
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermal- and salt-activated shape memory hydrogels based on a gelatin/polyacrylamide double network†
Fang Chen,
Kaixiang Yang,
Dinglei Zhao and
Haiyang Yang *
*
CAS Key Laboratory of Soft Matter Chemistry, School of Chemistry and Materials Science, University of Science and Technology of China, Hefei 230026, China. E-mail: yhy@ustc.edu.cn; Tel: +86-551-63607549
First published on 13th June 2019
Abstract
Shape memory hydrogels have been extensively studied in the past decades owing to their exceptionally promising potential in a wide range of applications. Here, we present a gelatin/polyacrylamide double network hydrogel with thermal- and salt-activated shape memory effect. The thermally activated behavior is attributed to the reversible triple helix transformation of gelatin, and the salt-activated performance can be ascribed to the formation of hydrophobic interaction domains under the Hofmeister effect. The hydrogel can memorize a temporary shape successfully through soaking with (NH4)2SO4 solution or decreasing temperature, and recovers its permanent shape by extracting ions with deionized water or increasing temperature. In particular, the hydrogel exhibits excellent shape fixity and recovery ratio. The presented strategy may enrich the construction as well as application of biopolymer based shape memory hydrogels.
Introduction
As typical representatives of “soft-wet” materials, hydrogels are widely used in fields ranging from actuators to biomaterials and tissue engineering.1–3 Among these hydrogels, the shape memory hydrogels (SMHs) have received intensive attention. These SMHs are capable of deforming and fixing one or more temporary shapes and subsequently recovering to its initial permanent shape under external stimuli, such as temperature,4 pH,5 light,6 solvents,7 ultrasound8 and so on. The combination of a strong network and a weak switchable network is the main concept of the SMHs.9 Shape memory effect (SME) can be achieved by reversibly changing the weak network for shape fixation and recovery.10 Therefore, stimuli responsive weak reversible cross-linking bonds have been introduced to endow hydrogels with shape memory properties, including dynamic covalent bonds,11 crystallization,12 metal–ligand coordination,13 hydrogen-bonding,14 peptide and protein self-assembly,15 host–guest interactions,16 etc.In recent years, many SMHs with weak reversible cross-linking networks in response to desire stimuli have been reported.17,18 Among various stimuli, temperature is the most widely employed.19 For example, Li et al.20 demonstrated a novel temperature responsive shape memory hydrogel based on the inclusion complexation between a star-shaped adamantyl-terminated 8-arm PEG and a star shaped PNIPAAm with a cyclodextrin core. In addition, salt-activated shape memory performance was also paid much attention.21 Chen and co-workers22 reported the polyacrylamide–chitosan (PAAm–CS) based shape memory hydrogel which was fabricated based on reversible physical interactions, microcrystal and chain-entanglement interactions. Here, chitosan can not only form a kind of chain-entangled structure in NaCl solution but also display a kind of microcrystalline structure in NaOH solution. Besides, there are also some SMHs triggered by orthogonal stimuli.23 We have previously reported the unusual thermal- and light-activated shape memory effects in an agarose/poly (acrylamide-co-acrylic acid) interpenetrating network hydrogel.24 The thermal-activated shape memory behavior is attributed to the reversible coil-helix transition of agarose macromolecules in response to temperature while the light-activated SME is a result of the photoreduction of Fe(III) to Fe(II) in the presence of reductive citric acid.
In comparison to the synthetic polymer, biopolymers are more attractive for fabricating biocompatible SMHs.25 In particular, natural gelatin, as a representative derivative of collagen, possesses distinctive biocompatibility.26 With temperature increasing, gelatin exhibits reversible triple helices transformation and decrease of mechanical properties.27 Recently, Wang groups28 reported that gelatin hydrogels which treated with (NH4)2SO4 solution had more excellent ductility and strength due to Hofmeister effect. Hofmeister effect was the interactions between the ions and the polymer chains, which can be easily found in the process of folding and precipitation of proteins.29 However, SMHs based on Hofmeister effect have been rarely reported.
In the present work, we presented a thermal- and salt-activated shape memory hydrogel featuring for the gelatin/polyacrylamide double network (gelatin/PAM DN). As depicted in Scheme 1, the gelatin/PAM hydrogel was prepared by free radical polymerization of AM in the presence of gelatin. The reversible configuration of the gelatin under different temperature endowed hydrogel with temperature responsive ability. Salt-activated performance was ascribed to the formation of reversible chain bundle and hydrophobic domains under Hofmeister effect. Thus, we here presented a new concept to fabricate biopolymer-based SMHs, which contributed to the development of smart soft materials.
