
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDimethylformamide-stabilised palladium nanoclusters catalysed coupling reactions of aryl halides with hydrosilanes/disilanes†
Tatsuki Nagataa,
Takeru Inouea,
Xianjin Lina,
Shinya Ishimotoa,
Seiya Nakamichia,
Hideo Okaa,
Ryota Kondoa,
Takeyuki Suzukib and
Yasushi Obora *a
*a
aDepartment of Chemistry and Materials Engineering, Faculty of Chemistry, Materials and Bioengineering, Kansai University, Suita, Osaka 564-8680, Japan. E-mail: obora@kansai-u.ac.jp
bComprehensive Analysis Center, The Institute of Scientific and Industrial Research (ISIR), Osaka University, 8-1 Mihogaoka, Ibaraki, Osaka 567-0057, Japan
First published on 3rd June 2019
Abstract
N,N-Dimethylformamide-stabilised Pd nanocluster (NC) catalysed cross-coupling reactions of hydrosilane/disilane have been investigated. In this reaction, the coupling reaction proceeds without ligands with low catalyst loading. N,N-Dimethylacetamide is a crucial solvent in these reactions. The solvent effect was considered by various techniques, such as transmission electron microscopy, X-ray photoelectron spectroscopy, and thermogravimetric analysis. The Pd NCs can be recycled five times under both hydrosilane and disilane reaction conditions.
Introduction
Arylsilanes have attracted a great deal of interest in various fields. Silicon-containing molecules have unique and specific properties and various uses, such as in medicine,1 in photonics,2 and as organic intermediates.3 Conventional methods for arylsilane synthesis are unsatisfactory because of byproducts and side reactions.4 Accordingly, considerable attention has focused on their selective synthesis.5 Transition metal catalysed cross-coupling reactions between aryl halides and hydrosilanes or disilanes have emerged as selective and easy to handle protocols. However, these reactions are generally achieved using transition metal complexes, such as Pd6 and Rh7 complexes, with 1–5 mol% of the catalyst. In addition, hydrosilanes act as weak reducing reagents8 and using them as coupling substrates inhibits reduction of aryl halides. Moreover, owing to the stability of the Si–Si bond, disilanes show low reactivity for silylation reagents, so the coupling reactions require bulky phosphorus ligands.9Metal nanoparticles (NPs) and nanoclusters (NCs) have unique physical and chemical properties.10 They are expected to show high catalytic performance owing to their stability, selectivity, activity, and recyclability.11,12 On the other hand, colloidal nanocatalysts require removal of capping agent to access catalytically active surface. Stabilizing agents such as thiolates, phosphines, surfactants, and polymers, behaves a protect shell for reactant.13 We have reported a surfactant-free preparation of dimethylformamide (DMF)-stabilised transition-metal (Au, Fe, Ir, Cu, and Pd) NPs by the DMF reduction method.14 Among these NPs, Pd NCs have proven to be highly effective in various palladium-catalysed cross-coupling reactions, such as Suzuki–Miyaura, Mizoroki–Heck,15a and Migita–Kosugi–Stille coupling reactions.15b DMF-stabilised Fe2O3 NPs also show high catalytic activity for hydrosilylation of unactivated terminal alkenes.16 The Pd–Pt–Fe2O3 nanocatalyst has recently been reported for coupling aryl halides with hydrosilanes. However, the Pd monometallic catalyst shows low reactivity and causes decomposition.17
In this paper, we describe DMF-stabilised palladium NCs catalysed coupling reactions of aryl halides with hydrosilanes/disilanes to give arylsilanes with moderate to good yields. These reactions proceed at low catalyst loading under ligand and additive free conditions. In addition, the Pd NCs catalysts in both the hydrosilane/disilane reaction systems can be recycled at least five times.
Results and discussion
Initially, we chose to study the cross-coupling reaction between iodobenzene (1a) and dimethylphenylsilane (2a) using LiOAc as a base in DMF at 100 °C. After 16 h, dimethyldiphenylsilane (3a) was obtained in moderate yield (Table 1, entry 1). When we changed N-dimethylacetamide (DMAc) solvent to the reaction, the yield increased to 80% (entry 2). The reaction was achieved by using low Pd loadings compared with the previously reported catalytic processes.6b,d During the course of the reaction, small amount of benzene (<10% yield) was observed as byproduct. The reaction was not accelerated in N-methyl-2-pyrrolidone (NMP) and toluene (entries 3 and 4). Various bases were tested. LiOAc gave the highest yield (entries 5–6). The use of KOtBu under these conditions gave the product in 8% yield along with intractable byproduct (by GC) (entry 7). The reaction also occurred in the absence of a base (entry 8). The use of PdCl2 under these conditions shows low catalytic activity (entry 9). To investigate the specific solvent effect, we prepared Pd NCs in DMF followed by a replacement of DMF on the Pd NCs by using DMAc by heating at 100 °C for 24 h. A TEM image and the particle size distribution of the DMAc-displaced Pd NCs are shown in Fig. 1. The average particle size of 2 nm is the same as that of the prepared Pd NCs (Fig. 1).
