubMed.
- M. H. Chisholm and A. M. Macintosh, Chem. Rev., 2005, 105, 2949–2976 CrossRef CAS PubMed.
- M. H. Chisholm, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2563–2570 CrossRef CAS PubMed.
- F. A. Cotton, N. S. Dalal, C. Y. Liu, C. A. Murillo, A. J. Micah North, X. Wang, F. A. Cotton, N. S. Dalal, C. Y. Liu, C. A. Murillo, J. M. North and X. Wang, J. Am. Chem. Soc., 2003, 125, 12945–12952 CrossRef CAS PubMed.
- M. H. Chisholm and B. J. Lear, Chem. Soc. Rev., 2011, 40, 5254 RSC.
- C. P. Kubiak, Inorg. Chem., 2013, 52, 5663–5676 CrossRef CAS PubMed.
- M. H. Chisholm, A. S. Dann, F. Dielmann, J. C. Gallucci, N. J. Patmore, R. Ramnauth and M. Scheer, Inorg. Chem., 2008, 47, 9248–9255 CrossRef CAS PubMed.
- F. A. Cotton, J.-Y. Jin, Z. Li, C. Y. Liu and C. A. Murillo, Dalton Trans., 2007, 2328–2335 RSC.
- W. Hsu, Y.-S. Li, H.-Y. He, K.-T. Chen, H.-S. Wu, D. M. Proserpio, J.-D. Chen and J.-C. Wang, CrystEngComm, 2014, 16, 7385–7388 RSC.
- X.-M. Cai, D. Höhne, M. Köberl, M. Cokoja, A. Pöthig, E. Herdtweck, S. Haslinger, W. A. Herrmann and F. E. Kühn, Organometallics, 2013, 32, 6004–6011 CrossRef CAS.
- Y. Ke, D. J. Collins and H.-C. Zhou, Inorg. Chem., 2005, 44, 4154–4156 CrossRef CAS PubMed.
- A. Y. Robin and K. M. Fromm, Coord. Chem. Rev., 2006, 250, 2127–2157 CrossRef CAS.
- F. J. Claire, S. M. Tenney, M. M. Li, M. A. Siegler, J. S. Wagner, A. S. Hall and T. J. Kempa, J. Am. Chem. Soc., 2018, 140, 10673–10676 CrossRef CAS PubMed.
- F. A. Cotton, L. M. Daniels, E. A. Hillard and C. A. Murillo, Inorg. Chem., 2002, 41, 2466–2470 CrossRef CAS PubMed.
- F. A. Cotton and J. G. Norman, J. Am. Chem. Soc., 1972, 94, 5697–5702 CrossRef CAS.
- A. S. Miehlich, H. S. Toll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef.
- B. G. Alberding, M. H. Chisholm, J. C. Gallucci, Y. Ghosh and T. L. Gustafson, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8152–8156 CrossRef CAS PubMed.
- F. E. Mabbs and D. Collison, Electron Paramagnetic Resonance of d Transition Metal Compounds, Elsiever Science Publishers, Amsterdam, The Netherlands, 1992 Search PubMed.
- F. A. Cotton, J. P. Donahue and C. A. Murillo, J. Am. Chem. Soc., 2003, 125, 5436–5450 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data table and crystal packing illustration, powder X-ray diffraction patterns from samples at different stir rates, Raman spectrum, BET nitrogen sorption isotherm, Fourier-transform infrared spectra, electron spray ionization mass spectrum, 1H NMR spectrum of acid-digested sample solution, thermogravimetric curve, and molecular orbital illustrations from DFT calculations. CCDC 1850472. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra03572a |
| ‡ These authors contributed equally. |
| § Current address: Department of Chemistry and Biochemistry, University of California, 607 Charles E. Young Drive East, Los Angeles, CA 90095, USA |
| This journal is © The Royal Society of Chemistry 2019 |