
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFe3O4/SO3H@zeolite-Y as a novel multi-functional and magnetic nanocatalyst for clean and soft synthesis of imidazole and perimidine derivatives
Mehdi Kalhor * and
Zohre Zarnegar
* and
Zohre Zarnegar
Department of Chemistry, University of Payame Noor, P. O. Box 19395-4697, Tehran, Iran. E-mail: mekalhor@gmail.com; Fax: +98 2537179170; Tel: +98 2537179170
First published on 20th June 2019
Abstract
In this study, SO3H@zeolite-Y was synthesized by the reaction of chlorosulfonic acid with zeolite-NaY under solvent-free conditions, which was then supported by Fe3O4 nanoparticles to give SO3H@zeolite-Y (Fe3O4/SO3H@zeolite-Y) magnetic nanoparticles. Several techniques were used to evaluate the physical and chemical characterizations of the zeolitic nanostructures. Fe3O4-loaded sulfonated zeolite was applied as a novel multi-functional zeolite catalyst for the synthesis of imidazole and perimidine derivatives. This efficient methodology has some advantages such as good to excellent yield, high purity of products, reusability of nanocatalyst, simple reaction conditions, environmental friendliness and an economical chemical procedure from the viewpoint of green chemistry.
1. Introduction
Nitrogen-containing heterocyclic compounds including imidazole and perimidine have attracted significant interest over the past decade due to their widespread applications.1–3 They are predominantly used as antimicrobial, antiulcer, anti-inflammatory, antifungal, antitumor, antibiotic, anti-thrombotic, and therapeutic agents.4–6 Some of the imidazole and perimidine molecules with biological activities are presented in Fig. 1.7–9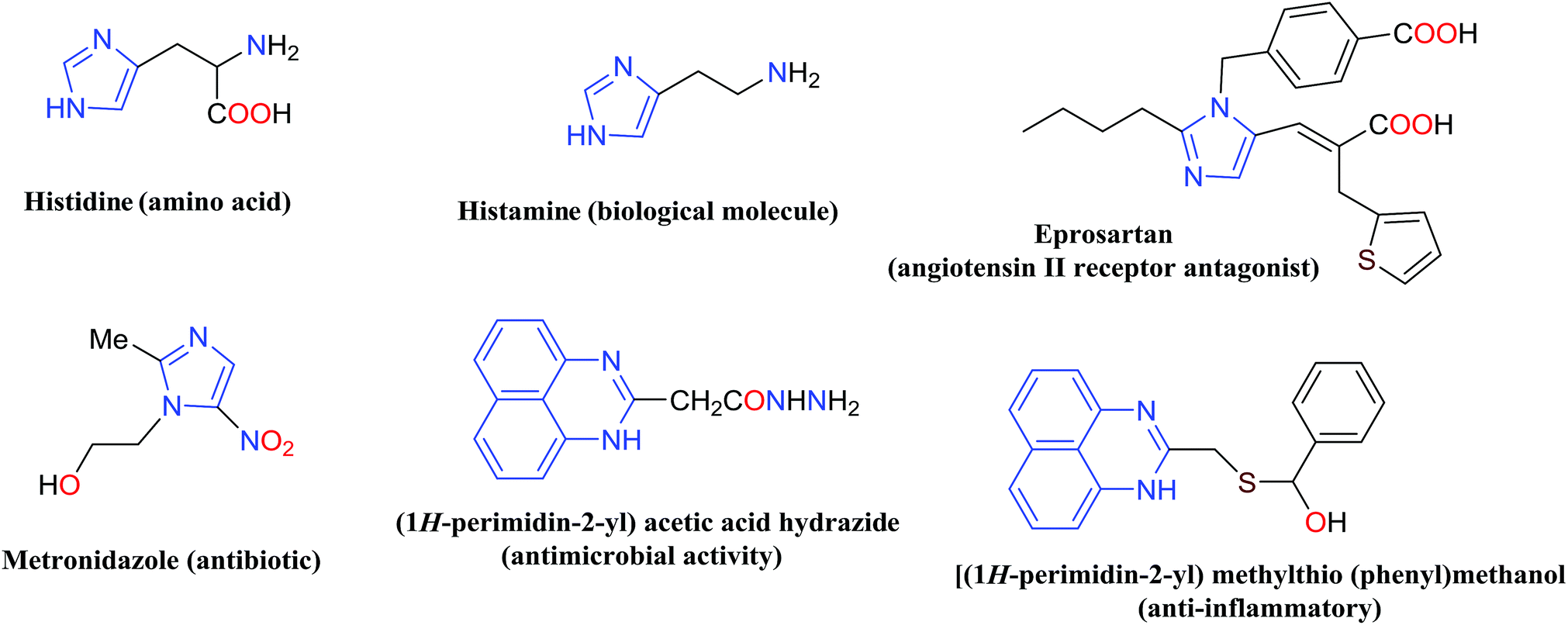 | ||
| Fig. 1 Some of the imidazoles and perimidines with medicinal properties and biological activities. | ||
Imidazole was first reported in 1858 by Heinrich Debus via cyclocondensation of glyoxal, formaldehyde, and ammonia.10 Various synthetic methods have already reported the synthesis of substituted imidazoles by varying the functional groups on reactants. The tri-substituted imidazoles have been synthesized via multicomponent reaction using 1,2-dicarbonyl compounds, different aldehydes, and a nitrogen source in the presence of acidic catalysts.11–18 Although these reported catalytic methods are very useful and effective in some aspects, they may have some limitations and difficulties such as strongly acidic conditions, occurrence of side reactions, long reaction time, and require harsh reaction conditions, and complex work-up and purification.
Perimidines or 1H-benzo[d,e]quinazolines as saturated N-heterocycles are valuable molecules in the pharmaceutical industry, agriculture, and synthetic chemistry. Perimidine derivatives are usually prepared through condensation reaction of 1,8-diaminonaphthalene with various carbonyl compounds.19–21 However, most of reported methods suffers from long reaction times, cumbersome work-up procedures, solvents and low yield reactions.20,21 Thus, the development of suitable and fantastic methods is still desirable for the synthesis of perimidines and highly substituted imidazoles especially by using highly active, robust, stable, and green catalysts.
Recently, magnetic separations have been introduced as optional approach for recovering and reusing catalysts in chemical catalytic procedures. Magnetic nanocatalysts are heterogeneous catalytic systems with advantages of simple recovery and reuse after completion of reactions. Among the various types of magnetic catalysts, the sulfonic acid functionalized heterogeneous catalyst based on the magnetic systems show excellent catalytic activities, high stability, easy preparation and reusability, enhanced selectivity, operational simplicity, and more economically viable processes. In this context, different types of magnetic SO3H-bearing organic and inorganic compounds used in catalytic reactions such as sulfonic acid magnetic graphene oxide for synthesis of indazolophthalazinetriones,22 Fe3O4@MCM-48–SO3H for the synthesis of benzo[f]chromeno[2,3-d]pyrimidinones,23 Fe3O4@PEG400-SO3H and its use in Paal–Knorr reaction,24 Fe3O4@SiO2–SO3H for the synthesis of amidoalkyl naphthols,25 cobalt ferrite chitosan sulfonic acid magnetic nanoparticle for one-pot, four-component synthesis of 2H-indazolo[2,1-b]phthalazine-triones,26 SO3H-functionalized magnetic-titania nanoparticles for the synthesis of benzimidazoquinazolinones and polyhydroquinolines,27 and etc.28 To the best of our knowledge, there is no report for the application of magnetic SO3H-functionalized zeolite in chemical organic reactions.
Over the past decade, the use of zeolites as heterogeneous catalysts or ideal supports for homogeneous catalysts have been employed for some catalytic processes due to their attractive properties such as high surface area, nanoporous crystalline structure, less or no corrosion, high thermostability, persistence in all organic solvents, no waste or disposal problems, easy set-up of continuous processes, etc.29,30 Although various SO3H-activated zeolites used for catalytic reactions,31 direct methanol fuel cell application,32 and as temperature tolerant proton conducting material,33 but there is no example of the use of magnetic SO3H-modified zeolite-NaY in order to design efficient a heterogeneous catalyst for the synthesis of N-containing heterocycles.
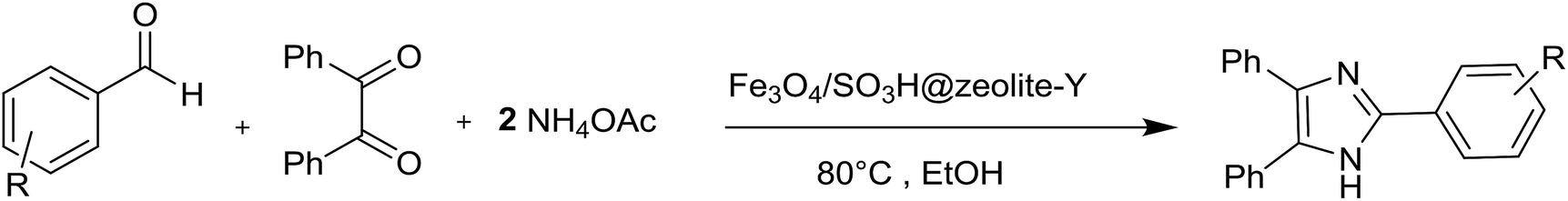
In the present research, an eco-friendly and simple synthetic method have been developed for the preparation of the imidazole and perimidine heterocycles by using of Fe3O4 magnetic nanoparticles supported on zeolosulforic acid (Fe3O4/SO3H@zeolite-Y) nanocomposite as a magnetic solid acid catalyst (Scheme 1).
