
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism of atom economical conversion of alcohols and amines to amides using Fe(II) pincer catalyst. An outer-sphere metal–ligand pathway or an inner-sphere elimination pathway?†
Bilal Ahmad Shiekh * and
Damanjit Kaur
* and
Damanjit Kaur
Department of Chemistry, UGC Sponsored Centre of Advanced Studies-I, Guru Nanak Dev University, Amritsar, India-143005. E-mail: bilal.ahmad459@gmail.com
First published on 4th June 2019
Abstract
In this present theoretical study, we investigated the reaction mechanism of atom-economical amide formation from alcohols and amines mediated by iron(II) hydride complex (iPrPNP)Fe(H)(CO) (iPrPNP = N[CH2CH2(PiPr2)]2) using state-of-the-art density functional theory. Two scenarios of mechanistic pathways were considered, the inner-sphere and the outer-sphere pathways. In former case, the reaction of encounter complex of formaldehyde with amine is the rate-determining step with ΔG298 K = 33.75 kcal mol−1 while as in latter case dehydrogenation from trans-hydride is the rate-determining step having ΔG298 K = 21.34 kcal mol−1. Both the mechanistic scenarios operate through stepwise ionic pathways. The assessment of computational results demonstrate that inner-sphere pathway is energetically demanding and thus rendering outer-sphere pathway to be the most plausible mechanism of amide formation. Ligand modifications reveal that electron-withdrawing groups like CF3 near N of PNP ligand reduce the catalytic efficiency of the catalyst. Furthermore, changing the isopropyl moiety of phosphine scaffold with CH3 has a minimal impact on catalytic activity of the catalyst. Overall, our computational results provide new insights for the design and development of new Fe(II) based pincer catalysts for atom economical amide formation from alcohols and amines.
Introduction
Amide bond is a prevalent linkage seen in both natural and synthetic molecules having widespread importance in pharmaceuticals and agrochemicals and is present in huge array of molecules such as proteins, synthetic polymers and numerous therapeutic drugs.1–4 Typically these types of molecules are prepared by the reaction of carboxylic acids with amines in presence of some organic or inorganic promoters.5 Alternatively they can also be synthesized by using transition metal catalyzed dehydrogenation of alcohols with amines which is doubtlessly an efficient and atom economical method of synthesis.6–19 The first transition metal catalyst (Ru-PNN pincer catalyst) for direct amide synthesis was proposed by Milstein which operates through metal–ligand cooperation by aromatization and dearomatization of the pyridine ring.8 Since then a lot of attention has been paid to direct amide formation from alcohols and amines.6,7,9–19 The atom economic strategy of amide synthesis faces lot of challenges such as limited substrate scope, long reaction timings and harsh reaction temperatures needed for the reaction to carry out. To circumvent these problems some efforts are being made to develop the transition-metal catalysts for the dehydrogenative coupling of alcohols with amines to form amides.20 In particular, Bernskoetter et al.21 described the use of iron(II) hydride complex (iPrPNP)Fe(H)(CO) (iPrPNP = N[CH2CH2(PiPr2)]2) for direct amide synthesis from alcohols and amines (eqn (1)) which shows greater TONs for secondary amines than some previously reported catalysts.22,23
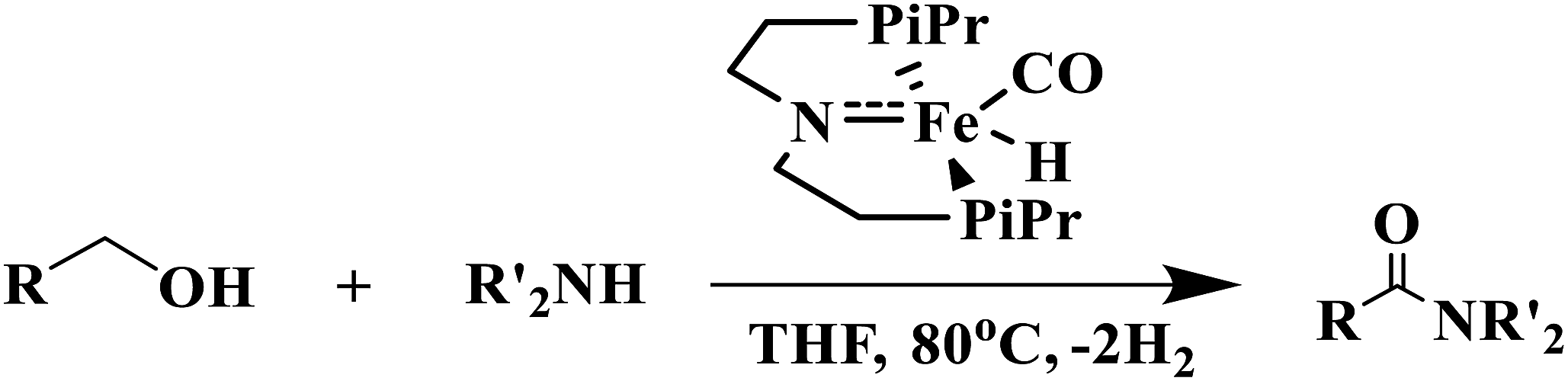 | (1) |
The mechanistic investigation of amide formation catalyzed by all homogeneous transition-metal catalysts is proposed to proceed through formation of carbonyl species that further reacts with amine to generate hemiaminal. The subsequent dehydrogenation of the hemiaminal results in the formation of amide.17,24–27 The same type of tentative mechanistic pathway was proposed by Bernskoetter et al.21 While trying to understand the mechanism of this catalytic reaction by varied temperature NMR studies, Bernskoetter et al.21 reported that a rapid equilibrium exists between the catalyst and the iron-methoxy species. However, the exact role of the iron-methoxy species formed by the reaction of methanol with the catalyst during the course of the reaction was not ascertained.
Despite the steady development of the homogenous transition metal catalysts for atom economical amide formation, a detailed computational study on catalytic mechanism is required to illuminate the precise path of the reaction (eqn (1)). In addition to this, the entitled mechanistic question need to be addressed for full understanding of the mechanistic pathways of the reaction. Though the mechanism of the catalytic alcohol dehydrogenation step has been studied previously by other research groups using density functional methods,27,28 however, an extensive catalytic mechanism involving more insights into elementary steps, energetics as well as alternate reaction pathways of amide formation using Fe(II) pincer catalyst (OFe) were not studied. Furthermore, the complete understanding of the reaction mechanism and the energetics of the reaction pathways will be advantageous to the researchers for the development and design of new catalysts. Being interested in catalyst designing and understanding their mechanistic aspects,29 our aim in the present work is to elucidate the underlying mechanism of the catalytic reaction and to study the effects of various ligand modifications on the catalytic activity of the catalyst using state-of-the-art density functional methodologies.
Computational details
All the complexes without truncations, were optimized with PBE-D3 (ref. 30 and 31) density functional theory (DFT) in combination with 6-31G** basis set for all atoms except Fe, for which LACVP**32,33 core potential and basis set was used. No symmetry constraints were employed during the geometry optimizations unless stated otherwise. The PBE is known to provide good results for the determination of thermochemistry and more recently it has been readily employed in metal catalyzed chemical reaction studies.34–37 Geometry optimizations were first carried out in gas phase and then in liquid phase using Poisson-Boltzmann implicit continuum solvation model (PBF) in THF with probe radius 2.52 and dielectric constant 7.6.38,39 The analysis of unscaled harmonic vibrational frequency data, computed on the optimized geometries confirmed the nature of complexes optimized. The intermediates were with no imaginary frequency while as the transition states were having single imaginary frequency along the reaction coordinate. The intrinsic reaction coordinate (IRC) calculations on the later were carried out to confirm that they were connected to minima. These types of calculations were also performed at PBE-D3/6-31G** level of theory. The single point energy calculations of the optimized structures were calculated at PBE-D3/6-311++G** with LACVP**++ basis set for Fe including the solvent effects.The free energy of the complexes was computed using the following equation
| G = Egas + ΔEsol + ZPE + (Htotal − TS) + ΔGgas→sol |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(22.4) = 1.9 kcal mol−1 is the correction for Gibbs energy from standard gas phase (1 atm) to liquid state (1 M). A correction of 2.6 or −2.6 kcal mol−1 has been employed in Gibbs energy for the conversion of one molecule to two or vice versa.40,41 All the calculations were performed at standard conditions of temperature (298.15 K) and not at the actual experimental conditions due to overestimation of entropic term which significantly overestimates the Gibbs energies.40,41 The natural population analysis along the IRC was performed using the method proposed by Weinhold et al.42 All the above said calculations have been performed by using Jaguar as implemented in Schrodinger suite of programs.43 The natural population analysis along the IRC were carried out by NBO 6.0 program42 as implemented in Jaguar.
ln(22.4) = 1.9 kcal mol−1 is the correction for Gibbs energy from standard gas phase (1 atm) to liquid state (1 M). A correction of 2.6 or −2.6 kcal mol−1 has been employed in Gibbs energy for the conversion of one molecule to two or vice versa.40,41 All the calculations were performed at standard conditions of temperature (298.15 K) and not at the actual experimental conditions due to overestimation of entropic term which significantly overestimates the Gibbs energies.40,41 The natural population analysis along the IRC was performed using the method proposed by Weinhold et al.42 All the above said calculations have been performed by using Jaguar as implemented in Schrodinger suite of programs.43 The natural population analysis along the IRC were carried out by NBO 6.0 program42 as implemented in Jaguar.
