
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCurrent and future functional imaging techniques for post-traumatic stress disorder
Alisha Prasad†
 a,
Ardalan Chaichi†a,
D. Parker Kelley†b,
Joseph Francis*b and
Manas Ranjan Gartia*a
a,
Ardalan Chaichi†a,
D. Parker Kelley†b,
Joseph Francis*b and
Manas Ranjan Gartia*a
aDepartment of Mechanical and Industrial Engineering, Louisiana State University, Baton Rouge, LA 70803, USA. E-mail: mgartia@lsu.edu
bComparative Biomedical Sciences, School of Veterinary Medicine, Louisiana State University, Baton Rouge, LA 70803, USA. E-mail: jfrancis@lsu.edu
First published on 8th August 2019
Abstract
Posttraumatic stress disorder (PTSD) is a trauma and stressor related psychiatric disorder associated with structural, metabolic, and molecular alternations in several brain regions including diverse cortical areas, neuroendocrine regions, the striatum, dopaminergic, adrenergic and serotonergic pathways, and the limbic system. We are in critical need of novel therapeutics and biomarkers for PTSD and a deep understanding of cutting edge imaging and spectroscopy methods is necessary for the development of promising new approaches to better diagnose and treat the disorder. According to the Diagnostic and Statistical Manual of Mental Disorders (DSM-V) criterion, all forms of traumatic stress-induced disorder are considered acute stress disorder for the first month following the stressor. Only after symptoms do not remit for one month can the disorder be deemed PTSD. It would be particularly useful to differentiate between acute stress disorder and PTSD during the one month waiting period so that more intensive treatments can be applied early on to patients with a high likelihood of developing PTSD. This would potentially enhance treatment outcomes and/or prevent the development of PTSD. Comprehension of the qualities and limitations of currently applied methods as well as the novel emerging techniques provide invaluable knowledge for fast paced development. Conventional methods of studying PTSD have proven to be insufficient for diagnosis, measurement of treatment efficacy, and monitoring disease progression. As the field currently stands, there is no diagnostic biomarker available for any psychiatric disease, PTSD included. Currently, emerging and available technologies are not utilized to their full capacity and in appropriate experimental designs for the most fruitful possible studies in this area. Therefore, there is an apparent need for improved methods in PTSD research. This review demonstrates the current state of the literature in PTSD, including molecular, cellular, and behavioral indicators, possible biomarkers and clinical and pre-clinical imaging techniques relevant to PTSD, and through this, elucidate the void of current practical imaging and spectroscopy methods that provide true biomarkers for the disorder and the significance of devising new techniques for future investigations. We are unlikely to develop a single biomarker for any psychiatric disorder however. As psychiatric disorders are incomparably complex compared to other medical diagnoses, its most likely that transcriptomic, metabolomic and structural and connectomic imaging data will have to be analyzed in concert in order to produce a dependable non-behavioral marker of PTSD. This can explain the necessity of bridging conventional approaches to novel technologies in order to create a framework for further discoveries in the treatment of PTSD.
1. Introduction
Post-traumatic stress disorder (PTSD) can be observed in about 20 to 30% of individuals exposed to extreme traumatic stressors like war, sexual assault, and life-threatening accidents. Despite the fact that about 70% of individuals experience a serious trauma in their lifetimes,1 the approximate lifetime occurrence for PTSD is reported to be 6.8% in the United States and 8% worldwide. The percentage of individuals that develop PTSD, as opposed to an acute stress response lasting 1–3 weeks is dependent on numerous elements including biological sex and sociocultural factors. An additional point is that according to a recent meta-analysis, unintentional traumas tend to result in a higher percentage of acute PTSD development than do intentional events acutely (30.1% vs. 11.8% respectively), but after 12 months, individuals exposed to intentional traumas show a higher percentage of chronic PTSD (14% vs. 23.3% respectively). This suggests an interpretive cognitive component by the patient.2–4 The likelihood of developing PTSD further depends on the intensity of the traumatic event, preexisting stressors experienced by an individual, the number of traumatic events a person is exposed to, and personal resilience and risk parameters.5According to the Diagnostic and Statistical Manual of Mental Disorders (DSM-V) the diagnosis of PTSD should be supported by five diagnostic categories. The first, around which the others must fall is (1) traumatic events: experience to real or vulnerable situations such as, injury, abuse, violence, or death. The DSM-V further requires that at least one symptom from each of the following categories be present for a PTSD diagnosis. (2) Intrusion symptoms: an involuntary re-experiencing of a memory of the event, repeated upsetting thoughts related to past events, or flashbacks associated with the event, intense and extended psychological agony of physiological feedbacks in response to in/external cues or contexts associated with the traumatic incident. (3) Avoidance symptoms: avoidance or efforts to avoid internal or external reminders of the event including distressing memories, feelings, thoughts, people or places associated with the traumatic event. (4) Negative alterations to cognition and mood: including the inability to recall imperative aspects of the trauma, determined and overstated negative theories about oneself, distorted cognitions related to the cause of the event, persistent negative emotional state, reduced interest or contribution in important events, emotional state of disinterest, and/or the inability to experience positive feelings, and (5) marked alterations and reactivity related with the traumatic event, including hypervigilance, irritable behavior and angry outburst, reckless behavior, exaggerated startle response, concentration problems, and sleep disturbances.6 PTSD is also associated with substance use disorders, chronic pain, and incapacitating anxiety.5,7,8
Traumatic stress can alter gene and therefore protein expression in the brain and periphery, causing downstream alterations to important molecular and neurochemical factors related to affective and cognitive processes, as well as systems involved in coping with and adapting to stress. In addition, stress can alter epigenetics, further modulating gene expression. PTSD is associated with altered immunological factors like increased inflammatory cytokines, particularly IL-1β, TNF-α, IL-6, and C-reactive proteins.9 PTSD is also generally associated with lower baseline levels of glucocorticoids (GC) and impaired hippocampal and prefrontal cortical functioning.10 Both psychological factors (cognitive reappraisal and optimism) and biological factors (neurotransmitter systems, single nucleotide polymorphisms (SNPs), epigenetic markers and endocrine factors) are important to the development, modulation, progression, and treatment of PTSD.11,12 Due to the currently limited diagnostic capacity and availability and efficacy of therapy for PTSD, most of the recent research is focused on the identification and detection of potential biomarkers for the disorder. The literature suggests that alterations in neurotransmitter signaling in various brain regions and changes to neuroendocrine signaling are potentially casually related to the development of PTSD and are therefore potential biomarkers for the disorder, but as of now they are limited clinically due to the impossibility of direct measurement in humans in vivo and the low reliability and specificity to PTSD respectively.11,13 Current research should therefore be concentrated on the ability to study these markers in vivo in PTSD patients using novel imaging and spectroscopy techniques, as well as on further elucidating novel cellular and behavioral indicators in animal models. Current studies are focused on identifying changes to receptor densities using positron emission tomography (PET) or single photon emission computed tomography (SPECT), evaluating the regional blood flow using functional magnetic resonance imaging (fMRI), or calculating the regional glucose consumption using PET in PTSD patients compared to healthy individuals or trauma exposed non-PTSD controls. Some brain regions that have been observed to differ in PTSD include, the prefrontal cortex (PFC), insula (Ins), hippocampus (Hipp), orbito-frontal cortex (OFC), parahippocampal regions (pHipp), the amygdala (Amy), the thalamus (Thal), anterior cingulate cortex (ACC), and the hypothalamus (HT).
In this review, we present the current state-of-art of the imaging techniques employed to study PTSD. We highlight the existing imaging and spectroscopy techniques and their advantages and limitations. We also present the emerging techniques and their potential to compensate the deficiencies of current approaches. Fig. 1 presents a schematic showing the organization and highlights of the review.
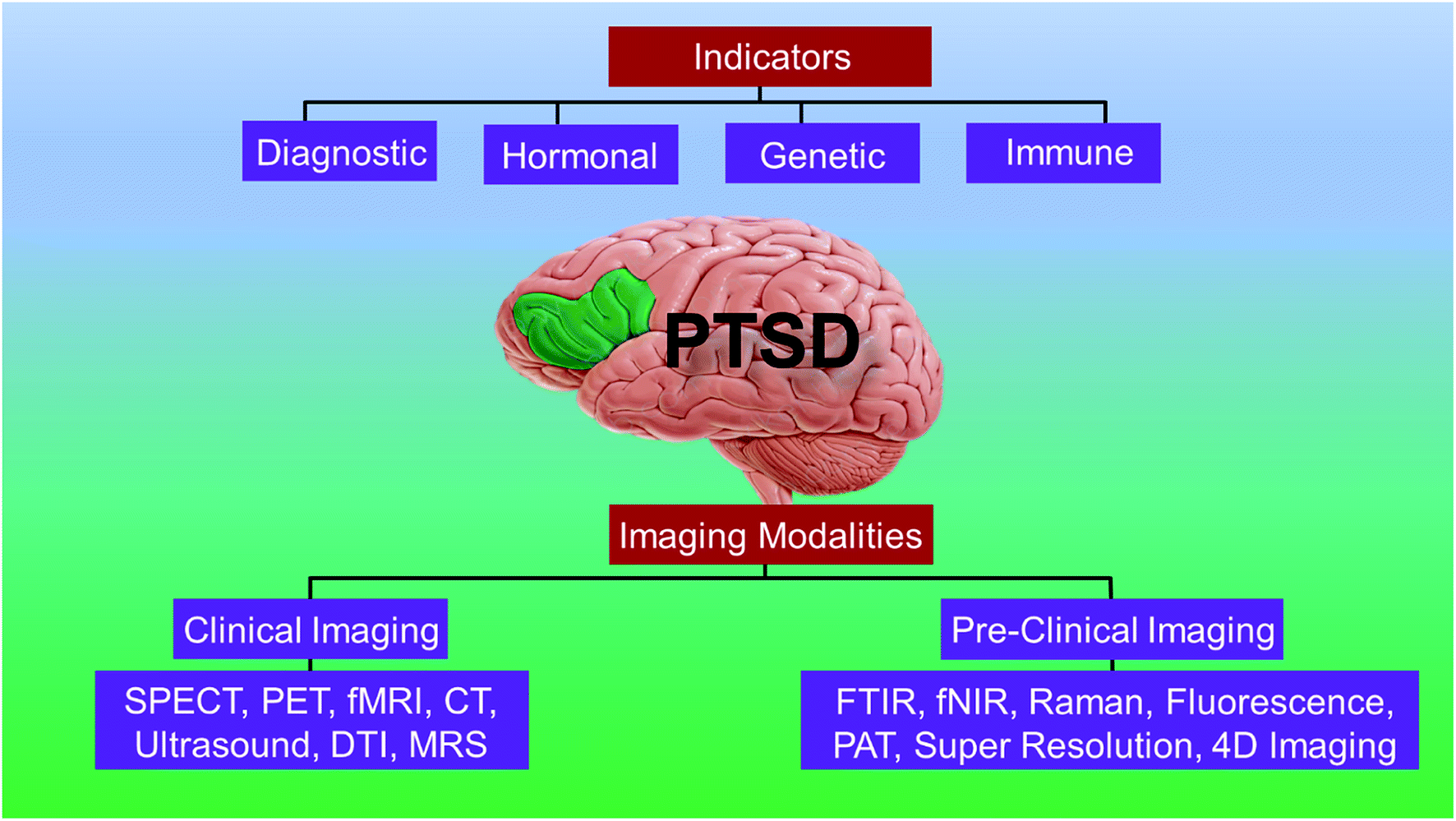 | ||
| Fig. 1 Schematic showing the organization and highlights of the review. | ||
2. Background
2.1. Molecular, cellular and behavioral indicators in PTSD
A short list of some currently available cellular and behavioral indicators of PTSD are listed in Fig. 2, and can be divided in four main categories, namely, diagnostic (DSM-V criterion), neuroendocrine, neurochemical and neurotropic indicators, genetic indicators, and immunological indicators respectively. The currently available indicators of PTSD can help in clinical diagnosis and therapy although they are currently not specific enough for effective diagnosis, medication choice, or therapy for PTSD based on the indicators alone.14 Hence, there is a need at present to find novel biomarkers for diagnosis and to determine which treatments will be most effective for treating PTSD.15 Resilience markers should also be taken into consideration due to the remarkable population of people who are exposed to traumatic stressors and do not develop PTSD.12,13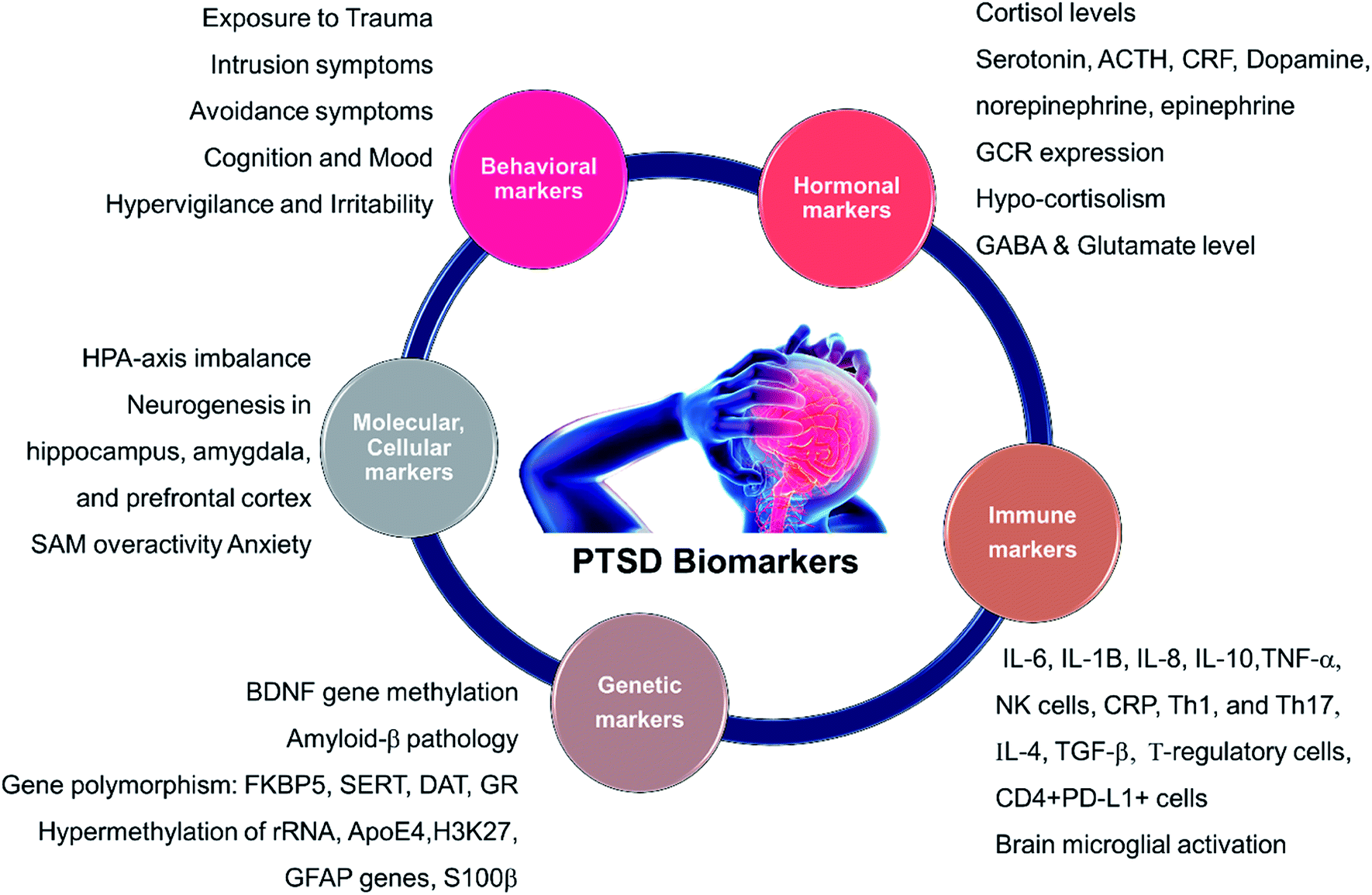 | ||
| Fig. 2 Different biomarkers of PTSD. HPA: hypothalamic pituitary adrenal; SAM: Synergistic Activation Mediator; ACTH: adrenocorticotropic hormone; CRF: corticotropin hormone releasing factor; GCR/GR: glucocorticoid receptor; BDNF: brain-derived neurotrophic factor; GABA: Gamma-Aminobutyric Acid; FKBP5 (protein); SERT: serotonin transporter or 5-HTT; DAT: dopamine transporter gene; rRNA: ribosomal ribonucleic acid; ApoE4: apolipoprotein E; H3K27: trimethylated histone H3 lysine 4 amino acid; GFAP: glial fibrillary acidic protein; IL: interleukin; TNF: tumor necrosis factor I; NK: Natural Killer cells; Th: T-helper cells. | ||
2.2. Neurotransmitters in PTSD
The only approved pharmacotherapies for PTSD are SSRI antidepressants. Particular selective serotonin reuptake inhibitors (SSRIs) like paroxetine and sertraline significantly improve symptoms in about 50–60% of PTSD patients, but this is not that much greater than the effect of placebo, which is about 40%. Furthermore, it must be acknowledged that a headache is not the absence of Advil, as the common saying goes. The partial efficacy of SSRIs in depression and PTSD does not necessarily suggest that there is a problem with serotonin (5-HT) in these disorders, only that altering 5-HT signaling can improve symptomology.16 Decades of study on 5-HT however suggest a role of 5-HT in several processes involved in PTSD, particularly in coping with or adapting to stress, sleep, memory, aggression, social bonding and general cognitive processes.17,18 It is currently impossible to measure 5-HT in the brain of live humans, although it can be measured indirectly through the measurement of 5-HT-related receptors, transporters, enzymes and by measuring peripheral 5-HT and 5-HT metabolites. These methods are limited however since clinical investigators cannot control PTSD development in a population of genetically similar individuals, as can be done in animal studies.Dr Joseph Francis' Lab at LSU used a predator exposure model of PTSD in rats to show that total 5-HT levels in the PFC and Hipp are decreased in PTSD animals compared to control animals. We also measured total norepinephrine (NE) levels and observed that NE levels were increased in the same regions.19 These data indicate that neurotransmitter changes in PTSD may be causally related to the disorder rather than being preexisting or predispositional. Future studies in this area could use molecular imaging techniques, like PET and SPECT imaging to investigate alterations to 5-HT and NE binding and receptor expression levels in military veterans before and after combat to determine if these changes exist in humans and to study their time course throughout the disorder and between combat exposed individuals that do and do not develop PTSD. Monoamine neurotransmitter levels themselves cannot be observed in living humans, but a proxy for extracellular neurotransmitter levels in humans could be the use of competitive binding studies with known 5-HT and NE agonists/antagonists bound to radionucleotides through PET or SPECT imaging.
