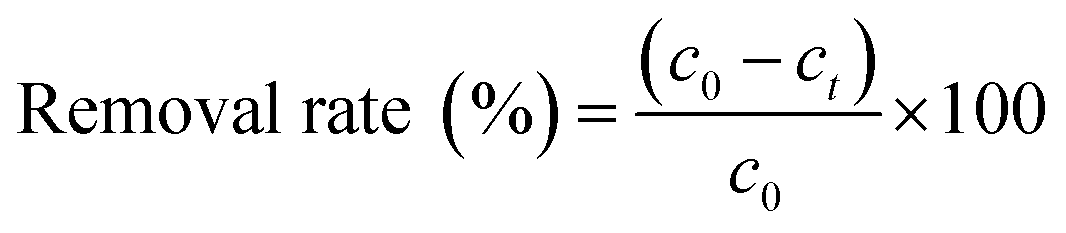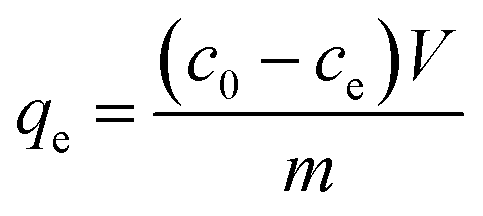DOI:
10.1039/C9RA02918D
(Paper)
RSC Adv., 2019,
9, 16258-16270
Microbial synthesis of hollow porous Prussian blue@yeast microspheres and their synergistic enhancement of organic pollutant removal performance†
Received
18th April 2019
, Accepted 18th May 2019
First published on 24th May 2019
Abstract
In this work, Prussian blue nanoparticles (PB NPs) were in situ grown on S. cerevisiae cells via one-step hydrothermal synthesis and the as-prepared Prussian blue@yeast (PB@yeast) hybrids exhibited synergistic adsorption and Fenton catalytic activities. FE-SEM, XRD and BET analysis of the prepared samples confirmed the successful formation of hollow porous structured PB@yeast microspheres, while FT-IR and XPS spectra indicated the fine structures were occupied by both functional adsorptive and catalytic sites. The experimental results of adsorption coupled Fenton reaction of PB@yeast hybrid microspheres revealed that the functional groups on the cell wall and the active iron sites in PB framework were fully utilized due to the triple synergistic effects of adsorption–Fenton catalysis–adsorption sites regeneration, thus endowing synergistically enhanced performance in removal of the selected cationic methylene blue (MB), anionic Methyl Orange (MO) and fluorescent brightener 71 (CXT) in aqueous solution. The high Fenton catalytic efficiency was related to the improvement of adsorption, in which the enrichment of contaminant molecules on the outer and inner surface of the hollow porous microspheres could lower mass transfer resistance and shorten charge transport pathways, thereby introducing more efficient Fenton catalytic activity than PB NPs.
1. Introduction
Heterogeneous Fenton catalysis employing iron-based nanoparticles has been extensively studied for removing bio-recalcitrant organic contaminants owing to their proven capability and pH resistance.1,2 Recently, Prussian blue nanoparticles (PB NPs) have received great attention as a heterogeneous Fenton catalyst in the field of organic pollution treatment.3–45 Compared to iron oxides, PB is a mixed valence hexacyanoferrate with high spin (S = 5/2) FeIII and low spin (S = 0) FeII atoms coordinated by –NC and –CN units respectively, which makes it more stable and easier to transfer charges in Fenton reaction.6,7 However, typical PB NPs prepared by nanoprecipitation have intrinsic problems of aggregation and dissolution in aqueous solution, especially in alkaline media,8,9 which limit their direct use in the large-scale environmental remediation. To meet the dilemma, PB NPs have been stabilized on a variety of materials, such as mesoporous silica,10 γ-Al2O3,11 chitin,12,13 CNTs,14,15 rGO,16 etc. Among these supports, the appropriate matrix with tunable structures and high-quality functions become very attractive since it can not only protect PB NPs against instability and agglomeration but also enable integration characteristics to extend potential of the hybrids.17 For instance, Edson Nossol and co-workers reported carbon nanotube/Prussian blue nanocomposite film as electrochemical and photo-electrochemical Fenton catalyst, which could degrade methyl orange dye efficiently.3 Unfortunately, the catalytic reaction can only occur on the outer surface of PB@support because of dense accumulation of PB NPs on the support, for which reason the accessibility of reactants is greatly decreased. Furthermore, due to the limited contact area between the PB NPs and the support, the inherent function of the valuable carrier is inhibited, which may reduce the coupling performance of the PB@support hybrids. Therefore, developing new PB-based hybrid materials to fully utilize the multifunctional activity of both the carrier and the PB NPs is still required to meet the practical applications.
Hollow-structured porous microsphere, because of the distinctive combination of the hollow interiors/voids with porous microsphere structure, has been widely investigated in varied application fields.18–21 In particular, these hollow porous systems can be well functioned as catalyst supports in the field of catalysis, in which the hollow surface provides sufficient spaces and anchored sites for locating the active guests, and the porous shell offers a short diffusion pathway for the transfer of both reactants and products into/out of the interior voids.22 Among the hollow structured microspheres, yeast cell are fascinating owing to their excellent mechanical and thermal stability, high availability, huge quantity and environmental friendliness.23,24 Yeast Saccharomyces cerevisiae (S. cerevisiae), known as a by-product of the brewing industry, is one sort of elliptical unicellular fungus with sturdy cell wall.25 It was found that the hollow porous structures were easy obtained at a low temperature (about 100 °C) as a result of the dead of S. cerevisiae cells and the escape of internal hydrated water.26 Most interestingly, the surface of these hollow microspheres is covered with numbers of functional groups, such as carboxyl, amide, hydroxyl, acyl amino, et al., which are capable of binding metal ions via coordination or electrostatic attraction, and also function as reactive adsorption/catalysis sites.27,28 Significantly, the inner surface of the cell wall can be also utilized efficiently when using these hollow microspheres as catalyst supports. Benefiting from these inherent advantages of S. cerevisiae cells, it is predicable thereby that S. cerevisiae will be an idea biological support to form metal containing multifunctional materials. Moreover, since the hollow microsphere is isolated by a permeable shell with relatively homogeneous internal surroundings, the hollow S. cerevisiae microsphere incorporated with catalytically active guest is expected to act as a microreactor for synergistic adsorption/catalysis.
Hence, inspired by the remarkable catalytic properties of PB NPs and novel hollow porous structure of inactivated S. cerevisiae cells, we believe that the combination of the two may find a synergistically enhanced performance in the application of organic pollution treatment. In the previous investigation, we had confirmed that the core–shell PB@yeast composites retained the adsorption-coupled Fenton catalytic activity.29 However, the as-prepared samples were synthesized by co-precipitation method, which involved multiple steps and several raw materials. Herein, we develop a one-step hydrothermal strategy for fabricating hollow porous PB@yeast microspheres at relatively low temperatures, in which the PB NPs were in situ grown on the functionalized S. cerevisiae cell walls. The as-prepared PB@yeast samples were characterized by FE-SEM, EDS, XRD, FT-IR, XPS and BET, respectively. Based on the results of characterization results, a detailed formation mechanism of the PB@yeast hollow microspheres was proposed. In addition, three typical organic pollutions including methylene blue (MB), anionic Methyl Orange (MO) and fluorescent brightener 71 (CXT) were selected as the model contaminants for bath experiments. The removal of MB, MO and CXT from aqueous solution using PB@yeast hollow microspheres as both adsorbent and heterogeneous Fenton catalyst was systematically investigated and a probable synergistic mechanism was further studied. In general, the in situ growth of PB on hollow-shelled S. cerevisiae cells not only extended the utilization of yeast cells and avoided aggregation and dissolution of PB NPs, but also achieved cooperative adsorption and catalytic activities, which showed better activity and stability than normal PB@support microspheres synthesized by chemical precipitation.
2. Experiments
2.1 Materials
Potassium ferricyanide (K4Fe(CN)6·3H2O), T-butanol (TBA) were acquired from Tianjin Kermel Chemical Reagent Co., Ltd, China. P-Benzoquinone and sodium azide were purchased from Aladdin Co., Ltd, China. Yeast powder was purchased from Angel Yeast Co., Ltd, China. Polyvinylpyrrolidone (PVP, KW = 30) was acquired from shanghai lanji technology Co., Ltd, China. Hydrochloric acid (HCl, 37.5%), sodium hydroxide (NaOH), hydrogen peroxide (H2O2, 30%), ethanol (C2H5OH, 95%) were purchased from Xi'an Chemical Agent Corp. Methyl orange, methylene blue and fluorescent brightener 71(CXT) were provided by Shaanxi research design institute of petroleum and chemical industry.
2.2 Synthesis of hollow PB@yeast hybrids
Preparation of PB nanoparticles (PB NPs). The PB NPs were prepared according to previous literature with a little modification.30 Typically, 7.60 g polyvinylpyrrolidone (PVP, K-30) and 0.22 g K4Fe(CN)6·3H2O were added into 50 mL HCl solution (0.1 M) under magnetic stirring. The mixed solution was stirred for 1 h at room temperature to produce a clear solution, followed by being transferred into a Teflon-lined stain-less-steel autoclave and heated at 100 °C for 24 h. The resulting precipitate was washed several times with deionized water and ethanol before drying in a vacuum oven at 60 °C.
