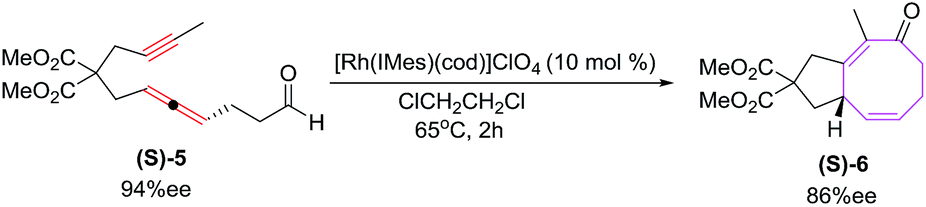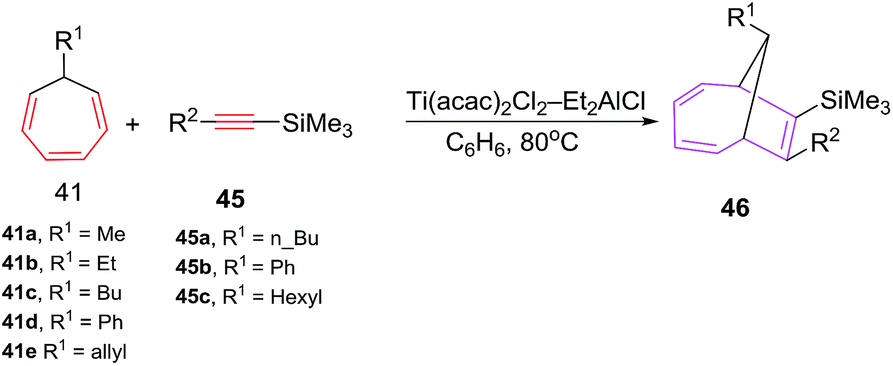DOI:
10.1039/C9RA02839K
(Review Article)
RSC Adv., 2019,
9, 25554-25568
Transition metal catalyzed [6 + 2] cycloadditions
Received
15th April 2019
, Accepted 2nd August 2019
First published on 15th August 2019
Abstract
The [6 + 2] cycloaddition reactions are one of the important synthetic transformations to construct eight membered carbo-/heterocyclic systems. The present review is an attempt to update readers on transition metal catalyzed [6 + 2] cycloaddition reactions of various 6π-contributing substrates such as cycloheptatrienes (CHT), cyclooctatetrenes (COTT), allenals, vinylcyclobutanones, fulvene etc. employing rhodium, cobalt, titanium, copper, platinum, ruthenium, rhenium and diphenylprolinolsilyl ethers etc. as catalysts. The transition metal catalyzed [6 + 2] cycloaddition reactions with a variety of functionalized substrates provide straightforward access to eight membered cyclic and/or 5/8, 6/8 etc. condensed carbo-/heterocyclic molecules in moderate to good yields.
Introduction
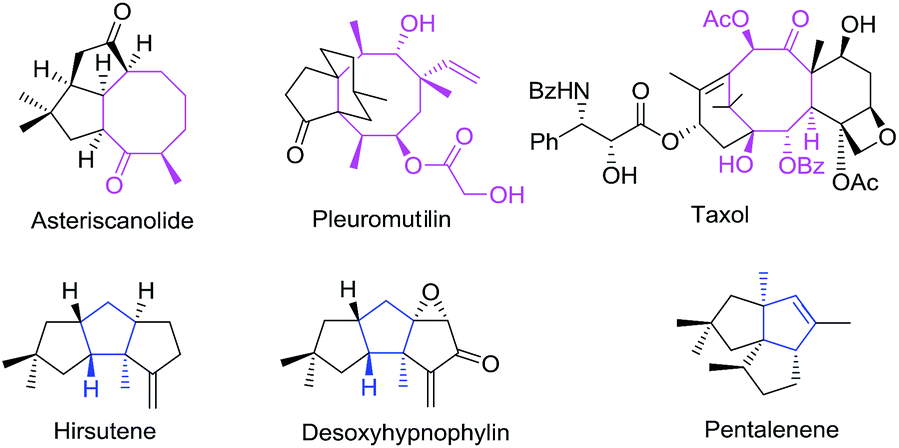
Cycloaddition reactions are considered to be one of the most powerful methods for the construction of cyclic skeletons1 with atom economy. There are ample reports on different variants of [4 + 2] and [3 + 2] cycloadditions which provide an easy access to a variety of five or six membered carbo-/heterocycles.2,3 However, the parallel methodologies such as [5 + 2],4 [6 + 2],5 [8 + 2]5b etc. for the synthesis of seven, eight or ten membered carbo-/heterocycles are still uncommon in the literature. Eight-membered rings are found in a wide variety of natural products and are useful ring systems available in variety of drugs, drug leads, or biological probes.6 Taxol, the well-known natural product in this family, is now among the most potent anticancer drug in clinical use. In 2008, another member of this family, pleuromutilin was approved for use as an antibiotic by Food and Drug Administration (FDA)7 (Fig. 1). Several methods8–12 including ring-closing metathesis and Cope rearrangement have been reported for the synthesis of eight-membered carbocycles.13 However, the intolerance to substrate substituent in conventional procedures along with complexities observed in natural products makes these techniques ineffective and instigated the development of alternate protocols. Recent years have seen a significant upsurge in this direction with relatively few reports appearing on the synthesis of eight membered carbocycles using [6 + 2]5,14 and [4 + 2 + 2]11 cycloaddition reactions. Concerted [6 + 2] cycloaddition reactions are forbidden thermally as per Woodward–Hoffmann rules, when the two reacting π-systems add in a suprafacial fashion. However, transition metal-catalyzed [6 + 2] cycloadditions constitute one such protocol for the synthesis of monocyclic or condensed functionalized cyclooctadienes. The present review summarizes various reports on the transition metal catalyzed inter- and intra-molecular [6 + 2] cycloaddition reactions appeared in literature since 2000.14
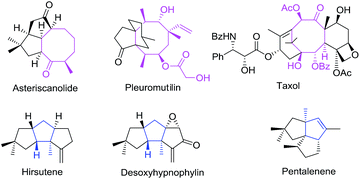 |
| | Fig. 1 Carbocycles in natural products. | |
Rhodium catalyzed [6 + 2] cycloadditions
Rhodium complexes plays a pivotal role in organic synthesis as major catalytic contributors to C–C bond formation reactions.15 A number of carbo-/heterocyclic systems have been generated employing rhodium complexes as efficient catalyst in a variety of cycloaddition/cyclization reaction16 Literature rationale suggests an inspiring advancement in this field and many research groups are extensively using rhodium complexes to efficiently conceive these reactions.17 Considering the importance of rhodium catalysed cycloadditions, there should have been a number of reports on generation of eight membered carbo-/heterocycles engaging rhodium complexes in [6 + 2] cycloadditions, however only a few reports are available which successfully comprehends [6 + 2] cycloadditions. Till now, different substrates such as cycloheptatriene, 2-vinylcyclobutanones, allenals, allenylcyclobutanes, cyclooctatrienes etc. have been reported as 6π-components in Rh(I) catalysed [6 + 2] cycloadditions involving variety of alkene alkyne, allene as 2π-components to afford monocyclic/condensed eight membered carbocycles/heterocycles.
Rh(I) catalysed [6 + 2] cycloadditions of 2-vinylcyclobutanones
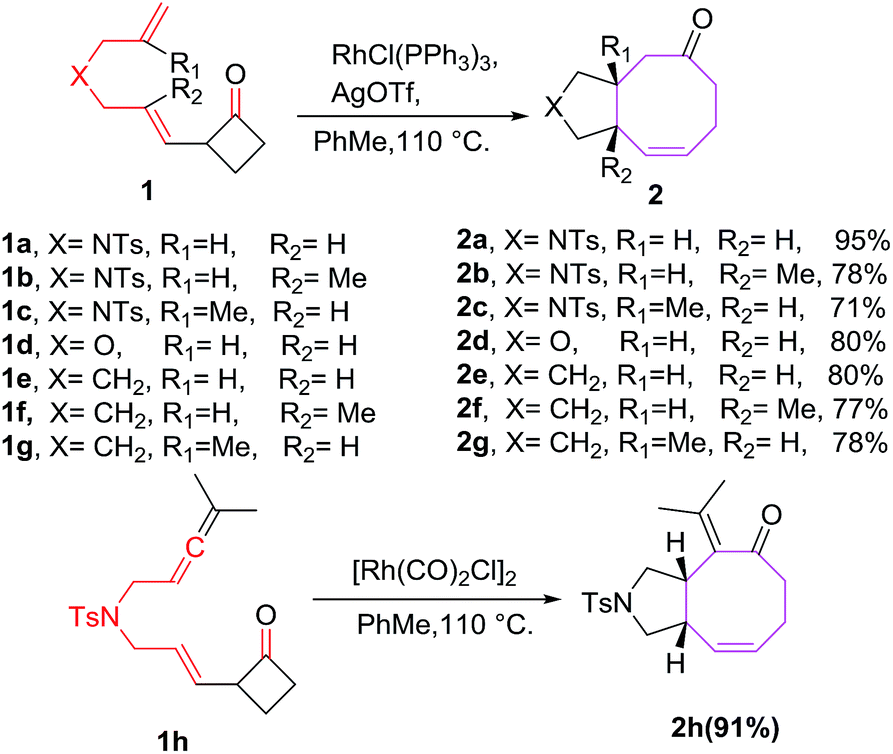
One of the earliest reports on Rh(I) catalyzed [6 + 2] cycloadditions of 2-vinylcyclobutanones as 6π-components is reported by Wender et al. It involves intramolecular [6 + 2] reactions of 2-vinylcyclobutanones 1a–h with tethered alkenes for the construction of eight-membered-ring systems 2a–h.18 The [6 + 2] reactions has diverse substrate compatibility and proceed for a variety of different tethers to produce cis-fused condensed cyclooctatrienes 2a–h in excellent yields (Scheme 1).