2.3. Genetic and epigenetic considerations
This section is offered as a partial guide to potential biomarkers in PTSD and to offer several recent findings on the subject which will inform future imaging investigations. These include a model of genetic predispositions to the development of PTSD and depression and the potential for an interaction with epigenetic changes associated with childhood trauma, possibly further tipping the scales toward susceptibility to psychiatric disorder. Beyond the description of an important aspect of the most recent PTSD literature, glucocorticoids, this section will also offer a jumping off point for future investigations that seek to integrate imaging findings with the most up to date findings in the molecular and genetic underpinnings of PTSD.Epigenetic alterations can result from experience or be inherited inter-generationally. In psychiatric disorders, epigenetic changes associated with early life adversities are of particular concern. Epigenetic changes can be measured directly through the analysis of methylation patterns on DNA or identified by modification of gene expression. Alterations to the transcriptome can be messenger RNA (mRNA) levels transcribed from these genes by PCR, in situ hybridization, or transcriptome sequencing. Transcriptomic changes in the brain can only be studied in animal models at the moment, but investigators also study changes to peripheral mRNA expression in search for relevant biomarkers in PTSD. The brain regions typically studied for epigenetic changes resulting from or predisposing one to PTSD are shown in Fig. 3(a).20
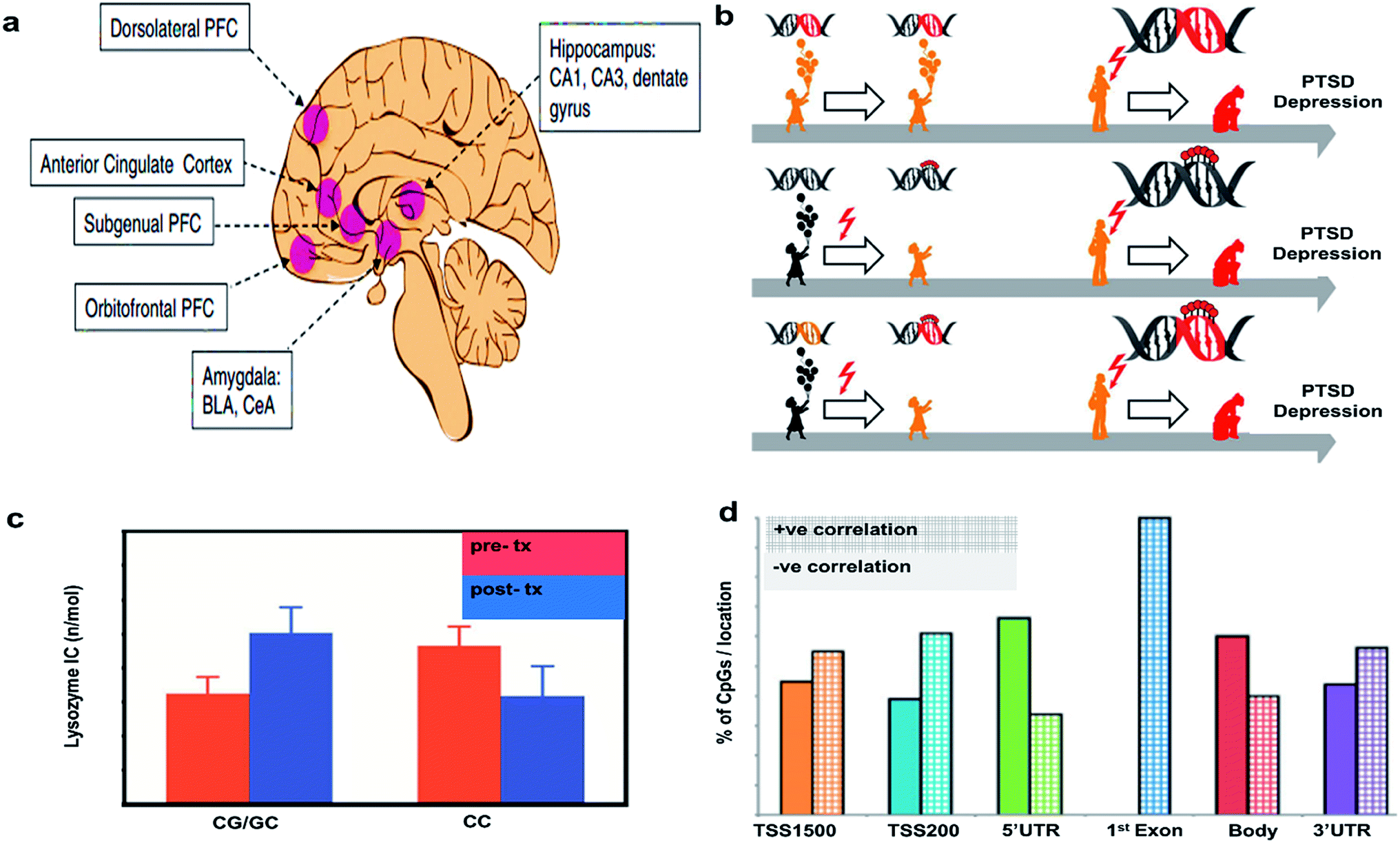 | ||
| Fig. 3 Epigenetic considerations. (a) Regions of brain involved in PTSD. Reproduced with permission from ref. 20. Copyright 2017, Society of Biological Psychiatry. (b) Mechanism of action of PTSD. Reproduced with permission from ref. 23. Copyright 2013, Raabe and Spengler. (c) Glucocorticoid sensitivity at pre- and post-treatment depending on GR BCLI polymorphism genotype (CG/GC or CC). Data were adjusted for age, gender and body mass index and are represented as mean ± SEM. Statistical significance was set at p < 0.05. Reproduced with permission from ref. 26. Copyright 2014, Published by the Royal Society. (d) Correlations of DNA methylation with gene expression. The bar graph shows the percent of CpGs with positive vs. negative correlations with gene expression. Reproduced with permission from ref. 31. Copyright 2013, National Academy of Sciences. | ||
Genetic and epigenetic alterations in PTSD are often evaluated in relation to the hypothalamic pituitary adrenal (HPA) axis. Corticotrophin Releasing Hormone (CRH) and Arginine Vasopressin (AVP) are two hormones that control the behavior and metabolism in response to stress. CRH, released from the hypothalamus in response to stress/trauma causes the release of adrenocorticotropin releasing hormone (ACTH) from the pituitary gland which in turn causes cortisol (CORT) release from the adrenal gland, which feeds back to the hypothalamus in a negative feedback loop, thus reducing CRH expression. CORT is a glucocorticoid (GC) and thought to be the major stress hormone in humans. GC signaling is complex, involving chaperon proteins, transcription factors and signaling cascades of accessory proteins which all help to restrict stress hormone expression by the negative feedback mechanism discussed above.21 GC help biological organisms to cope with the stress by facilitating resource utilization and reducing inflammation, but can also cause serious consequences if they persist at high levels for too long.
A greater understanding of the genetic and epigenetic risk factors associated with HPA axis function in PTSD and depression may help us elucidate causal mechanisms involved in these disorders. Fig. 3(b) shows a model to understand the epigenetic features responsible for depression and PTSD. The first arrow indicates that a predisposition to developing PTSD could be due to genetic mutations (red DNA double helix), which can later manifest as depression and/or PTSD development (red flash), but in the absence of trauma, they can still lead a healthy life as shown (orange colored child). The red flash on the second arrow indicates that childhood difficulties eventually leads to an altered translation of genes via DNA (de-)methylation, an epigenetic alteration, which may increase chances of PTSD development (red colored lolly on the helix). The third arrow indicates that early traumatic events can cause epigenetic alterations via DNA (de-) methylation, which can interact with existing mutations, possibly resulting in PTSD and depression (orange DNA becomes red DNA with red lolly). This model supports the concept that an infant carrying a stress allele is more likely to experience deleterious epigenetic changes associated with that gene. For example, the gene FKBP5 is associated with glucocorticoid signaling and the risk allele for that gene is associated with a PTSD diagnosis, as is childhood abuse.22 This model suggests that risk alleles, like FKBP5, could be triggered by childhood stress or trauma through epigenetic changes to gene expression and lead to the progression of PTSD and depression.23
Additionally, to their role in the stress response, GCs are also involved in the negative feedback mechanism controlling stress-associated immune activation, particularly through the inhibition of a transcription factor called NF-kB. Low baseline GCs expression is already known to be a potential biomarker for PTSD and PTSD is known to be associated with excessive immune activation.24,25 Furthermore, PTSD is associated with reduced sensitivity to GC. Recently, a study examined the role of GCs in PTSD by conducting a survey of 52 male and female veterans with PTSD who received either prolonged exposure (PE) treatment or a weekly minimal attention (MA) involvement for 12 consecutive weeks. The study did not show any effect of treatment on the clinician administered PTSD scale (CAPS). The lysozyme inhibition test was conducted with cultured peripheral blood mononuclear cells along with variable doses of dexamethasone (DEX), an anti-inflammatory synthetic steroid mimicking the effects of endogenous GC, to measure the GCs sensitivity (Fig. 3(c)). GCs sensitivity at pre-and post-treatment was dependent on the patient's glucocorticoid receptor gene GR BCLI genotype. Patients either had the wild-type (CC) or heterozygous genotype (CG/GC). Chi-squared analysis confirmed that patients were more likely to carry the GG/GC (∼71%) than the CC genotype (∼30%). GC sensitivity depended on the patient's genetic status and genetics predicted the treatment response such that those with GG/GC phenotype showed a decrease in GC sensitivity post treatment while those with CC genotype did not. These data suggest that genetics associated with the GC system can be helpful in understanding the mechanisms of action underlying symptom reduction and glucocorticoid sensitivity in PTSD.26
Previous reports have found that GC hormones are elevated in severely depressed patients27 and generally found to be reduced in PTSD, although there is some debate on the matter.28 Literature reveals that high GCs can reduce hippocampal neurogenesis and hinder the growth of neurons from stem cells in animal models.29,30
Elisabeth Binders Lab at the Max Plank Institute also studied the impact of childhood maltreatment by evaluating the epigenetic modifications in PTSD versus healthy patients. The DNA transcripts with CpG island were evaluated for genetic differences. It was found that 69.3% of transcripts with at least one CpG island were identified for PTSD due to genetic mutations at adolescence/childhood difficulties, whereas only 33.6% were identified for other forms of PTSD (Fig. 3(d)). Methylation in the CpGs island are more significant at 3′UTR region emphasizing the importance of epigenetic factors to differentiate the types of PTSD.31
Collectively these data highlight the importance of the GC system in PTSD and the related genetic and epigenetic differences that could impact disease development, treatment response, and outcome. Future imaging studies in this area should further investigate the functional (fMRI, PET, EEG) and molecular differences (PET, SPECT) in PTSD related brain regions, like the Hipp and pHipp, which have high levels of the glucocorticoid (GR) and mineralocorticoid receptors (MR), and determine if there is any association between these functional and molecular differences and a patient's genotype, childhood trauma, peripheral GC levels, and PTSD symptoms. These investigations should further utilize special experimental designs like twin studies and comparisons between PTSD patients and trauma exposed controls to further elucidate whether changes to the GC system precede or are caused by PTSD.
2.4. Imaging in PTSD: what do we know?
Although much of the underpinnings of PTSD are still to be uncovered, there do now exist many highly reported features of PTSD that are accessible through commonly available neuroimaging modalities. These features fall into several major categories including structural differences in various brain regions, functional differences within and between various brain regions and networks and molecular and neurochemical differences, which are further divided by the particular systems that those factors play a role in governing. Comparisons will be presented as either between PTSD patients and healthy controls or else between PTSD patients and combat exposed and/or trauma exposed entities who did not go on to develop PTSD.Five major brain regions represent the core of PTSD research, but that list is quickly expanding and each region is additionally being parsed into more and more granular subregions with distinct functionalities. We will predominantly review only the major regions here, but in some cases, we may include information on other regions if the weight of evidence is substantial enough to warrant it. These major regions are the hippocampus (Hipp) and parahippocampal regions (Hipp), known more generally as the medial temporal lobe (MTL), the ventromedial prefrontal cortex (vmPFC), the amygdala (Amy), the anterior cingulate cortex (ACC), and the insula (Ins).32–34
The purpose of this part of the communication is to provide a global perspective on the structural and functional basis of PTSD and to explicate the methods by which we may begin to understand the molecular basis of these functional changes.
Another consistent structural difference observed in PTSD is in the anterior cingulate cortex. The ACC is a cortical region layered above the corpus callosum (CC) and has two functionally distinct regions. The rostral-ventral region closest in proximity to the CC is implicated in emotional processing and is often called the limbic cortex. The dorsal component is the cognitive division and is involved in response inhibition to non-emotional stimuli. Strong evidence now exists suggesting the ACC plays a role in conflict monitoring, cognitive dissonance, and the subsequent adjustments of brain activity required for processing conflicting information.37,38 Numerous studies have exhibited reduced gray matter in the ACC in PTSD compared to both trauma exposed controls32,39 and healthy control participants.34
The amygdala, like the Hipp and ACC, is a component of the limbic system. The Amy is crucial for fear and stress-related memory formation including fear conditioning40 and generalization,41 and the resultant fear and emotionally related behaviors in response to reminders of the trauma.42 Reports on structural differences in the amygdala (Amy) in PTSD are mixed, some reporting reduced Amy volumes43 in PTSD patients and others suggesting either non-significantly increased volumes44 or no differences between PTSD patients and control participants.43 A meta-analysis including only a very homogenous group of participants in just three studies (N = 141) has however exhibited smaller Amy volumes in PTSD vs. healthy controls and traumatized non-PTSD controls.
If these structural differences in the limbic regions of PTSD patients do stand the test of time, it begs the question as to whether they precede or are caused by a traumatic event. Some of the most compelling evidence on this front has come from twin studies performed by the lab of Roger K. Pitman at Massachusetts General Hospital. These investigators compared combat exposed monozygotic twins who had either developed (N = 18) or did not develop (N = 23) PTSD in the Vietnam war with their combat-unexposed cotwins. Combat exposed twins with PTSD exhibited significantly reduced gray matter densities in the right Hipp, the pregenual ACC and both the left and right insula compared to their combat unexposed co-twins.45 This indicates that the differences are most likely caused by PTSD rather than the other way around. These data highlight the importance of between-participant controls on the measurement of structural changes associated with PTSD.
Network based neurobiological models of psychiatric disorders are the new frontier of integration between cognitive psychology and biomedical neuroscience. An important concept when considering imaging studies on psychopathology is functional connectivity. This refers to a statistical (Pearson correlation or Granger causality) coupling or association within or between brain regions of interest (ROI). Functional connectivity is essentially the basis of network based modeling. Recently, evidence is accumulating that suggests the brain is composed of functionally differentiated intrinsic connectivity networks (ICNs). These networks provide a broader framework in which to understand neurobiological dysfunction in PTSD rather than being limited to the interactions of discrete structures.35
There are 3 major networks commonly implicated in PTSD. These include the default mode network (DMN), also known as the task negative network, which is composed of the medial temporal lobe (MTL), the ventromedial prefrontal cortex (vmPFC), and the posterior cingulate cortex (PCC), the salience network (SN), also known as the ventral attention network, which is composed by the anterior cingulate cortex (ACC), the amygdala (Amy), and the insula (Ins) and the central executive network (CEN), also known as the fronto-parietal control network, which is composed of the dorsolateral prefrontal cortex (dlPFC) and the precuneous.35,46
The DMN is most active in normal control participants during task negative self-reflection. In PTSD, the DMN exhibits decreased coupling between each of these components at rest and this dysconnectivity correlates with symptom severity. Despite limited studies on the CEN in PTSD, there is some evidence of reduced functional connectivity in the CEN including the reduced ability of the CEN to recruit the precuneus during an autobiographical memory task.47 The salience network modulates attentional focus and the initiation of homeostatic responses to environmental events that are critical for survival. It does this through bottom up information integration and subsequent communication with higher order centers of cognition about the importance of environmental information. The SN is a sort of alarm system capable of triggering hypervigilance, irritability and aggression, a few diagnostic criterion of PTSD. The SN also plays a role in switching between the DMN and various task positive networks. In PTSD, the SN is typically over active both in its individual components across various tasks, but also in the functional connectivity between them.35,46 Overall, PTSD patients tend to exhibit increased salience processing, resulting in hypervigilance along with disrupted DMN integrity and an underactive CEN, resulting in poor top down control, poorly organized self-referential processing and dysregulated transitions between network-states. An extension of this literature would be a detailed mapping of particular PTSD symptom clusters to these intrinsic brain networks as is currently being performed by some labs.48 This sort of mapping could potentially be used in the future for diagnosis and confirmation of particular PTSD phenotypes and further articulated through a molecular imaging analysis of the underlying molecular changes which substantiate these functional differences.
