
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrospun silica nanofiber mats functionalized with ceria nanoparticles for water decontamination†
Ines Zucker *abc,
Nadir Dizgead,
Camrynn L. Fauseya,
Evyatar Shaulskya,
Meng Suna and
Menachem Elimelecha
*abc,
Nadir Dizgead,
Camrynn L. Fauseya,
Evyatar Shaulskya,
Meng Suna and
Menachem Elimelecha
aDepartment of Chemical and Environmental Engineering, Yale University, New Haven, Connecticut 06520-8286, USA
bPorter School of Environmental Studies, Tel Aviv University, Israel. E-mail: ineszucker@tauex.tau.ac.il; Tel: +972-3-640-8227
cSchool of Mechanical Engineering, Tel Aviv University, Israel
dDepartment of Environmental Engineering, Mersin University, Mersin, 33343, Turkey
First published on 20th June 2019
Abstract
This study demonstrates the fabrication of nanoceria-immobilized silica nanofibers for efficient water decontamination with easy reuse and regeneration properties. Commercially-available ceria (CeO2) nanoparticles were immobilized on electrospun silica nanofiber mats. Optimal conditions for both oxidation of model trace organic compounds (TrOCs) and hydroxyl-radical formation were determined in batch experiments with ceria nanoparticles. Kinetic experiments showed comparable oxidation rates of positively- and neutrally-charged TrOCs (95 and 98%, respectively) by the nanofiber composites at pH 2 in 0.5 mM H2O2. We demonstrated the reuse properties of the ceria nanofiber composites, finding less than a 15% reduction in oxidation performance after 10 cycles. In addition to this oxidative performance, the electrospun CeO2 nanofiber composites are also applicable for adsorbing the persistent contaminant perfluorooctanesulfonic acid (PFOS) through electrostatic interactions. Regeneration of the fibers was enabled through a one-hour heat treatment at 550 °C in air. Overall, results from this study show that the nanoceria-immobilized silica nanofibers can be used as efficient oxidative and adsorptive media to treat TrOCs in acidic waters, without use of an additional catalyst separation stage from solution.
1. Introduction
Contamination of water sources is a major global problem in both developing and industrialized countries.1 Of particular concern are organic pollutants that are toxic at trace quantities, such as pharmaceuticals, polyfluorinated compounds, hormones, and pesticides.2,3 The growing number of such trace organic compounds (TrOCs) and recent proposed regulations regarding their presence in water sources has created a need to develop more efficacious removal technologies to ensure contaminant-free and clean water.Conventional methods for water decontamination involve physicochemical separation and chemical oxidation. Specifically, advanced oxidation processes (AOPs) are frequently used to oxidize non-biodegradable, recalcitrant, and toxic organic pollutants through in situ production of reactive radicals.4 One of the earliest AOPs is the Fenton process, involving activation of hydrogen peroxide (H2O2) to generate hydroxyl (OH) radicals through a catalytic reaction with the dissolved Fe2+/Fe3+ redox pair.5 However, homogeneous Fenton reactions suffer from fundamental drawbacks such as operation at low pH values (below 4), slow reduction kinetics, and significant accumulation of undesired iron sludge after neutralization.
In recent years, nanotechnology-based approaches have been increasingly explored to enhance or replace traditional decontamination methods. Through manipulation of material size, morphology, and chemical structure, nanomaterials can gain exceptional adsorptive and catalytic properties useful for water decontamination.6 Among nano-assisted AOPs, heterogeneous iron-free Fenton-like processes frequently rely on nanoscale transition metal oxide catalysts to facilitate effective contaminant degradation, many times overcoming the limitations of traditional Fenton processes.7
Cerium oxide (CeO2 or ceria)-based materials are widely used in catalytic industrial applications, as well as in mechanical polishing, UV-shielding, and solid-oxide fuel cells.8 Specifically, catalytic redox reactions on the surface of ceria are mainly promoted by the formation of oxygen vacancy defects.9 The surface oxygen vacancy is generally accompanied by the reduction of Ce4+ ions to Ce3+ to balance the change in charge, resulting in both “oxygen buffering” behavior and potential catalytic reactivity of the ceria surface.10,11 Therefore, ceria nanoparticles (nanoceria) have gained considerable attention as a catalytic material due to their increase in effective surface concentration of surface Ce3+ species.12–14
Nanoceria is a versatile catalyst having both pro- and anti-oxidant actions. While the latter has been demonstrated as a strategy for scavenging reactive oxygen species (ROS) and eliminating oxidative stress in biological systems,15–18 nanoceria's intrinsic oxidation activity can be used to oxidize organics via the hydroxyl adduct route in environmental systems.19,20 Nanoceria usually exhibit strong oxidizing ability in the presence of an oxidizing agent (e.g., H2O2), but has also been reported to exhibit oxidative capabilities even in the absence of a strong oxidizing agent.17
Different approaches have been suggested to improve the catalytic activity of nanosized ceria, including changes in material design to enhance surface-to-volume ratio and increase the oxygen vacancy defect density, or by changes in operational design to maximize redox reactions. Material-based factors affecting nanoceria's redox reactions may include crystal structure, size distribution, and exposed facets.21 In liquid-phase applications, operational factors such as ion and redox agent presence, pH, and oxygen level can affect nanoceria's reactivity, even leading to crystallite dissolution and reprecipitation.22
Changes in nanoceria's colloidal stability and potential effects on living cells23–25 are limiting factors in using nanoceria in water treatment applications such as adsorption,26 photocatalysis,27 and oxidation processes.20 Another limiting parameter in decontamination using suspended nanoparticles is the challenge of separating and recycling the nanoparticles after use. To address this limitation, ceria micro/nanocomposite structures—as opposed to nanoceria in suspension—have been suggested for facile separation and recycling after use.26,28 For example, ceria flower-like particles and hollow fibers have been suggested as recoverable and regenerable structures for both antimicrobial and adsorptive purposes.26,28
In the context of nano-assisted water treatment technologies, electrospun nanofibers have attracted much attention as excellent nanomaterial carriers due to their high surface area, high porosity, and robust mechanical strength.29–31 Electrospun fibers can be tuned to achieve a desired chemical composition, structure, and stability, and can be post-treated to alter their chemical and structural properties.32 Affixing nanomaterials to the high surface area of electrospun fibers allows a high exposed surface area/mass ratio of the nanomaterial and, because the nanomaterial is irreversibly bound to the surface of the fibers, minimizes the risk of nanomaterial release to the environment.33 The exposed surface area/mass ratio of nanomaterial is especially important in decontamination because surface area, among other factors, can be related to functionalities like adsorption and oxidative capacity.34
In this study, we fabricated a reactive fibrous matrix by coating electrospun silica nanofibers with nanoceria for efficient water decontamination with easy reuse and regeneration properties. To optimize the oxidative operational conditions of ceria nanoparticles, they were analyzed both for their ability to oxidize of carbamazepine (CBZ) as a model TrOC as well as for their ability to generate OH-radicals. These studies were conducted in the presence and absence of hydrogen peroxide, as well as across pH values. Using the resultant optimal operational conditions, the synthesized ceria nanofiber composites were used for oxidation and adsorption of a set of emerging TrOCs, such as CBZ, propranolol (PRO) and perfluorooctanesulfonic acid (PFOS). Finally, the facile reuse and regeneration properties of the ceria nanofiber composites were elucidated, rendering the electrospun ceria/silica nanofiber composites as a promising material for removal of a myriad of water contaminants of different chemical characteristics.
2. Experimental
2.1. Fabrication of electrospun ceria/silica nanofiber composites
Tetraethyl orthosilicate (TEOS, Sigma-Aldrich), phosphoric acid (85%, Sigma-Aldrich), and deionized (DI) water (Milli-Q) were mixed together in the molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.01:11, respectively, for 6 h. An aqueous solution containing 10 wt% poly(vinyl alcohol) (PVA, Mw = 89000–98000, Sigma-Aldrich) was prepared by dissolving PVA powder in DI and mixing for 12 hours at 80 °C. Equal volumes of the TEOS gel and the PVA solution (5 mL of each) were mixed for 6 hours, and subsequently loaded in a 10 mL Luer-Lok Tip syringe (Becton Dickinson & Co., Franklin Lakes, NJ) fitted with an 21G × 1 inch PrecisionGlide needle (Becton Dickinson & Co.). After placing the syringe horizontally on the syringe pump (Harvard Apparatus, Plymouth Meeting, PA), an electrode of high-voltage supply (Gamma High Voltage Research Inc., Ormond Beach, FL) was connected to the syringe's needle tip. A grounded sheet of aluminum foil (10.5 cm × 38.5 cm) was used as a nanofiber collector and installed on a rotating drum moving at approximately 16 cm min−1. The needle-to-collector distance, flow rate, and voltage were 11 cm, 0.5 mL h−1, and 13 kV, respectively. The electrospinning was performed in a closed chamber under controlled temperature (30 °C) and relative humidity (30%). Fibers were collected for 10 hours, removed from the collector, and subsequently calcined in a furnace (Thermolyne 48000) at 700 °C for 2 hours.