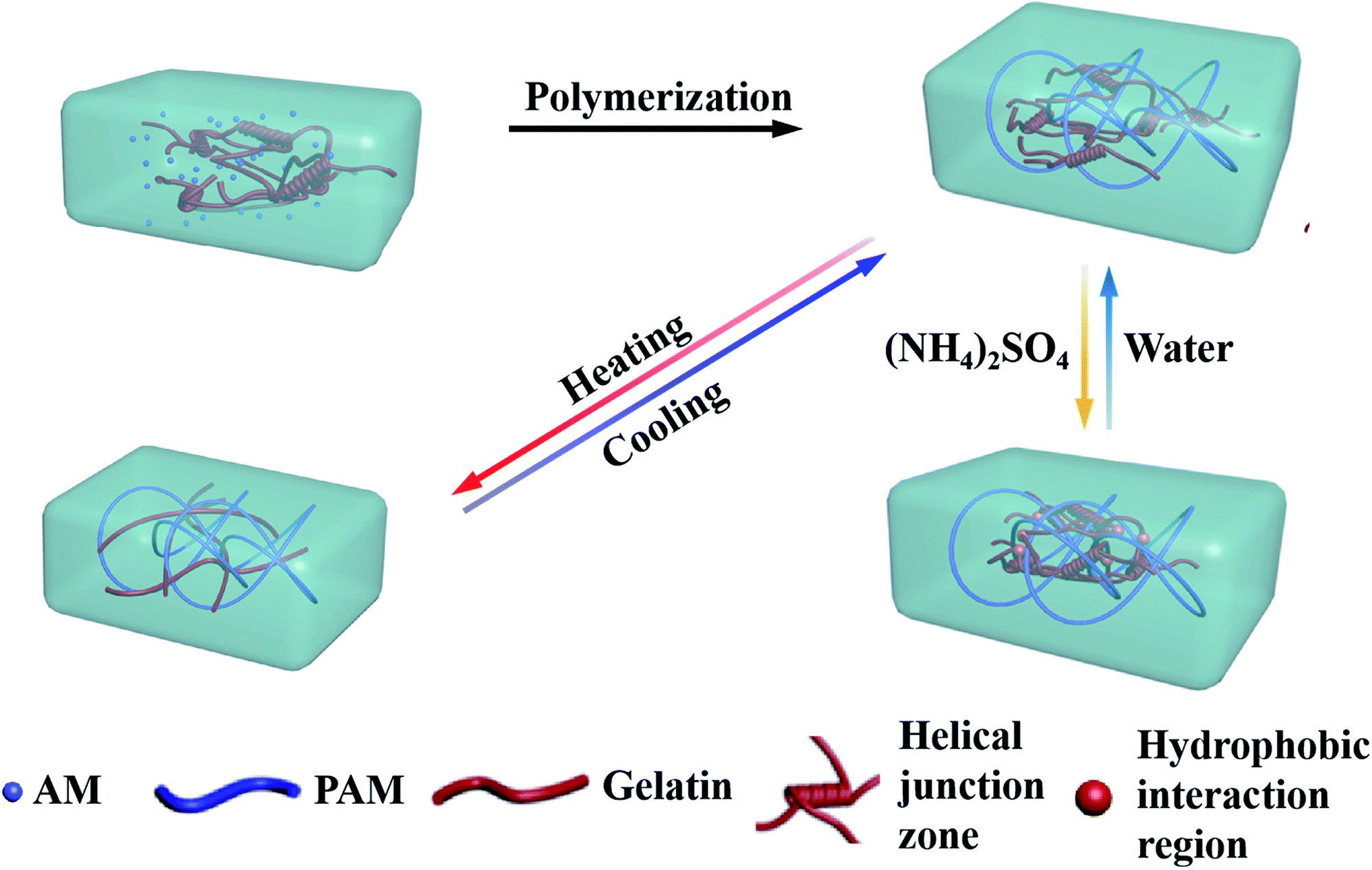 | ||
| Scheme 1 The mechanisms of the shape memory hydrogel. | ||
Experimental
Materials
Gelatin (Porcine skin, Type A, 300 Bloom) was purchased from Sigma-Aldrich Life Science & Technology Co., Ltd. Acrylamide (AM), ammonium sulfate, sodium carbonate, potassium carbonate, dipotassium hydrogenphosphate were obtained from Sinopharm Chemical Reagent Co., Ltd. N,N′-Methylene-bisacrylamide (MBA) and 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (IRGACURE 2959) were purchased from Energy Chemical. All materials were analytical reagents and used as received without further purification. Deionized water was used in all the experiments.Preparation of gelatin/PAM double network hydrogels
The synthesis of hydrogels was briefly described here. First, the suspension of gelatin in deionized water was heated at 50 °C for 30 min to obtain a transparent solution. Then, AM, MBA (chemical cross-linker) and IRGACURE 2959 as a photoinitiator were dissolved in the above solution. After removing all air bubbles by ultrasonication, the homogenous mixture was poured into molds to cool down for the formation of gel at 4 °C. Then it was polymerized under UV irradiation to form an gelatin/PAM DN hydrogel at room temperature for 6 hours. MBA was 0.02 mol% relative to the monomers and IRGACURE 2959 was 1 mol%. In order to investigating the influence of the contents of the gelatin and AM on the mechanical performance of the hydrogel, a series of hydrogels with the different contents of gelatin and AM were prepared. In the experiments, the as-prepared gelatin/PAM hydrogels were designated as GxAMy, in which “x” represents the mass contents of gelatin (g) and “y” represents the molarity of AM (M). Subsequently, the obtained gelatin/PAM hydrogels were soaked in (NH4)2SO4 solutions (the concentration of the solutions are 5, 10, 15, 20, 25, 30, 35, and 40 wt%, respectively) at room temperature for 6 hours to obtain the gelatin/PAM-A hydrogels.Mechanical properties tests
The mechanical properties of hydrogels were all tested on an electronic universal testing machine (EZTest, SHIMADZU) at room temperature. For the tensile and loading–unloading tests, the hydrogel samples were cut into dumbbell shape with length 13 mm, width 2 mm and thickness 1.2 mm. In the loading–unloading tests, in order to prevent water evaporating, all the samples were coated with silicone oil. The crosshead velocity was kept at 100 mm min−1. For the successive loading–unloading tests, the strain rate was fixed at 150 mm min−1. The dissipative energy was calculated from the integral area of the hysteretic circle of the extension-recovery curve.Rheological experiments
The rheological behavior of hydrogels were characterized with a rheometer (TA AR-G2, TA Instruments) equipped with 40 mm parallel plates. A solvent trap was filled with silicone oil to minimize water evaporation. The storage modulus (G′) and the loss modulus (G′′) were measured as a function of angular frequency from 100 to 0.1 rad s−1 at 25 °C. Temperature sweep data were obtained at a fixed frequency (ω = 6.28 rad s−1) and strain (γ = 0.1%) covering a range of 5-60-5 °C (heating and cooling rate: 2 °C min−1).Shape memory evaluation
The shape memory behaviors of the gelatin/PAM hydrogels were evaluated by bending tests at room temperature.30 For the thermal-activated SME, a cylindrical gel sample was immersed in 90 °C water for 20 s and then immersed in ice water for 30 s to fix the temporary shape “U”. The shape recover behavior was further observed by immersing hydrogels in 90 °C water. For the salt-activated SME, a cylindrical gel sample was immersed in (NH4)2SO4 solution for 6 h to fix the temporary shape “U”. The shape recover behavior was further observed by immersing hydrogels in deionized water. The shape fixity ratio (Rf) and shape recovery ratio (Rr) were defined by following equations:
 | (1) |
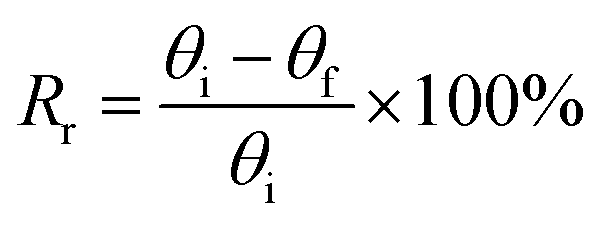 | (2) |
The shape recovery process was recorded using a digital camera. A series of photographs were picked out to demonstrate the transition from the temporary shape to the permanent shape.