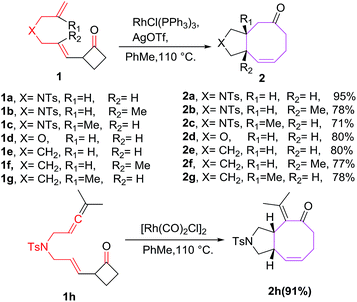 |
| | Scheme 1 Rh(I) catalysed [6 + 2] cycloaddition reactions of tethered alkenes with cyclobutanones. | |
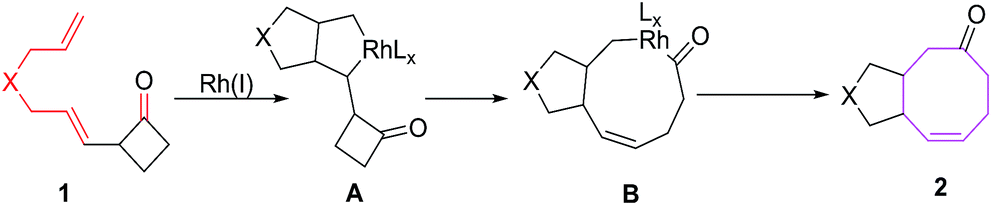
The [6 + 2] cycloadditions involve initial formation of a five membered metallacycle A, which undergoes ring expansion to nine-membered metallacycle B, demetallation of B affords eight-membered condensed carbocycles via reductive eliminations (Fig. 2).
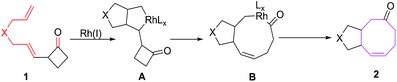 |
| | Fig. 2 Plausible mechanism for synthesis of eight-membered condensed carbocycles. | |
Rh(I)-catalyzed formal [6 + 2] cycloadditions of allenal
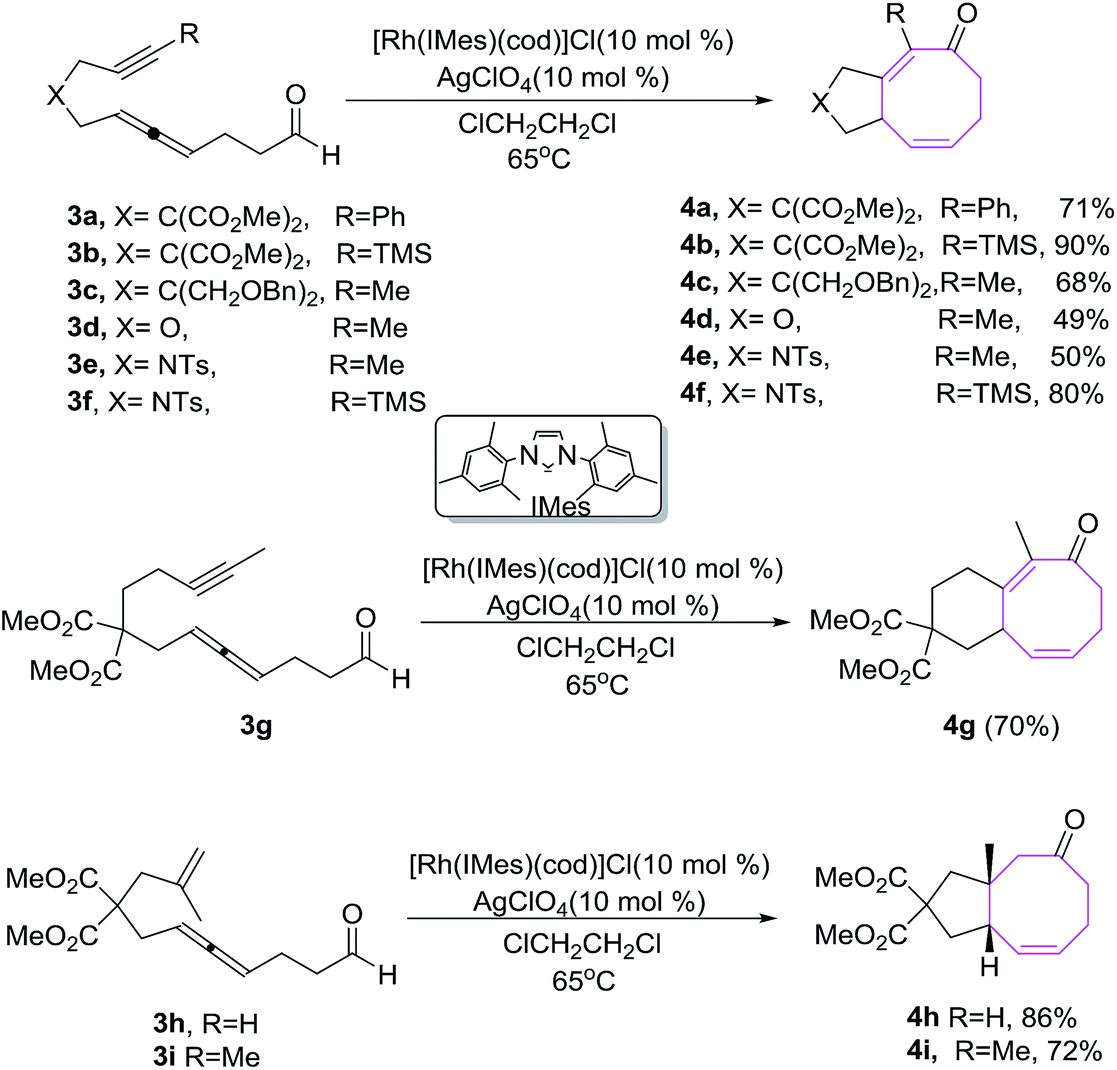
Sato et al. have developed an efficient methodology for the synthesis of fused bicyclic ketones 4. It involves the Rh(I)-catalyzed intramolecular [6 + 2] cycloadditions of alkyne/alkene tethered allenal 3a–j using Rh(I)-complex as catalyst. [Rh(IMes)(cod)]ClO4 is an effective catalyst in these [6 + 2] cycloadditions.19 The use of other Rh(I) complexes as catalyst such as [Rh(IiPr)(cod)]ClO4, [Rh(IPr)(cod)]ClO4 and [Rh(IMes)(cod)]ClO4 afford bicyclic ketones in lower yields and [Rh(dppe)]ClO4 is completely ineffective in conceiving [6 + 2] reactions. The [6 + 2] cycloadditions using RhCl(PPh3)3 afford bicyclic ketone 4 in relatively good yield (72%) but with longer time. The cyclizations of 3a and 3b having phenyl and TMS groups on the alkyne moiety provide 4a and 4b in 71% and 90% yields respectively. Substrate 3d–f containing a heteroatom in the chain afforded corresponding heterocycles 4d–f in good to excellent yields. Elongation of the tether between the alkyne and allene moieties by one carbon is well tolerated in the [6 + 2] cycloaddition reaction and afford 6/8 fused bicyclic ketone 4g in 70% yield as evident by the conversion of 3g to 4g. The cyclizations of substrates 3h–i having an tethered alkene also afford bicyclic ketones 4h–i in a stereoselectively (Scheme 2).
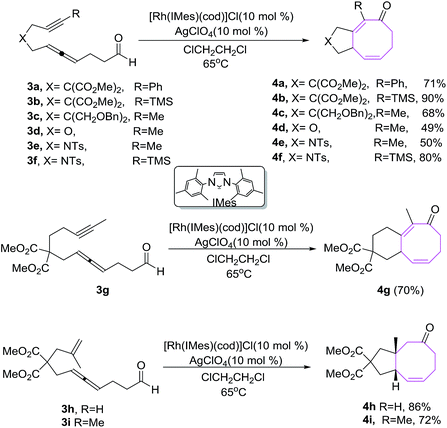 |
| | Scheme 2 Rh(I)-catalyzed [6 + 2] cycloaddition of alkene tethered allenal. | |
The [6 + 2] cycloaddition of enantiomerically enriched (S)-5 (94% ee) yields cyclic ketone (S)-6 in high yields with reasonable transfer of chirality (86% ee) as shown in Scheme 3.19
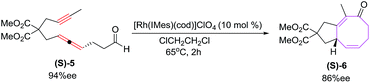 |
| | Scheme 3 Rh(I)-catalyzed [6 + 2] cycloaddition of alkyne tethered allenal. | |
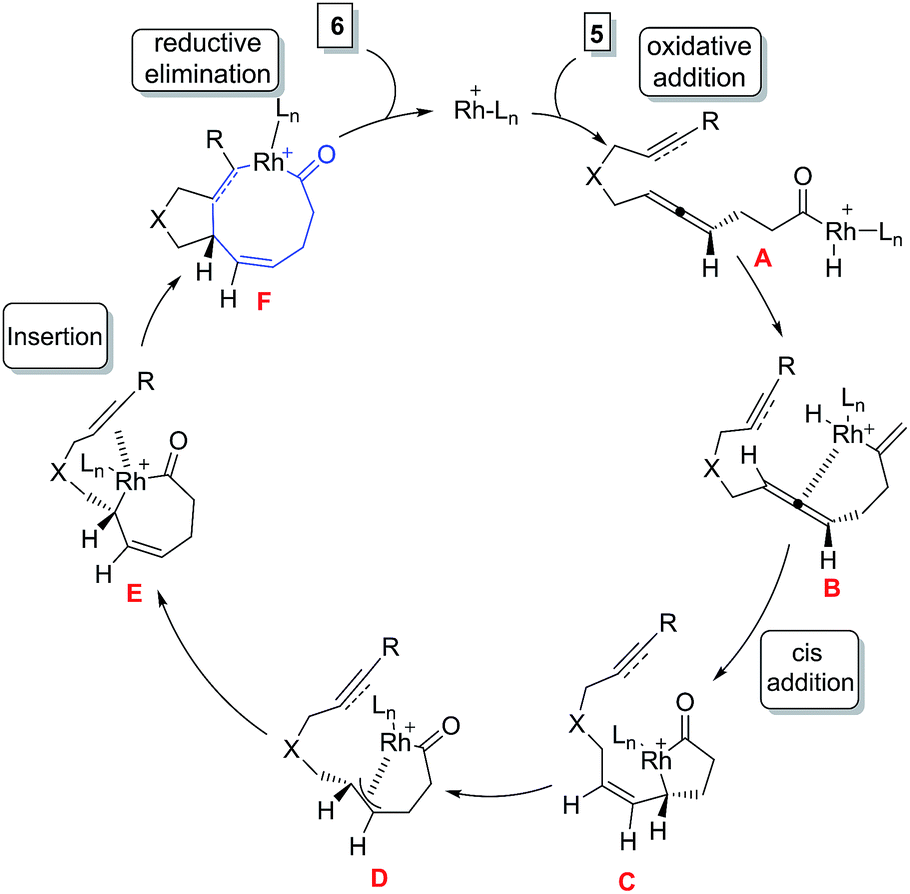
The possible mechanism for the formation of 6 involves the oxidative addition of Rh(I) complex to the C–H bond of the aldehyde 5 followed by insertion of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the allene to give oxo-rhodacycle C which undergoes isomerization to oxo-rhodacycle C via π-allylrhodium intermediate F. Insertion of an alkyne or alkene into the Rh–C bond followed by reductive elimination yields bicyclic ketone 4/6 (Fig. 3).
