
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltra-small palladium nano-particles synthesized using bulky S/Se and N donor ligands as a stabilizer: application as catalysts for Suzuki–Miyaura coupling†
Preeti Oswala,
Aayushi Aroraa,
Jolly Kaushala,
Gyandshwar Kumar Raob,
Sushil Kumar a,
Ajai K. Singhc and
Arun Kumar*a
a,
Ajai K. Singhc and
Arun Kumar*a
aDepartment of Chemistry, School of Physical Sciences, Doon University, Dehradun, 248012 India. E-mail: arunkaushik@gmail.com; akumar.ch@doonuniversity.ac.in
bDepartment of Chemistry, Amity School of Applied Sciences, Amity University Haryana, Gurgaon, Haryana 122413, India
cDepartment of Chemistry, India Institute of Technology Delhi, New Delhi, 110016, India
First published on 18th July 2019
Abstract
Two chalcogenated ligands L1 and L2 containing anthracene core and amine functionality have been synthesized. Both the ligands have been characterized using 1H and 13C{1H} NMR techniques. The structure of L1 has also been corroborated by single crystal X-ray diffraction. Application of L1 and L2 as stabilizers for palladium nano-particles (NPs) has been explored and six different types of NPs 1–6 have been prepared by varying the quantity of stabilizer. The nano-particles have been characterized by PXRD, EDX, and HRTEM techniques. The size of NPs has been found to be in the range of ∼1–2 nm, 2–3 nm, 4–6 nm, 1–2 nm, 1–2 nm and 3–5 nm for 1–6 respectively. The catalytic activities of 1–6 have been explored for Suzuki–Miyaura coupling of phenyl boronic acid with various aryl halides. These NPs showed good catalytic activity for various aryl chlorides/bromides at low catalyst loading (5 mg). Among 1–6, the highest activity has been observed for NPs 1, probably due to their relatively small size and high uniformity in the dispersion. The recyclability of the NPs upto 5 catalytic cycles is a distinct advantage.
Introduction
Palladium-catalyzed C–C coupling reactions are impressive synthetic methods in organic transformations.1 The tolerance of palladium compounds towards a wide range of functional groups and the easy interchange between Pd(0)/Pd(II) or Pd(II)/Pd(IV) oxidation states are mainly responsible for their versatility.2 Of the various C–C bond-forming reactions, Suzuki–Miyaura coupling is one of the most important methods for the syntheses of diversified and unsymmetrical biaryls (which have numerous applications in synthesis of drugs, natural products, agrochemicals, advanced material etc.)3 compared to other coupling reactions, mainly due to the low cost of reagents and ease in handling and removal of reagents and products. The highly efficient Suzuki–Miyaura catalysts include complexes of Pd(II) with phosphines,4,5 N-heterocyclic carbenes,6,7 and organochalcogens8,9 with an advantage of the ease with which they can be modified. In order to improve and expand the scope of Suzuki–Miyaura coupling, intensive research efforts are being carried out. In this regard, the development of new catalyst systems with high efficiency and recyclability is an important area that has received particular attention.10In the last few decades, nano-sized palladium particles stabilized with diverse agents, have drawn continuous attraction.11,12 Nanoparticles play remarkable roles in a variety of fields due to their large surface to volume ratio, reactive surface molecules, and the capability of tuning the properties of material etc.13–16 High surface to volume ratio of nanoparticles has constrained the researchers to stabilize them by utilizing appropriate stabilizers. The stabilizing ligands play an important role in dictating nanoparticles' size, shape, dispersion, and inter-particle spacing. These ligands also affect the catalytic efficiency, recyclability and solubility of synthesized NPs through ligation. For efficient organic synthesis, generally surfactants, organic ligands, dendrimers, and polymers have been used to stabilize metal nanoparticles.17–19 The stabilizer molecules generally have functional groups like amine, alcohol, carbonyl or hydroxyl, thiol and phosphine,20 which limits the catalytic activity because of strong chemisorption.21 Thus, the stabilizing ligand must be strong enough to stabilize nanoparticles. It must readily allow access of reactant to its surface so that they can be used for multiple reaction cycles. In the absence of a stabilizing agent, there is a possibility of aggregation or precipitation of nanoparticles which results in the loss of their catalytic activity. Therefore, it is worthy to explore new ligands as stabilizers suitable for ensuring minimum surface deactivation.
Sulphur and selenium donor groups may serve as good candidates for designing new ligand systems for stabilization of nanoparticles. The strong electron donating ability of sulphur and selenium and the advantage of their ligands for being air stable and moisture insensitive has made them attractive for designing catalyst frameworks. Such type of ligands have been reported in the recent past as an important building block for a variety of catalysts.22–29 These have shown good to excellent catalytic activity for the coupling of aryl halides.22–29 Presence of neutral chalcogen donor in stabilizers affects the catalytic efficiency, composition, size, recyclability, and dispersion of the nanoparticles to a great extent.30,31 The use of organochalcogen ligands in the development of molecular catalysts has been studied in a variety of cases.22–29 However, the use of such ligands in the stabilization of nanoparticles is limited.30,31 To the best of our knowledge, no such compound containing a combination of hard–hard or hard–soft donors have been reported for this purpose. Thus, in this article, we are reporting the synthesis (Scheme 1) and characterization of palladium nano-particles stabilized with protecting ligands (L1 an L2) containing a combination of sulphur/selenium and ‘N’ donors. The catalytic potential of these NPs has been explored for Suzuki–Miyaura coupling reaction and also found to be promising. Additionally, they have also been found to be recyclable up to five cycles.