Results and discussion
The preparation process for gelatin/PAM DN hydrogels was illustrated in Scheme 1. The physically cross-linking triple helices was formed by hydrogen bonds of gelatin at 4 °C. The chemically cross-linked PAM network was formed through the radical polymerization at a total monomer concentration of 1–3 M via UV light. FT-IR analysis was further carried out to verify gelatin/PAM DN hydrogels. As shown in Fig. 1, after polymerization, the absence of monomers was confirmed by the completely disappearance of the peak at 3100–3000 cm−1 corresponding to the carbon–carbon double bond.24 The gelatin/PAM curve has a broadband around 3410 cm−1 and a characteristic peak at 3210 cm−1 due to the stretching vibration of N–H, which were associated with AM and gelatin. The successful preparation of the gelatin/PAM DN hydrogels was thus confirmed by FTIR spectrum. Upon treatment with (NH4)2SO4, the intensity of the amide I bands (1650 cm−1) increased. A peak at 1400 cm−1 for the C–H bending vibrations and CH3 symmetrical deformation vibrations became a stronger absorption, which implying the occurrence of stronger hydrophobic interactions.28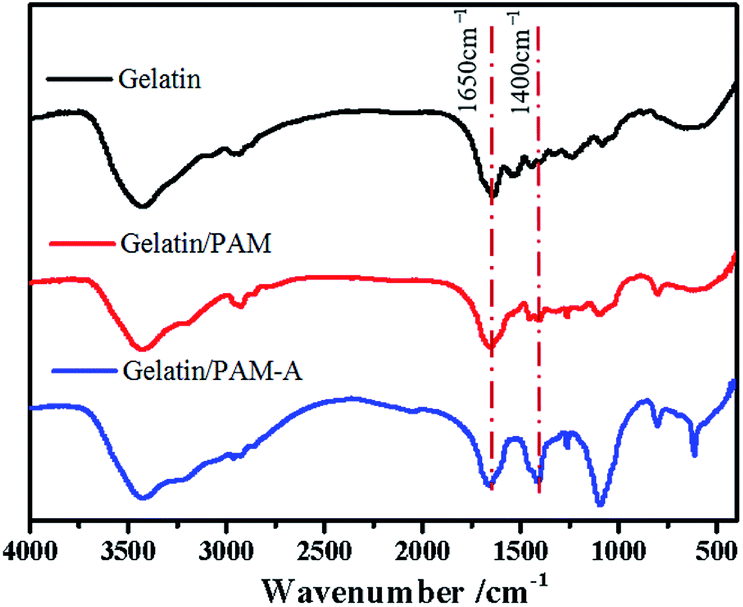 | ||
| Fig. 1 The FTIR spectra of the gelatin, gelatin/PAM hydrogels, and gelatin/PAM-A hydrogels. | ||
Then, the mechanical performance of hydrogels with different gelatin and AM concentrations have been tested at room temperature. From the Fig. 2, when the AM concentration was 1 M, both of the hydrogels (G5AM1 and G10AM1) exhibited poor mechanical properties. For example, under small tensile stress (16.21 kPa) and strain (142.99%), the G5AM1 hydrogel was easily ruptured with a small tensile stress (16.21 kPa) and strain (142.99%), as the gelatin contents increased, the maximum tensile stress (40.83 kPa), and strain (207.92%) increased little. When the concentration of AM was increased to 2 M or 3 M, all of samples show much better mechanical strength. Besides, the mechanical performance also influenced by the gelation concentration. In specifically, when the concentration of AM was set as 2 M, the tensile stress at fracture for the hydrogels was increased from 97.22 to 250.56 kPa and the failure strain was increased from 656.16% to 759.78% as the gelatin content changed from 5 wt% to 10 wt%. However, the elongation property showed opposite concentration dependent behavior. The fracture stress for G5AM3 was 237.02 kPa, while the G10AM3 sample exhibited a fracture stress of 346.08 kPa. Although G10AM3 was stronger than G5AM3, the fracture strain of G10AM3 (499.90%) was much smaller than G5AM3 (1171.15%). In a word, the G5AM3 hydrogel exhibited excellent mechanical property with tensile stress and strain of 237.02 kPa, and 1171.15%, respectively. Therefore, we chose G5AM3 hydrogel as the model sample and subjected to the following experiments.
