
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation of microparticles from amylose-grafted poly(γ-glutamic acid) networks obtained by thermostable phosphorylase-catalyzed enzymatic polymerization†
Jun-ichi Kadokawa *,
Saya Orio and
Kazuya Yamamoto
*,
Saya Orio and
Kazuya Yamamoto
Department of Chemistry, Biotechnology, and Chemical Engineering, Graduate School of Science and Engineering, Kagoshima University, 1-21-40 Korimoto, Kagoshima 890-0065, Japan. E-mail: kadokawa@eng.kagoshima-u.ac.jp
First published on 23rd May 2019
Abstract
Amylose is a natural polysaccharide with helical conformation, which spontaneously forms water-insoluble assemblies, such as double helixes and inclusion complexes, at ambient temperatures in aqueous media, whereas it is synthesized as a water-soluble single chain by thermostable phosphorylase-catalyzed enzymatic polymerization at elevated temperatures in aqueous buffer solvents. In this study, we investigated the enzymatic polymerization at 80 °C using a primer-grafted poly(γ-glutamic acid) (PGA) in the presence or absence of poly(L-lactic acid) (PLLA) as a guest polymer for inclusion by amylose. Consequently, the produced amylose-grafted PGAs formed microparticles by cooling the mixtures at room temperature after the enzymatic polymerization in either the presence or the absence of PLLA. The particle sizes, which were evaluated by SEM measurement, were dependent on the feed ratios of PLLA. Based on the characterization results by the powder X-ray diffraction, IR, and dynamic light scattering measurements, a mechanism for the formation of the microparticles in the present system is proposed.
Introduction
Biological polymers, such as polysaccharide and protein (polypeptide), are vital materials and exhibit specific in vivo functions in living systems, depending on not only their primary structures, but also regular higher-order structures, such as a controlled helical conformation.1–3 Amylose, which is a natural polysaccharide composed of glucose (G) repeating units through α(1→4)-glycosidic linkages, is known to form a regularly controlled double helix, owing to its left-handed helical conformation.4,5 Besides the double helix formation, amylose is a host molecule well-known to form inclusion complexes with guest molecules, typically with low molecular weight, mainly by hydrophobic interaction as the cavity inside the amylose helix is hydrophobic.6 However, amylose does not have sufficient ability to directly bind long chains of polymeric guests into the cavity inside the helix for the formation of amylose-polymer inclusion complexes.7–16Amylose with well-defined structure is synthesized by phosphorylase-catalyzed enzymatic polymerization using α-D-glucose 1-phosphate (G-1-P) and maltooligosaccharide as monomer and primer, respectively,17–21 as it has been well accepted that the enzymatic approach is a very useful tool to efficiently synthesize well-defined polysaccharides compared with general organic reactions.22–26 An initiation of the enzymatic polymerization occurs by a transfer reaction of a G residue from the monomer to the nonreducing end of the primer with the formation of an α(1→4)-glycosidic linkage. The consecutive transfer reactions then progress as the propagation with liberating inorganic phosphates (Pi), according to the following reversible reaction manner to produce amylose; [α(1→4)-G]n + G-1-P ⇄ [α(1→4)-G]n+1 + Pi. When the phosphorylase-catalyzed enzymatic polymerization is carried out under general conditions at around 40–45 °C, the produced amyloses spontaneously form an insoluble double helical assembly in aqueous media. On the other hand, when the enzymatic polymerization is conducted at higher temperatures such as at around 80 °C by using thermostable phosphorylase, which retains an activity at elevated temperatures, a water-soluble amylose with single chain fashion (single amylose) is present in the reaction media without the formation of the double helix. We have also developed the enzymatic approach for the inclusion complexation from the amylose host and polymeric guests by means of the phosphorylase-catalyzed enzymatic polymerization, which is performed in the presence of appropriate hydrophobic polymers as guests, such as poly(L-lactic acid) (PLLA), dispersed in aqueous buffer solution as polymerization solvent.27 The enzymatic elongation from the shorter maltooligosaccharide primer to the longer amylose chain by the polymerization is conceived to provide a sufficient dynamic field for the more efficient complexation with polymeric guests than the direct mixing the two molecules. Accordingly, as the propagation in this system likely images the way that the vines of plants grow with twining around a rod, we have proposed that this polymerization method for the construction of amylose-polymer inclusion complexes be called “vine-twining polymerization”.28–34
Owing to no participation of the reducing end (an opposite side of the propagating end) of the maltooligosaccharide primer into the reaction, the phosphorylase-catalyzed enzymatic polymerization also progresses using a primer-grafted polymer, where the reducing ends of the plural maltooligosaccharide primers are covalently attached on the polymeric chain, to produce amylose-grafted polymers, such as the following heteropolysaccharides; amylose-grafted cellulose, carboxymethyl cellulose, and chitin/chitosan.20,21,25,26,30,33,35–37 By means of this approach, an artificial saccharide–peptide conjugate, that is, an amylose-grafted poly(γ-glutamic acid) (PGA) was also synthesized.38 When the phosphorylase-catalyzed enzymatic polymerization from the primer-grafted PGA was conducted at 45 °C, the elongated amylose graft chains spontaneously formed double helical cross-linking points among the PGA main-chains, resulting in the formation of a hydrogel composed of amylose-grafted PGA networks. We have also carried out the vine-twining polymerization using the primer-grafted PGA in the presence of guest polymers, such as PLLA, poly(ε-caprolactone), and polytetrahydrofuran, to produce supramolecular networks composed of the amylose-polymer inclusion complexes among the PGA main-chains as cross-linking points. The resulting networks have formed different macroscopic structures, such as hydrogels and aggregates, in the reaction media depending on kinds of the guest polymers or guest polymer/primer feed ratios, because which strongly affected network sizes.39,40 In these systems, therefore, PGA acted as frameworks, whereas the roles of amylose and guest polymers were cross-linking agents in the network structures.
