
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAncistrobreveines A–D and related dehydrogenated naphthylisoquinoline alkaloids with antiproliferative activities against leukemia cells, from the West African liana Ancistrocladus abbreviatus†
Shaimaa Fayeza,
Doris Feineisa,
Laurent Aké Assi‡
b,
Ean-Jeong Seoc,
Thomas Efferthc and
Gerhard Bringmann *a
*a
aInstitute of Organic Chemistry, University of Würzburg, Am Hubland, D-97074 Würzburg, Germany. E-mail: bringman@chemie.uni-wuerzburg.de
bCentre National de Floristique, Conservatoire et Jardin Botanique, Université d`Abidjan, Abidjan 08, Ivory Coast
cInstitute of Pharmacy and Biochemistry, Department of Pharmaceutical Biology, University of Mainz, Staudinger Weg 5, D-55128 Mainz, Germany
First published on 20th May 2019
Abstract
A unique series of six biaryl natural products displaying four different coupling types (5,1′, 7,1′, 7,8′, and 5,8′) were isolated from the roots of the West African liana Ancistrocladus abbreviatus (Ancistrocladaceae). Although at first sight structurally diverse, these secondary metabolites all have in common that they belong to the rare group of naphthylisoquinoline alkaloids with a fully dehydrogenated isoquinoline portion. Among the African Ancistrocladus species, A. abbreviatus is so far only the second one that was found to produce compounds with such a molecular entity. Here, we report on four new representatives, named ancistrobreveines A–D (12–14, and 6). They were identified along with the two known alkaloids 6-O-methylhamateine (4) and ent-dioncophylleine A (10). The two latter naphthylisoquinolines had so far only been detected in Ancistrocladus species from Southeast Asia. All of these fully dehydrogenated alkaloids have in common being optically active despite the absence of stereogenic centers, due to the presence of the rotationally hindered biaryl axis as the only element of chirality. Except for ent-dioncophylleine A (10), which lacks an oxygen function at C-6, the ancistrobreveines A–D (12–14, and 6) and 6-O-methylhamateine (4) are 6-oxygenated alkaloids, and are, thus, typical ‘Ancistrocladaceae-type’ compounds. Ancistrobreveine C (14), is the first – and so far only – example of a 7,8′-linked fully dehydrogenated naphthylisoquinoline discovered in nature that is configurationally stable at the biaryl axis. The stereostructures of the new alkaloids were established by spectroscopic (in particular HRESIMS, 1D and 2D NMR) and chiroptical (electronic circular dichroism) methods. Ancistrobreveine C (14) and 6-O-methylhamateine (4) exhibited strong antiproliferative activities against drug-sensitive acute lymphoblastic CCRF-CEM leukemia cells and their multidrug-resistant subline, CEM/ADR5000.
Introduction
Ancistrocladus abbreviatus Airy Shaw (Ancistrocladaceae)1–3 is a West African scandent shrub abundantly occurring in wet evergreen forests, along roadsides, river banks, and islands. In the juvenile phase, the plants start growing as erect, self-supporting saplings with leaves arranged in terminal rosettes. Reaching maturity, they form climbing stems from elongated shoots that bear vegetative, often robust leaves. Adult specimens can reach a height of more than 6 m, supported by means of recurved to spiraling woody hooks, as typical of the Ancistrocladaceae.1,4Phytochemical studies on A. abbreviatus collected in the Parc National de Taï, Southwestern Côte d’Ivoire (Ivory Coast), resulted in the discovery of ca. 30 structurally most diverse naphthylisoquinoline alkaloids displaying four different coupling types (5,1′, 7,1′, 5,8′, and 7,8′).5–11 The metabolic pattern of this West African plant is strongly dominated by the presence of 5,1′- and 7,1′-linked naphthylisoquinolines. All of the alkaloids with a 5,1′-biaryl linkage constitute typical Ancistrocladaceae-type compounds5,12 like e.g., ancistrobrevine E (1) (Fig. 1), i.e., with S-configuration at C-3 and an oxygen function at C-6.5,6,10,11 Most of the 7,1′-coupled alkaloids of A. abbreviatus, by contrast, among them the two main constituents, N-methyldioncophylline A (9a) and its atropo-diastereomer 9b (Fig. 1), are 3R-configured and lack an oxygen substituent at C-6.5,9,10 They are thus categorized as Dioncophyllaceae-type compounds,5,12 because the only other plant family that likewise produces naphthylisoquinoline alkaloids, the Dioncophyllaceae,13 exclusively forms alkaloids with these two structural features.5,12,14 They are – similar to A. abbreviatus – endemic to West Africa.1–3,13
 | ||
| Fig. 1 Naphthylisoquinolines from the roots of the West African shrub Ancistrocladus abbreviatus displaying four different coupling types, among them a unique series of six metabolites belonging to the small subgroup of alkaloids with a fully dehydrogenated isoquinoline portion, the four new ancistrobreveines A (12), B (13), C (14), and D (6), and two previously known26,27 alkaloids, 6-O-methylhamateine (4) and ent-dioncophylleine A (10); ancistrobenomine B (5) and the ancistrolikokines G (7) and J2 (8) are examples of fully dehydrogenated naphthylisoquinoline alkaloids displaying strong antiproliferative activities against leukemia cells.21,30 | ||
Remarkably, only two 5,8′-coupled alkaloids, ancistrobrevine B (2) and its 5′-O-demethyl analog 3 (Fig. 1), have so far been detected in A. abbreviatus,6,11 whereas naphthylisoquinoline alkaloids of this coupling type are the most prevalent ones in Central African Ancistrocladus lianas.5,12,15–25 Ancistrocladus abbreviatus furthermore contains a substantial number of representatives (six compounds) of the otherwise rare group of 7,8′-linked naphthylisoquinoline alkaloids, one of them is ancistrobrevine I (11) (Fig. 1).10,11 All of the 5,8′- and 7,8′-coupled alkaloids are typical Ancistrocladaceae-type compounds.
The by far largest group of constituents of A. abbreviatus are naphthyltetrahydroisoquinolines,5–7,9–11 whereas only four of the alkaloids had a dihydroisoquinoline subunit, among them compound 11.8,11
Here, we describe the isolation and structural elucidation of an unprecedented series of minor metabolites possessing a fully dehydrogenated isoquinoline half. The series comprises four new compounds, named ancistrobreveines A–D (12–14, and 6) and the two previously known26,27 naphthylisoquinolines 6-O-methylhamateine (4) and ent-dioncophylleine A (10) (Fig. 1). Since they were devoid of any stereogenic centers, the only element of chirality in these compounds was the rotationally hindered biaryl axis.
Fully dehydrogenated naphthylisoquinoline alkaloids have as yet been found quite rarely in nature. Prior to this work, only 14 such compounds (out of a total of more than 250 known naphthylisoquinoline alkaloids) had been identified in Ancistrocladus lianas, most of them from Asian taxa.20,21,26–30 Only recently, phytochemical studies on the Congolese liana A. likoko has shown the presence of fully dehydrogenated naphthylisoquinolines for the first time in an African species, too.20,21
Each of the six alkaloids discovered in A. abbreviatus was obtained in an optically active form, initially leaving open whether they were enantiomerically pure or just scalemic. Analysis by HPLC chromatography on a chiral phase in combination with ECD spectroscopy revealed all of them to be enantiopure, except for one of the alkaloids, the new 7,8′-linked ancistrobreveine C (14). This compound was found to be produced as an atropo-enantiomeric mixture, with the P-enantiomer being the by far major one occurring in the plant. Likewise noteworthy, compounds 12–14 are the first 7,1′- and 7,8′-coupled Ancistrocladaceae-type alkaloids found in nature that possess a fully dehydrogenated isoquinoline subunit.
Some of the dehydrogenated alkaloids isolated from other Ancistrocladus species, such as ancistrobenomine B (5) and the ancistrolikokines G (7) and J2 (8) (Fig. 1), have attracted attention due to their strong antiproliferative activities against human drug-sensitive CCRF-CEM leukemia cells and their multidrug-resistant subline, CEM/ADR5000, similar to the effects induced by the established anticancer drug doxorubicin.21,30 These findings prompted us to evaluate the cytotoxic potential of the six alkaloids of A. abbreviatus presented in this paper. The results provide further insight into structural prerequisites for the activities and into the impact of axial chirality on the antileukemic potential of fully dehydrogenated naphthylisoquinoline alkaloids, in particular.
Results and discussion
Isolation and structural elucidation of ancistrobreveines A–D and related naphthylisoquinoline alkaloids
An HPLC-UV-MS-guided screening of root bark extracts of A. abbreviatus gave hints at the presence of minor constituents, with UV and MS profiles typical of naphthylisoquinoline alkaloids with a non-hydrogenated isoquinoline subunit. For the isolation of these compounds, air-dried ground material of the roots was exhaustively extracted with MeOH–H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), followed by liquid–liquid partitioning with n-hexane and fractionation of the crude methanolic extract by column chromatography on silica gel. The alkaloid-containing subfractions were directly subjected to preparative reversed-phase HPLC, which permitted isolation of six fully dehydrogenated naphthylisoquinoline alkaloids. Two of them, 6-O-methylhamateine (4) and ent-dioncophylleine A (10), were well known from previous phytochemical studies26,27 on related Ancistrocladus species from Southeast Asia.
:1, v/v), followed by liquid–liquid partitioning with n-hexane and fractionation of the crude methanolic extract by column chromatography on silica gel. The alkaloid-containing subfractions were directly subjected to preparative reversed-phase HPLC, which permitted isolation of six fully dehydrogenated naphthylisoquinoline alkaloids. Two of them, 6-O-methylhamateine (4) and ent-dioncophylleine A (10), were well known from previous phytochemical studies26,27 on related Ancistrocladus species from Southeast Asia.