Preparation of hollow PB@yeast microsphere. 1.0 g of pure yeast powder was washed comprehensively with distilled water/ethanol and dispersed in 50 mL of HCl solution (0.1 M) with agitated stirring at 30 °C. Then another 50 mL of HCl mixed with 3.80 g of PVP and 0.11 g of K4Fe(CN)6·3H2O were added into the above solution under magnetic stirring. The following steps were as the same as the preparation of PB NPs. The solution color changed from transparent to light blue, indicating the nucleation of PB nanoparticles. For comparison, PB@yeast microspheres were also synthesized by chemical precipitation, in which FeCl2 and K3Fe(CN)6 were used as precursors.
2.3 Characterization
Field emission scanning electron microscopy (FE-SEM) images were obtained on a HITACHI S4800. Detailed composition characterization was carried out with energy-dispersive spectroscopy (EDS) analysis (equipped with the FE-SEM). The X-ray diffraction (XRD) patterns were measured on a Bruker D8 Advanced X-ray Diffractometer (Cu Kα radiation λ = 0.15418 nm) at a scanning rate of 0.02 min−1. Fourier-transform infrared (FT-IR) spectra of samples were recorded on a Bio-Rad FTS135 spectrometer in the range 500–4000 cm−1 using a KBr wafer technique. The X-ray photoelectron spectra (XPS) were measured on PHI 5000 VersaProbe III. The specific surface area was analyzed by the Brunauer–Emmett–Teller (BET) N2 adsorption–desorption method on a Micromeritics Builder SSA-6000 instrument at 77 K and the pore size distributions were analyzed using the Barrett–Joyner–Halenda (BJH) method. The ZETA potential was tested by Malvern ZEN3690 potentiometer.
2.4 Adsorption coupled Fenton catalysis performance
The adsorption coupled Fenton behavior of PB@yeast were tested by the removal of MB, MO and CXT, where the initial concentration of contaminants was 25 mg L−1. The initial pH was adjusted to 3.0 for MB solution and 5.0 for the MO or CXT solution. Typically, 100 mL of contaminant solution was added to the beaker individually while adjusting to the designed pH value. 50 mg of PB@yeast was dispersed into the solution in dark under vigorously stirring. After equilibrium, the Fenton reaction began with adding amount of H2O2 (0.2 mol L−1) and simultaneously turning on UV light (TUV30W/G30T8, PHILIPS). During the reaction, samples from the liquid-phase were taken out at regular intervals, followed by centrifuging and measuring in the UV-vis spectrophotometer (UV-752, Shanghai). After each analysis, the samples were returned into the beaker immediately. To discern the role of individual components and possible synergistic effect, different situations including no catalyst, 10 mg of PB and 50 mg of yeast (after hydrothermal treatment at 100 °C) were studied respectively in removal of model pollutants.
The pH-dependent experiments were carried out in removal of CXT, in which the initial pH values were designed under varied conditions (3.0–9.0). After adsorption equilibrium, the Fenton reaction was investigated by adding certain amount of H2O2 with UV radiation. All the experiments were performed in beaker under magnetic stirring at ambient temperature. The pH values of the solutions were adjusted with 0.1 mol L−1 NaOH or 0.1 mol L−1 HNO3 using a pH meter (PHS-3C, Shanghai).
The removal efficiency is calculated by eqn (1).
| |
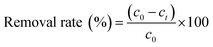 | (1) |
The equilibrium adsorption capacity is quantified by eqn (2).
| |
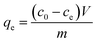 | (2) |
where
c0 (mg L
−1) and
ce (mg L
−1) represent the initial concentration and equilibrium concentration of contaminants;
m (mg) represents the mass of PB@yeast composites;
V (mL) is the solution volume.
2.5 Stability and reusability
The stability of PB@yeast was evaluated by PB release behavior and long-term stability. Typically, 10 mg of PB nanoparticles and 10 mg of PB@yeast hybrids were immersed in DI water respectively and shaken with a rotary shaker for designed time interval (6 h–3 month). The PB release was analyzed by UV-vis spectroscopy at 690 nm. The long-term stability and reusability of PB@yeast was tested by evaluating the degradation efficiency of CXT in consecutive runs. After each photo-Fenton reaction, the compounds were regenerated by filtration, washing, and over-dried for a new reaction cycle.
3. Results and discussion
3.1 Characterization of hollow PB@yeast microspheres
To clarify the successful assembly and the formation mechanism of PB@yeast hybrids, a number of techniques were applied to clarify the physiochemical properties of the prepared product. Firstly, XRD measurements were performed to validate the successful synthesis and the phase purity of the obtained products. Here, the XRD patterns of the original yeast cell, parallel PB NPs and PB@yeast sample are provided in Fig. 1a, respectively.
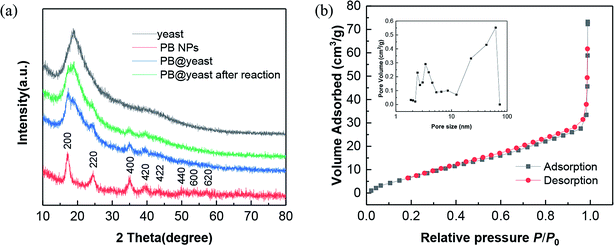 |
| | Fig. 1 (a) XRD patterns of yeast, PB NPs and PB@yeast. (b) N2 adsorption and desorption isotherms of PB@yeast. (Inset: corresponding pore-size distribution.) | |
As can be seen from the XRD pattern of yeast, a broad peak around 2θ = 20° appeared, indicating an amorphous phase of the yeast cell. For PB NPs curve, all of the diffraction peaks could match well with the standard values of face-centered cubic (fcc) phase of Fe4[Fe(CN)6]3 (JCPDS card no. 73-0687), which is consistent with other previous reports.28,31 Comparatively, the diffraction peaks of the PB@yeast sample were completely inherited the theoretical data of amorphous yeast and the fcc PB NPs, confirming that the PB NPs had been successfully formed on the yeast matrix through the proposed approach. Furthermore, no other diffraction peaks were detected, indicating the purity of the products.
In addition, the porosity of PB@yeast hybrid was assessed by N2 sorption measurements (Fig. 1b). The result displayed a type IV sorption with H3-hysteresis, indicating the presence of slit-like type porosity. The BET surface area of PB@yeast calculated from the adsorption isotherm curve was 28.38 m2 g−1 with a total pore volume of 0.067 cm3 g−1. The pore size distribution calculated by the BJH method (inset of Fig. 1b) confirmed the meso- and macroporous characteristics with pores ranging from a few nanometers to 70 nm. It is thus predictable that the hierarchical porous structures of PB@yeast microspheres may be beneficial for the transport of contaminant macromolecules.
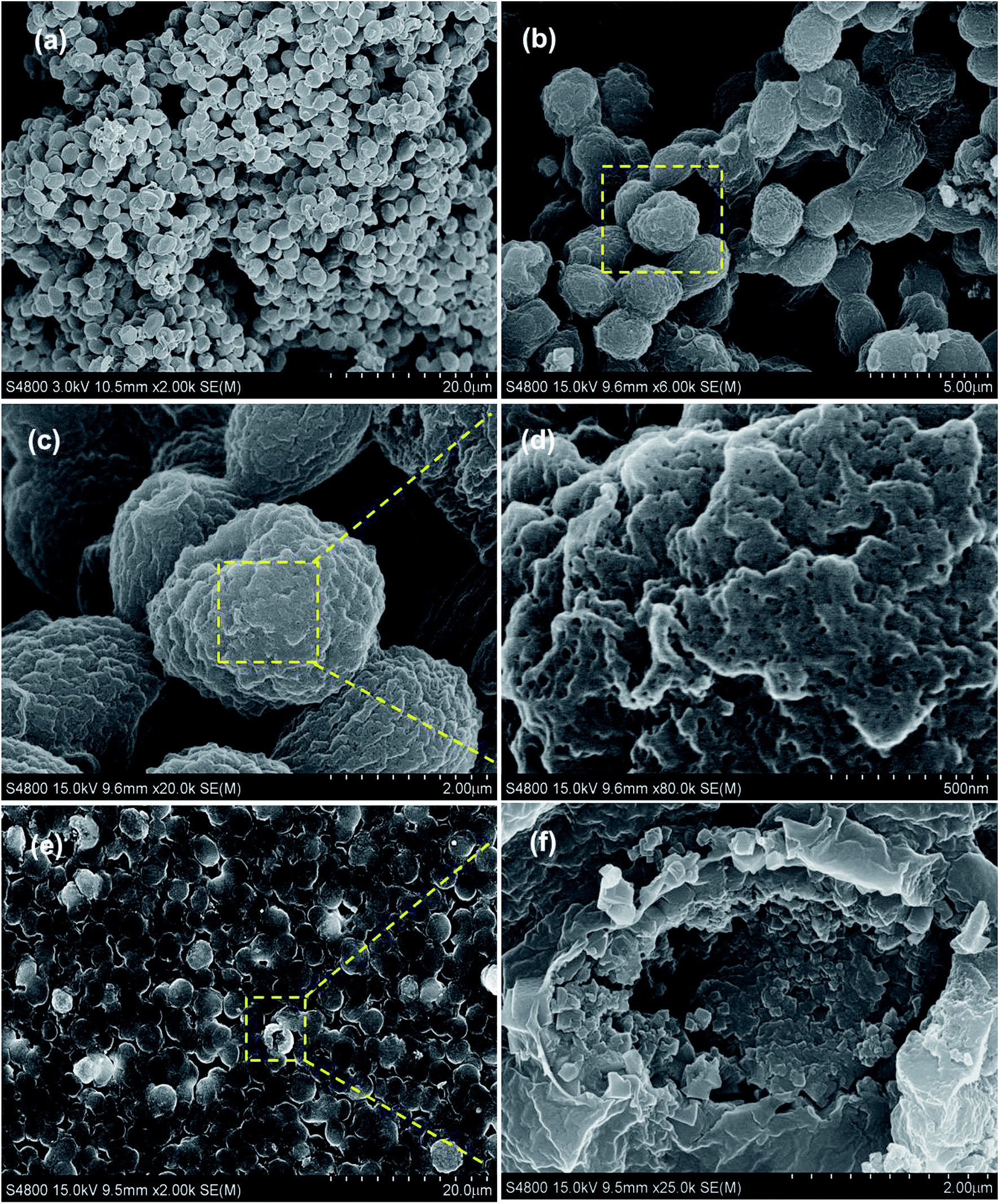
Afterwards, the morphology and structure of the PB@yeast hybrids were investigated by FE-SEM images (Fig. 2a–e).