|
||||
|---|---|---|---|---|
| Entry | Solvent | Base | Conv. (%) | Yieldb (%) |
| a Reaction conditions: 1a (1.0 mmol), 2a (3.0 mmol), Pd NCs (0.1 mol%), and base (1.0 mmol) in solvent (1.0 mL) at 100 °C for 16 h.b The conversion of 1a and yields were determined by gas chromatography (GC) analysis. The isolated yield is shown in parenthesis.c Not determined due to overlapping of GC peaks.d PdCl2 was used as catalyst precursor. | ||||
| 1 | DMF | LiOAc | >99 | 52 |
| 2 | DMAc | LiOAc | >99 | 80 (75) |
| 3 | NMP | LiOAc | —c | 41 |
| 4 | Toluene | LiOAc | 66 | 7 |
| 5 | DMAc | NaOAc | >99 | 68 |
| 6 | DMAc | KOAc | >99 | 57 |
| 7 | DMAc | KOtBu | >99 | 8 |
| 8 | DMAc | None | >99 | 52 |
| 9d | DMAc | LiOAc | 97 | 9 |

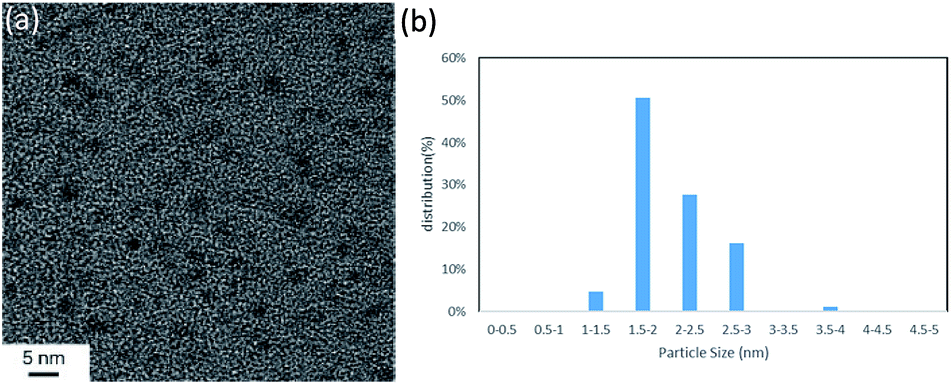 | ||
| Fig. 1 (a) TEM image (scale bar = 5 nm) and (b) nanoparticle size distribution of the DMAc-displaced Pd NCs. | ||
1H-NMR spectroscopy, Fourier transform-infrared (FT-IR) spectroscopy, thermogravimetric (TG) analysis, and X-ray photoelectron spectroscopy (XPS) were performed to analyse the DMAc-substituted Pd NCs. From 1H-NMR analysis, several peaks around 8 ppm in 1H-NMR assigned to formyl protons of DMF molecules on the Pd NCs.14f,15a–c The formyl group proton (δ = 8.14 ppm) disappears after solvent displacement (Fig. S1†). FT-IR analysis (Fig. S2†) shows a peak at 1670 cm−1, which is attributed to the n(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching vibration and indicates the presence of DMF and DMAc molecules. The thermal stability of the Pd NPs was investigated by TG analysis (Fig. 2). The TG curves in the range of 25 °C to 200 °C of the two Pd NCs-prepared by DMF (Pd NCs-DMF) and the Pd NCs covered by DMAc (Pd NCs-DMAc) indicated similar charts. A difference was observed in the TG curves of the both Pd nanoclusters from above 200 °C, suggesting that the two nanoparticles were protected by different molecules on the surface on the metal clusters (DMF and DMAc).
O) stretching vibration and indicates the presence of DMF and DMAc molecules. The thermal stability of the Pd NPs was investigated by TG analysis (Fig. 2). The TG curves in the range of 25 °C to 200 °C of the two Pd NCs-prepared by DMF (Pd NCs-DMF) and the Pd NCs covered by DMAc (Pd NCs-DMAc) indicated similar charts. A difference was observed in the TG curves of the both Pd nanoclusters from above 200 °C, suggesting that the two nanoparticles were protected by different molecules on the surface on the metal clusters (DMF and DMAc).
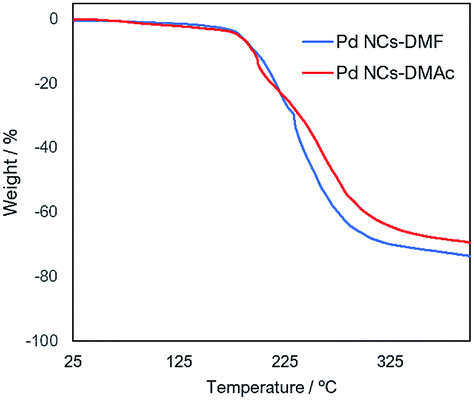 | ||
| Fig. 2 TG analysis of Pd NCs-DMF (blue) and Pd NCs-DMAc (red). | ||
The surface states of the as-prepared Pd NCs and solvent-displaced Pd NCs with DMAc were determined by XPS. The binding energy positions of Pd 3d5/2 and Pd 3d3/2 are shown in Fig. 3. Peak FWHMs are described in Table S1.† The other main peaks (C, O, and N) are shown in Fig. S4–S6.† The main peaks of Pd at 338.8, 337.2 eV of 3d5/2 and 344.0, 342.1 of 3d3/2 was higher than that of bulk Pd (Pd 3d5/2 335.1 eV and Pd 3d3/2 340.3 eV).18,19 The Pd 3d5/2 and 3d3/2 spectrum of Pd NCs with DMF is shown in Fig. 3a. The DMAc Pd NCs have a broad peak (Pd 3d5/2 343.7, 341.8 eV and Pd 3d3/2 338.5, 336.6 eV) (Fig. 3b). A significant peak shift in Pd NCs with DMAc by increase of the measurement time (Fig. 3d). In contrast, the as-prepared Pd NCs remain unchanged (Fig. 3c). The changes in the two samples are caused by removal of the protective molecules on the surface. With irradiation by X-ray and sample heating, the protective molecules (DMF rigid protecting layer) are removed as the number of measurements increases. With elimination of the protective surface molecules, surface palladium is reduced. These data are associated with the TG analysis results, where DMAc displacement of the Pd NCs removes the capping molecules and results in easy access to the active sites.
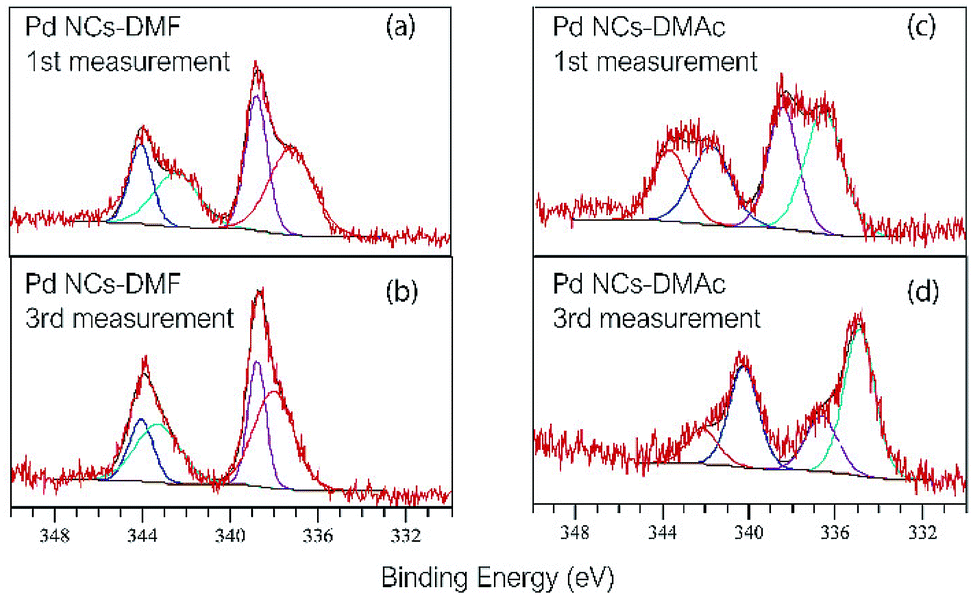 | ||
| Fig. 3 XPS spectra of Pd NCs with DMF (a) at first measurement, (b) at third measurement, and Pd NCs with DMA which substituted from DMF (c) at 1st and (d) at 3rd measurement. | ||
Next, we investigated aryl halides bearing both electron-donating and electron-withdrawing substituents (Scheme 1). Electron-donating substrates (4-methyl and 4-methoxy, and 4-tert-butyl) gave excellent yields (84%, 97%, and 82% for 3b, 3c, and 3d, respectively). Electron withdrawing substrates were less effective (34% and 42% for 3e and 3f, respectively). Various silanes were investigated. The corresponding arylsilanes diphenylmethylsilane (3g), triethylsilane (3h), and triethoxysilane (3i) were obtained in 38%, 32%, and 25% yield, respectively.