In this study, we attempted to fabricate new macroscopic structures from amylose-grafted PGA networks, which were obtained by the thermostable phosphorylase (from Aquifex aeolicus VF5)-catalyzed enzymatic polymerization using the primer-grafted PGA in the presence or absence of PLLA under the conditions for the production of the single amylose chains at 80 °C, followed by cooling at room temperature for the formation of the double helixes (Fig. 1). Consequently, we found that in either the presence or the absence of PLLA, the produced supramolecular networks formed the microparticles. Furthermore, their sizes were changed in accordance with the PLLA/primer feed ratios.
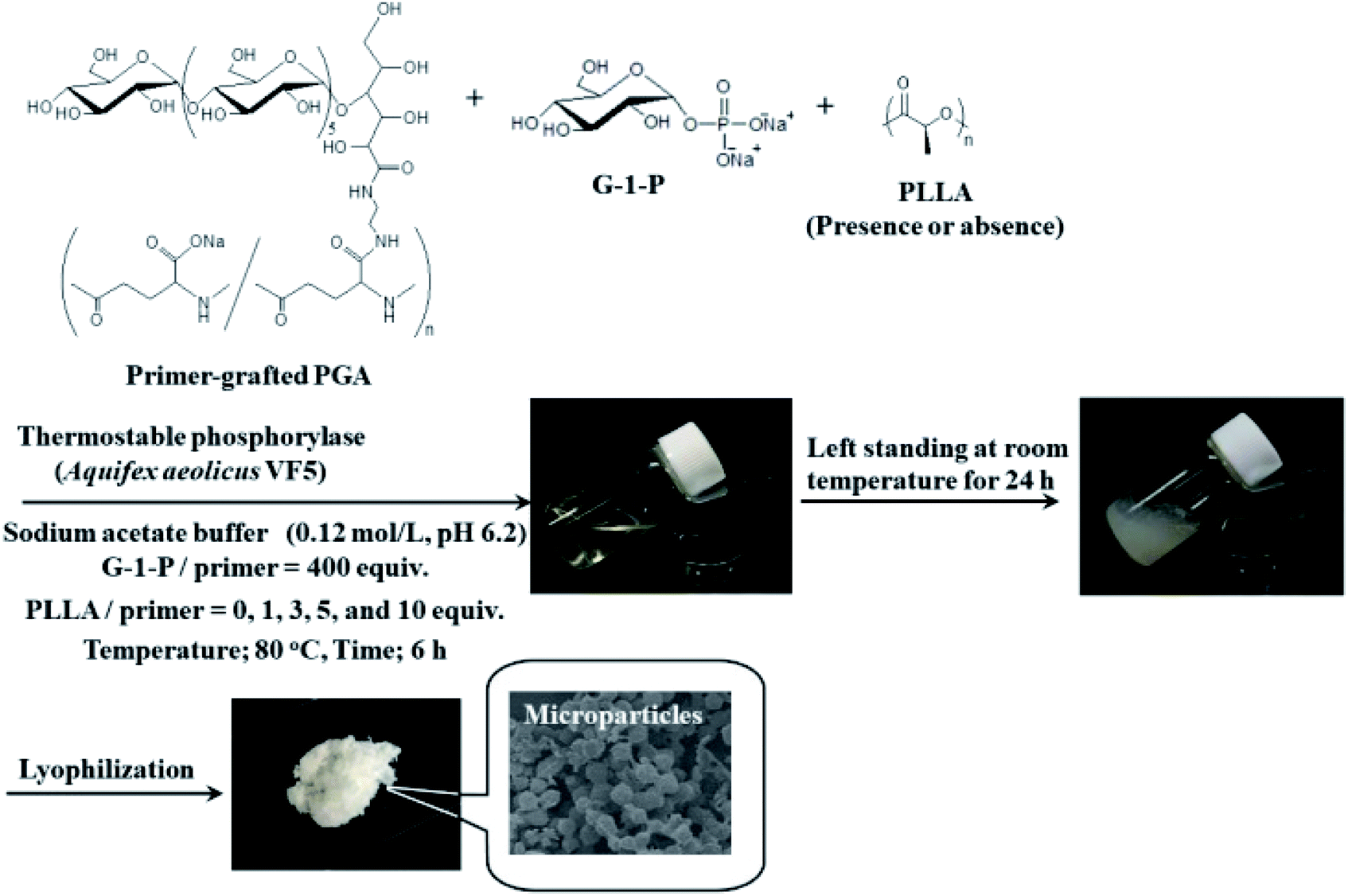 | ||
| Fig. 1 Formation of microparticles by thermostable phosphorylase-catalyzed enzymatic polymerization at 80 °C using primer-grafted PGA in the presence or absence of PLLA, followed by cooling at room temperature. | ||
Results and discussion
The thermostable phosphorylase-catalyzed enzymatic polymerization of G-1-P was conducted using primer (maltoheptaose)-grafted PGA (degree of substitution of primer on PGA = 70.2%) in the different PLLA/primer feed ratios (=0, 1, 3, 5, and 10, runs 1–5) dispersed in aqueous sodium acetate buffer under the conditions of G-1-P/primer feed ratio = 400 at 80 °C for 6 h (Fig. 1). Regardless of the PLLA/primer feed ratios, the reaction mixtures were transparent intact during the polymerization, suggesting the production of the water-soluble single amylose chains on the PGA main-chains. As suspended aggregates were slightly produced in the reaction mixture of run 5, the powder X-ray diffraction (XRD) measurement of their lyophilized sample was conducted, which did not show any diffraction peaks (Fig. 2a), supporting the formation of the single amylose chains with no crystalline structure. The reaction mixtures were then left standing at room temperature for 24 h to precipitate the products, which were isolated by centrifugation, washed with water and acetone, and lyophilized (Fig. 1). Based on weights of the recovered PLLA present in the acetone fractions, weight ratios (wt%) of PLLA present in the products were calculated, which increased in accordance with the PLLA/primer feed ratios (Table 1). Indeed, the intensities of carbonyl absorptions at 1750 cm−1 ascribed to ester linkage of PLLA in the IR spectra of the products increased with increasing the feed ratios (Fig. S1†).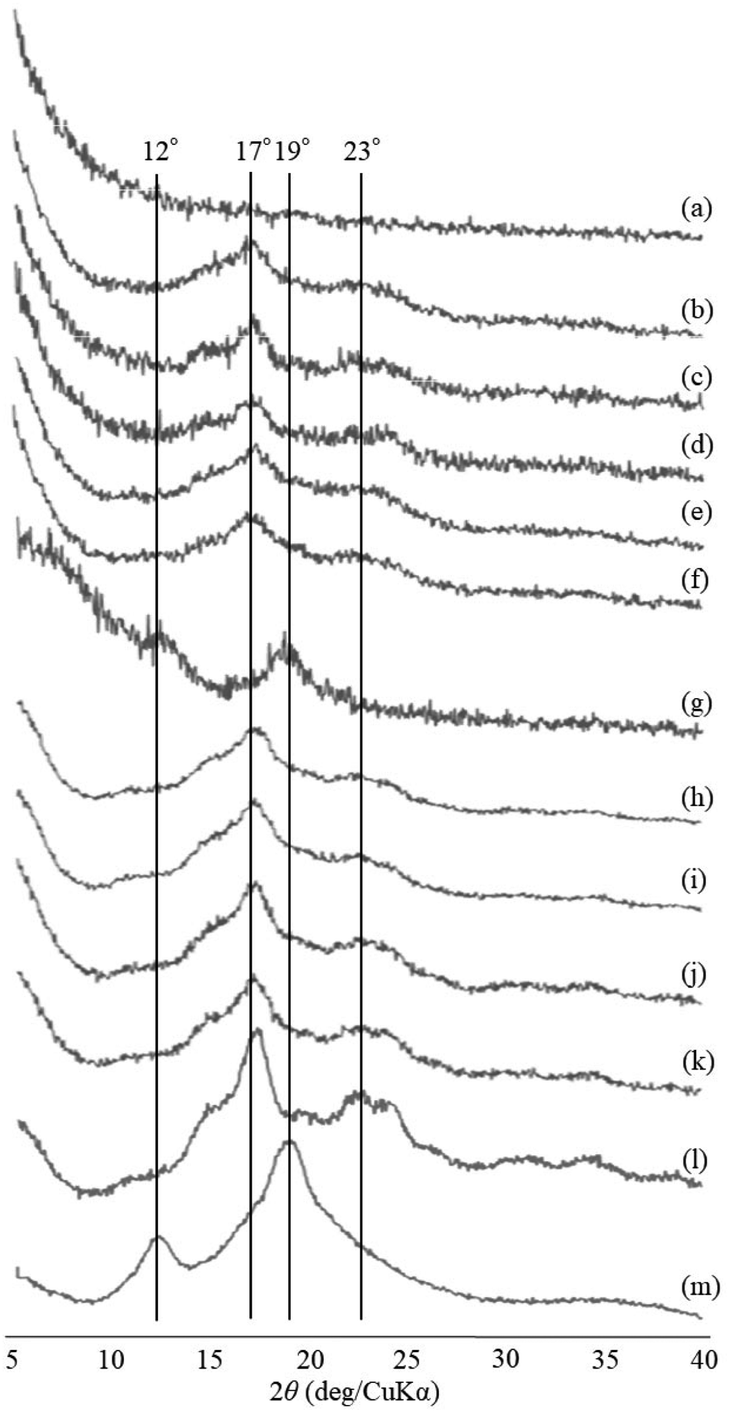 | ||