 | ||
| Scheme 1 Syntheses of ligands and nano-particles. | ||
Results and discussion
Syntheses and characterization
The syntheses of L1′ and L2′ have been carried out using a method similar to reported procedures.24 Ligands L1 and L2 have been synthesized using a methodology shown in Scheme 1. Ligands L1 and L2 have been characterized using 1H and 13C{1H} NMR techniques. The spectra were found to be consistent with the structures given in Scheme 1. The CH (imine) signal in the 1H NMR spectra of precursor Schiff bases L1′ and L2′ appeared at 9.4 and 9.31 ppm respectively, supporting the formation of![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond. This signal disappeared on their subsequent reduction to L1 and L2 and a new signal of –NCH2– emerged at 4.66 and 4.56 ppm respectively in case of L1 and L2. The needle-shaped light yellow single crystals of L1 were obtained by slow evaporation of its solution in ethanol. The molecular structure of L1 is given in Fig. 1 with 50% probability ellipsoids and with selected bond lengths and bond angles. Both L1 and L2 have been used to stabilize Pd(0) NPs 1–6 as summarized in Scheme 1. Some organic compounds have been used to develop NPs with good catalytic efficiency, their synthesis are very cumbersome along with the difficulty in separation, and consequently low yields.32–35 Only a few reports are available on the use of an organic ligand with neutral chalcogen donor in the stabilization and development of catalytically active Pd(0) NPs. The distinct advantage of the ligands in the present study is their simple and easy preparation in excellent yields.
N bond. This signal disappeared on their subsequent reduction to L1 and L2 and a new signal of –NCH2– emerged at 4.66 and 4.56 ppm respectively in case of L1 and L2. The needle-shaped light yellow single crystals of L1 were obtained by slow evaporation of its solution in ethanol. The molecular structure of L1 is given in Fig. 1 with 50% probability ellipsoids and with selected bond lengths and bond angles. Both L1 and L2 have been used to stabilize Pd(0) NPs 1–6 as summarized in Scheme 1. Some organic compounds have been used to develop NPs with good catalytic efficiency, their synthesis are very cumbersome along with the difficulty in separation, and consequently low yields.32–35 Only a few reports are available on the use of an organic ligand with neutral chalcogen donor in the stabilization and development of catalytically active Pd(0) NPs. The distinct advantage of the ligands in the present study is their simple and easy preparation in excellent yields.
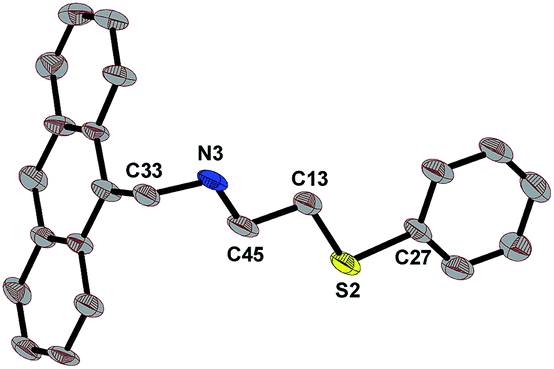 | ||
| Fig. 1 ORTEP of L1 (ellipsoid of 50% probability). Selected bond lengths (Å): S(2)–C(27) 1.765(6), S(2)–C(13) 1.826(6), N(3)–C(45) 1.436(8), N(3)–C(33) 1.492(7). Selected bond angles (°): C(27)–S(2)–C(13) 104.5(3), C(45)–N(3)–C(33) 113.9(4). | ||
The Pd(0) NPs 1–6 have been prepared (Scheme 1) using Na2PdCl4 as a palladium precursor and L1 or L2 as a stabilizer in varying ligand to metal ratio. The L1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd molar ratio is 1:1 for 1, 1:4 for 2, and 4:1 for 3. The L2:Pd molar ratio is 1:1 for 4, 1:4 for 5, and 4:1 for 6. The maximum quantity of ligand has been used for the purpose of stabilization in the case of 3 and 6. All the NPs have been subjected to powder XRD (Fig. 2), HRTEM (Fig. 3), and SEM-EDX (ESI Fig. S9–S13†) techniques for characterization. PXRD data of 1–6 showed the presence of a peak at 40.0 (2θ) corresponding to (111) plane of Pd(0) (d ∼ 2.26) (JCPDS 88-2335) with face-centered cubic structure. The peaks at 27.3, 31.7, 45.5, 56.4 and 66.2 which correspond to (111), (200), (220), (222), and (400) planes of the palladium oxide were observed in the PXRD pattern of NPs 2–4 (Fig. 2).36 Palladium oxide was also present in NPs 5, but the palladium(0) content was found to be higher. Small amount of palladium oxide was observed in the case of NPs 6 (Fig. 2). In the case of NPs 1, pure phase palladium was observed as revealed by PXRD (Fig. 2). The SEM-EDX studies have revealed that Pd:S ratio (atom%) in NPs 1–3 are 82:18, 76:24 and 79:21 respectively. The Pd:Se ratio in the case of NPs 4–6 has been found to be 69:31, 73:27 and 71:29 respectively.