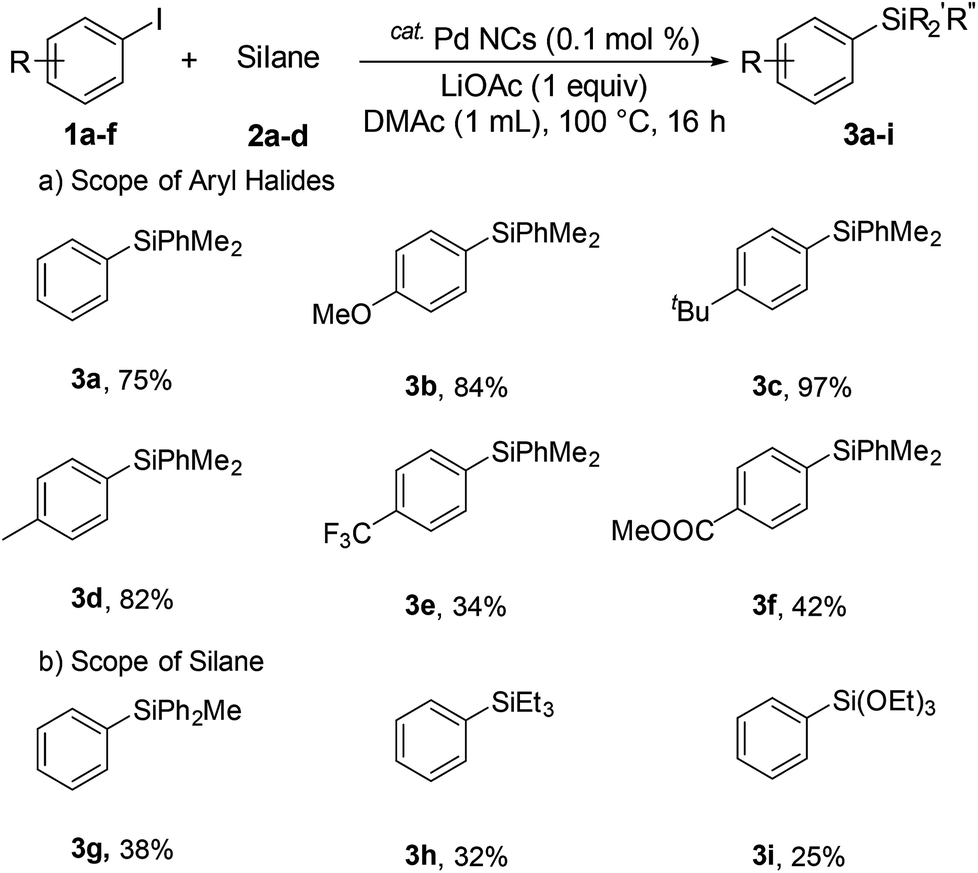 | ||
| Scheme 1 Scope of various substrates. | ||
We wanted to extend the coupling reaction to disilanes. Hexamethyldisilane is readily available as a Rochow direct process byproduct.20 The reaction of iodobenzene with hexamethyldisilane at 120 °C afforded 5a in 70% yield (Table 2, entry 1). Various bases were examined. NaOAc showed the best performance (entries 2–5). The same solvent effect was observed for DMF and DMAc (entry 6). The presence of a base was necessary for the reaction (entry 7). The reaction by using PdCl2 as catalyst precursor and gave the product in 38% (entry 8).
|
||||
|---|---|---|---|---|
| Entry | Solvent | Base | Conv. (%) | Yieldb (%) |
| a Reaction conditions: 1a (1.0 mmol), 4 (3.0 mmol), Pd NCs (0.1 mol%), and base (1.0 mmol) in solvent (1.0 mL) at 120 °C for 24 h.b The conversion of 1a and yields were determined by GC analysis. The isolated yield is shown in parenthesis.c PdCl2 was used as catalyst precursor. | ||||
| 1 | DMAc | LiOAc | >99 | 70 |
| 2 | DMAc | NaOAc | >99 | 83 (70) |
| 3 | DMAc | KF | >99 | 73 |
| 4 | DMAc | Na2CO3 | >99 | 78 |
| 5 | DMAc | KOtBu | 89 | 27 |
| 6 | DMF | NaOAc | >99 | 69 |
| 7 | DMAc | None | 58 | 32 |
| 8c | DMAc | NaOAc | 78 | 38 |
Iodobenzene derivatives bearing electron-donating groups, such as p-methyl, p-methoxy, and p-tert-butyl groups, gave the corresponding products in good yields (Table 3, entries 2–4). Electron-withdrawing groups were less effective (entries 5–7). However, the reaction of bromo/chlorobenzene under these conditions was sluggish.
|
||||
|---|---|---|---|---|
| Entry | X | R | Product | Yieldb (%) |
| a Reaction conditions: 1a (1.0 mmol), 2a (3.0 mmol), Pd NCs (0.1 mol%), and base (1.0 mmol) in DMAc (1.0 mL) at 120 °C for 24 h.b Isolated yield.c GC yield. | ||||
| 1 | I | H | 5a | 70 |
| 2 | p-Me | 5b | 65 | |
| 3 | p-OMe | 5c | 73 | |
| 4 | p-tBu | 5d | 86 | |
| 5 | p-COMe | 5e | 66 | |
| 6 | p-COOMe | 5f | 59 | |
| 7 | p-CF3 | 5g | 48 | |
| 8 | Br | H | 5a | 5c |
| 9 | Cl | H | 5a | Tracec |
Based on the results and those reported in the literature, the plausible reaction mechanisms are shown in Scheme S7.†21 Iodobenzene or hydrosilane oxidatively adds to the Pd NCs. The desired coupling product is obtained by the σ-bond metathesis reaction.