C bond of the allene to give oxo-rhodacycle C which undergoes isomerization to oxo-rhodacycle C via π-allylrhodium intermediate F. Insertion of an alkyne or alkene into the Rh–C bond followed by reductive elimination yields bicyclic ketone 4/6 (Fig. 3).
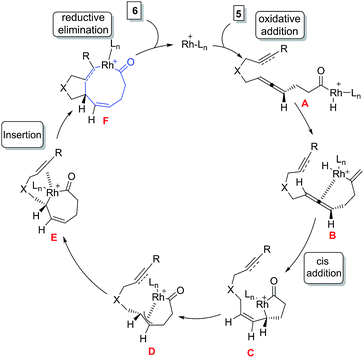 |
| | Fig. 3 Proposed mechanism for rhodium catalysed [6 + 2] cycloaddition reactions of allenals. | |
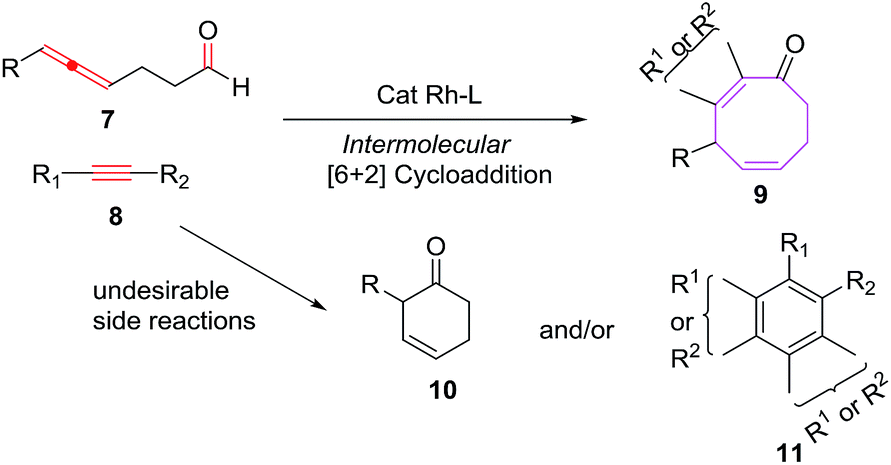
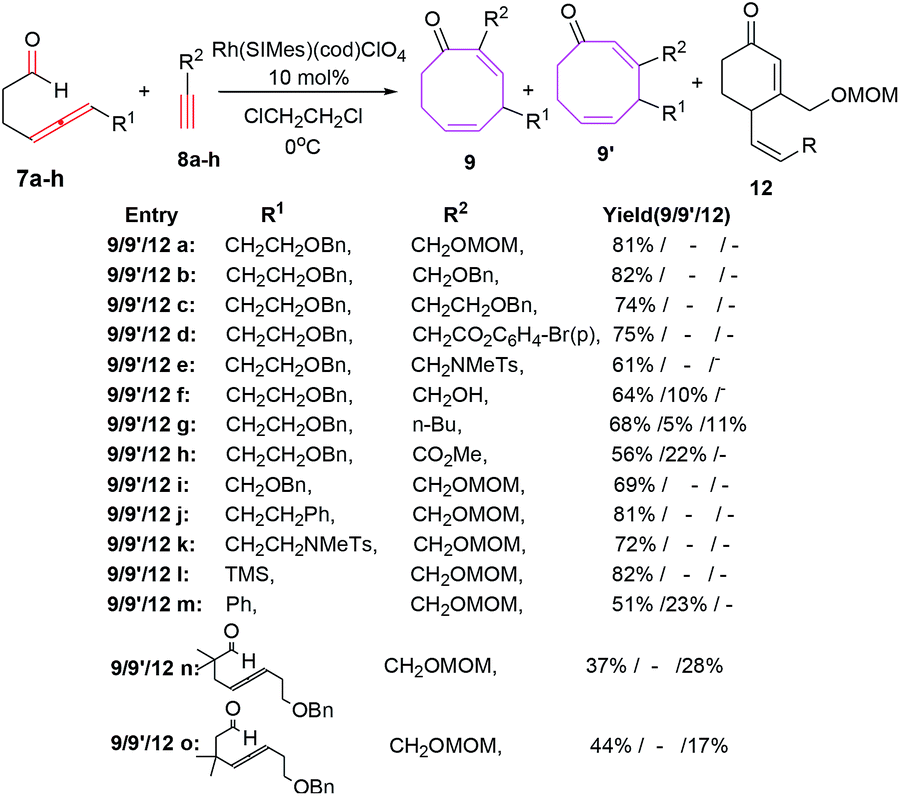
Sato et al. have also explored the intermolecular [6 + 2] cycloadditions of 4-allenal 7 and alkyne 8, for the synthesis of functionalised monocyclic cyclooctane derivatives 9.20 The intermolecular [6 + 2] cycloadditions suffer from unfavorable entropy disadvantage which resulted in decreased yield of the [6 + 2] cycloadducts with the isolation of side products 10 through hydroacylation of allenal 710c and formation of 11 by trimerization of alkyne 8 (Scheme 4).
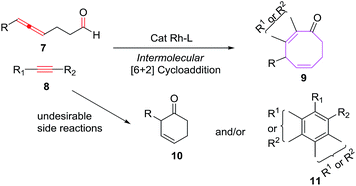 |
| | Scheme 4 Intermolecular [6 + 2] cycloadditions. | |
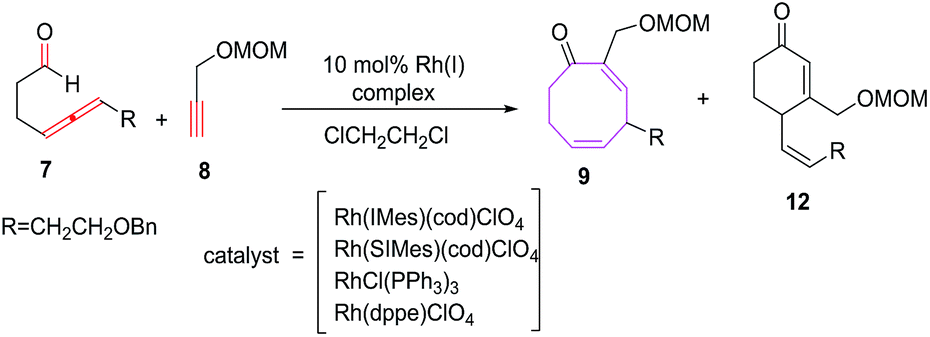
The [Rh(SIMes)(cod)]ClO4 have been found to be efficient in conceiving intermolecular reactions with the selective formation of eight membered carbocycle 9 (68%). The [Rh(IMes)(cod)]ClO4 as catalyst is non selective and afford mixture of eight membered carbocycle 9 (61%) and six-membered carbocycle 12 (19%). Other tested catalysts such as [RhCl(PPh3)3] and [Rh(dppe)]ClO4 do not promote the intermolecular [6 + 2] cycloaddition reactions (Scheme 5).
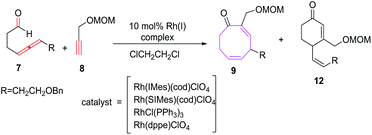 |
| | Scheme 5 Intermolecular [6 + 2] cycloadditions using various Rh(I) complexes. | |
The intermolecular [6 + 2] cycloadditions of 7a with various terminal alkynes possessing benzyloxy 8b–c, benzoate 8d and sulphonamide moiety 8e have also been reported to proceed in a stereoselective manner affording eight membered carbocycles 9b–e in high yields. The propargyl tethered alkyne moiety 8f, however, provide regiomeric cyclooctadienone 9f and 9f′ in yields of 64% and 10% respectively. Employing 1-hexyne have yielded a mixture of isomeric eight-membered rings 9g/9g′ in 68% and 5% yields along with six-membered carbocycles as side product 12g (11%). Electron-withdrawing carboxylate functionality on alkyne nucleus 8h have led to the regiomeric cyclooctadienones 9h/9h′ in comparatively lower yields (56% & 22% respectively). 4-Allenal substituted with benzyloxy 7a–b, benzyl 7c, sulphonamide 7d and TMS 7e afford regioselective octa cyclic compounds 9a, i–l in yields of 69%, 81%, 72% and 82% respectively. Phenyl substitution on allene however, retards the reaction rate and regioselectivity resulting in isolation of 9m/9m′ in 51% and 23% respectively.