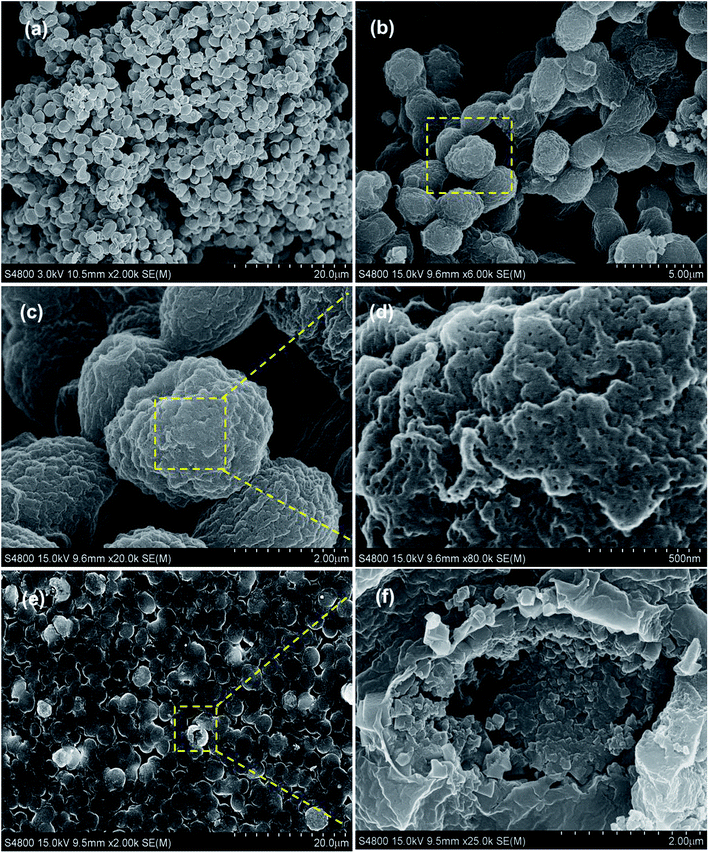 |
| | Fig. 2 FE-SEM micrographs of (a) the naked yeast and (b–f) PB@yeast under different magnification. | |
Fig. 2b shows the PB@yeast hybrids remained the ellipsoidal morphology with a size of about 3 μm on the long orientation axis. Close observation in Fig. 2c–d indicates that the surface of the microsphere was composed of closely packed nanoparticles with a size of several hundred nanometers, which is definitely different from naked yeast in Fig. 2a. As can been see, a rather rough surface and a hierarchical structure of PB@yeast microspheres were observed. Moreover, it can be seen in Fig. 2d that there were a large number of meso-pores with size below to 50 nm in the shell, indicating the porous feature of the prepared PB@yeast microspheres. Compared with the image of PB@yeast sample synthesized by chemical precipitation (see in Fig. S1†), a more uniform distribution of PB nanoparticles was observed, which may attributed to the guided nucleation growth of PB nanoparticles during mild hydrothermal procedures. After a vigorous sonication, some microspheres were broken and the hollow structure with closely packed PB shell could be clearly seen in a high magnification image (Fig. 2f), demonstrating the hollow hierarchical structure of the final PB@yeast products. The EDS analysis in Fig. S2† shows that Fe, C, N and O were the main elemental constituents, which were originated from both PB NPs and yeast matrix.
To understand the possible interactions between yeast and PB NPs in the formation of PB@yeast, FTIR spectroscopy was employed and the results are shown in Fig. 3a.
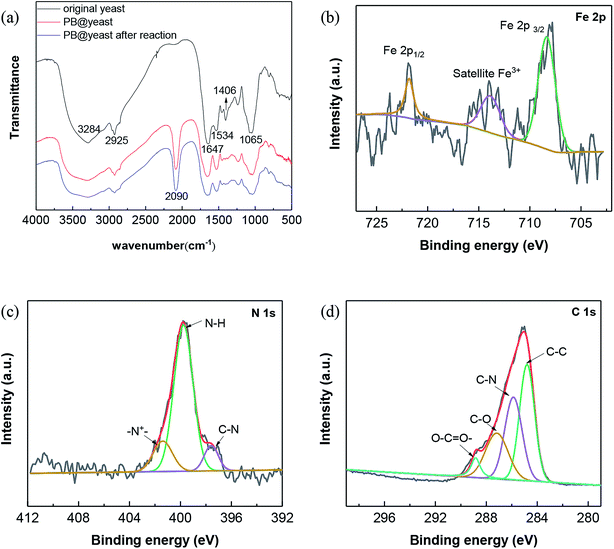 |
| | Fig. 3 (a) FT-IR spectra of yeast, PB@yeast before and after reaction; high resolution XPS spectra of PB@yeast: (b) Fe 2p, (c) N 1s and (d) C 1s. | |
For yeast, the broad and strong peak observed at 3284 cm−1 is oriented to hydroxyl stretching vibrations. The peaks at 2925, 1647, 1534, 1406, and 1240 cm−1 are assigned to CH2 asymmetric and symmetric stretching vibration, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in amide I, N–H in amide II, C–N in amide III, C–O in carboxylic acid groups, respectively.32,33 As to the FT-IR spectrum of the PB@yeast, it can be seen that besides the main stretching and deformation bands of yeast, a new IR adsorption peak was observed at 2090 cm−1, respecting to the CN stretching mode of PB.34,35 Notably, the stretching of –OH group at 3500–3200 cm−1 become a broad and intense peak probably due to inter and intramolecular hydrogen bonding interaction of compounds. The CO bond in carboxyl groups and the N–H in amide II were also shifted with growing shoulder bands. These changes suggested that the reactive functional groups such as carbonyls, hydroxyls, acid amides probably linked to PB NPs via electrostatic forces, hydrogen bonds and covalent bonds. In comparing the FT-IR spectra of the PB@yeast before and after the reaction, no significant changes were observed, indicating that this unique hybrid product could be stable during the reaction.
O in amide I, N–H in amide II, C–N in amide III, C–O in carboxylic acid groups, respectively.32,33 As to the FT-IR spectrum of the PB@yeast, it can be seen that besides the main stretching and deformation bands of yeast, a new IR adsorption peak was observed at 2090 cm−1, respecting to the CN stretching mode of PB.34,35 Notably, the stretching of –OH group at 3500–3200 cm−1 become a broad and intense peak probably due to inter and intramolecular hydrogen bonding interaction of compounds. The CO bond in carboxyl groups and the N–H in amide II were also shifted with growing shoulder bands. These changes suggested that the reactive functional groups such as carbonyls, hydroxyls, acid amides probably linked to PB NPs via electrostatic forces, hydrogen bonds and covalent bonds. In comparing the FT-IR spectra of the PB@yeast before and after the reaction, no significant changes were observed, indicating that this unique hybrid product could be stable during the reaction.