:0.01:11, respectively, for 6 h. An aqueous solution containing 10 wt% poly(vinyl alcohol) (PVA, Mw = 89000–98000, Sigma-Aldrich) was prepared by dissolving PVA powder in DI and mixing for 12 hours at 80 °C. Equal volumes of the TEOS gel and the PVA solution (5 mL of each) were mixed for 6 hours, and subsequently loaded in a 10 mL Luer-Lok Tip syringe (Becton Dickinson & Co., Franklin Lakes, NJ) fitted with an 21G × 1 inch PrecisionGlide needle (Becton Dickinson & Co.). After placing the syringe horizontally on the syringe pump (Harvard Apparatus, Plymouth Meeting, PA), an electrode of high-voltage supply (Gamma High Voltage Research Inc., Ormond Beach, FL) was connected to the syringe's needle tip. A grounded sheet of aluminum foil (10.5 cm × 38.5 cm) was used as a nanofiber collector and installed on a rotating drum moving at approximately 16 cm min−1. The needle-to-collector distance, flow rate, and voltage were 11 cm, 0.5 mL h−1, and 13 kV, respectively. The electrospinning was performed in a closed chamber under controlled temperature (30 °C) and relative humidity (30%). Fibers were collected for 10 hours, removed from the collector, and subsequently calcined in a furnace (Thermolyne 48000) at 700 °C for 2 hours.
To immobilize CeO2 nanoparticles on the surface of the calcined silica fibers, 1 mg mL−1 suspension of cerium(IV) oxide nanoparticles (30 nm particles in powder form, Sigma-Aldrich) at pH 2 (10 mM buffer acetate) was first sonicated in an ultrasonication bath (FS60, Fisher Scientific) for 15 min. Following sonication, 10 mL of the CeO2 nanoparticle suspension was poured over 10 mg of silica fibers in a Petri dish and mixed overnight on an orbital shaker (KJ-201BD, Kangjian). After CeO2 immobilization, the ceria/silica nanofiber composites were washed three times with 10 mL of deionized water and dried at 60 °C overnight. The CeO2 mass immobilized on the fibers' surface was estimated by weighing the dry fiber mats before and after dip-coating in the CeO2 suspension. A schematic illustration of the electrospun ceria/silica nanofiber composites fabrication is provided in Fig. S.1 in ESI.†
2.2. Characterization of ceria nanoparticles and electrospun ceria/silica nanofiber composites
Surface morphologies of commercial nanoceria and the nanofibers before and after modification with nanoceria were examined with a Hitachi scanning electron microscope (SEM) SU-70. Before SEM imaging, samples were dried at 60 °C overnight and then sputter-coated with an 8 nm coating of iridium (208HR, Cressington). Acceleration voltage of 5.0 kV was applied during SEM analysis for all samples. The distribution of fiber diameters was determined from a random sampling of ∼30 fibers from multiple SEM images of the same sample using ImageJ software (National Institutes of Health, Bethesda, MD).The specific surface areas of nanoceria particles, electrospun silica nanofibers, and nanoceria-immobilized silica nanofibers were determined using a Micromeritics 3Flex static volumetric system with Kr as the analysis adsorptive. To assure moisture-free samples, samples were dried overnight and degassed under vacuum at 110 °C for at least 10 hours.
To determine both dispersed aggregate size and zeta potential of the nanoceria, suspensions of the nanoparticles (0.5 μg mL−1) were prepared in 10 mM acetate buffer aqueous solution at pH 2 and bath-sonicated for 10 minutes. To determine aggregate size, samples were immediately analyzed after sonication by dynamic light scattering (DLS, ALV-GmbH) using a multi-detector light scattering unit with a neodymium-doped vanadate laser (Verdi V2, Coherent, 78 Inc., Santa Clara, CA) operating at a wavelength of 532 nm. DLS analysis was conducted at 90 °C for 12 runs, with 30 seconds of data collection for each run. To determine zeta potential, electrophoretic mobility measurements were taken using the NanoBrook Omni (Brookhaven Instruments, NY) with a 659 nm wavelength laser. The average and standard deviation of zeta potentials (using Smoluchowski approximation) were determined from 10 runs (30 cycles per run) for each sample.
The presence of nanoceria on the nanofiber composites was investigated by Fourier-transform infrared spectroscopy (FTIR) and Raman spectroscopy. FTIR spectra were recorded from 450 to 4000 cm−1 using a Thermo Nicolet 6700 spectrometer. Raman spectroscopy confirmed the binding of nanoceria to the fibers' surface, with spectra acquired using a 532 nm laser excitation on a Horiba Jobin Yvon HR-800 Raman spectrometer.
The hydrophilicity of the fibers was determined by the water contact angle using the sessile drop method and a goniometer (OneAttension, Biolin Scientific). A water droplet (5 μL) was placed on the fibers and photographed using a digital camera for 15 s. The left and right contact angles were analyzed from the digital images by post-processing software (OneAttension software) and averaged over six measurements (three contact angle measurements on two nanofiber samples).
Fiber porosity was estimated by gravimetric analysis. The oven-dried electrospun fibers were weighed and subsequently submerged in 2-propanol for one hour. After removal, the fibers were layered between nylon fabric (supplied by VWR), lightly pressed with Kimwipes for removal of excess 2-propanol and weighed. The porosity (φ) was estimated using
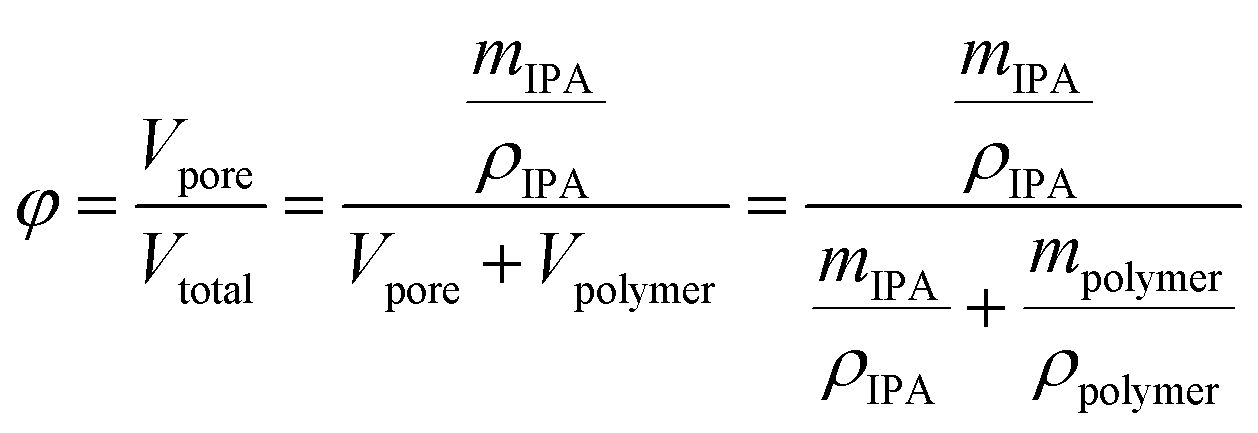 | (1) |
The pore-size distribution was evaluated using the wet/dry flow method with a custom-made porometer setup.31 The electrospun fibers were cut by a 1/2-inch diameter puncher and placed on a 13 mm stainless steel filter holder (Whatman, Maidstone, UK). Nitrogen gas pressure was controlled with a digital pressure regulator (Control Air Inc, Amherst, NH), gas flow across the membrane was monitored by a flow meter (Porter, Hatfield, PA), and pressure was monitored by a pressure transducer (Honeywell, Morris Plains, NJ).