Despite these substantial advances in understanding the functional variances among PTSD patients and trauma exposed or fit controls, we must always take into account that the underpinnings of these changes are molecular, neurochemical, and potentially neurotropic and structural. The observed differences in network integrity and function are based in molecular changes at the neuronal level that can currently only be measured in animal models and theoretically measured, albeit much more slowly, in human patients through molecular imaging. Molecular imaging through PET and SPECT are currently extremely limited in comparison to what can be done in animal models. In animal models, techniques like whole transcriptome RNA sequencing, metabolomics, proteomics, and lipidomics can give us a broad view of what changes are caused by trauma. One frailty of each of these techniques however is that they can only be performed in humans and animals post mortem. This makes it difficult to show causation within a single individual. Novel techniques, like Raman spectroscopy, that will be introduced later in this paper, can offer real time monitoring of molecular changes in the brain of animal models of PTSD and someday, this and/or similar techniques may be available for human research.
The rest of this paper will focus on the scientific basis of current and future imaging modalities applicable to PTSD, with a specific focus on molecular imaging in humans and other techniques, like Raman spectroscopy, and Fourier-transform infrared (FTIR) imaging by which we can investigate the molecular basis of these functional changes in animal models and ex vivo samples, setting the groundwork for the eventual technological progress necessary to monitor these changes non-invasively in human patients.
3. Imaging techniques
3.1. Clinical imaging techniques
Recently, functional neuroimaging methods have been utilized in neurological studies of various diseases to elucidate their mechanisms. SPECT, PET and fMRI are three fundamental techniques that measure physiological criteria like molecular factors (PET, SPECT), cerebral blood flow to specific regions of the brain over time (fMRI) and regional energy consumption via glucose utilization (PET), to investigate brain functionality. These techniques measure indirect neural activity based on the fact that neural activation requires blood flow, metabolism, and neurotransmission to occur. fMRI illustrates brain activity through Blood Oxygen Level Dependent (BOLD) changes. On other hand, PET and SPECT take advantage of regional cerebral blood flow (rCBF) and neuroreceptor concentrations to demonstrate patterns of brain activity and function.10,49 A summary of the imaging modalities currently used for PTSD study are provided in Table 1 and the main findings with the contrast agents used are provided in Table 2.| Technique | Depth | Spatial resolution | Temporal resolution | Strength | Limitation | Clinical use | Physical principle |
|---|---|---|---|---|---|---|---|
| a SPECT: Single Photon Emission Computed Tomography; PET: Positron Emission Tomography; CT: Computed Tomography; fMRI: functional Magnetic Resonance Imaging; EEG: electroencephalogram; MEG: magnetoencephalography; fNIR: functional Near-Infrared Imaging; VSDI: Voltage-Sensitive Dye Imaging; LSCI: Laser Speckle Contrast Imaging; FTIR: Fourier Transform Infrared Microscopy. | |||||||
| SPECT | Whole brain | 1–5 mm | Minutes | Non-invasive, lower cost compared to PET, longer half-life (∼6 h) for radio tracers, allow longer imaging time, sensitivity: 10−10 to 10−11 mol L−1 | Poor spatial resolution, radiation dose exposure, limited number of radionuclides (99mTc, 123I) | Yes | Low-energy γ-rays |
| PET | Whole brain | 1–5 mm | Seconds to minutes | High sensitivity compared to SPECT, radionuclides used in PET (11C, 13N, 8F) are abundant in the body and can be tailored for endogenous biomolecules such as carbohydrates, fats, nucleic acids and proteins, sensitivity: 10−11 to 10−12 mol L−1 | Short half-life (∼75 s) for the radio tracers and hence must be produced onsite before imaging, high-cost compared to SPECT, limited imaging time window | Yes | High-energy γ-rays |
| CT | Whole brain | 50 μm | Minutes | High spatial resolution, fast and cross-sectional images of the brain, sensitivity: 10−6 mol L−1 | Structure of the brain, not its function, low contrast | Yes | X-rays |
| fMRI | Whole brain | 25–100 μm | Minutes to hours | No radiation, structural and functional data, high spatial resolution, greater contrast for soft tissues, imaging agents with lower toxicity, sensitivity: 10−3 to 10−5 mol L−1 | High cost, long scanning time, sensitive to motion artifacts, relatively low sensitivity and low contrast | Yes | Radio waves |
| Ultrasound | 1-5 cm | 50–500 μm | Seconds to minutes | Low cost, no radiation, high speed, portable, sensitivity with microbubbles: 10−12 mol L−1 | Low contrast | Yes | High frequency sound waves |
| EEG | Scalp | 5–10 cm | Millisecond | Inexpensive, portable, high temporal resolution, electrical activity of brain | Low spatial resolution, prone to error due to environmental noise, localization of signal is difficult | Yes | Electrical |
| MEG | Scalp | 1-5 mm | Millisecond | High temporal resolution | Expensive | Yes | Electromagnetic |
| fNIR | < 1 cm | 2–3 mm | Seconds to minutes | No radiation, inexpensive, sensitivity: 10−9 to 10−12 mol L−1 | Scattering due to tissues may be a problem, low penetration depth | Preclinical | Near-infrared light |
| Photoacoustic | 0.6–5 cm | 10 μm to 1 mm | Seconds to minutes | No radiation, label-free, high spatial resolution and low cost compared to CT/PET, sensitivity: 10−6 to 10−12 mol L−1 | Distortion of acoustic signal due to skull, temperature dependent signal, weak absorption at shorter wavelengths | Preclinical | Pulsed laser and sound wave |
| VSDI | 1 mm | 50 μm | Millisecond | High temporal resolution | Invasive, prone to photobleaching of dye, toxic to cells | Preclinical | Voltage sensitive dye |
| Bioluminescence | 1-2 cm | 3–5 mm | Seconds-Minutes | No radiation, high sensitivity, inexpensive, sensitivity: 10−15 to 10−17 mol L−1 | Scattering due to tissues may be a problem, spatial resolution is low | Preclinical | Visible light |
| LSCI | 0.5–1 mm | 10 μm | Microseconds | Label-free, high temporal resolution | Invasive | Preclinical | Visible and near infrared laser |
| Two-photon | 1 mm | 1 μm | Microseconds | High spatial resolution | Invasive, photobleaching issues with dyes, scattering due to tissues | Preclinical | Infrared laser |
| FTIR | <1 cm | 5–12 μm | Seconds to minutes | Label-free method, short imaging time | High attenuation in liquid environment, difficult to distinguish closely related molecular structures | Preclinical | Infrared light |
| Raman | 5 mm | <1 μm | Minutes to days | Label-free analysis, high spatial resolution, can work with liquid environment | Complex statistical analysis may be required to separate analytes, long imaging time required for imaging large area at high resolution | Preclinical | Visible and near infrared laser |
| Technique | Contrast agent | Main findings | Sample size (n) | References | ||
|---|---|---|---|---|---|---|
| Measure | Change | Brain region | ||||
| a HMPAO: 99mTc hexamethyl-propyleneamine oxime; rCBF: regional cerebral blood flow; BZR: benzodiazepine receptors; AC: anterior cingulate; PC: posterior cingulate; mPFC/ACC: medial prefrontal cortex/anterior cingulated cortex; OFC: orbitofrontal cortex; FDG: fluorodeoxyglucose; GMB: glucose metabolism; BOLD: blood oxygen level-dependent signal; = : no change in activity; ↑: increase in activity; ↓: decrease in activity. | ||||||
| SPECT | HMPAO | rCBF | ↑ | Right hemisphere | 47 | 136 |
| HMPAO | rCBF | ↓ | Medial frontal gyrus | 30 | 137 | |
| rCBF | ↑ | Right cuneus | 30 | 137 | ||
| HMPAO | rCBF | ↑ | Cerebellum | |||
| ↓ | Right precentral, superior temporal, and fusiform gyri | 38 | 138 | |||
| HMPAO | rCBF | ↑ | Left hemisphere | 16 | 139 | |
| HMPAO | rCBF | ↑ | Prefrontal cortex | 24 | 140 | |
| HMPAO | rCBF | ↑ | Left amygdala, left nucleus accumbens | 28 | 141 | |
| HMPAO | rCBF | ↓ | Superior frontal cortex, right caudate | 31 | 142 | |
| [123I] Iomazenil | BZR | = | No difference in BZR levels between control and PTSD patient groups | 38 | 143 | |
| [123I] Iomazenil | BZR | ↓ | Prefrontal cortex | 26 | 144 | |
| HMPAO | rCBF | ↑ | Hippocampus, parahippocampus, amygdala | |||
| rCBF | ↓ | Dorsolateral prefrontal cortex (PFC) | 69 | 145 | ||
| HMPAO | rCBF | ↑ | Bilateral AC/PC, right temporal and parietal, right caudate/putamen, and left orbital and hippocampal | 25 | 146 | |
| HMPAO | rCBF | ↑ | Limbic | 87 | 145,147 | |
| HMPAO | rCBF | ↑ | Right superior parietal lobe | 38 | 148 | |
| rCBF | ↓ | Right thalamus | 38 | 148 | ||
| PET | [15O] H2O | rCBF | ↓ | mPFC/ACC | ||
| = | Amygdala | 29 | 149 | |||
| [O15] CO2 | rCBF | ↑ | Hippocampus, amygdala | 16 | 150 | |
| [15O] H2O | rCBF | ↓ | Hippocampus, mPFC/ACC, mPFC/OFC | 21 | 151 | |
| [15O] H2O | rCBF | ↓ | Hippocampus, mPFC/OFC | 22 | 152 | |
| 15Q-Butanol | rCBF | ↑ | Parahippocampus, amygdala | |||
| rCBF | ↓ | mPFC/ACC | 13 | 153 | ||
| [15O] H2O | rCBF | ↑ | Parahippocampus | 22 | ||
| rCBF | ↓ | mPFC/ACC | 154 | |||
| [15O] H2O | rCBF | ↓ | Hippocampus, mPFC/ACC, mPFC/OFC | |||
| rCBF | ↑ | Parahippocampus | 20 | 155 | ||
| [15O] CO2 | rCBF | ↑ | Parahippocampus, mPFC/OFC | |||
| rCBF | ↓ | mPFC/ACC | 16 | 156 | ||
| [15O] CO2 | rCBF | ↑ | Amygdala | 14 | 157 | |
| [18F] FDG | GMB | ↓ | Hippocampus, mPFC/OFC | |||
| = | Parahippocampus, amygdala, mPFC/ACC | 20 | 44 | |||
| [15O] H2O | rCBF | = | mPFC/ACC | 16 | 158 | |
| [15O] H2O | rCBF | ↑ | mPFC/OFC | 13 | 159 | |
| fMRI | BOLD | ↑ | Amygdala | |||
| ↓ | mPFC/ACC | 26 | 160 | |||
| BOLD | ↑ | Amygdala | 32 | 161 | ||
| BOLD | ↑ | Parahippocampus | ||||
| ↓ | mPFC/ACC | |||||
| = | Amygdala | 11 | 162 | |||
| BOLD | ↑ | Parahippocampus, amygdala | ||||
| ↓ | mPFC/OFC | 12 | 163 | |||
| BOLD | ↓ | Thalamus | 24 | 164 | ||
| BOLD | ↑ | Amygdala | 21 | 165 | ||
| BOLD | ↓ | Parahippocampus, thalamus, mPFC/ACC | 20 | 166 | ||
| BOLD | ↓ | Parahippocampus | ||||
| ↑ | mPFC/ACC | |||||
| = | Thalamus | 17 | 167 | |||
| BOLD | ↓ | Hippocampus, parahippocampus | ||||
| ↑ | Thalamus, mPFC/ACC | 16 | 160 | |||
| BOLD | ↓ | mPFC/ACC, mPFC/OFC, thalamus | ||||
| = | Amygdala | 18 | 168 | |||
| BOLD | ↑ | Amygdala | ||||
| = | mPFC/ACC | 16 | 169 | |||
The efficacy of SPECT is dependent on the biomolecule associated with the radio tracer. This allows SPECT to be utilized in numerous applications, only limited by the radiotracers available. In addition to 99mTc-HMPAO, 99mTc-ECD also measures cerebral blood flow. As opposed to large research projects, SPECT is also used to investigate individual cases in war torn areas like Iran. For example, SPECT imaging of a 54 year old male with PTSD was studied by Majid Assadi's Lab at the Kasra Hospital in Tehran, Iran, showing that hypoperfusion is observed in the left inferior temporal region of his brain.51
In a recent review by Raji et al. at UCSF52 the authors elaborated the ability of SPECT imaging to study a similar and often comorbid disease to PTSD, mild traumatic brain injury (mTBI), in human subjects. Accordingly, SPECT can be a promising method of investigating mTBI when equipped with quantitative analysis techniques. Their results indicate that when rCBF is measured in mTBI patients by SPECT imaging, abnormal frontal lobe perfusion was indicated in 94% of studies. Additionally, perfusion abnormalities have been detected by the SPECT approach in temporal (76%), parietal (71%), occipital (53%) and cerebellar (23%) regions of brain.52
In the characterization of a new SPECT ligand, Fabio et al. suggested SRX246, a vasopressin V1a antagonist has the ability to pass through the blood brain barrier of rats. Due to its high selectivity and permeability, SRX246 can be utilized in the radio labeling of V1a receptors in various stress related disorders. This new molecule remarkably improves characterization and visualization of SPECT imaging method for vasopressin receptors.53
In another clinical study, Hoexter et al. investigated the dopaminergic system in vivo in PTSD patients by means of SPECT imaging, using 99mTcTRODAT-1, a ligand for the dopamine transporter (DAT). They concluded that the density of striatal DAT is elevated in PTSD compared controlled subjects.54 Recently, thirty military subjects were studied by Harch et al. by means of clinical medicine and SPECT imaging. The main goal of their study was investigating the effectiveness and feasibility of hyperbaric oxygen treatments (HBOT) for PTSD and mild TBI persistent post-concussion syndrome (PPCS). SPECT results showed significant improvement after about six months of treatment for PTSD subjects. Moreover, 75% of abnormal areas were practically indistinguishable from control subjects after the treatment.55 These data suggest that SPECT imaging can be utilized to measure an increasing number of biomolecules in vivo, in human subjects, as well as regional blood flow, and has the capacity to produce important data for PTSD and mTBI.
Like any neuroimaging technique, this method also has some drawbacks, including the short half-life of radiotracers such that on-site cyclotron production of radiotracers is required, making PET much more expensive. Furthermore, PET also requires specialized chemists be present to bind these radiotracers to the bioactive molecules immediately after their creation. These fiscal factors should be taken into consideration as well when comparing imaging modalities. As a result, PET is not the most accessible device for neuroimaging. PET also has other drawbacks. The temporal resolution of this method is also limited (60–100 s) in comparison with fMRI technique which is less than 2 s. This is the main reason why fMRI and PET techniques are used in blocked and event-related designs or in tandem, in advanced PET-fMRI machines. PET also has no capacity to image targets not associated with the biomolecule employed. This makes it difficult to use PET alone when particular regional data is required. For this reason, PET often paired with computed tomography X-ray scans (CT), to provide additional structural data not available with PET alone. Most importantly, like SPECT, PET also requires the injection of radioactive materials into body which can be detrimental to the patient and combining PET with CT exacerbates this risk.10,56 Despite these limitations, PET is the most advanced imaging technique now available for imaging molecular factors in human patients and can be especially powerful when paired with fMRI, which does have the drawback of additional radiation exposure.
PET has already been employed in numerous studies on PTSD. A study conducted by Bremner et al. investigated the effects of mindful built stress reduction (MBSR) on combat PTSD veterans by means of PET imaging and measurements.57 These investigators used radiolabeled heavy water to measure regional neuronal activation. Patients were randomized into groups treated with either MBSR or present centered group therapy (PCGT). Exposure to combat related sounds and pictures resulted in hyper activation of temporal cortical and frontal brain areas in all PTSD patients. On the other hand, activation of cerebellum, insula and subcortical regions dropped considerably in PTSD, compared to control patients. MBSR treated PTSD patients not only showed a greater reduction in the clinician administered PTSD scale (CAPS) than the PCGT group, but also showed an increased activation of the right anterior cingulate, right inferior parietal lobe and decreased activation of the right insula and precuneus compared to the PCGT group when exposed to trauma associated stimuli. In the previously mentioned study by Holmes et al., investigators used [18F] FPEB, a radioligand for mGluR5 to measure this receptor by PET in human PTSD patients and observed it to be upregulated in several brain regions. Furthermore, post mortem brain samples were analyzed and an upregulation of SHANK-1 proteins which binds to mGluR5 was observed. Reduced expression of FKBP5 was also observed indicating altered glucocorticoid function in postmortem PTSD brains.58 This study exhibits the efficacy of combining molecular and imaging approaches to study particular molecular endpoints in PTSD. Additionally, in the previous study by Fabio et al. the aforementioned SPECT ligand, SRX246 is additionally useful as a radiolabeling candidate for PET imaging that can be used for PTSD related studies.53 Finally, in another study by S. E. Holmes, a PET tracer for TSPO, [(11C)(R)-PK11195] a translocator protein associated with activated microglia and commonly increased by inflammation was used to show increased inflammation in the anterior cingulate cortex in depressed patients.59 These data collectively show the efficacy of PET in measuring molecular targets associated with a wide range of systems involved in PTSD, but as with TSPO, have yet to be studied in PTSD in particular.
fMRI also has some important limitations. First, the temporal resolution of this device for rCBF changes is not perfect. There is a 5 second delay between neural activation and measurement, potentially causing temporal artifacts. This should be corrected in data analysis, but nonetheless adds noise to the data. Considering the high resolution of structural MRI and enhanced PET (modified with high resolution research tomography (HRRT)) which are less than 1 mm and 2 mm, respectively, fMRI provides lower resolution of 3 mm. Another serious disadvantage of fMRI is the inability of this technique to study patients with metallic implants and pacemakers, due to serious interactions of the powerful magnet with metals. The fMRI scan requires that a subject spend a significant amount of time in a relatively narrow tube in which the scan is taken, often causing anxiety and claustrophobia. The device also produces a considerable amount of noise that might either distract the patient from accomplishing a task or prohibits the investigation of medication effects by inducing anxiety in the patient, producing additional nose in the data through the activation of stress related pathways.10,60 Finally, unlike PET and SPECT, fMRI only able to study brain function and is unable to study molecular factors associated with disease conditions.