To better clarify the chemical composition of PB@yeast microspheres, XPS analysis was carried out and the corresponding results are presented in Fig. 3b–d. For Fe 2p spectra of the sample (Fig. 3c), the binding energies of Fe 2p3/2 and Fe 2p1/2 were observed at 713.8 and 721.8 eV, which were characteristics of Fe3+ in Fe4III[FeII(CN)6]3 (ref. 36 and 37) Besides, the peak at 708.3 eV can be assigned to Fe 2p3/2 (Fe2+) of [Fe(CN)6]4−.38 In the case of N 1s spectra (Fig. 3d), the peaks of N 1s spectra were fitted with three components at 397.6, 399.7 and 401.32 eV, suggesting the existence of C–N in PB NPs together with amine-like (–NH–) and positively charged nitrogen (N+) structure in yeast.39 In addition, the C 1s spectrum of PB@yeast also displayed the presence of C–C (284.8 eV), CO (287.2 eV), and O–CO (288.8 eV) species,40 which probably originated from the exposed surface of the yeast matrix. Based on the results of the above XPS analysis, it can therefore conclude that PB@yeast was successfully synthesized, in which the surface was fully modified by both PB NPs and yeast cell walls.
3.2 Formation mechanism of hollow PB@yeast microspheres
Based on the above characterizations, the possible formation mechanism of PB@yeast hollow microspheres can be described as follows (illustrated Scheme 1). In case of the S. cerevisiae, the cell is comprised of the cell wall, cell membrane, cytoplasm, nucleus, vacuoles and mitochondria.41 In addition, the bare cell wall of yeast consists mainly of phosphomannan, mannan, glucan, dextran, and a small fraction of protein, which produces multiple functional groups on the surface of cells, such as –COOH, –OPO3H2, –OH,–NH2, –CONH2, etc.24,42 According to previous work,43 these hydrophilic electrochemical groups can stimulate the electrochemical performance as pH changes. In particular, the –NH2 on yeast cell wall can be facilely converted into soluble protonate –NH3+ when the pH value was below its pKa (∼3.0) in aqueous solution. Hence, in this work when the initial yeast suspension pH value was adjusted to pH 2.0, the positively charged –NH3+ groups of S. cerevisiae cell wall were firstly combined with the negatively charged cations [Fe(CN)6]4− by electrostatic interaction. At the same time, the iron hydrates of potassium ferricyanide might further react with the hydroxyl groups via condensation reactions.44 Herein, as evidenced in FT-IR spectrum, the S. cerevisiae cell wall provided sufficient binding site to adsorb potassium ferricyanide precursors, which would be the oriented nucleation sites for the formation of PB NPs. It should be mentioned that the wall of S. cerevisiae is sensitive to the external temperature. Notably, the cell wall became loose and porous when the temperature was higher than 30 °C. Up to 100 °C, the S. cerevisiae cell was completely inactivated and hydration water inside the cell escaped from the porous cell wall, while the internal organics (nucleus, mitochondria, etc.) adhered to the inner wall of the cell membrane to form a hollow porous microsphere.23,45 Thereby, when the mixture was transferred into a Teflon-lined stain-less-steel autoclave and maintained at a hydrothermal temperature of 100 °C, some of the linked or free [Fe(CN)6]4− ions could enter to the interior of cell while others still being anchored outside the cell wall. Then, the nucleation and growth of PB nanocubes occurred not only on the surface of yeast cell, but also on the inner wall. At this point, the PB crystals were reasonably formed at the special positions by non-classical crystallization in the subsequent process.46 Besides, with the assistance of hydrochloride and PVP polymer, the generation rate of PB crystals was well controlled and the uniform structured hierarchical porous hybrid microspheres were final obtained.47 From the above analysis, it can be supposed that the yeast cell is essential for the formation of the hollow porous PB@yeast microsphere. On one hand, the reactive groups exposed on the surface of cell wall can act as binding sites for the potassium ferricyanide precursors and subsequently serve as targeted nucleation sites for in situ formation of PB NPs. In this way, PB NPs were evenly dispersed and away from agglomeration, which could address a favorable feature for the application of wastewater disposal. On the other hand, due to the unique hollow porous structure of the S. cerevisiae cell, the nucleation, growth and crystallization of PB NPs could be performed on both extracellular and intracellular cell walls, which significantly increased the utilization of the catalyst compared to the conventional loading of PB NPs on bulk supports. More importantly, since the hydrothermal treatment was carried out under a relatively low temperature, the functional groups of these tiny unicellular organisms were well preserved. Therefore, the in situ formation of PB NPs on hollow S. cerevisiae cell not only enhanced the stability of these nanoparticles, but also introduced new functionalities in the PB@yeast hybrid, which may find important applications in organic wastewater treatment.
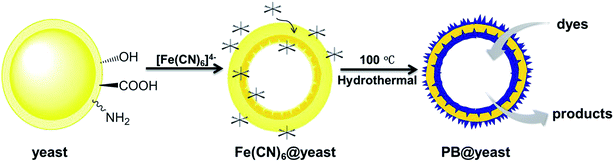 |
| | Scheme 1 Schematic illustration of PB@yeast hollow microspheres. | |
3.3 Adsorption coupled Fenton catalytic behavior
Dye MB and MO are known as typical recalcitrant organic pollutions that had been widely studies in the field of organic wastewater treatment. At the same time, CXT, one of the most prevalent organic assistant in machining process of washing, printing, plastic and textile, is considered to be toxic due to its chemical similarity to anionic direct dyes.48 The structures of the three molecules are displayed in Fig. S3.†
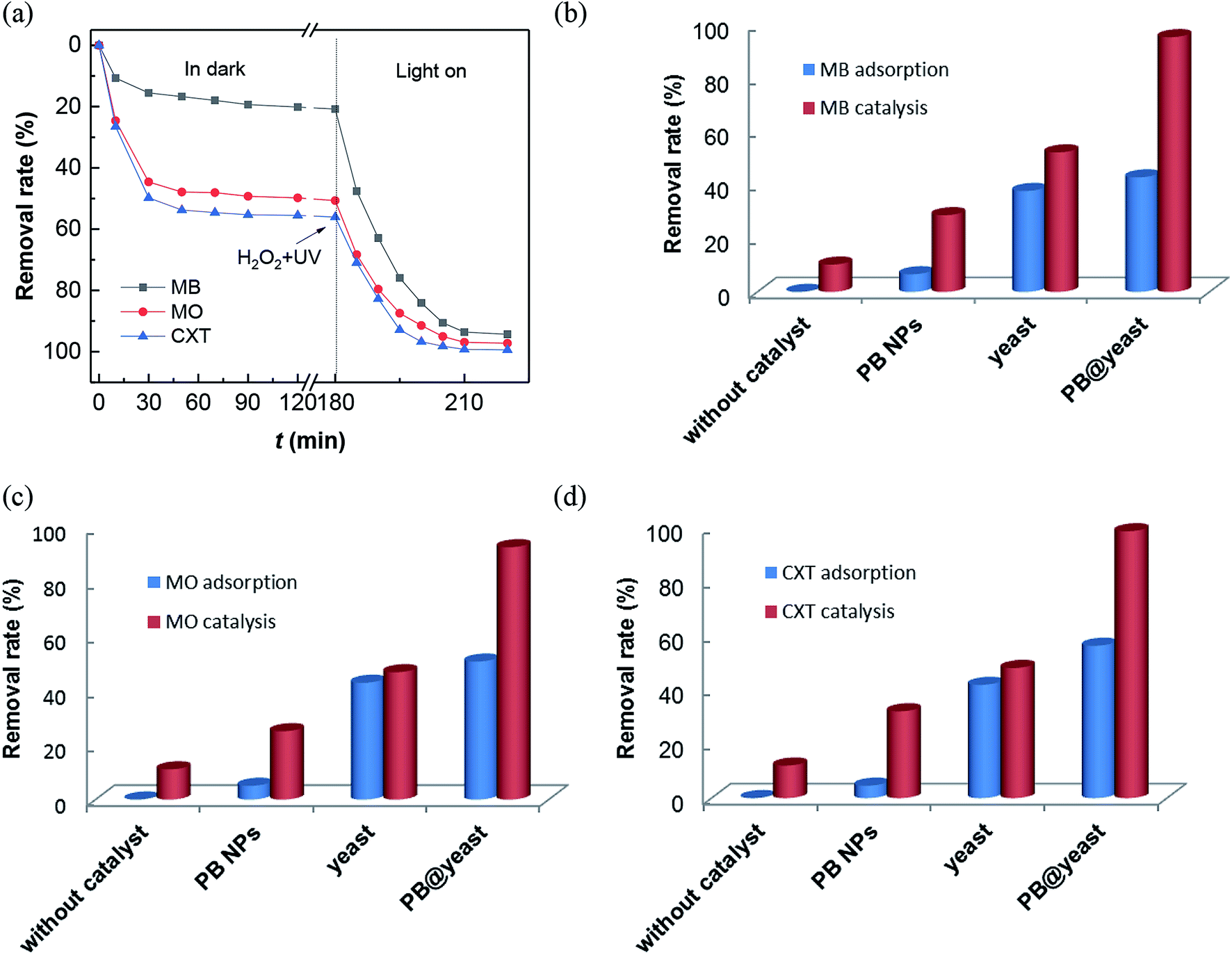
In this work, all of these three contaminants were used as pollutant molecules to evaluate the adsorption coupled Fenton catalytic behavior. The adsorption in dark followed by photo-Fenton performances for MB, MO and CXT removal are shown in Fig. 4a. It can be seen that the adsorption–desorption equilibrium is established within 120 min and the removal rates under dark conditions were approximately 43%, 51%, 56% respectively, which were mainly due to the adsorption ability of PB@yeast. After that, it was greatly increased to 97%, 96% and 99% over 40 min in the presence of H2O2 with the aid of UV irradiation, which was principally monitored by photo-Fenton catalysis. These findings confirmed that all of the three organic contaminants can be efficiently removed by adsorption coupled Fenton catalysis, indicating the universality of PB@yeast for the removal of both cationic and anionic organic contaminants. To understand the contribution of individual components and possible synergistic effect, a series of experiences under different conditions were also studied for MB, MO and CXT removal (Fig. 4b–d). It is worth to note that certain amounts of H2O2 was added after the adsorption–desorption equilibrium, and the equilibrium concentration was calculated as the initial concentration of the Fenton reaction. As is shown, little change was observed in the absence of catalysts, indicating that the self-degradation of CXT was negligible. Before the addition of H2O2, few target molecules were adsorbed onto the PB nanoparticles, and the removal rates were up to 29% of MB, 25% of MO and 32% of CXT after Fenton catalysis. Meanwhile, only about 10% of the three adsorbates were oxidized by H2O2 after 40 min. These phenomena suggested that neither H2O2 nor PB could removal MB, MO and CXT effectively. The adsorption of the hydrothermally treated yeast contributed to 38% of MB, 43% MO and 42% of CXT and increased to 52%, 47% and 48% in the addition of H2O2 with the UV radiation. It may attribute to the generation of a certain amount of hydroxyl radicals produced by H2O2 with UV irradiation. These radicals could act directly on organic pollutants and promote the regeneration and exposure of active sites on the yeast surface thereby increasing the removal efficiency, although this increase was small. The adsorption efficiency of PB@yeast for MB (43%), MO (51%) and CXT (56%) was slightly higher than that of yeast. However, these three organic compounds were rapidly removed within 30 min, suggesting the synergistically enhanced adsorption and Fenton catalysis of PB@yeast hybrids.