We investigated possible reuse of the catalyst.22 After the reaction, we performed annular dark field scanning transmission electron microscopy (ADF-STEM) and inductively coupled plasma-atomic emission spectroscopy (ICP-AES) analysis (Table S2†). The particle size remained the same (Fig. 4). Based on a previous study, the results suggest that the Pd NCs possess recyclability in the coupling reaction.
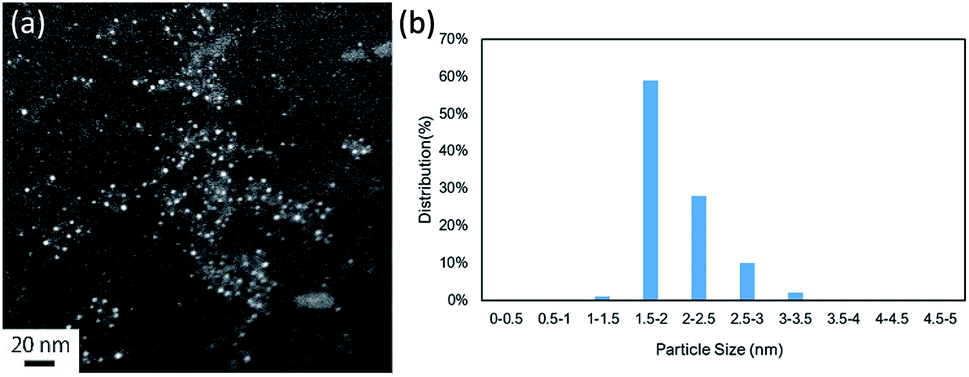 | ||
| Fig. 4 (a) ADF-STEM image (scale bar = 20 nm) and (b) particle size distribution of the Pd NCs after the reaction. | ||
We investigated the recyclability of the Pd NCs (Fig. 5 and 6). After performing the reaction with the disilane system (Table 2, entry 2), the hexane layer was extracted using 8 mL of hexane five times. The hexane layer (containing the starting materials and products) was removed. The DMAc was evaporated from the residual DMAc layer containing Pd NCs and the residue was reused as the catalyst for the reaction under the same conditions (Table 2, entry 2). The Pd NCs gave good yields five times. We also tested the coupling reaction of aryl halide with hydrosilane (reaction conditions as in Table 1, entry 2). In the hydrosilane coupling reaction, the extraction solvent was modified. Product 3a and the starting materials were extracted using a mixed solvent (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate = 95:5). The Pd NCs catalyst tolerated multiple cycles.
:ethyl acetate = 95:5). The Pd NCs catalyst tolerated multiple cycles.
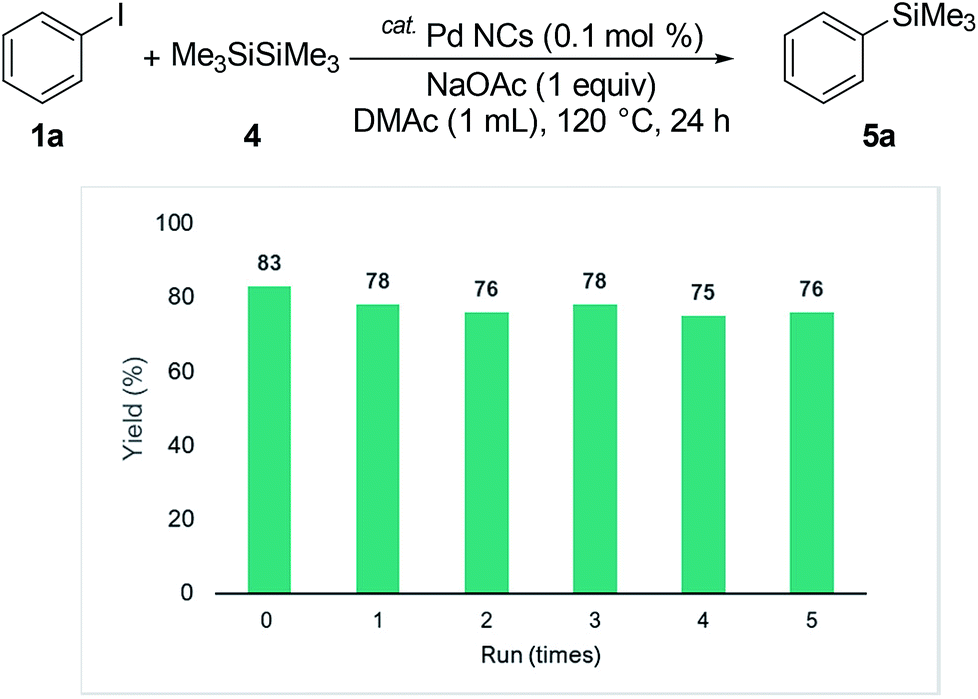 | ||
| Fig. 5 Multiple catalyst recycling for coupling of iodobenzene with disilane (conditions as in Table 2, entry 2). | ||
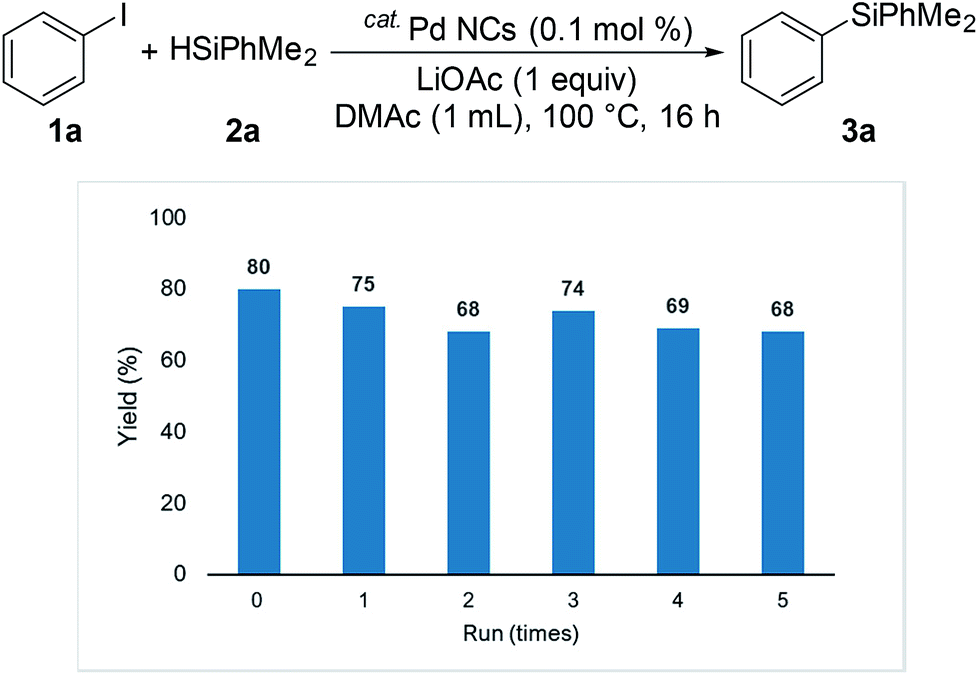 | ||
| Fig. 6 Multiple catalyst recycling for coupling of iodobenzene with hydrosilane (conditions as in Table 1, entry 2). | ||
Conclusions
We have developed Pd NCs catalytic system for silylation of aryl halides with hydrosilanes. The Pd NCs catalyst can be separated and reused at least five times without notable loss of the catalytic activity. The same catalyst can also be used for silylation of hexamethyldisilane. DMAs displacement enables easy access to the active sites of the Pd NCs. Further investigation of metal NPs as catalysts is underway.Experimental
1. General