| Fig. 2 XRD profiles of (a) suspended aggregates in the reaction mixture of run 5 at 80 °C for 6 h, (b–f) lyophilized products of runs 1–5, (g–k) lyophilized products by cooling the reaction mixture of run 5 at room temperature for 1, 2, 3, 6, and 12 h, (l) amylose, and (m) amylose–PLLA inclusion complex. | ||
| Run | PLLA/primer | PLLA content in productb (%) | Average diameter of microparticlesc (μm) | Average size of product in mixture after cooling for 1 hd (nm) |
|---|---|---|---|---|
| a Reaction was carried out at 80 °C for 6 h, followed by cooling at room temperature for 24 h. G-1 P/G7 feed ratio = 400.b Determined by weight of recovered PLLA.c Determined by SEM measurement.d Determined by DLS measurement. | ||||
| 1 | 0 | — | 1.19 | 7.73 |
| 2 | 1 | 4.2 | 1.82 | 12.9 |
| 3 | 3 | 6.2 | 2.50 | 14.9 |
| 4 | 5 | 9.5 | 4.28 | 17.3 |
| 5 | 10 | 16.2 | 5.37 | 32.6 |
The XRD measurement of the products was conducted to evaluate crystalline structures of the amylose graft chains. All the XRD profiles of the products exhibit a typical pattern mainly with peaks at 17 and 23° assignable to the amylosic double helix, indicating the formation of double helixes by the amylose graft chains during the cooling process at room temperature (Fig. 2b–f and l). The morphologies of the products at μm scale were then investigated by SEM measurement. All the SEM images of the products observe microparticle morphologies (Fig. 3a–e), where their interfacial areas are partly merged probably by the formation of double helixes from the amylose chains present on surfaces of the particles. The average particle diameters, which were calculated on the basis of the lengths of vertical and horizontal axis of fifty particles in each SEM image, increased with increasing the PLLA/primer feed ratios (Table 1). After the products of runs 1 and 5 were quickly treated with DMSO for the dissociation of double helixes present at interfacial areas, the SEM images of the spin-coated samples of the treated mixtures observe the morphologies of the individual microparticles (Fig. 3f and g). This result strongly supported that fusion among the microparticles has been caused by the double helix formation from the amylose chains at interfacial areas, because the amylosic double helix is known to dissociate in DMSO.
 | ||
| Fig. 3 SEM images of (a–e) lyophilized products of runs 1–5, (f and g) spin coated samples after treatment of products of runs 1 and 5 with DMSO, and (h–l) lyophilized products by cooling the reaction mixture of run 5 at room temperature for 1, 2, 3, 6, and 12 h. | ||
