
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of novel phenylaminopyrimidines with antiproliferative activity against colorectal cancer†
Hanan A. Henidia,
Ahmed M. Al-Abd *bc,
Fahad A. Al-Abbasia,
Hawazen A. BinMahfouza and
Ibrahim M. El-Deeb*de
*bc,
Fahad A. Al-Abbasia,
Hawazen A. BinMahfouza and
Ibrahim M. El-Deeb*de
aDepartment of Biochemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
bDepartment of Pharmaceutical Sciences, College of Pharmacy, Gulf Medical University, Ajman, UAE
cPharmacology Department, Medical Division, National Research Centre, Giza, Egypt. E-mail: ahmedmalabd@pharma.asu.edu.eg
dRoyal College of Surgeons in Ireland-Medical University of Bahrain, Bahrain
eInstitute for Glycomics, Griffith University, Gold Coast, Queensland, Australia. E-mail: i.el-deeb@griffith.edu.au
First published on 11th July 2019
Abstract
New phenylaminopyrimidine (PAP) derivatives have been designed and synthesised as potential tyrosine kinase inhibitors for the treatment of cancer. The synthesized compounds share a general structure and vary in the substitution pattern at position-2 of the pyridine ring. Several derivatives have demonstrated potent anticancer activities against HCT-116, HT-29 and LS-174T colorectal cancer cells. Furthermore, a number of hits showed good selectivity to Src-kinase. The cytotoxic mechanisms of these compounds were also investigated by studying their effects on cell-cycle distribution. Among all the compounds examined, compound 8b (with a terminal pyridin-3-yl moiety at the pyridine ring) showed the highest inhibitory selectivity towards src-kinase, which was coupled with cell cycle arrest, and apoptotic and autophagic interference, in colorectal cancer cells. This report introduces a novel category of PAP derivatives with promising kinase inhibitory and anticancer effects against colon cancer.
Introduction
Cancer is a major health problem worldwide and is the second leading cause of death.1 According to the Saudi Cancer Registry in 1994, the incidence of all cancers, including colorectal cancer, has been showing a steady state increase over the past two decades.2 In addition, colorectal cancer is the most common malignancy among males and the third most common cancer among females in Saudi Arabia, with an overall five-year survival rate of 44.6%.3Targeting oncogenic receptor tyrosine kinases (RTKs) has attracted considerable attention in development of novel cancer therapeutics due to their advantages over classical anticancer drugs such as selectivity, reduced toxicity, and enhanced permeation/retention in cancer tissues.4 In addition to cancer, several diseases' pathologic underlying reasons are connected to protein kinase dysfunction.5 Accordingly, the ability to modulate protein kinase activity represents an attractive therapeutic strategy for the treatment of many human illnesses.4,5 A considerable number of receptor tyrosine kinases was found to be involved in the pathologic background of many malignancies, and particularly colorectal cancer. Clinical studies revealed that the overexpression/deregulation of many receptor tyrosine kinases is of prognostic and tumour aggression predictive value in cancer patients.6
Phenylaminopyrimidines (PAPs) represent a large chemical group of anticancer kinase inhibitors. A representative example and a lead compound for such group is the Bcr-Abl tyrosine kinase inhibitor, imatinib (1, Gleevec, STI-571).7 Imatinib (Fig. 1), was the first kinase inhibitor approved for the treatment of cancer. After the successful approval of imatinib as a selective anticancer drug for treatment of chronic myeloid leukaemia (CML) with minimal side effects, in comparison to classical anticancer drugs, numerous kinase inhibitors targeting oncogenic kinases have been developed. A large number of these kinase inhibitors belong to the same chemical group of imatinib, PAPs. These compounds include inhibitors for several kinases, such as Bcr-Abl,8,9 CDK,10 c-KIT,11 EGFR,9,12 Lyn9 and Src family13 kinases. In addition to receptor tyrosine kinases, some anticancer PAPs modulate other13 receptors such as the angiopoietin Tie2 receptors.14
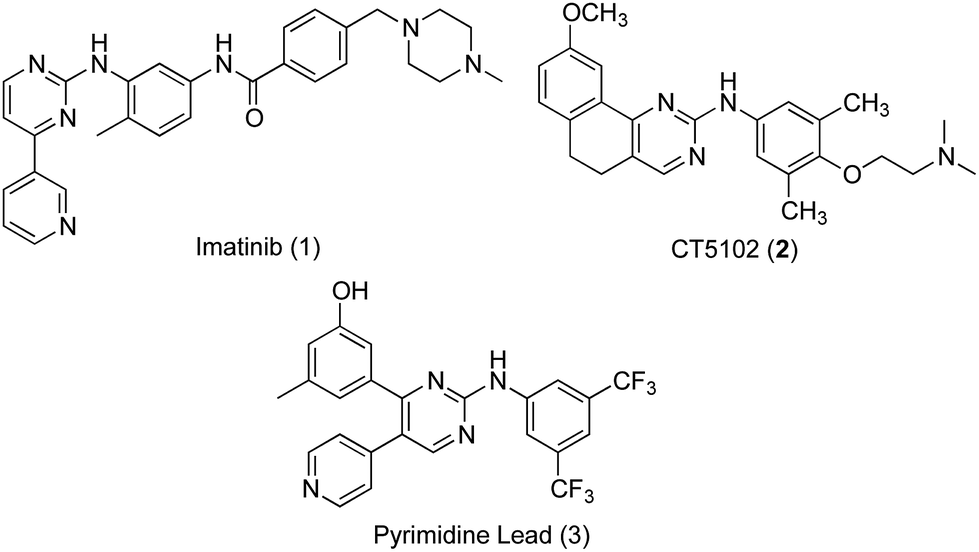 | ||
| Fig. 1 Structures of lead inhibitors, imatinib (1), CT5102 (2), and the PAP lead 3. | ||
Src subfamily of tyrosine kinases consists of several members such as c-Src, which is involved in colorectal cancer progression and metastasis. Several small molecules targeting Src kinases are under preclinical and clinical investigation for cancer treatment, including the PAP derivative CT5102, 2.13 The authors have previously reported the discovery of a new phenylaminopyrimidine (PAP) lead (3) with high potency and efficacy as an anticancer agent.15 This lead hit has showed good potency over a range of cancer cell lines, combined with remarkable efficacy expressed by low micromolar values for total tumour growth inhibition and 50% tumour regression. In this study, we further modified the structure of this anticancer lead through extended substitution at the pyridin-4-yl ring. A variety of substituted and unsubstituted aromatic structures have been attached to this point of the molecule through a series of Suzuki coupling reactions16,17 in order to properly evaluate the effect of this extended substitution on compound's activity (Fig. 2). The new inhibitors were tested for their capacity to inhibit the growth of colon cancer cells through inhibition of tyrosine kinase activity and the underlying mechanisms of their tumour cell-killing properties were also investigated.