The allenal 7g–h possessing alkyl substitution between allene and aldehyde have resulted in increased formation of hydroacylation product 12n–o in 28% and 17% yields along with intramolecular [6 + 2] cycloadducts 9n–o in 37% and 44% yields respectively (Scheme 6).
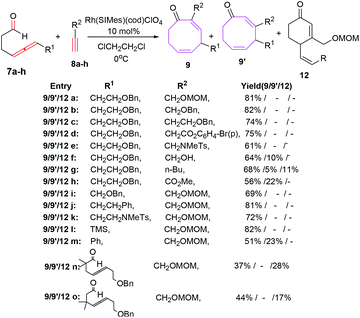 |
| | Scheme 6 Intermolecular [6 + 2] cycloadditions of allenals and alkynes. | |
Rh(I) catalysed [6 + 2] cycloaddition reactions of allenylcyclobutanes
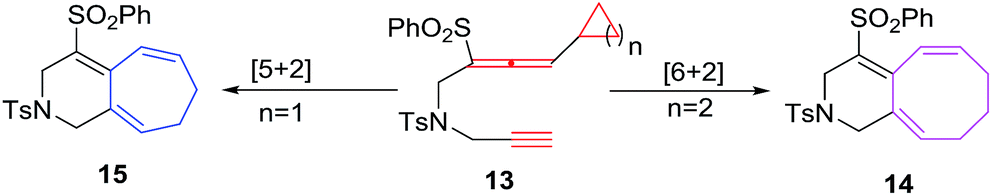
Mukai et al. have devised a cycloisomerisation methodology using rhodium(I) catalysed alkyne-allenyl unfunctionalised cyclopropane/butane 13 to afford bicyclic and compounds 15 and 14 through [5 + 2]21 and [6 + 2]22 cycloaddition respectively. The un-functionalized simple cyclobutane ring in [6 + 2] cycloadditions behaves as 6π-component and afford an easy construction of the bicyclo[6.3.0]undecatrienes 14 in good yields (Scheme 7).
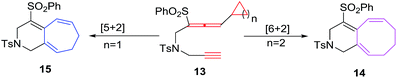 |
| | Scheme 7 Rh(I) catalysed [6 + 2] reactions of unfunctionalised alkyne-allenyl cyclopropane/butane. | |
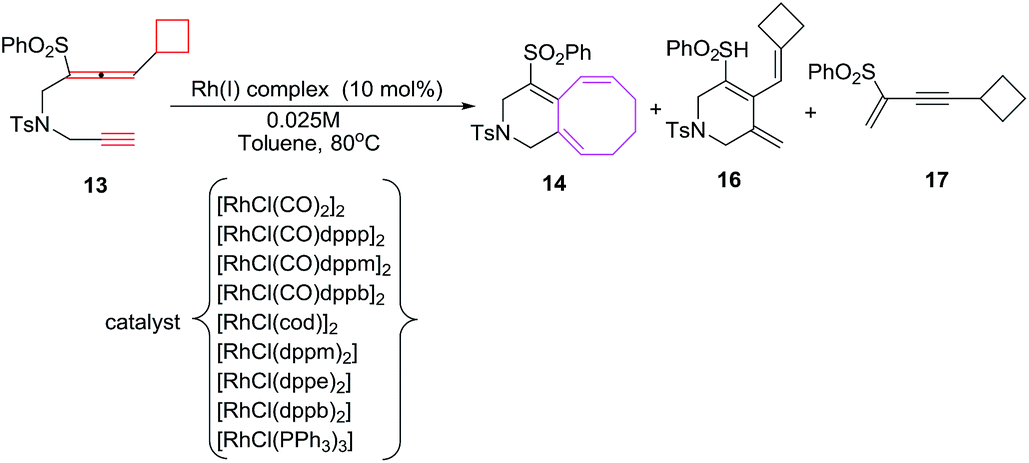
The [6 + 2] cycloaddition reactions are conceived by heating a solution of 13 in 1,2-dichloroethane at 80 °C in the presence of 10 mol% [RhCl(CO)2] as catalyst to afford the desired bicyclo[6.4.0]dodecatriene 14 in 40% yield along with monocyclic side product 16 (52%). Replacement of catalyst to [RhCl(CO)dppp]2 in toluene have witnessed a marked increase in yield of 6/8 bicyclic product 14 (80%). Other rhodium carbonyl catalysts are less effective in conceiving these [6 + 2] cycloadditions. Best results for [6 + 2] cycloaddition reactions are observed employing [RhCl(dppe)2] as catalyst without carbon monoxide as ligand. A decrease in catalyst loading to 5 mol% have observed a lowering in reaction yield of 14, however, using 5 mol% [RhCl(dppe)2] in 0.1 M dioxane solvent at 80 °C have led to the formation of bicyclo[6.4.0]dodecatrienes 14 (87%) (Scheme 8).
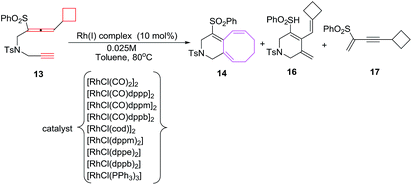 |
| | Scheme 8 Cycloadditions of alkyne-allenyl cyclobutane 13 in different Rh(I) complexes. | |
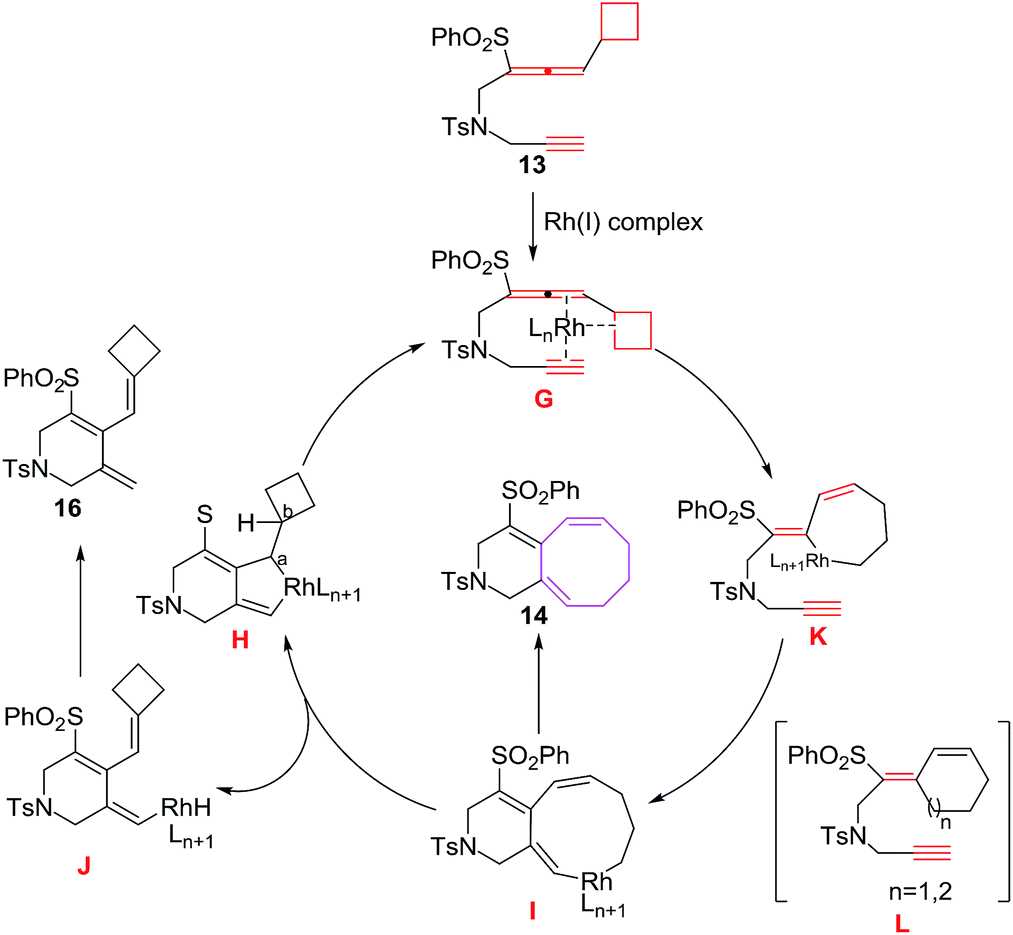
The proposed mechanism involves the coordination of 13 with Rh(I) complex through allenal double bond, C–C bond of the strained cyclobutane ring and alkyne to produce intermediate G, which opens up immediately to rhodabicyclo[4.3.0]nonadiene intermediate H followed by π-carbon elimination of the strained cyclobutane ring of H leading to the formation of nine membered intermediate I, which undergoes reductive elimination leading to bicyclo[6.4.0]dodecatrienes 14. However, π-hydride elimination instead of π-carbon elimination of I generates intermediate J which on reductive elimination yields monocyclic adduct 16. Alternatively, rhodacycle intermediate K,23–25 produced from ring opening of strained cyclobutane ring intermediate G, undergoes insertion of C–RhIII bond into the triple bond leading to the production of intermediate I22 (Fig. 4).