To investigate the formation process of the microparticles in the present system, the products obtained by different cooling times at room temperature, after the enzymatic polymerization in PLLA/primer feed ratio = 10 (run 5), were characterized. The SEM image of the product by cooling for 1 h exhibits irregular morphology (Fig. 3h). By prolonged cooling times, i.e., 2 and 3 h, network morphologies are gradually seen in the SEM images (Fig. 3i and j). The SEM images of the products by further cooling, such as for 6 and 12 h exhibit gradual growth of particle-like structures along the networks (Fig. 3k and l). After cooling for 24 h, consequently, the microparticle morphology is clearly seen in the SEM image as abovementioned (Fig. 3e). The XRD profile of the product by cooling for 1 h observes typical diffraction peaks at 12 and 19° assignable to the amylosic inclusion complex (Fig. 2g), as detected in that of an amylose–PLLA inclusion complex (Fig. 2m). On the other hand, a diffraction pattern ascribable to the amylosic double helix was detected in the XRD profiles of the other products by longer cooling times (Fig. 2h–k). These XRD results strongly indicate that a part of the single amyloses on the PGA main-chains, produced by the enzymatic polymerization at 80 °C, initially formed inclusion complexes with PLLA and the rest of the single amyloses then constructed double helixes. The Mn values of the amylose graft chains, produced by the enzymatic polymerization at 80 °C and then by cooling at room temperature for 24 h, were estimated from the λmax values in UV-vis spectra of violet solutions of complexes with iodine,41,42 which were comparable each other (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 and 15500, respectively, corresponding to ca. 24% monomer conversion). These data suggested that the enzymatic polymerization reached equilibrium for 6 h, and the produced single amylose chains participated into the formation of inclusion complexes and double helixes without enzymatic elongation at the cooling stage.
700 and 15500, respectively, corresponding to ca. 24% monomer conversion). These data suggested that the enzymatic polymerization reached equilibrium for 6 h, and the produced single amylose chains participated into the formation of inclusion complexes and double helixes without enzymatic elongation at the cooling stage.