 | ||
| Fig. 2 General structure of the novel PAP inhibitors derived from the structures of the lead inhibitor 3. | ||
Results
Synthesis of target compounds
The synthesis of the target compounds started with the key ester methyl 3-methoxy-5-methylbenzoate (4), which was prepared following reported procedures.16 As showed in Scheme 1, the benzoate ester 4 underwent a nucleophilic attack at its carboxylate carbon by the activated methylene group of 2-chloro-4-methylpyridine. The activation of this methyl group into an active methylene was achieved by the dropwise addition of lithium bis(trimethylsilyl)amide (LHMDS) in dry THF at room temperature. The resulted ketone 5 (obtained at 71% yield) was then converted to the required pyrimidin-2-amine derivative 6 through two successive steps. In the first step, compound 5 was heated with excess N,N-dimethylformamide dimethylacetal for 12 hours, and the resulted product was taken to the next step without further purification, where it was allowed to react with guanidine-hydrochloride in absolute ethanol and in the presence of sodium ethoxide, to undergo cyclization into the pyrimidine derivative 6 in an overall yield of 58% (over two steps).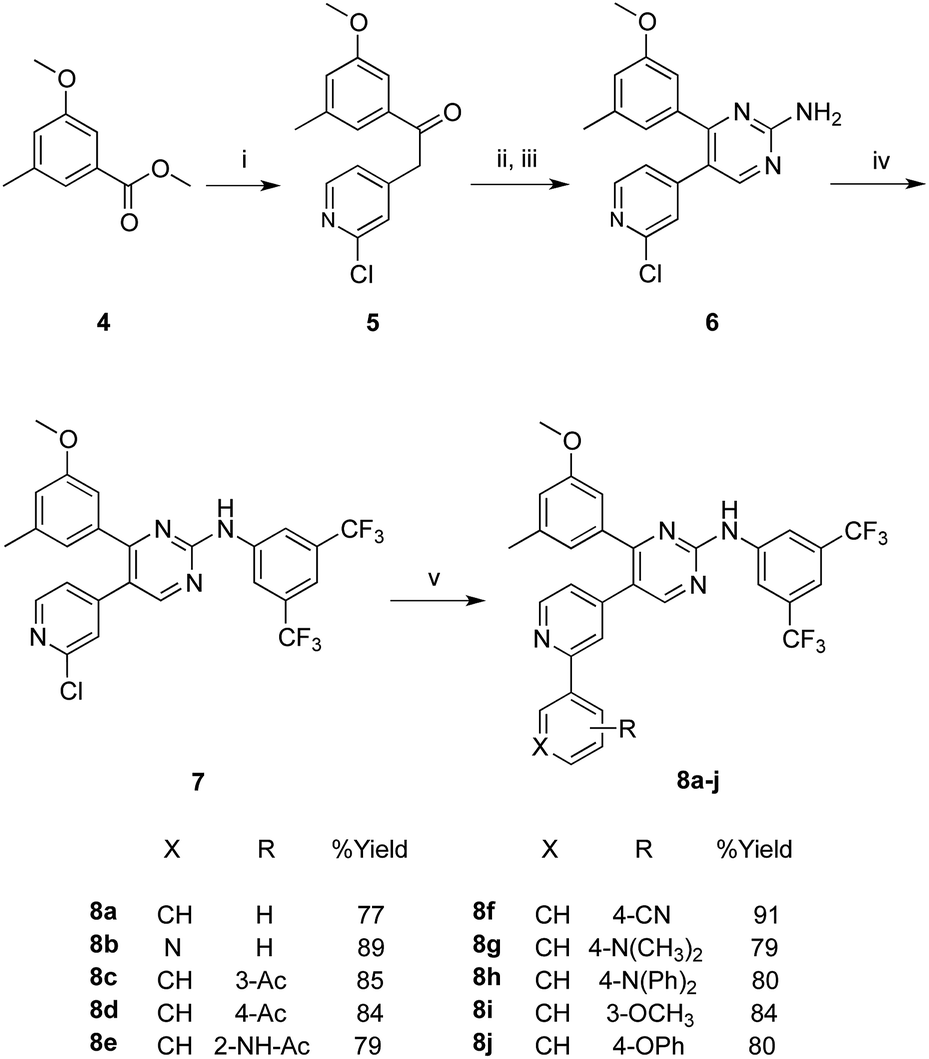 | ||
Scheme 1 Synthesis of target PAP derivatives 8a–j. Reaction conditions and yields: (i) 2-chloro-4-methylpyridine, LHMDS, THF, N2, rt, 18 h, 72%; (ii) DMF-DMA, reflux, 12 h; (iii) NaOEt, guanidine-HCl, abs. EtOH, reflux, 18 h, 58%; (iv) 3,5-bis(trifluoromethyl)phenyl bromide, Pd(PPh3)2Cl2, xantphos, NaOtBu, toluene, reflux, 12 h, 69%; (v) arylboronic acid, Pd(PPh3)2Cl2, K2CO3, N2, THF/H2O; (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 70 °C, 20 h. :1), 70 °C, 20 h. | ||
Arylation of the amino group of the resulted pyrimidine-2-amine (6) with 3,5-bis(trifluoromethyl)phenyl bromide, to obtain the key PAP intermediate 7 was achieved in a good yield (69%) applying optimized Buchwald–Hartwig amination protocol.18 The produced PAP intermediate 7 was then subjected to a series of Suzuki coupling reactions with a group of arylboronic acids, in the presence of dichloro bis(triphenylphosphine)Pd(II) and potassium carbonate, in a mixed solvent of THF and water in a (4:1) ratio at 70 °C, to yield the final PAP derivatives 8a–j in good yields.
In Scheme 2, the methoxy groups in the PAP derivatives 7 and 8a–j were demethylated under the effect of BF3–dimethylsulfide complex, in dichloromethane at room temperature, to produce the hydroxyl analogues 9 and 10a–j, respectively in good yields.