Results and discussion
Mechanistic aspects of the reaction
To explore the extensive mechanism of atom economical amide formation from alcohols and amines mediated by OFe pincer catalyst, two scenarios, the inner-sphere and the outer-sphere pathways were studied taking methanol and dimethyl amine as model substrates in our calculations. These two scenarios of amide formation have been illustrated in Scheme 1 and the detailed energy profiles are given in the ESI.†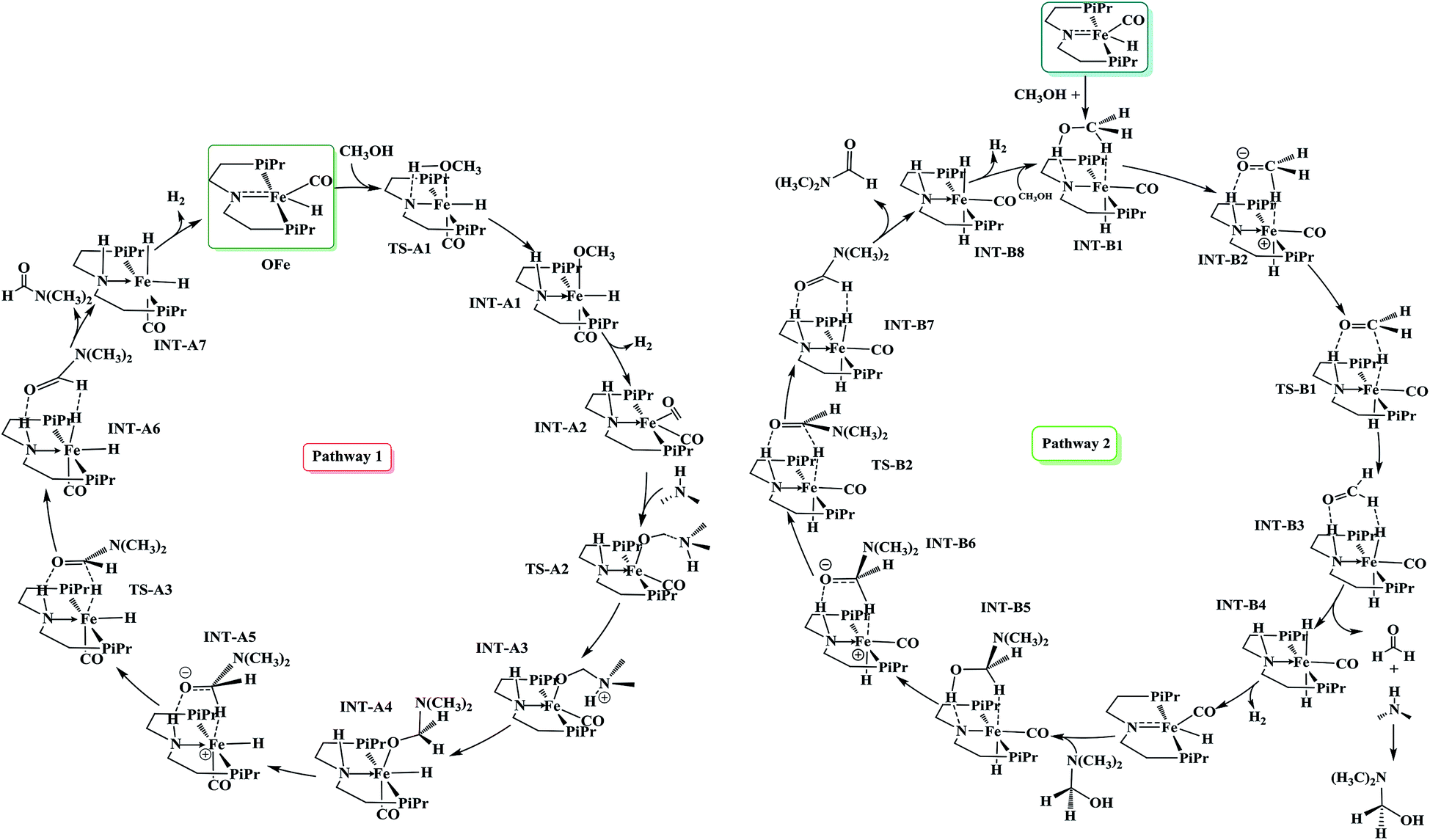 | ||
| Scheme 1 Stepwise mechanistic pathways for amide formation from methanol and N,N-dimethylamine mediated by OFe pincer catalyst. Pathway 1: inner-sphere, pathway 2: outer-sphere pathway. | ||
Inner-sphere pathway
This mechanistic pathway starts with the reaction of methanol along O–H bond with the catalyst in a concerted fashion forming a six coordinate iron-methoxy species, INT-A1 with a low barrier (TS-A1) equal to ΔG298 K = 9.76 kcal mol−1 as shown in Fig. 1. This step of reaction is accompanied by concomitant migration of methoxy group and hydrogen to the metal center and N of PNP ligand, respectively. In TS-A1, the O–H distance is 1.108 Å which is significantly higher from an initial value of 0.977 Å in free methanol. Furthermore, due to strong electron donation by the methoxy group to the metal center in INT-A1 the Fe–N charge transfer interaction decreases which results in the elongation of Fe–N bond distance from an initial value of 1.851 Å to a final value 2.105 Å (Fig. 2). The subsequent hydrogen transfer from the methoxy group to the metal center results in the elimination of H2 from INT-A1 thus forming an cationic (2+) encounter complex of formaldehyde with Fe metal center (INT-A2). The elimination of H2 is endergonic in nature with ΔG298.15 K = 12.60 kcal mol−1. In the next step, the substrate dimethyl amine reacts with the formaldehyde of encounter complex in a nucleophilic fashion to form INT-A3 via the transition state TS-A2. It is worth noting at this juncture that INT-A2 to INT-A3 is the only cationic elementary step found in inner-sphere mechanism and is the rate-determining step with energy barrier equal to 33.75 kcal mol−1. The migration of proton from the amine moiety to the metal center forms a re-coordinated complex, INT-A3 and the subsequent rearrangement forms a complex of cis-dihydride and hemiaminal type of species (INT-A5). There is a significant reduction in Fe–O bond length from 2.335 to 2.012 Å on going from INT-A3 to INT-A4 indicating a better coordination in the latter. The proton transfer to the metal center forms another encounter complex between the cis-dihydride and N,N-dimethylformamide, INT-A6 which exhibits a low barrier TS-A3 (22.74 kcal mol−1). The N,N-dimethylformamide is subsequently eliminated from the complex in an exergonic fashion leaving behind cis-dihydride (INT-A7) and the catalyst is regenerated in the last step from INT-A7 through the subsequent elimination of dihydrogen.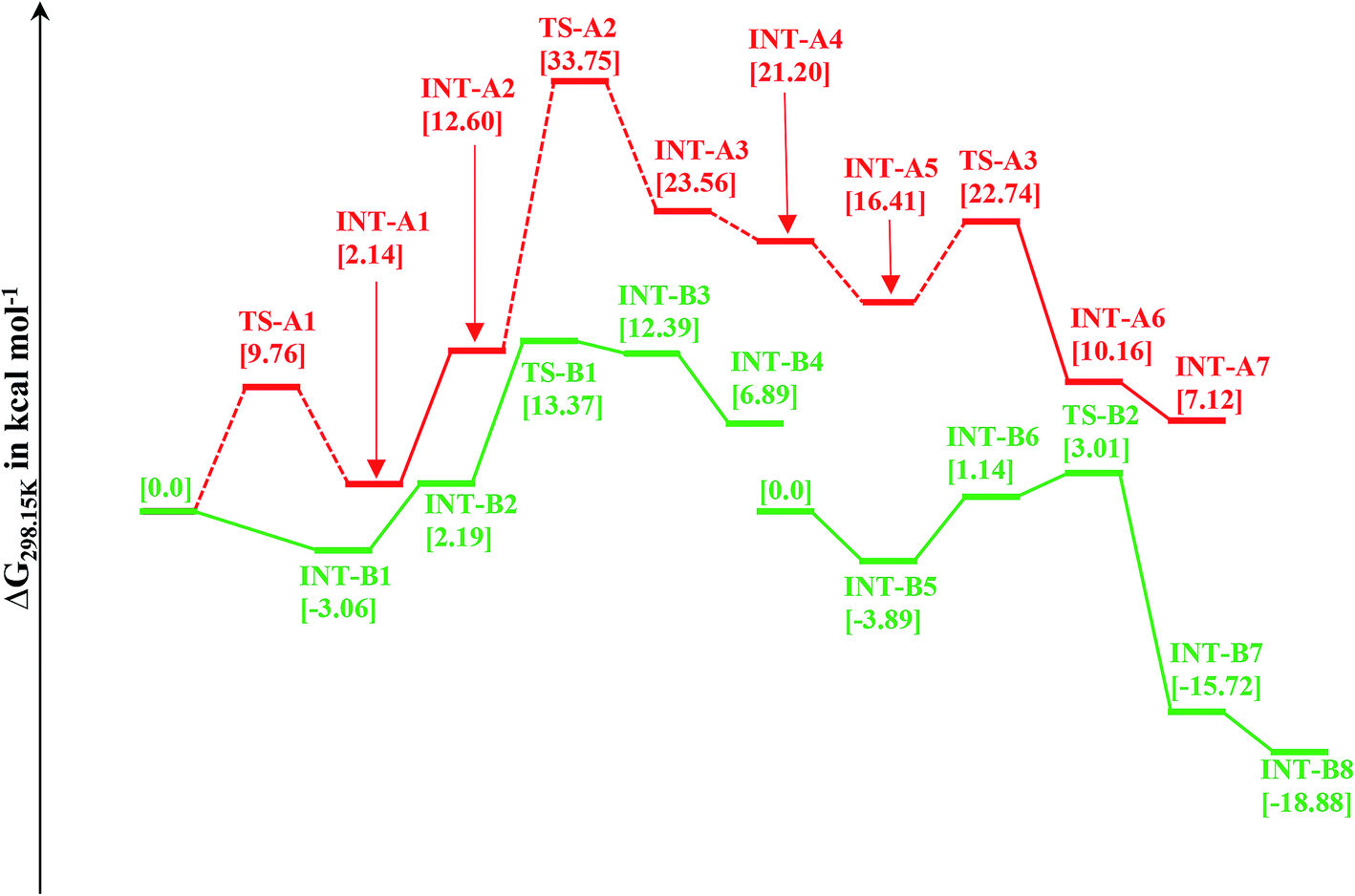 | ||
| Fig. 1 Relative Gibbs free energy for two mechanistic pathways, pathway 1 and pathway 2. | ||
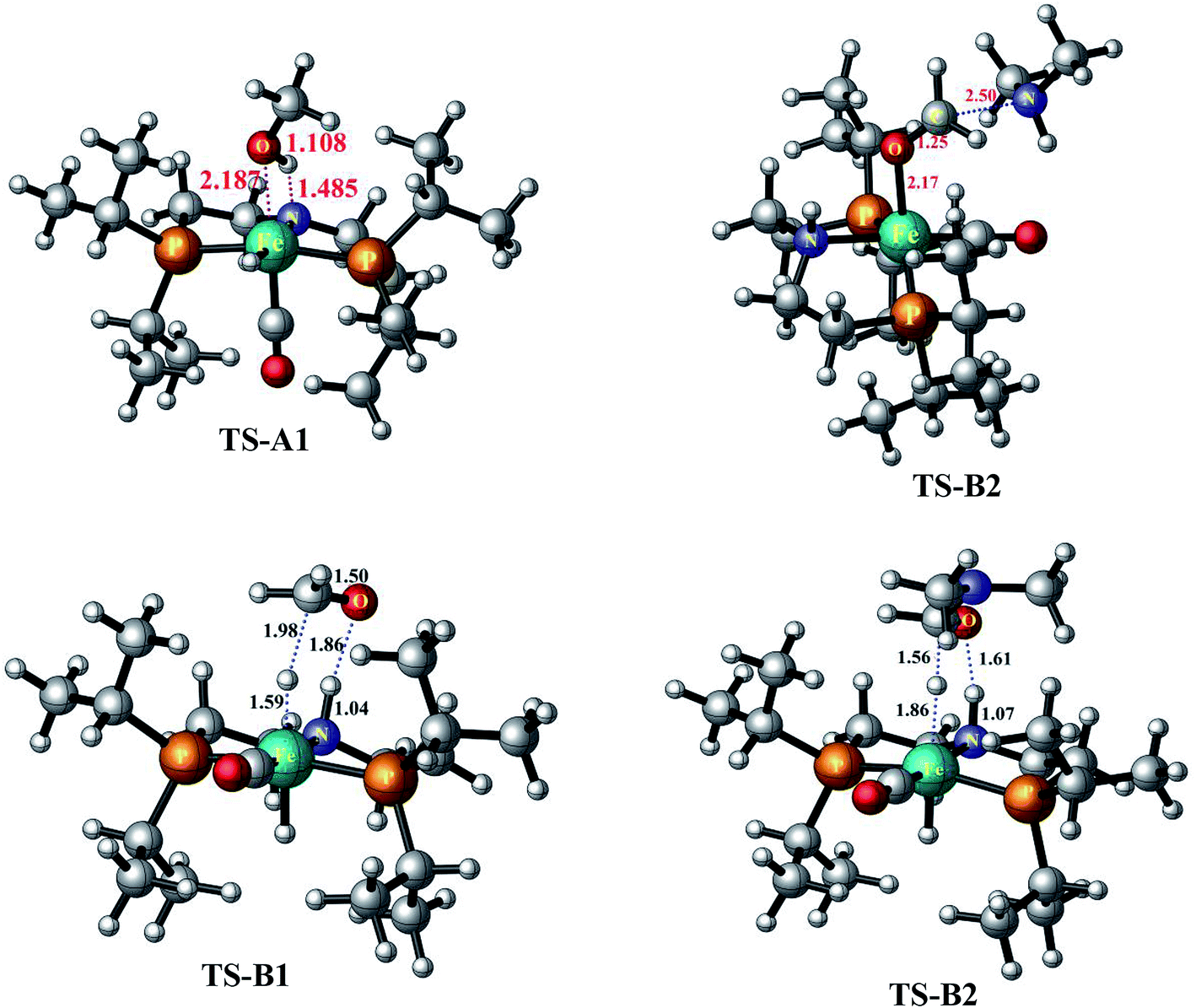 | ||
| Fig. 2 The optimized geometrical parameters of transition states computed at PBE-D3/6-31G**//LACVP** level of theory. All the mentioned distances are in angstroms. | ||
Indeed, from the above results it is clear that the inner-sphere mechanistic pathway of amide formation mediated by OFe pincer catalyst is an energetically demanding process and the infeasibility of this type of mechanistic pathway has been reported by other studies using some analogous complex species.44 However, a similar type of inner-sphere mechanism was also proposed by Gusev for amide formation mediated by PNN-Os catalyst using MeNH2.45
Outer-sphere pathway