:Pd molar ratio is 1:1 for 1, 1:4 for 2, and 4:1 for 3. The L2:Pd molar ratio is 1:1 for 4, 1:4 for 5, and 4:1 for 6. The maximum quantity of ligand has been used for the purpose of stabilization in the case of 3 and 6. All the NPs have been subjected to powder XRD (Fig. 2), HRTEM (Fig. 3), and SEM-EDX (ESI Fig. S9–S13†) techniques for characterization. PXRD data of 1–6 showed the presence of a peak at 40.0 (2θ) corresponding to (111) plane of Pd(0) (d ∼ 2.26) (JCPDS 88-2335) with face-centered cubic structure. The peaks at 27.3, 31.7, 45.5, 56.4 and 66.2 which correspond to (111), (200), (220), (222), and (400) planes of the palladium oxide were observed in the PXRD pattern of NPs 2–4 (Fig. 2).36 Palladium oxide was also present in NPs 5, but the palladium(0) content was found to be higher. Small amount of palladium oxide was observed in the case of NPs 6 (Fig. 2). In the case of NPs 1, pure phase palladium was observed as revealed by PXRD (Fig. 2). The SEM-EDX studies have revealed that Pd:S ratio (atom%) in NPs 1–3 are 82:18, 76:24 and 79:21 respectively. The Pd:Se ratio in the case of NPs 4–6 has been found to be 69:31, 73:27 and 71:29 respectively.
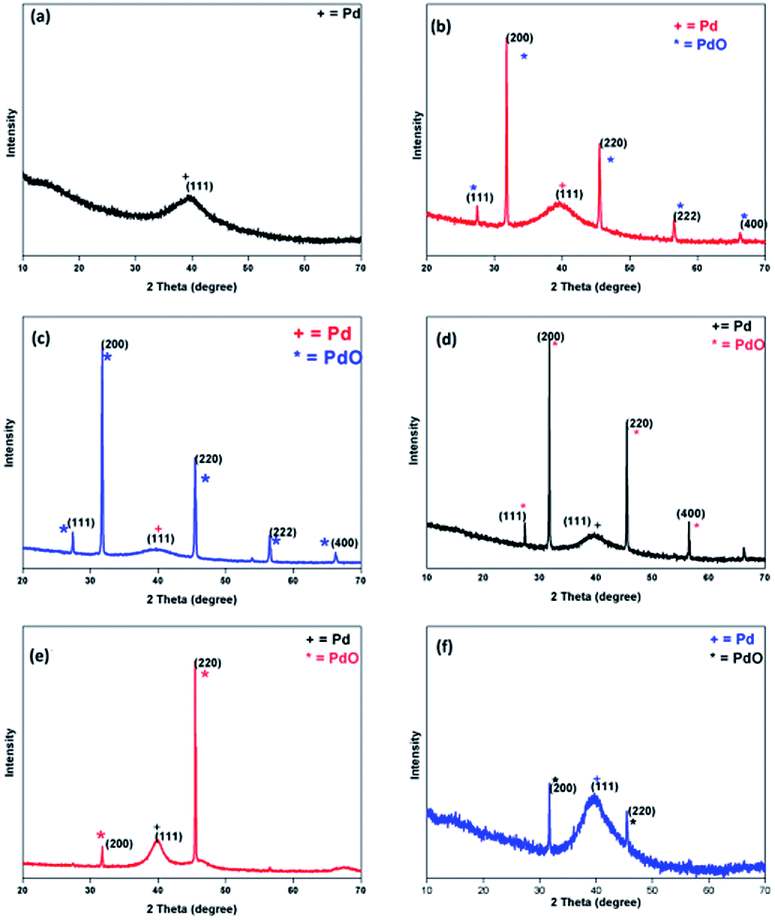 | ||
| Fig. 2 PXRD pattern of (a) NPs 1, (b) NPs 2, (c) NPs 3, (d) NPs 4, (e) NPs 5, (f) NPs 6. | ||
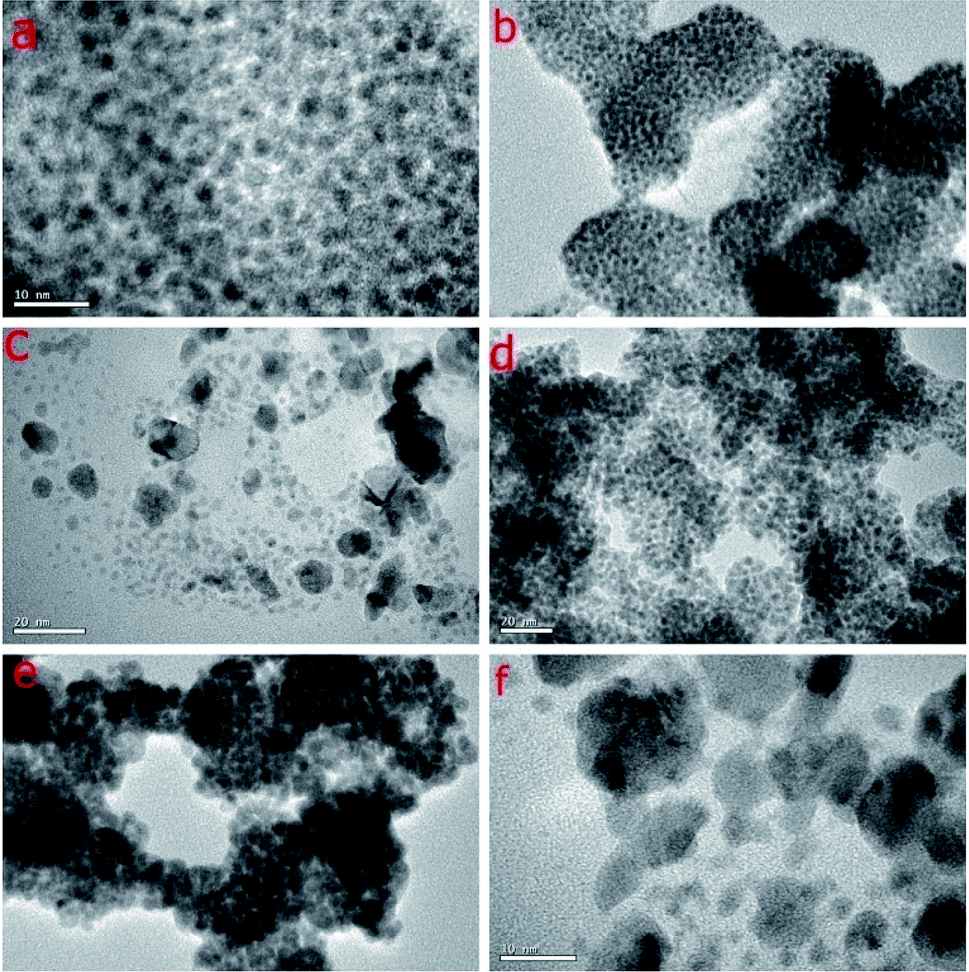 | ||
| Fig. 3 HRTEM images of Pd NPs. (a) 1 (scale bar 10 nm), (b) 2 (scale bar 50 nm), (c) 3 (scale bar 20 nm), (d) 4 (scale bar 20 nm), (e) 5 (scale bar 50 nm), (f) 6 (scale bar 10 nm). | ||
The HRTEM images of NPs 1–6 shown in Fig. 3, confirm the size and nature of dispersion of the nano-particles. The size of NPs of Pd have been found to be ultra-small which has been calculated to be ∼1–2 nm, 2–3 nm, 4–6 nm, 1–2 nm, 1–2 nm, 3–5 nm respectively for 1–6. The reason for small size and good dispersion of NPs 1–6 can be attributed to the fast reduction process by sodium borohydride used for the reduction of Pd(II) salt. The stabilizing ligands for Pd NPs containing bulky and soft donor sites have been reported to prevent their aggregation during growth.21 The slightly larger size in case of NPs 3 and 6 might be due to high ligand:Na2PdCl4 ratio. In these cases, possibly, the reduction is slowed down due to increase in the number of ligand molecules in the reaction mixture leading to an increase in the size as compared to NPs 1, 2, 4, 5.