2.3. Optimization of nanoceria oxidative and adsorptive performance
The oxidative and adsorptive performance of the nanoceria suspension toward carbamazepine (CBZ) and perfluorooctanesulfonic acid (PFOS) was tested in the presence of hydrogen peroxide (H2O2, Sigma-Aldrich) at different concentrations (up to 10 mM) and across a pH range (2–6). The pH was adjusted using sodium hydroxide (NaOH pellets, J. T. Baker) and hydrochloric acid (HCl, Sigma-Aldrich). In a typical experiment, environmentally-relevant concentrations of 1 mg L−1 model contaminant (i.e., CBZ or PFOS) was mixed with 1 mg mL−1 CeO2 nanoparticles in 10 mL of buffer acetate solution (10 mM) for one hour. Following this interaction, the mixed solution was centrifuged (13000 rpm for 10 minutes) to remove ceria nanoparticles, and the supernatant was analyzed to quantify CBZ or PFOS removal as described in the analytical quantification of TrOCs subsection.
In a set of parallel oxidation experiments, OH-radical formation was quantified using a tertiary butanol assay.35 Tertiary butanol was added in excess (100 mM) to a buffered 1 mg mL−1 CeO2 nanoparticle suspension (10 mM acetate buffer) to readily react with OH-radicals (rate constant of 6 × 108 M−1 s−1). The main product formed by this reaction is formaldehyde, which was quantified by the Hantzsch method.36 A reagent solution (2 M ammonium acetate, 0.05 M of acetic acid, and 0.02 M acetylacetone in water) was mixed with the tertiary butanol/CeO2 supernatant with a 1:1 volume-to-volume ratio. The mixture was heated for 10 minutes at 50 °C, and the change in color was measured spectrophotometrically at 412 nm. Finally, OH-radical yield could be calculated using
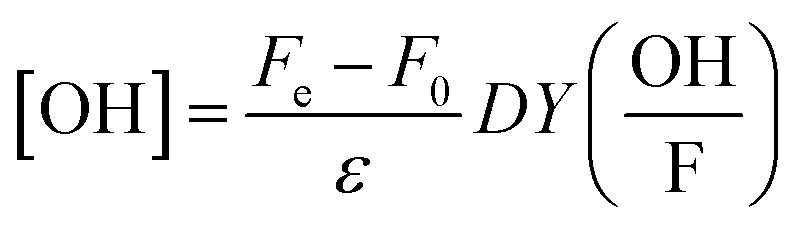 | (2) |
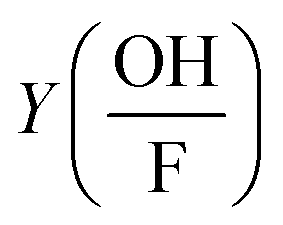 is the correction factor of hydroxyl radical to formaldehyde yields (∼2).35 This method is often used when fast competitive reactions with radicals are expected (e.g., during ozonation of wastewater),37 but can be used here to efficiently scavenge all OH-radicals formed during interaction.
is the correction factor of hydroxyl radical to formaldehyde yields (∼2).35 This method is often used when fast competitive reactions with radicals are expected (e.g., during ozonation of wastewater),37 but can be used here to efficiently scavenge all OH-radicals formed during interaction.
2.4. Oxidation experiments with electrospun CeO2 nanofiber composites
The oxidative performance of the electrospun CeO2 nanofiber composites toward three TrOCs was tested at pH 2 in the absence and presence (0.5 mM) of hydrogen peroxide. The TrOCs—CBZ, propranolol (PRO), and perfluorooctanesulfonic acid (PFOS)—were chosen based on their environmental relevance and differing charge in acidic solution. Chemical structures and properties of the TrOCs are shown in Table A.1.† Oxidation experiments were performed in triplicate with 1 mg L−1 CBZ, PRO, or PFOS (separately) at pH 2 in a buffered solution. CeO2 nanofiber composites were added to the solution, with final concentration approximately 1 mg CeO2 (on fibers) per 1 mL of TrOC solution. Samples were taken periodically, and the TrOC residual concentration was determined through HPLC (for CBZ and PRO) or HPLC-MS analysis (for PFOS). To quantify ceria release from the composite fibers during oxidation, cerium ion concentration in the final solution (following 2 hours of interaction) was measured by inductively coupled plasma mass spectrometry (ICP-MS, PerkinElmer, DRC-e) with a Meinhard nebulizer and argon plasma.2.5. Analytical quantification of TrOCs
Concentrations of CBZ and PRO were determined using HPLC (Shimadzu LC-20AD) equipped with an Eclipse Plus C18 column (1.8 μm, 4.6 × 50 mm, Agilent) and a UV/Vis detector (SPD-20AV). A sample volume of 100 μL was injected using an isocratic mobile phase of solvent comprising 35% acetonitrile and 65% water with 0.1% (v/v) phosphoric acid at a flow rate of 1.3 mL min−1. PFOS concentration was determined using HPLC (Agilent 1290 Infinity Series) coupled with a mass spectrometer (Agilent 6550A iFunnel Q-TOF MS). A sample volume of 1 μL was injected into an Eclipse Plus C18 column using a mobile phase gradient of solvent comprised of 40% methanol and 60% water with 20 mM ammonium acetate at a flow rate of 0.5 mL min−1. Methanol was increased from 40% to 98% in 5 min, held for 1 min, dropped back to 40%, and stabilized for 1 min. The Q-TOF MS was operated using an electrospray ionization (ESI) interface in negative mode.3. Results and discussion
3.1. Characteristics of ceria nanoparticles and electrospun ceria/silica nanofiber composites
SEM micrographs (Fig. 1A, top) illustrate that commercial ceria nanoparticles aggregate into large aggregates. Changes in ceria nanoparticle aggregate size were also analyzed in aqueous solutions at pH 2 and 6 (Fig. S.2 in ESI†). At pH 2, the ceria nanoparticles had a large zeta potential (45.1 mV, Table 1) and the aggregate size did not change over one hour (43.0 nm, Table 1), indicating colloidal stability at low solution pH. However, at pH 6, the ceria nanoparticles rapidly aggregated, which could be explained by reduced electrostatic repulsion between nanoparticles when solution pH is close to their point of zero-charge (∼6.5).38 As we discuss later, anchoring ceria nanoparticles to silica fiber mats could eliminate aggregation during water decontamination while enabling easy separation of the particles from treated solution. The specific surface area of ceria nanoparticles was 37.2 ± 0.2 m2 g−1, similar to previous measurements reported in literature.9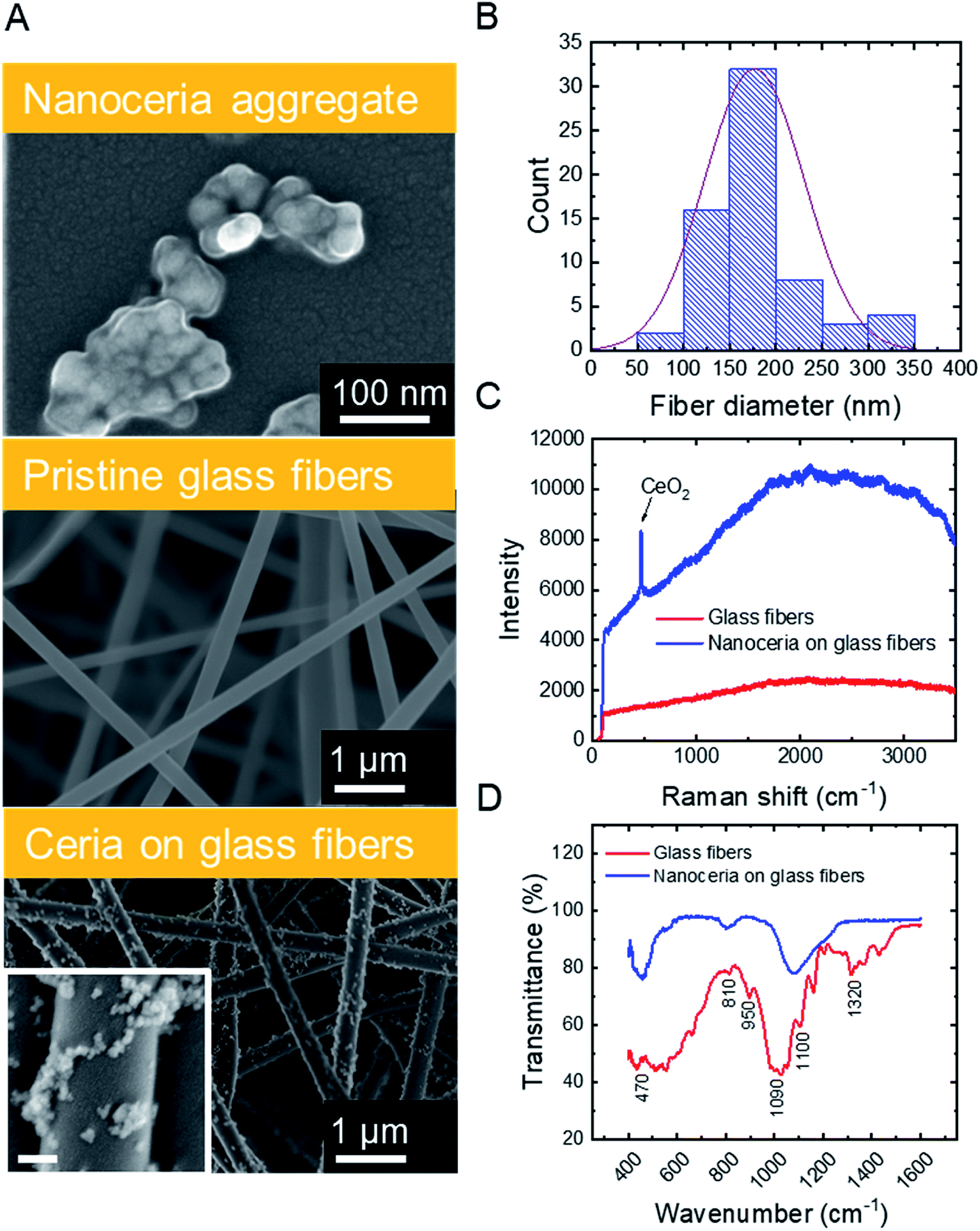 | ||