Since the inner-sphere mechanistic pathway is energetically demanding therefore a second pathway responsible for the amide formation must be pursued. In this alternate mode of catalysis, the substrate methanol reacts with the catalyst through O–H/C–H which leads to the formation of a precursor complex, INT-B1 and subsequent proton transfer from O–H group to N-atom of the PNP ligand generates intermediate INT-B2 and is ionic in nature. The initial anionic behavior of N-pincer ligand is thus neutralized by proton transfer and hence creates a positive charge over metal center. The negative charge over O-atom and positive charge over Fe-center favors the hydride transfer from C–H of methanol to the metal center forming an encounter complex of formaldehyde and trans-dihydride, INT-B3 via a very low barrier, TS-B1 with ΔG298.15 K = 13.37 kcal mol−1. It is noteworthy to mention at this juncture that we did not find the transition state which features simultaneous O–H/C–H hydrogen transfer to the catalyst, instead the one which involves the transfer of hydride from C–H of methanol to metal center. In fact, such type of transition state has been reported by Yang46 while studying dehydrogenation of ethanol using OFe pincer catalyst. However, Yang also reported a second transition state involving transfer of proton from O–H of methanol to PNP ligand which we were unable to locate. The INT-B3 thus formed is quite unstable and the exergonic dissociation of formaldehyde from the complex and the subsequent reaction of formaldehyde with secondary amine generates a hemiaminal. The trans-dihydride eliminates dihydrogen to regenerate the initial catalyst. In the next step, the hemiaminal thus formed undergoes dehydrogenation in a similar fashion as discussed above to close the catalytic cycle of amide formation. Like INT-B2, the INT-B6 is ionic in nature in which O-atom is negatively charged. In this overall catalytic cycle, the loss of dihydrogen from trans-dihydride is the rate-determining step. Intriguingly, methanol dehydrogenation is comparatively high in energy (ΔG298.15 K = 13.37 kcal mol−1) than hemiaminal dehydrogenation (ΔG298.15 K = 3.01 kcal mol−1) with respect to the Gibbs free energy regime. Furthermore, the INT-B1 is formed without any energy barrier while as formation of INT-A1 has uphill energy barrier equal to 2.14 kcal mol−1.Unlike other research groups,27,28 who propose a neutral concerted mechanism for dehydrogenation of alcohols we found a stepwise ionic pathway which is in good agreement with the DFT studies proposed by Yang.46 The free energy of transfer hydrogenation by methanol to the Fe-metal center in outer-sphere pathway is relatively in the similar range compared to other metal catalysts. The DFT studies at M06-L by Zhuofeng Ke et al.47 for sterically demanding dehydrogenation of phenethyl alcohols by PNN-Ru pincer catalyst also suggest an outer-sphere mechanism with ΔG#298.15 K = 11.6 kcal mol−1 for transfer hydrogenation step which is a bit low than OFe catalyst. The same free energy gap reported with DFT studies (B3PW91 functional) for dihydride complex formation produced by transfer hydrogenation of benzyl alcohols by Ir-based complex (Cp*Ir(bpyO)) is 23.6 kcal mol−1, higher than OFe and PNN-Ru.48 Similarly, the hydrides of Osmium, OsHCl(CO)[HN(C2H4PiPr2)2] and OsH2-(CO)[HN(C2H4PiPr2)2], are undoubtly versatile transfer hydrogenation catalysts with ΔG#298.15 K = 13.7 kcal mol−1 which is again in the similar range as that of OFe-complex.49
Comparison of dehydrogenation in cis and trans-dihydride complexes
The complex trans-dihydride is stable by 3.92 kcal mol−1 than cis-dihydride. The dehydrogenation of dihydride complexes is alcohol assisted as proposed by other studies,46,50 as it lowers the free energy of activation. In this fashion, the methanol reacts with two hydrogen atoms along the Fe–N bond of dihydride to form DhB complex with very low barrier in energy as displayed in Scheme 2. The proton transfer from methanol to Fe–H forms another intermediate DhC through cyclic transition, TS-Dh1. The loss of H2 from the intermediate DhC forms more stable DhD intermediate with barrier, TS-Dh2 and the subsequent loss of methanol regenerates the initial catalyst. The formation of cyclic transition state, TS-Dh1 from trans-dihydride is the rate-determining step of outer sphere mechanism with total barrier equal to 21.34 kcal mol−1. Schneider and co-workers50 reported relative free energy of this rate-determining step of methanol dehydrogenation equal to 25.1 kcal mol−1 using truncated form version of OFe catalyst employing RI-B3PW91-D3BJ/def2-QZVPP//B3LYP/def2-SVP level of theory at 393.15 K temperature. Moreover, Yang46 also reported the similar relative free energy (22.1 kcal mol−1) of ethanol dehydrogenation by OFe catalyst using M06 DFT functional. Thus, our computed results agree well with the literature. The formation of TS-Dh1 in both trans and cis-dihydride complexes is uphill in energy by almost ∼15 kcal mol−1. However, the loss of H2 from intermediate DhC in cis-dihydride is more energy demanding than its analogous trans-dihydride intermediate by almost 5 kcal mol−1 which is due to the strong trans-effect by hydride ligand in trans-dihydride.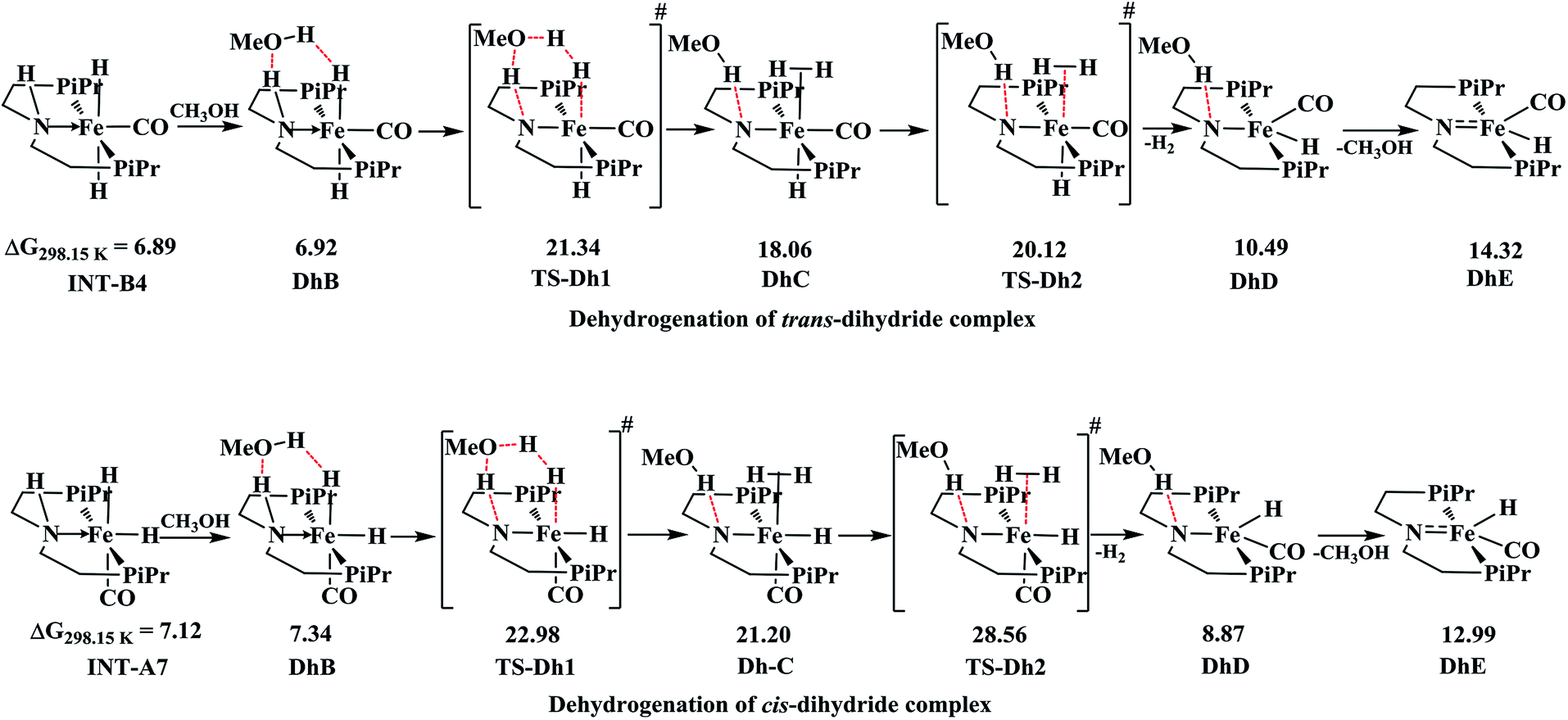 | ||