Based on the above findings, we proposed the following processes for the formation of the microparticles from the amylose-grafted PGAs (Fig. 4). The enzymatically elongated single amylose graft chains formed double helixes among the PGA main-chains in the absence of PLLA by cooling at room temperature to produce intermolecular network assemblies among the graft copolymers. During additionally cooling, the formation of the double helixes from the single amyloses among the networks progresses further to induce the growth of energetically stable spherical morphologies, giving rise to the microparticles (Fig. 4a). The similar process for the production of the microparticles is also conceived in the systems with PLLA. The single amylose graft chains are produced even in the presence of PLLA, but which do not form inclusion complexes, as revealed by the XRD profile in Fig. 2a. By cooling after the enzymatic polymerization, the network assemblies as nuclei of the particles are initially constructed by inclusion complexation of a part of the single amylose graft chains with PLLA among the PGA main-chains (Fig. 4b). The following double helix formation from the rest of the amylose graft chains among the nuclei results in the production of the microparticles. The dynamic light scattering (DLS) profiles of the mixtures of runs 1–5 by cooling for 1 h after the enzymatic polymerization at 80 °C exhibit the increase of average diameters in accordance with the PLLA/primer feed ratios (Fig. 5, Table 1). These results suggest that the larger numbers of the amylose-grafted PGAs initially assembled by the inclusion complexation with PLLA in accordance with the feed ratios to produce the larger nuclei. Accordingly, the sizes of the nuclei affected the formation of the larger microparticles, constructed by further assembly among the nuclei through the latter double helix formation process. The investigation on the more detailed formation process of the microparticles is now in progress in our group.
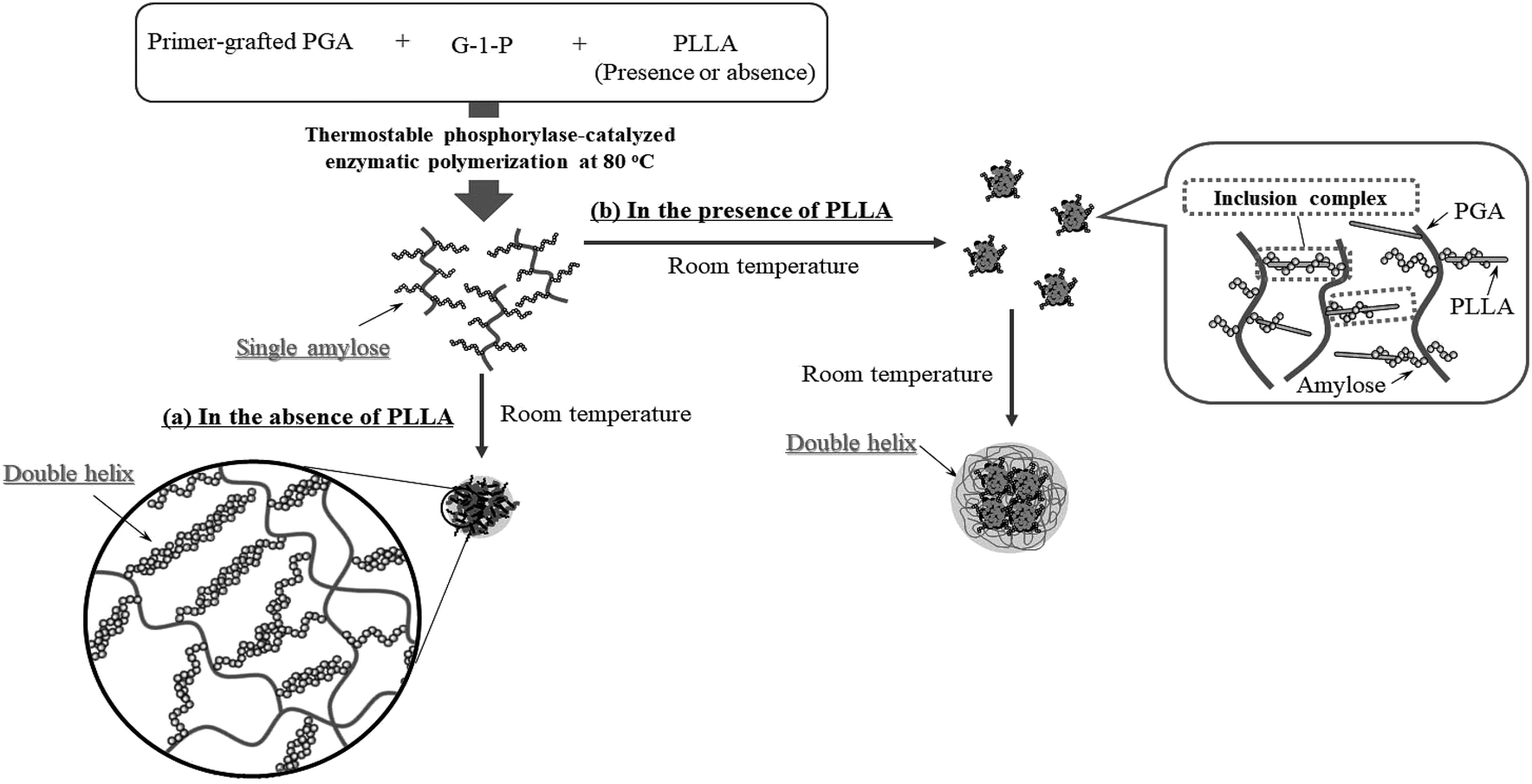 | ||
| Fig. 4 Plausible formation processes of microparticles by thermostable phosphorylase-catalyzed enzymatic polymerization at 80 °C (a) in the absence and (b) in the presence of PLLA, followed by cooling at room temperature. | ||
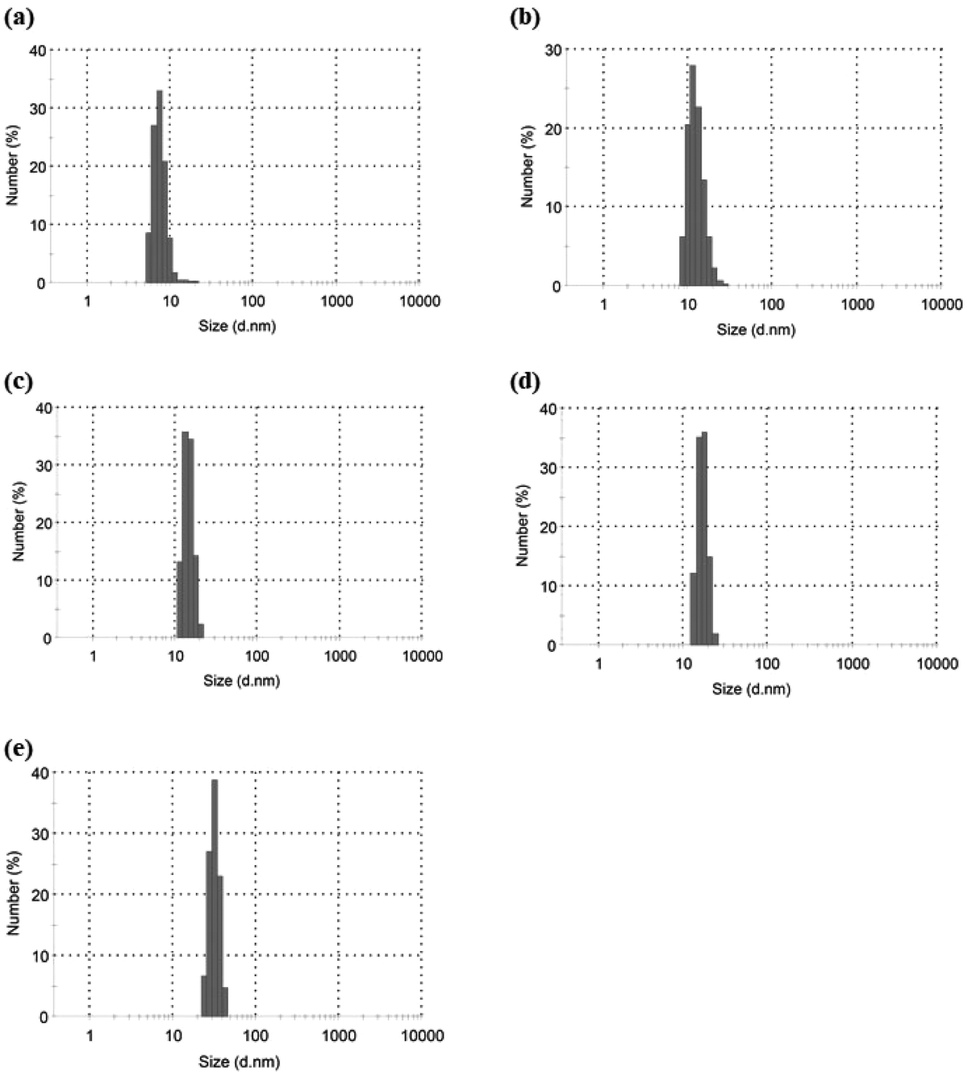 | ||
| Fig. 5 DLS profiles of (a–e) mixtures of runs 1–5 by cooling at room temperatures for 1 after enzymatic polymerization at 80 °C. | ||
Conclusions
This study achieved the formation of the microparticles by the thermostable phosphorylase-catalyzed enzymatic polymerization at 80 °C using the primer-grafted PGA, followed by cooling at room temperature. The enzymatic polymerization under such conditions produced the single amylose graft chains on the PGA main-chains, which formed double helixes among the graft copolymers at room temperature in the absence of PLLA, giving rise to the energetically stable microparticles. In the presence of PLLA, on the other hand, the single amylose graft chains partially