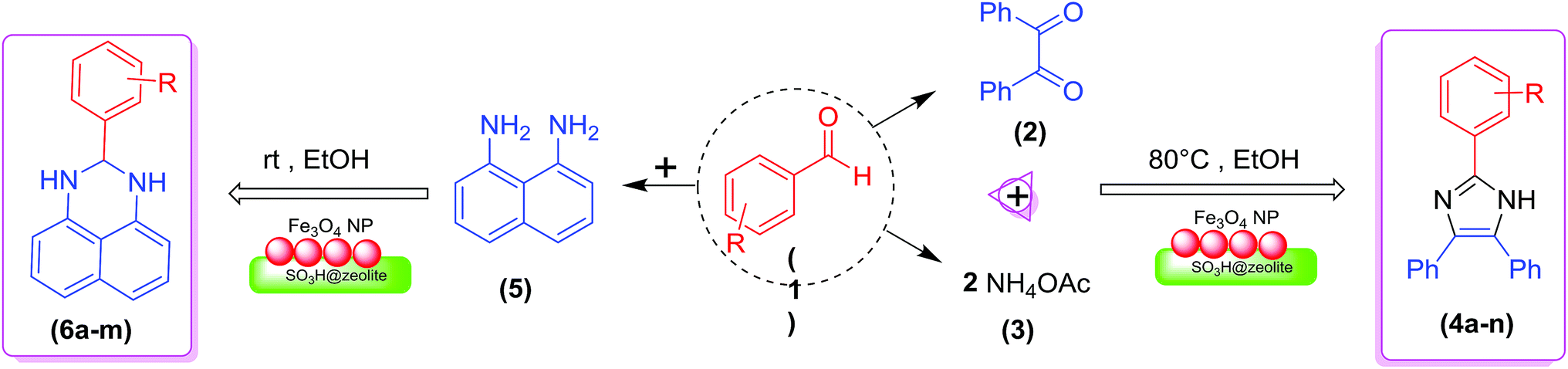 | ||
| Scheme 1 The synthetic pathway of imidazole and perimidine heterocycles using Fe3O4/SO3H@zeolite-Y. | ||
2. Results and discussion
2.1. Characterization of nanocatalyst
As shown in Scheme 2, zeolite-NaY was functionalized with SO3H groups, which has an affinity with positive ferrous and ferric ions. After the addition of ammonium hydroxide solution, Fe3O4 nanoparticles were formed on the surface of SO3H@zeolite-Y. A systematic study was carried out for the characterization of the synthesized Fe3O4/SO3H@zeolite-Y.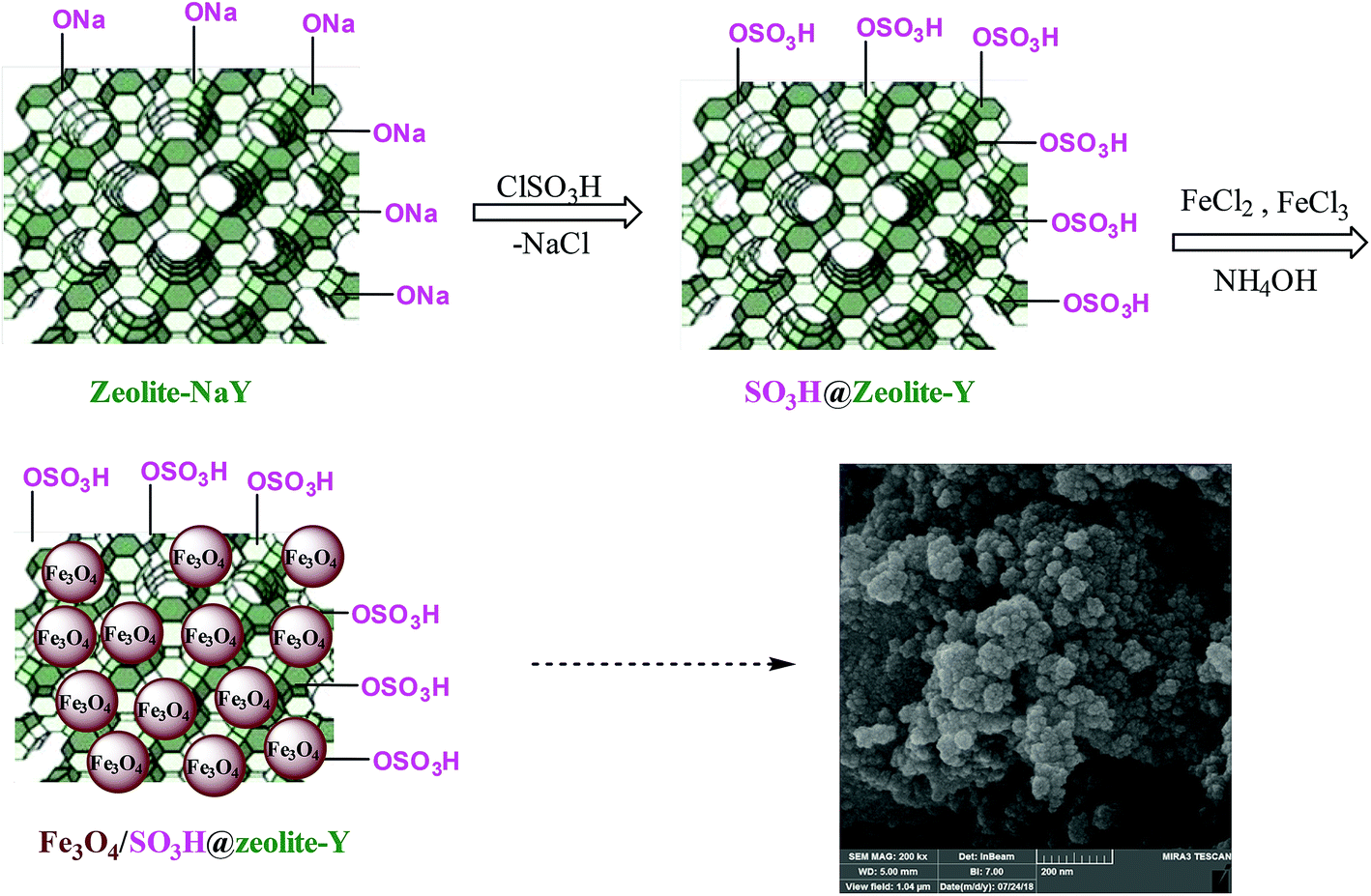 | ||
| Scheme 2 Simplified schematic representation of the preparation of Fe3O4/SO3H@zeolite-Y. | ||
Fig. 2 shows the SEM images of the zeolite-NaY, SO3H@zeolite-Y and Fe3O4/SO3H@zeolite-Y. In Fig. 2a, it is obvious that the zeolite-NaY has a crystalline structure. As Fig. 2b shows, the structure of SO3H@zeolite-Y is similar to that of the pristine zeolite with a rougher surface. Moreover, it can be observed from Fig. 2c that the surface of magnetic zeolite is rough after being coated with Fe3O4. Fig. 2d clearly shows, the Fe3O4 nanoparticles are distributed on the surface of SO3H@zeolite-Y. The above mention results indicate that zeolite was successfully coated with magnetic particles.
 | ||
| Fig. 2 FESEM photographs of (a) zeolite-NaY, (b) SO3H@zeolite-Y, (c) and (d) Fe3O4/SO3H@zeolite-Y. | ||
The composition of Fe3O4/SO3H@zeolite-Y was analyzed by energy dispersive X-rays (EDX) and the results are presented in Fig. 3. Elemental analysis results confirm the Fe element in the modified zeolite structure.
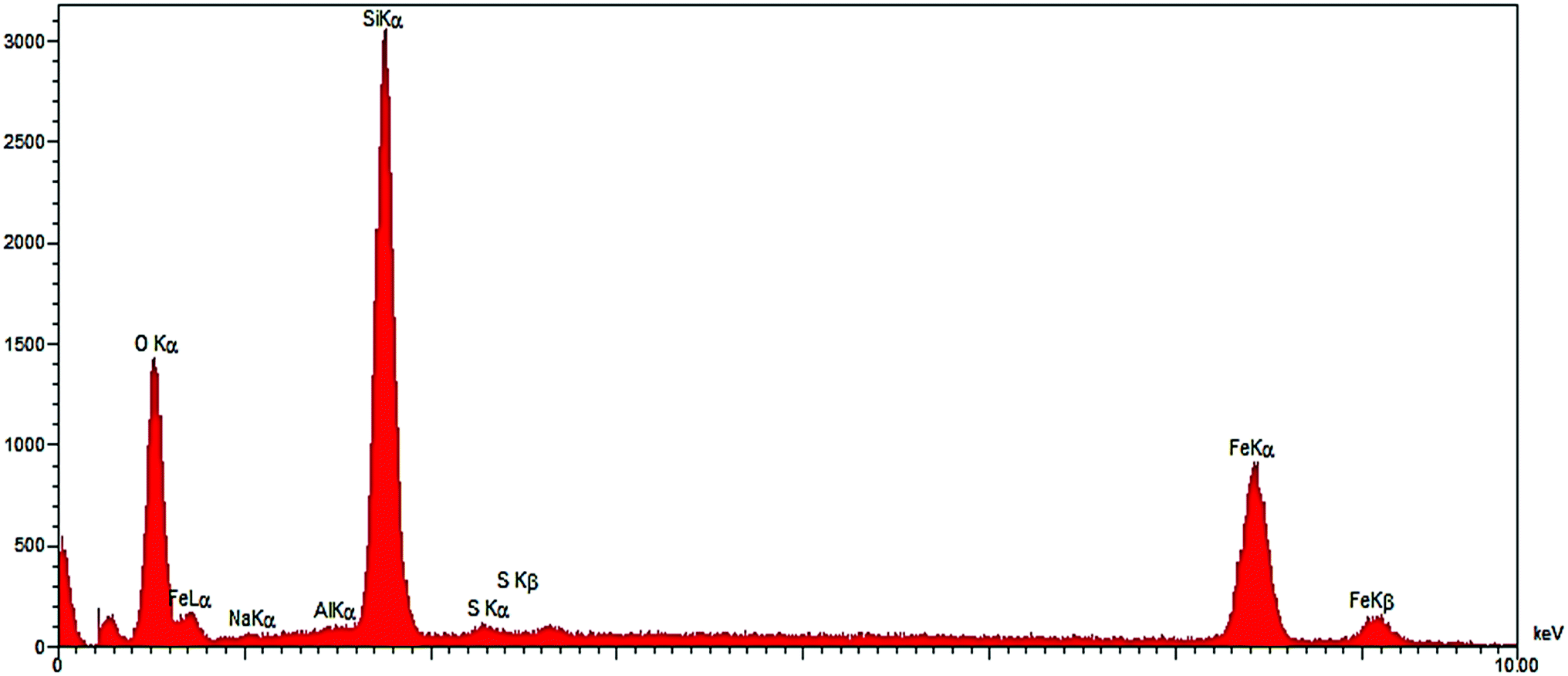 | ||
| Fig. 3 The EDX spectrum of Fe3O4/SO3H@zeolite-Y. | ||
The XRD determination was carried out to acquire crystalline structural information on Fe3O4/SO3H@zeolite-Y. The XRD patterns of magnetic zeolite supported SO3H is shown in Fig. 4. The main peaks at 2θ values of 30.56°, 35.96°, 43.66°, 53.94°, 57.59°, 63.20° and 74.97° are belong to crystal indexes of (220), (331), (400), (422), (511), (440) and (533), respectively. The obtained results are consistent with standard XRD data of Fe3O4 (JCPDS no. 01-1111), indicating that Fe3O4 has been successfully made. According to Scherrer's equation, the equivalent particle size could be calculated as about 10 nm.