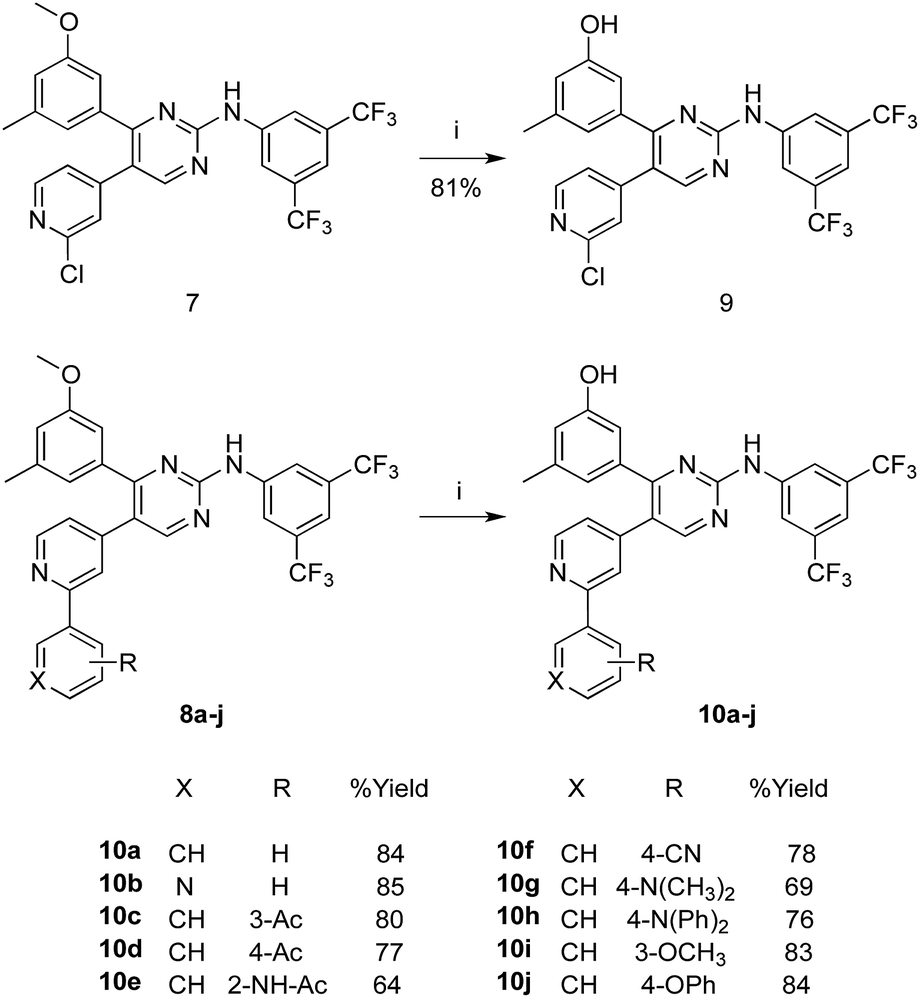 | ||
| Scheme 2 Synthesis of the hydroxyl analogues 9 and 10a–j. Reaction conditions and yields: (i) BF3S(CH3)2, CHCl3, N2, rt, 48 h. | ||
Tyrosine kinase inhibition assessment
The capacity of the synthesized compounds to inhibit protein kinases was assessed using a mixture of tyrosine kinases extracted from HT-29 cell line.At 10 μM concentration, compounds 7, 8a, 8b, 8g, 8j, 9, 10a, 10f, 10g and 10j showed multi-kinase inhibitory effects by 37.4 ± 3.7%, 23 ± 0.4%, 26.6 ± 2.4%, 33.1 ± 0.4%, 26 ± 0.5%, 30.4 ± 2.3%, 24.7 ± 4.6%, 23.9 ± 7.4%, 39 ± 0.4% and 30.2 ± 0.4%, respectively. The rest of the series did not show more than 16.5% overall inhibition against multi-kinase cellular extract (Fig. 3A).
 | ||
| Fig. 3 The effect of test compounds at 10 μM concentration against the activity of total intracellular tyrosine kinase (A) and purified Src kinase (B). Data are expressed as mean ± SD; n = 3. | ||
Further analysis for specific Src-kinase inhibitory potency was undertaken to determine compounds' specificity. Compounds 7, 8a, 8b, 8g, 8j, 9 and 10g showed considerable Src kinase inhibitions, at 10 μM concentration, by 42.3 ± 10.3%, 54 ± 0.6%, 77.8 ± 1.3%, 28.5 ± 1.2%, 54.9 ± 1.6%, 33.6 ± 3.3% and 51.3 ± 9.8% inhibitions, respectively. The rest of the compounds under investigation did not show more than 14.7% inhibition for Src kinase (Fig. 3B). As a measure for selectivity towards Src kinase, Src kinase selectivity index was used. Src kinase selectivity index is the ratio between Src kinase inhibitions to the non-specific multi-kinase inhibitions of compounds under investigation. Compounds 8a, 8b, 8j and 10g showed Src kinase indices of 2.4, 2.9, 2.1 and 3.1, respectively.
Detailed dose–response curves for compounds 7, 8b, 9, 10a, 10b, 10f and 10g as Src kinase inhibitors were plotted (Fig. 4). Compounds 8b and 10g showed potent src kinase inhibitions with EC50's of 1.3 ± 0.3 and 3.7 ± 0.7 μM, respectively. On the other hand, compounds 10a, 10b and 10f were completely inactive in inhibiting src kinase, while compounds 7 and 9 showed moderate inhibitory activity.
 | ||
| Fig. 4 Dose–response relationship of selected compounds against the activity of purified Src kinase enzyme. Data are expressed as mean ± SD; n = 3. | ||
Molecular modelling
In order to predict the potential binding mode of these inhibitors to kinases, inhibitor 8b was used as a model. A molecular docking simulation was applied for inhibitor 8b into the co-crystal structure of src kinase (PDB 4MXO19), using MOE.2008.10 software. The compound was docked over the ligand (bosutinib) atoms in the binding site using the software standard docking parameters, and the most stable docking pose of 8b was selected as showed in Fig. 5. | ||
| Fig. 5 Molecular docking simulation of compound 8b into src kinase co-crystal protein structure 4MXO. Upper panel: a 3D presentation of the most stable docking pose of 8b showing proposed HB interaction between the terminal pyridine nitrogen and glutamate 353. Lower panel: a 2D presentation for the docked structure. | ||
As showed in the figure, the more hydrophobic part of compound 8b skeleton (bis-3,5-(trifluoromethyl)phenyl moiety) was oriented towards the depth of the pocket, while the terminal pyridine ring (which represents the varied part of the skeleton within the whole series) points towards the protein surface. Interestingly, a hydrogen bonding (HB) interaction was predicted between the terminal pyridine nitrogen and glutamic acid residue 353.