GC analysis was performed with a flame ionisation detector using a 0.22 mm × 25 m capillary column (BP-5). The 1H and 13C NMR spectra were recorded at 400 and 100 MHz, respectively. The 1H and 13C NMR chemical shifts are reported in ppm with respect to the residual chloroform (1H 7.26 ppm, 13C 77 ppm). The products were characterised by 1H NMR, 13C NMR, and GC-MS. The TEM and STEM images were obtained with a JEOL JEM-ARM200F instrument at an accelerating voltage of 200 kV. Size distributions of nanoparticles were measured by counting 100 nanoparticles. ICP-AES was performed with a Shimadzu ICPS-8100 spectrometer. XPS analysis was performed with a ULVAC-PHI PHI5000 VersaProbe with Al Kα radiation. The measured spectra were calibrated by the C 1 s electron peak (284.5 eV). TG analysis was performed with a Thermo Plus EVO device (Rigaku, Japan) at a heating rate of 10 °C min−1 under nitrogen flow.All of the synthesised compounds (3a,23a 3b,23a 3c,23a 3d,23b 3e,23a 3f,23b 3g,23e 3h,23c 3i,23d 5a,24a 5b,24c 5c,24c 5d, 5e,24b 5f,24d and 5g24b) are known compounds and have been previously reported.
All of the starting materials were commercially available and used without further purification. The DMF-protected Pd NCs were prepared according to a previously reported method.15a
2. Typical procedure
3a dimethyldiphenylsilane: colorless liquid, 1H-NMR (400 MHz; CDCl3; CDCl3) δ: 7.57–7.54 (4H, m, Ph), 7.39–7.38 (6H, m, Ph), 0.59 (6H, s, Me); 13C-NMR (100 MHz; CDCl3; CDCl3) δ: 138.22 (C) 134.22 (CH), 129.11 (CH), 127.82 (CH), −2.42 (CH3); GC-MS (EI) m/z (relative intensity) 212(23) [M]+, 197(100), 198(19), 105(9).
3b dimethyl(phenyl)(p-tolyl)silane: colorless liquid, 1H-NMR (400 MHz; CDCl3; CDCl3) δ: 7.57–7.55 (2H, m, Ph), 7.48–7.46 (2H, m), 7.39–7.38 (3H, m, Ph), 7.23–7.21 (2H, m, Ph), 2.39 (3H, s, SiMe2), 0.58 (6H, s, Me); 13C-NMR (100 MHz; CDCl3; CDCl3) δ: 138.96 (C), 138.50 (C), 134.53 (C), 134.23 (CH), 134.15 (CH), 129.00 (CH), 128.65 (CH), 127.76 (CH), 21.45 (CH3), −2.32 (CH3); GC-MS (EI) m/z (relative intensity) 226(18) [M]+, 211(100), 212(21), 105(7).
3c (4-ethoxyphenyl)dimethyl(phenyl)silane: colorless liquid, 1H-NMR (400 MHz; CDCl3; CDCl3) δ: 7.53–7.44 (7H, m, Ph), 6.97–6.95 (2H, m, Ph), 3.85 (3H, s, OMe), 0.58 (6H, s, SiMe2); 13C-NMR (100 MHz; CDCl3; CDCl3) δ: 160.48 (C), 138.66 (C), 135.62 (CH), 134.11 (CH), 128.97 (CH), 127.75 (CH), 113.58 (CH), 54.97 (CH3), −2.32 (CH3); GC-MS (EI) m/z (relative intensity) 242(20) [M]+, 227(100), 135(4).
3d (4-(tert-butyl)phenyl)dimethyl(phenyl)silane: colorless liquid, 1H-NMR (400 MHz; CDCl3; CDCl3) δ: 7.21–7.01 (9H, m, Ph), 0.98 (9H, s, tBu), 0.21 (6H, s, SiMe2); 13C-NMR (100 MHz; CDCl3; CDCl3) δ: 151.98 (C), 138.46 (C), 134.59 (C), 134.15 (CH), 134.04 (CH), 128.99 (CH), 127.75 (CH), 124.77 (CH), 34.62 (C), 31.22 (CH3), −2.34 (CH3); GC-MS (EI) m/z (relative intensity) 268(16) [M]+, 253(100), 237(6), 105(10).