A recent PTSD related fMRI study was carried out by Garfinkel et al. regarding the impaired contextual modulation of memories in PTSD.61 These authors observed impaired extinction recall, higher amygdala activity and increased skin conductance in PTSD subjects compared to controls. An additional emerging benefit of fMRI is the ability to provide real time neurofeedback to patients, as a therapeutic technique or to enhance experimental paradigms. Neurofeedback allows patients to use real time data from their own brains to correct disease specific differences in neural activity associated with their disorder. Zotev et al. utilized fMRI neurofeedback in real time in order to help PTSD patients modify the interactions of the amygdala and prefrontal cortices.62 It was observed that PTSD subjects experienced significantly increased blood oxygenation levels (BOLD) action in the left amygdala while accomplishing happy emotion induction tasks. After treatment, 80% of patients experiencing fMRI neurofeedback saw significant reductions in their CAPS scores, while only 33% saw reduction in their CAPS scores in the sham group. This result showed that real-time fMRI neurofeedback procedure is capable of correcting the deficiencies in the functional connectivity of amygdala and prefrontal cortex that are specific to PTSD, at least for a short period of time after treatment. Gerin et al. also took advantage of real time neurofeedback to study this novel treatment in combat veterans with PTSD.63 fMRI analysis of participants after the treatment showed high resting-state functional connection (rsFC) patterns. Patients also showed reductions in CAPS scores, further illustrating that real time neural feedback is capable of altering resting state functional connectivity in PTSD patients that provides clinically significant changes in symptoms.
DTI has already been utilized to study mild traumatic brain injury (mTBI)70 and PTSD along with schizophrenia,71 depression72 and neurodegenerative disorders, like Alzheimer's disease.73 The literature suggests that PTSD is associated with disrupted white matter tractography in the cingulum angular bundle (CAB) as measured by fractional anisotropy (FA). The CAB connects the ACC to the MTL and Hipp, possibly underlying a deficit in emotional regulation.74,75 Other investigations reported lower mean diffusivity (MD) in the CAB, but not lower FA.76 In contrast, some reports have also exhibited elevated FA in the CAB and other relevant networks.77 Other studies did not observe differences in the CAB, but instead showed WMT changes in uncinate fasciculus, a white matter tract connecting the AMY to the vmPFC, a deficit possibly underlying reduced inhibitory control over the fear response.78 Other investigators have taken a networking approach and reported distinct differences in the white matter tracts of the salience network between PTSD and control participants.67,79
Overall, these studies are preliminary and few current literature reviews or metanalyses on this topic are available. Future investigations could benefit from pairing DTI investigations of WMT with fMRI data to determine the relationships between WMT and the observed functional changes, especially in the context of a network based perspective. Further investigations should also consider differences between PTSD patients and trauma exposed non-PTSD participants, which appear to be lacking in the DTI literature. Finally, future investigations in animal models and eventually humans could benefit substantially from the pairing of DTI in PTSD models with Raman spectroscopy and other molecular analyses, like lipidomics and proteomics to determine if correlations can be drawn between white matter integrity measured by DTI and a biochemical and molecular analysis of white matter tracts by the molecular techniques.
The literature suggests that PTSD is related with a reduction in the neuronal marker, NAA in left Hipp compared to fit control and trauma-exposed controls and in the right only against healthy controls. Cho was also shown to differ in the left hemisphere compared to healthy controls, but not trauma exposed controls. A study employing a Cho/Cr as the measure also indicated a reduction in Cho in the ACC. One new study published in 2017 used a powerful 7 T MRS was to study 43 PTSD and mTBI patients compared to 15 controls and the investigators made the observation that NAA/Ch levels significantly decreased in parts of the left hippocampus in patients with both PTSD and mTBI compared to PTSD patients alone.81
Like DTI, the literature on MRS is small and not very many large metanalyses are available. MRS has incredible potential, but is currently limited by the very small number of compounds available for measure and the requirement of a large magnet to obtain high quality results. Alternatively, MRS is the only way in which we can currently measure any neurotransmitters directly in vivo, in human patients and its possible that future technological advances could provide a more diverse set of analytes measurable by MRS, more regional specificity, and the capacity for more granular measures.
3.2. Pre-clinical imaging technologies
Quantitative and objective information is significant for the improvement of disease diagnosis and treatment in PTSD. Recent advances in the utilization of novel biomedical technologies make it possible to acquire such information from biological tissues. Various optical spectroscopy methods like Fourier transform infrared (FTIR), Raman spectroscopy, near infrared spectroscopy and fluorescence spectroscopy have been employed to achieve this goal. These methods can provide vital information about the chemical composition of biological tissues at the molecular level that is strikingly beneficial for PTSD studies. Some of the most promising emergent techniques in this area are discussed in the following.The distribution of particular chemicals can be illustrated in label-free chemical maps via the spectral data and absorbance spectrum in the region of interest. This technique can be utilized in various applications, from static systems to dynamic ones. Tissue biopsies,85 fixed cells,86 drug diffusion,87 protein crystallization,88 and live cell studies89 are some of the reported applications of the aforementioned method. The biomedical application, sample size, surrounding matrix, analyte concentration and some other variables can change in this type of imaging, relative to other techniques, allowing for different experimental designs. According to the desired application, various thermal sources as well as a synchrotron are employed to produce IR light for several FTIR imaging. Furthermore, several FTIR imaging modes such as macro imaging, micro imaging, transmission, transflection, and attenuated total reflection (ATR) methods can be utilized to accomplish imaging. Each of these modes are limited by either the image pixel size or the light diffraction. For instance, the dimension of the pixel is usually larger than the diffraction limit in macro-mode imaging in which pixel size is the determining factor for spatial resolution. On the other hand, diffraction of light is the significant limiting factor when the pixel dimension is smaller than the limit of diffraction. There have been several attempts to increase the spatial resolution of FTIR imaging due to the sub-micron nature of biological samples. In some cases, projected pixel size has been reported incorrectly as the limiting factor of spatial resolution. Meanwhile, projected pixel size becomes significant only in macro FTIR imaging and measurements.83,84
The use of a hemispherical lens above the specimen is a common method for enhancing the FTIR image quality. Initially, this method has been employed to eliminate optical aberrations caused by sandwiching the specimen between IR transparent windows in transmission FTIR imaging. Meanwhile, the numerical aperture of the system increases correspondingly by a factor of lens refractive index value due to installation of an additional hemispherical lens.83,84,90 ATR imaging mode can usually generate higher spatial resolution by not transmitting IR light through the specimen. Meanwhile, the internal reflection of IR light at the interface of specimen (low refractive index) and ATR (high refractive index) occurs within the system.83
The typical method of sample preparation includes fixing the sample in a medium, optimal cutting temperature compound (OCT), and mounting the sample on a glass substrate. Fixing usually takes place by means of paraffin and the thickness of sliced samples during OCT is about 5 mm to 10 mm. Due to the easy availability of sectioned samples which are already used for standard light microscopy, transmission mode is the most favored approach for FTIR imaging of tissues.83
This chemically specific, label free and non-destructive method is capable of being used in various aspects of biology, including protein detection in brain tissues. According to the literature, the chemical composition of biological samples can be investigated without the occurrence of any photo damage or disturbance to living tissues. This can be considered as a significant superiority over other methods like fluorescent labeling. However, the substantial drawbacks of FTIR imaging are low detection limit, limited spatial resolution and considerable background absorbance. These issues can be partially ameliorated by means of synchrotron sources of photons. Synchrotron sources have been proven to be a strong instrument, in order to conquer the limitations of FTIR imaging technique by increasing the signal to noise ration and spatial resolution in protein imaging.83,84
The literature reveals that FTIR has been utilized to investigate Lewy bodies (LBs) in the brain.91 The LBs are atypical deposits of alpha-synuclein protein (αSyn) and its fluctuations lead to problems with cognition, movement, behavior, and mood. Lewy body dementia (LBD) is a disease most commonly linked with Alzheimer's or Parkinson's disease (PD) but can also be involved in TBI, PTSD, and Dementia. A recent research reported by Mitchell Bermans' Lab at Mississippi State suggested that the αSyn gene polymorphism controls alcohol use a common PTSD symptom.92 Hence, the measurement of αSyn could be useful for understanding PTSD. The presence of high αSyn, a protein a biomarker of PD, causes harm to dopaminergic (DA) neurons. Missense mutations or gene polymorphism in αSyn gene can overproduce αSyn, causing rare inherited forms of PD. To understand αSyn at the cellular level, the adeno-associated virus (AAV) vector method was employed to inject foreign gene (hαSyn) stereotaxically into the SN of mouse brain (Fig. 4(a) left). The cells were immunostained as shown in Fig. 4(b) (right). hαSyn (green) and tyrosine hydroxylase (TH, red) (synthetic enzyme for DA) merged with hαSyn (yellow), nucleus staining by DAPI also merged (blue). From their study, it was concluded that the DA neuronal cell death was due to phosphorylation of αSyn at Ser129 residue.93 Studies like this could be streamlined with novel techniques like FTIR.
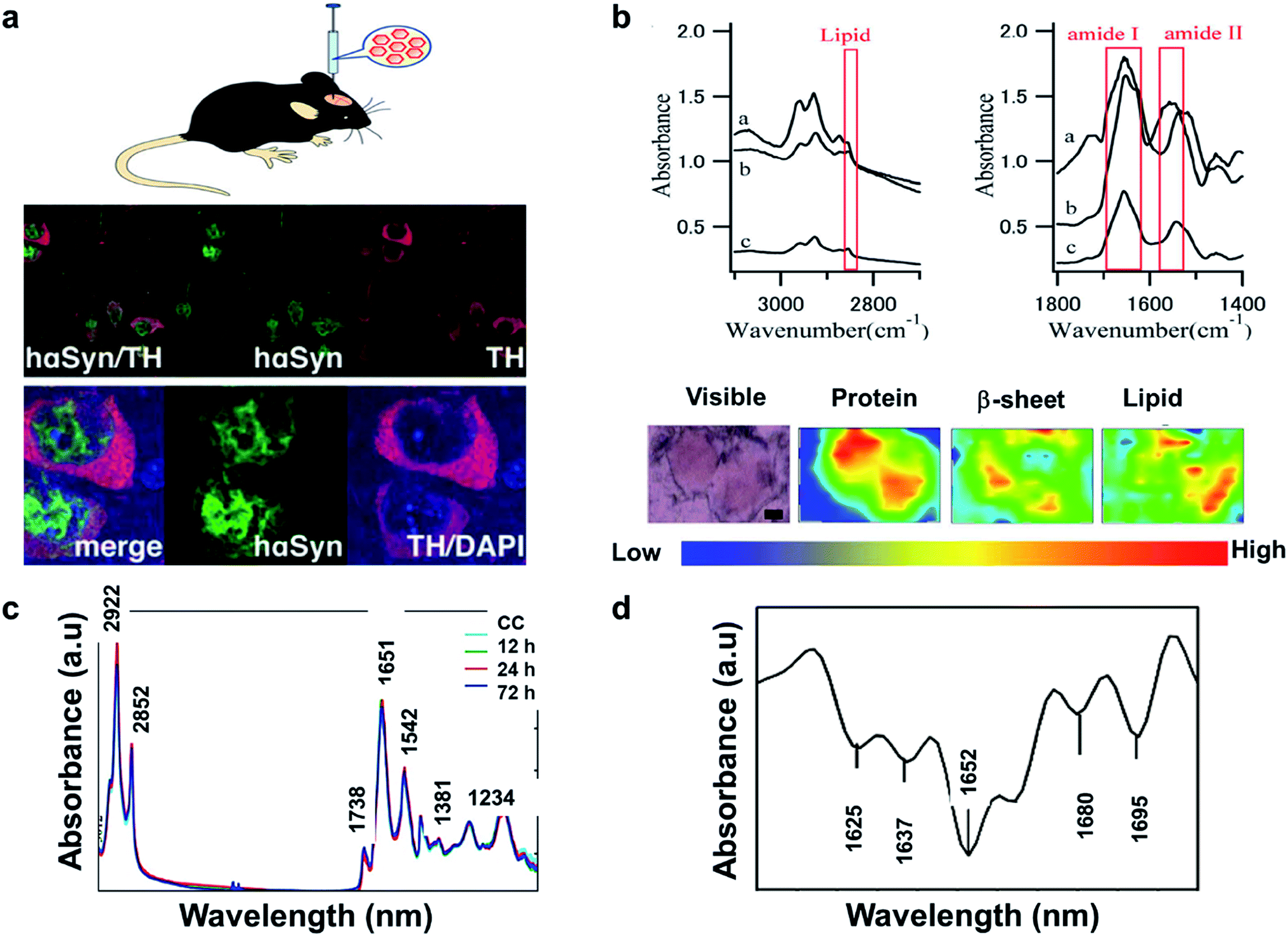 | ||
| Fig. 4 (a) AAV vector-mediated expression of foreign gene in mouse brain (left). Immunostained cells for AAV-hαSyn-injected mice (right). Reproduced with permission from ref. 93. Copyright 2012, Yasuda, Nakata, and Mochizuki. (b) FTIR spectra, a: core of an LB, b: halo of an LB, and c: normal brain tissue (top). FTIR imaging of brain showing total amount of proteins (from left to right, bottom). The color bar indicates low (blue) to high (red) contents. The area in the visible image was scanned with 5 μm steps (16 × 8 pixels = 80 × 40 μm2). Scale bar, 10 μm. Reproduced with permission from ref. 91. Copyright 2015, Macmillan Publishers Limited. (c) Secondary derivative infrared spectra of damaged and injured axons. Reproduced with permission from ref. 95. Copyright 2017, Zhang, Huang, Wang, and Dong. (d) TAI in the CC detected by b-APP staining following impact acceleration of TBI(top). A: No β-APP staining, B: β-APP-stained axons at 12 h, C: 24 h, D: 72 h (scale bar = 50 μm). Superimposition of all normalized FTIR spectra collected in the CC of rats at different post-injury groups (bottom). Reproduced with permission from ref. 94. Copyright 2015, John Wiley & Sons. | ||
In another study, synchrotron FTIR microspectroscopy was used for examination of LBs in PD subjects. This method is capable of identifying the proteins as well as investigating the changes to the secondary structure of the proteins. Fig. 4(b) (top) shows the FTIR spectra of the different sections of the protein. The spectral peaks at ∼1655, 1630 and 1645 cm−1 correspond to α-helix, β-sheet, and random coil respectively. Since LBs have low density, synchrotron radiation was used to conduct this study. Shown is a FTIR image of total proteins (specifically β-sheet) of an SP (Senile plaques used for amyloid detection) in the cerebral tissue derived from patient with Alzheimer's (Fig. 4(b): left to right, bottom). Congo red stained represented the protein-rich regions. The core of the plaque was found to be rich in β-sheets and lipids.91
The information about the protein secondary structure obtained from FTIR is helpful, as they correspond to the damaged areas. Researchers have also explored FTIR spectral analysis, comparing areas of damaged and normal axons. Peaks at 1625, 1637, 1652,1664, 1680 and at 1695 cm−1 corresponded to β-sheet, random coil, ordered α-helix, dis-ordered α-helix, β-turn, and anti-parallel β-sheets respectively (Fig. 4(c)). The changes were identified by principal component analysis (PCA). The change in amide-II band due to N–H and C–N stretch was significant in injured axons, which might be due protein conformational changes. β-APP (an amyloid precursor protein) has been known as an axonal injury biomarker and hence understanding of this using FTIR could be valuable.94
Traumatic axonal injury (TAI) was explored by another group of researchers in the corpus callosum (CC) of rats by β-APP staining post-injury at 0, 12, 24, and 72 hours' time intervals, respectively. The differences were significant between the 24 h and other groups and were mostly from lipids. At 24 h, there were higher intensities at 2922 and 2852 cm−1 corresponding to asymmetric and symmetric vibrations due to C–H vibrations from CH2, and a lower asymmetric vibration at 2958 cm−1 from CH3. Lower intensities for unsaturated lipids from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CH2 at 3012 cm−1 and band at 1738 cm−1 from lipid esters due to CO vibrations were also observed (Fig. 4(d) (bottom)). These findings were consistent with the histological examination as observed in Fig. 4(d) (top), wherein the biochemical changes associated with lipids and proteins become more prominent 24 h post injury. A disadvantage with FTIR spectroscopy is the difficulty to distinguish the different time points, depending on quantitation of the absorption peaks. Hence, statistical analysis is required to identify the subtle spectral variations.95