 | ||
| Fig. 2 Tensile stress–strain curves of the gelatin/PAM hydrogels with different gelatin and acrylamide concentrations. | ||
It has been mentioned31 that gelatin/PAM DN hydrogel can exhibit thermal-activated SME, and we briefly discuss below. Here, triple helices of gelatin were used as the thermal switching segments. Temperature sweep was conducted to investigate the temperature responsive behavior of the gelatin/PAM hydrogels system. Fig. 3a shows the storage modulus (G′) and loss modulus (G′′) of gelatin/PAM hydrogels under fixed frequency (6.28 rad s−1) during cooling–heating cycle. The G′′ was lower than the corresponding G′ in the entire temperature range, suggesting the elastic nature of the gelatin/PAM hydrogels. The G′ value decreased from 26.2 to 9.1 kPa under the temperature range from 5 to 45 °C, while the G′′ remained fundamentally unchanged in the entire range. When temperature exceeded 45 °C, G′ of the gelatin/PAM hydrogel also kept almost a constant in the heating and cooling process. This phenomenon was attributed to the presence of non-temperature responsive chemically crosslinked PAM network, leading to the steady cross-linking density. In essence, the decrease of G′ with the increasing of temperature contributed to the coil-helix transition of the gelatin in hydrogels. Upon cooling, G′ and G′′ went back to the original value due to the aggregation of triple helices, indicating thermoreversibility of the hydrogels. Fig. 3b shows the photos of the thermally triggered SME. A gelatin/PAM hydrogel was deformed into U-shape by cooling to 0 °C, and then recovered to a rod-shape within 5 s after immersing in a water bath at 90 °C. The given angle was 173°, while the final angle was 8°. The thermal-activated SME is attributed to the reversible coil to triple helices transformation of gelatin in response to temperature, and the chemically cross-linked of PAM network function as a fixing phase. Notably, the temporarily fixed shape slightly changed with time even though at freezing temperature (Fig. S1†).
 | ||
| Fig. 3 (a) Dependence of storage modulus (G′) and loss modulus (G′′) on temperature. (b) Images demonstrating the transition from the temporary shape to the permanent shape for the thermal-activated SME. | ||
We then focused on the salt effect on the mechanic behavior of the hydrogel. In order to investigate the effect of ions on the aggregation properties and the performance of gelatin/PAM hydrogels, we treated hydrogels with different ions. Considering that NH4+ and SO42− are strongly kosmotropic ions that has been examined in the Hofmeister series.28 Hence, we chose (NH4)2SO4 to act as the switchable agent. Hydrogels were cut into 10 mm parallel plates and then immersed in (NH4)2SO4 solutions at various concentrations (5, 10, 15, 20, 25, 30, 35, and 40 wt%). The graph in Fig. 5a indicated that hydrogels swelled to some extent at low salt concentration and but shrunk at high salt concentration. Interestingly, sample kept its original volume when the concentration of (NH4)2SO4 was 25 wt%. To gain more insight, we conducted tensile and rheological studies of gelatin/PAM and gelatin/PAM-A samples treated by 25 wt% (NH4)2SO4 solution. The tensile stress–strain and frequency data of the hydrogels were presented in Fig. 4b and c, respectively. Note that elongation at break of the gelatin/PAM hydrogel was 1171.15%, and the tensile stress was 237.02 kPa. After immersing in (NH4)2SO4 solution, the tensile stress and elongation at break were 498.41 kPa and 1071.92%, respectively (Fig. 4b). Obviously, the tensile stress of gelatin/PAM-A was apparently higher than the values of the gelatin/PAM hydrogels. Besides, compared to the original sample, elongation of gelatin/PAM-A hydrogel elongation at break decreased slightly. As expected, after immersing in (NH4)2SO4 solution, the reversible physical interaction assisted by Hofmeister effect in the hydrogel generated. As a result, the mechanical properties of the hydrogels were enhanced. The frequency dependence of the G′ and G′′ of hydrogels at 25 °C were shown in Fig. 4c. It can be seen that G′ and G′′ of the gelatin/PAM-A were higher than the corresponding storage G′ and G′′ of the gelatin/PAM in the entire frequency range (0.1–100 rad s−1). Gelatin/PAM-A showed an obvious enhancement in terms of G′ and G′′ compared to gelatin/PAM. Such changing tendency of the G′ and G′′ was attributed to the formation of the chain bundling and hydrophobic interaction domains which provided the tertiary network within the hydrogels.