3e dimethyl(phenyl)(4-(trifluoromethyl)phenyl)silane: 1H-NMR (400 MHz; CDCl3; CDCl3) δ: 7.64–7.61 (4H, m, Ph), 7.53–7.52 (2H, m, Ph), 7.41–7.38 (3H, m Ph), 0.60 (6H, s, SiMe2); 13C-NMR (100 MHz; CDCl3; CDCl3) δ: 143.36 (q, 4JC–F = 1.0 Hz, CH), 137.03 (CH), 134.41 (CH), 134.10 (CH), 130.99 (q, 2J C–F = 32.2 Hz, CH), 129.45 (CH), 127.97 (CH), 124.29 (q, 3JC–F = 3.7 Hz, CH), 124.19 (q, 1JC–F = 273.0 Hz, C), −2.63 (CH3); GC-MS (EI) m/z (relative intensity) 280(4) [M]+, 265(100), 184(15).
3f methyl 4-(dimethyl(phenyl)silyl)benzoate: colorless liquid, 1H-NMR (400 MHz; CDCl3; CDCl3) δ: 8.00 (2H, dd, J = 8.3, 0.7 Hz, Ph), 7.60 (2H, dd, J = 8.3, 0.7 Hz, Ph), 7.51–7.49 (2H, m, Ph), 7.38–7.37 (3H, m, Ph), 3.92 (3H, s, OMe), 0.58 (6H, d, J = 0.6 Hz, SiMe2); 13C-NMR (100 MHz; CDCl3; CDCl3) δ: 167.22 (C), 144.63 (C), 137.28 (C), 134.13 (CH), 134.11 (CH), 130.49 (C), 129.34 (CH), 128.51 (CH), 127.91 (CH), 52.12 (CH3), −2.61 (CH3); GC-MS (EI) m/z (relative intensity) 270(7) [M]+, 256(20), 255(100), 239(2).
3g methyltriphenylsilane: colorless liquid, 1H-NMR (400 MHz; CDCl3; CDCl3) δ: 7.56–7.36 (15H, m, Ph), 0.87 (3H, s, SiMe); 13C-NMR (100 MHz; CDCl3; CDCl3) δ: 136.06 (C), 135.25 (CH), 129.37 (CH), 127.83 (CH), −3.40 (CH3); GC-MS (EI) m/z (relative intensity) 274(5) [M]+, 259(100), 180(12).
3h triethyl(phenyl)silane: colorless liquid, 1H NMR (400 MHz; CDCl3; CDCl3): δ 7.50–7.35 (m, 2H, Ph), 7.34–7.25 (m, 3H, Ph), 0.96 (t, J = 8.0 Hz, 9H, Me), 0.79 (q, J = 7.2 Hz, 6H, SiCH2); 13C-NMR (100 MHz; CDCl3; CDCl3): δ 137.8 (C), 134.5 (CH), 129.0 (CH), 127.9 (CH), 7.7 (CH3), 3.6 (CH2); GC-MS (EI) m/z (relative intensity) 192(6) [M]+, 163(59), 135(100), 107(77).
3i triethoxy(phenyl)silane: colorless liquid, 1H NMR (400 MHz; CDCl3; CDCl3): δ 7.68 (d, J = 6.4 Hz, 2H, Ph), 7.34 (m, 3H, Ph), 3.87 (q, J = 7.2, 6H, OCH2), 1.25 (t, J = 6.8 9H, Me); 13C-NMR (100 MHz; CDCl3; CDCl3): δ 134.8 (C), 130.9 (CH), 130.4 (CH), 127.9 (CH), 58.7 (CH2), 18.2 (CH3); GC-MS (EI) m/z (relative intensity) 240(23) [M]+, 195(42), 145(100), 135(36).
5b trimethyl(p-tolyl)silane: colorless liquid, 1H-NMR (400 MHz; CDCl3; CDCl3): δ 7.45 (d, J = 8.0 Hz, 2H, Ph), 7.20 (d, J = 7.6 Hz, 2H, Ph), 2.37 (s, 3H, Me), 0.27 (s, 9H, SiMe3); 13C-NMR (100 MHz; CDCl3; CDCl3): δ 138.6 (C), 136.8 (C), 133.3 (CH), 128.6 (CH), 21.5 (CH3), −1.1 (CH3); GC-MS (EI) m/z (relative intensity) 164(13) [M]+, 149(100), 121(9).
5c (4-methoxyphenyl)trimethylsilane: colorless liquid, 1H-NMR (400 MHz; CDCl3; CDCl3): δ 7.20 (d, J = 8.8 Hz, 2H, Ph), 6.67 (d, J = 8.4 Hz, 2H, Ph), 3.56 (s, 3H, OMe), 0.00 (s, 9H, SiMe3); 13C-NMR (100 MHz; CDCl3; CDCl3): δ 162.2 (C), 135.7 (CH), 132.2 (C), 114.4 (CH), 55.9 (CH3), 0.00 (CH3); GC-MS (EI) m/z (relative intensity) 180(16) [M]+, 165(100), 135(9).
5d (4-(tert-butyl)phenyl)trimethylsilane: white solid, m.p. 77.5–78, 1H-NMR (400 MHz; CDCl3; CDCl3): δ 7.49 (d, J = 8.4 Hz, 2H, Ph), 7.41 (d, J = 8.0 Hz, 2H, Ph), 1.34 (s, 9H, Me), 0.27 (s, 9H, SiMe3); 13C-NMR (100 MHz; CDCl3; CDCl3): δ 151.7 (C), 136.9 (C), 133.2 (CH), 124.7 (CH), 34.6 (C), 31.2 (CH3), −1.06(CH3); GC-MS (EI) m/z (relative intensity) 206(12) [M]+, 191(100), 176(7).
5e 1-(4-(trimethylsilyl)phenyl)ethan-1-one: colorless liquid, 1H-NMR (400 MHz; CDCl3; CDCl3) δ: 7.92–7.91 (2H, m, Ph), 7.63–7.61 (2H, m, Ph), 2.60 (3H, s, COMe), 0.29 (9H, s, SiMe3); 13C-NMR (100 MHz; CDCl3; CDCl3) δ: 198.33 (C), 147.21 (C), 137.17 (C), 133.48 (CH), 127.19 (CH), 26.58 (CH3), −1.38 (SiMe3); GC-MS (EI) m/z (relative intensity) 192(14) [M]+, 177(100), 162(1), 119(7).