| Scheme 2 The computed free energy for alcohol assisted dehydrogenation of trans and cis-dihydride complexes formed during the outer and inner-sphere pathways, respectively. All the given values are in kcal mol−1. | ||
The inner-sphere pathway is disfavored over outer-sphere pathway because of several inherent properties. Firstly, the inner-sphere pathway operates through the cis-dihydride complex which is uphill in energy than trans-dihydride. Secondly, for inner-sphere pathway to occur metal-hydride bond pathway has to be in cis-position with respect to the reacting substrate attached at the metal center. Such type of cis-isomer is relatively less stable than trans-isomer as stronger π-acceptor CO ligand in such isomer reduces the possibility of creating a vacant site by H2 elimination by reducing the electron density at the metal center. On contrary, the CO-ligand in trans-form stabilizes the complex by strong π-back donation. Furthermore, in trans-form the hydride ligand favors the H2 elimination by strong trans-effect. Thus, all these factors favor outer-sphere pathway over inner-sphere pathway of amide formation.
Effect of spin state on catalysis by OFe
Since, the spin state of the Fe-center can have a great influence on the results,51–53 so we tried to find energetics of the triplet state for outer-sphere mechanism as well. As per the quantum mechanical calculations, the free energy difference between OFe in singlet and triplet states is 25.84 kcal mol−1. The free energy of INT-B1 and INT-B5 in triplet state is 5.98 and 3.47 kcal mol−1, respectively. However, transfer hydrogenation step in triplet state (TS-B1) is found to be energetically demanding with free energy equal to 23.31 kcal mol−1, which is ∼10 kcal mol−1 higher than singlet one. Therefore, from the DFT perspective, dehydrogenation of alcohols by OFe in singlet state takes dominance over triplet state.Stability of OFe-methanol species, NBO and NCI analysis
The charge transfer between the donor and acceptor orbitals plays an important role in stabilization of complexes and energy of such an interaction, according to second order perturbation theory54 is represented as:where 〈σ|
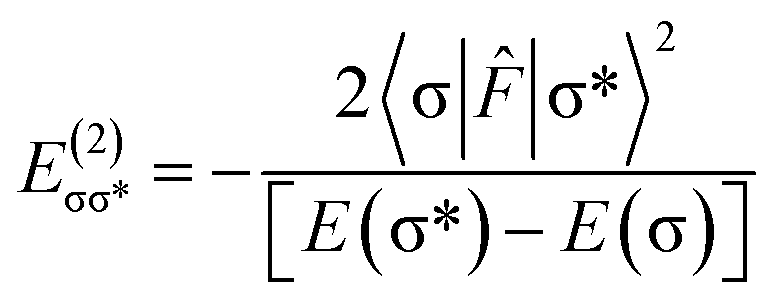
![[F with combining circumflex]](https://www.rsc.org/images/entities/i_char_0046_0302.gif) |σ*〉 represents the Fock matrix element with being Fock operator and [E(σ*) − E(σ)] is the orbital energy difference between the vacant σ* antibonding orbital and the filled σ bonding orbital. The second order stabilization energy,
|σ*〉 represents the Fock matrix element with being Fock operator and [E(σ*) − E(σ)] is the orbital energy difference between the vacant σ* antibonding orbital and the filled σ bonding orbital. The second order stabilization energy, 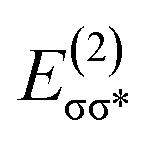 provides an estimation of bonding interaction and extent to which the charge is transferred from the donor to the acceptor orbitals in a complex. On the contrary NCI (non-covalent interaction) is used to visualize and quantify the noncovalent interactions by analyzing the graph plotted between electron density ρ(r) and reduced density gradient.55,56 The mode of interatomic interaction within a complex is determined by the sign of λ2 eigenvalue. The hydrogen bonding and ionic interactions appear in negative sign, the van der Waal interactions around zero and the repulsive regions appear far toward positive region. Thus, both NBO and NCI can be used to represent the chemical information and noncovalent interactions precisely within a complex.
provides an estimation of bonding interaction and extent to which the charge is transferred from the donor to the acceptor orbitals in a complex. On the contrary NCI (non-covalent interaction) is used to visualize and quantify the noncovalent interactions by analyzing the graph plotted between electron density ρ(r) and reduced density gradient.55,56 The mode of interatomic interaction within a complex is determined by the sign of λ2 eigenvalue. The hydrogen bonding and ionic interactions appear in negative sign, the van der Waal interactions around zero and the repulsive regions appear far toward positive region. Thus, both NBO and NCI can be used to represent the chemical information and noncovalent interactions precisely within a complex.