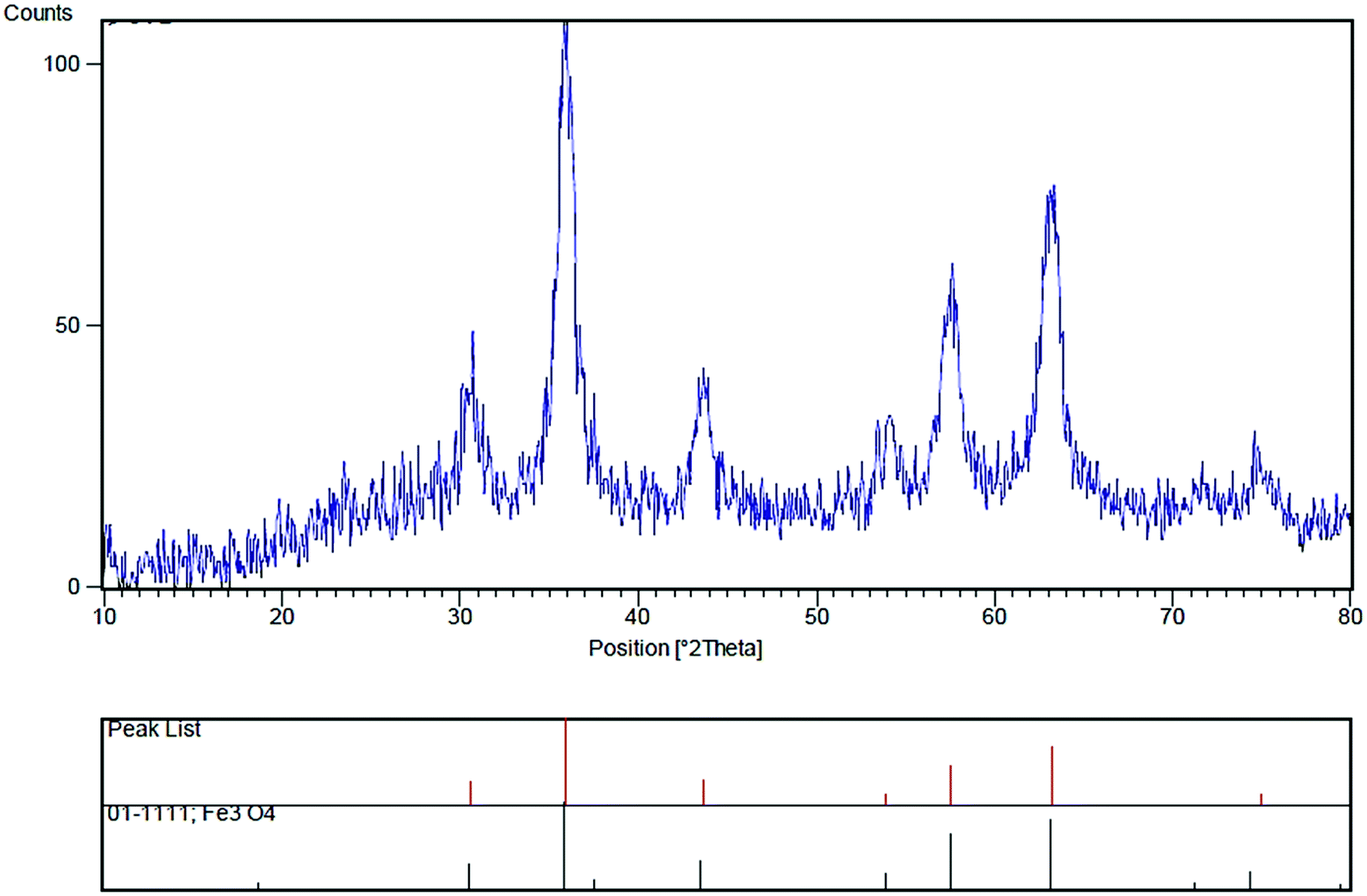 | ||
| Fig. 4 XRD pattern of Fe3O4/SO3H@zeolite-Y. | ||
A comparison is made between, FT-IR spectra of zeolite-NaY, SO3H@zeolite-Y and Fe3O4 zeolitic structures. In the spectrum of zeolite-NaY, in Fig. 5a, the broad absorption band in 3459 cm−1 region is related to the O–H stretching of hydrogen bonded internal silanol groups and hydroxyl stretching of water, while the peak at 1636 cm−1 corresponds to the O–H group bending mode of water. Moreover, the absorption bands 1021 and 792 cm−1 are attributed to the symmetric and asymmetric stretching vibrations of the Si–O–Si groups, respectively. In FT-IR spectrum of SO3H@zeolite (Fig. 5b), the broad band at 3420 cm−1 is related to the stretching vibrations of O–H groups. There is an adsorption band at 1635 cm−1 for OH bending vibration belonging to physically adsorbed H2O. The absorption band around 1087 cm−1 is related to the Si–O stretching vibration and the asymmetric and symmetric stretching of S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. The bands at 945 and 798 cm−1 maybe assigned to the S–O bond. The band at 455 cm−1 is attributed to the bending vibrations of Si–O–Si or Al–O–Si groups for the modified zeolite-NaY. FT-IR spectrum of Fe3O4/SO3H@zeolite-Y in Fig. 5c, reveals the presence of all adsorption band corresponding to SO3H@zeolite. Further, the existence of peak at 576 cm−1 corresponds to the stretching vibrations of Fe–O groups indicates that magnetic Fe3O4 nanoparticles is supported on sulfonated zeolite.
O bond. The bands at 945 and 798 cm−1 maybe assigned to the S–O bond. The band at 455 cm−1 is attributed to the bending vibrations of Si–O–Si or Al–O–Si groups for the modified zeolite-NaY. FT-IR spectrum of Fe3O4/SO3H@zeolite-Y in Fig. 5c, reveals the presence of all adsorption band corresponding to SO3H@zeolite. Further, the existence of peak at 576 cm−1 corresponds to the stretching vibrations of Fe–O groups indicates that magnetic Fe3O4 nanoparticles is supported on sulfonated zeolite.
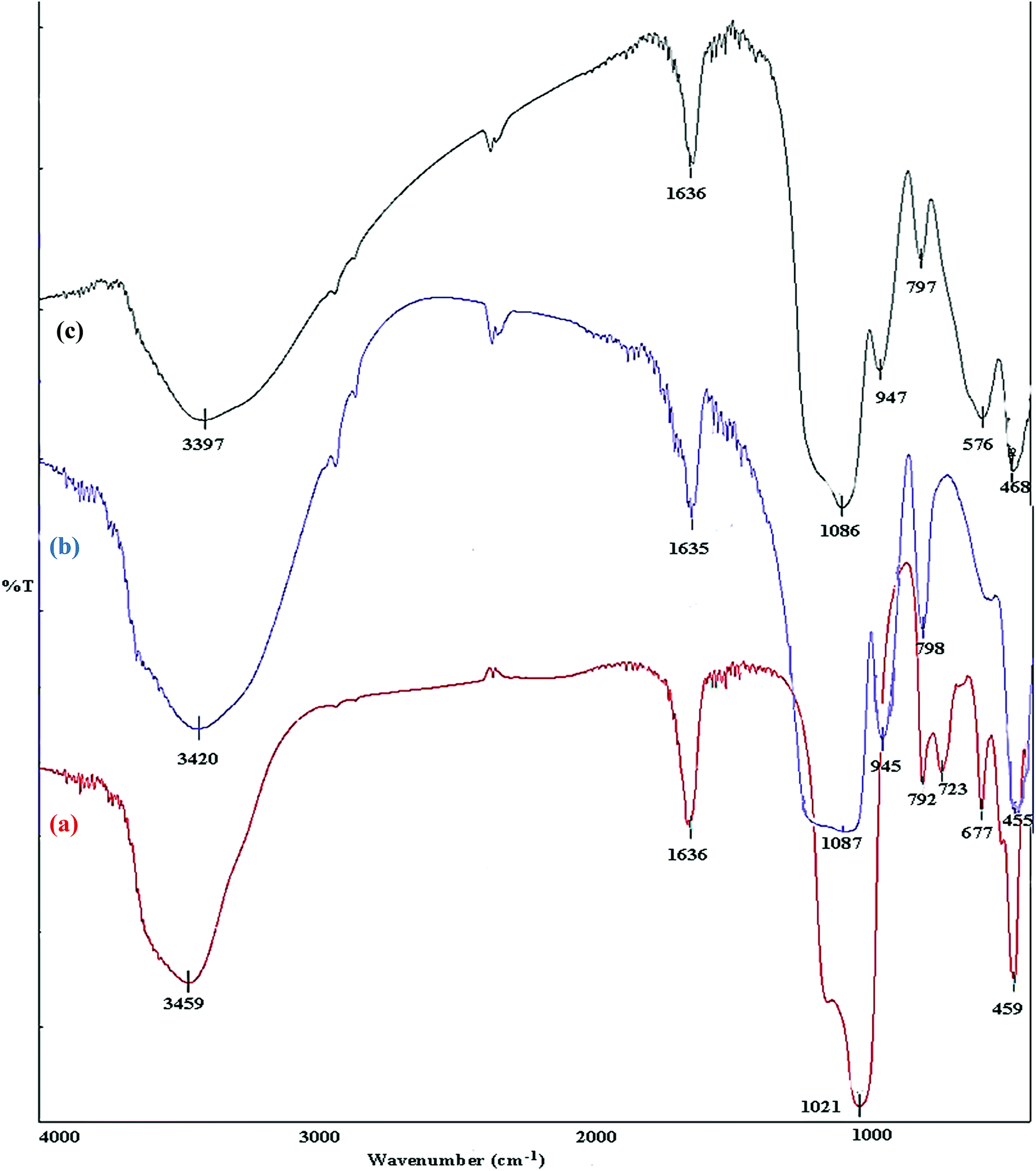 | ||
| Fig. 5 FTIR spectra of (a) zeolite-NaY, (b) SO3H@zeolite-Y and (c) Fe3O4/SO3H@zeolite-Y. | ||
Nitrogen adsorption–desorption isotherm analysis and BJH pore size distributions were carried out to evaluate the surface and structure properties of Fe3O4/SO3H@zeolite-Y. As shown in Fig. 6, the N2 adsorption–desorption isotherm of Fe3O4/SO3H@zeolite-Y displays an H1-type hysteresis loop and BJH pore distribution curve of type IV which is in agreement with typical mesostructure. Also, by looking at its hysteresis type, it can be seen that Fe3O4/SO3H@zeolite-Y has slits (layer and layer with high pores) structure and the initial nanostructure after the functionalize is still retained,35 The structure data of all these materials are summarized in Table 1. The BET surface area (205.68 m2 g−1) and average pore diameter (6.7767 nm) were recorded for Fe3O4/SO3H@zeolite-Y. The BET surface area is decreased relative to SO3H@zeolite-Y (284.84 nm). These results confirm successful supporting of Fe3O4 in the pores of SO3H functionalized zeolite-NaY.36 The Fe3O4/SO3H@zeolite-Y has a higher pore volume (6.77 nm) in comparison with sulfonated zeolite-Y (2.35 nm), which might be due to the presence of magnetic nanoparticles through the zeolite cavities.
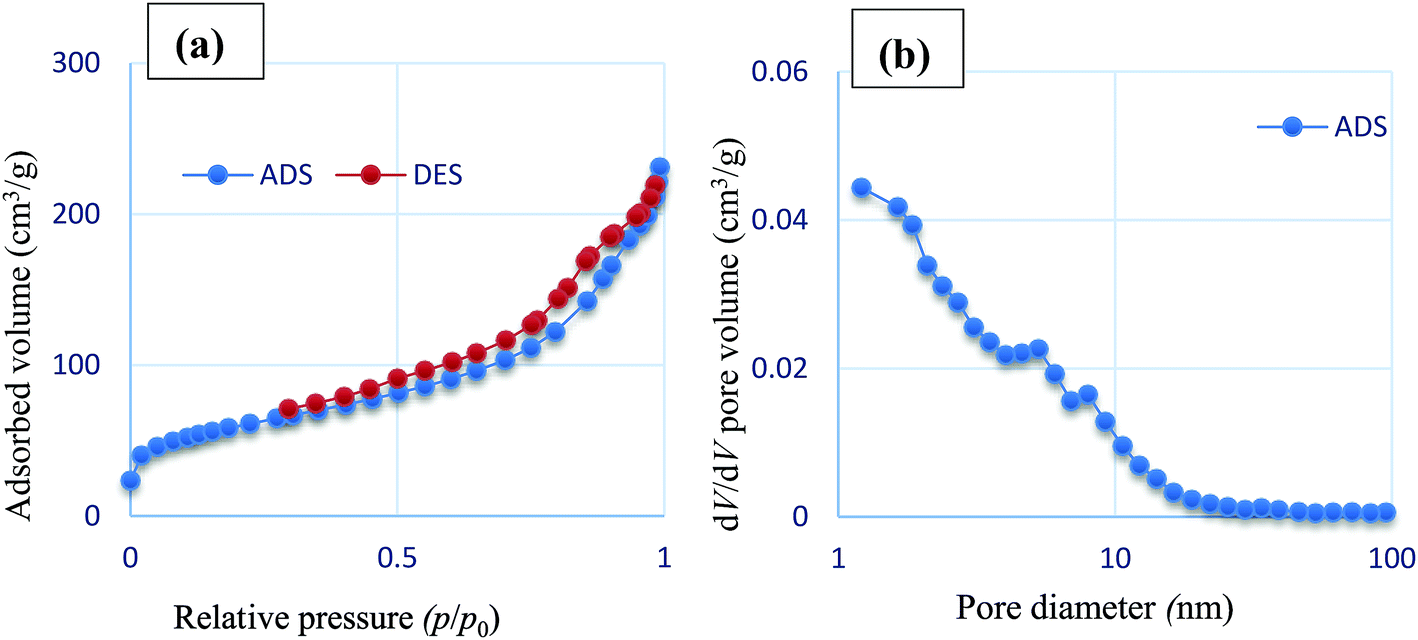 | ||
| Fig. 6 (a) Nitrogen adsorption–desorption isotherm and (b) BJH pore size distributions of Fe3O4/SO3H@zeolite-Y. | ||
| Samples | SBETa (m2 g−1) | VBJHb (cm3 g−1) | DBJHc (nm) | DAapd (nm) |
|---|---|---|---|---|
| a Specific surface area.b Pore volume.c Pore size (calculated from the adsorption branch).d Adsorption average pore diameter (4V/A) by BET. | ||||
| Zeolite-NaY | 619.66 | 0.0667 | 4.84 | 2.2358 |
| SO3H@zeolite-Y | 284.84 | 0.0532 | 5.15 | 2.3517 |
| Fe3O4/SO3H@zeolite-Y | 205.56 | 0.0348 | 2.42 | 6.7767 |
The magnetization curve of magnetic SO3H@zeolite nanocomposites is shown in Fig. 7. The saturation magnetization is found to be 21.30 emu g−1. With this saturation magnetization values, it could be rapidly separated from its liquid dispersions under a magnetic field. These results indicate that Fe3O4/SO3H@zeolite-Y exhibits enough magnetic response to meet the need of magnetic separation.