Antiproliferative/cytotoxicity assessment
Tyrosine kinase is an intracellular enzyme and inhibiting the enzyme in cell free system is not necessarily interpreted as a cytotoxic effect. Therefore, preliminary assessment of the potential antiproliferative/cytotoxic properties of the whole series under investigation is a key step in short-listing these compounds for more detailed biological assessment. Accordingly, HCT-116, HT-29 and LS-174T colorectal cancer (CRC) cell lines were exposed to 10 and 100 μM concentrations of the compounds under investigation for 72 h and viabilities were compared to control untreated cells. Only compounds 8h and 10h failed to show any considerable antiproliferative/cytotoxic effects even at 100 μM against all colorectal cancer cell lines used (Fig. 6A–C).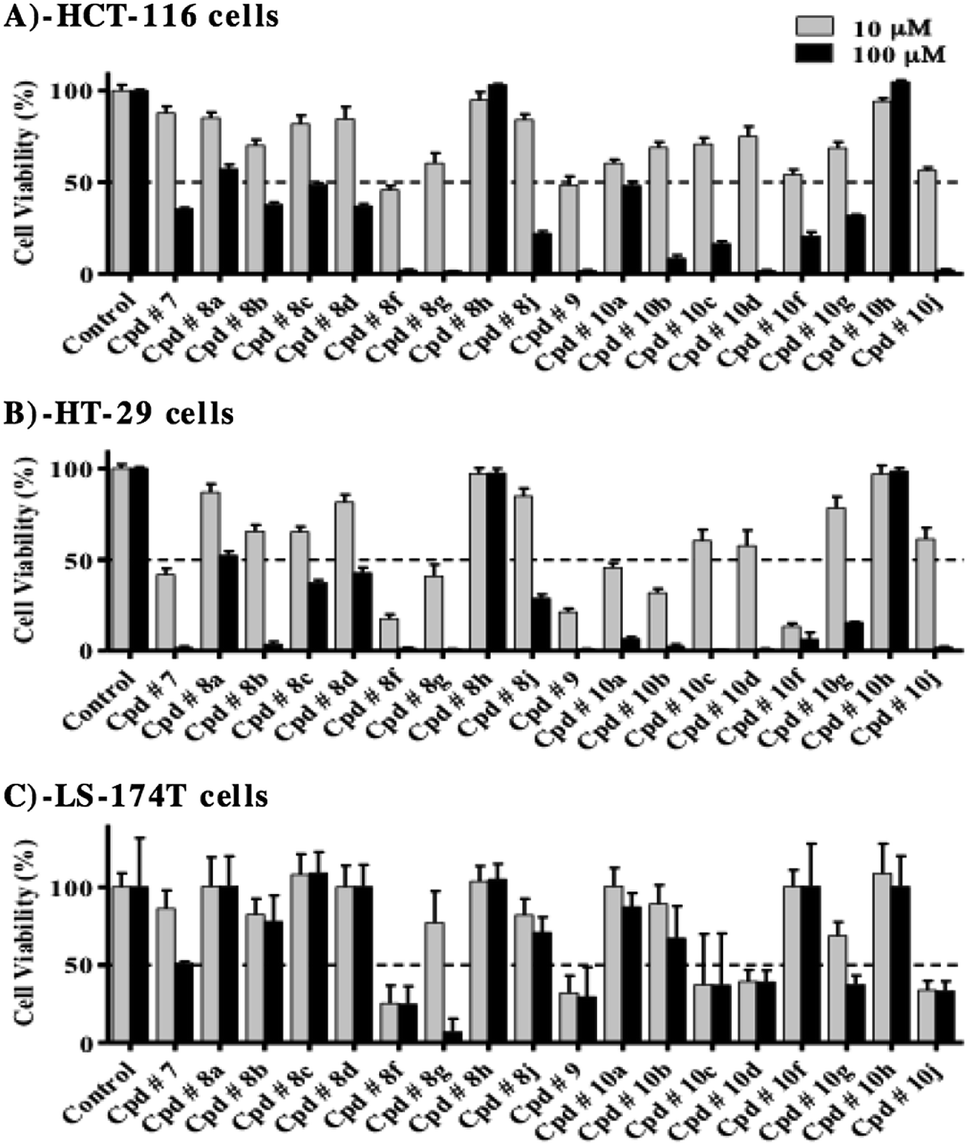 | ||
| Fig. 6 Effect of compounds under investigation on the viability of HCT-116 (A), HT-29 (B) and LS-174T colorectal cancer cells (C). Cells were treated with 10 μM and 100 μM of test compounds for 72 h and viability was determined using SRB assay. Data are expressed as mean ± SD; n = 6. | ||
In HCT-116 cells, all test compounds, except for 8h and 10h, induced viability drop by more than 50% at 100 μM, compared to control cells (Fig. 6A). Furthermore, compounds, 8f and 9 exhibited more than 50% drop in viability at 10 μM concentration, while at this concentration, compounds 8b, 8g, 10a, 10b, 10c, 10f, 10g and 10j showed viability drop by 30–45% compared to control cells.
In HT-29 cells, all compounds under investigation, except for 8h and 10h, reduced cell viability, at 100 μM concentration, by more than 50% compared to control cells (Fig. 6B). At 10 μM concentration, compounds, 7, 8f, 8g, 9, 10a, 10b and 10f induced more than 50% drop in viability, while compounds 8b, 8c, 10c, 10d and 10j reduced cell viability by 35–45%, compared to control cells.
LS-174T was the most resistant cell line among the colorectal cancer cells used for evaluation, where at 100 μM; only compounds 7, 8f, 8g, 10c, 10d, 10g and 10j induced viability drop by more than 50%, compared to control cells (Fig. 6C). In addition, only compounds 8f, 9, 10c, 10d and 10j were able to reduce viability by more than 50% at 10 μM concentration.