| Fig. 1 Characteristics of electrospun ceria/silica nanofiber composites. (A) SEM images of ceria aggregate (top), pristine fibers (center), and CeO2-immobilized fibers (bottom). Inset scale bar is 100 nm. (B) Diameter distribution of ceria/silica nanofiber composites. (C) Raman and (D) FTIR spectra of pristine fibers (red) and ceria/silica nanofiber composites (blue), indicating the presence of ceria nanoparticles on the surface of the nanofibers. | ||
| a Determined by dynamic light scattering (averaged over one hour of data collection).b Polydispersity index.c Based on pore-size distribution (Fig. A.3B in ESI). | ||
|---|---|---|
| Ceria nanoparticles | Dispersed aggregate sizea (PDI)b (nm, −) | 43.0 (0.24) |
| Zeta potential (mV) | 45.1 ± 0.5 | |
| Specific surface area (m2 g−1) | 37.2 ± 0.2 | |
| Glass fibers | Average fiber diameter (nm) | 176.6 |
| Isoelectric point | ∼2 (ref. 39) | |
| Water contact angle (°) | <10 | |
| Fiber mat mean pore sizec (μm) | 1.59 | |
| Specific surface area (m2 g−1) | 27.1 ± 0.2 | |
| Fiber mat porosity (%) | 96.5 ± 0.4 | |
Silica nanofibers were chosen as the substrate material for catalytic nanoceria because of their thermal stability (allowing facile thermal regeneration), negative surface charge39 (attracting positively charged nanoceria), and outstanding mechanical properties. Representative characteristics of silica fiber mats are outlined in Table 1. SEM micrographs (Fig. 1A, center) illustrate the cylindrical structure of electrospun glass fibers which were devoid of beads or branched structures. The fiber diameters follow a typical Gaussian distribution (Fig. 1B) with an average fiber diameter of 176.6 nm. The silica nanofiber mats are porous (Fig. S.3 in ESI†) and hydrophilic with a contact angle of <10° (Table 1), consistent with previous studies on electrospun silica fibers.40
Surface modification of the silica nanofibers with ceria nanoparticles was characterized through SEM, Raman spectroscopy, and FTIR. SEM micrographs for electrospun silica nanofibers (Fig. 1A, center) and ceria/silica nanofiber composites (Fig. 1A, bottom) illustrate nanoceria bound to the surface of the silica fiber mats. Using ImageJ image processing software, an average nanoceria aggregate size of 52 ± 21 nm was determined using at least 100 ceria nanoparticles from three representative SEM images. Using a mass balance of six sets of dry fiber mats, the typical loading was 18.0 ± 3.7 mg CeO2 per 35.0 ± 1.2 mg of fibers. No significant change was seen in the nanocomposite specific surface area (27.2 ± 0.1 m2 g−1) as compared to that of pristine silica nanofibers (27.1 ± 0.2 m2 g−1). This minimal change in surface area after surface functionalization with ceria nanoparticles is mainly due to the moderate ceria loading (∼34% by weight), and because ceria clusters (rather than nanoparticles) are bound to the surface of the fibers.
The functionalization of the silica fibers with ceria nanoparticles was also confirmed by Raman spectroscopy (Fig. 1C). For the pristine electrospun silica mats, no prominent bands between 200–3500 cm−1 were observed. However, upon modification with nanoceria, a sharp single peak at ∼465 cm−1 appeared, representing the optical Raman F2g mode attributed to the symmetric breathing of oxygen atoms in the CeO2 lattice.41
FTIR was used to demonstrate the bonding of ceria to the active functional groups (Si–OH and Si–O−) on the silica nanofibers, both before and after ceria deposition (Fig. 1D). The peaks at 1100 cm−1 and 1090 cm−1 in the silica fiber sample illustrate asymmetric stretching and deformation of Si–O and Si–O–Si bonds, respectively,42,43 while the peak at 950 cm−1 indicates the stretching of Si–OH bonds and SiO− groups. The peaks at 810 cm−1 and 470 cm−1 are related to the symmetrical stretching and in-plane bending of the Si–O–Si modes, respectively. After addition of CeO2 to the fibers, the Ce–O and Ce–Si peaks become distinguishable in the range between 400 and 450 cm−1.44,45 Corroborating this evidence of ceria on the surface of the fibers is the decrease in the peak at 950 cm−1, which arises from dangling oxygen atoms, as well as a shift in the Si–O–Si asymmetrical stretching at 1100 cm−1; both changes are associated with electropositive cerium ions bound to electronegative oxygen in the silica nanofibers.46
3.2. Optimization of operational conditions for TrOC removal by ceria nanoparticles
To optimize the oxidative and adsorptive conditions for TrOC removal, pristine CeO2 nanoparticles were tested for removal of a model compound, carbamazepine (CBZ), across pH values (2–6) and H2O2 concentrations (up 10 mM). As shown in Fig. 2 (blue open circles), higher CBZ removal was associated with higher OH-radical formation, suggesting that CBZ was oxidized by the formed radicals rather than adsorbed to the ceria nanoparticles. Appearance of secondary peaks in HPLC chromatographs strengthen this claim, as they indicate possible formation of CBZ transformation products during oxidation by the ceria nanoparticles (Fig. S.4 in ESI†).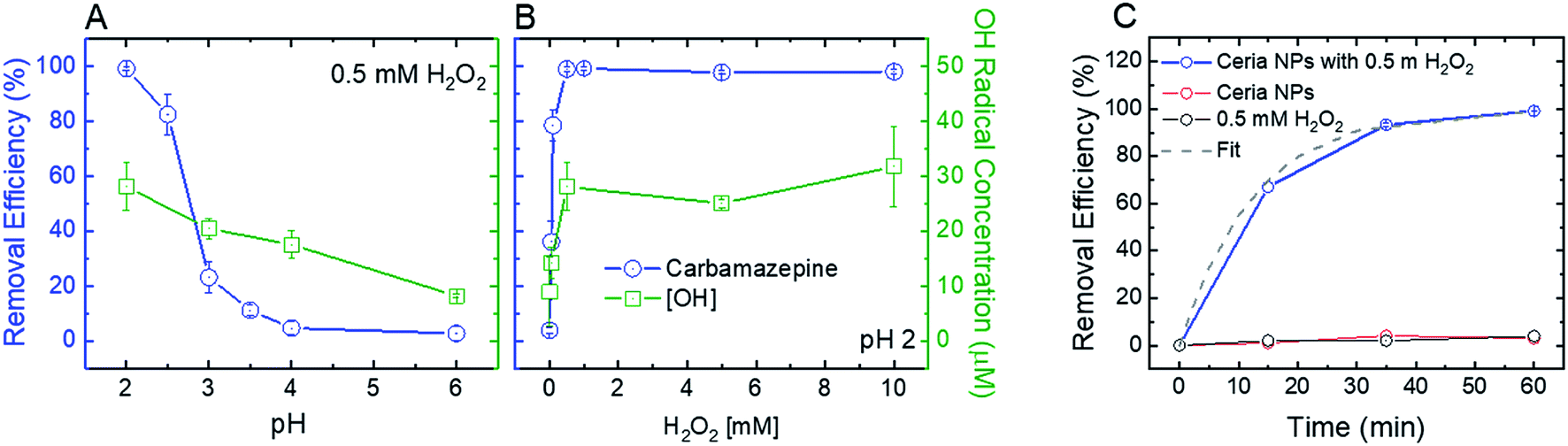 | ||