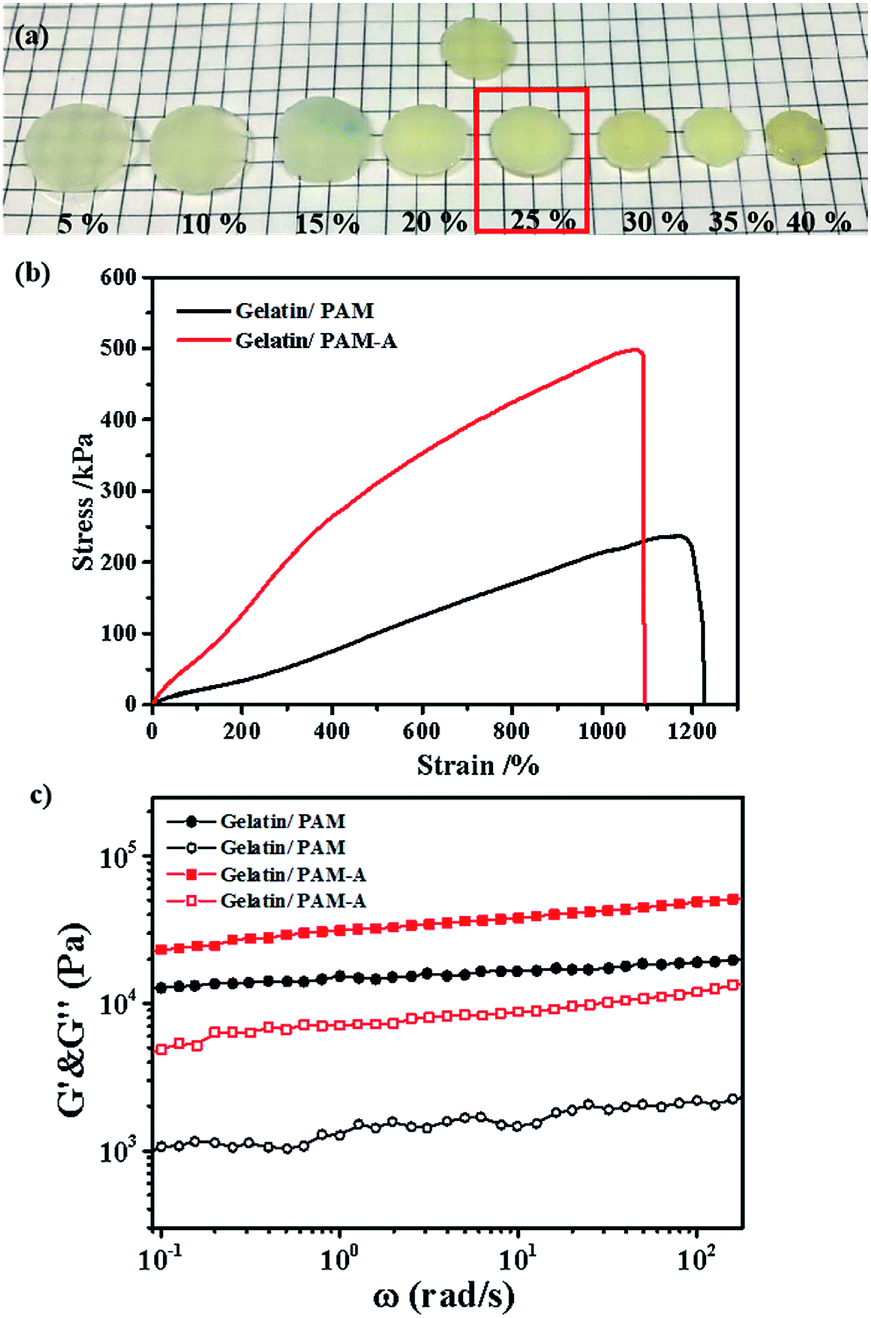 | ||
| Fig. 4 (a) Photographs of the gelatin/PAM hydrogels after immersion in (NH4)2SO4 solutions with various concentrations. (b) Tensile stress–strain curves of the gelatin/PAM and gelatin/PAM-A hydrogels. (c) Frequency sweep data of the gelatin/PAM and gelatin/PAM-A hydrogels in terms of (filled symbols) G′ and (open symbols) G′′. | ||
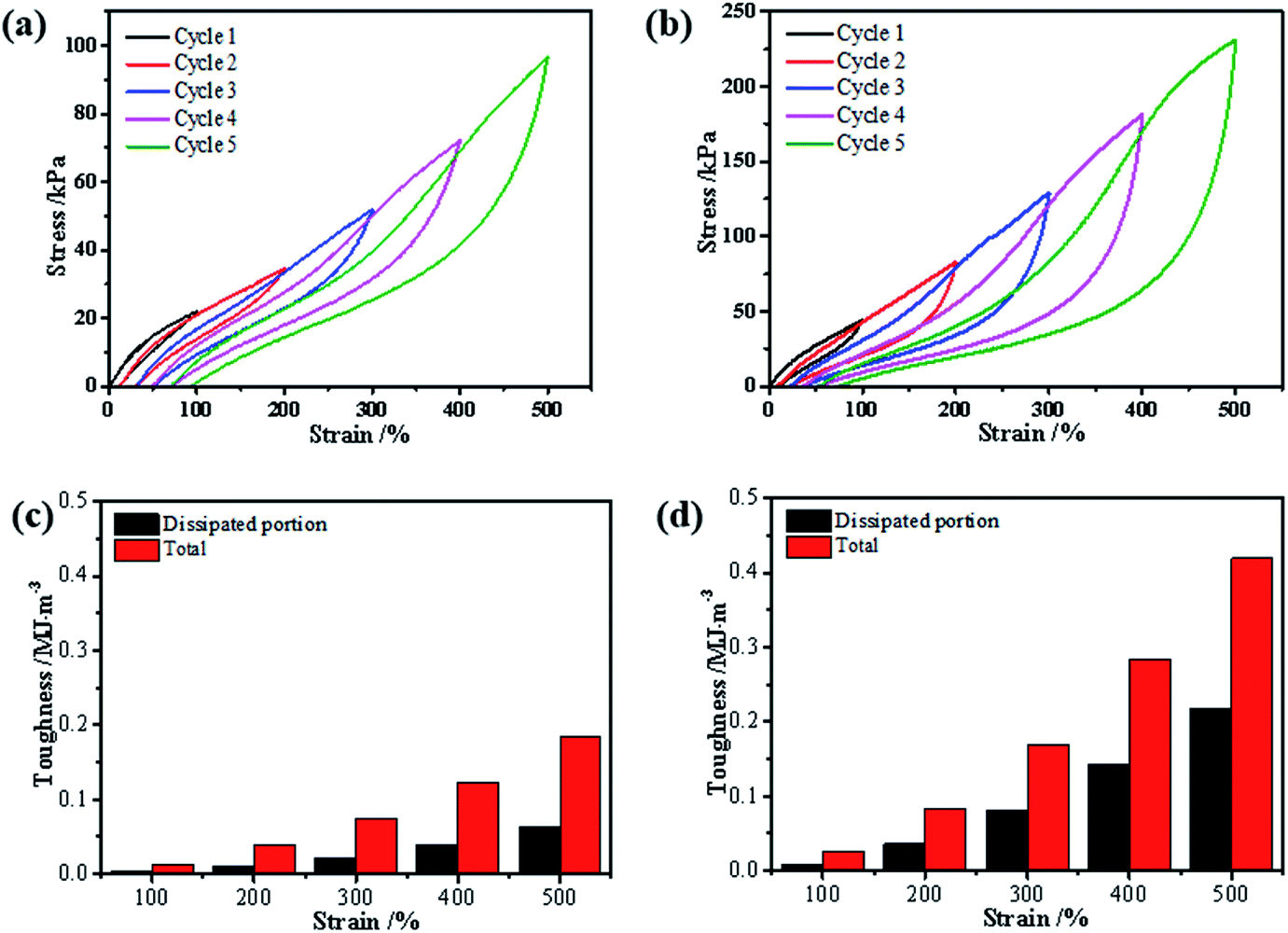 | ||
| Fig. 5 Loading–unloading tests of the (a) gelatin/PAM hydrogels and (b) gelatin/PAM-A hydrogels with varying maximum tension strains; the calculated total and dissipated toughnesses (curve area) of the (c) gelatin/PAM hydrogels and (d) gelatin/PAM-A hydrogels during the loading–unloading cycles with varying maximum tension strains. | ||
In addition, the formation of triple helices and physical interaction was also supported by loading–unloading tests. The loading–unloading cycles of the hydrogel before and after (NH4)2SO4 treatment were tested and the stress–strain curves were displayed. Apparently, all of these hydrogels showed a hysteresis loop in the extension-recovery cycles. For gelatin/PAM, Fig. 5 showed the increase of the dissipated energy from 0.003 MJ m−3 (cycle 1) to 0.06 MJ m−3 (cycle 5). Similarly, it increased from 0.01 MJ m−3 (cycle 1) to 0.22 MJ m−3 (cycle 5) for gelatin/PAM-A. For each cycle, the dissipated energy of gelatin/PAM-A was higher than that of gelatin/PAM. The hysteresis indicated the energy dissipated during the loading–unloading cycles.32 Based on the energy dissipation mechanism, the chemical cross-linking was used to act as a skeleton to maintain structure, while reversible cross-linking served as the sacrificial bonds to dissipate energy.33 The aforementioned results suggested that triple helices in gelatin/PAM functioned as the sacrificial bonds; by comparison, not only triple helices but also physical interaction domains assisted by Hofmeister effect in gelatin/PAM-A served as the sacrificial bonds. Consequently, physical interaction domains formed by soaking with (NH4)2SO4 solutions could enhance the mechanical properties and apply as temporary cross-links.