Detailed dose–response relationship against colorectal cancer cells
After evaluation of multi-tyrosine kinase inhibition, selective Src kinase inhibition and the preliminary antiproliferative/cytotoxic effects of the synthesized compounds, it was important to construct dose–response curves of the most potent compounds 7, 8b, 9, 10a, 10b, 10f and 10g to calculate accurate IC50 values against HCT-116 and HT-29 cell lines. Furthermore, detailed cytotoxic parameters were also deduced. Compounds 9, 10a and 10f showed the most potent cytotoxic effects against both cell lines with IC50's ranging from 7.2 ± 1.1 μM to 10.3 ± 0.6 μM and resistance fraction less than 1% (Table 1, Fig. 7C, D and F).| HCT-116 | HT-29 | |||
|---|---|---|---|---|
| IC50 (μM) | R (%) | IC50 (μM) | R (%) | |
| Cpd # 7 | 83.3 ± 5.8 | N/A | 9.4 ± 0.5 | 3.9 ± 1.3 |
| Cpd # 8b | 32.4 ± 2.8 | 3.7 ± 0.9 | 11.3 ± 0.5 | <1 |
| Cpd # 9 | 9.4 ± 1.2 | <1 | 7.5 ± 0.5 | <1 |
| Cpd # 10a | 10.3 ± 0.6 | <1 | 9.9 ± 0.3 | <1 |
| Cpd # 10b | 25.9 ± 1.3 | <1 | 6.4 ± 0.5 | <1 |
| Cpd # 10f | 9.2 ± 0.8 | <1 | 7.2 ± 1.1 | <1 |
| Cpd # 10g | 9.4 ± 0.9 | 41.6 ± 3.2 | 24.2 ± 2.1 | 12.9 ± 3.9 |
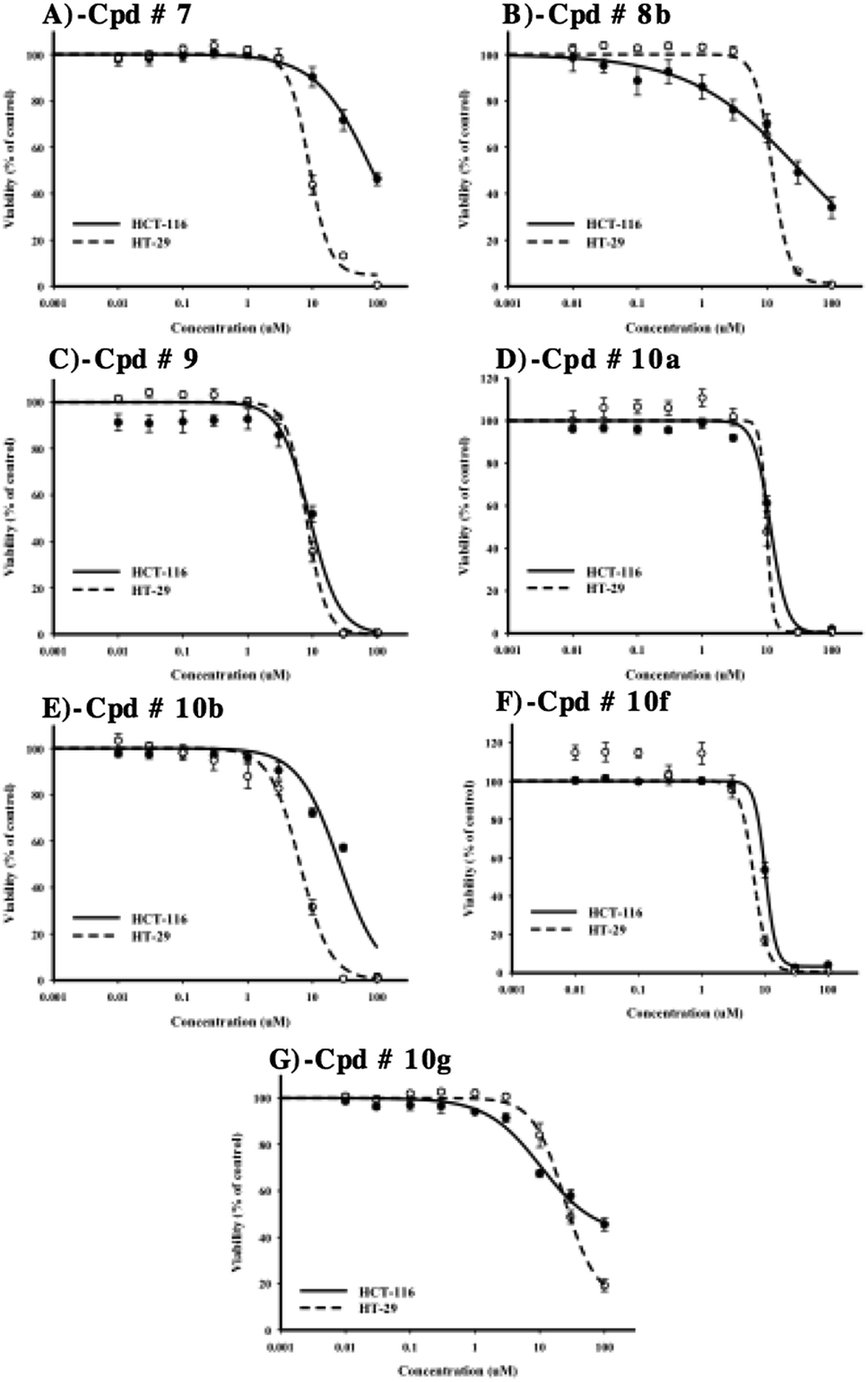 | ||
| Fig. 7 Dose–response assessment for compounds 7 (A), 8b (B), 9 (C), 10a (D), 10b (E), 10f (F) and 10g (G) against HCT-116 (solid lines) and HT-29 cells (dotted lines). Cells were treated with test compounds for 72 h and viability was determined using SRB assay. Data are expressed as mean ± SD; n = 6. | ||
Compounds 7, 8b and 10b have also showed good antiproliferative/cytotoxic effects in HT-29 with IC50's of 9.4 ± 0.5, 11.3 ± 0.5 and 6.4 ± 0.5 μM, respectively (Fig. 7A, B and E). The resistance fractions of HT-29 cells towards these compounds were less than 5% (Table 1). On the other hand, compounds 7, 8b and 10b showed significantly weaker antiproliferative/cytotoxic effects against HCT-116 cells with IC50's higher than 25 μM (Fig. 7A, B and E). Interestingly, compound 10g showed the weakest antiproliferative/cytotoxic profile among the selected compounds with both high IC50 value against HT-29 cells (24.2 ± 2.1 μM) and very high resistance fraction (41.6 ± 3.2%) within HCT-116 cells (Fig. 7F) (Table 1).
Influence of test compounds on cell cycle distribution of colorectal cancer cells
In order to inspect in further details the potential antiproliferative properties, cell cycle distribution was assessed after exposure of HCT-116 and HT-29 cells to 10 μM of test compounds for 48 h. For this test and the following tests, the six compounds that showed the most potent inhibitions on both cell lines (compounds 7, 8b, 9, 10a, 10b and 10f) were used.In HCT-116 cells, only compound 8b induced significant antiproliferative effect in terms of accumulating cells in G0/G1-phase (50.2 ± 1.0%) with reciprocal decrease of cells entering S-phase (22.8 ± 2.3%) compared to 39.5 ± 4.6% and 36.4 ± 4.1% of control cells in G0/G1-phase and S-phase, respectively (Fig. 8A).
 | ||
| Fig. 8 The effect of compounds 7, 8b, 9, 10a, 10b, and 10f on the cell cycle distribution of HCT-116 (A) and HT-29 (B) cells. Cells were exposed to 10 μM of test compounds for 48 h and cell cycle distribution was determined using DNA cytometry analysis and different cell phases were plotted as percentage of total events. Data is presented as mean ± SD; n = 3. (*): Significantly different from control group. | ||
In HT-29 cells, both compounds 8b and 9 induced significant antiproliferative effects, increasing cell population in G0/G1-phase from 53.4 ± 2.7% to 58.8 ± 1.8% and 59.7 ± 0.73%, respectively. Reciprocally, compounds 8b and 9 significantly decreased cells in S-phase from 27.3 ± 0.6% to 21.0 ± 0.9% and 22.0 ± 1.2%, respectively. Compound 10a induced mild but significant cell cycle arrest in S-phase and G2/M-phase increasing their cell populations from 27.3 ± 0.6% and 19.3 ± 1.6% to 29.8 ± 0.6% and 24.8 ± 0.4%, respectively. This cell cycle arrest induced by compound 10a resulted in reciprocal decrease in cell population in G0/G1-phase to 45.4 ± 0.6% (compared to 53.4 ± 2.7% in control). Compound 10b selectively arrested cells in G2/M-phase increasing its cell population from 19.3 ± 1.6% to 25.1 ± 0.6%; which resulted in reciprocal decrease of S-phase cells from 27.3 ± 0.6% to 23.0 ± 0.5% (Fig. 8B).