| Fig. 2 Effect of (A) pH and (B) H2O2 concentration on the removal efficiency of 1 mg L−1 carbamazepine (blue) and OH-radical formation (green) by 1 mg mL−1 CeO2 nanoparticles in 10 mM buffer acetate solution after 1 hour of mixing. Experiments were carried out under ideal conditions for maximum removal efficiency and OH radical generation: pH 2 and 0.5 mM H2O2. (C) Removal kinetics of 1 mg L−1 carbamazepine by 1 mg L−1 ceria nanoparticles (NPs) at pH 2 in the presence of 0.5 mM H2O2 (blue line) and best-fit line (pseudo-first order, dashed grey line). Isolated effect of H2O2 (black line) without ceria NP addition and ceria NPs in the absence of H2O2 (red line) on carbamazepine removal was determined as well. | ||
Decreasing solution pH resulted in higher removal efficiency of CBZ as well as higher OH-radical formation (Fig. 2A), suggesting that ceria nanoparticles act as an oxidizing heterogeneous catalyst in acidic conditions. When H2O2 was added to a pristine CeO2 nanoparticle suspension, the color of the suspension promptly changed from nearly colorless to yellow, providing a visible indication of formation of surface peroxide complex.47 A hydrogen peroxide concentration of 0.5 mM maximizes both removal efficiency of CBZ and OH-radical formation (Fig. 2B), indicating that ceria nanoparticles catalyze a Fenton-like reaction with hydrogen peroxide.11 Higher concentrations of hydrogen peroxide did not produce a significantly higher concentration of OH-radicals or increase CBZ removal efficacy.
Control kinetic experiments suggest that exposure to either H2O2 (i.e., without ceria addition) or ceria nanoparticles (i.e., without H2O2 addition) is not sufficient to induce CBZ removal (Fig. 2C). These results are similar to those of Abdelkader et al., who found a synergistic relationship between a cerium oxide polishing powder and H2O2 in the oxidation of neutral red dye.48 In their study, a homogeneous H2O2 system achieved 12.58% oxidation of the dye and the cerium oxide powder achieved 67.36% oxidation, each after 60 minutes. However, a heterogeneous system of both the cerium oxide powder and H2O2 achieved 85.9% oxidation after 60 minutes, illustrating a synergetic effect from increased OH-radical production. Similarly, our control experiments at low solution pH (2) show negligible CBZ oxidation by H2O2 and ceria nanoparticles (3–4% in both experiments, Fig. 2C) after 60 minutes, while heterogeneous system of both the ceria nanoparticles and H2O2 achieved over 99% oxidation after 60 minutes. The reaction rate for oxidation of 1 mg L−1 CBZ (0.08 min−1) is similar to that achieved by homogenous Fenton (∼0.1 min−1).49
Cerium has been illustrated as a redox-cycling metal capable of generating highly-reactive radicals in the presence of peroxide.50 This catalytic behavior was directly related to the ceria's ability to cycle between the 3+ and 4+ states at oxygen vacancy sites. However, as Ce3+ is an intermediate valence state of ceria that could be rapidly oxidized to Ce4+ in the presence of the H2O2,9,51 we could only observe the 4+ state on the surface of ceria nanoparticles by XPS analysis prior to and following addition of 0.5 mM H2O2 (Fig. S.5 in ESI†). Interestingly, ceria-based materials have also shown promise as antioxidants and radical scavengers in both cell culture models and animal studies.15–17,52 These seemingly contradictory phenomena are likely due to differences in media (i.e., biological and environmental media) and applied dosages of ceria.
In addition to their oxidative performance, positively-charged CeO2 nanoparticles have the potential to adsorb negatively-charged persistent TrOCs through electrostatic attractions, as demonstrated in experimentation with perfluorooctanesulfonic acid (PFOS) as a representative compound (Fig. 3). Adsorption of PFOS was evaluated at several pH values and H2O2 concentrations. Not surprisingly, adsorption of PFOS was significantly higher at low solution pH, far from the CeO2 point of zero-charge (∼6.5).38 Due to the very low pKa value of PFOS (−3.27), electrostatic attraction is likely to be responsible for its adsorption even at low pH.53,54
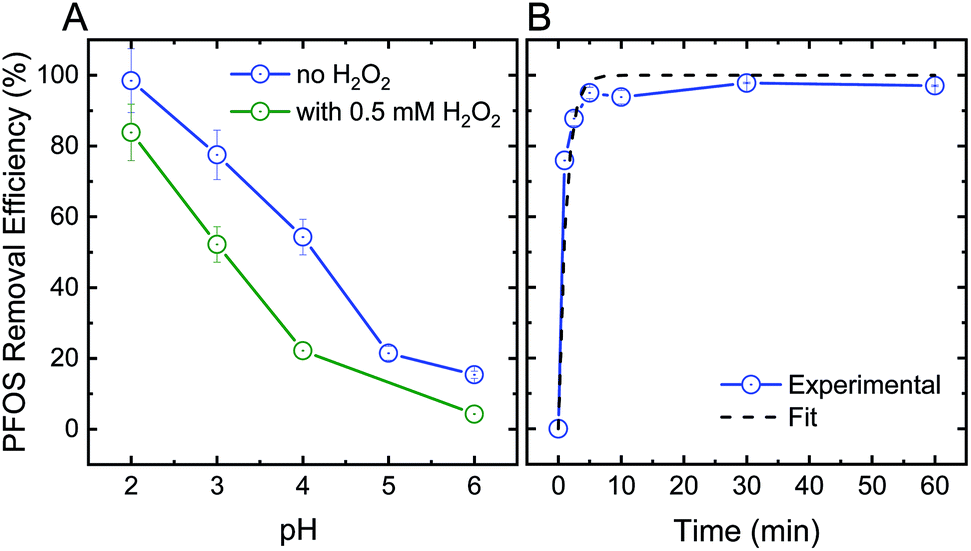 | ||
| Fig. 3 (A) Effect of pH on the adsorption efficiency of 1 mg L−1 perfluorooctanesulfonic acid (PFOS) by 1 mg mL−1 ceria nanoparticles after 1 hour of mixing in the absence (blue) and presence (green) of 0.5 mM H2O2. (B) PFOS adsorption kinetics and best-fit line (pseudo-first order) at pH 2. | ||
Addition of hydrogen peroxide to the CeO2 adsorption matrix decreased the removal of PFOS at all pH values tested (Fig. 3A). This decrease in PFOS adsorption upon addition of hydrogen peroxide is possibly due to competition over reactive sites on the ceria nanoparticle surface. The kinetics of sorption (Fig. 3B) illustrate a rapid and significant decrease in PFOS concentration in the supernatant (i.e., following separation from nanoparticles) after ten minutes of adsorption, which resulted in 98% removal of PFOS by the ceria nanoparticles.