In outer-sphere pathway, there are two modes of interactions between the catalyst and methanol, the one interacting along O–H and the second one interacting through O–H as well as C–H of methanol as portrayed in Fig. 3. In former case, the NBO analysis depict a significant charge transfer from the π orbital of Fe–N (ψπFe−N) to the σ* antibonding orbital of O–H 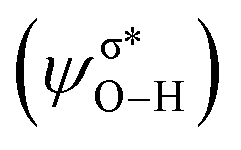 of methanol with
of methanol with 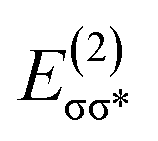 equal to 22.88 kcal mol−1. NCI displays this mode of interaction as a blue disc between the N-atom of PNP ligand and the O–H of methanol. In the latter case, the lone pair (lp) of N (ψlpN) interacts with σ* antibonding orbital of O–H
equal to 22.88 kcal mol−1. NCI displays this mode of interaction as a blue disc between the N-atom of PNP ligand and the O–H of methanol. In the latter case, the lone pair (lp) of N (ψlpN) interacts with σ* antibonding orbital of O–H 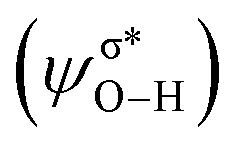 with
with 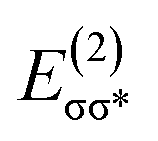 of 20.03 kcal mol−1. Furthermore, the σ bonding orbital of C–H (ψσC−H) interacts with Fe-dxz orbital
of 20.03 kcal mol−1. Furthermore, the σ bonding orbital of C–H (ψσC−H) interacts with Fe-dxz orbital 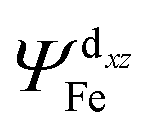 with second order stabilization equal to 4.09 kcal mol−1. This mode of interaction is clearly visible by two isosurface discs between the methanol and the catalyst. Careful inspection of the above results suggest that stepwise mode of interaction of methanol with OFe is favored over concerted mode of interaction.
with second order stabilization equal to 4.09 kcal mol−1. This mode of interaction is clearly visible by two isosurface discs between the methanol and the catalyst. Careful inspection of the above results suggest that stepwise mode of interaction of methanol with OFe is favored over concerted mode of interaction.
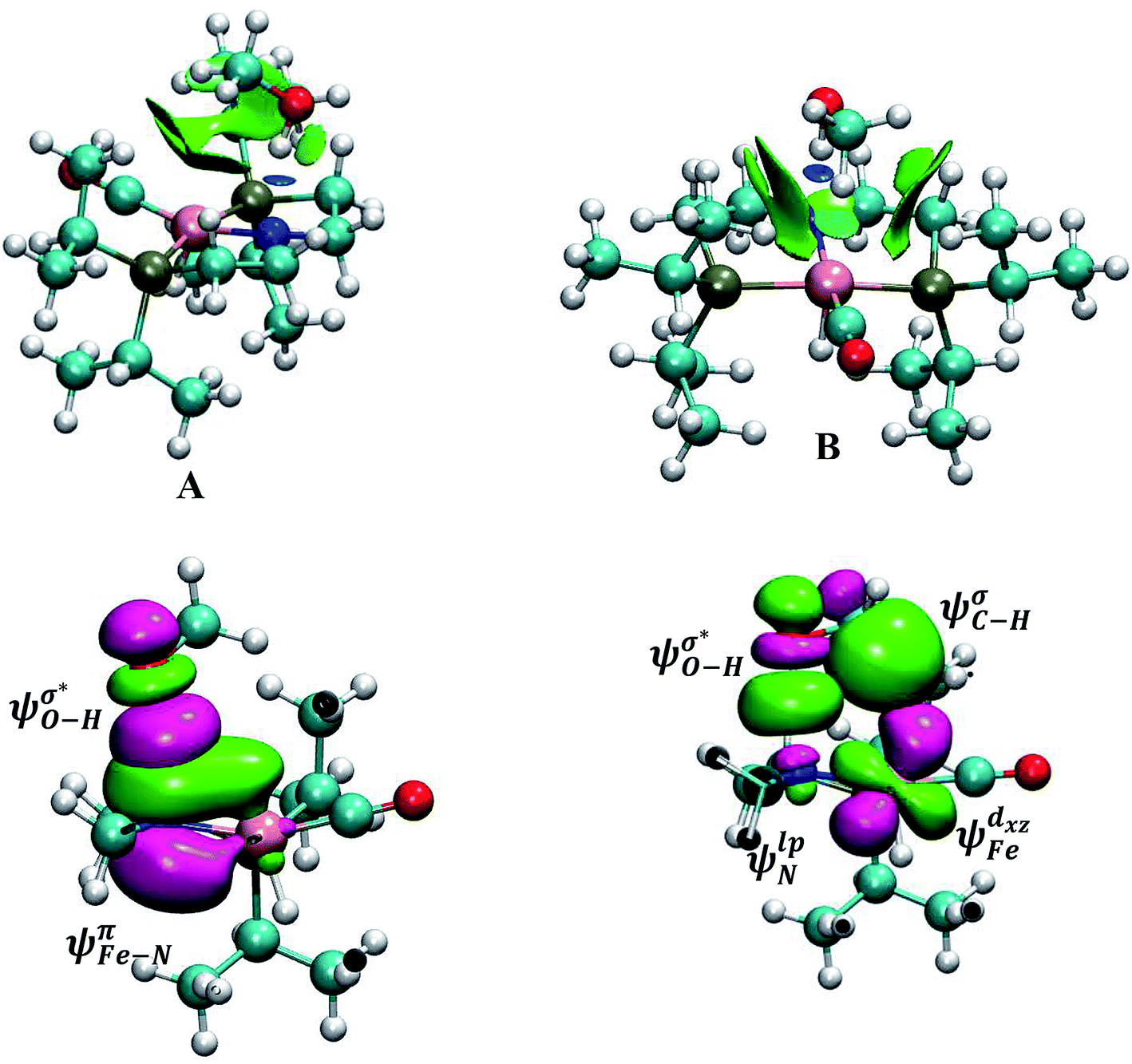 | ||
| Fig. 3 Two modes of charge transfer interactions between the catalyst and the methanol in outer-sphere mechanistic pathway: (A) only O–H is interacting with PNP-ligand (B) both O–H and C–H are interacting with the catalyst. Isosurface value of Lp of N increased for the sake of clarity. NCI isosurface value 0.07 a.u. | ||
Electronic distribution changes in TS-A1 and TS-B1
In order to fully understand the role of substituents in amide formation mediated by bifunctional OFe catalyst, the NBO analysis along intrinsic reaction coordinate (IRC) was carried out in TS-A1 and TS-B1 as shown in Fig. 4 and 5. As discussed above, the inner-sphere pathway of methanol dehydrogenation is accompanied by simultaneous migration of OCH3 and H groups to the metal center and PNP ligand, respectively. However, in outer-sphere pathway there is proton transfer from O–H and hydride transfer from C–H of methanol to PNP ligand and metal center, respectively. The population changes accompanied during these geometrical alterations along the IRC are quite large and thus we will discuss these changes briefly here.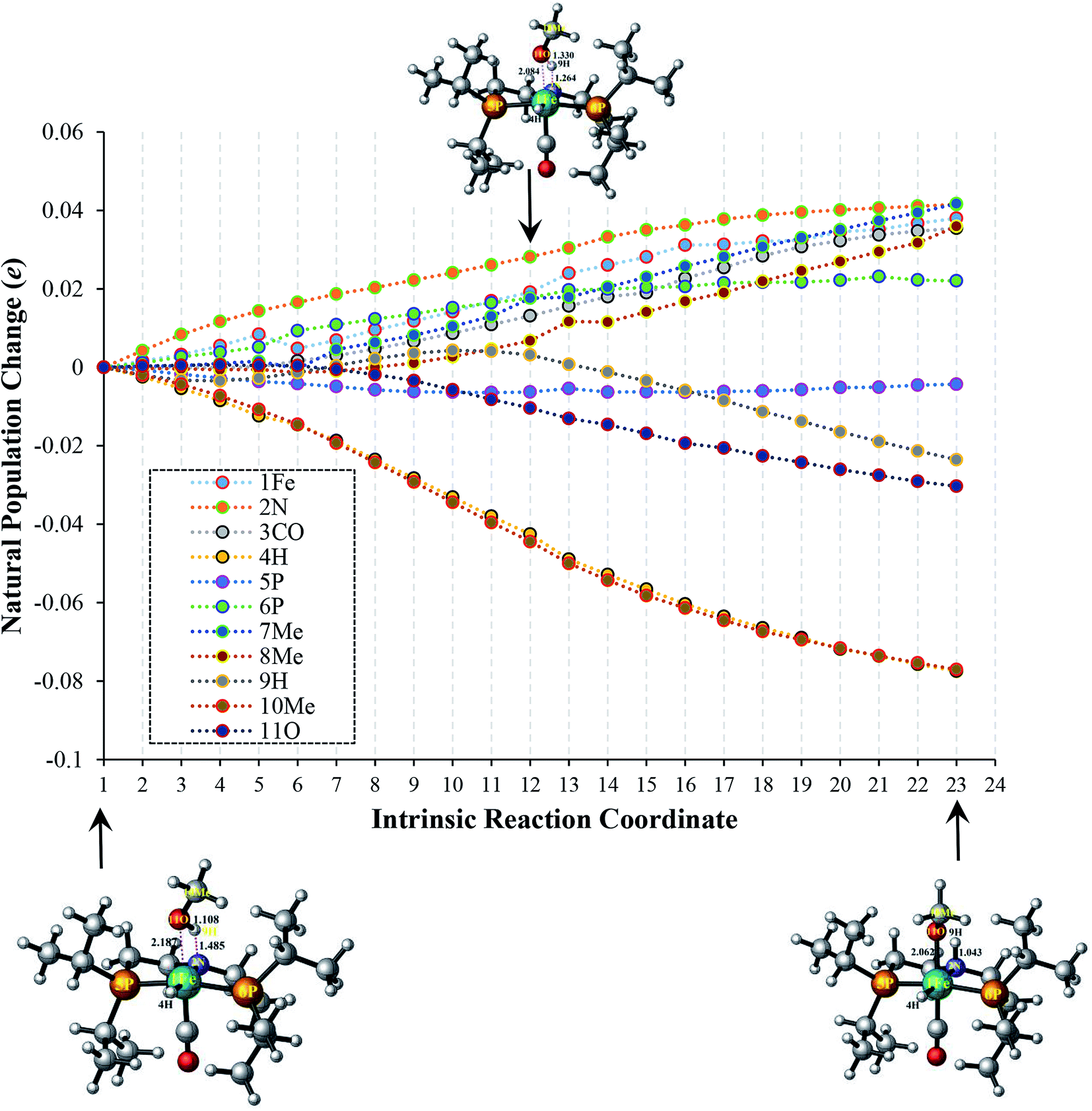 | ||