formed inclusion complexes among the PGA main-chains at room temperature to produce the nuclei. The rest of the amylose graft chains then constructed double helixes among the nuclei to obtain microparticles, where sizes increased in accordance with the feed ratios of PLLA. The resulting microparticles composed of the amylose-grafted PGA networks will have potential to be employed in practical applications as new functional bio-based materials with controlled micromorphology in the future. The additional properties and applications of the present microparticles will be reported in forthcoming paper.Experimental
Materials and methods
PGA (MW = 1.5–2.5 × 106) was purchased from Wako Pure Chemicals, Tokyo, Japan. Thermostable phosphorylase from Aquifex aeolicus VF5 was supplied from Ezaki Glico Co. Ltd., Osaka, Japan.19,43,44 The primer-grafted PGA was synthesized according to the literature procedure.38 1H NMR (D2O) δ 1.86–2.01, 2.01–2.18 (br, β-CH2 of PGA), 2.30–2.42 (br, γ-CH2 of PGA), 3.40–4.08 (br, sugar protons of H2–H6), 4.12–4.25 (br, α-CH of PGA), 5.18, 5.41 (br s, H1 of primer). The DS for the grafting was determined by the integrated ratio of the H1 signal of the primer to the γ-CH2 signal of PGA to be 70.2%. The guest polymer, PLLA, was synthesized by ring-opening polymerization of L-lactide initiated with L-lactic acid.45 The Mn value was calculated by the integrated ratio of the main-chain signal to the terminal signal to be 1540. Other reagents and solvents were available commercially and used without further purification. 1H NMR spectra were recorded on JEOL ECX400 spectrometer. XRD measurements were performed using a PANalytical X'Pert Pro MPD diffractometer with Ni-filtered Cu Kα radiation (λ = 0.15418 nm). UV-vis measurements were conducted using a Jasco V-650Q1 spectrometer. The DLS measurements were obtained on a Zetasizer Nano ZS (Malvern Instruments). IR spectra were recorded on a PerkinElmer Spectrum Two spectrometer.Enzymatic polymerization in the absence of PLLA (run 1)
The primer-grafted PGA (0.0013 g, 0.0014 unit mmol, primer; 1.0 μmmol) was dissolved in an aqueous sodium acetate buffer solution (0.2 mol L−1, pH 6.2, 2.4 mL) and G-1-P disodium salt (0.12 g, 0.4 mmol, 400 equiv. with primer) was added to the solution. After thermostable phosphorylase (12.6 units) was added to this solution, the mixture was maintained at 80 °C for 6 h. The resulting mixture was left standing at room temperature for 24 h. The mixture was then subjected to centrifugation, and washed with water and acetone in turn, three times for purification, which was then lyophilized to give the product (37.0 mg).Enzymatic polymerization in the presence of PLLA