Application in Suzuki–Miyaura coupling reaction
The NPs 1–6 have been investigated for the Suzuki–Miyaura coupling reaction of several aryl halides including aryl iodides, bromides, and chlorides. To optimize the reaction conditions, the coupling reaction of 4-bromobenzaldehyde with phenylboronic acid in the presence of NPs 1 was studied (Table 1) using various bases and solvents. K2CO3 has been found to be the most appropriate base as the yield of cross-coupled product is higher than that obtained with the use of other alternative bases (Table 1, entry no 4). Of the various solvents investigated, a mixture of DMF and water was found to be the best solvent system for the reaction (Table 1, entry no 4) (Scheme 2).| S. no. | Solvent | Base | Yield (%) |
|---|---|---|---|
| a Reaction conditions: 4-bromobenzaldehyde (1.0 mmol), phenylboronic acid (1.1 mmol), base (2.0 mmol), solvent (5.0 mL), temperature; 100 °C, time; 6 h. | |||
| 1 | DMF:water (3:2) |
NaOH | 42 |
| 2 | DMF:water (3:2) |
Cs2CO3 | 85 |
| 3 | DMF:water (3:2) |
CH3COONa | 52 |
| 4 | DMF:water (3:2) |
K2CO3 | 95 |
| 5 | DMF | K2CO3 | 79 |
| 6 | Toluene | K2CO3 | 38 |
| 7 | THF | K2CO3 | 47 |
| 8 | EtOH:water (3:1) |
K2CO3 | 68 |
 | ||
| Scheme 2 Suzuki–Miyaura reaction. | ||
The results of investigations on catalytic activities of NPs 1–6 under optimized parameters have been summarized in Tables 2 and 3. In the coupling reaction of 4-bromobenzaldehyde under optimum conditions for 6 hours at 100 °C in presence of NPs 1, 2, 3 and 4, the cross-coupled product biphenyl-4-carboxaldehyde was obtained in more than 99% yield (Table 2: entry 7 and Table 3: entry 6). 1-Bromo-4-nitrobenzene was successfully converted in to 4-nitrobiphenyl in presence of NPs 1, 2, 3, 5 as a catalyst in 99%, 99%, 71% and 99% respectively (Table 2: entry 3, Table 3: entry 7). More than 99% yield has been observed for 4-bromotoluene, 4-bromoaniline and 4-bromobenzoic acid with NPs 4 in 6 hours (Table 2: entry 1, 2 and 3). It has also been assessed that among 1, 2, 3 and 5, NPs 1 (Pd:L = 1:1) showed maximum efficiency in the coupling of 4-bromophenol (Table 2: entry 5), a very less reactive substrate.