C–CH2 at 3012 cm−1 and band at 1738 cm−1 from lipid esters due to CO vibrations were also observed (Fig. 4(d) (bottom)). These findings were consistent with the histological examination as observed in Fig. 4(d) (top), wherein the biochemical changes associated with lipids and proteins become more prominent 24 h post injury. A disadvantage with FTIR spectroscopy is the difficulty to distinguish the different time points, depending on quantitation of the absorption peaks. Hence, statistical analysis is required to identify the subtle spectral variations.95
The most prevalent types of near infrared spectrometers are categorized as frequency domain, time domain and continuous wave NIRS. The frequency domain approach includes a head which shines with amplitude modulated NIR light. In this method, alternations of amplitude and phase are measured in the back scattered light. On the other hand, a time domain NIRS spectrometer generates short pulses of NIR and transmitted scattered photons are detected in accordance with the arrival time. However, continuous wave mode is the most ubiquitous technique in which constant amplitude and frequency are employed to illuminate the samples. In this mode, light attenuation through the sample is the only parameter that is measured. Depending on the required information, any of the aforementioned techniques can be utilized. The source of the NIR light is another significant factor that can strongly affect the final results. Generally, both light emitting diodes and fiber optic lasers are used as the source in fNIRS. However, fiber optic lasers have better maneuverability that can be applied in different sample types and positions. Required wavelength, intensity and study area are some influential factors in NIR source selection and the source detector distance (which is typically 3 cm).97,98
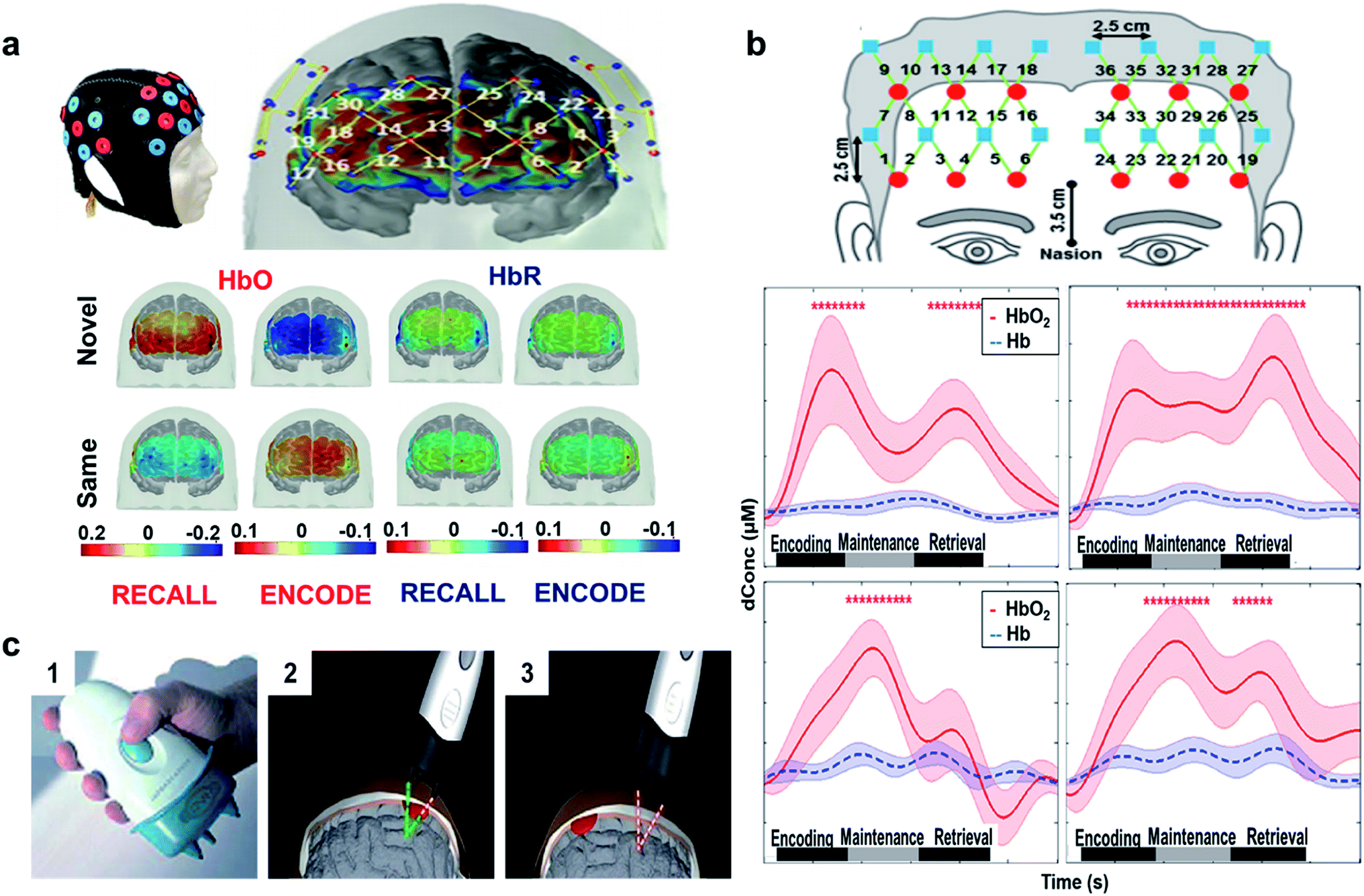
fNIRS have been demonstrated to measure the cerebral activity in memory encoding and recovery. fNIRS utilizes in near-infrared light source to non-invasively monitor changes in the oxy (HbO-increase in blood supply) and deoxy (HbR-decrease in blood supply)-hemoglobin content in the cerebrum. HbO and HbR spatial and temporal concentrations are generally interrelated with the blood oxygen level dependent signals (BOLD) to track the changes in the brain.99
In another study, the temporal, parietal, and prefrontal regions were monitored to examine paired-associate learning task (PAL).100 PAL is used to evaluate memory performance/impairment, which requires use of associative memory to pair one item to another item to which it was previously associated. A person's ability on this task depends on the individual to build a link among the paired groups. Fig. 5(a) (top) shows the designed probe used for the study and the points corresponding to the prefrontal cortex.101 The results revealed that novel face encoding required activation of left inferior frontal cortex and inactivation of medial-to-superior frontal cortices which is shown to be significant from the color bar as shown in Fig. 5(a) (bottom left). These results were in accordance with fMRI and PET studies showing the left frontal gyrus activation during encoding.96,102 Fig. 5(a) (bottom right) shows that novel face recalling activated the inferior, middle, and right frontal cortices. Inactivation in middle frontal cortex through recall of the same faces might be due to the lack of attention or inactivation of a default memory network (DMN). The colored bar suggests (Fig. 5(a) bottom) participation of neural activity throughout recall, which might be due to more attentional resources. Another reason might be that harder tasks requires both high cerebral initiation and participation in comparison to easy tasks. Since these data were substantiated by other imaging techniques, fNIRS may serve as a substitute biomarker to assess encoding and retrieval memory responses in order to track a likely associated disease. In comparison to PET and fMRI, fNIRS is portable, low cost, and is non-invasive.
 | ||
| Fig. 5 (a) fNIRS designed probe. Location of sources (red circles) and detectors (blue circles) on the cap (top left). Sensitivity profile for the probe. The channel numbers are in yellow. The color scale spans the sensitivity logarithmically from 0.01 (blue) to 1 (red). Group mean HbO and HbR hemodynamic responses overlayed over the brain surface for: encoding (bottom left) and recalling (bottom right) of novel and same faces. The displayed concentration changed was averaged from t = 25 to 50 s. The color bar indicates the scale of the concentration change in μM units. Reproduced with permission from ref. 101. Copyright 2017, Nature Publishing Group. (b) Configuration (top) of the fNIRS probe. Red circles represent light sources, blue squares represent detectors, and green lines represent the nearest source–detector pairs (channels) to measure the brain activities. Mean task-evoked prefrontal hemodynamic responses in the: control group (middle) forward task (middle left) and backward task (middle right), PTSD group (bottom) forward task (bottom left) and backward task (bottom right). In both panels, the solid lines represent the mean time courses of HbO2, the dotted lines represent the mean time courses of Hb, the shaded regions represent the standard errors, the * symbols indicate the period of significant HbO2 changes (one-sample t-test, p < 0.01) from the baseline, and the gray bars in the bottom indicate the three phases of the task. It is noted that the actual retrieval time was slightly variable among participants. Therefore, the retrieval phases labeled in both panels are schematic and approximate. Reproduced with permission from ref. 104. Copyright 2014, Tian et al. Published by Elsevier. (c) Handheld NIR device. Bluetooth equipped Full device (part 1) intra (part 2) and extra (part 3) – axial hemorrhagic haematomas acquisition. Reproduced with permission from ref. 105. Copyright 2010, Taylor & Francis. | ||
The phases of working memory (encoding, maintenance, and retrieval) are mediated by prefrontal activation (central executive function). A similar fNIRS study was conducted in veterans with PTSD versus healthy controls. Results obtained from statistical analysis of task-evoked hemo-dynamic responses showed that PTSD veterans had irregular prefrontal responses in comparison to healthy subjects, suggesting deficits in prefrontal control of working memory. Healthy subjects had equal activations in both hemispheres throughout all the three phases (Fig. 5(b) (middle)), while PTSD veterans exhibited initiations in the left dorsolateral prefrontal cortex (DLPFC) during encoding and maintenance (Fig. 5(b) (bottom)). In healthy controls, HbO2 had approximately 0.58 μM peak (middle right) higher than the retrieval phase with approximately 0.4 μM peak (middle left). For PTSD group, HbO2 changes in the forward task (bottom left) and backward task (middle right) were dominant in the maintenance phase with a discrete decline in the retrieval phase. The results were consistent with studies from Schiffer et al.103 that confirmed variances in the right and left hemispheres between the healthy controls and trauma patients. The study also showed that the right frontal cortex processed negative reactions, while the left frontal cortex processed positive and neutral reactions.100 Although, fNIRS is an established technique for brain imaging, most of the literature is concentrated on veterans with comorbid stage PTSD and hence the future work must be directed towards broader diagnoses covering all stages of PTSD from mild to severe, as well as PTSD derived from different types of trauma.104 Recently, a handheld NIR device was attached to the scalp through optical fibers and utilized to scan brain haematomas (Fig. 5(c)).105
Generally, biological tissues exhibit several bands (usually 10 cm−1 to 20 cm−1 wide) in their spectra. The intensity of each peak can also demonstrate quantitative information about different chemicals in a tissue or cell. Therefore, Raman spectroscopy can be utilized as a powerful tool for either quantitative or qualitative studies of biological substances by providing distinctive fingerprints of tissues and cells. Despite the long history of Raman spectroscopy in chemical analysis of materials, the application of this technique in biology and biomedical sciences has only begun in recent years. Through technological improvements in detectors and lasers, Raman spectroscopy and microscopy has improved. Meanwhile, the current challenge in this sphere is the appropriate data analysis of Raman spectra in biological applications, which require advanced techniques of data analysis like principal component analysis (PCA). These types of analysis should focus more on information content of Raman spectra rather than only the band positions to fully take advantage of Raman data. Statistical, chemical, and morphological issues substantially control the data analysis quality in Raman spectroscopy and microscopy. Moreover, in terms of technological limitations, the distal tip of the probe requires more studies and improvements to be done in order to enhance the quality of data acquisition. A fully calibrated probe–tissue interface can greatly improve the acquisition of information in different depths of biological samples. Depth ranging studies of biological samples is usually limited by the natural turbidity of them caused by the elastic scattering of these media. So far, depth resolution and light collection issues have been the significant drawbacks of Raman spectroscopy and imaging in biomedical applications. These problems occur due to the weak penetration of light into samples (a few millimeters) even at low absorption conditions. At the moment, attempting to eliminate the elastic scattering and/or converting it to an advantage are being scrutinized as alternatives in designing new Raman probes. There is a great interest in the literature to provide a method in which Raman signals can penetrate much deeper into tissues to study brain injury through the skull, in PTSD patients for example. By devising a solution for probe issues in Raman microscopy, other interesting applications can also be introduced like functional imaging. Employing Raman imaging in applications such as endoscopic configurations, can generate morphological and chemical images of live tissues. For a comprehensive study of PTSD patients, Raman spectroscopy and imaging can be utilized alongside other approaches like magnetic resonance imaging, intravascular ultra sound, computed tomography scan and different types of X-ray analyses. Such studies provide complementary morphological and chemical Raman information as well as tissue density information achieved by other methods.107,108
Aforementioned methods can be used for brain imaging, but only Raman and FTIR imaging can be utilized for minimally invasive tracking of tissue biochemistry. Raman spectroscopy (RS) has also been reported to acquire label-free spectra in TBI mice. The left parietotemporal cortex of the mice brain was injured, and they were excised 7th day post TBI, and the spectra was acquired at several spatial locations namely, contusion core (CC), contralateral (CL); pericontusional (PC), ipsilateral tissue, and distant from contusion core (ID). The spectra showed elevated levels of lipids, proteins, and cholesterol in comparison to sham control mice (Fig. 6(a) (bottom)). To locate the source of the changing lipid signal observed, RS was acquired for cholesterol, cholesteryl nonanoate, cholesteryl oleyl carbonate, intralipid, and albumin respectively. The spectra for these species in brain samples are: bands at 426, 701, 801, and 1128 cm−1 corresponding to cholesterol peaks, 1301 cm−1 for fatty acids observed in cholesterol derivates, 1440 cm−1 due to CH stretch, 1462 & 1660 cm−1 from CC stretching associated with lipids (Fig. 6(b)). Few other peaks lie within the boundary line of these peaks, which might be due to changes in protein and lipid composition post injury and trauma. Hence, RS can be used as a potential probe that is sensitive to detect pathology of TBI and can also be applied to PTSD. A disadvantage with RS is the difficulty of data interpretation and analysis to isolate the origin component, and hence requires rigorous statistical or PCA analysis.109 Recently, handheld contact Raman spectroscopy probes have gained huge attention, and have been applied for real time acquisition in human subjects (Fig. 6(c)).110
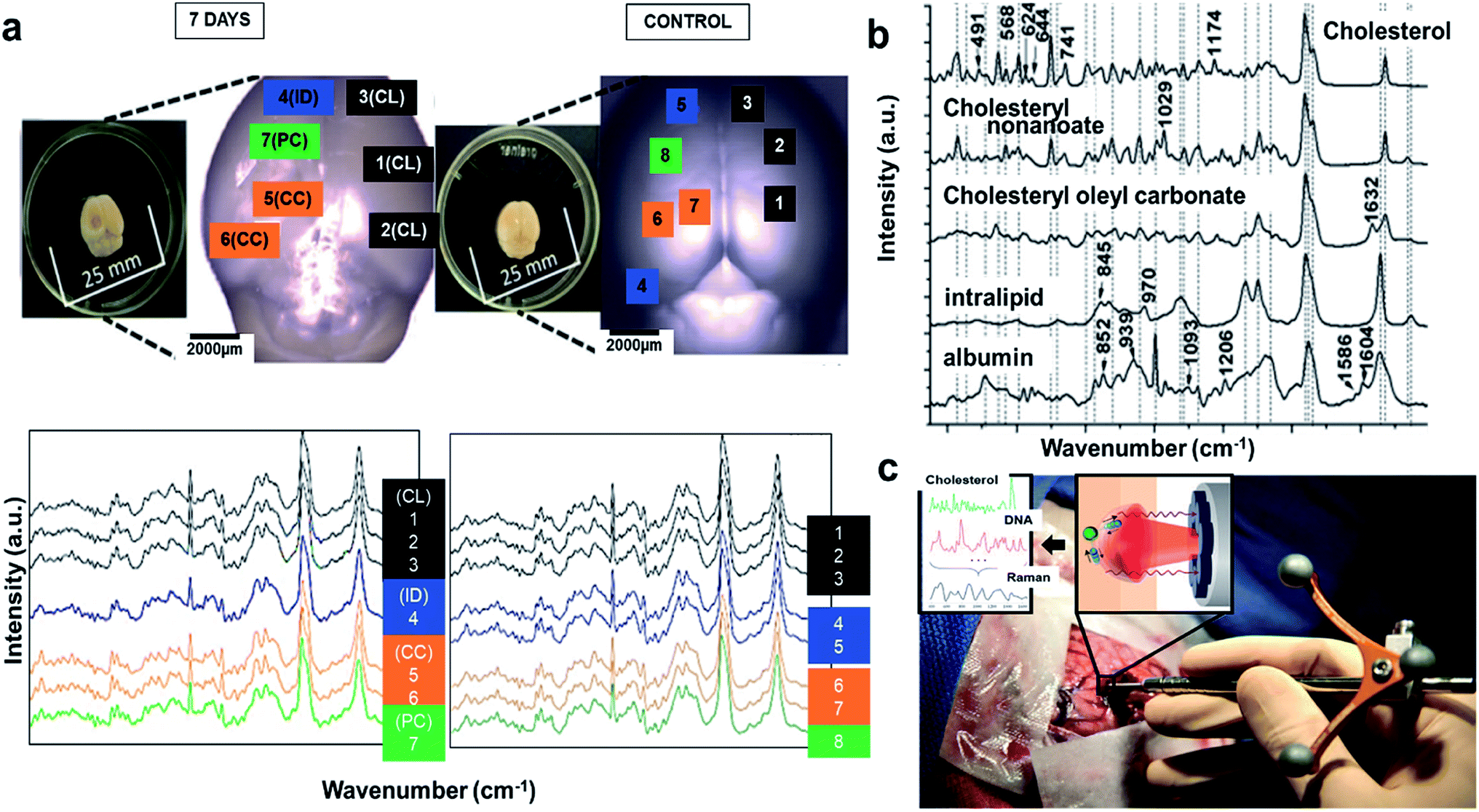 | ||
| Fig. 6 Raman spectroscopy to detect tissue biochemistry. (a) Bright field microscopy of murine brains at 7th day and control (inset photographs show sample preparation). Average Raman spectra shown were acquired from the areas indicated in matching colors in the photos. CL = contralateral; ID = ipsilateral distant from contusion core; PC = pericontusional tissue; and CC = contusion core. Spectra were acquired at 785 nm, with 1 s exposure and 10 accumulations; the average is performed over 30 separate spatial points (bottom). (b) Raman spectral of the cholesterol, cholesteryl nonanoate, cholesteryl oleyl carbonate, and intralipid (no structure was shown as mixed lipids). Protein albumin was shown for reference. Spectra were acquired at 785 nm, with 1 s exposure and 10 accumulations at 175 mw; the average is performed over 10 separate spatial points. Reproduced with permission from ref. 109. Copyright 2017, Published by The Royal Society of Chemistry. (c) Image of a handheld contact Raman spectroscopy probe. Reproduced with permission from ref. 110. Copyright 2015, American Association for the Advancement of Science. | ||
A great strength of RS over currently available techniques is the potential to track tissue biochemistry changes in real time. Numerous studies, including recent investigations in an animal model of PTSD by Dr Francis Lab, suggest the possibility of stress induced oxidative and immune-mediated24 damage to cellular proteins, lipids, cholesterol that could be measured in real time by RS. These data could give us an idea of how quickly these changes occur after a traumatic incident and offer a measure in relation to which we could introduce therapeutics to help attenuate these stress-induced alterations to tissue biochemistry and molecular function.