A typical experimental procedure was as follows (run 5). A solution of primer-grafted PGA (0.0013 g, 0.0014 unit mmol, primer; 1.0 μmmol) and G-1-P disodium salt (0.12 g, 0.4 mmol, 400 equiv. with primer) in an aqueous sodium acetate buffer solution (0.2 mol L−1, pH 6.2, 2.4 mL) was added to a solution of PLLA (15.4 mg, 10 μmol) in acetone (0.06 mL) and the resulting mixture was ultrasonicated. After thermostable phosphorylase (12.6 units) was added to this solution, the mixture was maintained at 80 °C for 6 h. The resulting mixture was left standing at room temperature for 24 h. The mixture was then subjected to centrifugation, and washed with water and acetone in turn, three times for purification, which was then lyophilized to give the product (43.4 mg). By evaporating the acetone fraction, the un-complexed PLLA was recovered (8.4 mg).Treatment of microparticles with DMSO
The microparticle products (2 mg), obtained by the above enzymatic polymerization was mixed with DMSO (0.2 mL). The mixture was immediately spin-coated for the SEM measurement.Determination of Mn values of amylose graft chains
The Mn value of the amylose graft chains was estimated by UV-vis analysis of the complex with iodine.41,42 A standard iodine-iodide solution was first prepared by dissolving potassium iodide (26 mg, 0.157 mmol) and iodine (26 mg, 0.100 mmol) in water (50 mL). The standard iodine-iodide solution (1.0 mL) was added to the enzymatic polymerization mixture (200 μL) or a solution of microparticles (1.0 mg) in DMSO (200 μL) and the resulting solution was diluted with water (10 mL). The violet solution was then characterized by UV-vis spectroscopy to determine the Mn value.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by a Grant-in-Aid for Scientific Research from Ministry of Education, Culture, Sports, and Technology, Japan (No. 17K06001). The authors acknowledge the supplement of thermostable phosphorylase from Ezaki Glico Co. Ltd., Osaka, Japan.References
- C. Schuerch, in Encyclopedia of Polymer Science and Engineering, ed. H. F. Mark, N. Bilkales andC. G. Overberger, John Wiley & Sons, New York, 2nd edn, 1986, vol. 13, pp. 87–162 Search PubMed.
- S. Kasapis, I. T. Norton and J. B. Ubbink, Modern Biopolymer Science: Bridging the Divide between Fundamental Treatise and Industrial Application, Academic Press, San Diego, 2009 Search PubMed.
- A. Miller and J. Tanner, Essentials of Chemical Biology: Structure and Dynamics of Biological Macromolecules, John Wiley & Sons, Chichester, England, Hoboken, NJ, 2008 Search PubMed.
- W. Hinrichs, G. Buttner, M. Steifa, C. Betzel, V. Zabel, B. Pfannemuller and W. Saenger, Science, 1987, 238, 205–208 CrossRef CAS.
- F. Eisenhaber and W. Schulz, Biopolymers, 1992, 32, 1643–1664 CrossRef CAS.
- J. A. Putseys, L. Lamberts and J. A. Delcour, J. Cereal Sci., 2010, 51, 238–247 CrossRef CAS.
- R. L. Shogren, R. V. Greene and Y. V. Wu, J. Appl. Polym. Sci., 1991, 42, 1701–1709 CrossRef CAS.
- R. L. Shogren, Carbohydr. Polym., 1993, 22, 93–98 CrossRef CAS.
- A. Star, D. W. Steuerman, J. R. Heath and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 2508–2512 CrossRef CAS.
- M. Ikeda, Y. Furusho, K. Okoshi, S. Tanahara, K. Maeda, S. Nishino, T. Mori and E. Yashima, Angew. Chem., Int. Ed., 2006, 45, 6491–6495 CrossRef CAS PubMed.
- T. Kida, T. Minabe, S. Okabe and M. Akashi, Chem. Commun., 2007, 1559–1561 RSC.