| Ar–X + PhB(OH)2 → Ar–Ph (X = Cl or Br) | ||||
|---|---|---|---|---|
| Entry no. | Aryl halide | Yield% | ||
| NPs 1 | NPs 2 | NPs 3 | ||
| a Reaction conditions: catalyst, 5 mg; time 6 h; aryl halide 1 mmol; K2CO3, 2 mmol; phenylboronic acid, 1.1 mmol; time 6 h; aqueous DMF, 5.0 mL.b Time 12 h. NT, reaction not tested. | ||||
| 1b | 4-Chlorobenzophenone | 90% | NT | NT |
| 2b | 4-Chlorobenzaldehyde | 75% | 68% | NT |
| 3 | 1-Bromo-4-nitrobenzene | 99% | 99% | 71% |
| 4 | 4-Bromobenzonitrile | 99% | 99% | NT |
| 5 | 4-Bromophenol | 91% | 50% | 29% |
| 6 | 4-Bromobenzophenone | 88% | NT | 86% |
| 7 | 4-Bromobenzaldehyde | 99% | 99% | 75% |
| 8b | 4-Chlorotoluene | 71% | NT | NT |
| Ar–X + PhB(OH) → Ar–Ph (X = Cl or Br) | ||||
|---|---|---|---|---|
| Entry no. | Aryl halide | Yield% | ||
| NPs 4 | NPs 5 | NPs 6 | ||
| a Reaction conditions: catalyst, 5 mg; time 6 h; aryl halide 1 mmol; K2CO3, 2 mmol; phenylboronic acid, 1.1 mmol; time 6 h; aqueous DMF, 5.0 mL.b Time 12 h. NT, reaction not tested. | ||||
| 1 | 4-Bromotoluene | 99% | NT | NT |
| 2 | 4-Bromoaniline | 99% | NT | 61% |
| 3 | 1-Bromobenzoic acid | 99% | NT | NT |
| 4b | 4-Chlorotoluene | 62% | NT | 68% |
| 5b | 4-Chlorobenzaldehyde | 84% | NT | 71% |
| 6 | 4-Bromobenzaldehyde | 99% | NT | NT |
| 7 | 1-Bromo-4-nitrobenzene | NT | 99% | NT |
| 8 | 4-Bromobenzophenone | NT | 99% | NT |
| 9 | 4-Bromophenol | NT | 66% | NT |
| 10 | 4-Bromobenzonitrile | NT | 99% | NT |
| 11b | 4-Chlorobenzophenone | NT | NT | 72% |
| 12b | 4-Chlorobenzonitrile | NT | NT | 76% |
| 13 | 4-Iodoacetophenone | NT | 99% | NT |
| 14 | 4-Iodoanisole | NT | 99% | NT |
Success has also been achieved in the coupling of chloroarenes (least reactive halides), and that too at the low catalyst loading in 12 hours. For instance, 4-chlorobenzophenone is converted to 4-phenyl benzophenone in the presence of NP's 1 with 90% yield in 6 hours (Table 2: entry 1). In addition to that, 4-chlorotoluene is also successfully coupled to yield 4-methylbiphenyl in 71%, 62%, 68% with NPs 1, 4 and 6 respectively (Table 2: entry 8, Table 3: entry 4). Interestingly, NPs 1 have been found to be more efficient than NPs 2, 4 and 6 in converting 4-chlorobenzaldehyde to biphenyl-4-carboxaldehyde in 75% yield (Table 2: entry 2). NPs 6 were also explored for 4-chlorobenzonitrile and 76% conversion was achieved under standard reaction conditions (Table 3, entry 12).
The results of catalysis corroborate on the observation that ligand:metal ratio used in the synthesis of NPs affect their catalytic activity. The reason for good efficiency of each of NPs 1, 2, 4 and 5 may be attributed to the large surface to volume ratio and greater shielding of NPs by weakly coordinating ligand molecules which results in the development of well-dispersed NPs with high catalytic activity. The size of the nanoparticles 1, 2, 4 and 5 is in the range of 1–5 nm. NPs 1 were found to be the most catalytically active owing to the fact that they have uniformity in their size and dispersion and the complete conversion of Pd(II) salt to Pd(0) NPs without the formation of the PdO NPs as evident from XRD data (Fig. 2). The NPs 3 and 6 showed lower catalytic efficiency, which may be due to their large size as evident from TEM images (Fig. 3) and high ligand ratio in comparison to palladium. It might be possible that the lower number of active catalytic sites is available for coupling reaction since most of them are blocked by an excess of ligand molecules.
The recyclability of the NPs is one of the distinct advantages which was explored for 1 and 3. For this purpose, Suzuki coupling of 4-bromobenzaldehyde for 1 and 1-bromo-4-nitro benzene for 3 was carried out under optimized reaction conditions. After carrying out the reaction, the NPs were centrifuged, washed with methanol and dried in vacuo. These were reused for further catalytic runs and it was found that NPs do not lose their catalytic efficiency even after 5 runs. The results are summarized in Table 4.
| Entry no. | Aryl bromide | Reaction cycle | |||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |||
| a Reaction conditions: catalyst, 5 mg; time 6 h; aryl halide 1 mmol; K2CO3, 2 mmol; phenylboronic acid, 1.1 mmol; time 6 h; aqueous DMF, 5.0 mL.b NPs 1.c NPs 3. | |||||||
| 1b | 4-Bromobenzaldehyde | t (h) | 6 | 6 | 6 | 6 | 6 |
| Yield (%) | 99 | 99 | 99 | 95 | 95 | ||
| 2c | 4-Bromonitrobenzene | t (h) | 6 | 6 | 6 | 6 | 6 |
| Yield (%) | 99 | 99 | 95 | 92 | 85 | ||
Some Pd(0) NPs stabilized with mono dentate organochalcogen ligands30,31 are reported earlier. These NPs showed excellent activity towards the coupling of aryl bromides and iodides. However, the Suzuki–Miyaura coupling of aryl chlorides is less explored. In the present study, Pd NPs have been found to show the efficient catalytic activity for the coupling of a wide range of aryl chlorides substrates in addition to aryl bromides and iodides.