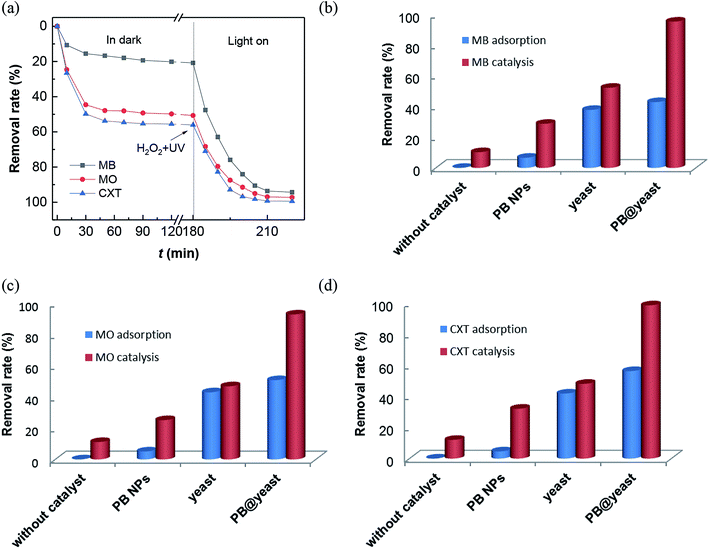 |
| | Fig. 4 (a) Removal rates of MB, MO and CXT by PB@yeast under dark and UV radiation; comparison of adsorption/Fenton catalysis for removal of (b) MB, (c) MO and (d) CXT in different reaction systems. | |
In view of the performance of enhanced activity of PB@yeast in the three contaminants, the following hypothesis was formulated. Generally, the remarkable adsorption activity of the hollow porous hybrids contributed to many advantages such as lower mass transfer resistance, shortened charge transport pathways, which allowed the existing target molecules to easily enter immobilized active sites. Subsequently, these locked organic molecules tended to be especially attacked by formed hydroxyl radicals and thus rapidly degraded in Fenton oxidation.49 As a novel class of hierarchical porous architectures, the hollow-shelled S. cerevisiae with high specific surface and functional bonds can protect PB nanoparticles from aggregation, what would help to protect the active sites and achieve improved Fenton catalytic efficiency. Scanning electron micrographs (Fig. 2b) had given a certain support that the PB nanoparticles were dispersed on the entire surface of yeasts with high degree of homogeneity. Also, the interaction between PB and yeast had been demonstrated via hydrogen bond, electrostatic attraction and coordinate bond as mentioned in FT-IR analysis (Fig. 3a). It rendered high stability of the catalyst, what is really important in Fenton reaction.50 What's more, owing to decomposition of organic pollutants and UV irradiation, the certain adsorption sites on the surface of yeasts regenerated, that further enhanced the adsorption coupled Fenton activity. In conclusion, the possible synergistic effect primarily involved three steps: (a) bio-adsorption from S. cerevisiae cells, (b) Fenton catalysis from PB NPs, and (c) regeneration of adsorptive sites from target pollutants degradation. These three behaviors also had synergistic effect in the reaction. In a word, the synergistically strengthened PB@yeast can provide remarkable adsorption coupled Fenton performance, leading to high efficiency for selected contaminants removal.
3.4 pH-dependent performances of PB@yeast microspheres
It has been evidenced that the PB@yeast hybrids combined the adsorption and Fenton catalytic properties, which could remove these three selected contaminants synergistically. Nevertheless both adsorption and Fenton reactions are pH-dependent.51,52 Here, CXT was used to assess the pH tolerance at pH 3.0–9.0. To investigate the possible mechanism of CXT removal, several kinetic studies were applied to evaluate the adsorption process. The corresponding kinetic models were also applied to estimate the pH-dependent adsorption and Fenton catalysis respectively. For adsorption, the pseudo second order equation was used to fit the data, which is expressed in eqn (3).| |
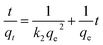 | (3) |
The initial adsorption rate h was calculated additionally according to eqn (4).53
where
qe (mg g
−1) and
qt (mg g
−1) are the amount of pollutions adsorbed at equilibrium and time
t (min);
k2 (g (mg min)
−1) is the kinetic rate constant for the pseudo second order kinetic model; and
h (mg g
−1 min
−1) is the initial adsorption rate.
For Fenton reaction, the pseudo first order kinetic law (eqn (5)) was utilized in the reactions.54
| |
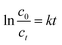 | (5) |
where
c0 (mg L
−1) is the initial concentration;
ct is the concentration of CXT at the time
t;
k is the relevant constant.
Fig. 5 shows the initial pH influence on CXT adsorption and degradation, respectively.
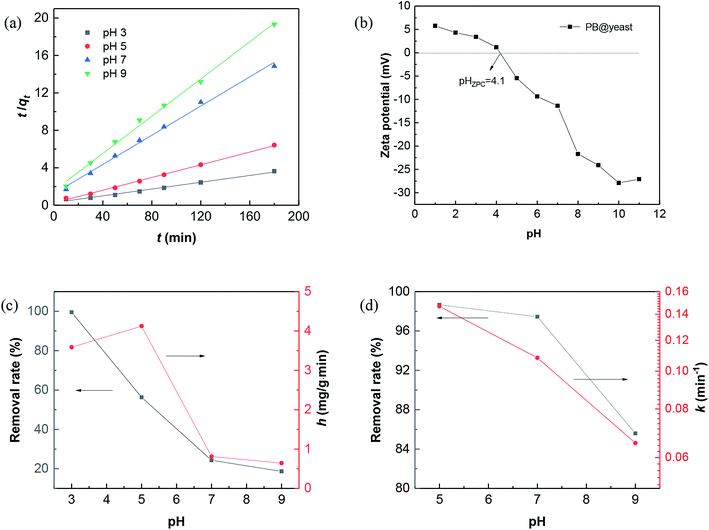 |
| | Fig. 5 (a) Fitting data for pseudo-second-order kinetics of CXT adsorption; (b) zeta potentials of PB@yeast; initial pH influence on (c) CXT adsorption and (d) CXT degradation. Reaction conditions: [CXT] = 25 mg L−1, catalyst = 50 mg, and [H2O2] = 2.0 mol L−1. | |
For the effect of pH on CXT adsorption, plotting t/qt against t displayed a well linear relationship when adjusting pH value from 3.0 to 9.0 (Fig. 5a) with the correlation coefficients were 0.9881, 0.9975, 0.9945 and 0.9952 (Table 1), which indicated that the CXT adsorption at pH 3–9 were well fitted by the pseudo-second-order kinetic model. From this inference, it is easy to verdict that the adsorption was probably a physicochemical process, which was carried out by sharing or exchanging electrons between the adsorbent and organic contaminant.55 Hence, completely different adsorption activities occurred when it referred to organic contaminants with different charges. The results of initial pH influence on CXT adsorption in dark is shown in Fig. 5c. As shown, the CXT solution was completely adsorbed in 120 min at pH 3.0, while the adsorption rates were approximately 56% at pH 5.0 and 24% pH 7.0 then fell to 18% at pH 9.0. It seems that low pH circumstances were in favor of the CXT sorption better. The initial adsorption rate h showed the same tendency roughly, which is primarily responsible for the pH-dependent absorption of yeast. Considering the phenomenon, the ζ-potential values of PB@yeast at different pH values were investigated. As shown in Fig. 5b, the zero point of zeta potentials (pHZPC) for PB@yeast composites was at pH 4.1. It perfectly explains the causes that complete adsorption of CXT at pH 3.0. Here, the surfaces of PB@yeast are protonated and thus become positively charged; it could strong attract the anionic CXT molecules through electrostatic attraction. As the pH value increased above the pHZPC, however, deprotonation of the surface occurred, resulting in negative charges.56 In this case, some functional groups such as carboxyl, phosphate, and sulfonyl started to ionize with the consumption of protons, leading to decreased efficiencies of CXT adsorption. Simultaneously, it provides a certain interpretation on Fig. 5b that low sorption of cationic MB at low pH value. Still, the adsorption of anionic CXT and cationic MB were significant affected by electrostatic forces.