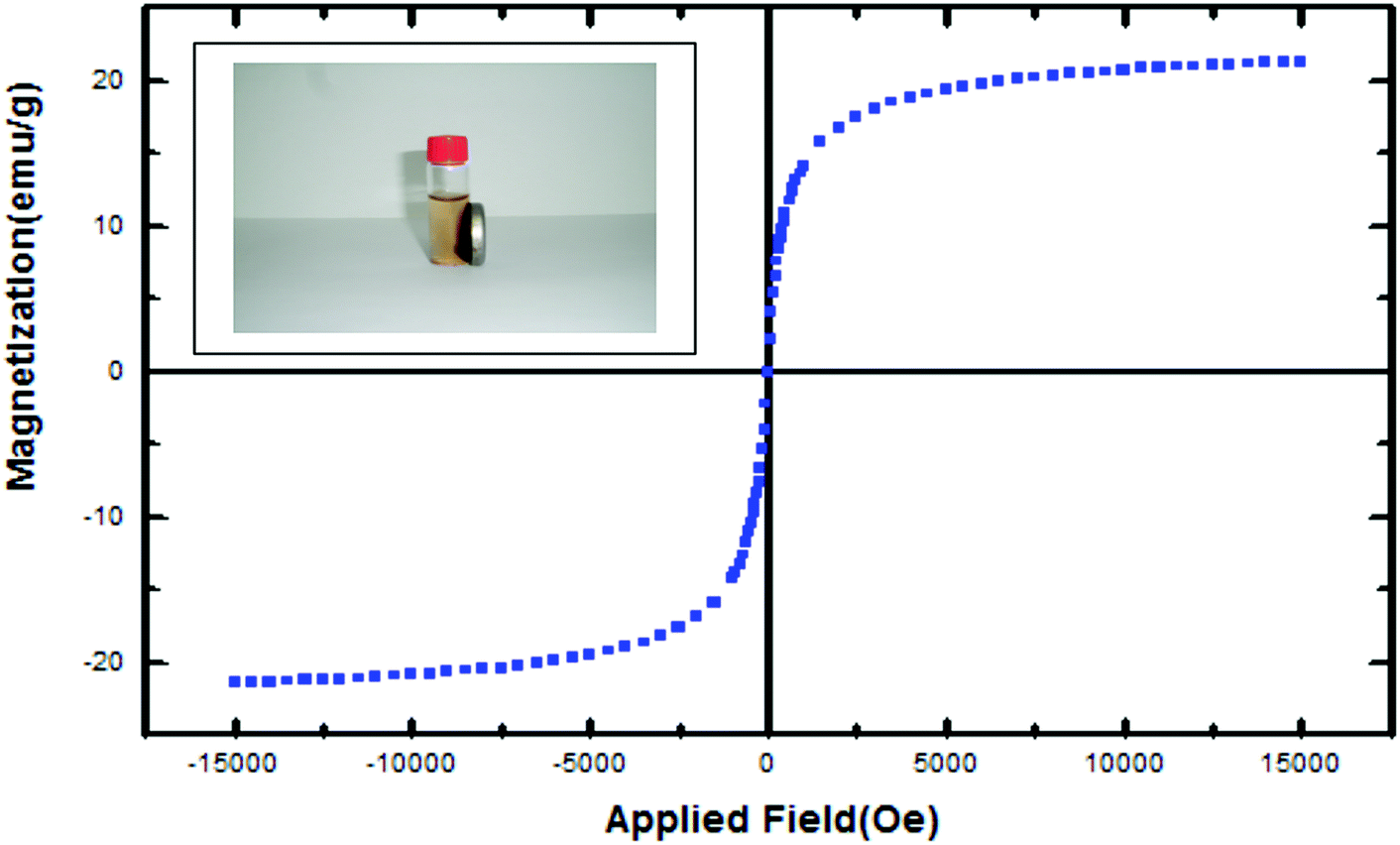 | ||
| Fig. 7 Field-dependent magnetization curve measured at room temperature for Fe3O4/SO3H@zeolite-Y. | ||
2.2. The catalytic activity of Fe3O4/SO3H@zeolite-Y in the synthesis of imidazole and perimidine derivatives
At first, the catalytic ability of Fe3O4/SO3H@zeolite-Y has been investigated for the synthesis of various substituted imidazoles. To optimize the reaction conditions for the synthesis of 2,4,5-triarylsubstituted imidazoles, the reaction of the benzaldehyde (1 mmol), benzil (1 mmol) and ammonium acetate (2 mmol) was considered as a model reaction. To find the optimized nanocatalyst ratio, different amounts of the magnetic zeolite were evaluated in EtOH solvent. The obtained results were listed in Table 2. The best quantity of the Fe3O4/SO3H@zeolite-Y for the preparation of tri-substituted imidazoles was 0.02 g (Table 2, entry 3). By increasing the amount of the nanocatalyst the reaction yield decreased (Table 2, entries 4 and 5). In the next step, the catalytic reaction was carried out in the various solvents like EtOH, MeOH, CH3CN, H2O, EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1), DMSO, and CH2Cl2 under reflux conditions (Table 2, entries 3 and 6–11). The best yield was obtained in EtOH solvent. The same reaction in the presence of 0.02 g of zeolitic catalyst was carried out at different temperatures to assess the effect of temperature on the reaction yield. The yield increased as the reaction temperature was raised. At 80 °C, the desired product was obtained in high yield within 45 min (Table 1, entry 3). A further increase in temperature and time did not improve the product yield (Table 2, entry 12). Although SO3H@zeolite-Y showed a good catalytic effect for this reaction (Table 2, entry 16), but centrifugation had required in order to separate the nanocatalyst. Modern catalytic methods offer a great potential for future developments in the synthesis of heterogeneous catalysts based on magnetic nanomaterials. Magnetic separation is a simpler, faster, and cleaner way to recover the catalyst. The presence of magnetite nanoparticles also improved catalytic effect and decreased slightly at reaction time. Therefore, the Fe3O4/SO3H@zeolite-Y was chosen was chosen to improve the catalytic path. Also, this condensation was performed under catalyst-free conditions. The yield of the product was 50% after 4 h (Table 2, entry 17). Therefore, the presence of the nanocatalyst was necessary for a shorter time and higher efficiency.
:H2O (1:1), DMSO, and CH2Cl2 under reflux conditions (Table 2, entries 3 and 6–11). The best yield was obtained in EtOH solvent. The same reaction in the presence of 0.02 g of zeolitic catalyst was carried out at different temperatures to assess the effect of temperature on the reaction yield. The yield increased as the reaction temperature was raised. At 80 °C, the desired product was obtained in high yield within 45 min (Table 1, entry 3). A further increase in temperature and time did not improve the product yield (Table 2, entry 12). Although SO3H@zeolite-Y showed a good catalytic effect for this reaction (Table 2, entry 16), but centrifugation had required in order to separate the nanocatalyst. Modern catalytic methods offer a great potential for future developments in the synthesis of heterogeneous catalysts based on magnetic nanomaterials. Magnetic separation is a simpler, faster, and cleaner way to recover the catalyst. The presence of magnetite nanoparticles also improved catalytic effect and decreased slightly at reaction time. Therefore, the Fe3O4/SO3H@zeolite-Y was chosen was chosen to improve the catalytic path. Also, this condensation was performed under catalyst-free conditions. The yield of the product was 50% after 4 h (Table 2, entry 17). Therefore, the presence of the nanocatalyst was necessary for a shorter time and higher efficiency.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (g) | Solvent | Temperature (°C) | Time (min) | Yieldb (%) |
| a Reaction conditions: benzil (1 mmol), benzaldehyde (1 mmol), ammonium acetate (2 mmol) and catalyst in solvent.b Isolated yield. | |||||
| 1 | 0.005 | EtOH | 80 | 90 | 70 |
| 2 | 0.010 | EtOH | 80 | 60 | 82 |
| 3 | 0.020 | EtOH | 80 | 45 | 98 |
| 4 | 0.030 | EtOH | 80 | 45 | 95 |
| 5 | 0.040 | EtOH | 80 | 45 | 93 |
| 6 | 0.020 | MeOH | 65 | 60 | 90 |
| 7 | 0.020 | DMSO | 120 | 60 | 85 |
| 8 | 0.020 | CH3CN | 80 | 60 | 55 |
| 9 | 0.020 | H2O | 100 | 90 | 20 |
| 10 | 0.020 | EtOH:H2O (1:1) |
100 | 90 | 35 |
| 11 | 0.020 | CH2Cl2 | 40 | 90 | 58 |
| 12 | 0.020 | EtOH | 50 | 90 | 60 |
| 13 | 0.020 | EtOH | 60 | 60 | 75 |
| 14 | 0.020 | EtOH | 70 | 45 | 92 |
| 15 | Zeolite-NaY (0.02) | EtOH | 80 | 120 | 15 |
| 16 | SO3H@zeolite-Y (0.02) | EtOH | 80 | 55 | 90 |
| 17 | — | EtOH | 80 | 240 | 50 |
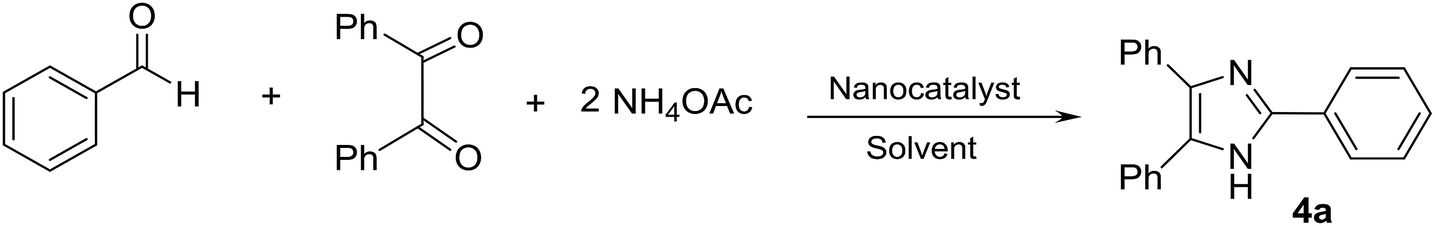
The scope and generality of the synthesis of 2,4,5-triaryl substituted imidazoles were studied by using various aromatic aldehydes bearing both electron-withdrawing and electron-donating groups. As the results clearly show (Table 3), this organic transformation in the presence of the zeolitic nanocomposite provided the corresponding products in high yields and appropriate reaction times (Table 3, 4a–n).