To further understand the catalysis by these Pd(0) NPs, a mercury poisoning, PPh3 poisoning and hot filtration tests were conducted. The mercury poisoning test was performed for a coupling reaction of 1-bromo-4-nitrobenzene with phenylboronic acid using NPs 1 as catalyst under optimum conditions. The biaryl product was obtained in 99% yield even in the presence of an excess of mercury. It suggests that the NPs are protected by ligand and Hg does not have access to interact with them and catalysis is proceeding via a homogeneous pathway. For the PPh3 test, coupling reaction of 1-bromo-4-nitrobenzene was carried out with phenylboronic acid using NPs 1 as a catalyst. There was only a slight and insignificant decrease in the yield of the cross-coupled product corroborating the aforementioned homogeneous nature of the catalysis. These results are also concluding the assumption that the steric effect of stabilizing ligands does play a significant role and prohibit Hg or PPh3 to reach the surface of NPs of palladium. Hot filtration test for the coupling of 4-bromobenzaldehyde with phenylboronic acid using NPs 1 as catalyst under optimum conditions was carried out in order to understand the leaching of palladium. The reaction mixture was hot filtered after 10 minutes of reaction and half of the reaction mixture was quenched and subjected to NMR. The other half was stirred for another 3 h under optimum reaction conditions and subjected to NMR. It was found that the 84% of 4-bromobenzaldehyde was converted into the product in 10 minutes and after 3 h a maximum conversion of 92% was obtained. It suggests that the leaching of Pd is very low as they are efficiently stabilized by ligands and hence the reaction in the filtrate continued at an insignificant rate.
Experimental section
Physical measurements
The 1H and 13C{1H} spectra were recorded on a JNM ECX-500 NMR spectrometer at 500 and 125 MHz respectively. The chemical shifts are reported in ppm relative to the internal standard tetramethylsilane. Single crystal X-ray diffraction studies were performed on Supernova X-ray diffractometer system at 150 K using Mo Kα radiation (0.710 Å). CrysalisPro Software (online version) was used for data collection. The structure was solved by direct methods using olex2, SHELXS-97 and refined by full matrix least-squares with SHELXL-97, refining on F2. The image was created using the program Diamond.TEM studies were carried out with an FEI Tecnai G2-S-twin electron microscope operated at 200 kV. The specimen for TEM was prepared by dispersing the powder in methanol by ultrasonic treatment, dropping slurry onto a porous carbon film supported on a copper grid, and then drying in air. Elemental composition of NPs was analyzed by using an EDX system (model: JSM 6100). The samples were scanned in different regions. Powder X-ray diffraction (PXRD) studies were carried out on Panalytical XPert diffractometer with Cu filtered radiation using a scan speed of 2 degrees per min and scan step of 0.02 degree. The products of catalytic reactions were authenticated by matching their NMR spectral data with those reported in the literature. The commercial nitrogen gas was used as received. Nitrogen atmosphere if required was created using Schlenk techniques. All reactions were carried out in glassware dried in an oven, under ambient conditions.
Chemicals
2-(Phenylthio)ethyl amine and 2-(phenylseleno)ethyl amine was synthesized by reported methods.37–40 Diphenyldisulphide, diphenyl diselenide, 2-chloroethylamine hydrochloride, anthracene carboxaldehyde, sodium tetrachloropalladate, aryl bromides, iodides and chlorides, phenyl boronic acid, K2CO3 and NaBH4 were procured from Sigma-Aldrich (USA) and used as received. The solvents were dried and distilled before use by standard procedures.Synthesis of L1 and L2
The ligand L1′ (0.345 g, 1.0 mmol)/L2′ (0.387 g, 1.0 mmol) was stirred in 20 mL dry ethanol for 30 minutes at 70 °C. NaBH4 (0.079 g, 2.1 mmol) was added and the mixture then refluxed for 10 h. The solvent was removed under reduced pressure on a rotary evaporator. The residue was extracted with chloroform and distilled water (100 mL) followed by drying over anhydrous sodium sulphate. The solvent was evaporated off under reduced pressure on a rotary evaporator to obtain the product L1 and L2 as light yellow oil.Syntheses of NPs 1–6
The Na2[PdCl4] (0.147 g, 0.5 mmol) was dissolved in 50 mL of methanol. The solution of ligand L1 [0.173 g, 0.5 mmol (L:Pd = 1:1)] and L2 [0.194 g, 0.5 mmol (L:Pd = 1:1)] for NP's 1 and 4; [0.689 g, 2 mmol (L:Pd = 1:4)] of L1 and [0.778 g, 2 mmol of L2 (L:Pd = 1:4)] for NPs 2 and 5; [0.0865 g, 0.125 mmol (L:Pd = 4:1)] of L1 and [0.097 g, 0.125 mmol (L:Pd = 4:1)] of L2 for NPs 3 and 6 made in 50 mL dichloromethane was added to above solution with vigorous stirring. The mixture was further stirred for 1 h. A solution of NaBH4 (2 equivalents) made in methanol (5 mL) was added as quickly added to the reaction mixture and was further stirred for 2 h. The solvent was reduced to 15 mL on a rotary evaporator. The residue was centrifuged and washed three times with methanol. The resulting nanoparticles 1–6 were dried in vacuo.