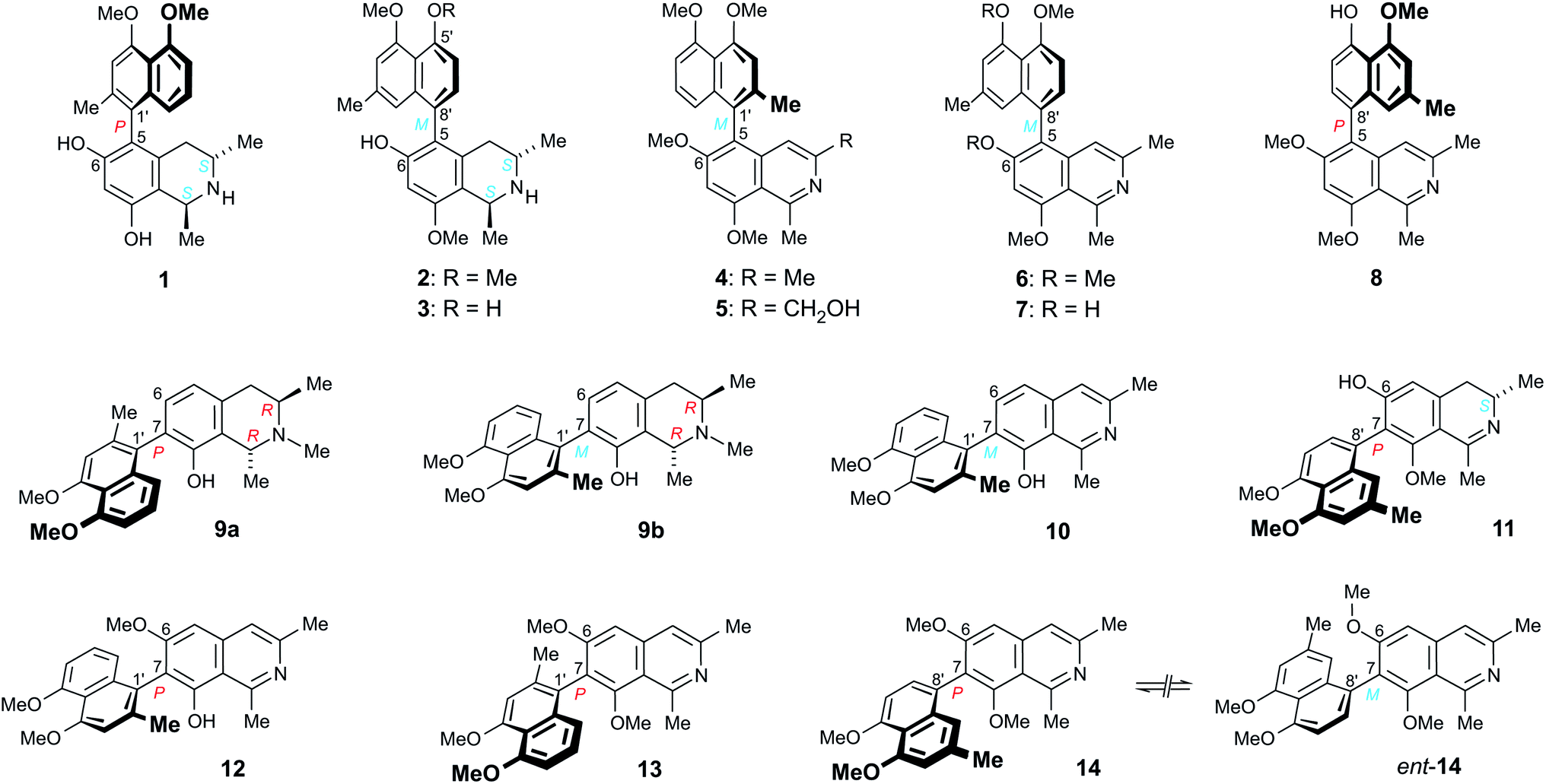
The 5,1′-linked 6-O-methylhamateine (4) had so far been identified only in the leaves of the Vietnamese species A. cochinchinensis, but its enantiomeric purity was never analyzed.26 HPLC analysis of the material of 4 isolated from the roots of A. abbreviatus on a chiral phase (Lux Cellulose-1) resulted only in one sharp peak (Fig. 2, left), thus revealing this fully dehydrogenated naphthylisoquinoline to occur in this West African species in an enantiomerically pure form. The ECD spectra recorded at different positions of the peak (e.g., at the left or right slope) were all identical, and virtually opposite to the ECD curve of ancistrocladeine (15)31 (Fig. 2, right), a known, likewise fully dehydrogenated and 5,1′-coupled, but P-configured (and 6-O-demethylated) naphthylisoquinoline, previously obtained by semi-synthesis from ancistrocladine.31,32
 | ||
| Fig. 2 HPLC analysis of 6-O-methylhamateine (4) on a chiral phase (Lux Cellulose-1) evidencing that 4 is produced by A. abbreviatus in an enantiomerically pure form, and comparison of the ECD spectrum of 4 with that of the known,31,32 likewise 5,1′-coupled, but P-configured ancistrocladeine (15). | ||
With its oxygen function at C-6, 6-O-methylhamateine (4) had the constitution of an Ancistrocladaceae-type naphthylisoquinoline, whereas the constitution of the second known compound isolated from the roots of A. abbreviatus, was categorized as a Dioncophyllaceae-type alkaloid, since it lacked an oxygen group at C-6. Within this classification, the presence or absence of the 6-oxygen function is given priority over the – here not applicable – absolute configuration at C-3.
The second fully dehydrogenated naphthylisoquinoline was readily identified as the 7,1′-linked ent-dioncophylleine A (10), which had previously been discovered in the leaves of the Malaysian highland liana A. benomensis.27 In that species, 10 had been found to be produced in a scalemic form, as a 93:7-mixture of 10 and its P-configured enantiomer dioncophylleine A.5,27,32
The material of 10 isolated from the roots of A. abbreviatus, by contrast, was determined to be enantiomerically pure, as obvious from HPLC analysis on a chiral Lux Cellulose-1 column, giving rise to only one peak (see ESI†), the ECD spectra measured in intervals at different sections of the peak were all identical.
The other four metabolites isolated from the root bark extracts of A. abbreviatus were as yet undescribed.
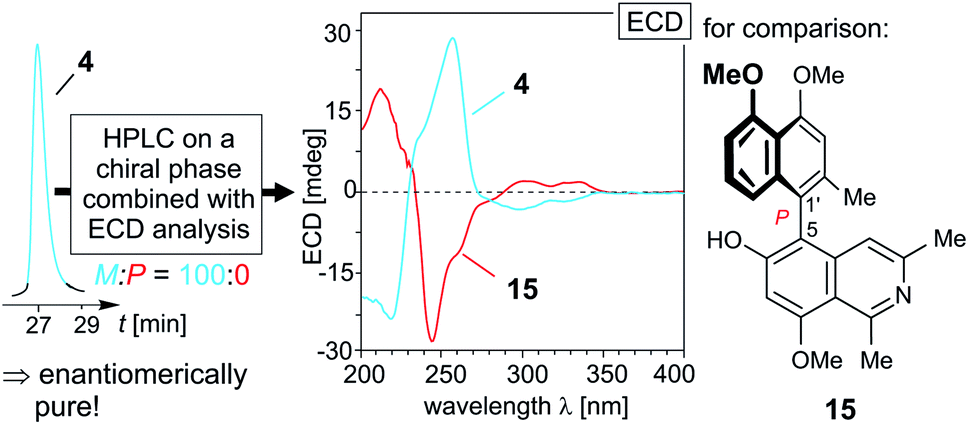
The first of these new alkaloids, compound 12, obtained as a yellow amorphous solid, had a molecular formula of C25H25NO4, as deduced from its monoprotonated molecular ion, [M + H]+, at m/z 404.1861 by HRESIMS. This, together with the UV spectrum displaying a second maximum around 258 nm, suggested it to be yet another fully dehydrogenated naphthylisoquinoline alkaloid. This assumption was corroborated by the appearance of an aromatic singlet at δH 7.77 in the 1H NMR spectrum (Table 1), instead of the signals of two diastereotopic geminal protons at C-4 usually observed for alkaloids with a tetra- or dihydroisoquinoline subunit. This finding was in accordance with the lack of resonances for protons at C-1 and C-3, i.e., with the missing quartet of H-1 around δH 4.20 and the absence of the multiplet of H-3 in the region between δH 3.20 and 4.00. Likewise typical of fully dehydrogenated naphthylisoquinolines was the significant downfield shift of the three-proton singlet of Me-1 (δH 3.20) in the 1H NMR spectrum and the likewise down-field-shifted 13C NMR resonances of C-1 (δC 158.4), C-3 (δC 143.7), and C-4 (δC 121.3) (Table 1). The new alkaloid 12 possessed three methoxy functions, two of them (δH 3.92 and 3.98) located at C-4′ and C-5′ in the naphthalene part, as obvious from NOESY correlations with H-3′ (δH 6.94) and H-6′ (δH 6.87) (Fig. 3A). In the isoquinoline half, the position of the remaining methoxy group was established to be at C-6 as deduced from a NOESY interaction between OMe-6 (δH 3.81) and H-5 (δH 7.15), leaving the fourth oxygenated carbon atom, C-8 (δC 158.0), to bear a free hydroxy function. In the aromatic region of the 1H NMR spectrum, the appearance of a spin system of three neighboring protons giving rise to two doublets (δH 6.87 and 6.73) and one pseudo-triplet (δH 7.18) indicated that the coupling site of the naphthalene moiety was either C-1′ or C-3′. The latter possibility was excluded by two NOESY correlation sequences, {Me-2′ ↔ H-3′ ↔ OMe-4′} and {OMe-5′ ↔ H-6′ ↔ H-7′ ↔ H-8′}, and by the high-field shift of the signal of Me-2′ (δH 2.11). Consequently, the biaryl axis had to be located at C-1′ (δC 158.4). This was also confirmed by HMBC long-range interactions from H-8′ (δH 6.73) and H-3′ to C-1′ (Fig. 3A). In the isoquinoline part, the coupling position was deduced to be at C-7 (δC 117.9), based on the NOESY correlation sequence {OMe-6 ↔ H-5 ↔ H-4} and on an HMBC interaction from H-5 to C-7. In conclusion, the new compound was a 7,1′-coupled alkaloid with a constitution similar to that of ent-dioncophylleine A (10), except for the additional methoxy group at C-6.27 The absolute configuration of this new Ancistrocladaceae-type naphthylisoquinoline 12 at the biaryl axis was attributed to be P, due to the fact that the new metabolite and 10 displayed nearly identical ECD spectra (Fig. 3B, left); for formal reasons, however, the two alkaloids have opposite descriptors, according to the Cahn–Ingold–Prelog denotion. The new fully dehydrogenated naphthylisoquinoline 12 thus possessed the stereostructure presented in Fig. 1. It was named ancistrobreveine A.