In fluorescence microscopy, lateral and axial resolution are generally confined to 250 nm and 500 nm, respectively. However, some super resolution devices provide higher spatial resolution than the typical diffraction limits. According to Nyquist condition, a N-fold resolution rise in D-dimension needs a N × D-fold reduction in the magnitude of pixels. Therefore, performing super high resolution imaging requires almost eight times higher signal collection rate to maintain the same SNR of conventional devices.111,112
Multicolor fluorescence imaging provides an advantage of monitoring multiple sections after tagging it with specific fluorophores. Multi-imaging was demonstrated in a cryoinjured mice dosed with both PSS-794 (for detection of apoptotic cells) and Tracer-653 (deep-red fluorescent vascular imaging agent) for optical imaging of blood brain barrier (BBB). A 60 s cryo-brain injury was delivered to 3-cohorts of hairless mice followed by single dose injection of PSS-794 at 0, 3 and 7th day respectively. Fig. 7(a) (left) shows pictures obtained after fluorescence staining using PSS-794 (apoptotic cells), Tracer-794 (vasculature), and Annexin-Vivo 750 (cellular protein for early stages of apoptosis) in a cryoinjured mouse model. Tracer-794 are seen after probe injection at the site of cryolesion but it disappears later. Extensive accumulation of Annexin-Vivo 750 was observed in the kidneys. After 5 h, the mice were also injected with Tracer-653 and after 1 h, they were sedated, sacrificed, and the brain were removed and positioned on an epifluorescence station for ex vivo imaging. The PSS-794 images in Fig. 7(a) (left) reveals that PSS-794 accumulation decreased with the age of the injury and the amount of cell death decreased due to removal by the mice immune system after Day 3, as indicated by the MPI (mean pixel intensities) for a cryolesion section, which was 6-fold lower on Day 3 than on Day 0. In contrast, Tracer-653 pictures showed that BBB repair slowed with the age of injury. The BBB was porous after Day 7 than before the injury.113 In another similar study, TBI versus control results were acquired from confocal imaging of the brain vasculature. Fig. 7(b) (top) exhibited substantial damage of vessels at the perilesional region in TBI in comparison to sham control mice. Apart from the cortical imaging, several injured vascular region of interest were also measured (Fig. 7(b) (bottom)). The results showed a decrease in vessel number and length in TBI mice. From quantitative analysis, it was found that the vessel number decreased by 34.4% in TBI mice in comparison to sham controls.114 These data show how brain injury and BBB permeability can be imaged in models of TBI. Since PTSD exhibits similar characteristics to TBI, these methods could also improve the study of PTSD in animal models. Recently, a portable and light weight fluorescence endoscope was assembled for imaging both cells and tissues. Fig. 7(c), shows the computer aided design (CAD) (part 1) of the two-photon micro-endoscope, with the essential components such as the endoscope probe (part 3), piezo actuator, the excitation and the collection fiber (part 4). This device weighed ∼3.9 g making it applicable for visualization of blood vessels in the hippocampus of live mice.115 Recently, a 3 mm outer diameter multiphoton raster scanning endoscope was demonstrated for imagining unstained tissues (Fig. 7(d)).116
 | ||
| Fig. 7 (a) Representative in vivo near-infrared fluorescence assortments of PSS-794, Tracer-794, and Annexin-Vivo 750 accumulations in a brain cryoinjury mouse model. A precooled metal cylinder was applied to the head of each mouse for 60 s followed by intravenous injection of either PSS794 (3.0 mg kg−1), Tracer-794 (3.0 mg kg−1), or Annexin-Vivo 750. Images were acquired at the indicated time points after probe injection. N = 5. (left) Multicolor fluorescence ex vivo imaging of cell death and blood brain-barrier disruption in cryoinjured brains (right). Reproduced with permission from ref. 113. Copyright 2012, American Chemical Society. (b) Confocal images of vessel painted vasculature at the perilesional region for Sham and TBI. Cal bar = 200 μm (top). Classical vascular analysis using AngioTool imaging software. Axial vessels are displayed (red) and junctions (blue). Reproduced with permission from ref. 114. Copyright 2017, Nature Publishing Group. (c) Two-photon Fluorescence Micro endoscope. Computer aided design (CAD) (part 1), top view of entire setup (part 2), internal components (part 3), mechanism of light collection (part 4). Reproduced with permission from ref. 115. Copyright 2005, The Optical Society. (d) Image of a 3 mm outer diameter multiphoton endoscope. Reproduced with permission from ref. 116. Copyright 2011, National Academy of Sciences. | ||
Photoacoustic imaging (PAT). Photoacoustic imaging (PAT) is another emerging technique which has been demonstrated in literature recently. The acoustic/sound waves are generated from specimens underneath the skin by exposing to either radio frequency waves or by absorption of electromagnetic waves. Fig. 8(a) fig.9 (part 1) shows the schematics of PAT working along with images of the mouse brain vasculature (part 2) and the cortical vasculature of part 2 with scalp removed (part 3). Although PAT is promising, it is limited due to the low penetration depth resulting in low resolution, and hence is less applicable.117 Laser speckle contrast imaging (LSCI) is another emerging technique that have been utilized for acquiring the spatiotemporal dynamics of a vasculature. It is a simple technique based on the tissue contrast from dynamic scattered light. Fig. 8(b) shows the integration of the lenses for LSCI imaging. The advantage of this technique is that due to its simple configuration, it can be utilized for the cortical imaging of a moving mouse without tethering it as shown in Fig. 8(b) (right). This technique is limited due to its temporal resolution.118 Voltage-sensitive dye imaging (VSDi) is another technique that have been reported for visualizing neural networks with a high temporal resolution (∼1 ms). Fig. 8(c) shows the schematics of the VSDi working. It is largely dependent on the ratio of absorbance and fluorescence w.r.t the applied membrane potential. This technique is limited by applied voltage.119
 | ||
| Fig. 8 Other optical imaging techniques. (a) Schematics of Photoacoustic Tomography (PAT). Reproduced with permission from ref. 117. Copyright 2014, National Academy of Sciences. (b) Schematics of the assembled Laser speckle contrast imaging (LSCI) with microscope showing incident and reflected light paths in blue and green (left), and imaging of free moving mice (right). Reproduced with permission from ref. 118. Copyright 2009, Elsevier B.V. (c) Demonstration of voltage sensitive dye imaging (VSDi) system. Schematics shows localization of dye in the membranes and a VSDi optical image acquired from the assembled setup. Reproduced with permission from ref. 119. Copyright 2013, Elsevier B.V. | ||
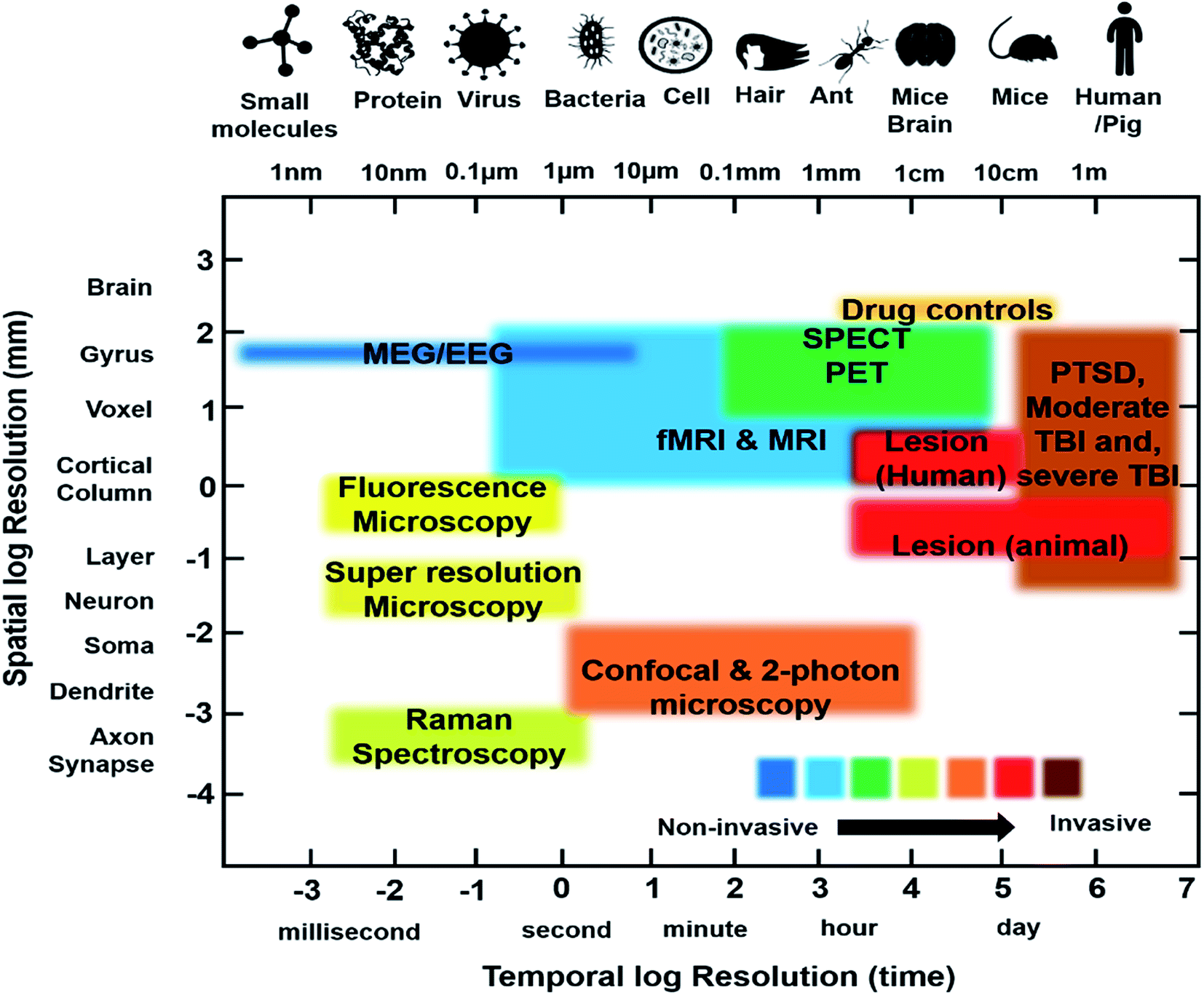 | ||
| Fig. 9 Comparison of temporal and spatial resolution for conventional and emerging methods. | ||
Super resolution imaging. In order to overcome the diffraction limit of light microscopy methods, various novel techniques have been introduced to enhance the resolution of microscopes. Although electron microscopy also provides a nanometric resolution for biological imaging, lack of large volume imaging, labeling of multi-targets and the need for fixation of samples are serious challenges that restrain its applications.120 As a result, more advanced light microscopy methods such as stimulated emission depletion (STED), stochastic optical reconstruction microscopy (STORM) (also known as photo-activated localization microscopy (PALM)) and structured illumination microscopy (SIM) have attracted so much interest in neurobiological field. Lateral and axial resolution of normal light microscopes are limited to 200 and 600 nm, respectively.121 However, super resolution techniques overcome this limitation by utilizing near and far field mechanisms of electromagnetic fields.122 In STED method, the spatial resolution in the focal plane is enhanced by using a quenching laser. Although an extremely high spatial resolution of 6 nm has been reported in the literature,123 the achievable resolution with this method is commonly between 30–70 nm.124 The capability of this method to obtain images from live cell sub-cellular structures is the main reason of its popularity in the field of neurobiology. Thick tissue imaging is another interesting feature of STED which has been utilized for dendritic spines morphological dynamics studies of brain samples.125 Furthermore, combination of STED with neurobiological techniques has revealed strong connections between electrical properties of synapses and morphological dynamics of dendritic spines.126 On the other hand, STORM technique utilizes super resolved imaging of each fluorochrome molecule to overcome the diffraction limit of light. By this method, nanometric resolution can be achieved via calculation of the center of mass for individual fluorochromes.127 However, the fluorochromes should be activated sparsely in order to prevent overlapping. Furthermore, this approach heavily depends on image processing/analysis techniques and temporal resolution is also limited due to the huge number of acquired images (∼1000) which are needed to be taken to form the final high-resolution image. Because of the wide field nature of STORM, it is rarely used for thick tissue slices. Instead, it is usually employed for membrane protein and intracellular studies.128 In SIM, heterodyne detection method is used to continuously excite the sample in a wide field mode. In this imaging technique, higher spatial frequency signal is converted into a detectable bandwidth to form a fluorescence image. Although the contrast is usually low for thicker samples in this method, it provides a fast imaging (∼10 Hz) rate due to the use of wide field imaging mode.129
4D imaging. Another novel imaging technique in neurobiology studies uses ultra sound scanning to generate a 4D image of brain. The differences in reflection of ultra sound from various regions of brain produces contrast to form an image. Wave-return time and the strength of the returned wave constructs the image. While 3D imaging provides structural information from various parts of the brain, 4D imaging also generates live streaming of the region of interest. Therefore, 4D imaging is actually similar to 3D imaging plus providing live streaming capabilities.130 Despite the advantages of this method for live studies of brain activities, its application is mainly limited due to the low resolution (∼100 μm) of this method compared to other emerging techniques.131 The equipment used for 4D ultrasound imaging is also extremely costly which makes it an unaffordable option for many cases. As a result, very few studies are available which has been utilized this approach. In a recent study, 4D ultrasound microvascular imaging was performed to visualize the brain hemodynamics without usage of contrast agents.132 They have showed the successful employment of this technique for whole brain imaging that can be used in various brain conditions and disorders. Other 4D imaging techniques were also rarely used for brain-related studies like 4D-CT angiography (CTA),133 4D electrical capacitance volume tomography (ECVT)134 and 4D two-photon inverted selective plane illumination microscopy (2PE-iSPIM).135
4. Conclusion & future perspectives
This review summarized the literature regarding conventional and emerging imaging and spectroscopy techniques in PTSD related studies. Fig. 9 illustrates the spatial and temporal resolution of the methods discussed in this article at a glance. Information from animal models of the disease as well as some of the most important potential biomarkers being studied were also included for a deeper comprehension of this topic and to provide example endpoints that may be important for future imaging studies. Conventional methods of investigating PSTD like SPECT, PET and fMRI were discussed along with their advantages and limitations to provide a basis for our current capacity to study PTSD. Furthermore, novel approaches for detection of PTSD biomarkers and their recent improvements and drawbacks were mentioned to provide an overview of the current literature and to create a guideline for future improvements. Conventional methods of PTSD studies are not able to detect a unique biomarker for this disease, or any psychiatric disease for that matter. All of the revealed cellular and behavioral indicators for PSTD are common to other disorders and this makes the diagnosis and treatment dramatically difficult. On the other hand, novel approaches like FTIR and Raman spectroscopy may be capable of introducing unique identifiers of this disease. However, some enhancements need to be made in the design of current devices to make them suitable in order to reach the aforementioned goal. Finally, emerging methods should be employed as complementary tools for conventional approaches to achieve the best results in the shortest timeline. Ultimately, a true biomarker for PTSD may be a composite formed from the analysis of several biomarkers in tandem, within the context of computational models that integrate functional and molecular imaging as well as genetic and peripheral markers of the disease. This may be the only way to develop a truly diagnostic and predictive capacity of the sort required for a comprehensive diagnosis and assessment of PTSD that could support adequate treatment plans for treatment resistant patients.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
M. R. G. acknowledges the support from LSU start-up fund, Louisiana Board of Regents Support Fund (RCS Award Contract Number: LEQSF (2017-20)-RD-A-04), and LaSPACE (LEQSF (2015-18)-LASPACE, GR-4216). A. C. was supported by Economic Development Assistantship (EDA) grant. A. P. was supported by the National Science Foundation (NSF Award Number: 1660233).References
- C. Benjet, E. Bromet, E. Karam, R. Kessler, K. McLaughlin, A. Ruscio, V. Shahly, D. Stein, M. Petukhova and E. Hill, Psychol. Med., 2016, 46, 327–343 CrossRef CAS PubMed.
- R. C. Kessler, A. Sonnega, E. Bromet, M. Hughes and C. B. Nelson, Arch. Gen. Psychiatry, 1995, 52, 1048–1060 CrossRef CAS PubMed.