|
|||||
|---|---|---|---|---|---|
| Entry | R | Product | Time (min) | Yieldb (%) | mprep/mplit. (°C) |
| a Reaction conditions: benzil (1 mmol), aldehyde (1 mmol), ammonium acetate (2 mmol) and Fe3O4/SO3H@zeolite-Y (0.020 g) in EtOH at 80 °C.b Isolated yield. | |||||
| 1 | H | 4a | 45 | 98 | 270–272 (271–273)11 |
| 2 | 2-NO2 | 4b | 65 | 97 | 226–228 (226–227)37 |
| 3 | 3-NO2 | 4c | 20 | 85 | 268–270 (267–268)11 |
| 4 | 3-OH, 4-OMe | 4d | 45 | 93 | 215–217 (215–217)11 |
| 5 | 2-OMe | 4e | 65 | 97 | 200–202 (209–211)38 |
| 6 | 3,4-OMe | 4f | 45 | 98 | 214–216 (214–216)11 |
| 7 | 3-OH | 4g | 35 | 97 | 259–261 (259–260)11 |
| 8 | 4-OMe | 4h | 60 | 95 | 226–228 (230–231)11 |
| 9 | 2-Cl | 4i | 65 | 95 | 168–170 (196–199)37 |
| 10 | 2,3-Cl | 4j | 50 | 98 | 254–256 (194–197)37 |
| 11 | 2,4-Cl | 4k | 50 | 98 | 168–170 (175–178)37 |
| 12 | 4-Cl | 4l | 35 | 97 | 260–262 (260–262)11 |
| 13 | 3-Br | 4m | 30 | 98 | 301–303 (302–303)11 |
| 14 | 4-Me | 4n | 45 | 96 | 231–233 (230–232)11 |
A plausible mechanism for the formation of 2,4,5-triaryl-1H-imidazoles in the presence of Fe3O4/SO3H@zeolite-Y catalyst is shown in Scheme 3. Fe3O4/SO3H@zeolite-Y as heterogeneous multi-functional nanocatalyst facilitates this cyclocondensation due to the existence of both Lewis acid (Fe3O4) and Brønsted acidic (SO3H) sites. On the basis of the literature and substrates chemistry, the SO3H Brønsted acid supported on Fe3O4 Lewis acid polarizes the carbonyl group of aldehyde. Then, hydroxylamine intermediate (1a) is formed by condensation reaction of an aromatic aldehyde (1) and NH3 (2), which is then dehydrated to imine (2a). The addition of the second mole of NH3 produces diamine intermediate (A). Condensation of diamine with benzil (3) followed by cyclization, dehydration, and then rearrangement through the imino intermediate (B) yielded the substituted imidazole product (4).11
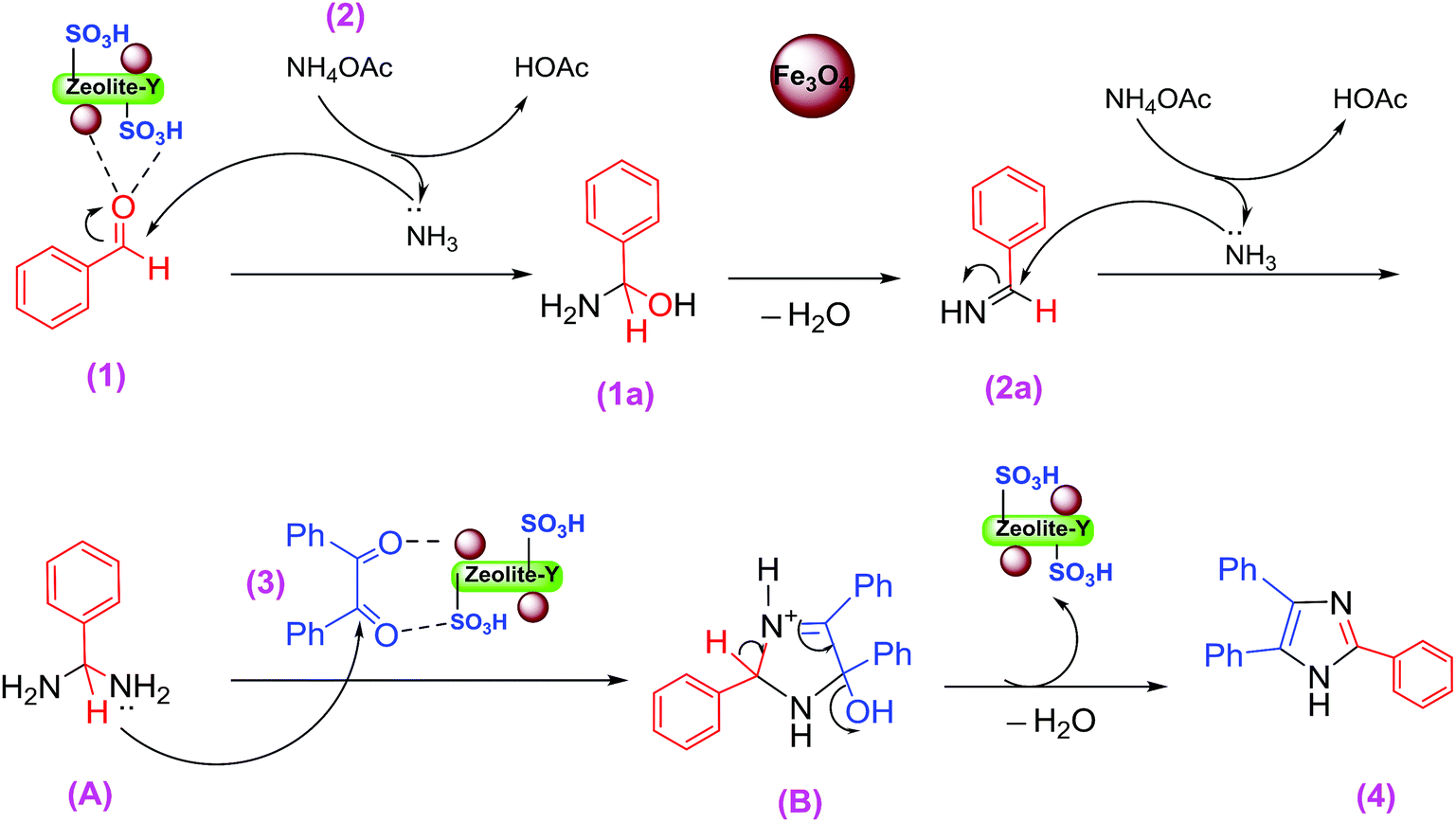 | ||
| Scheme 3 The suggested mechanism of tri-substituted imidazoles synthesis. | ||
In another synthesis procedure, the catalytic performance of Fe3O4/SO3H@zeolite-Y was investigated for the synthesis of dihydroperimidine derivatives (Table 4, entries 1–12). In order to optimize the reaction conditions, some parameters such as solvent, catalyst amount, and temperature, were probed for the condensation of benzaldehyde with 1,8-diaminonaphthalene as a model reaction in the presence of Fe3O4/SO3H@zeolite-Y. The optimum reaction conditions were found to be use of 0.008 g of Fe3O4/SO3H@zeolite-Y in the EtOH solvent at 4 min with 98% yield (Table 4, entry 2). It can be concluded that the nano-size and large surface area of the support makes the heterogenized SO3H catalyst readily available for substrates. Further, the catalytic performance of Fe3O4 (as a Lewis acid) supported on the zeolite for the reaction is attributed to the large surface area of the nanocatalyst and support, which facilitate the appropriate interaction of the reactant and product molecules. Moreover, this technique makes catalyst separation from aqueous solutions more effective and simple (as a significant advantage in comparison with the SO3H@zeolite-Y catalyst). The catalyst free reaction has no good results for the synthesis of 2-substituted 2,3-dihydro-1H-perimidines (Table 4, entry 12).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (g) | Solvent | Temperature (°C) | Time (min) | Yieldb (%) |
| a Reaction conditions: benaldehyde (1 mmol), 1,8-diaminonaphthalene (1 mmol), catalyst, in solvent at 25 °C.b Isolated yield. | |||||
| 1 | 0.0047 | EtOH | rt | 4 | 81 |
| 2 | 0.008 | EtOH | rt | 4 | 98 |
| 3 | 0.016 | EtOH | rt | 4 | 70 |
| 4 | 0.008 | MeOH | rt | 4 | 80 |
| 5 | 0.008 | DMSO | rt | 4 | 75 |
| 6 | 0.008 | CH3CN | rt | 10 | 63 |
| 7 | 0.008 | CH2Cl2 | rt | 15 | 51 |
| 8 | 0.008 | H2O | rt | 18 | 46 |
| 9 | 0.008 | EtOH:H2O (1:1) |
rt | 10 | 81 |
| 10 | Zeolite-Y (0.008) | EtOH | rt | 120 | 20 |
| 11 | SO3H@zeolite-Y (0.008) | EtOH | rt | 8 | 89 |
| 12 | — | EtOH | rt | 120 | 10 |
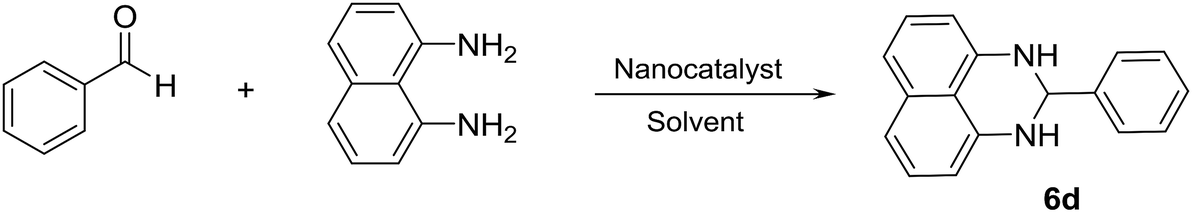
To extend the scope of this study, the above optimized conditions, were employed for a cyclocondensation reaction between a variety of aromatic aldehydes and 1,8-diaminonaphthalene over the Fe3O4/SO3H@zeolite-Y catalyst (Table 5, 6a–m). Based on these results, both electron-donating and electron-withdrawing groups proceed smoothly with very good efficiency to form the desired products in high yields within the very short time.
|
|||||
|---|---|---|---|---|---|
| Entry | R | Product | Time (min) | Yieldb (%) | mprep/mplit. (°C) |
| a Reaction conditions: aldehyde (1 mmol), 1,8-diaminonaphthalene (1 mmol), Fe3O4/SO3H@zeolite-Y (0.008 g), in EtOH at 25 °C.b Isolated yield.c 5-Nitrofuran-2-yl. | |||||
| 15 | H | 6a | 4 | 98 | 104–106 (104–106)20 |
| 16 | 2-NO2 | 6b | 4 | 98 | 180–192 (192–194)20 |
| 17 | 3-NO2 | 6c | 4 | 90 | 200–202(183–185)39 |
| 18 | 4-NO2 | 6d | 3 | 98 | 224–226 (201–202)20 |
| 19 | 4-OMe | 6e | 4 | 90 | 160–162 (161–163)20 |
| 20 | 3,4-OMe | 6f | 5 | 89 | 214–216 (205–207)20 |
| 21 | 2-OH | 6g | 5 | 90 | 180–182 (192–193)40 |
| 22 | 2-OH, 5-Br | 6h | 4 | 95 | 148–150 (165–166)20 |
| 23 | 2-OH, 4-OMe | 6i | 6 | 89 | 204–206 (168–169)20 |
| 24 | 3-Cl | 6j | 4 | 95 | 144–146 (145–146)20 |
| 25 | 4-Cl | 6k | 4 | 98 | 171–173 (173–174)20 |
| 26 | 2,3-Cl | 6l | 4 | 95 | 130–132 (131–133)21b |
| 27 | 5-NO2c | 6m | 6 | 88 | 340–342 (350)20 |
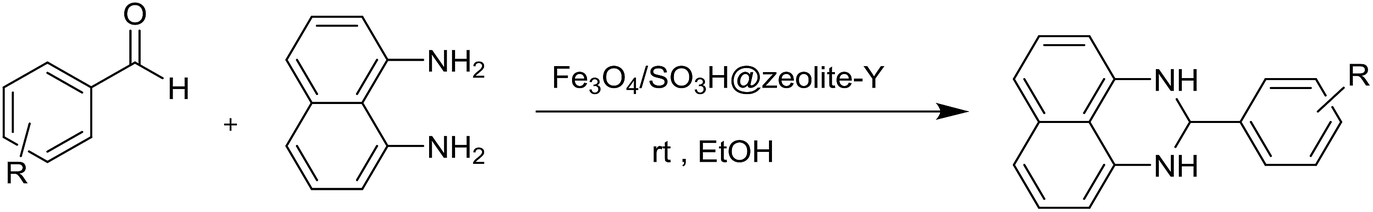
A possible mechanism (Scheme 4) is proposed for the synthesis of 2-aryl perimidines which is consistent with the literature.21 The nucleophilic attack of diamino aromatic ring to Fe3O4/SO3H@zeolite-Y-activated aldehyde generated the imine intermediate with the removal of H2O molecule. Subsequently, nucleophilic attack of the second amino group to nanocatalyst-activated imine intermediate is followed by cyclization process to give the desired product.