| Position | 12 | 13 | 14 | 6 | ||||
|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| a Overlapped with the methanol peak. | ||||||||
| 1 | 158.4 | 157.7 | 157.5 | 158.6 | ||||
| 3 | 143.7 | 144.0 | 143.8 | 141.8 | ||||
| 4 | 7.77, s | 121.3 | 7.90, s | 122.0 | 7.89, s | 122.0 | 6.86, s | 119.3 |
| 5 | 7.15, s | 98.9 | 7.40, s | 102.5 | 7.37, s | 102.3 | 116.9 | |
| 6 | 166.7 | 166.9 | 167.0 | 166.3 | ||||
| 7 | 117.9 | 126.5 | 128.1 | 7.20, s | 97.8 | |||
| 8 | 158.0 | 160.4 | 160.7 | 164.1 | ||||
| 9 | 114.5 | 118.0 | 118.0 | 114.2 | ||||
| 10 | 142.2 | 142.8 | 142.8 | 142.4 | ||||
| 1′ | 119.8 | 121.8 | 6.68, s | 118.2 | 6.42, s | 118.0 | ||
| 2′ | 138.8 | 137.4 | 138.7 | 138.2 | ||||
| 3′ | 6.94, s | 110.2 | 6.93, s | 109.8 | 6.79, s | 109.7 | 6.78, s | 109.9 |
| 4′ | 159.0 | 158.8 | 158.7 | 158.9 | ||||
| 5′ | 158.9 | 158.8 | 159.2 | 158.8 | ||||
| 6′ | 6.87, d (7.4) | 107.2 | 6.87, d (7.3) | 107.0 | 6.96, d (8.1) | 106.1 | 6.97, d (8.0) | 106.5 |
| 7′ | 7.18, dd (7.8, 8.3) | 128.0 | 7.21, dd (7.8, 8.4) | 128.0 | 7.26, d (8.0) | 130.7 | 7.14, d (7.9) | 131.0 |
| 8′ | 6.73, d (7.5) | 118.2 | 6.78, dd (0.9, 8.5) | 118.9 | 122.7 | 124.2 | ||
| 9′ | 138.0 | 137.6 | 137.4 | 137.6 | ||||
| 10′ | 118.0 | 117.5 | 117.2 | 117.5 | ||||
| 1-Me | 3.20, s | 23.3 | 3.20, s | 22.5 | 3.20, s | 22.5 | 3.23, s | 23.6 |
| 3-Me | 2.69, s | 18.5 | 2.74, s | 18.5 | 2.74, s | 18.4 | 2.40, s | 18.5 |
| 2′-Me | 2.11, s | 20.4 | 2.16, s | 20.8 | 2.28, s | 22.0 | 2.19, s | 21.9 |
| 6-OMe | 3.81, s | 57.0 | 3.85, s | 57.2 | 3.31a | 57.2 | 3.92, s | 56.9 |
| 8-OMe | 3.30a | 61.1 | 3.85, s | 61.6 | 4.25, s | 57.2 | ||
| 4′-OMe | 3.98, s | 56.7 | 3.98, s | 56.7 | 3.93, s | 56.8 | 3.93, s | 57.1 |
| 5′-OMe | 3.92, s | 56.9 | 3.93, s | 56.8 | 3.97 s | 56.6 | 3.98, s | 56.7 |
 | ||
| Fig. 3 (A) Selected 1H and 13C NMR data (in methanol-d4, δ in ppm), HMBC (single green arrows), and NOE interactions (double red arrows) of ancistrobreveines A (12) and B (13); the values of 13 that are different from those of 12 are given in {}; (B) HPLC-ECD analysis on a chiral phase (Lux Cellulose-1) giving only one peak, revealing that in A. abbreviatus 12 occurs in an enantiopure form (left), and assignment of the absolute axial configuration of 12 (center) and 13 (right) by comparison of the ECD spectrum of 12 with that of the 7,1′-coupled known27 M-configured ent-dioncophylleine A (10; for its structure, see Fig. 1), and by comparison of the ECD spectrum of 13 with that of 12. | ||
Like in the case of ent-dioncophylleine A (10) isolated from A. abbreviatus, ancistrobreveine A (12) was found to be enantiomerically pure by HPLC-ECD analysis on a chiral phase (Lux Cellulose-1), yielding only one peak (Fig. 3B, left). The ECD chromatogram monitored for one single wavelength (here at 258 nm, where 12 has a strong negative ECD signal) resulted in one single – and negative – peak. The online ECD spectra obtained from different peak positions were all identical.
HRESIMS analysis of the second new alkaloid, compound 13, gave a molecular formula of C26H27NO4, which thus had 14 mass units more than ancistrobreveine A (12). Similar to the latter, the new compound 13 again displayed NMR signals (see Table 1) typical of a fully dehydrogenated 7,1′-coupled naphthylisoquinoline. This metabolite had nearly the same constitution as 12, but possessed a methoxy function (δH 3.92) at C-8 (instead of a hydroxy group as in 12) (Fig. 3A). Its nearly mirror-image-like ECD spectrum compared to that of ancistrobreveine A (12) (Fig. 3B, right) revealed the isolated metabolite to exhibit an opposite axial stereo-array. For formal reasons, however, the two compounds had the same P-descriptor according to the Cahn–Ingold–Prelog denotion. Consequently, the new alkaloid had the full stereostructure 13 as presented in Fig. 1. It was named ancistrobreveine B. Similar as in the case of 12, HPLC analysis on a chiral phase coupled to ECD spectroscopy revealed 13 to occur in the plant in an enantiomerically pure form.
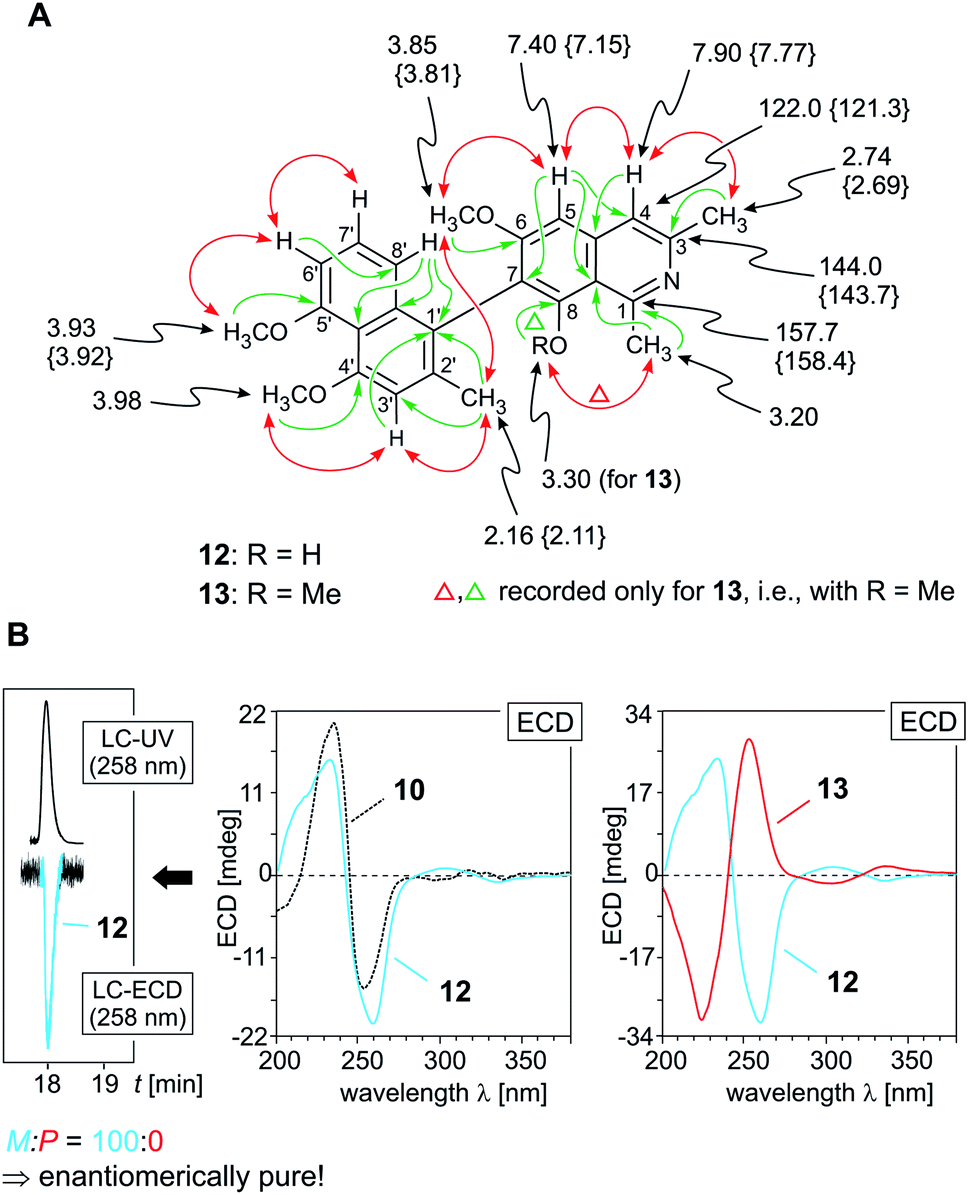
According to HRESIMS and 13C NMR, the third new alkaloid, compound 14, likewise a minor metabolite of the root bark extract, corresponded to a molecular formula of C26H27NO4, identical to that of ancistrobreveine B (13). NMR spectroscopic data (Table 1) again suggested the presence of a fully dehydrogenated naphthylisoquinoline alkaloid equipped with four methoxy functions, resonating at δH 3.30, 3.85, 3.93, and 3.97. Different from 13, the new compound 14 displayed a normal, not high-field shifted signal of Me-2′ (δH 2.28) and two NOESY correlation sequences, {H-1′ ↔ Me-2′ ↔ H-3′ ↔ OMe-4′} and {OMe-5′ ↔ H-6′ ↔ H-7′}, which evidenced the naphthalene portion to be connected to the isoquinoline part via its methyl-free ring, establishing C-8 to be the coupling position. This was further confirmed by HMBC interactions from H-1′ (δH 6.68) and H-6′ (δH 6.96) to the quaternary carbon atom C-8′ (δC 160.7). In the isoquinoline moiety, C-7 was determined to be the axis-bearing carbon atom, as obvious from NOESY interactions in the series {H-4 ↔ H-5 ↔ OMe-6} and from HMBC cross peaks to C-7 (δC 128.1), observed for both, the signals of H-5 (δH 7.37) and H-7′ (δH 7.26). In conclusion, the new alkaloid 14 was a 7,8′-coupled naphthylisoquinoline with the constitution shown in Fig. 4A.