- N. Breslau and G. C. Davis, Am. J. Psychiatry, 1992, 149, 671 CrossRef CAS PubMed.
- P. N. Santiago, R. J. Ursano, C. L. Gray, R. S. Pynoos, D. Spiegel, R. Lewis-Fernandez, M. J. Friedman and C. S. Fullerton, PLoS One, 2013, 8, e59236 CrossRef CAS PubMed.
- P. R. Zoladz and D. M. Diamond, Neurosci. Biobehav. Rev., 2013, 37, 860–895 CrossRef PubMed.
- American Psychiatric Association, Diagnostic and statistical manual of mental disorders (DSM-5®), American Psychiatric Pub, 2013 Search PubMed.
- P. J. Ebenezer, C. B. Wilson, L. D. Wilson, A. R. Nair and J. Francis, PLoS One, 2016, 11, e0160923 CrossRef PubMed.
- N. Gilpin and J. J. G. Weiner, Brain Behav., 2017, 16, 15–43 CAS.
- V. Michopoulos, A. Powers, C. F. Gillespie, K. J. Ressler and T. Jovanovic, Neuropsychopharmacology, 2017, 42, 254 CrossRef CAS PubMed.
- V. Francati, E. Vermetten and J. Bremner, Depression Anxiety, 2007, 24, 202–218 CrossRef CAS PubMed.
- U. Schmidt, S. F. Kaltwasser and C. T. Wotjak, Dis. Markers, 2013, 35, 43–54 CrossRef PubMed.
- H. J. Kang, S. Yoon and I. K. Lyoo, Exp. Neurobiol., 2015, 24, 186–196 CrossRef PubMed.
- L. Zhang, H. Li and R. J. Ursano, in Heat shock proteins and whole body physiology, Springer, 2010, pp. 179–192 Search PubMed.
- R. Yehuda, N. P. Daskalakis, F. Desarnaud, I. Makotkine, A. Lehrner, E. Koch, J. D. Flory, J. D. Buxbaum, M. J. Meaney and L. M. Bierer, Front. Psychiatry, 2013, 4, 118 Search PubMed.
- R. D. Marshall, K. L. Beebe, M. Oldham and R. Zaninelli, Am. J. Psychiatry, 2001, 158, 1982–1988 CrossRef CAS PubMed.
- J. G. Hensler, in Handbook of Behavioral Neuroscience, Elsevier, 2010, vol. 21, pp. 367–378 Search PubMed.
- C. A. Lowry and M. W. Hale, in Handbook of Behavioral Neuroscience, Elsevier, 2010, vol. 21, pp. 379–397 Search PubMed.
- A. C. Linthorst and J. M. Reul, in Handbook of behavioral neuroscience, Elsevier, 2010, vol. 21, pp. 475–491 Search PubMed.
- C. B. Wilson, P. J. Ebenezer, L. D. McLaughlin and J. Francis, PLoS One, 2014, 9, e89104 CrossRef PubMed.
- M. J. Girgenti and R. S. Duman, Biol. Psychiatry, 2017 Search PubMed.
- E. R. De Kloet, M. Joëls and F. Holsboer, Nat. Rev. Neurosci., 2005, 6, 463 CrossRef CAS PubMed.
- D. A. Young, S. S. Inslicht, T. J. Metzler, T. C. Neylan and J. A. Ross, Psychiatry Res., 2018, 270, 961–966 CrossRef CAS PubMed.
- F. J. Raabe and D. Spengler, Front. Psychiatry, 2013, 4, 80 Search PubMed.
- C. B. Wilson, L. D. McLaughlin, A. Nair, P. J. Ebenezer, R. Dange and J. Francis, PLoS One, 2013, 8, e76146 CrossRef CAS PubMed.
- K. Speer, D. Upton, S. Semple and A. McKune, J. Inflammation Res., 2018, 11, 111 CrossRef PubMed.
- R. Yehuda, L. C. Pratchett, M. W. Elmes, A. Lehrner, N. P. Daskalakis, E. Koch, I. Makotkine, J. D. Flory and L. M. Bierer, Interface Focus, 2014, 4, 20140048 CrossRef PubMed.
- C. Anacker, P. A. Zunszain, L. A. Carvalho and C. M. Pariante, Psychoneuroendocrinology, 2011, 36, 415–425 CrossRef CAS PubMed.
- M.-L. Meewisse, J. B. Reitsma, G.-J. De Vries, B. P. Gersons and M. Olff, Br. J. Psychiatry, 2007, 191, 387–392 CrossRef PubMed.
- C. Anacker, P. A. Zunszain, A. Cattaneo, L. A. Carvalho, M. J. Garabedian, S. Thuret, J. Price and C. M. Pariante, Mol. Psychiatry, 2011, 16, 738 CrossRef CAS PubMed.
- C. Anacker, A. Cattaneo, K. Musaelyan, P. A. Zunszain, M. Horowitz, R. Molteni, A. Luoni, F. Calabrese, K. Tansey and M. Gennarelli, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8708–8713 CrossRef CAS PubMed.
- D. Mehta, T. Klengel, K. N. Conneely, A. K. Smith, A. Altmann, T. W. Pace, M. Rex-Haffner, A. Loeschner, M. Gonik and K. B. Mercer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8302–8307 CrossRef CAS PubMed.
- A. Karl, M. Schaefer, L. S. Malta, D. Dörfel, N. Rohleder and A. Werner, Neurosci. Biobehav. Rev., 2006, 30, 1004–1031 CrossRef PubMed.
- D. C. O'Doherty, K. M. Chitty, S. Saddiqui, M. R. Bennett and J. Lagopoulos, Psychiatry Res., Neuroimaging, 2015, 232, 1–33 CrossRef PubMed.
- S. H. Woodward, D. G. Kaloupek, C. C. Streeter, C. Martinez, M. Schaer and S. Eliez, Biol. Psychiatry, 2006, 59, 582–587 CrossRef PubMed.
- T. J. Akiki, C. L. Averill and C. G. Abdallah, Curr. Psychiatry Rep., 2017, 19, 81 CrossRef PubMed.
- R. Yehuda, J. A. Golier, L. Tischler, P. D. Harvey, R. Newmark, R. K. Yang and M. S. Buchsbaum, J. Psychiatr. Res., 2007, 41, 435–445 CrossRef PubMed.
- V. Van Veen and C. S. Carter, Physiol. Behav., 2002, 77, 477–482 CrossRef CAS PubMed.
- K. Izuma, M. Matsumoto, K. Murayama, K. Samejima, N. Sadato and K. Matsumoto, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22014–22019 CrossRef CAS PubMed.
- S. L. Rauch, L. M. Shin, E. Segal, R. K. Pitman, M. A. Carson, K. McMullin, P. J. Whalen and N. Makris, NeuroReport, 2003, 14, 913–916 CrossRef PubMed.
- K. S. LaBar, J. C. Gatenby, J. C. Gore, J. E. LeDoux and E. A. Phelps, Neuron, 1998, 20, 937–945 CrossRef CAS PubMed.
- J. E. Dunsmoor, S. E. Prince, V. P. Murty, P. A. Kragel and K. S. LaBar, NeuroImage, 2011, 55, 1878–1888 CrossRef PubMed.
- R. J. Dolan, Philos. Trans. R. Soc., B, 2007, 362, 787–799 CrossRef CAS PubMed.
- T. V. Gurvits, M. E. Shenton, H. Hokama, H. Ohta, N. B. Lasko, M. W. Gilbertson, S. P. Orr, R. Kikinis, F. A. Jolesz and R. W. McCarley, Biol. Psychiatry, 1996, 40, 1091–1099 CrossRef CAS PubMed.
- J. D. Bremner, R. B. Innis, C. K. Ng, L. H. Staib, R. M. Salomon, R. A. Bronen, J. Duncan, S. M. Southwick, J. H. Krystal and D. Rich, Arch. Gen. Psychiatry, 1997, 54, 246–254 CrossRef CAS PubMed.
- K. Kasai, H. Yamasue, M. W. Gilbertson, M. E. Shenton, S. L. Rauch and R. K. Pitman, Biol. Psychiatry, 2008, 63, 550–556 CrossRef PubMed.
- P. R. Szeszko and R. Yehuda, Psychiatry Res., 2019, 277, 52–57 CrossRef PubMed.
- P. L. S. Jacques, P. A. Kragel and D. C. Rubin, Cogn. Affect. Behav. Neurosci., 2013, 13, 554–566 CrossRef PubMed.
- M. Tursich, T. Ros, P. Frewen, R. Kluetsch, V. D. Calhoun and R. A. Lanius, Acta Psychiatr. Scand., 2015, 132, 29–38 CrossRef CAS PubMed.
- P. V. Bayly, E. H. Clayton and G. M. Genin, Annu. Rev. Biomed. Eng., 2012, 14, 369–396 CrossRef CAS PubMed.
- P. Garrigue, L. Giacomino, C. Bucci, V. Muzio, M. A. Filannino, F. Sabatier, F. Dignat-George, P. Pisano and B. Guillet, Int. J. Stroke, 2016, 11, 117–126 CrossRef PubMed.
- M. T. Asl, R. Nemati, F. Yousefi, H. Salimipour, I. Nabipour and M. Assadi, Iran. J. Nucl. Med., 2017, 25, 1–14 Search PubMed.
- C. A. Raji and T. A. Henderson, Neuroimaging Clin., 2018, 28, 67–82 CrossRef PubMed.
- K. Fabio, C. Guillon, C. J. Lacey, S.-f. Lu, N. D. Heindel, C. F. Ferris, M. Placzek, G. Jones, M. J. Brownstein and N. G. Simon, Bioorg. Med. Chem., 2012, 20, 1337–1345 CrossRef CAS PubMed.
- M. Q. Hoexter, G. Fadel, A. C. Felício, M. B. Calzavara, I. R. Batista, M. A. Reis, M. C. Shih, R. K. Pitman, S. B. Andreoli and M. F. Mello, Psychopharmacology, 2012, 224, 337–345 CrossRef CAS PubMed.
- P. G. Harch, S. R. Andrews, E. F. Fogarty, J. Lucarini and K. W. Van Meter, Med. Gas Res., 2017, 7, 156 CrossRef CAS PubMed.
- C. Wang, M. S. Placzek, G. C. Van de Bittner, F. A. Schroeder and J. M. Hooker, ACS Chem. Neurosci., 2016, 7, 484–489 CrossRef CAS PubMed.
- J. D. Bremner, S. Mishra, C. Campanella, M. Shah, N. Kasher, S. Evans, N. Fani, A. J. Shah, C. Reiff, L. L. Davis, V. Vaccarino and J. Carmody, Front. Mol. Psychiatry, 2017, 8, 157 CrossRef PubMed.
- S. E. Holmes, M. J. Girgenti, M. T. Davis, R. H. Pietrzak, N. DellaGioia, N. Nabulsi, D. Matuskey, S. Southwick, R. S. Duman and R. E. Carson, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8390–8395 CrossRef CAS PubMed.
- S. E. Holmes, R. Hinz, S. Conen, C. J. Gregory, J. C. Matthews, J. M. Anton-Rodriguez, A. Gerhard and P. S. Talbot, Biol. Psychiatry, 2018, 83, 61–69 CrossRef CAS PubMed.
- A. R. Mayer, P. S. Bellgowan and F. M. Hanlon, Neurosci. Biobehav. Rev., 2015, 49, 8–18 CrossRef PubMed.
- S. N. Garfinkel, J. L. Abelson, A. P. King, R. K. Sripada, X. Wang, L. M. Gaines and I. Liberzon, J. Neurosci., 2014, 34, 13435–13443 CrossRef CAS PubMed.
- V. Zotev, R. Phillips, M. Misaki, C. K. Wong, B. E. Wurfel, F. Krueger, M. Feldner and J. Bodurka, NeuroImage Clin., 2018, 19, 106–121 CrossRef PubMed.
- M. I. Gerin, H. Fichtenholtz, A. Roy, C. J. Walsh, J. H. Krystal, S. Southwick and M. Hampson, Front. Mol. Psychiatry, 2016, 7, 111 Search PubMed.
- P. J. Basser, J. Mattiello and D. LeBihan, Biophys. J., 1994, 66, 259–267 CrossRef CAS PubMed.
- P. J. Basser and C. Pierpaoli, Magn. Reson. Med., 1998, 39, 928–934 CrossRef CAS PubMed.
- C. Pierpaoli, P. Jezzard, P. J. Basser, A. Barnett and G. Di Chiro, Radiology, 1996, 201, 637–648 CrossRef CAS PubMed.
- Y. Assaf and O. Pasternak, J. Mol. Neurosci., 2008, 34, 51–61 CrossRef CAS PubMed.
- S. Pajevic and C. Pierpaoli, Magn. Reson. Med., 1999, 42, 526–540 CrossRef CAS PubMed.
- M. Catani, R. J. Howard, S. Pajevic and D. K. Jones, NeuroImage, 2002, 17, 77–94 CrossRef PubMed.
- A. Klimova, M. S. Korgaonkar, T. Whitford and R. A. Bryant, Biol. Psychiatry Cogn. Neurosci. Neuroimaging, 2019, 4, 81–90 CrossRef PubMed.
- M. Kubicki, R. McCarley, C.-F. Westin, H.-J. Park, S. Maier, R. Kikinis, F. A. Jolesz and M. E. Shenton, J. Psychiatr. Res., 2007, 41, 15–30 CrossRef PubMed.
- Y. Liao, X. Huang, Q. Wu, C. Yang, W. Kuang, M. Du, S. Lui, Q. Yue, R. C. Chan and G. J. Kemp, J. Psychiatry Neurosci., 2013, 38, 49 CrossRef PubMed.
- A. R. Hoy, M. Ly, C. M. Carlsson, O. C. Okonkwo, H. Zetterberg, K. Blennow, M. A. Sager, S. Asthana, S. C. Johnson and A. L. Alexander, PLoS One, 2017, 12, e0173982 CrossRef PubMed.
- C. L. Averill, L. A. Averill, K. M. Wrocklage, J. C. Scott, T. J. Akiki, B. Schweinsburg, S. M. Southwick, J. H. Krystal and C. G. Abdallah, Mol. Neuropsychiatry, 2018, 4, 75–82 CrossRef PubMed.
- N. Fani, T. Z. King, T. Jovanovic, E. M. Glover, B. Bradley, K. Choi, T. Ely, D. A. Gutman and K. J. Ressler, Neuropsychopharmacology, 2012, 37, 2740 CrossRef PubMed.
- L. M. Bierer, I. Ivanov, D. M. Carpenter, E. W. Wong, J. A. Golier, C. Y. Tang and R. Yehuda, Psychoneuroendocrinology, 2015, 51, 567–576 CrossRef PubMed.
- K. Aschbacher, S. H. Mellon, O. M. Wolkowitz, C. Henn-Haase, R. Yehuda, J. D. Flory, L. M. Bierer, D. Abu-Amara, C. R. Marmar and S. G. Mueller, Brain Imaging Behav., 2018, 12, 989–999 CrossRef PubMed.
- S. B. Koch, M. Van Zuiden, L. Nawijn, J. L. Frijling, D. J. Veltman and M. Olff, J. Psychiatry Neurosci., 2017, 42, 331 CrossRef PubMed.
- Z. Long, X. Duan, B. Xie, H. Du, R. Li, Q. Xu, L. Wei, S.-x. Zhang, Y. Wu and Q. Gao, J. Affective Disord., 2013, 150, 798–806 CrossRef PubMed.
- D. Soares and M. Law, Clin. Radiol., 2009, 64, 12–21 CrossRef CAS PubMed.
- A. Karl and A. Werner, Neurosci. Biobehav. Rev., 2010, 34, 7–22 CrossRef PubMed.
- Z. Movasaghi, S. Rehman and D. I. Ur Rehman, Appl. Spectrosc. Rev., 2008, 43, 134–179 CrossRef CAS.
- K. A. Chan and S. G. Kazarian, Chem. Soc. Rev., 2016, 45, 1850–1864 RSC.
- A. C. S. Talari, M. A. G. Martinez, Z. Movasaghi, S. Rehman and I. U. Rehman, Appl. Spectrosc. Rev., 2017, 52, 456–506 CrossRef CAS.
- S. M. Levine and D. L. B. Wetzel, Appl. Spectrosc. Rev., 1993, 28, 385–412 CrossRef CAS.
- P. Bassan, H. J. Byrne, J. Lee, F. Bonnier, C. Clarke, P. Dumas, E. Gazi, M. D. Brown, N. W. Clarke and P. Gardner, Analyst, 2009, 134, 1171–1175 RSC.
- M. Hartmann, B. D. Hanh, H. Podhaisky, J. Wensch, J. Bodzenta, S. Wartewig and R. H. Neubert, Analyst, 2004, 129, 902–905 RSC.
- M. J. Hackett, J. Lee, F. El-Assaad, J. A. McQuillan, E. A. Carter, G. E. Grau, N. H. Hunt and P. A. Lay, ACS Chem. Neurosci., 2012, 3, 1017–1024 CrossRef CAS PubMed.
- R. Zhao, L. Quaroni and A. Casson, Analyst, 2010, 135, 53–61 RSC.
- M. J. Baker, J. Trevisan, P. Bassan, R. Bhargava, H. J. Butler, K. M. Dorling, P. R. Fielden, S. W. Fogarty, N. J. Fullwood and K. A. Heys, Nat. Protoc., 2014, 9, 1771–1791 CrossRef CAS PubMed.
- K. Araki, N. Yagi, Y. Ikemoto, H. Yagi, C.-J. Choong, H. Hayakawa, G. Beck, H. Sumi, H. Fujimura and T. Moriwaki, Sci. Rep., 2015, 5, 17625 CrossRef CAS PubMed.