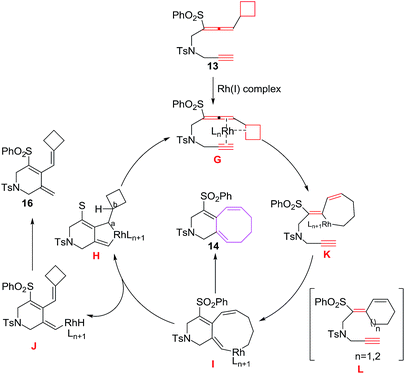 |
| | Fig. 4 Proposed mechanism for the formation of 14. | |
Rh(I) catalysed [6 + 2] cycloadditions of cyclooctatrienes
Conjugated cyclic trienes viz. cycloheptatrienes (CHT), cyclooctatrienes (COTT) etc. are excellent participants in [6 + 2] cycloadditions as the embedded trienes in the favored cisoid confirmations acts as 6 π components with different alkenes, alkynes and dienes acting as 2 π component. The use of metal catalysts in these cycloaddition reactions have found an extensive place in literature. Metal catalysts such as titanium,26 ruthenium,27 molybdenum,28 cobalt,29 chromium30 and iron31 complexes have been widely used to perceive such reactions. Mach et al. have engaged Ziegler catalyst (TiCl4/Et2AlCl) in achieving these [6 + 2] cycloadditions.32 Rigby et al. have employed carbonylchromium(0) complex bearing labile ligand to effectively achieve [6 + 2] cycloadditions.10a–d Buono et al. have recently established [6 + 2] cycloadditions of CHT with terminal alkynes.33 Also, cobalt(I) catalysed protocol has been applied to [6 + 2] cycloadditions of COTT and internal and terminal akynes34 and CHT to substituted allenes.35
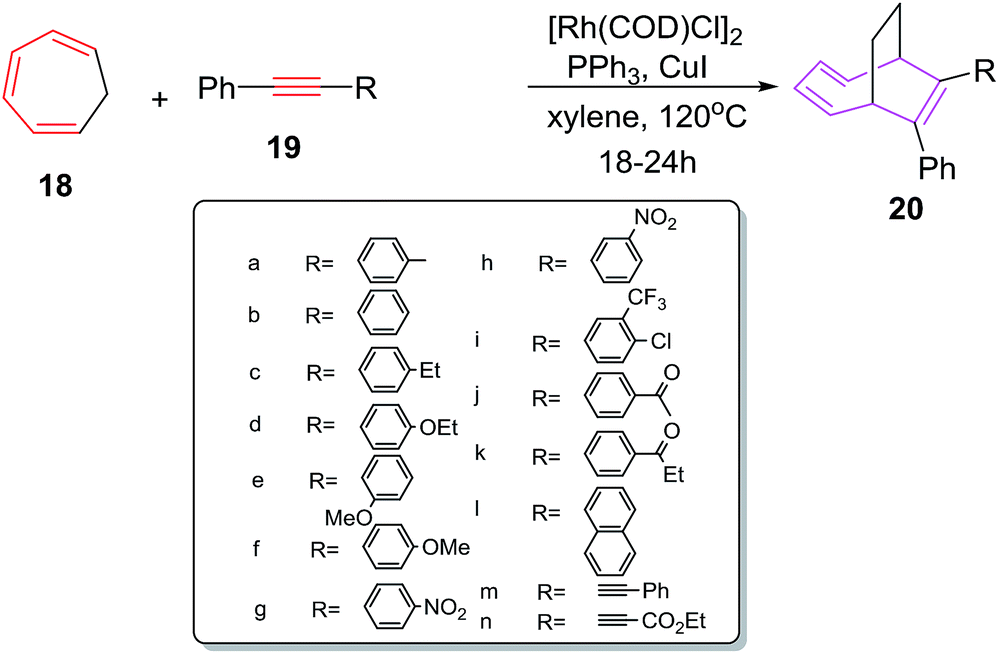
Wang et al. have explored [6 + 2] cycloadditions of cycloheptatriene 18 with internal alkynes 19 employing different rhodium(I) catalysts. The [6 + 2] cycloadditions do not occur using PPh3RhCl as catalyst at 60 °C.36 The [6 + 2] cycloadditions at elevated temperature (120 °C) witness the formation of cycloadduct (64%). Further increase in reaction temperature causes decrease in yield of 20 (53%). Introducing additives such as CuI (10 mol%) which acts as phosphine cleavage reagent37 have increased the yield of 20 (75%). Exchanging the catalyst to [Rh(COD)Cl]2 steers the reaction to better results (71%). Best results for [6 + 2] cycloadditions (88%) are witnessed using [Rh(COD)Cl]2 as catalyst along with PPh3 (10 mol%) and CuI (10 mol%) at 120 °C. A number of terminal alkynes have been tried by Wang et al. The electron donating groups at para position of phenyl acetylene such as 19a, 19c, 19d and 19f have afforded better yields of [6 + 2] cycloadducts (88%, 85%, 87% and 88% respectively). Conversely, electron withdrawing substituents on alkyne such as 19g–h, 19i, 19j, 19k resulted in lowering of reaction yields to 70%, 73%, 70%, 72% and 67% respectively demonstrating the importance of electron donating substituents. No substitution on alkyne nucleus 19b have led to 81% isolation of 20b. Introducing bulky groups as in 19l–n have resulted in poor isolation of 20l–n in 48%, 40% and 30% yields owing to the strong detrimental steric effect played by these groups (Scheme 9).
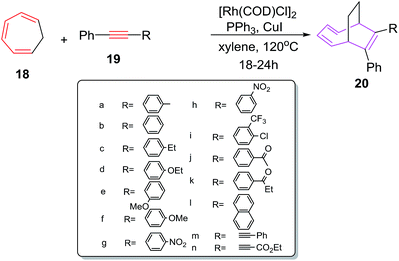 |
| | Scheme 9 [6 + 2] cycloaddition of CHT with various alkynes. | |
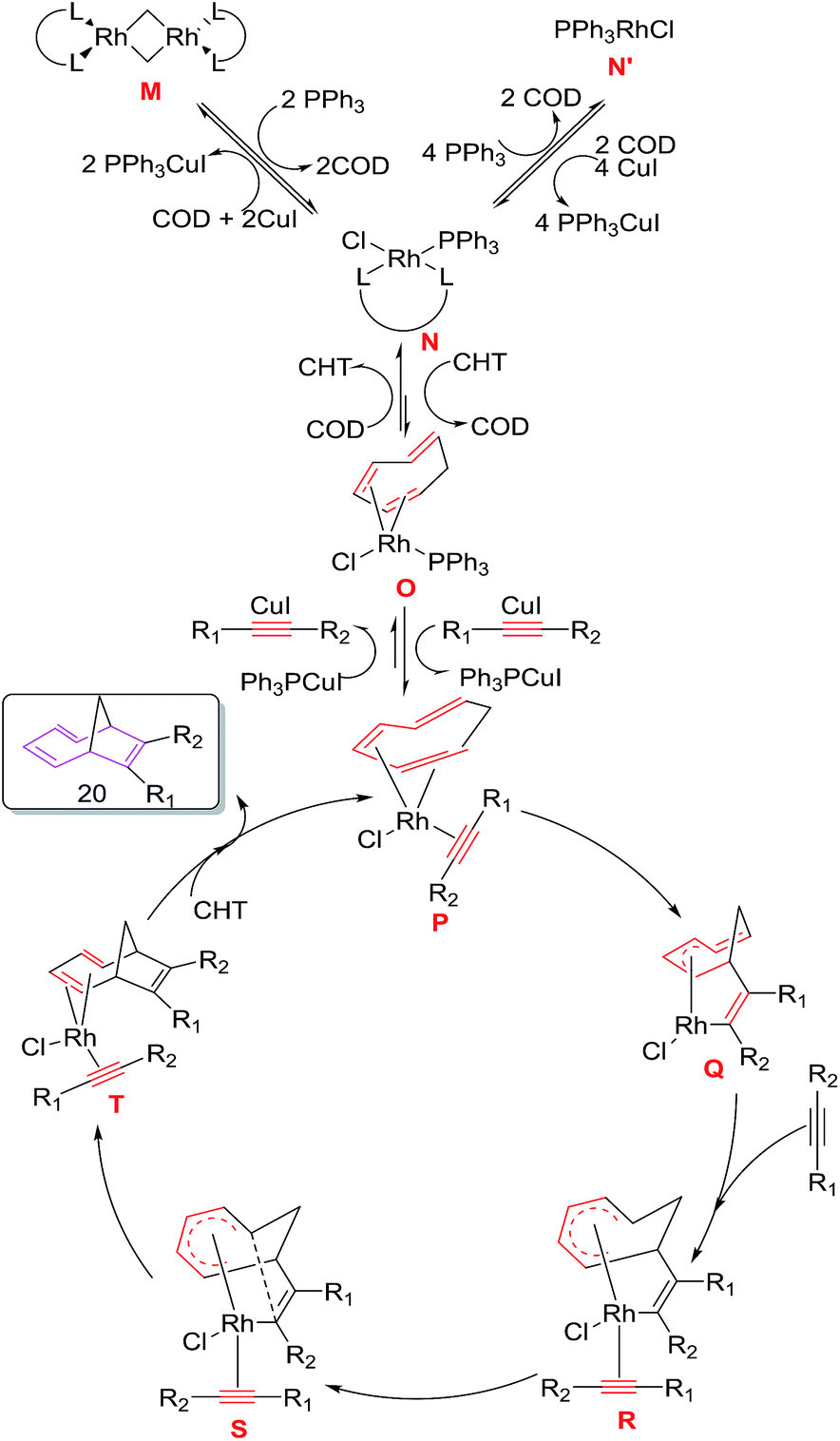
The mechanism for [6 + 2] cycloadditions involves the initial conversion of catalytic precursor [Rh(COD)Cl2] to (Ph3P)Rh(COD)Cl N, followed by exchange of COD ligand with CHT generating intermediate O. Replacement of PPh3 with alkyne in the presence of CuI leads to intermediate P which immediately undergoes oxidative cyclometalation to intermediate Q. Addition of alkyne to Q produces σ,π-allyl complex R which undergoes reductive elimination through transition state S to intermediate T. Ligand exchange of T with CHT delivers cycloadduct 20 and regenerates rhodium complex P to renter the catalytic cycle. DFT calculations employing M06 function taking Rh(I) model as catalyst also confirms the proposed reaction mechanism with the observation of strong additive effect for the CuI in these [6 + 2]reactions (Fig. 5).