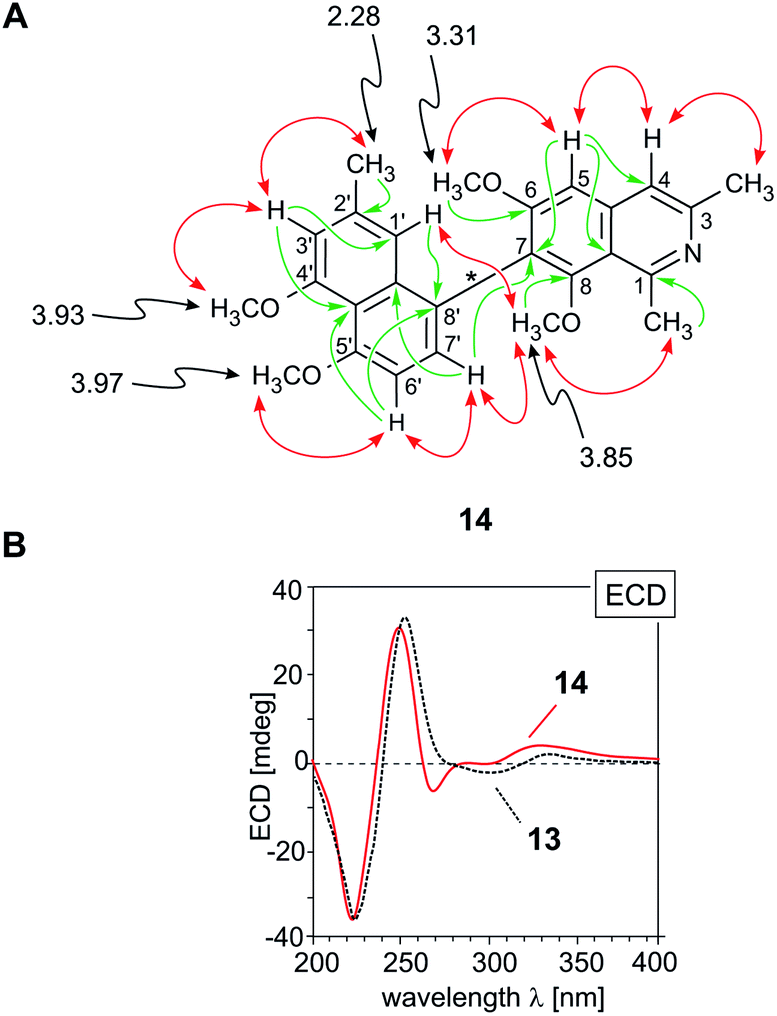 | ||
| Fig. 4 (A) Selected 1H NMR data (in methanol-d4, δ in ppm), HMBC (single green arrows), and NOE interactions (double red arrows) of ancistrobreveine C (14); (B) assignment of the absolute axial configuration of 14, by comparison of its ECD spectrum with that of the structurally related likewise P-configured, but 7,1′-coupled ancistrobreveine B (13, for its structure, see Fig. 1). | ||
Its absolute axial configuration was deduced by comparison of its ECD spectrum with those of ent-dioncophylleine A (10)27 (see ESI†) and ancistrobreveine B (13) (Fig. 4B), the latter differing from the new alkaloid 14 only by the position of the methyl group on the naphthalene moiety. The influence of that methyl group on the ECD behavior can be assumed as small compared with that of the large naphthalene chromophore. The ECD spectrum of the new metabolite 14 was nearly identical to that of 13 and virtually opposite to that of 10, which evidenced the biaryl axis of the isolated 7,8′-linked alkaloid to be P-configured. The new compound 14 thus had the full stereostructure presented in Fig. 1; it was named ancistrobreveine C.
HPLC-UV analysis of 14 on a chiral phase (Lux Cellulose-1) resulted in a nearly unresolved chromatographic peak (Fig. 5A, left), but the ECD trace of an HPLC run at 255 nm showed a small negative signal at the rising slope of the UV-detected peak and a large positive one on the descending side (see ESI†), thus suggesting that in the plant ancistrobreveine C (14) occurs in a not entirely enantiopure form. This was further confirmed by full LC-ECD spectra recorded online, in stopped-flow mode, directly taken at the left slope of the UV peak and at the right one, giving mirror-imaged ECD curves (Fig. 5A).
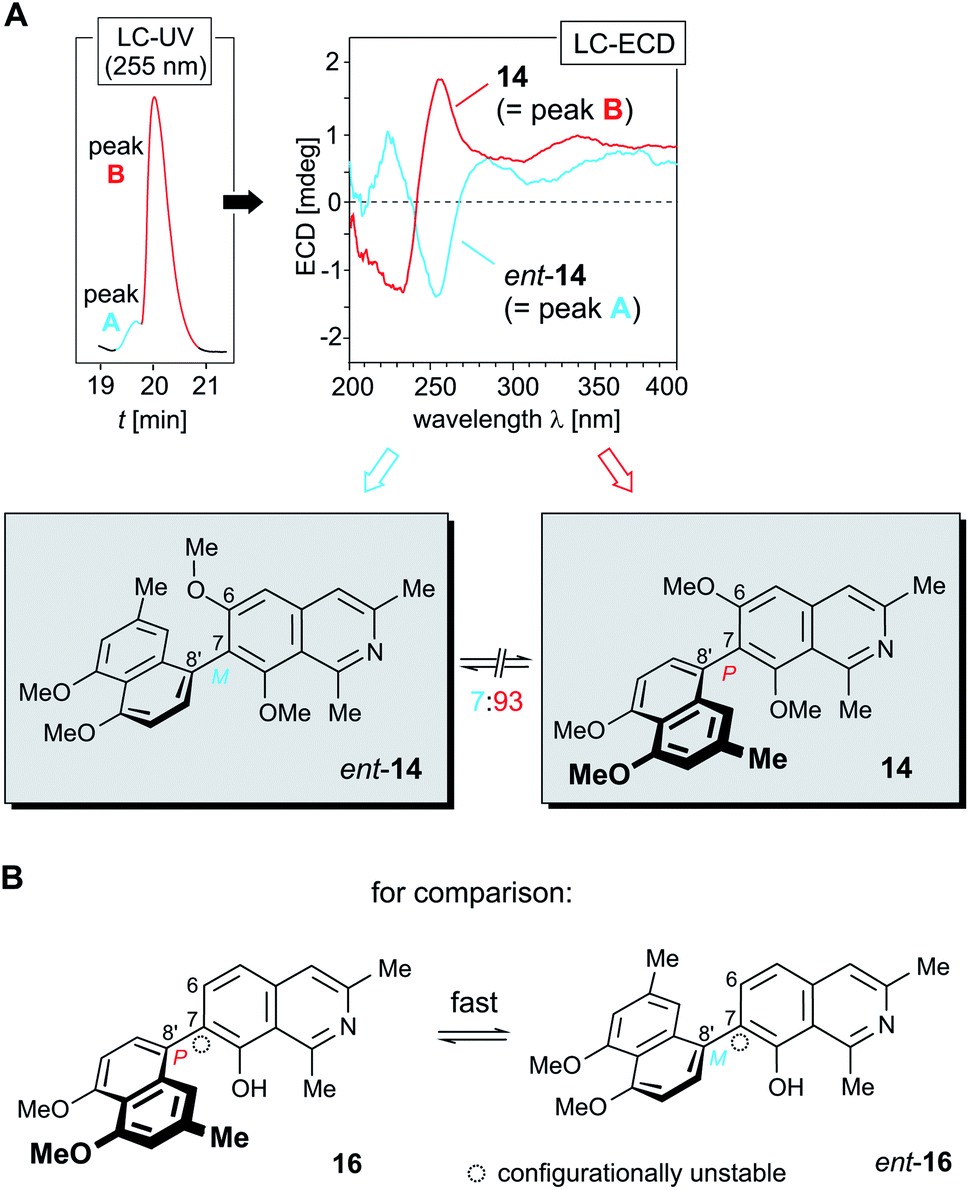 | ||
| Fig. 5 (A) HPLC-ECD analysis on a chiral phase (Lux Cellulose-1), revealing that in A. abbreviatus 14 occurs as a mixture of its two enantiomers 14 and ent-14 in a ratio of ca. 93:7; (B) the structurally closely related, likewise 7,8′-coupled dioncophylleine D (16),27 which lacks a methyl group ortho to its biaryl axis: this structural feature results in the occurrence of 16 as a racemic mixture of its rapidly interconverting enantiomers. | ||
Consequently, one enantiomer indeed eluted faster than the other one, but the interactions of the two enantiomers with the adsorbent material were not different enough to achieve a baseline resolution of these two compounds. The more rapidly eluting minor peak (peak A, Fig. 5A, left) was easily identified to correspond to the M-configured enantiomer of ancistrobreveine C, ent-14, as obvious from its ECD curve, which was fully opposite to that of the prevalent enantiomer 14 (peak B, Fig. 5B, right). The ratio of the two enantiomers, 14:ent-14, was determined to be ca. 93:7.
As in numerous earlier cases,33–38 this finding again demonstrated the usefulness of the hyphenation of HPLC with ECD spectroscopy. This analytical device permitted to reliably distinguish between the two enantiomers of 14, although they exhibited only slightly different retention times and were present in quite different concentrations.