Table 1 Kinetics model for CXT adsorption and degradation at different pH value
| pH value |
For adsorption |
For Fenton catalysis |
| qe,exp mg g−1 |
qe,cal mg g−1 |
k2 g mg−1 min−1 |
h mg g−1 min−1 |
R2 |
k min−1 |
R2 |
| 3 |
49.50 |
55.12 |
0.0012 |
3.588 |
0.9881 |
— |
— |
| 5 |
28.02 |
29.43 |
0.0048 |
4.122 |
0.9975 |
0.1467 |
0.9953 |
| 7 |
12.11 |
12.82 |
0.0049 |
0.8102 |
0.9945 |
0.1082 |
0.9989 |
| 9 |
9.32 |
10.06 |
0.0064 |
0.6433 |
0.9952 |
0.0654 |
0.9864 |
After reaching adsorption–desorption equilibrium, amount of H2O2 was added in each system with UV light turning on synchronously. From Fig. 5d, it can be seen the removal rate of CXT was up to 99% at pH 5.0 and to 97% at pH 7.0 in 30 min. Even at pH 9.0, it could be still reached to 86% markedly. The values of k decreased slightly within the specified pH value, suggesting that PB@yeast could degrade CXT under a wide pH range, which is greatly consistent with heterogeneous Fenton oxidation reaction.57,58 Given all this, it can be affirmed that PB@yeast had the strong ability to remove CXT by integrating adsorption and Fenton catalysis over a wide pH range. The notable Fenton activity of PB@yeast could week the limitation that the adsorption is subject to pH value, and this bifunctional PB@yeast with pH-tolerant property could bring a promising application in organic wastewater treatment.
3.5 Synergistically enhanced reaction mechanism
It is currently accepted that ·OH radicals play significant role in Fenton reaction. However, some controversies still exist with regard to the formation of different oxidants.59,60 In this study, a popular hydroxyl radical scavenger t-butanol was employed to catching ·OH produced by Fenton reaction for CXT degradation. As shown in Fig. 6a, the removal rates of CXT decreased distinctly with the participation of t-butanol. This result demonstrated that ·OH was the major oxidant in the degradation of CXT, the possible mechanism can be proposed as following eqn (6)–(9).61,62| |
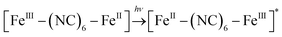 | (6) |
| | |
[FeII–(NC)6–FeIII]* + H2O2 → [FeIII–(NC)6–FeIII] + ·OH + OH−
| (7) |
| |
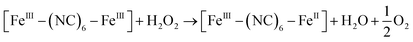 | (8) |
| | |
·OH + organics → intermediates → CO2 + H2O
| (9) |
Here, the inter valence charge-transfer reaction of PB firstly occurred under UV irradiation (eqn (6)). The excited PB was metastable, it can be easy to transfer electron from FeII to hydrogen peroxide to form active hydroxyl radicals (eqn (7)). Afterwards, the resulting product Berlin green (FeIII(NC) FeIII), with a high potential 1.3 V versus NHE,63 can oxidize hydrogen peroxide to dioxygen (eqn (8)), thus promoting the catalytic reaction cycle. It should be mentioned that with the assistance of UV light, the PB nanoparticles could capture the conduction band and be easy to deoxidize FeIII to FeII, which can increase the Fenton reaction recycle (eqn (10)).64| | |
[FeIII–(NC)6–FeIII] + e− → [FeIII–(NC)6–FeII]
| (10) |
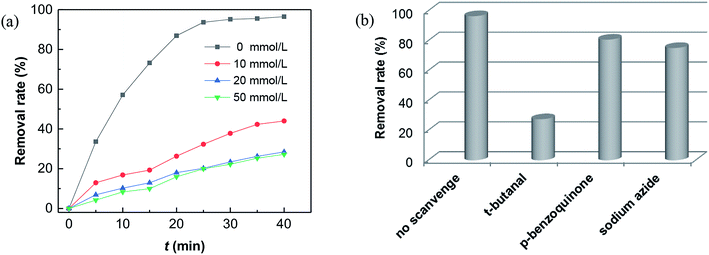 |
| | Fig. 6 Influence of (a) t-butanol scavenger at different dosage; (b) various radical scavengers in Fenton reaction. Reaction conditions: [CXT] = 40 mg L−1, [H2O2] = 2.0 mol L−1, catalyst = 50 mg, [scavengers] = 50 mmol L−1 and UV lamp: 30 W. | |
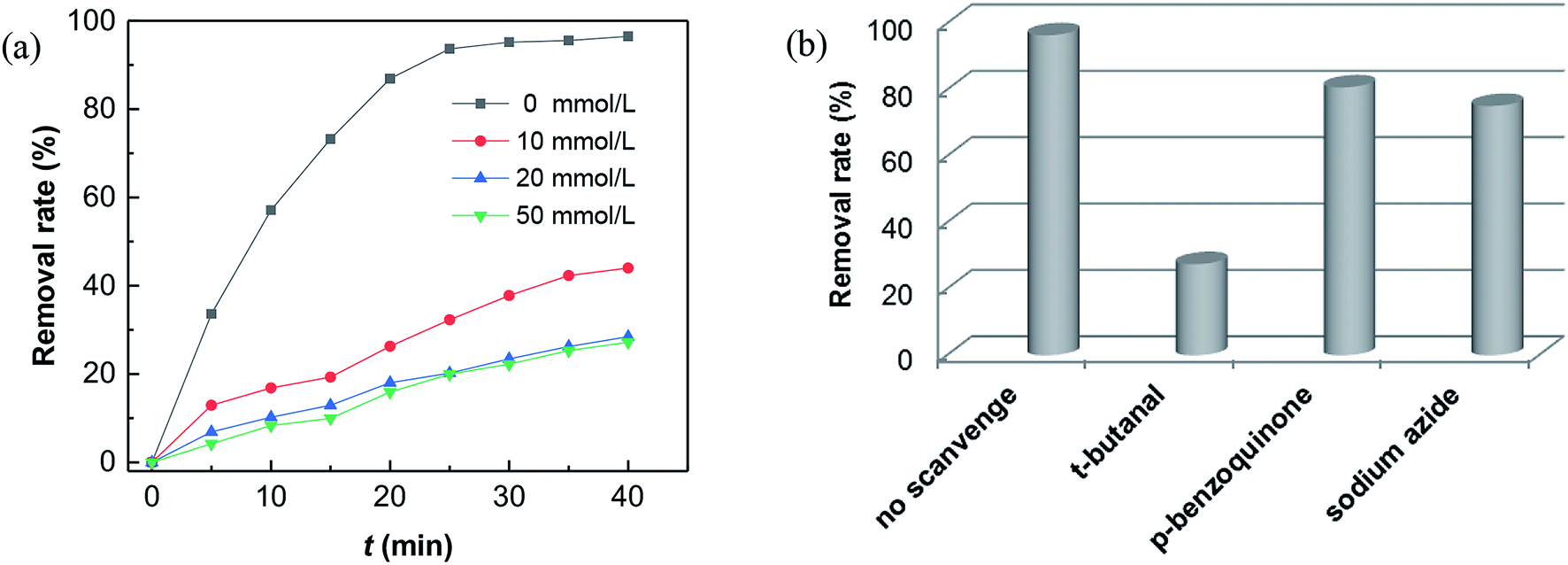
What's interesting, the CXT degradation efficiency could still reach to 27% when the t-butanol concentration was up to 50 mM, which inferred that other reaction mechanism may exist in CXT degradation. Considering the statement that ·OH is more likely attack organic molecules nearby,65,66 an important aspect to be deliberated in CXT degradation is the adsorption of anionic CXT molecules from substrate to PB active site. It had been assessed that the pHZPC was about 4.1, which means a certain degree of electrostatic forces including electrostatic attraction and repulsion occurred between the PB@yeast surface and the CXT molecules at pH 5.0 which had a great impact on mass transfer resistance. In this case, the lower the concentration of CXT molecules near the surface of PB@yeast, the weaker the reaction of ·OH would carry out. In the study of Li et al.,67 except ·OH, other radicals such as HOO˙ and 1O2 may also be active radicals intermediate involved in the Fenton reaction of Prussian blue analogues. Based on this, p-benzoquinone and sodium azide were employed in the reaction. The result presented in Fig. 6b indicated that HOO˙ and 1O2 also played role in CXT degradation in this condition. The relevant radical reactions expressed as eqn (11)–(17).