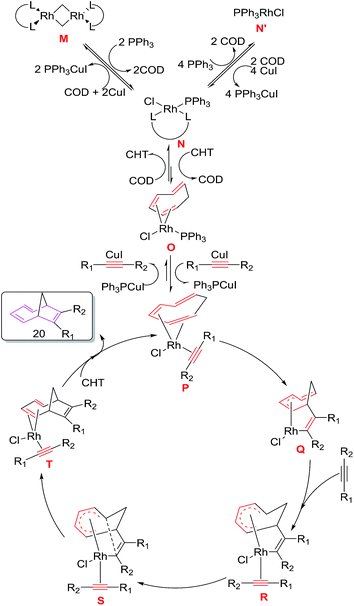 |
| | Fig. 5 Proposed Mechanism for rhodium catalysed [6 + 2] cycloaddition reaction of CHT with internal alkyne. | |
Cobalt catalyzed [6 + 2] cycloadditions
Co(I) complexes have been observed an excellent catalyst for [6 + 2] cycloadditions of cycloheptatrienes, cyclooctatetraenes, dicobalt acetylene complexes as 6p-components with alkyne, allene enol silyl ethers as 2p-components to yield variety of monocyclic or condensed eight membered carbocycles. All the reported studies involve the intermolecular [6 + 2] variants of these cycloadditions and Co(I) catalysed intramolecular [6 + 2] cycloadditions are scarcely available in literature.
[6 + 2] cycloadditions of cycloheptatriene (CHT) and alkynes
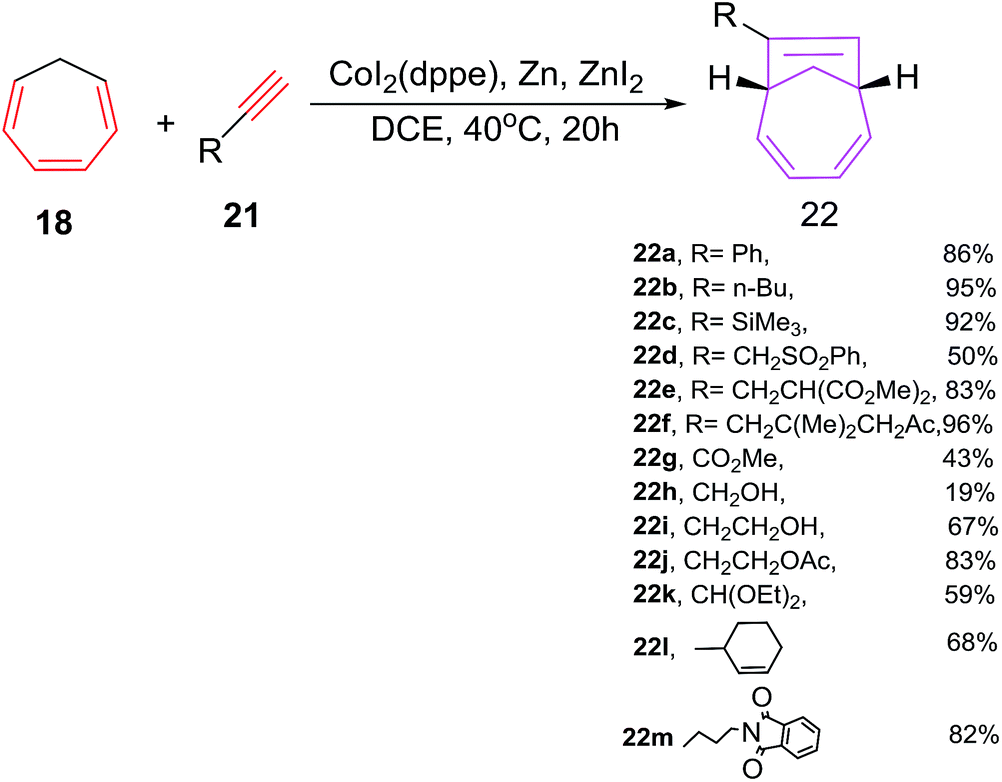
Buono et al. have reported the first cobalt(I) catalyzed [6 + 2] cycloadditions of cycloheptatriene (CHT) 18 with alkynes 21 to afford 7-alkyl-bicyclo[4.2.1]nona-2,4,7-trienes 22 (Scheme 10).38 Various cobalt(II) catalytic systems39 have been tested for [6 + 2] cycloadditions. Best results are witnessed using cobalt(II)iodide/1,2-bis(diphenylphosphino)ethane (dppe) as catalyst, zinc metal as reducing agent, zinc iodide as a Lewis acid in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:2 molar ratio in 1,2-dichloroethane as solvent. The catalytic combination of CoI2(dppe)/Zn/ZnI2 have also proven excellent tolerance to various functional groups such as nitrile, alcohol, imide, sulfone, ketone, ester, ketal or ethers substituted alkyne 21a–m in their [6 + 2] cycloaddition reactions with CHT 18. Alkynes having remotely substituted functional groups to the triple bond such as 21f, 21j, 21m afford cycloadducts 22f, 22j and 22m in excellent yields. The alkynes have substitution propargylic carbon 21d, 21k provide low yield of [6 + 2] cycloadducts. Electron poor alkynes such as methyl propiolate 21g have afforded [6 + 2] cycloadduct 22g with poor yield (21%) due to the preferential coordination of cobalt with 21g initiating its cyclotrimerisation process. The efficacy of cobalt catalysed [6 + 2] cycloadditions are bettered using syringe pump controlled slow addition of the alkyne and lowering the catalyst loading (2.5 mol%) (Scheme 10).
:3:2 molar ratio in 1,2-dichloroethane as solvent. The catalytic combination of CoI2(dppe)/Zn/ZnI2 have also proven excellent tolerance to various functional groups such as nitrile, alcohol, imide, sulfone, ketone, ester, ketal or ethers substituted alkyne 21a–m in their [6 + 2] cycloaddition reactions with CHT 18. Alkynes having remotely substituted functional groups to the triple bond such as 21f, 21j, 21m afford cycloadducts 22f, 22j and 22m in excellent yields. The alkynes have substitution propargylic carbon 21d, 21k provide low yield of [6 + 2] cycloadducts. Electron poor alkynes such as methyl propiolate 21g have afforded [6 + 2] cycloadduct 22g with poor yield (21%) due to the preferential coordination of cobalt with 21g initiating its cyclotrimerisation process. The efficacy of cobalt catalysed [6 + 2] cycloadditions are bettered using syringe pump controlled slow addition of the alkyne and lowering the catalyst loading (2.5 mol%) (Scheme 10).