 | ||
| Scheme 4 The possible reaction mechanism for the synthesis of 2-aryl perimidines in the presence of Fe3O4/SO3H@zeolite-Y. | ||
The reusability and recyclability of Fe3O4/SO3H@zeolite-Y catalyst was investigated in the model reactions for the preparation of tri-substituted imidazoles and 2-aryl perimidines (Fig. 8). In both of these reactions, the nanocatalyst was easily recycled at least five times without significant loss of activity. In this regard, after completion of the reaction, the catalyst was separated by an external magnet, washed with ethyl acetate and dried at 50 °C to be ready for the subsequent runs. The FTIR and XRD data of the recycled catalyst shows no change in its structure (Fig. 9 and 10).
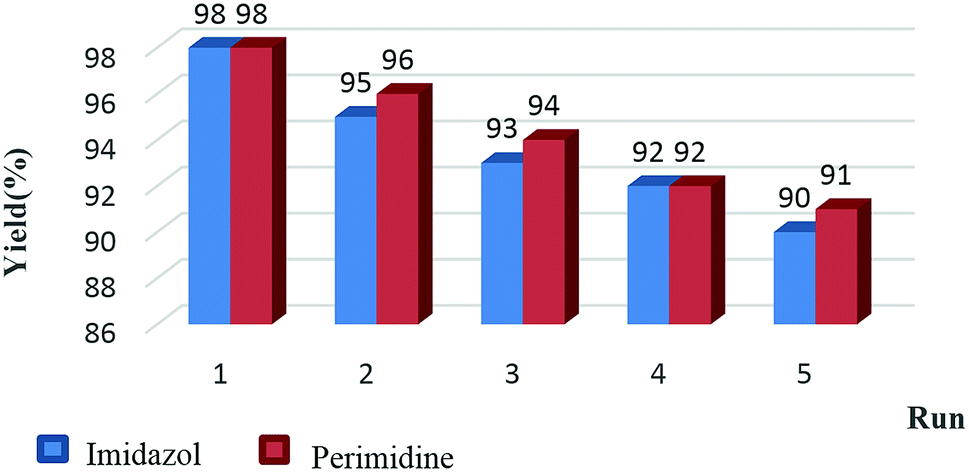 | ||
| Fig. 8 Examination of Fe3O4/SO3H@zeolite-Y reusability in the synthesis of imidazoles and perimidines. | ||
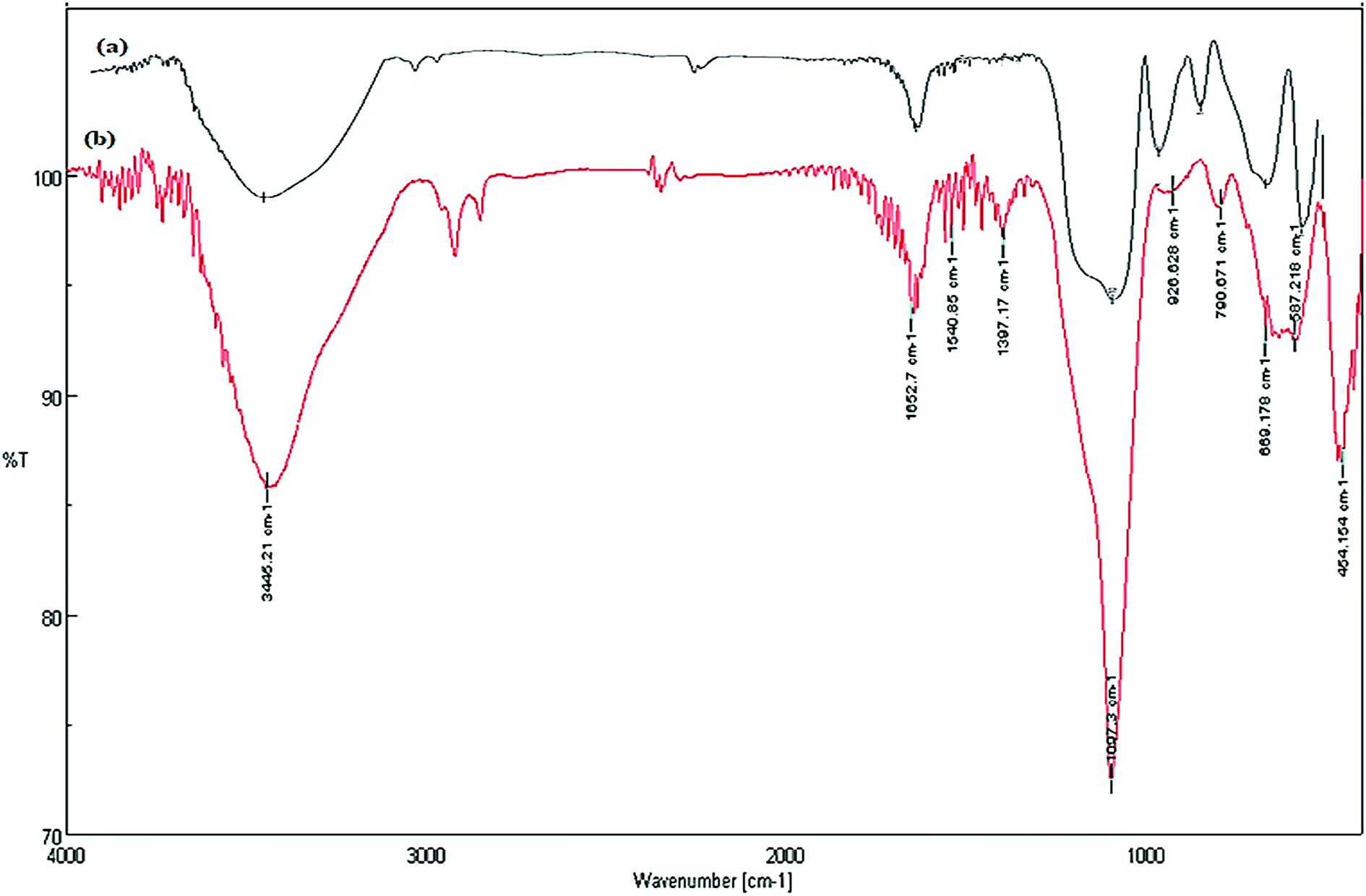 | ||
| Fig. 9 FTIR spectra of (a) fresh and (b) recovered Fe3O4/SO3H@zeolite-Y. | ||
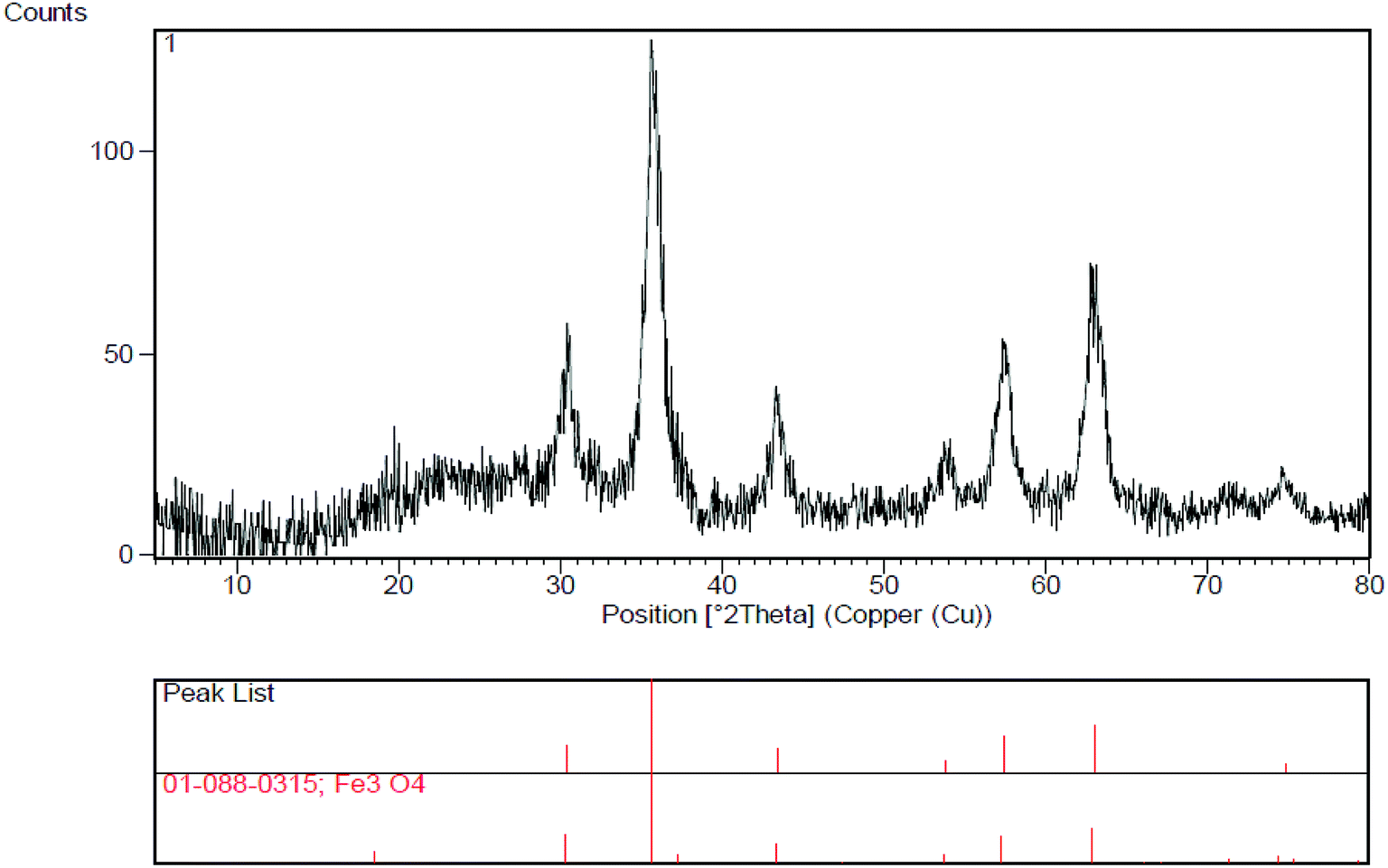 | ||
| Fig. 10 XRD pattern of recovered Fe3O4/SO3H@zeolite-Y. | ||
Also, to show the advantages of using Fe3O4/SO3H@zeolite-Y nanocatalyst in the synthesis of 2,4,5-triaryl-1H-imidazoles (Table 4, entries 1–8) and 2,3-dihydro-1H-perimidines (Table 4, entries 9–16), this catalytic method was compared with literature resulted in reports of using various catalysts for the model reactions. As shown in Table 6, in the presence of this new magnetic nanocomposite [with Lewis acid (Fe3O4) and Brønsted–Lowry acid (SO3H)], the results were better than other catalysts.