After confirming the salt enhanced mechanic property, we then studied the salt-activated SMH shape memory phenomenon. The shape fixity ratio (Rf) and shape recovery ratio (Rr) of the hydrogels were obtained by bending experiments performed on U-shaped samples. Here, the Rf described the efficiency of programming and Rr characterized the ability of the material to memorize the permanent shape. As shown in Fig. 6, a straight strip of gelatin/PAM hydrogel was applied with a continuous external force to bend its shape to a U-shape. Then, it was soaked in the 25 wt% (NH4)2SO4 solutions to fix shape with Rf = 94.4%. These physically cross-linking domains assisted by Hofmeister effect could act as temporary cross-links to stabilize the temporary shape. The hydrogel was sequentially placed in deionized water to remove the (NH4)2SO4. It was found that the resultant hydrogel recovered to its initial shape within about 120 min and the Rr reached to 89% (Fig. 6c). As the physical interaction disappeared, the shape gradually recovered to permanent network as a result of extraction of ions with deionized water. In addition, as presented in Fig. S2,† the shape memory behavior of G10AM3 performed the best in fracture stress. G10AM3 sample had a larger modulus and yield stress, which indicated lower fixed ratio (Rf = 89.4%). As show in Fig. S2b,† Rr of G10AM3 hydrogel reached to 85.6% after 150 min. To examine the repeatability of the shape memory capability, the cycles of shape fixing and recovery were repeated for 5 times (Fig. 6d). The cyclicity was evaluated by measuring Rf and Rr quantitatively for each cycle.34 The hydrogel almost remained the same Rf (∼94%) and could recover to its original shape after five cycles. During each cycle, the Rf and Rf were similar to each other. The reversible physical interaction domains endowed the hydrogel with good reproducible shape memory ability. As a result, the gelatin/PAM hydrogels exhibited excellent shape memory behaviors.