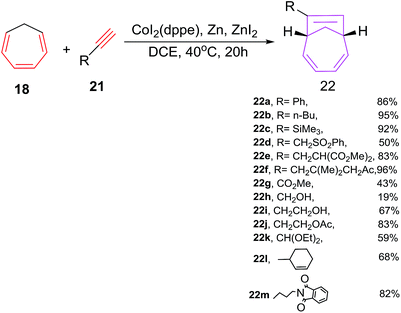 |
| | Scheme 10 Cobalt(II) catalyzed [6 + 2] cycloaddition reactions of CHT with terminal alkynes. | |
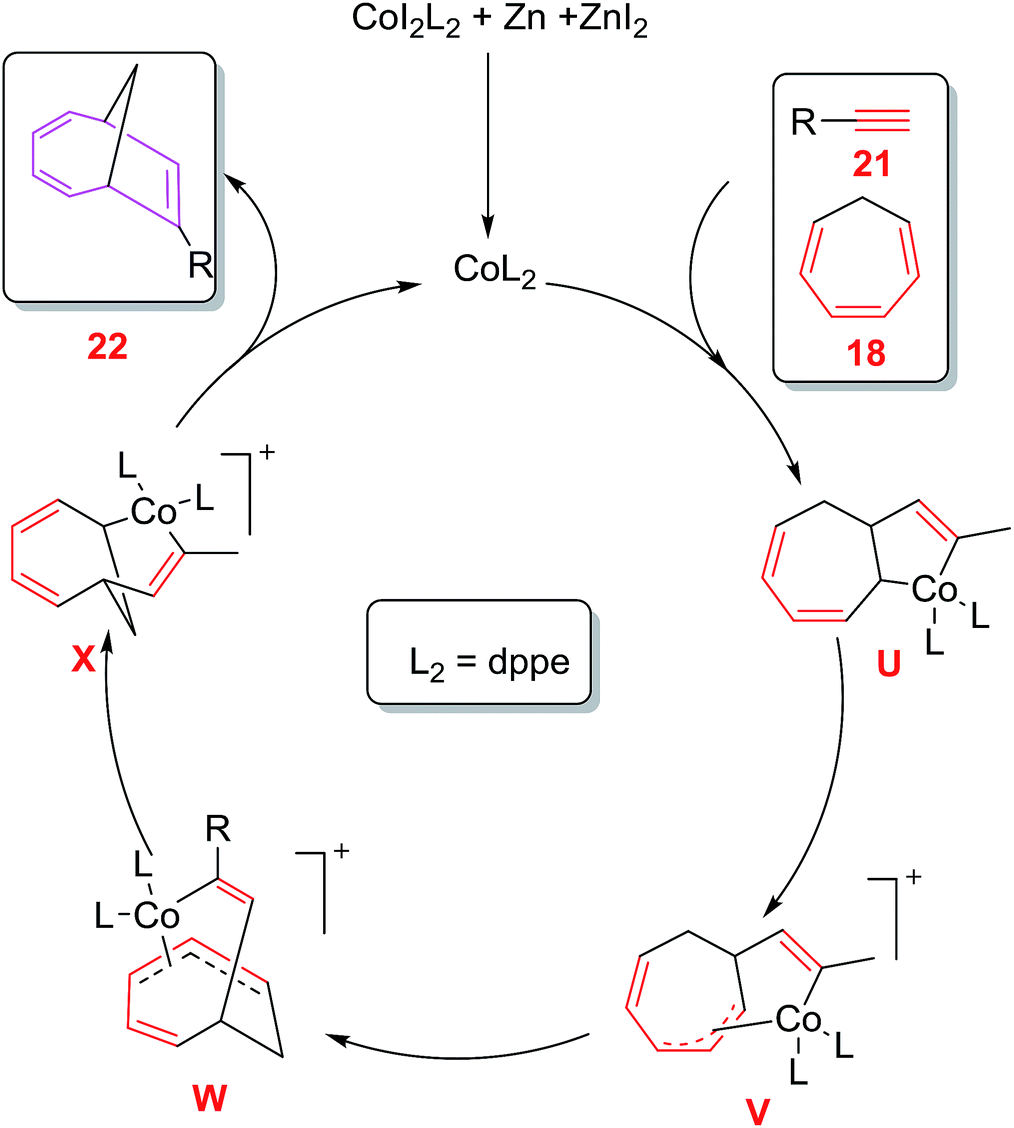
A plausible mechanism involves the ZnI2 reduction of CoL2I2 by zinc metal to cationic [Co(I)L2]+complex which coordinates with alkyne 21 and CHT 18 followed by oxidative cyclometalation to produce cobalta-cyclopentene intermediate U.1,5-migration of the C(sp3)–Co bond of U through consecutive σ,π-allyl complexes V and W leads to bicyclo cobalta-cycle X which on reductive elimination generates cycloadduct 22 and regenerates cationic intermediate [Co(I)L2]+to return to the catalytic cycle38 (Fig. 6).
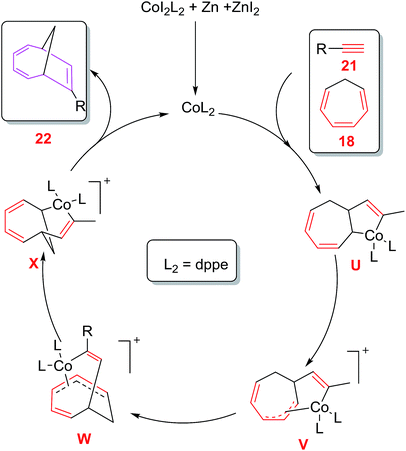 |
| | Fig. 6 Proposed Mechanism for cobalt catalysed [6 + 2] cycloadditions of CHT with terminal alkynes. | |
An enantioselective version of the [6 + 2] reactions has also been explored using phosphoramidite ligand as chirality inductors with improved yields and good enantioselectivity.
[6 + 2] cycloadditions of cycloheptatriene (CHT) and allene
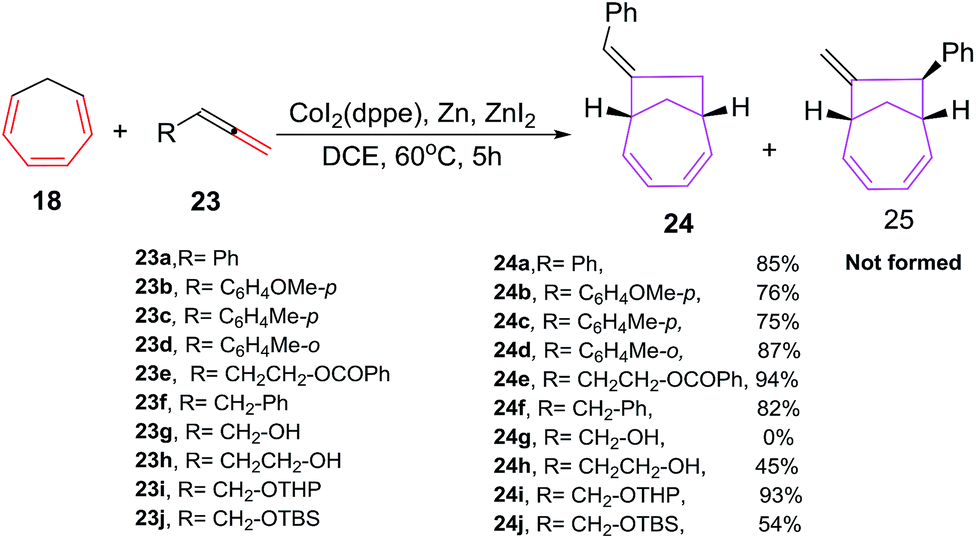
Buono et al. have also reported cobalt catalyzed [6 + 2] cycloadditions of cycloheptatriene (CHT) 18 with variety of allenes 23 to afford bicyclic 7-bicyclo[4.2.1]nona-2,4-dienes 24 with an improved yield, enhanced regio- and geometrical selectivity (Scheme 11). The treatment of CHT 18 with phenylallene 23a under CoI2(dppe)/Zn powder/ZnI2 as catalytic system have resulted in an exclusive formation of E-isomer of 7-benzylidenebicyclo[4.2.1]nona-2,4-diene 24a without the isolation of even traces of the other geometrical or regioisomer 25. Zinc metal plays a pivotal role in these cycloaddition reactions, absence of which leads to non-isolation of [6 + 2] cycloadduct. Also, ligand plays an equally important role as bis(diphenylphosphino)methane (dppm) and 1,3 bis(diphenylphosphino)propane (dppp) were ineffective in conceiving these reactions. Optimal reaction conditions leading to best results in terms of yield of 24 is witnessed using CoI2(dppe) catalytic system with Zn/ZnI2 reducing agent in 1,2-dichloroethane as solvent.40
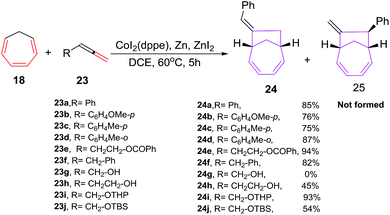 |
| | Scheme 11 Cobalt catalysed [6 + 2] reactions of CHT with substituted allenes. | |
The [6 + 2] cycloaddition reactions of monosubstituted allenes 23 are also studied using Co(II) complexed as catalysts. The methoxy or methyl substitution at ortho- or para- position of phenylallene 23a–d, is well accepted and lead to the exclusive formation of E-isomers 24a–d in good yields. [6 + 2] cycloadditions of benzylallene 23f with CHT 18 also leads to the exclusive formation of E-7-benzylidenebicyclo[4.2.1]nona-2,4-diene 24f in 82% yield. α-allenol 23g and β-allenol 23h are non reactive under these reaction conditions. With tetrahydropyranyl and benzoate protected α-allenols 23i and 23e, excellent yields of 93% and 94% are achieved for cycloadducts 24i and 24e respectively. Conversely, tert-butyl dimethylsilyl protected α-allenols 23j leads to relatively low yields of [6 + 2] cycloadducts 24j (54%) due to the degradation of allene during the catalytic cycle. The [6 + 2] cycloadditions of disubstituted allenes, require high temperature and greater reaction time to manage similar results both in terms of yield and selectivity40 (Scheme 11).
[6 + 2]-cycloadditions of cyclooctatetraene and terminal allenes
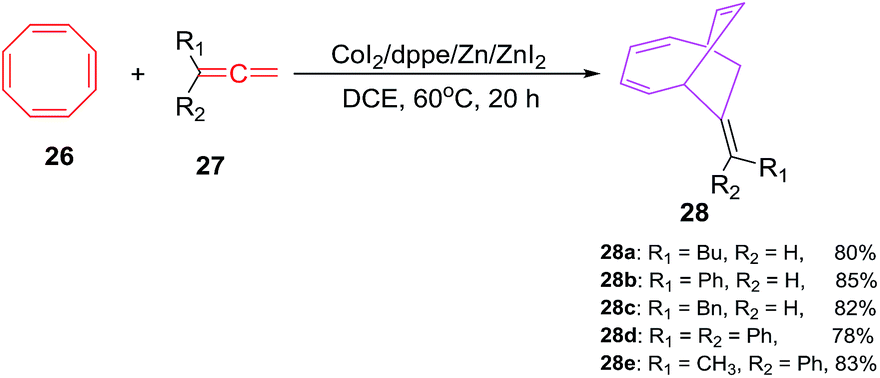
D'yakonov et al. have accomplished the Co(I) catalyzed [6 + 2]-cycloadditions of 1,3,5,7-cyclooctatetraene 26 (COTT) with functionalized terminal allenes 27a–e in the presence of CoI2/dppe/Zn/ZnI2 catalytic system using 1,2-dichloroethane as solvent at 60 °C to afford (E)-bicyclo[4.2.2]deca-2,4,7-trienes 28a–e in good yields (78–85%; Scheme 12).41 The reactions are gauged using varied reaction and catalytic system. It is observed that CoI2 or ZnI2 alone do not conceive the reactions even at elevated temperature (100 °C).