| Fig. 4 Change in population (e) of various substituents along intrinsic reaction coordinate in TS-A1 transition state of inner-sphere pathway. 7Me and 8Me represent the isopropyl groups including CH2–CH2 moiety around the 5P and 6P, respectively. | ||
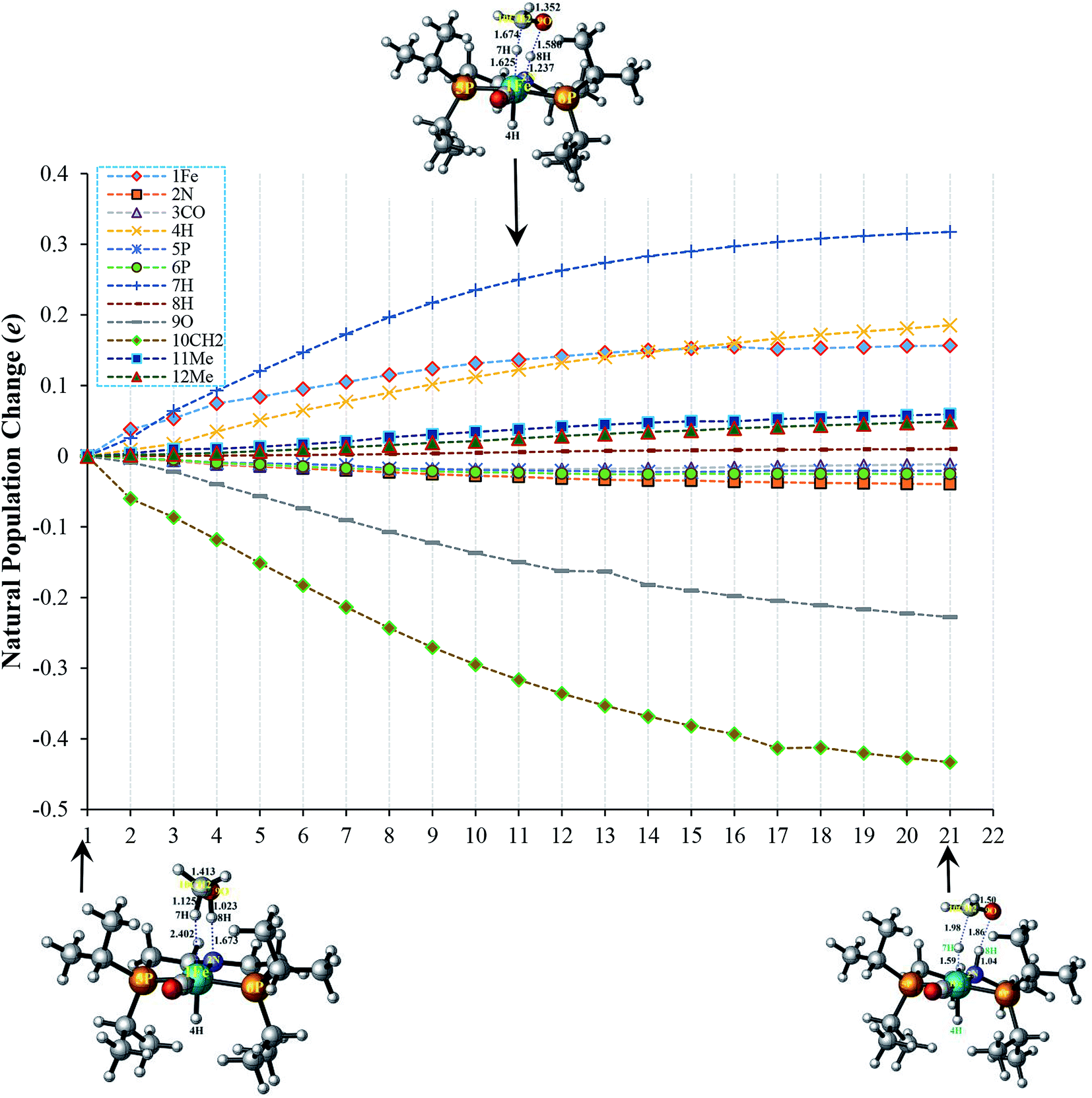 | ||
| Fig. 5 Change in population (e) of substituents along intrinsic reaction coordinate in TS-B1 transition state of outer-sphere pathway. 11Me and 12Me represent the isopropyl groups including CH2–CH2 moiety around the 5P and 6P, respectively. | ||
In TS-A1, there is continuous increase in population of 1Fe by 0.04e due to charge transfer to the metal center by OCH3 moiety as we proceed from reaction step 1 to 23. Such transfer of charge from OCH3 continuously oppresses the charge transfer from 3CO and 2N of PNP ligand to the metal center along IRC thereby enhancing their population as portrayed in Fig. 4. Simultaneously, the 4H also undergoes decrease in population indicating the enhancement of its protonic-character. The population of 9H initially increases as it starts to dissociate from the OCH3 due to increase in its charge density by OCH3 and N. However after reaching 11, the population decreases as it starts to separate from OCH3. The electron populations of 7Me as well as 8Me also increases during the course of the reaction. The atomic population of 5P almost remains constant, however, 6P shows an early increase in its population. This can be partially rationalized due to the steric interaction between the OCH3 and the iPr groups of 6P that results in poor overlap with metal center and thus increasing its population.
In TS-B1, the atomic population of 1Fe initially increases slightly for few steps along IRC due to initial charge transfer from electron-donating 4H ligand, thereafter it tends to remain constant. Concomitantly, 2N also undergoes a small decrease in its population. Such a change in its population is attributed to the bond formation with proton transferred from methanol (8H). Similarly, the population of 5P and 6P increases a little. Unlike TS-A1, the electron population of 3CO in TS-B1 remains almost constant probably because of lesser charge transfer ability of 7H compared to OCH3 in TS-A1. The 9O as well as 10CH2 undergoes a large reduction in their populations as portrayed in Fig. 5.
From the above results, it is clear that the CH2–CH2 groups around N of PNP in both transition states (TS-A1 and TS-B1) receive the electronic population during the initial steps of the reaction. Thus, introducing electron-withdrawing groups in these positions must destabilize these transition states thereby reducing the catalytic activity of the catalyst, which is indeed the case as discussed in the next section.