Ancistrobreveine C (14) is the as yet only example of a 7,8′-linked fully dehydrogenated naphthylisoquinoline alkaloid that is optically active. The only other known alkaloid of this coupling type with a similar molecular framework, dioncophylleine D (16)27 (Fig. 5B), a Dioncophyllaceae-type alkaloid isolated from the Malaysian liana A. benomensis, was found to occur as a racemic mixture of its two rapidly interconverting atropo-enantiomers, which are configuratively unstable due to the lack of an oxygen function at C-6.27
HRESIMS analysis of the fourth new metabolite (m/z 418.1925 [M + H]+), compound 6, again gave a molecular formula of C26H27NO4. Likewise similar to the ancistrobreveines B (13) and C (14), its 1H NMR spectrum (Table 1) showed the chemical shifts of a fully dehydrogenated naphthylisoquinoline with four O-methyl groups (δH 3.92, 3.93, 3.98, and 4.25), but its proton pattern in the aromatic region was distinctly different from those of the 7,1′- and 7,8′-coupled alkaloids described above.
In the naphthalene part, two meta-coupled protons (δH 6.42 and 6.78) were observed, together with an AB spin system (δH 6.97 and 7.14), indicating that either C-8′ or C-6′ was the axis-bearing carbon atom. The latter possibility was excluded as one of the protons of the AB spin system, the doublet signal at δH 6.97, was located at C-6′, as obvious from a COSY correlation between H-6′ and H-7′ (δH 7.14). NOESY cross-peaks in the series {H-1′ ↔ Me-2′ ↔ H-3′ ↔ OMe-4′} and joint HMBC interactions from H-1′ (δH 6.42) and H-6′ to C-8′ assigned the coupling position in the naphthalene part to be at C-8′ (δC 124.2) (Fig. 6A).
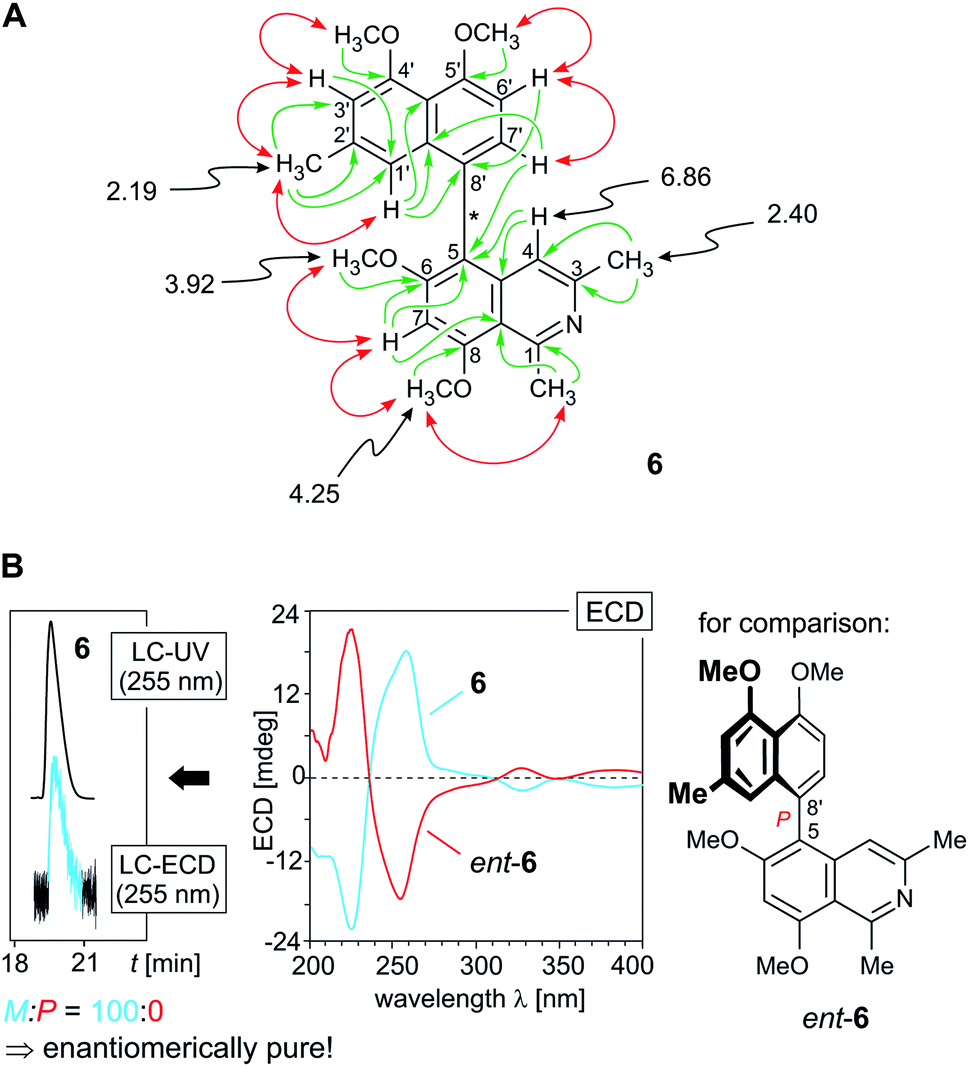 | ||
| Fig. 6 (A) Selected 1H NMR data (in methanol-d4, δ in ppm), HMBC (single green arrows), and NOE interactions (double red arrows) of ancistrobreveine D (6); (B, right) assignment of the absolute axial configuration of 6 by comparison of its ECD spectrum with that of the known,21 likewise 5,8′-coupled, but P-configured ancistrolikokine J3 (ent-6), earlier isolated from A. likoko, which is the enantiomer of ancistrobreveine D; (B, left) HPLC-ECD analysis on a chiral phase (Lux Cellulose-1) giving only one peak, revealing that 6 occurs in A. abbreviatus in an enantiopure form. | ||
In the isoquinoline subunit, 3J HMBC correlations from H-7 (δH 7.20) and H-7′ to C-5 (δC 116.9) established the biaryl axis to be located at C-5, which was confirmed by the NOESY correlation sequence {Me-1 ↔ OMe-8 ↔ H-7 ↔ OMe-6}. The new alkaloid 6 was hence 5,8′-coupled and possessed the constitution as presented in Fig. 6A. Its absolute axial configuration was deduced to be M, since it displayed a virtually mirror-imaged ECD spectrum (Fig. 6B, center) compared to that of the constitutionally identical ancistrolikokine J3 (ent-6),21 which is P-configured at the biaryl axis. This alkaloid has only recently been identified in the Congolese liana A. likoko and represents the enantiomer of this 5,8′-linked compound 6 isolated from A. abbreviatus. The new alkaloid 6 presented here thus had the full absolute stereostructure shown in Fig. 1. In continuation of the series of fully dehydrogenated metabolites discovered in the roots of A. abbreviatus, it was named ancistrobreveine D.
Investigations on the enantiomeric purity of 6 by chromatography on a chiral phase coupled to ECD spectroscopy resulted in only one peak. The online ECD spectra, which were monitored in intervals at the left and the right slopes of the peak, were all identical. In the plant, ancistrobreveine D (6) was thus present in an enantiopure form, similar to its P-configured enantiomer ancistrolikokine J3 (ent-6), which was likewise found to be produced stereochemically pure in A. likoko.21
Antileukemic activities of the ancistrobreveines A–D and of related fully dehydrogenated naphthylisoquinoline alkaloids
Multi-drug resistance (MDR) leading to recurrent and refractory leukemia is one of the major problems in the treatment of this severe malignancy.39,40 Overexpression of P-glycoprotein (P-gp), an ATP-dependent drug pump, is considered to be the most important factor for the development of MDR, because it results in an increased drug efflux and, thus, in a dramatic reduction of intracellular drug concentrations of the antileukemic compounds in the cells.41–43 Therefore, the search for novel therapeutic agents to overcome MDR44,45 and to efficiently target leukemia is still an urgent task.More recently, some representatives of naphthylisoquinolines possessing a fully dehydrogenated isoquinoline portion were found to strongly inhibit the viability of drug-sensitive CCRF-CEM leukemic cells and their multidrug-resistant P-gp-overexpressing subline, CEM/ADR5000.21,30 In particular, the 5,1′-coupled ancistrobenomine B (5)30 (Fig. 1) from the Chinese liana A. tectorius, additionally equipped with a hydroxymethylene function at C-3, and the 5,8′-linked ancistrolikokine G (7)20,21 (Fig. 1), a minor metabolite of the Congolese liana A. likoko, attracted attention, since they inhibited cell proliferation at a low micromolar range, revealing only a small degree of cross-resistance, or, as in the case of ancistrolikokine J2 (8)21 (Fig. 1), likewise from A. likoko, even collateral sensitivity.46 The alkaloid 8 displayed a more pronounced growth-retarding activity towards the CEM/ADR5000 cells as compared to that against the parental CCRF-CEM cell line (Table 2).21 This finding indicated that these alkaloids may bear a substantial therapeutic potential.
| Compound | IC50a [μM] | ||
|---|---|---|---|
| CCRF-CEM | CEM/ADR5000 | Degree of resistanceb | |
| a The lymphoblastic leukemia cells were treated with different concentrations of 4, 6, 10, and 12–14 or with doxorubicin. Cell viability was assessed by the resazurin assay. Mean values and standard deviation of three independent experiments with each six parallel measurements are shown.b The degrees of resistance were calculated by division of the IC50 values of the compounds for CEM/ADR5000 cells by the corresponding IC50 values for CCRF/CEM cells.c Values reported earlier, see ref. 21 and 30. | |||
| Doxorubicin | 0.017 ± 0.002 | 30.07 ± 11.81 | 1769 |
| 4 | 3.948 ± 1.667 | 5.523 ± 1.756 | 1.40 |
| 5c | 3.501 ± 0.069 | 21.38 ± 1.302 | 6.11 |
| 6 | 24.27 ± 1.398 | 29.51 ± 4.297 | 1.21 |
| 7c | 4.731 ± 0.690 | 7.731 ± 0.992 | 1.63 |
| 8c | 23.34 ± 1.630 | 20.06 ± 2.231 | 0.86 |
| 10 | 26.88 ± 0.285 | 31.41 ± 3.550 | 1.17 |
| 12 | 24.78 ± 3.910 | >100 | — |
| 13 | 39.94 ± 6.864 | 50.64 ± 7.465 | 1.27 |
| 14 | 12.44 ± 1.661 | 12.64 ± 2.290 | 1.02 |
Here, we report on the evaluation of the antileukemic effects of 6-O-methylhamateine (4), ent-dioncophylleine A (10), and the four new ancistrobreveines A–D (12–14, and 6) (Table 2), especially focussing on the influence of the position of the biaryl linkage in these fully dehydrogenated naphthylisoquinoline alkaloids and on the impact of axial chirality on the cytotoxic activities of these compounds.