General procedure for catalysis of Suzuki–Miyaura C–C coupling
To a 100 mL round bottom flask equipped with a reflux condenser, aryl halide (1.0 mmol), phenylboronic acid (1.1 mmol, 0.133 g), K2CO3 (2.0 mmol, 0.276 g), aqueous dimethylformamide (5.0 mL) and 5 mg of catalyst were added. The reaction mixture was refluxed at 100 °C and the reaction progress was monitored with TLC. After the maximum conversion of aryl halide into the product has been accomplished, the reaction mixture was allowed to cool to room temperature. It was mixed with water and extracted with diethyl ether. The organic phase was dried over anhydrous Na2SO4. The solvent was removed on a rotary evaporator under reduced pressure and product was subjected to 1H NMR studies. In the case of impure product, further purification was carried out by column chromatography on silica gel using an ethyl acetate and hexane mixture as the eluent.Catalytic recyclability
An oven-dried round bottom flask was charged with 4-bromobenzaldehyde (1 mmol), phenylboronic acid (1.1 mmol, 0.133 g), K2CO3 (2.0 mmol, 0.276 g) and aqueous DMF (5.0 mL). Nano-particles (10 mg of 1), were added to the reaction mixture and the temperature was maintained at 100 °C. The progress of the reaction was monitored with TLC. After completion of the reaction, water and ethyl acetate were added and the mixture was centrifuged. The black residue was collected and washed with methanol in order to remove the organic content and base. The resulting nanoparticles were dried in vacuo. The organic layer from water–ethyl acetate was separated and the solvent was evaporated off. The residue was subjected to 1H NMR for conversion estimation. The NPs separated were reused for the next cycle and this procedure was repeated for five times.Hg poisoning test
To carry out the Hg poisoning test, catalyst (NPs 1; 5 mg) was stirred with an excess of Hg (Hg:Pd; 500:1) in an oven dried flask for 15 minutes before the addition of the coupling reactants. Thereafter, 4-bromobenzaldehyde (1.0 mmol, 0.185 g), phenylboronic acid (1.1 mmol, 0.133 g) and K2CO3 (2 mmol) were added to the flask and reaction was carried out under optimum conditions. Reaction progress was monitored with TLC. The reaction was further continued for 10 hours at 100 °C. The used mercury was collected and stored safely.
PPh3 poisoning test
PPh3 (5 equivalents) was added under optimum conditions to the catalyst (1 equivalent) and stirred to 15 minutes before the addition of the coupling substrates 4-bromobenzaldehyde (1.0 mmol, 0.185 g) and phenylboronic acid (1.1 mmol, 0.133 g). After 10 hours, the cross-coupled product was obtained in 99% yield.Hot filtration test
Hot filtration test carried out for the Suzuki coupling of phenylboronic acid (1.1 mmol), 4-bromobenzaldehyde (1 mmol) in the presence of NPs 1 under optimum reactions conditions. After 10 minutes of reaction, the hot reaction mixture was filtered through G4 crucible containing Celite. Half of the reaction mixture was quenched and the product conversion was monitored by 1H NMR. The other half of reaction mixture was further stirred for additional 3 hours, and then the product conversion was then the conversion was estimated using 1H NMR. The cross coupled product was obtained in 84% and 92% after 10 minutes and 3 h of reaction respectively.Conclusions
In summary, ligands L1 and L2 have been synthesized and characterized by 1H and 13C{1H} NMR spectroscopy. The structure of L1 has also been supported by single crystal X-ray diffraction studies. The ligand L1 and L2 were further used as stabilizers for syntheses of catalytically active and recyclable palladium NPs 1–6. The powder X-ray diffraction (PXRD), HRTEM, and SEM-EDX techniques were used to authenticate NPs 1–6. HRTEM study of NPs 1–6 suggests the formation of ultra-small nanoparticles of size between 1–6 nm. These NPs have been explored as catalysts for Suzuki–Miyaura cross coupling reaction and found to be efficient for coupling of aryl bromides and chlorides. NPs 1 stabilized by thioether ligand have shown maximum catalytic efficiency amongst all due to the relatively small size (∼1–2 nm), uniformity in the dispersion and high purity. The NPs stabilized by secondary amine containing sulphur donor atom are slightly more efficient than that of selenium analogue. The distinct advantage of these nanoparticles as a catalyst is their recyclability. The catalytic pathway as suggested by the Hg and PPh3 poisoning tests is proceeding via homogeneous mode. These tests also suggest that the anthracene unit being bulky in nature stabilize the NPs so efficiently that Hg/PPh3 does not reach the catalyst surface to poison them. At the same time, they allow the reactant molecules to reach the NPs surface.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge SAIF, PU for EDS facility. P. O. acknowledge DST for INSPIRE Fellowship [DST/INSPIRE Fellowship/2017/IF170491]. A. A. and A. K. also acknowledge DST SERB [ECR/2016/001549] for fellowship and research grant. A. K. also thanks University Grants Commission for financial support in the form of UGC-BSR-Start-Up Research Grant Vide Letter No. F.30-371/2017 (BSR), dated 10 July 2017. SK would like to thank DST for INSPIRE Faculty Award [DST/INSPIRE/04/2015/002971].Notes and references
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 29, 4442–4489 CrossRef.