5f methyl 4-(trimethylsilyl)benzoate: colorless liquid, 1H-NMR (400 MHz; CDCl3; CDCl3) δ: 7.99 (2H, d, J = 8.3 Hz, Ph), 7.59 (2H, d, J = 8.2 Hz, Ph), 3.92 (3H, s, OMe), 0.29 (9H, s, SiMe3); 13C-NMR (100 MHz; CDCl3; CDCl3) δ: 167.28 (C), 146.84 (C), 133.27 (CH, s), 130.19 (C), 128.44 (CH), 52.09 (CH3), −1.34 (CH3); GC-MS (EI) m/z (relative intensity) 208(5) [M]+, 193(100), 133(14).
5g trimethyl(4-(trifluoromethyl)phenyl)silane: 1H-NMR (400 MHz, CDCl3; CDCl3): δ 7.64 (d, J = 7.6 Hz, 2H, Ph), 7.59 (d, J = 8.0 Hz, 2H, Ph), 0.30 (s, 9H, SiMe3); 13C-NMR (100 MHz; CDCl3; CDCl3): δ 145.6 (C), 133.8 (CH), 130.8 (q, J = 31.6 Hz, C), 124.3 (q, J = 270.2 Hz, C), 124.2 (q, J = 3.9 Hz CH), −1.41(CH3) GC-MS (EI) m/z (relative intensity) 218(6) [M]+, 203(100), 189(0.60).
3. Preparation of the TEM sample
:ethyl acetate (99:1), 5 mL, 5 times] to remove 5a. DMAc was removed by evaporation under high vacuum and then redispersed in ethanol.4. Recycling experiment
:ethyl acetate = 95:5, 5 mL, ten times, upper phase containing 3a). The Pd NCs remained in the DMAc solvent (bottom phase). Conversion of the substrate and the yield of the product were calculated from their GC peak areas based on an internal standard (n-decane). The DMAc solution containing the Pd NCs was filtered through a cotton plug to remove the Pd NCs from the DMAc solution. DMAc was removed under vacuum and the residue was redissolved in DMAc (1 mL). Pd NCs in DMF were used for the next catalytic reaction under the reaction conditions given in Table 1, entry 2 by adding iodobenzene (1a, 109 mg, 1.0 mmol), dimethylphenylsilane (2a, 409 mg, 3.0 mmol), and LiOAc (67 mg, 1.0 mmol).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the “Development of Innovative Catalytic Processes for Organosilicon Functional Materials” project (PL: K. Sato, AIST) from the New Energy and Industrial Technology Development Organization (NEDO). We thank the members of the Comprehensive Analysis Center, SANKEN (ISIR), Osaka University, for TEM, ICP-AES analyses.Notes and references
- (a) J. Wang, C. Ma, Y. Wu, R. A. Lamb, L. H. Pinto and W. F. Degrado, J. Am. Chem. Soc., 2011, 133, 13844 CrossRef CAS PubMed; (b) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388 CrossRef CAS PubMed.
- (a) M. Shimizu, K. Mochida and T. Hiyama, Angew. Chem., Int. Ed., 2008, 47, 9760 CrossRef CAS PubMed; (b) X. M. Liu, C. He, J. Huang and J. Xu, Chem. Mater., 2005, 17, 434 CrossRef CAS; (c) G. W. Kim, D. R. Yang, Y. C. Kim, H. I. Yang, J. G. Fan, C. H. Lee, K. Y. Chai and J. H. Kwon, Dyes Pigm., 2017, 136, 8 CrossRef CAS; (d) H. Inubushi, Y. Hattori, Y. Yamanoi and H. Nishihara, J. Org. Chem., 2014, 79, 2974 CrossRef CAS PubMed; (e) N. You, C. G. An, J. J. Kim and Y. P. Soo, J. Org. Chem., 2007, 72, 6241 CrossRef PubMed.
- (a) P. Somfai and B. Seashore-Ludlow, Organosilicon Reagents: Vinyl-, Alkynyl-, and Arylsilanes, Elsevier Ltd., 2014 Search PubMed; (b) T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631 CrossRef CAS; (c) A. S. Manoso and P. DeShong, J. Org. Chem., 2001, 66, 7449 CrossRef CAS PubMed.
- (a) S. Luliński and J. Serwatowski, J. Org. Chem., 2003, 68, 9384 CrossRef PubMed; (b) A. S. Manoso, C. Ahn, A. Soheili, C. J. Handy, R. Correia, W. M. Seganish and P. DeShong, J. Org. Chem., 2004, 69, 8305 CrossRef CAS PubMed.
- Z. Xu, W. S. Huang, J. Zhang and L. W. Xu, Synthesis, 2015, 47, 3645 CrossRef CAS.
- (a) H. Guo, X. Chen, C. Zhao and W. He, Chem. Commun., 2015, 51, 17410 RSC; (b) Y. Yamanoi, J. Org. Chem., 2005, 70, 9607 CrossRef CAS PubMed; (c) N. Iranpoor, H. Firouzabadi and R. Azadi, J. Organomet. Chem., 2010, 695, 887 CrossRef CAS; (d) M. Murata, K. Suzuki, S. Watanabe and Y. Masuda, J. Org. Chem., 1997, 62, 8569 CrossRef CAS PubMed.
- (a) M. Murata, M. Ishikura, M. Nagata, S. Watanabe and Y. Masuda, Org. Lett., 2002, 4, 1843 CrossRef CAS PubMed; (b) Y. Yamanoi and H. Nishihara, Tetrahedron Lett., 2006, 47, 7157–7161 CrossRef CAS; (c) Y. Yamanoi and H. Nishihara, J. Org. Chem., 2008, 73, 6671 CrossRef CAS PubMed; (d) M. Murata, H. Yamasaki, K. Uogishi, S. Watanabe and Y. Masuda, Synthesis, 2007, 2944 CrossRef CAS.
- (a) J. Y. Corey, Chem. Rev., 2011, 111, 863 CrossRef CAS PubMed; (b) R. Boukherroub, C. Chatgilialoglu and G. Manuel, Organometallics, 2002, 15, 1508 CrossRef.
- (a) T. Iwasawa, T. Komano, A. Tajima, M. Tokunaga, Y. Obora, T. Fujihara and Y. Tsuji, Organometallics, 2006, 25, 4665 CrossRef CAS; (b) J. Jang, S. Byun, B. M. Kim and S. Lee, Chem. Commun., 2018, 54, 3492 RSC; (c) E. Shirakawa, T. Kurahashi, H. Yoshida and T. Hiyama, Chem. Commun., 2000, 1895 RSC; (d) E. McNeill, T. E. Barder and S. L. Buchwald, Org. Lett., 2007, 9, 3785 CrossRef CAS PubMed.