The lymphoblastic leukemia cells were treated with different concentrations of the respective naphthylisoquinolines in a range from 0.001 to 100 μM or with the reference drug doxorubicin. Cell viability was assessed by the resazurin assay.
Within this series of six naphthylisoquinolines exhibiting four different coupling types (5,1′, 5,8′, 7,1′, and 7,8′), the 5,1′-linked 6-O-methylhamateine (4) was by far the most potent compound (Table 2). Dose–response curves of 4 (Fig. 7) revealed excellent half-maximum inhibitory concentrations in the low micromolar range towards CCRF-CEM cells (IC50 = 3.948 μM), and against the resistant CEM/ADR5000 subline (IC50 = 5.523 μM). The MDR leukemia cell line CEM/ADR5000, which is known to be highly resistant to the standard drug doxorubicin (ca. 1.770-fold compared to CCRF-CEM), showed only a low degree of cross-resistance for 6-O-methylhamateine (4) (1.4-fold) (Table 2), thus indicating that 4 may be a strong and efficient inhibitor also for other drug-sensitive cancer cells.
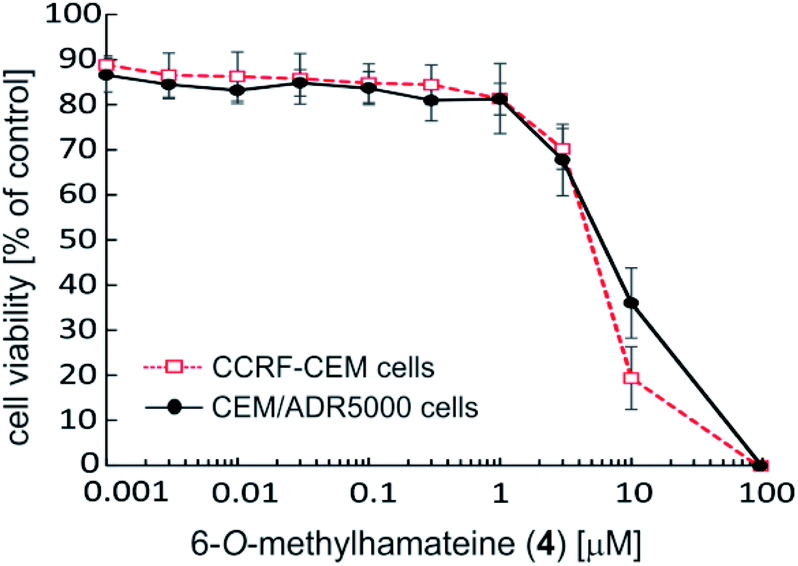 | ||
| Fig. 7 Cytotoxic activity of 6-O-methylhamateine (4) against parental drug-sensitive CCRF-CEM leukemia cells and their multi-drug resistant subline, CEM/ADR5000. The compound was dissolved in DMSO (<1%) and cell culture medium at concentrations of 0.001, 0.003, 0.01, 0.03, 0.1, 0.3, 1, 3, 10, or 100 μM. Cell viability was assessed by the resazurin assay. Mean values and standard deviation of three independent experiments with each six parallel measurements are shown. | ||
Considerable antiproliferative activities against the drug-sensitive CCRF-CEM and the multidrug-resistant CEM/ADR5000 leukemia cells were also observed for the 7,8′-coupled ancistrobreveine C (14) (Table 2). Its growth-inhibitory potential was distinctly lower than that of 4, with an IC50 value of 12.44 μM determined in CCRF-CEM cells, showing that, in comparison to 4, the cytotoxicity of 14 was reduced down to ca. one third. No cross-resistance was observed for ancistrobreveine C (14). Its IC50 values were nearly the same in both cell lines, thus inhibiting the cell viability of sensitive and multidrug-resistant leukemia cells with similar efficacies (Table 2).
The three 7,1′-coupled alkaloids ancistrobreveines A (12) and B (13), and ent-dionocophylleine A (10), structurally closely related to the 7,8′-linked ancistrobreveine C (14), by contrast, showed only moderate to weak antiproliferative activities against leukemia cancer cells. As outlined in Table 2, their IC50 values ranged from 24.8 to 50.6 μM. Ancistrobreveine A (12) even did not display any considerable cytotoxic effect at all against the multidrug-resistant CEM/ADR5000 subline. Similar to ancistrobreveine C (14), two of the 7,1′-coupled compounds, 10 and 13, showed growth inhibitory activities that were nearly the same in both cell lines, thus they did not exhibit any cross-resistances.
The previously investigated 5,8′-coupled fully dehydrogenated ancistrolikokine G (7),21 with its particular OH/OMe-substitution pattern in the naphthalene and isoquinoline portions, had displayed strong cytotoxic effects against CCRF-CEM and CEM/ADR5000 leukemia cells, with IC50 values in the low micromolar range (Table 2). The now isolated O-permethylated ancistrobreveine D (6), by contrast, exhibited only moderate growth-retarding activities: compared to the IC50 values of ancistrolikokine G (7),21 the growth-inhibitory potential of compound 6 was reduced by a factor of ca. 5.1 for the drug-sensitive CCRF-CEM cells, and by a factor of ca. 3.8 for the multi-drug resistant subline CEM/ADR5000. These test results showed that the degree of O-methylation in the two molecular portions may have a distinct impact on the antileukemic activities of 5,8′-coupled naphthylisoquinoline alkaloids.
The position of the biaryl axis seemed to be of crucial importance, too, as seen from the fact that the 5,1′-linked 6-O-methylhamateine (4) strongly inhibited the cell viability of leukemia cells (Fig. 7), whereas the related 5,8′-coupled ancistrobreveine D (6), which, in other words, differed from 4 only by the position of its methyl group in the naphthalene subunit, was only moderately active.
In summary, from previous studies21,30 and the results presented here, 5,1′- and 5,8′-coupled alkaloids were the most potent ones among the fully dehydrogenated naphthylisoquinolines tested. Most of these alkaloids were found to inhibit drug-sensitive CCRF-CEM and multidrug-resistant CEM/ADR5000 leukemia cells with nearly similar efficacies, revealing only minimal (ca. 1.2- to 6-fold) or even no cross-resistances. With respect to these findings, the compounds might also serve as promising agents for the treatment of cancer cells unresponsive towards chemotherapeutics routinely used in clinical treatment.
Experimental
General experimental procedures
All organic solvents were of analytical-grade quality. Ultra-pure water was obtained from an Elga Purelab Classic system. Optical rotations were measured on a Jasco P-1020 polarimeter operating with a sodium light source (λ = 589 nm). UV spectra were recorded on a Shimadzu UV-1800 spectrophotometer. IR spectra were taken on a Jasco FT/IR-410 spectrometer. ECD spectra were acquired on a Jasco J-715 spectropolarimeter at room temperature, using a 0.1 cm standard cell and spectrophotometric-grade MeOH. The ECD data were processed using SpecDis.47,48 1D and 2D NMR measurements were performed on a Bruker DMX 600 instrument using methanol-d4, with the 1H or 13C signals of methanol (1H, δ 3.31 ppm, 13C, δ 49.0 ppm) in the deuterated solvent as the internal reference. Chemical shifts (δ) are reported in parts per million (ppm), and coupling constants (J) are given in Hertz (Hz). NMR signal multiplicities are denoted as singlet (s), doublet (d), doublet of doublets (dd), quartet (q), or multiplet (m). LC-MS investigations were performed on an Agilent 1100 system equipped with a photodiode array (PDA) detector and an Esquire 3000 Plus ion-trap mass spectrometer using an electrospray ionization interface. HRESIMS measurements were acquired in positive mode on a Bruker Daltonics micrOTOF-focus mass instrument. Preparative HPLC was carried out on a Jasco System (PU-1580 Plus) in combination with UV/Vis detection at 200–680 nm (Jasco MD-2010 Plus diode array detector) at room temperature. For the isolation and purification of the constituents of the root bark extracts, a SymmetryPrep™ C18 column (19 × 300 mm, 7 μm, Waters) was used; mobile phases: (A) 90% H2O with 10% MeCN (0.05% trifluoroacetic acid) and (B) 90% MeCN with 10% H2O (0.05% trifluoroacetic acid). For further purification, a Waters XSelect HSS PFP column (10 × 250 mm, 5 μm) was applied; mobile phases: (A′) 90% H2O with 10% MeOH (0.05% trifluoroacetic acid) and (B′) 90% MeOH with 10% H2O (0.05% trifluoroacetic acid). The enantiomeric purities of the fully dehydrogenated compounds were analyzed by HPLC-ECD employing a chiral Lux Cellulose-1 column (250 × 4.6 mm, 5 μm, Phenomenex), with H2O (0.05% trifluoroacetic acid) (A′′) and MeCN (0.05% trifluoroacetic acid) (B′′) as the eluents.Plant material
Roots of Ancistrocladus abbreviatus Airy Shaw (Ancistrocladaceae) were collected by late Prof. L. Aké Assi (Centre National de Floristique, Université d’Abidjan) in May 1996, in the Parc National de Taï, in the South-Western region of the Ivory Coast. A voucher specimen (no. 3) has been deposited at the Herbarium Bringmann, Institute of Organic Chemistry, University of Würzburg.Extraction and isolation
Air-dried, powdered root bark material (ca. 420 g) of A. abbreviatus was repeatedly extracted for 1 h with MeOH (2 × 3 L) at 40 °C, assisted by ultrasonication. The extract was filtered and then concentrated in vacuo. The solid residue was dissolved in MeOH/H2O (9:1) and repeatedly washed with n-hexane to remove the non-polar impurities. The methanolic layer was dried under reduced pressure to yield ca. 17.5 g of a viscous alkaloid-rich solution. It was directly subjected to silica gel column chromatography (CC) using a linear solvent system of MeOH in CH2Cl2 (10 → 80%).