3.3. Oxidation and adsorption of TrOCs by electrospun ceria/silica nanofiber composites
Optimal batch conditions of pH 2 and 0.5 mM H2O2 were used to establish removal kinetics of PRO, CBZ, and PFOS using the electrospun ceria/silica nanofiber composites (Fig. 4). In the absence of hydrogen peroxide, no PRO and CBZ were removed, suggesting that the nanofiber composites had negligible adsorptive capacity toward these TrOCs. However, addition of 0.5 mM H2O2 resulted in almost-complete oxidation of the positively- and neutrally-charged TrOCs (95 and 98%, respectively) following two hours of interaction with similar pseudo-first order kinetics (0.03 min−1). Control experiments showed that pristine nanofibers had minor effect on PRO and CBZ removal (Fig. S.6 in ESI†) under optimal oxidation conditions (i.e., pH 2 and 0.5 mM H2O2), indicating oxidation by the immobilized ceria as the sole removal mechanism. The removal kinetics by the nanofiber composite (i.e., when ceria was immobilized on the composite) was significantly lower than that of the nanoparticles alone (Fig. 2C) due to a decrease in both available reactive nanoparticle surface area and probability of interaction with pollutants.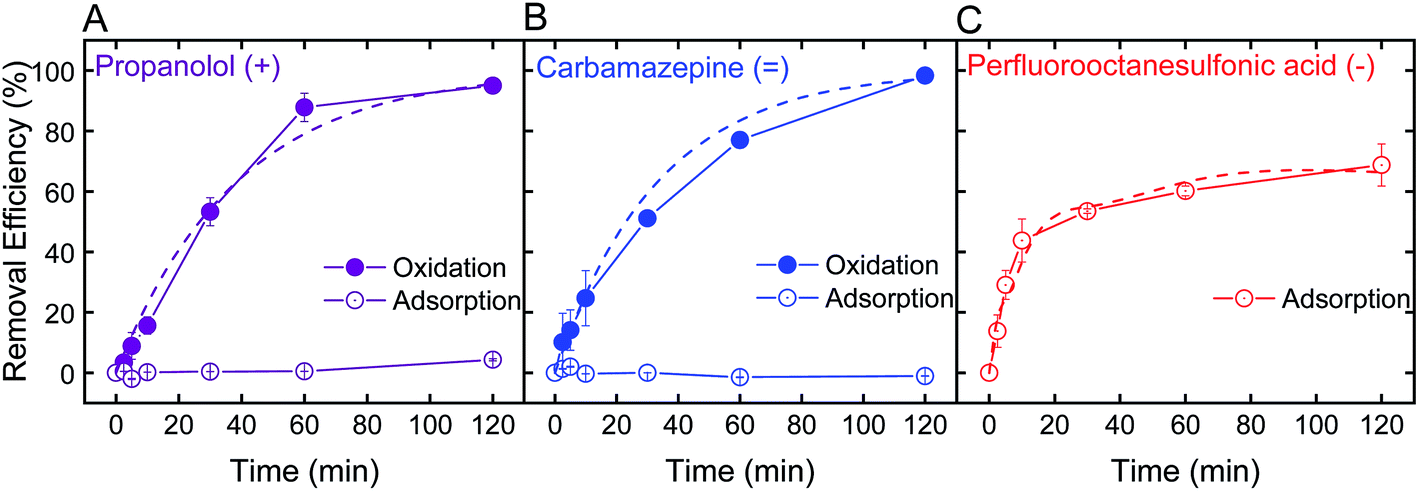 | ||
| Fig. 4 Removal kinetics and best-fit line (pseudo-first order) of 1 mg L−1 (A) propranolol (purple), (B) carbamazepine (blue), and (C) perfluorooctanesulfonic acid (red) by electrospun ceria/silica nanofiber composites for a final concentration of 1 mg nanoceria per mL of treated solution, through oxidation (solid circles) at pH 2 in 0.5 mM H2O2 and adsorption (open circles) at pH 2 and no H2O2. | ||
We note that even when ceria nanoparticle aggregation was prevented through immobilization onto the silica nanofibers, experiments at higher pH (e.g., pH 6) in the presence of hydrogen peroxide showed negligible removal of both PRO and CBZ by the ceria/silica nanofibers (Fig. S.6 in ESI†). This limited oxidation performance at higher solution pH is indicative of the inherent catalytic characteristics of nanoceria, which rely on the existence of oxygen vacancies on the surface of ceria nanoparticles to allow for redox mechanisms in catalysis as well as activation by oxygen containing compounds (e.g., H2O2).19,20
Removal of PFOS reached 69% after two hours of interaction with the ceria/silica nanofiber composite (Fig. 4C). Control experiments showed that pristine nanofibers adsorb approximately 19% of the PFOS after 2 hours, illustrating that ceria does not account for all adsorption achieved by the nanocomposite. Because the fibers adsorb PFOS, it can be inferred that the ceria nanoparticles on the fibers are responsible for less than 69% removal of PFOS (the total amount removed by the nanocomposite), which is a significant decrease in PFOS adsorption achieved by the same loading of ceria nanoparticles in suspension (98%, Fig. 3B).
This decrease in adsorption performance of the ceria/fiber nanofiber composite as compared to the rapid and full PFOS adsorption with suspended nanoceria (Fig. 3B) could possibly be explained by limitations in both ceria surface area and contaminant diffusion toward ceria's adsorptive sites. Ceria nanoparticles alone have a surface area of 37.2 ± 0.2 m2 g−1, but when added to the surface of the fibers, they impart a negligible change in the nanocomposite surface are (which, as mentioned previously, increases from 27.1 ± 0.2 m2 g−1 to 27.2 ± 0.1 m2 g−1 after ceria immobilization). In addition, by anchoring ceria on the fibers, ceria's contact with contaminants is limited because the ceria nanoparticles now cannot move freely in suspension, limiting their number of interactions with contaminants of concern.
Our results collectively suggest that although immobilized ceria nanoparticles can serve as an efficient oxidative media in acidic conditions, they have limited adsorption performance. For practical application, ceria nanoparticles could be coupled with a conventional adsorbent (e.g., activated carbon), strategically imparting oxidative capabilities on materials typically known for their singular adsorptive capability.
Both oxidative and adsorptive performances of the electrospun ceria/silica nanofiber composites are maximized in acidic conditions, limiting the applicability for water decontamination. However, such a system should be considered not only when pH can be adjusted for optimal treatment, but also for niche applications, such as treatment of contaminated acidic waters. For example, wastewaters that may benefit from decontamination at low pH are acid mine drainage (with mostly inorganic contamination and some secondary organic pollution)55,56 as well as some industrial wastes such as metal plating57 and dairy wastes58 laden with veterinary antibiotics59,60 and detergents.61 To increase the feasibility and potential of these fibers in treating a variety of contaminants (as opposed to only organic contaminants, as studied herein), future studies are recommended in the use and regeneration of these fibers in the context of inorganic pollutant removal. The concentration of H2O2 (i.e., 0.5 mM) needed to oxidize pollutants (at a concentration of ∼5 μM) is within the lower range of concentrations used for a typical H2O2/UV advanced oxidation process, which is dictated by the low molar absorption coefficient of H2O2 as well as the need to overcome scavenging effects by constituents in the treated waters.62
3.4. Reuse and regeneration of electrospun ceria/silica nanofiber composites
Using CBZ as a model TrOC, experiments were conducted to determine the oxidative performance of the composite fibers after multiple oxidation cycles and regeneration. The ceria/silica nanofiber composites could be reused for oxidation many times, with only an approximate 15% drop in oxidative performance after 10 cycles (Fig. 5A). The fibers were then regenerated by facile heating at 550 °C in air for one hour. After regeneration, the fibers regained their initial oxidative performance (99% recovery). This performance is quite impressive, as multiple oxidation cycles cause very little drop in performance (allowing much more treatment of water before eventual regeneration) and the fibers immediately return to their initial performance after regeneration. Because of the high energy requirements for heating the fibers to 550 °C, other regeneration options should be considered, such as pH change, as has been illustrated by desorption of cationic organics from negatively-charged silicate surfaces.63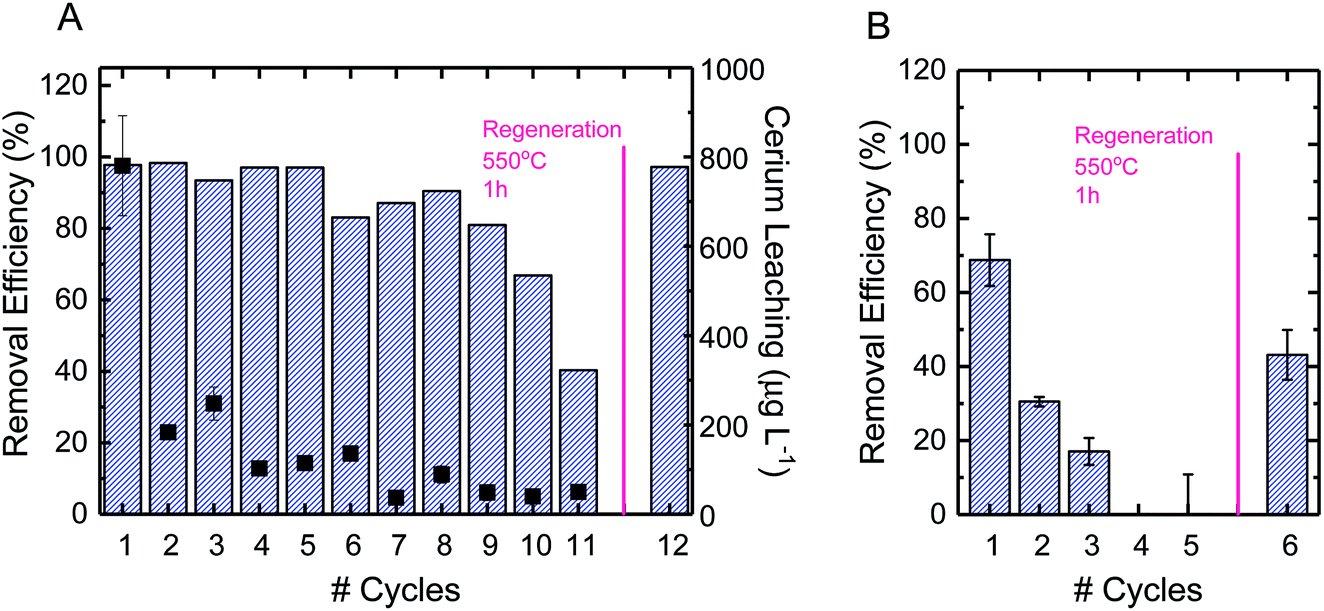 | ||