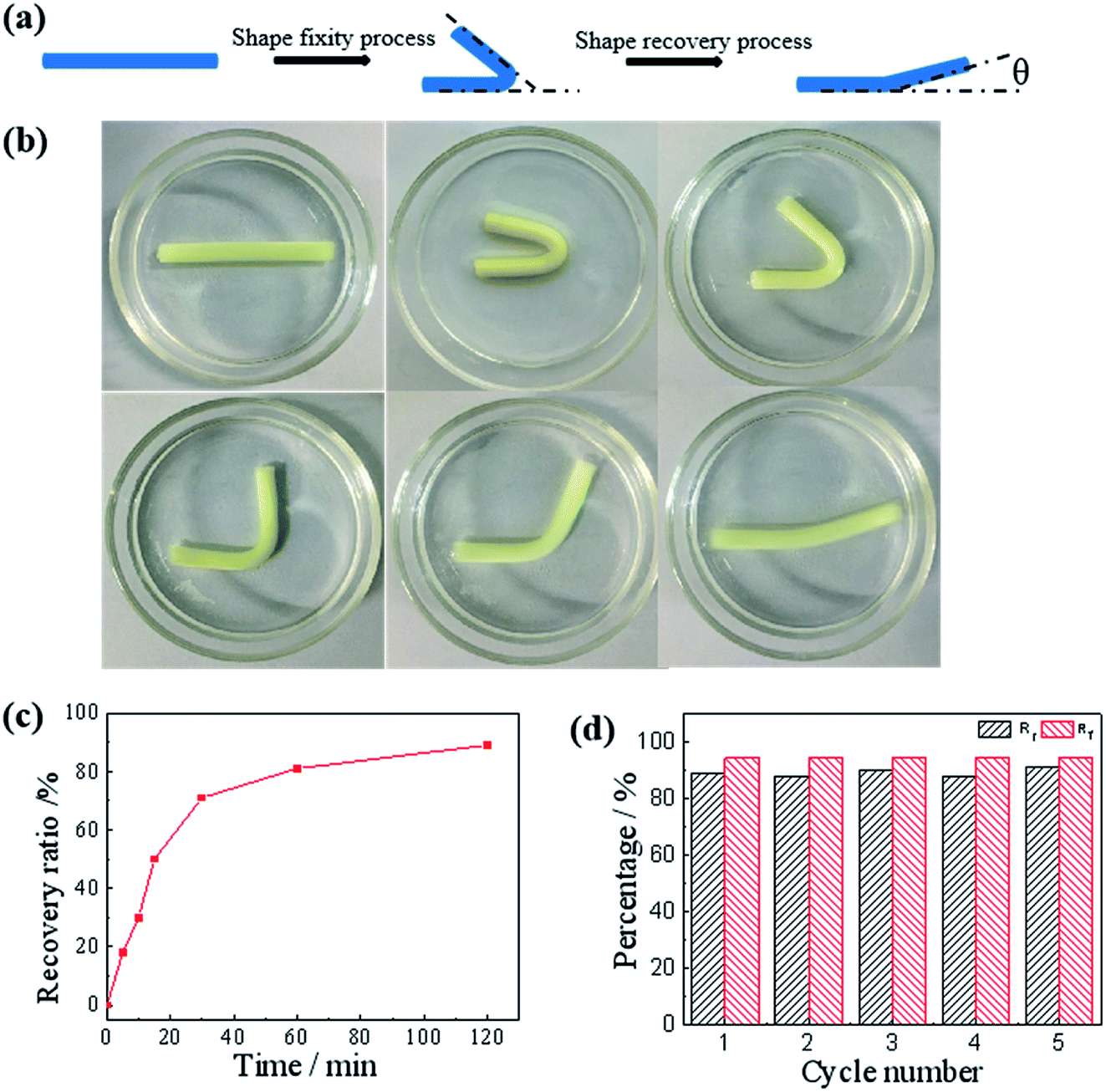 | ||
| Fig. 6 (a) Schematic illustrations of the shape memory effect. (b) Images demonstrating the transition from the temporary shape to the permanent shape for the salt-activated SME. (c) Rr for the gelatin/PAM hydrogels shown as a function of time at room temperature. (d) Evaluation of the gelatin/PAM hydrogels shape memory cycles at room temperature. | ||
Moreover, considering ions in Hofmeister series would have different effect on aggregation properties, we hypothesized that various kosmotropes ions could also induce SME. The typical order of the anion series is: CO32− > SO42− > S2O32− > H2PO4− > F− > CH3COO− > Cl− > Br− > NO3− > I− > ClO4− > SCN−.35 The ions on the left are referred as kosmotropes, and those on the right side are called chaotropes. As shown in Fig. S3 and S4,† strong kosmotropic ions induced SME with high fracture stress and Rf while chaotropes ions weaken the fracture stress and couldn't fix temporary shape. Anions were reported to have more pronounced effect than cations.28 As show in Fig. S5,† K2CO3 and Na2CO3 exhibited similar influence on fracture stress or Rf (92.2% and 93.3%, respectively). The recovering process was achieved by immersing hydrogels into deionized water by extracting ions from hydrogels, leading to the higher Rf ∼90% (Fig. S4b and S5b†). The outstanding shape fixed capacities of hydrogels make it possible to deform complicated temporary shapes like spiral and helix. We selected three kinds of salts containing strongly kosmotropes ions for inducing SME. As can be seen from Fig. 7, three straight gelatin/PAM hydrogels were deformed to spiral, wavy and helix shapes. The resulted shapes were fixed by soaking with Na2CO3, K2CO3, and K2HPO4 solutions, respectively. When the hydrogels immersed into deionized water, the temporary shapes could recover to their original shapes. These results suggested that gelatin/PAM hydrogels can be easily fixed to any desirable form by immersing into saline solution, which would extend extending the great potential application range.
 | ||
| Fig. 7 Photos showing various (a) initial shape, (b) fixed shape and (c) permanent shape at room temperature treated by Na2CO3, K2CO3, K2HPO4 solution. | ||
Conclusions
In summary, a type of gelatin/PAM DN hydrogel was successfully synthesized through polymerization of AM in the presence of gelatin. We demonstrate the thermal and salt-activated shape memory effects in such biopolymer based hydrogels for the first time. The PAM network serves as a permanent network. The gelatin not only endows gelatin/PAM DN hydrogel with temperature-responsiveness but also brings the hydrogel with salt-responsiveness. Thus, we believe that the facile strategy could provide new opportunities with regard to the practical application. In addition, the presented strategy could broaden the list of SMHs and apply to other collagen systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science and Technology Major Project of the Ministry of Science and Technology of China (2016ZX05016), the National Science and Technology Major Project of the Ministry of Science and Technology of China (2016ZX05046), and the National Natural Science Foundation of China (Grant no. 51273189).Notes and references
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- X. Le, W. Lu, J. Zhang and T. Chen, Adv. Sci., 2019, 6, 1801584 CrossRef PubMed.