Apoptosis/necrosis assessment
Annexin V-FITC/PI staining coupled with flow cytometry was used to differentially assess proportion of cells dying via necrosis versus cells undergoing apoptosis in colorectal cancer cell lines. HCT-116 and HT-29 cells were treated with 10 μM of selected test compounds for 48 h prior to apoptosis/necrosis differential quantification by flow cytometry. In HCT-116 cells, compounds 8b, 9, 10a, 10b and 10f induced significant apoptosis, increasing apoptotic cell population by 3.8, 2.1, 7.5, 4.7 and 15.2 folds, respectively.In addition, compounds, 7, 8b, 10a, 10b and 10f induced significant necrosis, increasing necrotic cell population by 3.4, 2.9, 4.0, 2.4 and 3.7 folds, respectively. This explains the cell killing effects attributed to these compounds against HCT-116 cells; and shows that the calculated IC50 values of compounds 7, 8b, 9, 10a, 10b and 10f are indicative of cytotoxic effects with/without moderate antiproliferative activity. In addition, the cytotoxic effects of compounds 9, 10a, 10b and 10f were found to be attributed more to apoptosis rather than non-specific necrosis (Fig. 9A).
 | ||
| Fig. 9 Apoptosis/necrosis assessment in HCT-116 (A) and HT-29 (B) cells after exposure to test compounds. Cells were exposed to 10 μM of compounds 7, 8b, 9, 10a, 10b, and 10f for 48 h and were stained with annexin V-FITC/PI. Different cell populations were plotted as percentage of total events. Data is presented as mean ± SD; n = 3. (*): Significantly different from control group. | ||
With respect to HT-29 cells, compounds 8b, 10b and 10f were found to induce apoptosis, increasing apoptotic cell population by 2.5, 2.3 and 1.7 folds, respectively. The rest of the tested compounds induced apparent apoptotic cell population increase; but this increase was not significant. On the other hand, none of these compounds induced any significant non-specific necrosis. In other words, cell-killing effects of these compounds against HT-29 cells are mainly attributed to apoptosis rather than non-specific necrosis (Fig. 9B).
Autophagy assessment
Besides apoptosis, autophagy represents a big research controversy; whether it is a programmed cell death mechanism or an escape-death mechanism. Herein, we further investigated the effect of test compounds on autophagy process within colorectal cancer cell lines using Cyto-ID autophagy detection dye coupled with flow cytometry.In HCT-116, compounds 8b, 10b and 10f significantly induced autophagic signal higher than control cells by 54%, 30% and 99%, respectively. Positive control treatment (chloroquine) increased autophagic signal in HCT-116 cells by 65% (Fig. 10A). In contrast to what was observed at HCT-116, compounds 8b, 9 and 10a inhibited autophagy in HT-29 cells by 21%, 15% and 24%, respectively. On the other hand and in a similar fashion to what was observed with HCT-116 cells, positive control treatment (chloroquine) induced autophagy in HT-29 cells (Fig. 10B).
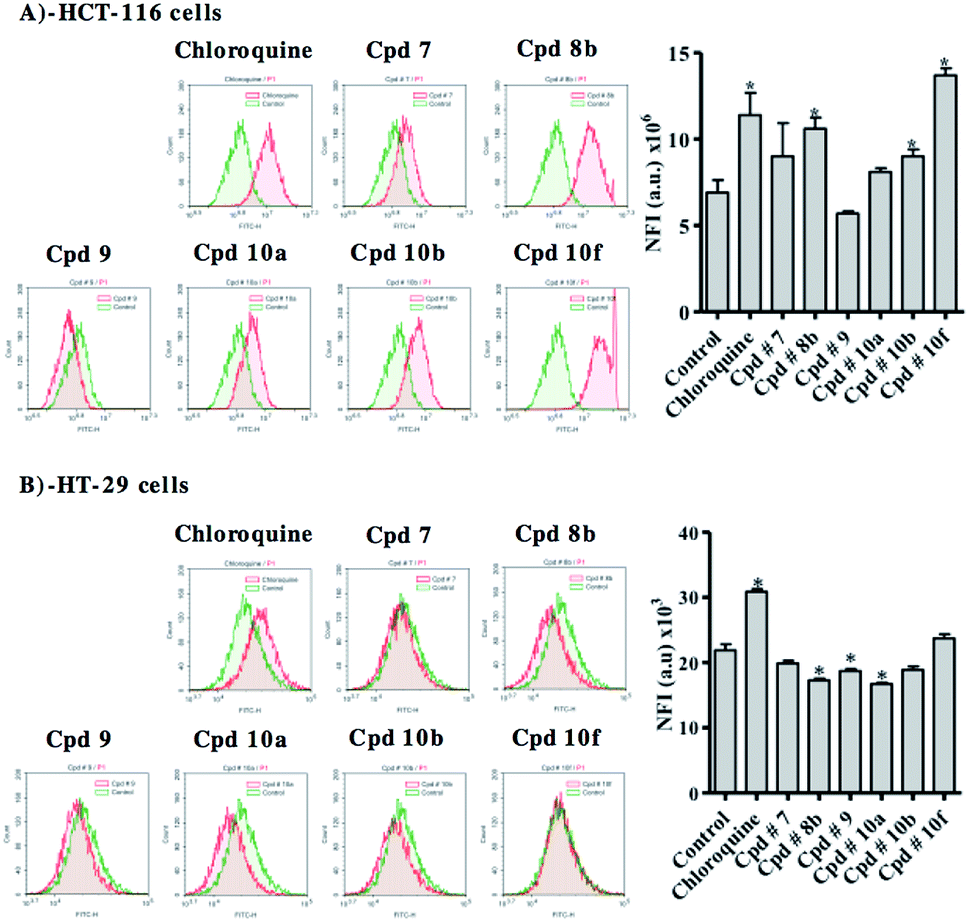 | ||
| Fig. 10 Autophagic cell death assessment in HCT-116 (A) and HT-29 (B) cells after exposure to test compounds. Cells were exposed to 10 μM of compounds 7, 8b, 9, 10a, 10b, and 10f for 48 h. Cells were stained with Cyto-ID autophagosome tracker. Net fluorescent intensity (NFI) were plotted and compared to basal fluorescence of control group. Data is presented as mean ± SD; n = 3. (*): Significantly different from control. | ||
Discussion
The design of the new series aimed at extending the modifications at the terminal pyridine ring in the potent PAP inhibitor 3 to yield the methoxy series (7 and 8a–j) and their hydroxy analogues (9 and 10a–j). The initial screening of the synthesized series over multiple kinases and on different colorectal cancer cells demonstrated that this new design was successful in producing potent anticolorectal cancer agents that are capable of targeting and inhibiting protein kinases. From the whole synthesized series, several compounds showed considerable non-specific multi-tyrosine kinase inhibitory activity. However, compounds 8a, 8b and 8j showed the highest src-kinase selectivities. On the other hand, compounds 10a and 10f did not exert any significant src-kinase inhibition despite their considerable multi-kinase inhibition. Additionally, compounds 7, 9 and 10b non-selectively inhibited the multi-tyrosine kinases panel at an equivalent level to the inhibition of src-kinase.These results show that the new skeleton provides a good template for the development of novel kinase inhibitors. However, the substitution pattern plays a critical role in defining the kinase inhibition selectivity profile of the inhibitor. At the terminal pyridine ring, the attachment of an unsubstituted phenyl (as in 8a) or 3-pyridyl (as in 8b) resulted in good inhibitory selectivities towards src kinase. On the other hand, decorating the phenyl ring with either electron donating or electron withdrawing groups resulted in disturbance of this kinase selectivity profile. Surprisingly, the only substitution that proved to be effective in maintaining the selectivity towards src kinase was that with a bulky 4-phenoxy group at the terminal phenyl ring (compound 8j). The effect of the terminal aryl substituent on inhibitor's src kinase inhibition corresponds with the molecular modelling simulations carried out on compound 8b. The docking simulations have showed that the terminal pyridine moiety of 8b orients towards the surface of the binding pocket where it engages, through its heterocyclic nitrogen, in HB interaction with GLU353. This hydrogen bonding interaction can account for the higher selectivity and potency of 8b towards src kinase, relative to other inhibitors within the series. This observed orientation could also explain why excessive bulkiness at this part of the skeleton can result in decreased potency of the compound (as in inhibitors 8h and 10h), where excessive bulkiness may result in clashing with certain surface residues in the protein, such as GLU 353.