Difference in catalytic activity by altering PNP ligand
In order to delineate the extent to which catalytic activity of the catalyst is influenced, we modified the PNP ligand with methyl (–CH3) and trifluoromethyl (–CF3) groups as shown in Scheme 3. The pictorial diagram of relative Gibbs free energy of such modified catalysts is portrayed in Fig. 6. Computational assessment of the alteration of ligands with CH3 and CF3 at 298.15 K suggest that transfer hydrogenation steps are more endergonic compared to OFe by 3.56 and 14.19 kcal mol−1. Apparently, as one can visualize from the figure, that TS-B1 (OFe) transition state lies well below the transition state of CF3-substituted catalyst (CFe). Furthermore, there is a little difference in relative Gibbs free energy between the OFe and MFe as previously suggested by various research groups.50 The Fig. 6 clearly depicts that electron-withdrawing groups like CF3 near N-atom of PNP ligand decrease the catalytic activity of the iron catalysts by making alcohol dehydrogenation difficult while as changing iPr-groups of PNP-ligand with CH3 don not have large effect. The reason for decrease in catalytic activity in CFe has already been mentioned in previous section. In addition to this, the electron-withdrawing groups like CF3 make N-atom electron-deficient in nature thereby decreasing its basicity. Furthermore, CF3 being bulky increase the steric crowding around N thereby reducing the overlap between N and methanol.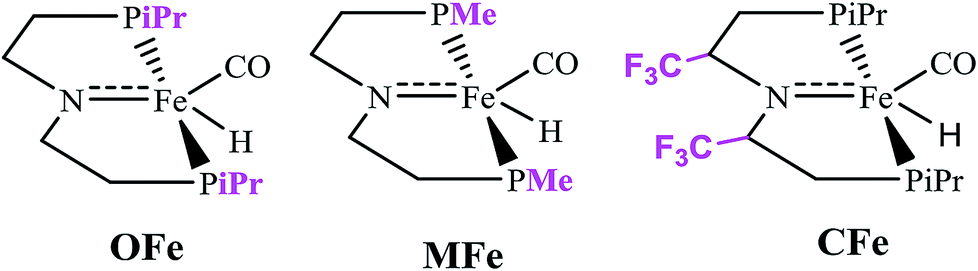 | ||
| Scheme 3 Catalysts examined by altering PNP ligands. | ||
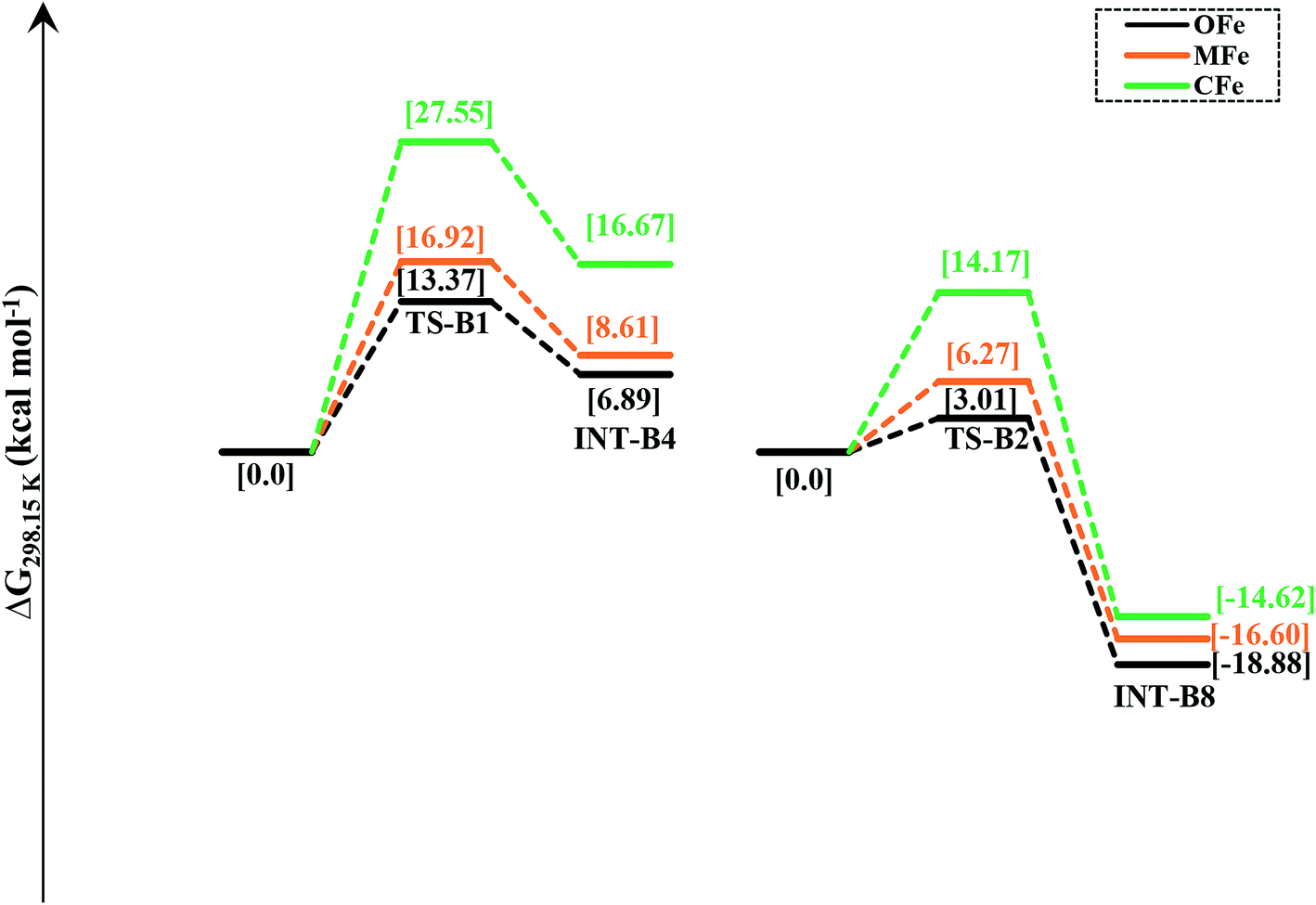 | ||
| Fig. 6 The effect on relative Gibbs free energy due to alteration of PNP ligand in OFe. OFe is having low relative Gibbs free energy on contrary to CFe and MFe, thus being highly active catalyst for amide formation. | ||
In order to gain in depth understanding of the effect of ligand modifications on the catalytic activity, we carried out molecular orbital analysis (Fig. 7) of all the complexes and methanol. The electronic distribution across Fe–N of the complexes is an important factor as it has to interact with the C–H/O–H σ-orbitals of CH3OH. The molecular orbital study shows that the HOMO of all complexes represent the Fe–N bonding orbital. The LUMO orbital of CH3OH having energy equal to 0.01 eV interacts with the HOMO of Fe–N bonding orbital which results in the dehydrogenation of methanol. The dehydrogenation of CH3OH is more feasible when the energy gap between HOMO of complexes and LUMO of methanol is lowest. These HOMO–LUMO gaps between the catalysts and methanol agrees well with the computed reaction Gibbs free energies. The most active catalyst is OFe with relative free energy equal to 13.37 kcal mol−1 is indeed having lowest HOMO–LUMO gap of 5.12 eV. The least active catalyst CFe with relative free energy equal to 27.55 kcal mol−1 is having maximum HOMO–LUMO gap (5.58 eV).
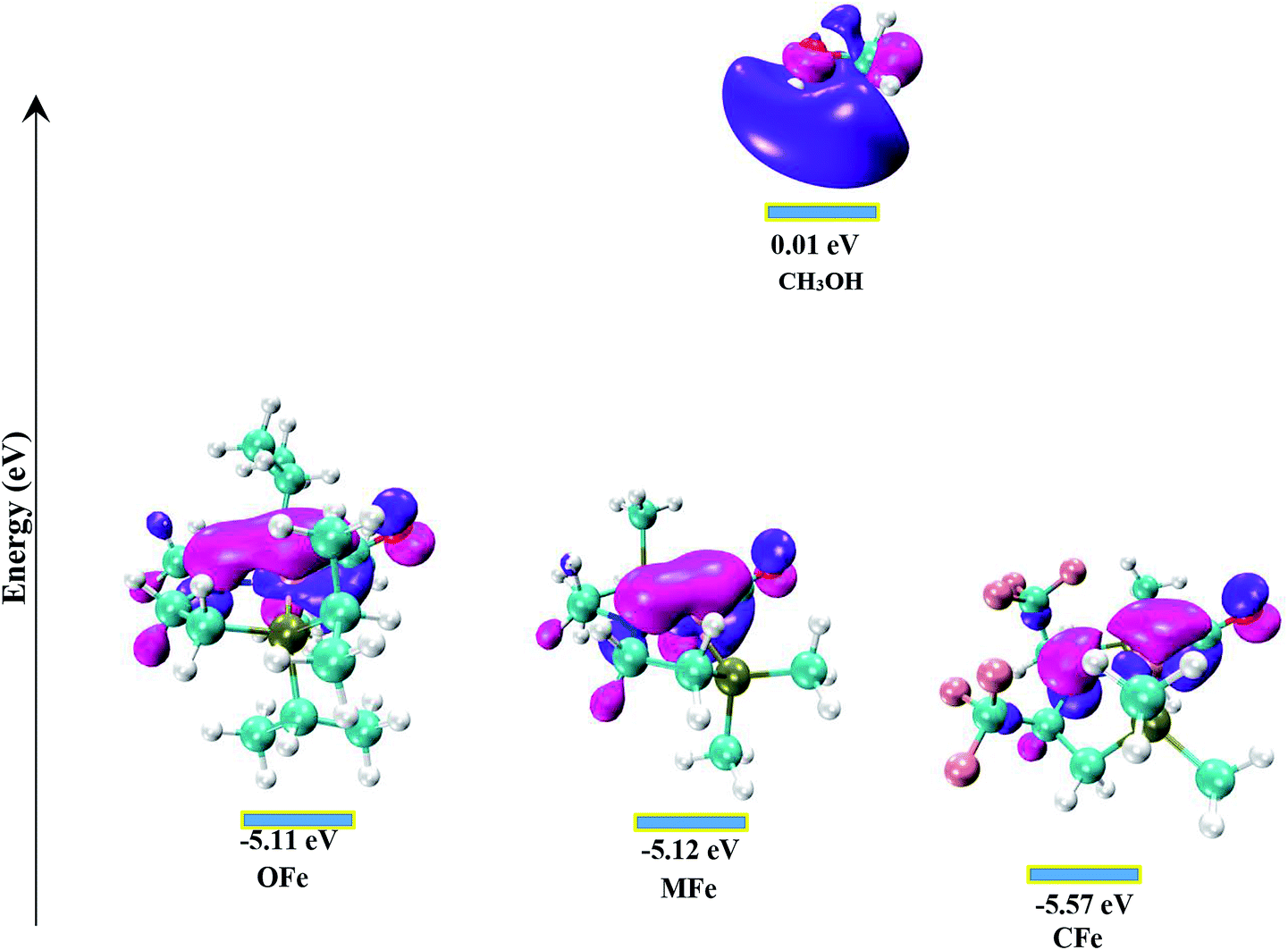 | ||
| Fig. 7 The energy difference between the LUMO of CH3OH and the HOMO of complexes. | ||
Thus, from the above discussion it is clear that OFe is highly active catalyst compared to its MFe and CFe analogous variants for the direct amide formation from alcohols and amines.