- J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527–2571 CrossRef CAS.
- R. Rossi, F. Bellina and M. Lessi, Adv. Synth. Catal., 2012, 354, 1181–1255 CrossRef CAS.
- A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020–4028 CrossRef CAS.
- X. Bei, H. W. Turner, W. H. Weinberg and A. S. Guram, J. Org. Chem., 1999, 64, 6797–6803 CrossRef CAS.
- C. Fleckenstein, S. Roy, S. Leuthaeusser and H. Plenio, Chem. Commun., 2007, 2870–2872 RSC.
- A. I. Moncada, J. M. Tanski and L. M. Slaughter, J. Organomet. Chem., 2005, 690, 6247–6251 CrossRef CAS.
- A. Kumar, M. Aggarwal and A. K. Singh, J. Organomet. Chem., 2008, 693, 3533–3545 CrossRef CAS.
- A. Kumar, M. Aggarwal, A. K. Singh and R. J. Butcher, Inorg. Chim. Acta, 2009, 362, 3208–3218 CrossRef CAS.
- I. D. Kostas, B. R. Stelle, A. Terzis, S. V. Amosova, A. V. Martynov and N. A. Makhaeva, Eur. J. Inorg. Chem., 2006, 2642–2646 CrossRef CAS.
- M. Cao, J. Lin, H. Yang and R. Cao, Chem. Commun., 2010, 46, 5088–5090 RSC.
- A. Fihri, M. Bouhrara, B. Nekoueishahraki, J. M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181–5203 RSC.
- M. N. Nadaguada, V. Polshettiwar and R. S. Varma, J. Mater. Chem., 2009, 19, 2026–2031 RSC.
- V. Polshettiwar, M. N. Nadaguada and R. S. Varma, Chem. Commun., 2008, 6318–6320 RSC.
- R. Narayanan and M. A. El-Sayed, Langmuir, 2005, 21, 2027–2033 CrossRef CAS PubMed.
- V. Polshettiwar, D. Cha, X. Zhang and J. M. Basset, Angew. Chem., Int. Ed., 2010, 49, 9652–9656 CrossRef CAS.
- E. Filippo, A. Serra and D. Manno, Sens. Actuators, B, 2009, 138, 625–630 CrossRef CAS.
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757–3778 CrossRef CAS.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS.
- L. M. Rossi, J. L. Fiorio, M. A. Garcia and C. P. Ferraz, Dalton Trans., 2018, 47, 5889–5915 RSC.
- V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588–4589 CrossRef CAS.
- Q. Yao, E. P. Kinney and C. Zheng, Org. Lett., 2004, 17, 2997–2999 CrossRef PubMed.
- G. K. Rao, A. Kumar, S. Kumar, U. B. Dupare and A. K. Singh, Organometallics, 2013, 32, 2452–2458 CrossRef CAS.
- P. Dubey, S. Gupta and A. K. Singh, Dalton Trans., 2018, 47, 3764–3774 RSC.
- P. Dubey, S. Gupta and A. K. Singh, Organometallics, 2019, 38, 944–961 CrossRef CAS.
- S. I. Aizawa, A. Majumder, Y. Yokoyama, M. Tamai, D. Maeda and A. Kitamura, Organometallics, 2009, 20, 6067–6072 CrossRef.
- M. Shi, L. P. Liu and J. Tang, J. Org. Chem., 2005, 25, 10420–10425 CrossRef.
- D. Das, G. K. Rao and A. K. Singh, Organometallics, 2009, 20, 6054–6058 CrossRef.
- F. Saleem, G. K. Rao, A. Kumar, S. Kumar, M. P. Singh and A. Singh, RSC Adv., 2014, 99, 56102–56111 RSC.
- S. Kumar, G. K. Rao, A. Kumar, M. P. Singh, F. Saleem and A. K. Singh, RSC Adv., 2015, 26, 20081–20089 RSC.
- G. K. Rao, A. Kumar, B. Kumar and A. K. Singh, Dalton Trans., 2012, 41, 4306–4309 RSC.
- M. Cao, J. Lin, H. Yang and R. Cao, Chem. Commun., 2010, 46, 5088–5090 RSC.
- R. Tatumi, T. Akita and H. Fujihara, Chem. Commun., 2006, 3349–3351 RSC.
- K. R. Gopidas, J. K. Whitesell and M. A. Fox, Nano Lett., 2003, 3, 1757–1760 CrossRef CAS.
- L. Wu, Z.-W. Li, F. Zhang, Y.-M. He and Q.-H. Fan, Adv. Synth. Catal., 2008, 350, 846–862 CrossRef CAS.
- J. Kumar and R. Saxena, J. Less-Common Met., 1989, 147, 59–71 CrossRef CAS.
- V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588–4589 CrossRef CAS.
- A. R. Katritzky, Y. J. Xu, H. Y. He and S. Mehta, J. Org. Chem., 2001, 66, 5590–5594 CrossRef CAS.
- A. Habtemariam, B. Watchman, B. S. Potter, R. Palmer, S. Parsons, A. Parkin and P. J. Sadler, J. Chem. Soc., Dalton Trans., 2001, 8, 1306–1318 RSC.
- A. K. Singh and V. Srivastava, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 47, 471–475 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. Spectral data of L1 and L2; single crystal data of L1 (CCDC 1887878), SEM-EDS data of NPs 1–6. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra03498f |
| This journal is © The Royal Society of Chemistry 2019 |