Another important observation is the effect of the methoxy group in the (3-methoxy-5-methylphenyl) ring on src-kinase inhibitory selectivity in these three compounds (8a, 8b and 8j). Removal of this O-methyl group from these inhibitors to yield a free phenolic OH in their hydroxy counterparts (10a, 10b and 10j, respectively) resulted in a drop in src-kinase inhibitory potency, which emphasized the importance of this O-methyl group in maintaining good inhibitory potency for src-kinase.
Further assessment of antiproliferative/cytotoxicity activity showed that LS-174T cells were the most resistant to the compounds under investigation. The other two colorectal cancer (CRC) cell lines (HCT-116 and HT-29) showed considerable susceptibilities to the tyrosine kinase inhibitors under investigations. It is worthy to mention that LS174T and HCT-15 cells were reported to show resistance to the selective MEK1/2 tyrosine kinase inhibitor (selumetinib).20 In contrast to the effect of the methoxy group in maintaining good src-kinase inhibition selectivity, series 10 (compounds with a free phenolic OH) demonstrated in general more antiproliferative/cytotoxic effects than their methoxy group counterpart (series 8). This observation suggests that the overall cytotoxic effects of these compounds might be attributed in part to kinase inhibition, but does not exclude the involvement of other potential underlying mechanisms. In studying the effect of substitution at the terminal pyridine ring on the overall antiproliferative effect of the compound, it was interesting to see that compounds 8h and 10h, with the bulkiest substituent on the pyridine ring (N,N-diphenylamino group) have showed the least anticancer activities on all the tested cell-lines. This indicates that there is a size limit for the extended modifications within this new series of compounds, and that this particular modification has exceeded this limit, resulting in completely diminishing the anticancer activity of the compound.
To further dissect the antiproliferative/cytotoxic effects of the tyrosine kinase inhibitors under investigation, the influence of these compounds on cell cycle distribution and apoptosis/autophagy induction was examined. In this testing, only compound 8b induced significant antiproliferative effect and accumulation of both HCT-116 and HT-29 cells in G0/G1-phase. This could be explained on the basis of selective src-kinase inhibition, which is usually coupled with antiproliferative activity and accumulation of cells in G0/G1-phase. Several reports showed that interference with receptor tyrosine kinases such as src-kinase results in cell cycle arrest at G0/G1-phase.21,22 This arrest is partly attributed to interrupting the function of cyclin-dependent kinases (CDK) and other cell cycle controlling kinases.23 On the other hand, the non-src-kinase inhibitors (compounds 10a and 10b) resulted in accumulation of HT-29 cells in S-phase and G2/M-phase. This was manifested in the form of blocking DNA replication and eventually resulted in altered cell morphology and induction of apoptosis.24 Furthermore, compounds 8b (selective src-kinase inhibitor), 10b and 10f induced significant apoptosis in both HCT-116 and HT-29 cells. Apoptosis induction by compound 8b might be attributed to accumulation of cells at G0/G1-phase and failure of transition to S-phase.21,22 Yet, it is reported that defects in G2-M-phase checkpoint might allow a damaged cell to enter mitosis and undergo apoptosis.25 In addition, cell cycle arrest can explain proliferation inhibition and is known to trigger apoptosis in cancer cells.26 On the other hand, compounds 7, 8b, 9, 10a, 10b and 10f induced programmed as well as non-programmed necrotic cell death in HCT-116 cells.
Autophagy is another programmed cell death mechanism. It is highly controversial and complicated to conclude whether it is a pro-death mechanism or an escape-death mechanism in cancer cell death.27 Herein, HCT-116 cells underwent autophagy in response to treatment with compounds 8b, 10b and 10f; while autophagy was suppressed in HT-29 cells treated with compounds 8b, 9 and 10a. Putting the killing profile of these compounds (Table 1) in consideration, we suggest that, HCT-116 and HT-29 CRC cells are escaping apoptosis via autophagy shunting. In other words and similar to what was observed in our previous study on breast cancer cells,28 autophagy is considered here to be an apoptosis escape shelter.27,29,30
Conclusions
In conclusion, a novel series of small molecular anticancer agents has been developed in this study. The new template allows for structural modifications at various parts of the skeleton and these modifications were found to be critical in defining both compound's potency and the underlying mechanism(s) of its anticancer activities. A number of hits within the series showed multiple-kinase inhibitions, and a number of these hits showed good selectivities toward src-kinase, a commonly encountered overexpressing kinase in CRC. Among all the compounds examined, compound 8b (with a pyridine-3-yl moiety at the terminal pyridine ring) showed the most selective src-kinase inhibitory activity, which is coupled with cell cycle arrest, apoptotic and autophagic interference potential in colorectal cancer cells. Another interesting compound is 10f (with a 4-cyanophenyl moiety at the terminal pyridine ring) that showed potent anticancer effects against both HCT-116 and HT-29 CRC cells, where it strongly induced apoptosis and significantly increased the apoptotic cell population.The controversial structure activity relationship made it difficult to correlate the anticancer potencies of this new series of compounds to kinase inhibition alone, and suggests the presence of other underlying mechanisms that contribute together with kinase inhibition to the overall anticancer effect of these agents. Overall, this is a new promising category of anticancer compounds that worth to run through further developments to yield more potent derivatives and to be studied further to fully understand their underlying anticancer mechanisms.