| | |
FeII + H2O2 → FeIII + HO˙ + OH−
| (11) |
| | |
FeIII + H2O2 → FeII + HOO˙ + H+
| (12) |
| | |
HO˙ + H2O2 → HOO˙ + H2O
| (13) |
| |
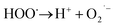 | (14) |
| |
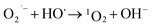 | (15) |
| |
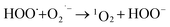 | (16) |
| | |
HOO˙ + HOO˙ → 1O2 + H2O2
| (17) |
As far as the results under the selected conditions were concerned, it can be clearly demonstrated that the Fenton reaction mechanism may be closely related to ·OH, 1O2 and HOO˙ radicals, which were significantly dominated by the adsorption of PB@yeast. In this respect, the adsorption by electrostatic attraction should contribute to attracting the CXT substrate and bringing it close to the active site of catalyst. Furthermore, the regeneration of adsorption sites with the degradation of CXT molecules probably stimulate a synergistic effect which was supported in Fig. 6a that the removal rates increased slightly as the reaction time prolonged, even if t-butanol in range of 10–50 mmol L−1 was added. On the other hand, conversely, the existed electrostatic repulsion might weaken the transfer of the substrate to some extent, thus opening a gate to 1O2 and HOO˙ radicals in Fenton reaction. Table S1† shows a comparison of the performance of PB-based Fenton catalysts in dealing with dye wastewater. A higher relevant rate constant was obtained in the CXT degradation over PB@yeast composites, suggesting that the Fenton activity of PB@yeast is even higher than those of the PB-based catalysts reported in previous literatures. It is probably related to the excellent adsorption-coupled Fenton properties of the PB@yeast microspheres. In general, the finding that an adsorption-controlled reaction mechanism involved in CXT degradation on multi-functional PB@yeast hybrids would develop an attractive perspective for the synergistic enhancement mechanism of PB@yeast.
3.6 Stability and reusability of PB@yeast microspheres
The high efficiency and easy separation of Fenton catalysts are essential to meet their practical applications. Generally, a suspended-state catalyst provides more effects than the settled one since it can facilitate the contact area between the catalyst and the contaminant molecules in aqueous solution. However, the separation and reusability of nano-sized catalyst in suspension remains a challenge. In this work, it was observed that hollow structured PB@yeast microspheres were suspended in the reactor only with slight agitation or vibration. It was mainly attributed to low density of PB@yeast microspheres, in which the wet density of the yeast support (1.09 ± 0.008 g cm−3) was almost equal to the density of water.68 Benefiting from this advantage of PB@yeast microspheres, the synergistic adsorption and Fenton reaction were further enhanced by a mildly stirred suspended-state reaction system. Moreover, due to the relatively large micronized particles in the suspension, the PB@yeast catalyst can be easily separated by conventional filtration for reuse. The reusability of PB@yeast was tested by evaluating the removal efficiency of CXT in consecutive runs. From Fig. 7a, the catalyst can be efficiently effectively operated in four consecutive runs for CXT removal without significant drop (99–94%). Moreover, the XRD pattern of the hybrids after 4 cycles was similar to the fresh and no impurity peak was detected (displayed in Fig. 1a), suggesting the chemical stability of PB@yeast.
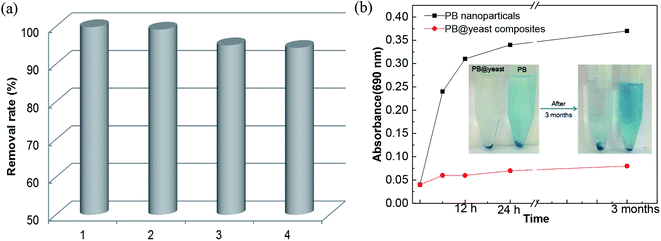 |
| | Fig. 7 (a) Reusability of PB@yeast in 4 cycles and (b) the PB release behavior (inset: images of PB release over time). | |
Fig. 7b shows the change in PB release behavior over time. Compared to PB nanoparticles, few changes were detected by UV-vis spectroscopy, indicating that the PB@yeast was stable with scarce release of PB nanoparticles. It is mainly attributed to the protection of yeast where the strong hydrogen bonds interaction and amide moieties of yeast linking to PB nanoparticles that strengthened the stability of PB@yeast composites. The inset photograph of Fig. 7b displayed that no distinct release of PB from PB@yeast was observed even after 3 months, suggesting high stability of PB@yeast composites in solution, which was more stable than PB@yeast synthesized by chemical precipitation in previous study. In summary, these results implied that PB@yeast held excellent stability and recyclability in aqueous solution, which would make a great contribution to the adsorption coupled Fenton catalytic performance.
4. Conclusion
In this work, hollow porous Prussian blue@yeast (PB@yeast) microspheres were fabricated from a facile hydrothermal synthesis, in which Prussian blue nanoparticles (PB NPs) were in situ grown on the multifunctional S. cerevisiae cells. Benefiting from the unique hollow porous structure and integrated dual function, the newly PB@yeast hybrid microspheres exhibited synergistic adsorption and Fenton catalytic activity in removal of the selected cationic methylene blue (MB), anionic Methyl Orange (MO) and fluorescent brightener 71 (CXT) in aqueous solution. Synergistic mechanism study implied that the enhanced performance contained triple behaviors as adsorption–Fenton catalysis–adsorption sites regeneration and the three also had synergistic effect in the progress. In brief, the main Fenton reaction involved ·OH, 1O2 and HOO˙ radicals, which were significantly improved by appropriate adsorption of PB@yeast. The pH tolerance analysis and final stability results indicated the newly developed PB@yeast could be operated efficiently over a wide pH range (3.0–9.0) and maintain high efficiency in four consecutive runs without significant drops (99–94%). Based on these findings, it can be concluded that the design in this work proposed a promising approach for exploiting the synergistically strengthened PB@yeast hybrids, which would give considerably practical significance in the treatment of organic wastewater.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Innovation Platform for the Development and Construction of Special Project of Key Laboratory of Tibetan Medicine Research of Qinghai Province (No. 2017-ZJ-Y11) and Qinghai Provincial Science Foundation (2017-SF-A8).
References
- Y. J. Shih, C. H. Ho and Y. H. Huang, Water Environ. Res., 2013, 85, 340–345 CrossRef CAS PubMed.
- M. Gui, V. Smuleac, L. E. Ormsbee, D. L. Sedlak and D. Bhattacharyya, J. Nanopart. Res., 2012, 14, 1–16 CrossRef.
- E. Nossol, A. B. S. Nossol, A. J. G. Zarbin and A. M. Bond, RSC Adv., 2013, 3, 5393–5400 RSC.
- G. Salierno, Int. J. Chem. Eng. Appl., 2013, 4, 295–300 Search PubMed.
- N. X. D. Mai, J. Yoon, J. H. Kim, I. T. Kim, H. B. Son, J. Bae and J. Hur, Sci. Adv. Mater., 2017, 9, 1484–1487 CrossRef CAS.
- N. A. Sitnikova, M. A. Komkova, I. V. Khomyakova, E. E. Karyakina and A. A. Karyakin, Anal. Chem., 2014, 86, 4131–4134 CrossRef CAS PubMed.
- M. Zhang, C. Hou, A. Halder, J. Ulstrup and Q. Chi, Biosens. Bioelectron., 2017, 89, 570–577 CrossRef CAS PubMed.
- A. A. Karyakin, E. E. Karyakina and L. Gorton, Electrochem. Commun., 1999, 1, 78–82 CrossRef CAS.
- D. Iveković, M. Japec, M. Solar and N. Živković, Int. J. Electrochem. Sci., 2012, 7, 3252–3264 Search PubMed.
- T. Sangvanich, V. Sukwarotwat, R. J. Wiacek, R. M. Grudzien, G. E. Fryxell, R. S. Addleman, C. Timchalk and W. Yantasee, J. Hazard. Mater., 2010, 182, 225–231 CrossRef CAS PubMed.
- L. Doumic, G. Salierno, M. Cassanello, P. Haure and M. Ayude, Catal. Today, 2015, 240, 67–72 CrossRef CAS.
- C. Vincent, Y. Barré, T. Vincent, J. M. Taulemesse, M. Robitzer and E. Guibal, J. Hazard. Mater., 2015, 287, 171–179 CrossRef CAS PubMed.
- T. Vincent, C. Vincent, Y. Barré, Y. Guari, G. L. Saout and E. Guibal, J. Mater. Chem. A, 2014, 2, 10007–10021 RSC.
- T. Li, F. He and Y. D. Dai, J. Radioanal. Nucl. Chem., 2016, 310, 1–7 CrossRef.
- G. Liang, L. Zheng, S. P. Bao, B. Fei, H. Y. Gao, F. M. Zhu and Q. Wu, Macromolecules, 2015, 48, 4115–4121 CrossRef CAS.
- S. Manivannan, I. Kang and K. Kim, Langmuir, 2016, 32, 1890–1898 CrossRef CAS PubMed.