- C. R. Guillot, J. R. Fanning, T. Liang, A. M. Leventhal and M. E. Berman, J Anxiety Disord., 2015, 30, 41–47 CrossRef PubMed.
- T. Yasuda, Y. Nakata and H. Mochizuki, Mol. Neurobiol., 2013, 47, 466–483 CrossRef CAS PubMed.
- J. Zhang, F. Niu, H. Dong, L. Liu, J. Li and S. Li, J. Forensic Sci., 2015, 60, 759–763 CrossRef CAS PubMed.
- J. Zhang, P. Huang, Z. Wang and H. Dong, Biosci. Rep., 2017, 37, BSR20170720 CrossRef CAS PubMed.
- S. Wijeakumar, T. J. Huppert, V. A. Magnotta, A. T. Buss and J. P. Spencer, NeuroImage, 2017, 147, 204–218 CrossRef PubMed.
- D. R. Patel, Investigation of Prefrontal Cognition Responses to Transcranial Laser Stimulation in PTSD Using Functional Near Infrared Spectroscopy, MS thesis, University of Texas at Arlington, 2016.
- M. A. Yücel, J. J. Selb, T. J. Huppert, M. A. Franceschini and D. A. Boas, Curr. Opin. Biomed. Eng., 2017, 4, 78–86 CrossRef PubMed.
- T. J. Huppert, R. D. Hoge, S. G. Diamond, M. A. Franceschini and D. A. Boas, NeuroImage, 2006, 29, 368–382 CrossRef CAS PubMed.
- F. Schiffer, M. H. Teicher, C. Anderson, A. Tomoda, A. Polcari, C. P. Navalta and S. L. Andersen, Behav. Brain Funct., 2007, 3, 13 CrossRef PubMed.
- S. Jahani, A. L. Fantana, D. Harper, J. M. Ellison, D. A. Boas, B. P. Forester and M. A. Yücel, Sci. Rep., 2017, 7, 9533 CrossRef PubMed.
- M. K. Sidhu, J. Stretton, G. P. Winston, S. Bonelli, M. Centeno, C. Vollmar, M. Symms, P. J. Thompson, M. J. Koepp and J. S. Duncan, Brain, 2013, 136, 1868–1888 CrossRef PubMed.
- F. Schiffer, M. H. Teicher and A. C. Papanicolaou, J. Neuropsychiatry Clin. Neurosci., 1995, 7(2), 169–175 CrossRef CAS PubMed.
- F. Tian, A. Yennu, A. Smith-Osborne, F. Gonzalez-Lima, C. S. North and H. Liu, NeuroImage Clin., 2014, 4, 808–819 CrossRef PubMed.
- J. Leon-Carrion, J. M. Dominguez-Roldan, U. Leon-Dominguez and F. Murillo-Cabezas, Brain Inj., 2010, 24, 1193–1201 CrossRef PubMed.
- A. Chaichi, A. Prasad and M. Gartia, Biosensors, 2018, 8, 107 CrossRef PubMed.
- E. Hanlon, R. Manoharan, T. W. Koo, K. Shafer, J. Motz, M. Fitzmaurice, J. Kramer, I. Itzkan, R. Dasari and M. Feld, Phys. Med. Biol., 2000, 45, R1 CrossRef CAS PubMed.
- D. Fu, Curr. Opin. Chem. Biol., 2017, 39, 24–31 CrossRef CAS PubMed.
- J. M. Surmacki, L. Ansel-Bollepalli, F. Pischiutta, E. R. Zanier, A. Ercole and S. E. Bohndiek, Analyst, 2017, 142, 132–139 RSC.
- M. Jermyn, K. Mok, J. Mercier, J. Desroches, J. Pichette, K. Saint-Arnaud, L. Bernstein, M.-C. Guiot, K. Petrecca and F. Leblond, Sci. Transl. Med., 2015, 7, 274ra219 Search PubMed.
- P. W. Winter and H. Shroff, Curr. Opin. Chem. Biol., 2014, 20, 46–53 CrossRef CAS PubMed.
- R. Yuste, Nat. Methods, 2005, 2, 902–904 CrossRef CAS PubMed.
- B. A. Smith, B.-W. Xie, E. R. van Beek, I. Que, V. Blankevoort, S. Xiao, E. L. Cole, M. Hoehn, E. L. Kaijzel and C. W. Löwik, ACS Chem. Neurosci., 2012, 3, 530–537 CrossRef CAS PubMed.
- A. Obenaus, M. Ng, A. M. Orantes, E. Kinney-Lang, F. Rashid, M. Hamer, R. A. DeFazio, J. Tang, J. H. Zhang and W. J. Pearce, Sci. Rep., 2017, 7, 239 CrossRef PubMed.
- B. A. Flusberg, J. C. Jung, E. D. Cocker, E. P. Anderson and M. J. Schnitzer, Opt. Lett., 2005, 30, 2272–2274 CrossRef PubMed.
- D. R. Rivera, C. M. Brown, D. G. Ouzounov, I. Pavlova, D. Kobat, W. W. Webb and C. Xu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(43), 17598–17603 CrossRef CAS PubMed.
- M. Nasiriavanaki, J. Xia, H. Wan, A. Q. Bauer, J. P. Culver and L. V. Wang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 21–26 CrossRef CAS PubMed.
- K. Murari, E. Greenwald, R. Etienne-Cummings, G. Cauwenberghs and N. Thakor, Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Minneapolis, September, 2009 Search PubMed.
- L.-D. Liao, V. Tsytsarev, I. Delgado-Martínez, M.-L. Li, R. Erzurumlu, A. Vipin, J. Orellana, Y.-R. Lin, H.-Y. Lai and Y.-Y. Chen, Biomed. Eng. Online, 2013, 12, 38 CrossRef PubMed.
- R. Chéreau, J. Tønnesen and U. V. Nägerl, Methods, 2015, 88, 57–66 CrossRef PubMed.
- A. N. Boettiger, B. Bintu, J. R. Moffitt, S. Wang, B. J. Beliveau, G. Fudenberg, M. Imakaev, L. A. Mirny, C.-t. Wu and X. Zhuang, Nature, 2016, 529, 418 CrossRef CAS PubMed.
- F. Huang, G. Sirinakis, E. S. Allgeyer, L. K. Schroeder, W. C. Duim, E. B. Kromann, T. Phan, F. E. Rivera-Molina, J. R. Myers and I. Irnov, Cell, 2016, 166, 1028–1040 CrossRef CAS PubMed.
- E. Rittweger, K. Y. Han, S. E. Irvine, C. Eggeling and S. W. Hell, Nat. Photonics, 2009, 3, 144 CrossRef CAS.
- J. Tønnesen, V. K. Inavalli and U. V. Nägerl, Cell, 2018, 172, 1108–1121 CrossRef PubMed.
- U. V. Nägerl, K. I. Willig, B. Hein, S. W. Hell and T. Bonhoeffer, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18982–18987 CrossRef PubMed.
- K. Takasaki and B. L. Sabatini, Front. Neuroanat., 2014, 8, 29 Search PubMed.
- A. Hainsworth, S. Lee, P. Foot, A. Patel, W. Poon and A. Knight, Neuropathol. Appl. Neurobiol., 2018, 44, 417–426 CrossRef CAS PubMed.
- J. Tønnesen and U. V. Nägerl, Exp. Neurol., 2013, 242, 33–40 CrossRef PubMed.
- L. Schermelleh, P. M. Carlton, S. Haase, L. Shao, L. Winoto, P. Kner, B. Burke, M. C. Cardoso, D. A. Agard and M. G. Gustafsson, Science, 2008, 320, 1332–1336 CrossRef CAS PubMed.
- D. Smythe, 3D/4D ultrasound imaging system, US Pat. 6,702,745 B1, 2004.
- J. Sauvage, J. Porée, C. Rabut, G. Férin, M. Flesch, B. Rosinski, A. Nguyen-Dinh, M. Tanter, M. Pernot and T. Deffieux, IEEE International Ultrasonics Symposium (IUS), Kobe, Japan, October, 2018 Search PubMed.
- C. Demené, E. Tiran, L.-A. Sieu, A. Bergel, J. L. Gennisson, M. Pernot, T. Deffieux, I. Cohen and M. Tanter, NeuroImage, 2016, 127, 472–483 CrossRef.
- P. W. Willems, P. Taeshineetanakul, B. Schenk, P. A. Brouwer, K. G. Terbrugge and T. Krings, Neuroradiology, 2012, 54, 123–131 CrossRef PubMed.
- W. P. Taruno, M. R. Baidillah, R. I. Sulaiman, M. F. Ihsan, S. E. Fatmi, A. H. Muhtadi, F. Haryanto and M. Aljohani, IEEE 10th International Symposium on Biomedical Imaging, San Francisco, USA, April, 2013 Search PubMed.
- Z. Lavagnino, G. Sancataldo, M. d'Amora, P. Follert, D. D. P. Tonelli, A. Diaspro and F. C. Zanacchi, Sci. Rep., 2016, 6, 23923 CrossRef CAS.
- M. Pagani, G. Högberg, D. Salmaso, B. Tarnell, A. Sanchez-Crespo, J. Soares, A. Åberg-Wistedt, H. Jacobsson, T. Hällström and S. A. Larsson, Eur. Arch. Psychiatry Clin. Neurosci., 2005, 255, 359–365 CrossRef.
- R. J. Lindauer, J. Booij, J. B. Habraken, H. B. Uylings, M. Olff, I. V. Carlier, G. J. den Heeten, B. L. van Eck-Smit and B. P. Gersons, Biol. Psychiatry, 2004, 56, 853–861 CrossRef.
- O. Bonne, A. Gilboa, Y. Louzoun, D. Brandes, I. Yona, H. Lester, G. Barkai, N. Freedman, R. Chisin and A. Y. Shalev, Biol. Psychiatry, 2003, 54, 1077–1086 CrossRef.
- S. Mirzaei, P. Knoll, A. Keck, B. Preitler, E. Gutierrez, H. Umek, H. Köhn and M. Pecherstorfer, Neuropsychobiology, 2001, 43, 260–264 CrossRef CAS.
- J.-K. Zubieta, J. A. Chinitz, U. Lombardi, L. M. Fig, O. G. Cameron and I. Liberzon, J. Psychiatr. Res., 1999, 33, 259–264 CrossRef CAS.
- I. Liberzon, S. F. Taylor, R. Amdur, T. D. Jung, K. R. Chamberlain, S. Minoshima, R. A. Koeppe and L. M. Fig, Biol. Psychiatry, 1999, 45, 817–826 CrossRef CAS.
- J. Lucey, D. C. Costa, G. Adshead, M. P. Deahl, G. Busatto, S. Gacinovic, M. Travis, L. S. Pilowsky, P. J. Ell and I. M. Marks, Br. J. Psychiatry, 1997, 171, 346–350 CrossRef CAS PubMed.
- M. Fujita, S. M. Southwick, C. C. Denucci, S. S. Zoghbi, M. S. Dillon, R. M. Baldwin, A. Bozkurt, A. Kugaya, N. P. L. Verhoeff and J. P. Seibyl, Biol. Psychiatry, 2004, 56, 95–100 CrossRef CAS PubMed.
- J. D. Bremner, R. B. Innis, S. M. Southwick, L. Staib, S. Zoghbi and D. S. Charney, Am. J. Psychiatry, 2000, 157, 1120–1126 CrossRef CAS.
- Y. A. Chung, S. H. Kim, S. K. Chung, J.-H. Chae, D. W. Yang, H. S. Sohn and J. Jeong, Clin. Neurophysiol., 2006, 117, 637–642 CrossRef PubMed.
- N. Sachinvala, H. Von Scotti, M. McGuire, L. Fairbanks, K. Bakst, M. McGuire and N. Brown, J. Nerv. Ment. Dis., 2000, 188, 818–823 CrossRef CAS PubMed.
- T. C. Buckley, E. B. Blanchard and W. T. Neill, Clin. Psychol. Rev., 2000, 20, 1041–1065 CrossRef CAS PubMed.
- S. Kim, I. Lyoo, Y. Lee, J. Kim, M. Sim, S. Bae, H. Kim, J. Y. Lee and D. U. Jeong, Acta Psychiatr. Scand., 2007, 116, 145–153 CrossRef CAS PubMed.
- J. C. Britton, K. L. Phan, S. F. Taylor, L. M. Fig and I. Liberzon, Biol. Psychiatry, 2005, 57, 832–840 CrossRef PubMed.
- L. M. Shin, S. P. Orr, M. A. Carson, S. L. Rauch, M. L. Macklin, N. B. Lasko, P. M. Peters, L. J. Metzger, D. D. Dougherty and P. A. Cannistraro, Arch. Gen. Psychiatry, 2004, 61, 168–176 CrossRef PubMed.
- J. D. Bremner, M. Vythilingam, E. Vermetten, S. M. Southwick, T. McGlashan, A. Nazeer, S. Khan, L. V. Vaccarino, R. Soufer and P. K. Garg, Am. J. Psychiatry, 2003, 160, 924–932 CrossRef PubMed.
- J. D. Bremner, M. Vythilingam, E. Vermetten, S. M. Southwick, T. McGlashan, L. H. Staib, R. Soufer and D. S. Charney, Biol. Psychiatry, 2003, 53, 879–889 CrossRef PubMed.
- W. E. Semple, P. F. Goyer, R. McCORMICK, B. Donovan, R. F. Muzic Jr, L. Rugle, K. McCutcheon, C. Lewis, D. Liebling and S. Kowaliw, Psychiatry, 2000, 63, 65–74 CrossRef CAS PubMed.
- J. D. Bremner, M. Narayan, L. H. Staib, S. M. Southwick, T. McGlashan and D. S. Charney, Am. J. Psychiatry, 1999, 156, 1787–1795 CAS.
- J. D. Bremner, L. H. Staib, D. Kaloupek, S. M. Southwick, R. Soufer and D. S. Charney, Biol. Psychiatry, 1999, 45, 806–816 CrossRef CAS PubMed.
- L. M. Shin, R. J. McNally, S. M. Kosslyn, W. L. Thompson, S. L. Rauch, N. M. Alpert, L. J. Metzger, N. B. Lasko, S. P. Orr and R. K. Pitman, Am. J. Psychiatry, 1999, 156, 575–584 CAS.
- L. M. Shin, S. M. Kosslyn, R. J. McNally, N. M. Alpert, W. L. Thompson, S. L. Rauch, M. L. Macklin and R. K. Pitman, Arch. Gen. Psychiatry, 1997, 54, 233–241 CrossRef CAS PubMed.
- W. E. Semple, P. F. Goyer, R. McCormick, B. Compton-Toth, E. Morris, B. Donovan, G. Muswick, D. Nelson, M. L. Garnett and J. Sharkoff, Psychiatry Res., Neuroimaging, 1996, 67, 17–28 CrossRef CAS.
- W. E. Semple, P. Goyer, R. McCormick, E. Morris, B. Compton, G. Muswick, D. Nelson, B. Donovan, G. Leisure and M. Berridge, Biol. Psychiatry, 1993, 34, 115–118 CrossRef CAS.
- L. M. Shin, C. I. Wright, P. A. Cannistraro, M. M. Wedig, K. McMullin, B. Martis, M. L. Macklin, N. B. Lasko, S. R. Cavanagh and T. S. Krangel, Arch. Gen. Psychiatry, 2005, 62, 273–281 CrossRef PubMed.
- X. Protopopescu, H. Pan, O. Tuescher, M. Cloitre, M. Goldstein, W. Engelien, J. Epstein, Y. Yang, J. Gorman and J. LeDoux, Biol. Psychiatry, 2005, 57, 464–473 CrossRef PubMed.
- P. Yang, M.-T. Wu, C.-C. Hsu and J.-H. Ker, Neurosci. Lett., 2004, 370, 13–18 CrossRef CAS PubMed.
- M. Driessen, T. Beblo, M. Mertens, M. Piefke, N. Rullkoetter, A. Silva-Saavedra, L. Reddemann, H. Rau, H. J. Markowitsch and H. Wulff, Biol. Psychiatry, 2004, 55, 603–611 CrossRef PubMed.
- R. A. Lanius, P. C. Williamson, M. Densmore, K. Boksman, R. Neufeld, J. S. Gati and R. S. Menon, Am. J. Psychiatry, 2004, 161, 36–44 CrossRef PubMed.
- T. Hendler, P. Rotshtein, Y. Yeshurun, T. Weizmann, I. Kahn, D. Ben-Bashat, R. Malach and A. Bleich, NeuroImage, 2003, 19, 587–600 CrossRef PubMed.
- R. A. Lanius, P. C. Williamson, J. Hopper, M. Densmore, K. Boksman, M. A. Gupta, R. W. Neufeld, J. S. Gati and R. S. Menon, Biol. Psychiatry, 2003, 53, 204–210 CrossRef.
- R. A. Lanius, P. C. Williamson, K. Boksman, M. Densmore, M. Gupta, R. W. Neufeld, J. S. Gati and R. S. Menon, Biol. Psychiatry, 2002, 52, 305–311 CrossRef.
- R. A. Lanius, P. C. Williamson, M. Densmore, K. Boksman, M. A. Gupta, R. Neufeld, J. S. Gati and R. S. Menon, Am. J. Psychiatry, 2001, 158, 1920–1922 CrossRef CAS PubMed.
- S. L. Rauch, P. J. Whalen, L. M. Shin, S. C. McInerney, M. L. Macklin, N. B. Lasko, S. P. Orr and R. K. Pitman, Biol. Psychiatry, 2000, 47, 769–776 CrossRef CAS PubMed.
Footnote |
| † Equal contribution authors. |
| This journal is © The Royal Society of Chemistry 2019 |