Author contributions
HAA performed the biological assessment, data analysis and wrote the manuscript; AMA supervised the biological assessment, data analysis, wrote the manuscript and corresponds the submission; FAA supervised the biological assessment and revised the manuscript; HAM shared in the biological assessment; IME synthesized the inhibitors, wrote the manuscript and corresponds the submission.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
This research was supported by the Royal College of Surgeons in Ireland-Medical University of Bahrain (Grant number BR00063). The authors are grateful for Dr Marc Devocelle and Ms Siobhán O'Flaherty for their assistance in characterising the synthesized compounds using spectroscopy facility in RCSI (Royal College of Surgeons in Ireland).Notes and references
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2017, 67, 7–30 CrossRef.
- S. Bazarbashi, H. Al Eid and J. Minguet, Asian Pac. J. Cancer Prev., 2017, 18, 2437–2444 Search PubMed.
- N. Alsanea, A. S. Abduljabbar, S. Alhomoud, L. H. Ashari, D. Hibbert and S. Bazarbashi, Ann. Saudi Med., 2015, 35, 196–202 CrossRef.
- K. S. Bhullar, N. O. Lagarón, E. M. McGowan, I. Parmar, A. Jha, B. P. Hubbard and H. P. V. Rupasinghe, Mol. Cancer, 2018, 17, 48 CrossRef.
- H. Patterson, R. Nibbs, I. McInnes and S. Siebert, Clin. Exp. Immunol., 2014, 176, 1–10 CrossRef CAS.
- G. Finocchiaro, L. Toschi, L. Gianoncelli, M. Baretti and A. Santoro, Ann. Transl. Med., 2015, 3, 83 Search PubMed.
- R. Capdeville, E. Buchdunger, J. Zimmermann and A. Matter, Nat. Rev. Drug Discovery, 2002, 1, 493–502 CrossRef CAS.
- D. L. Deremer, C. Ustun and K. Natarajan, Clin. Ther., 2008, 30, 1956–1975 CrossRef CAS PubMed.
- T. Niwa, T. Asaki and S. Kimura, Anal. Chem. Insights, 2007, 2, 93–106 Search PubMed.
- M. Steegmaier, M. Hoffmann, A. Baum, P. Lenart, M. Petronczki, M. Krssak, U. Gurtler, P. Garin-Chesa, S. Lieb, J. Quant, M. Grauert, G. R. Adolf, N. Kraut, J. M. Peters and W. J. Rettig, Curr. Biol., 2007, 17, 316–322 CrossRef CAS PubMed.
- A. Fernandez, A. Sanguino, Z. Peng, E. Ozturk, J. Chen, A. Crespo, S. Wulf, A. Shavrin, C. Qin, J. Ma, J. Trent, Y. Lin, H. D. Han, L. S. Mangala, J. A. Bankson, J. Gelovani, A. Samarel, W. Bornmann, A. K. Sood and G. Lopez-Berestein, J. Clin. Invest., 2007, 117, 4044–4054 CrossRef CAS PubMed.
- B. Nuijen, M. Bouma, R. E. Henrar, U. Brauns, P. Bette, A. Bult and J. H. Beijnen, Int. J. Pharm., 2000, 194, 261–267 CrossRef CAS.
- S. Rapecki and R. Allen, J. Pharmacol. Exp. Ther., 2002, 303, 1325 CrossRef CAS.
- P. A. Harris, A. Boloor, M. Cheung, R. Kumar, R. M. Crosby, R. G. Davis-Ward, A. H. Epperly, K. W. Hinkle, R. N. Hunter 3rd, J. H. Johnson, V. B. Knick, C. P. Laudeman, D. K. Luttrell, R. A. Mook, R. T. Nolte, S. K. Rudolph, J. R. Szewczyk, A. T. Truesdale, J. M. Veal, L. Wang and J. A. Stafford, J. Med. Chem., 2008, 51, 4632–4640 CrossRef CAS.
- I. M. El-Deeb, D. K. Han, I. T. Kim and S. H. Lee, Bull. Korean Chem. Soc., 2010, 31, 1848–1858 CrossRef CAS.
- I. M. El-Deeb and S. H. Lee, Bioorg. Med. Chem., 2010, 18, 3961–3973 CrossRef CAS.
- I. M. El-Deeb and S. H. Lee, Bioorg. Med. Chem., 2010, 18, 3860–3874 CrossRef CAS.
- I. M. El-Deeb, J. C. Ryu and S. H. Lee, Molecules, 2008, 13, 818–830 CrossRef CAS.
- N. M. Levinson and S. G. Boxer, Nat. Chem. Biol., 2013, 10, 127 CrossRef.
- T. Troiani, L. Vecchione, E. Martinelli, A. Capasso, S. Costantino, L. P. Ciuffreda, F. Morgillo, D. Vitagliano, E. D'Aiuto, R. De Palma, S. Tejpar, E. Van Cutsem, M. De Lorenzi, M. Caraglia, L. Berrino and F. Ciardiello, Br. J. Cancer, 2012, 106, 1648–1659 CrossRef CAS.
- S. K. Mitra and D. D. Schlaepfer, Curr. Opin. Cell Biol., 2006, 18, 516–523 CrossRef CAS.
- P. Blume-Jensen and T. Hunter, Nature, 2001, 411, 355–365 CrossRef CAS PubMed.
- M. E. Noble, J. A. Endicott and L. N. Johnson, Science, 2004, 303, 1800–1805 CrossRef CAS.
- X. Xu, F. Hamhouyia, S. D. Thomas, T. J. Burke, A. C. Girvan, W. G. McGregor, J. O. Trent, D. M. Miller and P. J. Bates, J. Biol. Chem., 2001, 276, 43221–43230 CrossRef CAS.
- R. S. DiPaola, Clin. Cancer Res., 2002, 8, 3311–3314 CAS.
- X. Xu, Y. Zhang, D. Qu, T. Jiang and S. Li, J. Exp. Clin. Cancer Res., 2011, 30, 33 CrossRef CAS.
- M. M. Hippert, P. S. O'Toole and A. Thorburn, Cancer Res., 2006, 66, 9349–9351 CrossRef CAS PubMed.
- H. A. Bashmail and A. A. Alamoudi, Sci. Rep., 2018, 8, 11674 CrossRef.
- R. U. Janicke, Breast Cancer Res. Treat., 2009, 117, 219–221 CrossRef.
- S. Jin and E. White, Autophagy, 2007, 3, 28–31 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental procedures, characterisation data, 1H and 13C NMR spectra of new intermediates and final compounds. See DOI: 10.1039/c9ra03359a |
| This journal is © The Royal Society of Chemistry 2019 |