Conclusions
In this present work, the reaction mechanism of atom economical amide formation from alcohols and amines mediated by (iPrPNP)Fe(H)(CO) hydride complex reported by Bernskoetter and co-workers was investigated by DFT analysis. Two mechanistic scenarios were studied, the inner-sphere and the outer-sphere pathways. In former case, the methanol interacts through O–H bond while as in latter case the methanol interacts along C–H/O–H with the catalyst. We also studied the substituent effects and various electronic processes along IRC in first transition state formed in these two reported mechanistic pathways.The inner-sphere pathway operates through several steps: dissociation of methanol along O–H, dehydrogenation of iron-methoxy species, reaction of formaldehyde encounter complex with amine, formation of cis-dihydride complex and the elimination of dihydrogen. In first step of the reaction methanol reacts with the catalyst and thus forms a six coordinate iron-methoxy species. The second step involve dehydrogenation of iron-methoxy and thus forms an encounter complex of formaldehyde with metal center. Third step involves reaction of formaldehyde with amine. Subsequent rearrangement of the complex forms a cis-dihydride, which undergoes dehydrogenation in the final step. The reaction of INT-A2 with amine is the rate determining step with ΔG298.15 K = 33.75 kcal mol−1. On the contrary, outer-sphere pathway involves three major steps: (i) dehydrogenation of methanol to formaldehyde (ii) formation of hemiaminal from formaldehyde and amine (iii) amide formation by dehydrogenation of hemiaminal. In first step of catalysis, methanol undergoes dehydrogenation and thus forms formaldehyde. Second step involves subsequent reaction of formaldehyde to form hemiaminal, which enters the second catalytic cycle to form amide in the final step of the catalytic reaction. The dehydrogenation of methanol is high in energy than dehydrogenation of hemiaminal. In this mechanistic pathway, the loss of dihydrogen from trans-dihydride is the rate-determining step with ΔG298.15 K = 21.34 kcal mol−1. Both the mechanistic pathways are ionic in nature and the energetics of the reaction suggest that inner-sphere pathway of amide formation is unlikely. Our computational results of methanol dehydrogenation don not agree with neutral stepwise mechanism but instead the one involving ionic species which was proposed by Yang.46
Our computational results suggest that CH2–CH2 groups around N of PNP ligand receive electronic population and introducing EWD groups at these sites reduce the catalytic activity in OFe and thus making its analogous variant (CFe) less effective for amide formation. These results are supported by molecular orbital analysis, which suggest that HOMO–LUMO energy gap is lowest for OFe and highest for CFe.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financially supported by University Grants Commission (UGC), Govt. of India under UGC-BSR scheme as SRF (Senior Research Fellow) vide notification number, No. F.25-1/2013-14(BSR)/5-27/2007(BSR).References
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55–68 CrossRef CAS PubMed.
- T. J. Deming, Prog. Polym. Sci., 2007, 32, 858–875 CrossRef CAS.
- J. W. Bode, Curr. Opin. Drug Discovery Dev., 2006, 9, 765–775 CAS.
- B. S. Jursic and Z. Zdravkovdki, Synth. Commun., 1993, 23, 2761–2770 CrossRef CAS.
- B. Saha, G. Sengupta, A. Sarbajna, I. Dutta and J. K. Bera, J. Organomet. Chem., 2014, 771, 124–130 CrossRef CAS.
- A. J. A. Watson, R. J. Wakeham, A. C. Maxwell, M. J. Jonathan and J. M. J. Williams, Tetrahedron, 2014, 70, 3683–3690 CrossRef CAS.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed.
- S. W. Krabbe, V. S. Chan, T. S. Franczyk, S. Shekhar, J. G. Napolitano, C. A. Presto and J. A. Simanis, J. Org. Chem., 2016, 81, 10688–10697 CrossRef CAS PubMed.
- X. Xie and H. V. Huynh, ACS Catal., 2015, 5, 4143–4151 CrossRef CAS.
- T. T. Nguyen and K. L. Hull, ACS Catal., 2016, 6, 8214–8218 CrossRef CAS.
- S. Chakraborty, U. Gellrich, Y. Diskin-Posner, G. Leitus, L. Avram and D. Milstein, Angew. Chem., Int. Ed., 2017, 56, 4229–4233 CrossRef CAS PubMed.
- N. Ortega, C. Richter and F. Glorius, Org. Lett., 2013, 15, 1776–1779 CrossRef CAS PubMed.
- C. Chen, Y. Zhang and S. H. Hong, J. Org. Chem., 2011, 76, 10005–10010 CrossRef CAS PubMed.
- P. Hu, E. Fogler, Y. Diskin-Posner, M. A. Iron and D. Milstein, Nat. Commun., 2015, 6, 6859 CrossRef CAS PubMed.
- Y. Zhang, C. Chen, S. C. Ghosh, Y. Li and S. H. Hong, Organometallics, 2010, 29, 1374–1378 CrossRef CAS.
- C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC.
- C. Cheng Chen and S. H. Hong, Org. Biomol. Chem., 2011, 9, 20 RSC.
- C. Chen, F. Verpoort and Q. Wu, RSC Adv., 2016, 6, 55599 RSC.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- E. M. Lane, K. B. Uttley, N. Hazari and W. Bernskoetter, Organometallics, 2017, 36, 2020–2025 CrossRef CAS.
- D. Srimani, E. Balaraman, P. Hu, Y. Ben-David and D. Milstein, Adv. Synth. Catal., 2013, 355, 2525–2530 CrossRef CAS.
- N. J. Oldenhuis, V. M. Dong and Z. Guan, Tetrahedron, 2014, 70, 4213–4218 CrossRef CAS PubMed.
- G. Zeng and S. Li, Inorg. Chem., 2011, 50, 10572–10580 CrossRef CAS PubMed.
- I. S. Makarov, P. Fristrup and R. Madsen, Chem. - Eur. J., 2012, 18, 15683–15692 CrossRef CAS PubMed.
- H. Li, X. Wang, F. Huang, G. Lu, J. Jiang and Z. Wang, Organometallics, 2011, 30, 5233–5247 CrossRef CAS.
- E. A. Bielinski, M. Förster, Y. Zhang, W. H. Bernskoetter, N. Hazari and M. Holthausen, ACS Catal., 2015, 5, 2404–2415 CrossRef CAS.
- S. Chakraborty, P. O. Lagaditis, M. Förster, E. A. Bielinski, N. Hazari, M. C. Holthausen, W. D. Jones and S. Schneider, ACS Catal., 2014, 4, 3994–4003 CrossRef CAS.
- B. A. Shiekh, D. Kaur and S. K. Godara, Catal. Commun., 2019, 124, 19–23 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Kreig, J. Chem. Phys., 2010, 132, 154104–154119 CrossRef PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- S. Biswas, Z. Huang, Y. Choliy, D. Y. Wang, M. Brookhart, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2012, 134, 13276–13295 CrossRef CAS PubMed.
- D. Y. Wang, Y. Choliy, M. C. Haibach, J. F. Hartwig, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2016, 138, 149–163 CrossRef CAS PubMed.
- J. Jover, M. García-Ratés and N. López, ACS Catal., 2016, 6, 4135–4143 CrossRef CAS.
- S. Siek, D. B. Burks, D. L. Gerlach, G. Liang, J. M. Tesh, C. R. Thompson, F. Qu, J. E. Shankwitz, R. M. Vasquez, N. Chambers, G. J. Szulczewski, D. B. Grotjahn, C. E. Webster and E. T. Papish, Organometallics, 2017, 36, 1091–1106 CrossRef CAS PubMed.
- B. Marten, K. Kim, C. Cortis, R. A. Friesner, R. B. Murphy, M. N. Ringnalda, D. Sitkoff and B. Honig, J. Phys. Chem., 1996, 100, 11775–11788 CrossRef CAS.
- D. J. Tannor, B. Marten, R. Murphy, R. A. Friesner, D. Sitkoff, A. Nicholls, M. Ringnalda, W. A. Goddard and B. J. Honig, J. Am. Chem. Soc., 1994, 116, 11875–11882 CrossRef CAS.
- T. Fan, F. K. Sheong and Z. Lin, Organometallics, 2013, 32, 5224–5230 CrossRef CAS.
- F. Schoenebeck and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 2496–2497 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- Jaguar, Schrodinger, Inc., New York, NY, 2014 Search PubMed.
- E. A. Bielinski, M. Förster, Y. Zhang, W. H. Bernskoetter, N. Hazari and M. C. Holthausen, ACS Catal., 2015, 5, 2404–2415 CrossRef CAS.
- D. G. Gusev, ACS Catal., 2016, 6, 6967–6981 CrossRef CAS.
- X. Yang, ACS Catal., 2013, 3, 2684–2688 CrossRef CAS.
- C. Hou, Z. Zhang, C. Zhao and Z. Ke, Inorg. Chem., 2016, 55, 6539–6551 CrossRef CAS PubMed.
- G. Zeng, S. Sakaki, K. Fujita, H. Sano and R. Yamaguchi, ACS Catal., 2014, 4, 1010–1020 CrossRef CAS.
- M. Bertoli, A. Choualeb, A. J. Lough, B. Moore, D. Spasyuk and D. G. Gusev, Organometallics, 2011, 30, 3479–3482 CrossRef CAS.
- S. Chakraborty, P. O. Lagaditis, M. Förster, E. A. Bielinski, N. Hazari, M. C. Holthausen, W. D. Jones and S. Schneider, ACS Catal., 2014, 4, 3994–4003 CrossRef CAS.
- Y. Kang, X. Li, K. Cho, W. Sun, C. Xia, W. Nam and Y. Wang, J. Am. Chem. Soc., 2017, 139, 7444–7447 CrossRef CAS PubMed.
- A. K. Sharma, W. M. C. Sameera, M. Jin, L. Adak, C. Okuzono, T. Iwamoto, M. Kato, M. Nakamura and K. Morokuma, J. Am. Chem. Soc., 2017, 139, 16117–16125 CrossRef CAS PubMed.
- W. Lee, J. Zhou and O. Gutierrez, J. Am. Chem. Soc., 2017, 139, 16126–16133 CrossRef CAS PubMed.
- M. R. Sundberg, R. Uggla, C. Viñas, F. Teixidor, S. Paavola and R. Kivekäs, Inorg. Chem. Commun., 2007, 10, 713–716 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J. –P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03309b |
| This journal is © The Royal Society of Chemistry 2019 |