| Fig. 5 (A) Reuse cycles of electrospun ceria/silica nanofiber composites for carbamazepine oxidation in the presence of 0.5 mM H2O2 (bars). The right axis shows leaching of cerium from the nanofiber composites to the solution (solid squares). (B) Reuse cycles of nanofiber composites for perfluorooctanesulfonic acid adsorption in the absence of H2O2 (bars). In each cycle, 1 mg L−1 pollutant solution at pH 2 (10 mM buffer acetate) was added to nanofiber composites for a final concentration of 1 mg nanoceria per mL of treated solution for 2 hours of mixing. The figure also shows nanofiber composite recovery following a thermal regeneration at 550 °C in air for one hour. | ||
Using PFOS as a model TrOC, experiments were also conducted to determine the reuse of the composite fibers over multiple cycles of adsorption as well as after regeneration. As opposed to the multiple cycles of oxidative use before necessary regeneration, only three cycles of adsorption were possible before regeneration was necessary (Fig. 5B). This difference in time required before regeneration is due to the consumption of adsorption sites on ceria nanoparticles. When oxidation is the predominant mechanism of removal, as opposed to adsorption, the reactive sites continue to remain effective after use. However, when a contaminant sorbs to an adsorption site, that sorption site cannot be used in future adsorption. This explains the decrease in adsorption by approximately 50% after each subsequent cycle. Regeneration of the fibers after adsorption, however, can be accomplished similarly to regeneration after oxidation (i.e., heating to 500 °C in air for one hour).
Leaching of cerium ions from the fibers was also evaluated over the course of multiple oxidative cycles of reuse. The first cycle of oxidation incurred a leaching of 800 μg L−1, which was approximately 5% of the total ceria on the surface of the composite fibers (Fig. 5A). There are currently no federal standards for cerium leaching,64,65 so further studies are recommended to elucidate the effect of this level of leaching. After repeated cycles, however, the leaching per cycle decreased to less than 1%. In practice, a possible way to avoid the higher ceria leaching of the first cycle is to soak the fresh composite fibers in DI water before beginning oxidative cycles.
4. Conclusions
This study proposes the design of regenerable nanoceria/silica nanofibers for decontamination of water through both Fenton-like oxidation reactions and adsorption. In ideal batch conditions (determined using ceria nanoparticles alone), the nanocomposite fibers achieve 95% and 98% oxidation of positively- and neutrally-charged model trace organic compounds, respectively. Over the course of 10 oxidation cycles using the nanoceria/silica nanofibers, performance drops by less than 15%, and after heating, is brought back to the levels of initial performance. Using a different mechanism, i.e. through adsorption, these fibers also achieve more than 65% removal of a negatively-charged model trace organic compound (perfluorooctanesulfonic acid) through electrostatic attraction. After multiple adsorptive cycles, the same regenerative process as with oxidation can be used to reclaim the fibers for additional adsorption cycles. These fibers therefore prove themselves as multifunctional, regenerable nanocomposites, able to both oxidize and/or adsorb multiple trace organic compounds of concern.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Science Foundation (NSF) Nanosystems Engineering Research Center for Nanotechnology-Enabled Water Treatment (Grant EEC-1449500). Facility use was supported by Facility for Light Scattering, YINQE, and West Campus Analytical Core. The authors would like to thank Dr Chiheng Chu, Prof. Jea-Hong Kim, and Dr Igal Gozlan for their support with HPLC analysis and Dr Amanda Lounsbury and Danielle Lee for their advice and help with data analysis. BET measurements were conducted at the Particle Testing Authority in Munich by Dr Torben Sick as well as Adi Ben-Yaakov and Omer Shenkman, both representatives of Dr Golik Scientific Solutions. We also acknowledge the YIBS Postdoctoral Fellowship and Tel Aviv University Presidential Postdoctoral Fellowship awarded to I. Z. and the Scientific and Technological Research Council of Turkey (TUBITAK) Department of Science Fellowships and Grant Programs (1059B191501137) for financially supporting N. D.Notes and references
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Mariñas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef CAS PubMed.
- Y. Luo, W. Guo, H. H. Ngo, L. D. Nghiem, F. I. Hai, J. Zhang, S. Liang and X. C. Wang, Sci. Total Environ., 2014, 473–474, 619–641 CrossRef CAS PubMed.
- USEPA, Integrated Risk Information System (IRIS), online database, 2014 Search PubMed.
- M. M. Huber, S. Canonica, G. Y. Park and U. von Gunten, Environ. Sci. Technol., 2003, 37, 1016–1024 CrossRef CAS PubMed.
- A. Babuponnusami and K. Muthukumar, J. Environ. Chem. Eng., 2014, 2, 557–572 CrossRef CAS.
- M. S. Mauter, I. Zucker, F. Perreault, J. R. Werber, J. Kim and M. Elimelech, Nat. Sustain., 2018, 1, 166–175 CrossRef.
- M. Aleksić, H. Kušić, N. Koprivanac, D. Leszczynska and A. L. Božić, Desalination, 2010, 257, 22–29 CrossRef.
- P. Ni, X. Wei, J. Guo, X. Ye and S. Yang, RSC Adv., 2015, 5, 97512–97519 RSC.
- E. Mamontov, T. Egami, R. Brezny, M. Koranne and S. Tyagi, J. Phys. Chem. B, 2000, 104, 11110–11116 CrossRef CAS.
- S. Deshpande, S. Patil, S. V. Kuchibhatla and S. Seal, Appl. Phys. Lett., 2005, 87, 1–3 CrossRef.
- Y. Zhou, Nanostructured cerium oxide based catalysts: synthesis, physical properties, and catalytic performance, PhD thesis, University of Nebraska, 2015.
- E. Grulke, K. Reed, M. Beck, X. Huang, A. Cormack and S. Seal, Environ. Sci.: Nano, 2014, 1, 429–444 RSC.
- K. Reed, A. Cormack, A. Kulkarni, M. Mayton, D. Sayle, F. Klaessig and B. Stadler, Environ. Sci.: Nano, 2014, 1, 390–405 RSC.
- A. Kumar, S. Das, P. Munusamy, W. Self, D. R. Baer, D. C. Sayle and S. Seal, Environ. Sci.: Nano, 2014, 1, 516–532 RSC.
- S. S. Lee, W. Song, M. Cho, H. L. Puppala, P. Nguyen, H. Zhu, L. Segatori and V. L. Colvin, ACS Nano, 2013, 7, 9693–9703 CrossRef CAS.
- B. Nelson, M. Johnson, M. Walker, K. Riley and C. Sims, Antioxidants, 2016, 5, 15 CrossRef.