- Y. Kaneko, T. Kyutoku, N. Shimomura and J. Kadokawa, Chem. Lett., 2011, 40, 31–33 CrossRef CAS.
- R. Rachmawati, A. J. J. Woortman and K. Loos, Biomacromolecules, 2013, 14, 575–583 CrossRef CAS PubMed.
- K. Kumar, A. J. J. Woortman and K. Loos, Biomacromolecules, 2013, 14, 1955–1960 CrossRef CAS PubMed.
- R. Rachmawati, A. J. J. Woortman and K. Loos, Macromol. Biosci., 2013, 13, 767–776 CrossRef CAS.
- R. Rachmawati, A. J. J. Woortman and K. Loos, Macromol. Biosci., 2014, 14, 56–68 CrossRef CAS.
- G. Ziegast and B. Pfannemuller, Carbohydr. Res., 1987, 160, 185–204 CrossRef CAS.
- K. Ohdan, K. Fujii, M. Yanase, T. Takaha and T. Kuriki, Biocatal. Biotransform., 2006, 24, 77–81 CrossRef CAS.
- M. Yanase, T. Takaha and T. Kuriki, J. Sci. Food Agric., 2006, 86, 1631–1635 CrossRef CAS.
- J. Kadokawa, Polymers, 2016, 8, 138 CrossRef PubMed.
- J. Kadokawa, Curr. Org. Chem., 2017, 21, 1192–1204 CrossRef CAS.
- S. Kobayashi, H. Uyama and S. Kimura, Chem. Rev., 2001, 101, 3793–3818 CrossRef CAS PubMed.
- S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353 CrossRef CAS PubMed.
- J. Kadokawa and S. Kobayashi, Curr. Opin. Chem. Biol., 2010, 14, 145–153 CrossRef CAS PubMed.
- J. Kadokawa, Chem. Rev., 2011, 111, 4308–4345 CrossRef CAS PubMed.
- S. Shoda, H. Uyama, J. Kadokawa, S. Kimura and S. Kobayashi, Chem. Rev., 2016, 116, 2307–2413 CrossRef CAS PubMed.
- Y. Kaneko, K. Ueno, T. Yui, K. Nakahara and J. Kadokawa, Macromol. Biosci., 2011, 11, 1407–1415 CrossRef CAS PubMed.
- Y. Kaneko and J. Kadokawa, Chem. Rec., 2005, 5, 36–46 CrossRef CAS PubMed.
- Y. Kaneko and J. Kadokawa, J. Biomater. Sci., Polym. Ed., 2006, 17, 1269–1284 CrossRef CAS PubMed.
- Y. Kaneko and J. Kadokawa, in Modern Trends in Macromolecular Chemistry, ed. J. N. Lee, Nova Science Publishers, Inc., Hauppauge, NY, 2009, ch. 8, pp. 199–217 Search PubMed.
- J. Kadokawa, Polymers, 2012, 4, 116–133 CrossRef CAS.
- J. Kadokawa, Biomolecules, 2013, 3, 369–385 CrossRef PubMed.
- J. Kadokawa, Pure Appl. Chem., 2014, 86, 701–709 CAS.
- S. Orio, K. Yamamoto and J. Kadokawa, Polymers, 2017, 9 DOI:10.3390/polym9120729.
- J. Kadokawa, in Biobased Monomers, Polymers, and Materials, ed. P. B. Smith and R. A. Gross, ACS Symposium Series 1105; American Chemical Society, Washington, DC, 2012, ch. 15, pp. 237–255 Search PubMed.
- J. Kadokawa, in Green Polymer Chemistry: Biocatalysis and Materials II, ed. H. N. Cheng, R. A. Gross and P. B. Smith, ACS Symposium Series 1144; American Chemical Society, Washington, DC, 2013, ch. 11, pp. 141–161 Search PubMed.
- T. Nishimura and K. Akiyoshi, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, c1423 Search PubMed.
- T. Shouji, K. Yamamoto and J. Kadokawa, Int. J. Biol. Macromol., 2017, 97, 99–105 CrossRef CAS PubMed.
- J. Kadokawa, T. Shoji and K. Yamamoto, Polymer, 2018, 140, 73–79 CrossRef CAS.
- S. Orio, T. Shoji, K. Yamamoto and J. Kadokawa, Polymers, 2018, 10 DOI:10.3390/polym10111277.
- W. Banks, C. T. Greenwood and K. M. Khan, Carbohydr. Res., 1971, 17, 25–33 CrossRef CAS.
- K. Kobayashi, S. Kamiya and N. Enomoto, Macromolecules, 1996, 29, 8670–8676 CrossRef CAS.
- S. H. Bhuiyan, A. A. Rus’d, M. Kitaoka and K. Hayashi, J. Mol. Catal. B: Enzym., 2003, 22, 173–180 CrossRef CAS.
- M. Yanase, H. Takata, K. Fujii, T. Takaha and T. Kuriki, Appl. Environ. Microbiol., 2005, 71, 5433–5439 CrossRef CAS PubMed.
- Y. J. Fan, G. P. Chen, J. Tanaka and T. Tateishi, Biomacromolecules, 2005, 6, 3051–3056 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02999k |
| This journal is © The Royal Society of Chemistry 2019 |