Resolution of fraction F1 (2.1 g) on a SymmetryPrep C18 column, using the solvent systems A and B, with a linear gradient (0 min 26% B, 27 min 65% B), at a flow rate of 10 mL min−1, furnished some alkaloid-containing subfractions, which were further purified by semi-preparative HPLC on an XSelect HSS PFP column. Applying isocratic solvent systems of the mobile phases A′ and B′ (v/v), containing 57 or 59% of B′, at a flow rate of 4 mL min−1, provided pure ancistrobreveine A (12) (1.4 mg) (retention time 15.4 min), ancistrobreveine B (13) (0.9 mg) (retention time 16.0 min), ancistrobreveine C (14) (2.3 mg) (retention time 22.5 min), 6-O-methylhamateine (4) (0.5 mg) (retention time 23.2 min), ent-dioncophylleine A (10) (0.6 mg) (retention time 23.4 min), and ancistrobreveine D (6) (1.1 mg) (retention time 26.2 min).
ε): λmax 234 (2.6) and 258 (2.1) nm; ECD (c 0.1, MeOH) λmax (Δε): 218 (+10.5), 223 (+12.7), 231 (+14.9), 258 (−19.1), 302 (+0.9), and 334 (−0.8) nm; IR (ATR) νmax: 3363, 2925, 1668, 1612, 1199, and 1131 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS: m/z 404.1861 [M + H]+ (calcd for C25H26NO4, 404.1856).ε): λmax 228 (3.2), 257 (2.8), 305 (2.6), and 334 (2.5) nm; ECD (c 0.1, MeOH) λmax (Δε): 223 (−5.0), 252 (+4.6), 299 (−0.3), and 335 (+0.2) nm; IR (ATR) νmax: 3362, 2979, 1670, 1461, 1385, 1258, 1200, 1132, and 947 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS: m/z 418.2015 [M + H]+ (calcd for C26H28NO4, 418.2013).ε): λmax 236 (3.8) and 258 (3.4) nm; ECD (c 0.1, MeOH) λmax (Δε): 223 (−36.2), 250 (+30.1), 269 (−6.9), 286 (−0.7), 303 (−0.6), and 331 (+3.3) nm; IR (ATR) νmax: 3383, 2900, 1666, 1585, 1375, 1195, and 1112 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS: m/z 418.2013 [M + H]+ (calcd for C26H28NO4, 418.2013).ε): λmax 234 (2.8) and 258 (2.1) nm; ECD (c 0.1, MeOH) λmax (Δε): 207 (−13.9), 223 (−24.6), 257 (+19.8), 274 (+2.1), 326 (−1.8), and 380 (−0.9) nm; IR (ATR) νmax: 3349, 2945, 1673, 1349, 1198, and 1015 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS: m/z 418.1925 [M + H]+ (calcd for C26H28NO4, 418.2013).Known alkaloids isolated
The two fully dehydrogenated naphthylisoquinoline alkaloids 6-O-methylhamateine (4) and ent-dioncophylleine A (10) (Fig. 1), well known from previous phytochemical studies on A. benomensis and A. cochinchinensis, now isolated for the first time from root bark extracts of A. abbreviatus, were found to be identical in their spectroscopic, physical, and chromatographic behavior with the data reported previously.26,27Antileukemic assay
The cytotoxic effects (Table 2) of ancistrobreveines A (12), B (13), C (14), and D (6), 6-O-methylhamateine (4), and ent-dioncophylleine A (10) on drug-sensitive leukemia CCRF-CEM and multidrug-resistant P-glycoprotein-overexpressing CEM/ADR5000 cells49–51 were monitored by the resazurin assay as previously described.52,53 Doxorubicin (Sigma Aldrich, Munich, Germany) was applied as the positive control, while DMSO, used to dissolve the compounds, was employed as the negative control. The highest concentration of DMSO was less than 1.0%. Fluorescence was measured on an Infinite M2000 Pro™ plate reader (Tecan, Crailsheim, Germany), using an excitation wavelength of 544 nm and an emission wavelength of 590 nm. All experiments were performed in triplicate. The viability was evaluated based on the comparison with untreated cells. IC50 values represent the concentrations of the compounds required to inhibit 50% of cell proliferation. They were calculated from a calibration curve by linear regression using Microsoft Excel.54,55Conclusions
The alkaloid pattern of the West African liana Ancistrocladus abbreviatus is characterized by a remarkable structural diversity, comprising naphthylisoquinoline alkaloids of four different coupling types (5,1′, 5,8′, 7,1′, and 7,8′),5–11 among them Ancistrocladaceae-type compounds,5,6,10,11 which are 6-oxygenated and 3S-configured, but also Dioncophyllaceae-type alkaloids5,9,10 were found, which are deoxygenated at C-6 and have the R-configuration at C-3. Most of them represent naphthyltetrahydroisoquinolines,5–7,9–11 whereas compounds possessing a dihydroisoquinoline subunit have been isolated much less frequently.8,11 The newly discovered series of six naphthylisoquinoline alkaloids, presented in this paper, are all equipped with a fully dehydrogenated isoquinoline portion, comprising compounds displaying the four coupling types mentioned above. Four of these metabolites, named ancistrobreveines A–D (12–14, and 6), were identified for the first time in nature, whereas 6-O-methylhamateine (4) and ent-dioncophylleine A (10) had been known from previous phytochemical studies on related Ancistrocladus species.26,27 Ancistrobreveine C (14) is the first example of a 7,8′-linked fully dehydrogenated naphthylisoquinoline that is configurationally stable at the biaryl axis. It was found not to occur in an enantiopure form, but accompanied by 7% of its atropo-enantiomer.The occurrence of such a large series of fully dehydrogenated naphthylisoquinoline alkaloids in a distinct species is remarkable since so far only a total of 14 representatives with such a structural entity had been identified in Ancistrocladaceae lianas,20,21,26–30 with very few examples from African species in particular. Only the Malaysian highland liana A. benomensis was likewise found to produce such a high number of fully dehydrogenated compounds.27 Of all of the Ancistrocladus plants investigated so far,5,12,56 among them 16 accepted5,15–17,19–21,23,57–59 and six as yet botanically undescribed18,22,24,25,60,61 species, only four taxa were found to contain naphthylisoquinolines with a fully dehydrogenated isoquinoline subunit.20,21,26–30
Furthermore, 6-O-methylhamateine (4) and ancistrobreveine C (14) have proven to be potent bioactive compounds displaying good to strong inhibitory effects against drug-sensitive (CCRF-CEM) and multidrug-resistant (CEM/ADR5000) leukemia cells. Since the efficiency of many recommended drugs routinely used for the treatment of malignant disorders is threatened by the increasing emergence of resistance,39–45 the development of new potent agents preventing the overexpression of Pgp is an important goal. The promising antiproliferative activities of fully dehydrogenated naphthylisoquinoline alkaloids warrant more-in-depth studies regarding their potential as effective MDR suppressors. This work is in progress.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (Individual Research Grant Br699/14-2 “Molecular Phylogeny and Chemotaxonomy of the Ancistrocladaceae Plant Family”; SFB 630 “Agents against Infectious Diseases”, project A2), and by grants from the German Academic Exchange Service (Deutscher Akademischer Austauschdienst, DAAD) to one of us (S. F.). The publication of the manuscript was funded by the DFG and the University of Würzburg within the funding programme Open Access Publishing. We acknowledge experimental assistance from Dr M. Büchner and Mrs J. Adelmann (MS), and from Dr Grüne and Mrs P. Altenberger (NMR), all from the University of Würzburg.Notes and references
- C. M. Taylor, R. E. Gereau and G. M. Walters, Ann. Mo. Bot. Gard., 2005, 92, 360–399 Search PubMed.
- H. K. Airy Shaw, Kew Bull., 1949, 4, 68–69 Search PubMed.
- H. K. Airy Shaw, Kew Bull., 1950, 5, 147–150 CrossRef.
- M. Cheek, Kew Bull., 2000, 55, 871–882 CrossRef.
- G. Bringmann and F. Pokorny, The naphthylisoquinoline alkaloids, in The Alkaloids, ed. G. A. Cordell, Academic Press Inc, New York, 1995, vol. 46, pp. 127–271 Search PubMed.
- G. Bringmann, R. Zagst, H. Reuscher and L. Aké Assi, Phytochemistry, 1992, 31, 4011–1014 CrossRef CAS.
- G. Bringmann, R. Weirich, D. Lisch and L. Aké Assi, Planta Med., 1992, 58, A703–A704 CrossRef.
- G. Bringmann, F. Pokorny, M. Stäblein, M. Schäffer and L. Aké Assi, Phytochemistry, 1993, 33, 1511–1515 CrossRef CAS.
- G. Bringmann, D. Lisch, H. Reuscher, L. Aké Assi and K. Günther, Phytochemistry, 1991, 30, 1307–1310 CrossRef CAS.
- G. Bringmann, D. Koppler, D. Scheutzow and A. Porzel, Magn. Reson. Chem., 1997, 35, 297–301 CrossRef CAS.
- S. Fayez, D. Feineis, L. Aké Assi, M. Kaiser, R. Brun, S. Awale and G. Bringmann, Fitoterapia, 2018, 131, 245–259 CrossRef CAS PubMed.
- G. Bringmann, C. Günther, M. Ochse, O. Schupp and S. Tasler, in Progress in the Chemistry of Organic Natural Products, ed. W. Herz, H. Falk, G. W. Kirby and R. E. Moore, Springer, Wien, New York, 2001, vol. 82, pp. 111–123 Search PubMed.
- H. K. Airy Shaw, Kew Bull., 1951, 6, 327–347 CrossRef.
- G. Bringmann, G. François, L. Aké Assi and J. Schlauer, Chimia, 1998, 52, 18–28 CAS.
- Y. F. Hallock, K. P. Manfredi, J. W. Blunt, J. H. Cardellina II, M. Schäffer, K. P. Gulden, G. Bringmann, A. Y. Lee, J. Clardy, G. François and M. R. Boyd, J. Org. Chem., 1994, 59, 6349–6355 CrossRef CAS.