- N. Annabi, A. Tamayol, J. A. Uquillas, M. Akbari, L. E. Bertassoni, C. Cha, G. Camci-Unal, M. R. Dokmeci, N. A. Peppas and A. Khademhosseini, Adv. Mater., 2014, 26, 85–124 CrossRef CAS PubMed.
- J. Hao and R. A. Weiss, ACS Macro Lett., 2013, 2, 86–89 CrossRef CAS.
- W. Guo, C. H. Lu, R. Orbach, F. Wang, X. J. Qi, A. Cecconello, D. Seliktar and I. Willner, Adv. Mater., 2015, 27, 73–78 CrossRef CAS PubMed.
- W. Feng, W. Zhou, S. Zhang, Y. Fan, A. Yasin and H. Yang, RSC Adv., 2015, 5, 81784–81789 RSC.
- T. Zhao, M. Tan, Y. Cui, C. Deng, H. Huang and M. Guo, Polym. Chem., 2014, 5, 4965 RSC.
- G. Li, Q. Yan, H. Xia and Y. Zhao, ACS Appl. Mater. Interfaces, 2015, 7, 12067–12073 CrossRef CAS PubMed.
- Y. N. Chen, L. Peng, T. Liu, Y. Wang, S. Shi and H. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 27199–27206 CrossRef CAS PubMed.
- C. Lowenberg, M. Balk, C. Wischke, M. Behl and A. Lendlein, Acc. Chem. Res., 2017, 50, 723–732 CrossRef PubMed.
- H. Meng, J. Zheng, X. Wen, Z. Cai, J. Zhang and T. Chen, Macromol. Rapid Commun., 2015, 36, 533–537 CrossRef CAS PubMed.
- C. Bilici and O. Okay, Macromolecules, 2013, 46, 3125–3131 CrossRef CAS.
- N. Li, G. Chen, W. Chen, J. Huang, J. Tian, X. Wan, M. He and H. Zhang, Carbohydr. Polym., 2017, 178, 159–165 CrossRef CAS PubMed.
- B. Xu, Y. Zhang and W. Liu, Macromol. Rapid Commun., 2015, 36, 1585–1591 CrossRef CAS PubMed.
- P. J. Skrzeszewska, L. N. Jong, F. A. de Wolf, M. A. Cohen Stuart and J. van der Gucht, Biomacromolecules, 2011, 12, 2285–2292 CrossRef CAS PubMed.
- K. Miyamae, M. Nakahata, Y. Takashima and A. Harada, Angew. Chem., Int. Ed., 2015, 54, 8984–8987 CrossRef CAS PubMed.
- W. Lu, X. Le, J. Zhang, Y. Huang and T. Chen, Chem. Soc. Rev., 2017, 46, 1284–1294 RSC.
- B. Gyarmati, B. Á. Szilágyi and A. Szilágyi, Eur. Polym. J., 2017, 93, 642–669 CrossRef CAS.
- C. Bilici, V. Can, U. Nöchel, M. Behl, A. Lendlein and O. Okay, Macromolecules, 2016, 49, 7442–7449 CrossRef CAS.
- Z. X. Zhang, K. L. Liu and J. Li, Angew. Chem., Int. Ed., 2013, 52, 6180–6184 CrossRef CAS PubMed.
- X. Li, S. Xu, Y. Wang, Z.-Y. Hu and R. Wang, Colloid Polym. Sci., 2019, 297, 503–512 CrossRef CAS.
- H. Xiao, C. Ma, X. Le, L. Wang, W. Lu, P. Theato, T. Hu, J. Zhang and T. Chen, Polymers, 2017, 9, 138 CrossRef PubMed.
- X. Le, W. Lu, J. Zheng, D. Tong, N. Zhao, C. Ma, H. Xiao, J. Zhang, Y. Huang and T. Chen, Chem. Sci., 2016, 7, 6715–6720 RSC.
- K. Peng, K. Yang, Y. Fan, A. Yasin, X. Hao and H. Yang, Macromol. Chem. Phys., 2017, 218, 1700170 CrossRef.
- L. J. Del Valle, A. Diaz and J. Puiggali, Gels, 2017, 3, 27 CrossRef PubMed.
- K. Ullah, S. Ali Khan, G. Murtaza, M. Sohail, Azizullah, A. Manan and A. Afzal, Int. J. Pharm., 2019, 556, 236–245 CrossRef CAS PubMed.
- J. Huang, L. Zhao, T. Wang, W. Sun and Z. Tong, ACS Appl. Mater. Interfaces, 2016, 8, 12384–12392 CrossRef CAS PubMed.
- Q. He, Y. Huang and S. Wang, Adv. Funct. Mater., 2018, 28, 1705069 CrossRef.
- X. Chen, S. C. Flores, S. M. Lim, Y. Zhang, T. Yang, J. Kherb and P. S. Cremer, Langmuir, 2010, 26, 16447–16454 CrossRef CAS PubMed.
- W. Nan, W. Wang, H. Gao and W. Liu, Soft Matter, 2013, 9, 132–137 RSC.
- N. Li, W. Chen, G. Chen and J. Tian, Carbohydr. Polym., 2017, 171, 77–84 CrossRef CAS PubMed.
- Z. Wang, W. Li, X. Yang, J. Cao, Y. Tu, R. Wu and W. Wang, Mater. Today Commun., 2018, 17, 246–251 CrossRef CAS.
- T. Nakajima, H. Sato, Y. Zhao, S. Kawahara, T. Kurokawa, K. Sugahara and J. P. Gong, Adv. Funct. Mater., 2012, 22, 4426–4432 CrossRef CAS.
- Y. Fan, W. Zhou, A. Yasin, H. Li and H. Yang, Soft Matter, 2015, 11, 4218–4225 RSC.
- Y. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02842k |
| This journal is © The Royal Society of Chemistry 2019 |