- J. M. Perez, A. Asati, S. Nath and C. Kaittanis, Small, 2008, 4, 552–556 CrossRef CAS PubMed.
- C. Xu and X. Qu, NPG Asia Mater., 2014, 6, 1–16 Search PubMed.
- F. Chen, X. Shen, Y. Wang and J. Zhang, Appl. Catal., B, 2012, 121–122, 223–229 CrossRef CAS.
- M. A. Asati, D. S. Santra, M. C. Kaittanis, D. S. Nath and P. J. M. Perez, Angew. Chem., 2009, 48, 2308–2312 CrossRef PubMed.
- M. Taguchi, S. Takami, T. Naka and T. Adschiri, Cryst. Growth Des., 2009, 9, 5297–5303 CrossRef CAS.
- P. Yu, S. A. Hayes, T. J. O'Keefe, M. J. O'Keefe and J. O. Stoffer, J. Electrochem. Soc., 2006, 153, C74–C79 CrossRef CAS.
- F. Rollin-Genetet, C. Seidel, E. Artells, M. Auffan, A. Thiéry and C. Vidaud, Chem. Res. Toxicol., 2015, 28, 2304–2312 Search PubMed.
- V. Shah, S. Shah, H. Shah, F. J. Rispoli, K. T. McDonnell, S. Workeneh, A. Karakoti, A. Kumar and S. Seal, PLoS One, 2012, 7, e47827 CrossRef CAS PubMed.
- A. Asati, S. Santra, C. Kaittanis and J. M. Perez, ACS Nano, 2010, 4, 5321–5331 CrossRef CAS PubMed.
- L. S. Zhong, J. S. Hu, A. M. Cao, Q. Liu, W. G. Song and L. J. Wan, Chem. Mater., 2007, 19, 1648–1655 CrossRef CAS.
- J. M. Coronado, A. Javier Maira, A. Martínez-Arias, J. C. Conesa and J. Soria, J. Photochem. Photobiol., A, 2002, 150, 213–221 CrossRef CAS.
- Q. Cui, X. Dong, J. Wang and M. Li, J. Rare Earths, 2008, 26, 664–669 CrossRef.
- S. Homaeigohar and M. Elbahri, Materials, 2014, 7, 1017–1045 CrossRef CAS PubMed.
- Z. M. Huang, Y. Z. Zhang, M. Kotaki and S. Ramakrishna, Compos. Sci. Technol., 2003, 63, 2223–2253 CrossRef CAS.
- E. Shaulsky, S. Nejati, C. Boo, F. Perreault, C. O. Osuji and M. Elimelech, J. Membr. Sci., 2017, 530, 158–165 CrossRef CAS.
- G. Andreas and W. H. Joachim, Angew. Chem., 2007, 46, 5670–5703 CrossRef PubMed.
- A. F. de Faria, F. Perreault, E. Shaulsky, L. H. Arias Chavez and M. Elimelech, ACS Appl. Mater. Interfaces, 2015, 7, 12751–12759 CrossRef CAS PubMed.
- L. Zhi, W. Zuo, F. Chen and B. Wang, ACS Sustainable Chem. Eng., 2016, 4, 3398–3408 CrossRef CAS.
- R. Flyunt, A. Leitzke, G. Mark, E. Mvula, E. Reisz, R. Schick and C. von Sonntag, J. Phys. Chem. B, 2003, 107, 7242–7253 CrossRef CAS.
- T. Nash, Biochem. J., 1953, 55, 416–421 CrossRef CAS PubMed.
- T. Nöthe, H. Fahlenkamp and C. von Sonntag, Environ. Sci. Technol., 2009, 43, 5990–5995 CrossRef.
- K. M. Buettner, C. I. Rinciog and S. E. Mylon, Colloids Surf., A, 2010, 366, 74–79 CrossRef CAS.
- G. V. Franks, J. Colloid Interface Sci., 2002, 249, 44–51 CrossRef CAS PubMed.
- Z. V. Feng, W. S. Chen, K. Keratithamkul, M. Stoick, B. Kapala, E. Johnson, A. C. Huang, T. Y. Chin, Y. W. Chen-Yang and M. L. Yang, Int. J. Nanomed., 2016, 11, 729–741 CAS.
- D. Mukherjee, B. G. Rao and B. M. Reddy, Top. Catal., 2017, 60, 1673–1681 CrossRef CAS.
- M. C. Matos, L. M. Ilharco and R. M. Almeida, J. Non-Cryst. Solids, 1992, 147–148, 232–237 CrossRef CAS.
- K. M. S. Khalil, H. A. Mahmoud and T. T. Ali, Langmuir, 2008, 24, 1037–1043 CrossRef CAS PubMed.
- A. Singh and C. A. Hogarth, J. Mater. Sci., 1988, 23, 1090–1097 CrossRef CAS.
- C. Ho, J. C. Yu, T. Kwong, A. C. Mak and S. Lai, Chem. Mater., 2005, 4514–4522 CrossRef CAS.
- P. Munusamy, S. Sanghavi, T. Varga and T. Suntharampillai, RSC Adv., 2014, 4, 8421–8430 RSC.
- Y. Lin, C. Xu, J. Ren and X. Qu, Angew. Chem., 2012, 51, 12579–12583 CrossRef CAS PubMed.
- E. Abdelkader, L. Nadjia, B. Naceur, B. Boukoussa, A. Mohamed, Advances in Science, Technology & Innovation, 2018, pp. 129–132 Search PubMed.
- C. Dai, X. Zhou, Y. Zhang, Y. Duan, Z. Qiang and T. C. Zhang, Environ. Technol., 2012, 33, 1101–1109 CrossRef CAS PubMed.
- E. G. Heckert, S. Seal and W. T. Self, Environ. Sci. Technol., 2008, 42, 5014–5019 CrossRef CAS PubMed.
- M. Sun, G. Zhang, Y. Qin, M. Cao, Y. Liu, J. Li, J. Qu and H. Liu, Environ. Sci. Technol., 2015, 49, 9289–9297 CrossRef CAS PubMed.
- C. Korsvik, S. Patil, S. Seal and W. T. Self, Chem. Commun., 2007, 1056–1058 RSC.
- Q. Yu, R. Zhang, S. Deng, J. Huang and G. Yu, Water Res., 2009, 43, 1150–1158 CrossRef CAS PubMed.
- D. Brooke, A. Footitt, T. Nwaogu and G. Britain, Environmental risk evaluation report: Perfluorooctanesulphonate (PFOS), 2004 Search PubMed.
- K. K. Kefeni, T. A. M. Msagati and B. B. Mamba, J. Cleaner Prod., 2017, 151, 475–493 CrossRef CAS.
- P. Gao, Y. Ding, H. Li and I. Xagoraraki, Chemosphere, 2012, 88, 17–24 CrossRef CAS PubMed.
- M. Singh and S. Verghese, Int. J. Eng. Sci. Res. Technol., 2016, 5, 150–159 CAS.
- A. K. Slavov, Food Technol. Biotechnol., 2017, 55, 14–28 CAS.
- M. O. Aust, F. Godlinski, G. R. Travis, X. Hao, T. A. McAllister, P. Leinweber and S. Thiele-Bruhn, Environ. Pollut., 2008, 156, 1243–1251 CrossRef CAS PubMed.
- N. Wang, X. Guo, J. Xu, X. Kong, S. Gao and Z. Shan, J. Environ. Sci. Health, Part B, 2014, 49, 468–479 CrossRef CAS PubMed.
- J. R. Danalewich, T. G. Papagiannis, R. L. Belyea, M. E. Tumbleson and L. Raskin, Water Res., 1998, 32, 3555–3568 CrossRef CAS.
- H. Juretić, G. Smoljanić and M. Barta, Holistic Approach Environ., 2015, 5, 135–149 Search PubMed.
- C. Karaguzel and Z. Xu, Physicochem. Probl. Miner. Process., 2017, 53, 750–757 CAS.
- J. G. Clar, W. E. Platten, E. J. Baumann, A. Remsen, S. M. Harmon, C. L. Bennett-Stamper, T. A. Thomas and T. P. Luxton, Sci. Total Environ., 2018, 613–614, 714–723 CrossRef CAS.
- EPA, Toxicological review of cerium oxide and cerium compounds, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03467f |
| This journal is © The Royal Society of Chemistry 2019 |