| Entry | Catalyst/conditions | Time (h) | Yielda (%) |
|---|---|---|---|
| a Isolated yield. | |||
| 1 | Fe3O4g-C3N4 (0.02 g), EtOH, 78 °C | 2 | 95 (ref. 41) |
| 2 | Fe3O4@chitosan (0.05 g), EtOH, reflux | 2 | 95 (ref. 42) |
| 3 | Benzethonium chloride (10 mole%), EtOH:H2O, 70 °C |
0.75 | 95 (ref. 43) |
| 4 | Sodium dodecyl-sulfate (0.02 g), H2O, 80 °C | 1 | 90 (ref. 44) |
| 5 | Fe3O4@SiO2·HM·SO3H (0.04 g), solvent-free, 110 °C | 0.25 | 93 (ref. 37) |
| 6 | Fe3O4@SiO2·HM·SO3H (0.060 g), solvent-free, MWI | 0.13 | 77 (ref. 45) |
| 7 | Sulfamic acid-Fe3O4 NPs (0.1 g), EtOH, reflux | 2 | 90 (ref. 46) |
| 8 | Fe3O4/SO3H@zeolite-Y (0.02 g), EtOH, 80 °C | 0.75 | 98 (this work) |
| 9 | CuY-zeolite (0.002 g), EtOH, rt | 0.42 | 81 (ref. 20) |
| 10 | NaY zeolite (0.2 g), EtOH, rt | 45–50 | 70 (ref. 47) |
| 11 | FePO4 (10 mol%), EtOH, rt | 0.22 | 80 (ref. 48) |
| 12 | Ytterbium(III) trifluoromethanesulfonate (0.1 equiv.), MeCN, rt | 24 | 88 (ref. 49) |
| 13 | Fe3O4/SiO2/(CH2)3N+Me3Br3− (0.007), solvent-free, 80 °C | 12 | 93 (ref. 50) |
| 14 | Nano-γ-Al2O3/SbCl5 (0.1 g), solvent-free, rt | 0.25 | 95 (ref. 21) |
| 15 | Cu(NO3)2 (5 mole%), 2H2O, EtOH, rt | 0.17 | 83 (ref. 51) |
| 16 | Fe3O4/SO3H@zeolite-Y (0.008 g), EtOH, rt | 0.07 | 98 (this work) |
3. Conclusions
In conclusion, magnetic SO3H@zeolite-Y nanocomposite supported nano Fe3O4 was successfully synthesized and used for the synthesis of 2,4,5-trisubstituted imidazoles and 2-aryl perimidines under mild conditions. The most interesting features of the present method are simple preparation, the use of non-toxic nature of the nanocatalyst, easy separation of the catalyst after reaction, and substantial improvements in the reaction time and yield. The nanocatalyst was readily recycled by an external permanent magnet and could be reused five times without any significant loss of its performance. Further applications of multi-functional nanocatalysts based on the zeolie-NaY to other green reactions are currently under investigation.4. Experimental
4.1. Chemicals and apparatus
All chemicals were purchased from the Merck and Fluka Chemical Companies. The products were identified by comparing the physical data with those of known samples or by their spectral data. FT-IR spectra were obtained with KBr disc on a galaxy series FT-IR 5000 spectrometer. 1H NMR and 13C NMR spectra were recorded with a Bruker DRX-400 spectrometer at 400 and 100 MHz respectively. The surface morphology of zeolite and modified structures were analyzed by field emission scanning electron microscopy (FESEM) (EVO LS 10, Zeiss, Carl Zeiss, Germany). The magnetic measurement of samples was carried out in a vibrating sample magnetometer (VSM) (4 inch, NDKF, Kashan, Iran) at room temperature. Diffuse reflectance spectra were acquired on an X-ray photoelectron spectrometer (Escalab 250Xi). Nitrogen adsorption and desorption isotherms (BET analysis) were measured at 196 °C by a Japan Belsorb II system after the samples were vacuum dried at 150 °C overnight.4.2. Synthesis of Fe3O4/SO3H@zeolite-Y
For preparation of SO3H@zeolite-Y, a 500 mL flask was equipped with a constant pressure dropping funnel containing 3 mL of ClSO3H. Then, 1.5 g of zeolite-NaY was charged into the flask and ClSO3H was added dropwise manner to zeolite-NaY over a period of 30 min. at 0 °C. After the addition was complete, the mixture was stirred for 45 min. Afterwards, the mixture was washed with CH2Cl2 and H2O under sonication and dried at 50 °C. In continue, nano magnetic zeolosulforic acid (NMSZ) was prepared by a chemical co-precipitation process as described by Mesdaghinia et al.34 FeCl2·4H2O (2.3 mmol, 0.45 g) and FeCl3·6H2O (4.6 mmol, 1.23 g) precursors (1:2, molar ratio) were dissolved in 25 mL of water and 1 g of SO3H@zeolite-Y were dispersed in this aqueous solution under N2 atmosphere bubbling for 1 h. Then, 20 mL of ammonium hydroxide (25%) was added to the mixture. The mixture was stirred at 90 °C for 30 minutes. The black precipitate was extracted by an external magnet and washed with HCl solution (0.1 M), double-distilled water and dried at 70 °C in vacuum oven to obtain the Fe3O4/SO3H@zeolite-Y nanocomposite.
4.3. General procedure for the synthesis of 2,4,5-triaryl substituted imidazoles
A mixture of benzil (1 mmol, 0.210 g), aldehyde (1 mmol), ammonium acetate (2 mmol, 0.154 g) and Fe3O4/SO3H@zeolite-Y (0.020 g) in EtOH was stirred at 80 °C. After completion of the reaction (TLC monitoring using n-hexane and ethyl acetate, 9:1), the nanocatalyst was separated by magnetic decantation and the solvent was evaporated. The crude product was either recrystallized from aqueous-acetone and air dried. The products were characterized by comparing the results of physical and spectroscopic data with those of the authentic samples. Characterization data for some of the imidazole compounds are given below.
C), 1524 (CN), 14.89 (NO), 1348 (N–O); 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 7.14–7.16 (m, 5H, Ar–H), 7.38–7.44 (m, 5H, Ar–H), 7.47 (d, 1H, 3J = 7.6 Hz, Ar–H), 8.00 (d, 1H, 3J = 7.6 Hz, Ar–H), 8.46 (d, 1H, 3J = 7.6 Hz, Ar–H), 8.48 (d, 1H, 3J = 7.6 Hz, Ar–H), 8.91 (s, 1H, Ar–H), 9.8 (s, 1H, N–H).C), 1499 (CN); 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.77 (s, 3H, CH3), 6.75 (d, 1H, 3J = 8.4 Hz, Ar–H), 7.14–716 (m, 6H, Ar–H), 7.35–7.39 (m, 5H, Ar–H), 7.49 (s, 1H, Ar–H), 7.53 (s, 1H, OH), 7.8 (s, 1H, N–H).C), 1479 (CN); 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 6.98 (t, 2H, 3J = 8 Hz, Ar–H), 7.06–7.10 (m, 4H, Ar–H), 7.11–7.16 (m, 5H, Ar–H), 7.41–7.43 (m, 4H, Ar–H), 7.46 (s, 1H, OH), 8.8 (s, 1H, N–H).C), 1455 (CN); 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 7.34–7.35 (m, 7H, Ar–H), 7.51–7.56 (m, 5H, Ar–H), 7.89 (s, 1H, Ar–H), 8.13 (s, 1H, Ar–H), 9.9 (s, 1H, N–H).C), 1497 (CN); 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.30 (s, 3H, CH3), 7.14–7.20 (d, 3H, 3J = 7.2 Hz, Ar–H), 7.21–7.30 (m, 5H, Ar–H), 7.49–7.50 (m, 4H, Ar–H), 7.92 (d, 2H, 3J = 7.2 Hz, Ar–H), 10.0 (s, 1H, N–H).4.4. General procedure for the preparation of 2-substituted 2,3-dihydro-1H-perimidines
A mixture of aldehyde (1 mmol), 1,8-diaminonaphthalene (1 mmol, 0.158 g), Fe3O4/SO3H@zeolite-Y (0.008 g), in EtOH was stirred at 25 °C. After completion of the reaction, (TLC monitoring using hexane and ethyl acetate, 3:1), the nanocatalyst was separated by magnetic field. A mixture of ice and water was added to the filtrate and the precipitate product was obtained by filtration and washed thoroughly with water–ethanol mixture. The product was purified by recrystallization in ethanol–water and identified by physical and spectroscopic techniques. Characterization data for some of the compounds are given below.
C), 1165, 1125, 1031 (C–N), 826, 758 cm−1; 1H NMR (300 MHz, DMSO-d6): δH 8.03 (d, J = 8.11 Hz, 1H, H–Ar), 7.88 (d, J = 7.92 Hz, 1H, H–Ar), 7.75–7.64 (m, 2H, H–Ar), 7.18 (t, J = 8.10 Hz, 2H, H–Ar), 7.03 (d, J = 8.25 Hz, 2H, H–Ar), 6.82 (br, 2H, NH), 6.53 (d, J = 7.5 Hz, 2H, H–Ar), 5.82 (s, 1H, N–CH–N) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 149.3, 142.5, 136.9, 134.6, 134.0, 130.4, 130.0, 127.5, 124.4, 116.2, 112.5, 105.2, 61.4 ppm.C), 1257, 1107 (C–O), 1028 (C–N), 811, 759, 418 cm−1; 1H NMR (300 MHz, DMSO-d6): δH 9.70 (s, 1H, OH), 7.35 (d, J = 8.82 Hz, 1H, H–Ar), 7.22–7.11 (m, 2H, H–Ar), 6.96 (d, J = 8.05 Hz, 2H, H–Ar), 6.52–6.43 (m, 6H, H–Ar and NH), 5.57 (s, 1H, N–CH–N), 3.70 (s, 3H, OCH3) ppm; 13C NMR (75 MHz, DMSO-d6): δC 160.6, 156.9 (C–O), 143.9, 134.8, 129.8, 127.2, 127.2, 120.0, 115.7, 112.9, 104.9, 101.5, 61.1 (N–C–N), 55.4 (OCH3) ppm.C), 1182, 1161, 1043 (C–N), 835, 787, 757 (C–Cl), 603 cm−1; 1H NMR (500 MHz, DMSO-d6): δH 7.69 (d, J = 8.15 Hz, 2H, H–Ar), 7.45 (t, J = 7.90 Hz, 1H, H–Ar), 7.19 (t, J = 7.78 Hz, 2H, H–Ar), 7.04 (d, J = 8.15 Hz, 2H, H–Ar), 6.83 (s, 2H, NH), 6.52 (d, J = 7.35 Hz, 2H, H–Ar), 7.83 (s, 1H, N–CH–N) ppm; 13C NMR (125 MHz, DMSO-d6): δC 142.7, 141.8, 134.7, 132.1, 131.1, 130.7, 128.8, 128.6, 127.3, 116.1, 112.5, 105.0, 63.5 (N–CH–N) ppm.C), 1520, 1360 (–NO2), 1020 (C–N), 808, 601 cm−1; 1H NMR (300 MHz, DMSO-d6): δH 8.25 (d, J = 7.00 Hz, 2H, H–Ar), 7.69 (s, 2H, H–Ar), 7.52–7.27 (m, 6H, H–Ar and 2NH), 5.25 (s, 1H, N–CH–N) ppm; 13C NMR (75 MHz, DMSO-d6): δC 161.3, 150.2, 137.0, 135.5, 130.1, 128.4, 124.4, 119.2, 116.1, 114.6, 69.7 (N–C–N) ppm.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to the Payame Noor University for the partial support of this work.References
- L. Zhang, X. M. Peng, G. L. Damu, R. X. Geng and S. H. Zhou, Med. Res. Rev., 2014, 34, 340–437 CrossRef CAS PubMed.