- M. R. Boyd, Y. F. Hallock, J. H. Cardellina II, K. P. Manfredi, J. W. Blunt, J. B. McMahon, R. W. Buckheit Jr, G. Bringmann, M. Schäffer, G. M. Cragg, D. W. Thomas and J. G. Jato, J. Med. Chem., 1994, 37, 1740–1745 CrossRef CAS.
- Y. F. Hallock, K. P. Manfredi, J. R. Dai, J. H. Cardellina II, R. J. Gulakowski, J. B. McMahon, M. Schäffer, M. Stahl, K. P. Gulden, G. Bringmann, G. François and M. R. Boyd, J. Nat. Prod., 1997, 60, 677–683 CrossRef CAS PubMed.
- B. K. Lombe, D. Feineis, V. Mudogo, R. Brun, S. Awale and G. Bringmann, RSC Adv., 2018, 8, 5243–5254 RSC.
- G. Bringmann, C. Günther, W. Saeb, J. Mies, A. Wickramasinghe, V. Mudogo and R. Brun, J. Nat. Prod., 2000, 63, 1333–1337 CrossRef CAS.
- S. Fayez, D. Feineis, V. Mudogo, S. Awale and G. Bringmann, RSC Adv., 2017, 7, 53740–53751 RSC.
- S. Fayez, D. Feineis, V. Mudogo, E.-J. Seo, T. Efferth and G. Bringmann, Fitoterapia, 2018, 129, 114–125 CrossRef CAS PubMed.
- G. Bringmann, B. K. Lombe, C. Steinert, K. Ndjoko Ioset, R. Brun, F. Turini, G. Heubl and V. Mudogo, Org. Lett., 2013, 15, 2590–2593 CrossRef CAS PubMed.
- D. T. Tshitenge, D. Feineis, V. Mudogo, M. Kaiser, R. Brun, E.-J. Seo, T. Efferth and G. Bringmann, J. Nat. Prod., 2018, 81, 918–933 CrossRef CAS PubMed.
- B. K. Lombe, T. Bruhn, D. Feineis, V. Mudogo, R. Brun and G. Bringmann, Org. Lett., 2017, 19, 1342–1345 CrossRef CAS PubMed.
- B. K. Lombe, T. Bruhn, D. Feineis, V. Mudogo, R. Brun and G. Bringmann, Org. Lett., 2017, 19, 6740–6743 CrossRef CAS PubMed.
- N. H. Anh, A. Porzel, H. Ripperger, G. Bringmann, M. Schäffer, R. God, T. V. Sung and G. Adam, Phytochemistry, 1997, 45, 1287–1291 CrossRef.
- G. Bringmann, M. Dreyer, H. Kopff, H. Rischer, M. Wohlfarth, H. A. Hadi, R. Brun, H. Meimberg and G. Heubl, J. Nat. Prod., 2005, 68, 686–690 CrossRef CAS PubMed.
- G. Bringmann, M. Dreyer, H. Rischer, K. Wolf, H. A. Hadi, R. Brun, H. Meimberg and G. Heubl, J. Nat. Prod., 2004, 67, 2058–2062 CrossRef CAS PubMed.
- G. Bringmann, B. Hertlein-Amslinger, I. Kajahn, M. Dreyer, R. Brun, H. Moll, A. Stich, K. Ndjoko Ioset, W. Schmitz and L. H. Ngoc, Phytochemistry, 2011, 72, 89–93 CrossRef CAS PubMed.
- G. Bringmann, R. Seupel, D. Feineis, M. Xu, G. Zhang, J. Wu, M. Kaiser, R. Brun, E.-J. Seo and T. Efferth, Fitoterapia, 2017, 121, 76–85 CrossRef CAS PubMed.
- T. R. Govindachari, K. Nagarajan, P. C. Parthasarathy, T. G. Rajagopalan, H. K. Desai, G. Kartha, S. M. L. Chen and K. Nakanishi, J. Chem. Soc., Perkin Trans. 1, 1974, 1413–1417 RSC.
- J. Fleischhauer, A. Koslowski, B. Kramer, E. Zobel, G. Bringmann, K. P. Gulden, T. Ortmann and B. Peter, Z. Naturforsch., B: J. Chem. Sci., 1993, 48, 140–148 CAS.
- G. Bringmann, J. Mühlbacher, M. Reichert, M. Dreyer, J. Kolz and A. Speicher, J. Am. Chem. Soc., 2004, 126, 9283–9290 CrossRef CAS PubMed.
- G. Bringmann, S. Tasler, H. Endress, J. Kraus, K. Messer, M. Wohlfarth and W. Lobin, J. Am. Chem. Soc., 2001, 123, 2703–2711 CrossRef CAS PubMed.
- X. Yang, T. A. M. Gulder, M. Reichert, C. Tang, C. Ke, Y. Ye and G. Bringmann, Tetrahedron, 2007, 63, 4688–4694 CrossRef CAS.
- G. Bringmann, T. Gulder, M. Reichert and F. Meyer, Org. Lett., 2006, 8, 1037–1040 CrossRef CAS PubMed.
- G. Bringmann, S. Rüdenauer, D. C. G. Götz, T. A. M. Gulder and M. Reichert, Org. Lett., 2006, 8, 4743–4746 CrossRef CAS PubMed.
- A. Goel, F. V. Singh, V. Kumar, M. Reichert, T. A. M. Gulder and G. Bringmann, J. Org. Chem., 2007, 72, 7765–7768 CrossRef CAS PubMed.
- G. Damia and S. Garattini, Cancer Treat. Rev., 2014, 40, 909–916 CrossRef CAS PubMed.
- C. Holohan, S. Van Schaeybroeck, D. B. Longley and P. G. Johnston, Nat. Rev. Cancer, 2013, 13, 714–726 CrossRef CAS.
- M. M. Gottesman, T. Fojo and S. E. Bates, Nat. Rev. Cancer, 2002, 2, 48–58 CrossRef CAS PubMed.
- A. Dlugosz and A. Janecka, Curr. Pharm. Des., 2016, 22, 4705–4716 CrossRef CAS.
- C. Q. Xia and P. G. Smith, Mol. Pharmacol., 2012, 82, 1008–1021 CrossRef CAS PubMed.
- J. Wang, N. Seebacher, H. Shi, Q. Kan and Z. Duan, Oncotarget, 2017, 8, 84559–84571 Search PubMed.
- Z. Binkhathlan and A. Lavasanifar, Curr. Cancer Drug Targets, 2013, 13, 326–346 CrossRef CAS PubMed.
- M. D. Hall, M. D. Handley and M. M. Gottesman, Trends Pharmacol. Sci., 2009, 30, 546–556 CrossRef CAS PubMed.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Pescitelli, SpecDis, Version 1.71, Berlin, Germany, 2017, http://www.specdis-software.jimdo.com Search PubMed.
- A. Kimmig, V. Gekeler, M. Neumann, G. Frese, R. Handgretinger, G. Kardos, H. Diddens and D. Niethammer, Cancer Res., 1990, 50, 6793–6799 CAS.
- T. Efferth, A. Sauerbrey, A. Olbrich, E. Gebhart, P. Rauch, H. O. Weber, J. G. Hengstler, M. E. Halatsch, M. Volm, K. D. Tew, D. D. Ross and J. O. Funk, Mol. Pharmacol., 2003, 64, 382–394 CrossRef CAS PubMed.
- J. Gillet, T. Efferth, D. Steinbach, J. Hamels, F. de Longueville, V. Bertholet and J. Remacle, Cancer Res., 2004, 64, 8987–8993 CrossRef CAS PubMed.
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Eur. J. Biochem., 2000, 267, 5421–5426 CrossRef.
- V. Kuete, P. D. Tchakam, B. Wiench, B. Ngameni, H. K. Wabo, M. F. Tala, M. L. Moungang, B. T. Ngadjui, T. Murayama and T. Efferth, Phytomedicine, 2013, 20, 528–536 CrossRef CAS PubMed.
- V. Kuete, H. K. Wabo, K. O. Eyong, M. T. Feussi, B. Wiench, B. Krusche, P. Tane, G. N. Folefoc and T. Efferth, PLoS One, 2011, 6, e21762 CrossRef CAS PubMed.
- J. P. Dzoyem, A. H. Nkuete, V. Kuete, M. F. Tala, H. K. Wabo, S. K. Guru, V. S. Rajput, A. Sharma, P. Tane, I. A. Khan, A. K. Saxena, H. Laatsch and N. H. Tan, Planta Med., 2012, 78, 787–792 CrossRef CAS PubMed.
- S. R. M. Ibrahim and G. A. Mohamed, Fitoterapia, 2015, 106, 194–225 CrossRef CAS PubMed.
- G. Bringmann, C. Steinert, D. Feineis, V. Mudogo, J. Betzin and C. Scheller, Phytochemistry, 2016, 128, 71–81 CrossRef CAS PubMed.
- J. Li, R. Seupel, D. Feineis, V. Mudogo, M. Kaiser, R. Brun, D. Brünnert, M. Chatterjee, E.-J. Seo, T. Efferth and G. Bringmann, J. Nat. Prod., 2017, 80, 443–458 CrossRef CAS PubMed.
- G. Bringmann, M. Dreyer, J. H. Faber, P. W. Dalsgaard, D. Stærk, J. W. Jaroszewski, H. Ndangalasi, F. Mbago, R. Brun, M. Reichert, K. Maksimenka and S. Brøgger Christensen, J. Nat. Prod., 2003, 66, 1159–1165 CrossRef CAS PubMed.
- G. Bringmann, G. Zhang, T. Büttner, G. Bauckmann, T. Kupfer, H. Braunschweig, R. Brun and V. Mudogo, Chem.–Eur. J., 2013, 19, 916–923 CrossRef CAS PubMed.
- S. M. Kavatsurwa, B. K. Lombe, D. Feineis, D. F. Dibwe, V. Maharaj, S. Awale and G. Bringmann, Fitoterapia, 2018, 130, 6–16 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectroscopic data include NMR (1H, 13C, 1H, 1H–COSY, HSQC, HMBC, NOESY, and ROESY), HRESIMS, IR, and ECD spectra of compounds 4, 6, 12, 13, and 14. See DOI: 10.1039/c9ra03105g |
| ‡ Deceased on January 14, 2014. |
| This journal is © The Royal Society of Chemistry 2019 |