- (a) Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594 RSC; (b) L. Zhang and E. Wang, Nano Today, 2014, 9, 132 CrossRef CAS; (c) A. Mathew and T. Pradeep, Part. Part. Syst. Charact., 2014, 31, 1017 CrossRef CAS.
- (a) S. Campisi, M. Schiavoni, C. Chan-Thaw and A. Villa, Catalysts, 2016, 6, 185 CrossRef; (b) Y. Dai, Y. Wang, B. Liu and Y. Yang, Small, 2015, 11, 268 CrossRef CAS PubMed; (c) T. Nagata, Y. Adachi and Y. Obora, Synlett, 2018, 29, 2655 CrossRef CAS; (d) Y. Adachi, H. Kawasaki, T. Nagata and Y. Obora, Chem. Lett., 2016, 45, 1457 CrossRef CAS.
- (a) A. Bej, K. Ghosh, A. Sarkar and D. W. Knight, RSC Adv., 2016, 6, 11446 RSC; (b) D. Roy and Y. Uozumi, Adv. Synth. Catal., 2018, 360, 602 CrossRef CAS; (c) C. Deraedt and D. Astruc, Acc. Chem. Res., 2014, 47, 494 CrossRef CAS PubMed.
- (a) W. Huang, Q. Hua and T. Cao, Catal. Lett., 2014, 144, 1355 CrossRef CAS; (b) Z. Niu and Y. Li, Chem. Mater., 2014, 26, 72 CrossRef CAS.
- (a) H. Kawasaki, Nanotechnol. Rev., 2013, 2, 5 CAS; (b) I. Pastorizo-Santos and L. M. Liz-Marzán, Adv. Funct. Mater., 2009, 19, 679 CrossRef; (c) H. Oka, K. Kitai, T. Suzuki and Y. Obora, RSC Adv., 2017, 7, 22869 RSC; (d) K. Oikawa, S. Itoh, H. Yano, H. Kawasaki and Y. Obora, Chem. Commun., 2017, 53, 1080 RSC; (e) Y. Isomura, T. Narushima, H. Kawasaki, T. Yonezawa and Y. Obora, Chem. Commun., 2012, 48, 3784 RSC; (f) H. Yamamoto, H. Yano, H. Kouchi, Y. Obora, R. Arakawa and H. Kawasaki, Nanoscale, 2012, 4, 4148 RSC.
- (a) M. Hyotanishi, Y. Isomura, H. Yamamoto, H. Kawasaki and Y. Obora, Chem. Commun., 2011, 47, 5750 RSC; (b) H. Yano, Y. Nakajima and Y. Obora, J. Organomet. Chem., 2013, 745–746, 258 CrossRef CAS; (c) K. Onishi, K. Oikawa, H. Yano, T. Suzuki and Y. Obora, RSC Adv., 2018, 8, 11324 RSC; (d) S. Asada, A. Nito, Y. Miyagi, J. Ishida, Y. Obora and F. Sanda, Macromolecules, 2017, 50, 4083 CrossRef CAS.
- R. Azuma, S. Nakamichi, J. Kimura, H. Yano, H. Kawasaki, T. Suzuki, R. Kondo, Y. Kanda, K. I. Shimizu, K. Kato and Y. Obora, ChemCatChem, 2018, 10, 2378 CrossRef CAS.
- J. Jang, S. Byun, B. M. Kim and S. Lee, Chem. Commun., 2018, 54, 3492 RSC.
- (a) M. G. Mason, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 27, 748 CrossRef CAS; (b) G. K. Wertheim, S. B. DiCenzo and D. N. E. Buchanan, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 5384 CrossRef CAS.
- (a) M. Chiba, M. N. Thanh, Y. Hasegawa, Y. Obora, H. Kawasaki and T. Yonezawa, J. Mater. Chem. C, 2015, 3, 514 RSC; (b) M. Ren, Y. Jin, W. Chen and W. Huang, J. Phys. Chem. C, 2015, 119, 27588 CrossRef CAS.
- (a) E. G. Rochow, J. Am. Chem. Soc., 1945, 67, 963 CrossRef CAS; (b) W. J. Ward, A. Ritzer, K. M. Carroll and J. W. Flock, J. Catal., 1986, 100, 240 CrossRef CAS.
- J. Cao, Y.-M. Cui, Z. Xu, J. Zhang, L.-W. Xu, Z.-J. Zheng and J.-Z. Xu, Chem.–Asian J., 2017, 12, 1749 CrossRef PubMed.
- (a) Á. Molnár, Chem. Rev., 2011, 111, 2251 CrossRef PubMed; (b) Á. Molnár and A. Papp, Coord. Chem. Rev., 2017, 349, 1 CrossRef.
- (a) M. Uchiyama, Y. Kobayashi, T. Furuyama, S. Nakamura, Y. Kajihara, T. Miyoshi, T. Sakamoto, Y. Kondo and K. Morokuma, J. Am. Chem. Soc., 2008, 130, 472 CrossRef CAS PubMed; (b) H. Guo, X. Chen, C. Zhao and W. He, Chem. Commun., 2015, 51, 17410–17412 RSC; (c) N. Iranpoor, H. Firouzabadi and R. Azadi, J. Organomet. Chem., 2010, 695, 887 CrossRef CAS; (d) M. Murata, H. Yamasaki, T. Ueta, M. Nagata, M. Ishikura, S. Watanabe and Y. Masuda, Tetrahedron, 2007, 63, 4087 CrossRef CAS; (e) N. Tsukada and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 5022 CrossRef CAS PubMed.
- (a) H. Inubushi, H. Kondo, A. Lesbani, M. Miyachi, Y. Yamanoi and H. Nishihara, Chem. Commun., 2013, 49, 134 RSC; (b) M. Tobisu, Y. Kita, Y. Ano and N. Chatani, J. Am. Chem. Soc., 2008, 130, 15982 CrossRef CAS PubMed; (c) A. P. Lothian, C. A. Ramsden, M. M. Shaw and R. G. Smith, Tetrahedron, 2011, 67, 2788 CrossRef CAS; (d) M. Tobisu, Y. Kita and N. Chatani, J. Am. Chem. Soc., 2006, 128, 8152 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization and spectra. See DOI: 10.1039/c9ra02895a |
| This journal is © The Royal Society of Chemistry 2019 |