- (a) M. Zendehdel, A. Mobinikhaledi, H. Alikhani and N. Jafari, J. Chin. Chem. Soc., 2010, 57, 683–689 CrossRef CAS; (b) A. Mobinikhaledi, M. A. Bodaghi Fard, F. Sasani and M. A. Amrollahi, Bulg. Chem. Commun., 2013, 45, 353–356 CAS.
- Y. L. Fan, X. H. Jin, Z. P. Huang, H. F. Yu, Z. G. Zeng, T. Gao and L. S. Feng, Eur. J. Med. Chem., 201, 15, 347–365 Search PubMed.
- (a) K. Undheim and C. Benneche, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C.W. Rees and E. F. Scriven, Pergamon, Oxford, 1996 Search PubMed; (b) A. F. Pozharskii and V. V. Dalnikovskaya, Russ. Chem. Rev., 1981, 50, 816–835 CrossRef; (c) X. Bu, L. W. Deady, G. J. Finlay, B. C. Baguley and W. A. Denny, J. Med. Chem., 2001, 44, 2004–2014 CrossRef CAS PubMed; (d) J. M. Herbert, P. D. Woodgate and W. A. Denny, J. Med. Chem., 1987, 30, 2081–2086 CrossRef CAS PubMed; (e) D. R. Luthin, A. K. Rabinovich, D. R. Bhumralkar, K. L. Youngblood, R. A. Bychowski, D. S. Dhanoa and J. M. May, Bioorg. Med. Chem. Lett., 1999, 9, 765–770 CrossRef CAS PubMed; (f) F. A. Bassyouni, S. M. Abu-Bakr, K. H. Hegab, W. El-Eraky, A. A. El Beih and M. E. Abdel Rehim, Res. Chem. Intermed., 2012, 38, 1527–1528 CrossRef CAS.
- (a) J. G. Lambardino and E. H. Wiseman, J. Med. Chem., 1974, 17, 1182–1188 CrossRef; (b) A. Puratchikody and M. Doble, Bioorg. Med. Chem. Lett., 2007, 15, 1083–1090 CrossRef CAS PubMed; (c) J. G. Lombardino and E. H. Wiseman, J. Med. Chem., 1974, 17, 1182–1188 CrossRef CAS PubMed; (d) J. G. Lombardino, US Pat., 3772441, 1973; (e) T. Maier, R. Schmierer, K. Bauer, H. Bieringer, H. Buerstell and B. Sachse, US Pat., 4820335, 1989; (f) U. Ucucu, I. Iskdag and B. Cakir, Gazi Univ. Eczacilik Fak. Derg., 1988, 5, 161–165 CAS; (g) S. E. Wolkenberg, D. D. Wisnoski, W. H. Leister, Y. Wang, Z. Zhao and C. W. Lindsley, Org. Lett., 2004, 6, 1453–1456 CrossRef CAS PubMed.
- K. V. Belyaeva, L. V. Andriyankova, L. P. Nikitina, A. G. Mal'kina, A. V. Afonin and B. A. Trofimov, Tetrahedron Lett., 2012, 53, 7040–7043 CrossRef CAS.
- (a) T. A. Farghaly, M. A. Abdallah and Z. A. Muhammad, Res. Chem. Intermed., 2015, 41, 3937–3947 CrossRef CAS; (b) F. A. Bassyouni, S. M. Abu-Bakr, K. H. Hegab, W. El-Eraky, A. A. E. Beih and M. E. A. Rehim, Res. Chem. Intermed., 2012, 38, 1527–1550 CrossRef CAS.
- J. L. Davis, M. G. Papich and M. C. Heit, Antifungal and Antiviral Drugs, in Veterinary Pharmacology and Therapeutics, ed. J. E. Riviere and M. G. Papich, Wiley-Blackwell, 9th edn, 2009, ch. 39, vol. 1019–1020, ISBN 978-0-8138-2061-3 Search PubMed.
- (a) N. Xi, Q. Huang and L. Liu, in comprehensive heterocyclic chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier science, Oxford, 2008, vol 4, pp. 143–348 Search PubMed; (b) L. De Luca, Curr. Med. Chem., 2006, 13, 1–23 CAS.
- H. Debus, Ann. Chem. Pharm., 1858, 107, 199–208 CrossRef.
- Z. Zarnegar and J. Safari, New J. Chem., 2014, 38, 4555–4565 RSC.
- K. F. Shelke, S. B. Sapkal, S. S. Sonar, B. R. Madje, B. B. Shingate and M. S. Shingare, Bull. Korean Chem. Soc., 2009, 30, 1057–1060 CrossRef CAS.
- S. D. Jadhave, N. D. Kokare and S. D. Jadhave, J. Heterocycl. Chem., 2009, 45, 1461–1464 CrossRef.
- A. Shaabani, A. Rahmati, E. Farhangi and Z. Badri, Catal. Commun., 2007, 8, 1149–1152 CrossRef CAS.
- H. Weinmann, M. Harre, K. Koeing, E. Merten and U. Tilestam, Tetrahedron Lett., 2002, 43, 593–595 CrossRef CAS.
- J. Liu, J. Chen, J. Zhao, Y. Zhao, L. Li and H. Zhang, Synthesis, 2003, 2661–2666 CAS.
- M. M. Khodaei, K. Bahrami and I. Kavianinia, J. Chin. Chem. Soc., 2007, 54, 829–833 CrossRef CAS.
- N. D. Kokare, J. N. Sangshetti and D. B. Shinde, Synthesis, 2007, 2829–2834 CAS.
- (a) V. Paragamian, M. B. Baker, B. M. Puma and J. Reale, J. Heterocycl. Chem., 1968, 5, 591–597 CrossRef CAS; (b) A. Shaabani and A. Maleki, Chem. Pharm. Bull., 2008, 56, 79–81 CrossRef CAS PubMed; (c) J. B. Hendrickson and M. S. Hussoin, J. Org. Chem., 1987, 52, 4137–4139 CrossRef; (d) J. J. Vanden Eynde, F. Delfosse, A. Mayence and Y. V. Haverbeke, Tetrahedron, 1995, 51, 5813–5818 CrossRef CAS; (e) I. Yavari, H. Mostafavi, D. Tahmassebi and R. Hekmat-Shoar, Monatsh. Chem., 1997, 128, 675–679 CrossRef CAS; (f) V. A. Ozeryanskii, E. A. Filatova, V. I. Sorokin and A. F. Pozharskii, Russ. Chem. Bull., 2001, 50, 846–853 CrossRef CAS.
- M. Kalhor and N. Khodaparast, Res. Chem. Intermed., 2015, 41, 3235–3242 CrossRef CAS.
- (a) A. Bamoniri, B. B. F. Mirjalili and S. Saleh, RSC Adv., 2018, 8, 6178–6182 RSC; (b) M. Kalhor, F. Rezaee-Baroonaghi, A. Dadras and Z. Zarnegar, Appl. Organomet. Chem., 2019, e4784 CrossRef.
- E. Doustkhah and S. Rostamnia, J. Colloid Interface Sci., 2016, 478, 280–287 CrossRef CAS PubMed.
- H. Kefayati, M. Golshekan, S. Shariati and M. Bagheri, Chin. J. Catal., 2015, 36, 572–578 CrossRef CAS.
- F. Bonyasi, M. Hekmati and H. Veisi, J. Colloid Interface Sci., 2017, 496, 177–187 CrossRef CAS PubMed.
- J. Safari and Z. Zarnegar, J. Mol. Catal. A: Chem., 2013, 379, 269–276 CrossRef CAS.
- X. N. Zhao, G. F. Hu, M. Tang, T. T. Shi, X. L. Guo, T. T. Li and Z. H. Zhang, RSC Adv., 2014, 4, 51089–51097 RSC.
- E. Tabrizian and A. Amoozadeh, RSC Adv., 2016, 6, 96606–96615 RSC.
- C. Zhang, H. Wang, F. Liu, L. Wang and H. He, Cellulose, 2013, 20, 127–134 CrossRef CAS.
- G. Perot and M. Guisnet, J. Mol. Catal., 1990, 61, 173–196 CrossRef CAS.
- M. Kalhor, S. Banibairami and S. A. Mirshokraie, Green Chem. Lett. Rev., 2018, 11, 334–344 CrossRef CAS.
- R. Estevez, I. Iglesias, D. Luna and F. M. Bautista, Molecules, 2017, 22, 2206–2217 CrossRef PubMed.
- N. Krathumkhet, K. Vongjitpimol, T. Chuesutham, S. Changkhamchom, K. Phasuksom, A. Sirivata and K. Wattanakul, Solid State Ionics, 2018, 319, 278–284 CrossRef.
- H. Nur, G. L. Kee, H. Hamdan, T. M. I. Mahlia, J. Efendi and H. S. C. Metselaar, Int. J. Hydrogen Energy, 2012, 37, 12513–12521 CrossRef CAS.
- A. Mesdaghinia, A. Azari, R. N. Nodehi, K. Yaghmaeian, A. K. Bharti, S. Agarwal, V. K. Gupta and K. Sharafi, J. Mol. Liq., 2017, 233, 378–390 CrossRef CAS.
- F. Rouquerol, J. Rouquerol and K. S. W. Sing, Adsorption by powders and porous solids, Academic Press, 1999, pp. 1–25 Search PubMed.
- M. M. Khakzad Siuki, M. Bakavoli and H. Eshghi, Appl. Organomet. Chem., 2018, 32, e4290 CrossRef.
- H. Naeimi and D. Aghaseyedkarimi, Dalton Trans., 2016, 45, 1243–1253 RSC.
- K. Nikoofar, M. Haghighi, M. Lashanizadegan and Z. Ahmadvand, J. Taibah Univ. Sci., 2015, 9, 570–578 CrossRef.
- M. A. Bodaghifard, S. Asadbegi and Z. Bahrami, J. Iran. Chem. Soc., 2017, 14, 365–376 CrossRef CAS.
- O. Maloshitskaya, J. Sinkkonen, V. V. Ovcharenko, K. N. Zelenin and K. Pihlaja, Tetrahedron, 2004, 60, 6913–6921 CrossRef CAS.
- T. S. Ahooie, N. Azizi, I. Yavari and M. M. Hashemi, J. Iran. Chem. Soc., 2018, 15, 855–862 CrossRef CAS.
- Z. Zarnegar and J. Safari, RSC Adv., 2014, 4, 20932–20939 RSC.
- D. Parthiban and R. J. Karunakaran, Orient. J. Chem., 2018, 34, 3004–3015 CrossRef CAS.
- R. Bansal, P. K. Soni and A. K. Halve, J. Heterocycl. Chem., 2018, 55, 1308–1312 CrossRef CAS.
- H. Naeimi and D. Aghaseyedkarimi, New J. Chem., 2015, 39, 9415–9421 RSC.
- J. Safari and Z. Zarnegar, Ultrason. Sonochem., 2013, 20, 740–746 CrossRef CAS PubMed.
- A. Mobinikhaledi and N. Foroughifar, Turk. J. Chem., 2009, 33, 555–560 CAS.
- F. K. Behbahani and F. M. Golchin, J. Taibah Univ. Sci., 2017, 11, 85–89 CrossRef.
- C. K. Wu, T. J. Liou, H. Y. Wei, P. S. Tsai and D. Y. Yang, Tetrahedron, 2014, 70, 8219–8225 CrossRef CAS.
- A. Farrokhi, K. Ghodrati and I. Yavari, Catal. Commun., 2015, 63, 41–46 CrossRef CAS.
- A. Mobinikhaledi and P. Steel, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2009, 39, 133–135 CAS.
| This journal is © The Royal Society of Chemistry 2019 |