- W. Xi, G. Yan, Z. Lang, Y. Ma, H. Tan, H. Zhu, Y. Wang and Y. Li, Small, 2018, 1802204 CrossRef PubMed.
- Y. Chen, H. R. Chen and J. L. Shi, Acc. Chem. Res., 2014, 47, 125–137 CrossRef CAS.
- S. H. Wu, C. Y. Mou and H. P. Lin, Chem. Soc. Rev., 2013, 42, 3862 RSC.
- C. Li, B. Jiang, M. Imura, V. Malgrasa and Y. Yamauchi, Chem. Commun., 2014, 97, 15337–15340 RSC.
- G. Yan, X. Zhang and L. Xiao, J. Mater. Sci., 2019, 6315, 1–9 Search PubMed.
- Y. Li and J. Shi, Adv. Mater., 2014, 26, 3176–3205 CrossRef CAS PubMed.
- W. He, S. Yan, Y. Wang, X. Zhang, W. Zhou, X. Tian, X. Sun and X. Han, J. Alloys Compd., 2009, 477, 657–660 CrossRef CAS.
- X. Tian, W. He, J. Cui, X. Zhang, W. Zhou, S. Yan, X. Sun, X. Han, S. Han and Y. Yue, J. Colloid Interface Sci., 2010, 343, 344–349 CrossRef CAS PubMed.
- D. Ni, L. Wang, Y. Sun, Z. Guan, S. Yang and K. Zhou, Angew. Chem., 2010, 49, 4223–4227 CrossRef CAS PubMed.
- X. Fan, X. Song, X. Yang and L. Hou, Mater. Res. Bull., 2011, 46, 1315–1319 CrossRef CAS.
- P. Magnelli, J. F. Cipollo and C. Abeijon, Anal. Biochem., 2002, 301, 136–150 CrossRef CAS PubMed.
- J. Margaret and J. S. Pierce, J. Inst. Brew., 2013, 70, 307–315 Search PubMed.
- C. Si, B. Bo, W. Hong-lun and S. You-rui, Environ. Sci., 2018, 8, 3759–3766 Search PubMed.
- L. Zhang, H. B. Wu, S. Madhavi, H. H. Hng and X. W. Lou, J. Am. Chem. Soc., 2012, 134, 17388–17391 CrossRef CAS PubMed.
- L. Zhang, H. B. Wu and X. W. Lou, J. Am. Chem. Soc., 2013, 135, 10664–10672 CrossRef CAS PubMed.
- W. He, Z. Li, Y. Wang, X. Chen, X. Zhang, H. Zhao, S. Yan and W. Zhou, J. Mater. Sci.: Mater. Med., 2010, 21, 155–159 CrossRef CAS PubMed.
- M. Beekes, P. Lasch and D. Naumann, Vet. Microbiol., 2007, 123, 305–319 CrossRef CAS PubMed.
- S. C. Jang, S. B. Hong, H. M. Yang, K. W. Lee, J. K. Moon, B. K. Seo, Y. S. Huh and C. Roh, Nanomaterials, 2014, 4, 894–901 CrossRef PubMed.
- J. Li, J. Qiu, J. Xu, H. Chen and X. Xia, Adv. Funct. Mater., 2010, 17, 1574–1580 CrossRef.
- L. Zhang, A. Zhang, D. Du and Y. Lin, Nanoscale, 2012, 4, 4674–4679 RSC.
- Y. Hou, T. Huang, Z. Wen, S. Mao, S. Cui and J. Chen, Adv. Energy Mater., 2014, 4, 1400337 CrossRef.
- K. B. Yatsimirskii, V. V. Nemoshkalenko, Y. P. Nazarenko, V. G. Aleshin, V. V. Zhilinskaya and N. A. Tomashevsky, J. Electron Spectrosc. Relat. Phenom., 1977, 10, 239–245 CrossRef CAS.
- Z. Yang, X. Zheng and J. Zheng, Ind. Eng. Chem. Res., 2016, 55, 12161–12166 CrossRef CAS.
- L. Cao, Y. Liu, B. Zhang and L. Lu, ACS Appl. Mater. Interfaces, 2010, 2, 2339–2346 CrossRef CAS PubMed.
- E. Nevoigt, Microbiol. Mol. Biol. Rev., 2008, 72, 379–412 CrossRef CAS PubMed.
- E. Cabib, D. H. Roh, M. Schmidt, L. B. Crotti and A. Varma, J. Biol. Chem., 2001, 276, 19679–19682 CrossRef CAS PubMed.
- L. Chen and B. Bai, Ind. Eng. Chem. Res., 2013, 52, 15568–15577 CrossRef CAS.
- W. He, J. Cui, Y. Yue, X. Zhang, X. Xia, H. Liu and S. Lui, J. Colloid Interface Sci., 2011, 354, 109–115 CrossRef CAS PubMed.
- Y. N. Li, J. Su, X. Y. Lv, Y. F. Long and Y. X. Wen, Electrochim. Acta, 2015, 182, 596–603 CrossRef CAS.
- M. Hu, J. S. Jiang, R. P. Ji and Y. Zeng, CrystEngComm, 2009, 11, 2257–2259 RSC.
- M. Hu, N. L. K. Torad, Y. D. Chiang, K. C. W. Wu and Y. Yamauchi, CrystEngComm, 2012, 14, 3387–3396 RSC.
- W. C. Shu and W. H. Ding, J. Chromatogr. A, 2005, 1088, 218–223 CrossRef CAS.
- S. P. Sun, X. Zeng and A. T. Lemley, J. Mol. Catal. A: Chem., 2013, 371, 94–103 CrossRef CAS.
- H. Ramirez, M. A. Vicente and L. M. Madeira, Appl. Catal., B, 2010, 98, 10–26 CrossRef.
- M. López-García, P. Lodeiro, R. Herrero, J. L. Barriada, C. Reycastro and C. David, Bioresour. Technol., 2013, 139, 181–189 CrossRef PubMed.
- Y. M. Zheng, T. Liu, J. Jiang, L. Yang, Y. Fan, A. T. Wee and J. P. Chen, J. Colloid Interface Sci., 2011, 356, 741–748 CrossRef CAS PubMed.
- Q. Cao, F. Huang, Z. Zhuang and Z. Lin, Nanoscale, 2012, 4, 2423–2430 RSC.
- S. Q. Liu, S. Cheng, L. Luo, H. Y. Cheng, S. J. Wang and S. Lou, Environ. Chem. Lett., 2011, 9, 31–35 CrossRef CAS.
- L. Tang, Y. Fang, Y. Pang, G. Zeng, J. Wang, Y. Zhou, Y. Deng, G. Yang, Y. Cai and J. Chen, Chem. Eng. J., 2014, 254, 302–312 CrossRef CAS.
- T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 1519–1527 CrossRef CAS PubMed.
- S. R. Pouran, A. A. Abdul Raman and W. M. A. Wan Daud, J. Cleaner Prod., 2014, 64, 24–35 CrossRef.
- W. J. Li, H. Liu, A. Guo, L. Chang, X. Y. Yao, B. Z. Sun, J. L. Meng, Y. Li and W. J. Ruan, RSC Adv., 2016, 6, 68227–68230 RSC.
- C. Minero, M. Lucchiari, V. Maurino and D. Vione, RSC Adv., 2013, 3, 26443–26450 RSC.
- J. A. Donadelli, L. Carlos, A. Arques and F. S. G. Einschlag, Appl. Catal., B, 2018, 231, 51–61 CrossRef CAS.
- S. Q. Liu, S. Cheng, L. R. Feng, X. M. Wang and Z. G. Chen, J. Hazard. Mater., 2010, 182, 665–671 CrossRef CAS PubMed.
- M. Luo, L. Lv, G. Deng, W. Yao, Y. Ruan, X. Li and A. Xu, Appl. Catal., B, 2014, 469, 198–205 CrossRef CAS.
- Y. Ding, Y. Hu, G. Gu and X. Xia, J. Phys. Chem. C, 2009, 113, 14838–14843 CrossRef CAS.
- X. Li, J. Wang, A. Rykov, V. Sharma, H. Wei, C. Jin, X. Liu, M. Li, S. Yu and C. Sun, Catal. Sci. Technol., 2014, 5, 504–514 RSC.
- X. Hu, B. Liu, Y. Deng, H. Chen, S. Luo, C. Sun, P. Yang and S. Yang, Appl. Catal., B, 2011, 107, 274–283 CrossRef CAS.
- L. Gu, N. Zhu, H. Guo, S. Huang, Z. Lou and H. Yuan, J. Hazard. Mater., 2013, 246, 145–153 CrossRef PubMed.
- X. Li, J. Liu, A. I. Rykov, H. Han, C. Jin, X. Liu and J. Wang, Appl. Catal., B, 2015, 179, 196–205 CrossRef CAS.
- B. Bai, N. Quici, Z. Li and G. L. Puma, Chem. Eng. J., 2011, 170, 451–456 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02918d |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bcd,
Xiaohui Xuab,
Na Hucd,
Honglun Wangcd and
Yourui Suocd
*bcd,
Xiaohui Xuab,
Na Hucd,
Honglun Wangcd and
Yourui Suocd