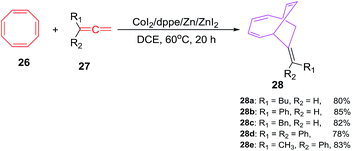 |
| | Scheme 12 Cobalt(II) catalysed [6 + 2] cycloaddition reactions of COTT with terminal allenes. | |
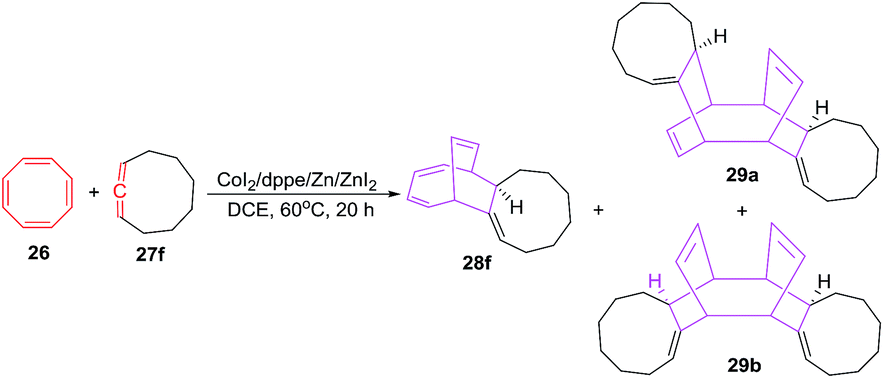
The [6 + 2] cycloadditions of COTT 26 with 1,2-cyclononadiene 27f under similar reaction conditions afford tricyclo[9.4.2.0]heptadeca-2,12,14,16-tetraene 28f in 65% yield. Interestingly, the reactions also proceed further to produce regio- and strereomeric bis-adduct mixture 29a and 29b in total yield of 30%41 (Scheme 13).
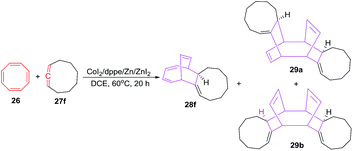 |
| | Scheme 13 Cobalt(II) catalysed [6 + 2] cycloaddition reactions of COTT with cyclic allene. | |
[6 + 2] cycloadditions of cyclooctatetraene with alkynes
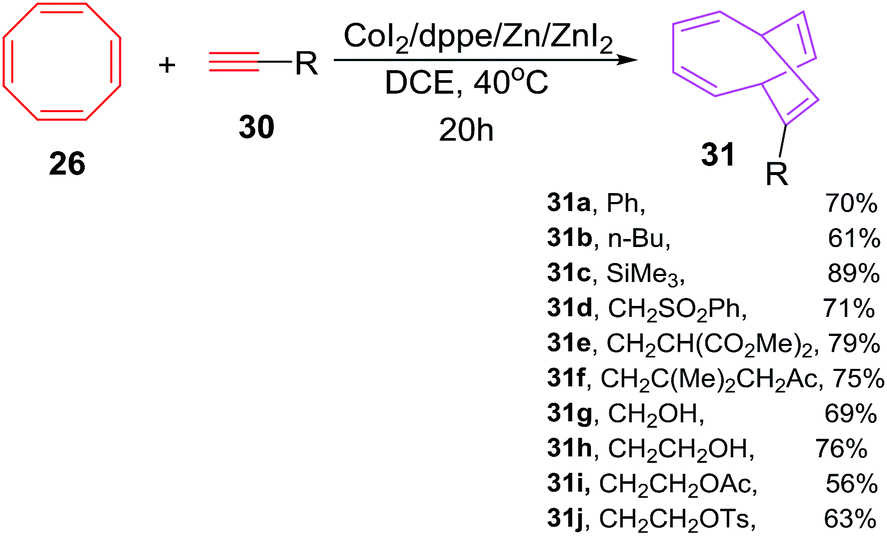
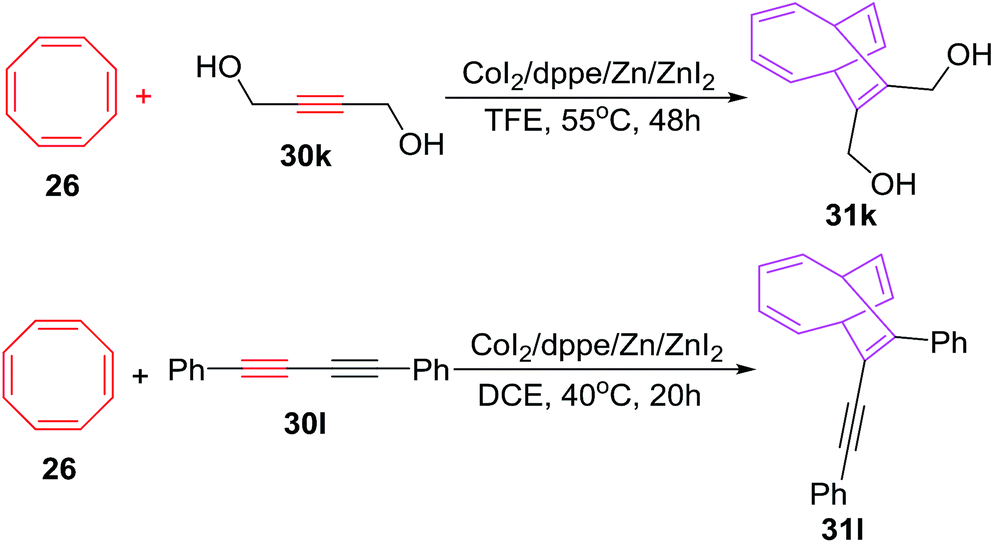
Buono et al. have also explored the cobalt-catalyzed [6 + 2] cycloadditions of cyclooctatetraene 26 with alkynes 30a–j for the synthesis of monosubstituted bicyclo[4.2.2]-deca-2,4,7,9-tetraenes 31a–j in fair to good yields (56–94%). The reaction of phenylethyne 30a with COTT 26 using CoI2(dppe) as catalyst/ligand and ZnI2/Zn as Lewis acid/reducing agent couple in 1,2-dichloroethane at 40 °C have resulted in the isolation of 7-phenylbicyclo[4.2.2]deca-2,4,7,9-tetraene 31a in 70% yield. Alternatively employing CoI2(dppe), Bu4NBH4/ZnI2 catalytic system have led to the isolation of 31a in 66% yield. The reaction protocol had proven to be of wide spread applicability as various functionalities on alkyne nucleus viz. ester, amide, nitrile, sulphonate, ketone, sulphones and trimethylsilyl are very well accepted as evident from moderate to good yields of [6 + 2] cycloadducts 31a–j (56–94%; Scheme 14).42
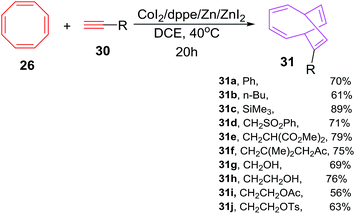 |
| | Scheme 14 Cobalt-catalyzed [6 + 2] cycloadditions of cyclooctatetraene with terminal alkynes. | |
The [6 + 2] cycloadditions encountered similar success when COTT 26 is made to react with symmetrical alkynes 30k–l for synthesis of cycloadducts 31k–l in yields of 78% and 77% respectively. The [6 + 2] cycloadditions however have failed for diphenylethyne or 3-hexyne (Scheme 15).42
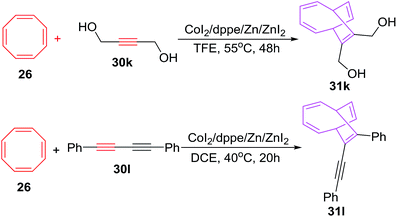 |
| | Scheme 15 Cobalt-catalyzed [6 + 2] cycloadditions of cyclooctatetraene with symmetrical alkynes. | |
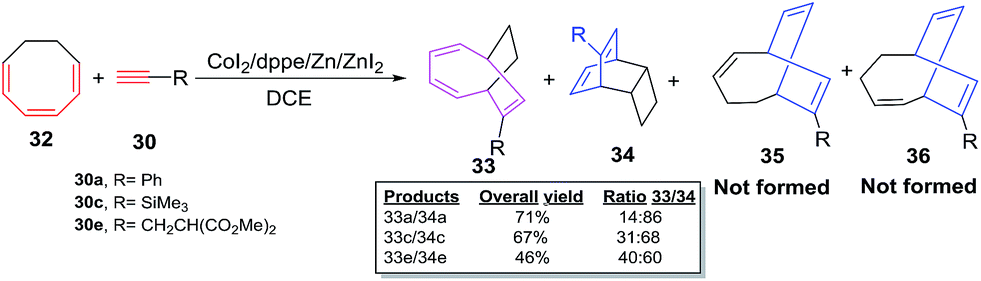
The cobalt-catalyzed cycloadditions of cyclooctatrienes (COT) 32 with various alkynes 30a–e afford a mixture of desired [6 + 2] cycloadducts 33a–e and tricyclic adduct 34a–e. The absence of cycloadducts of type 35 and 36 from the product range rules out the possible [4 + 2] cycloaddition pathway for COTT 26 and COT 32 (Scheme 16).
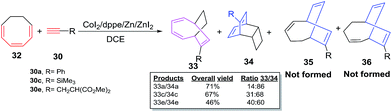 |
| | Scheme 16 Cobalt-catalyzed [6 + 2] cycloadditions of cyclooctatriene (COT) with terminal alkynes. | |
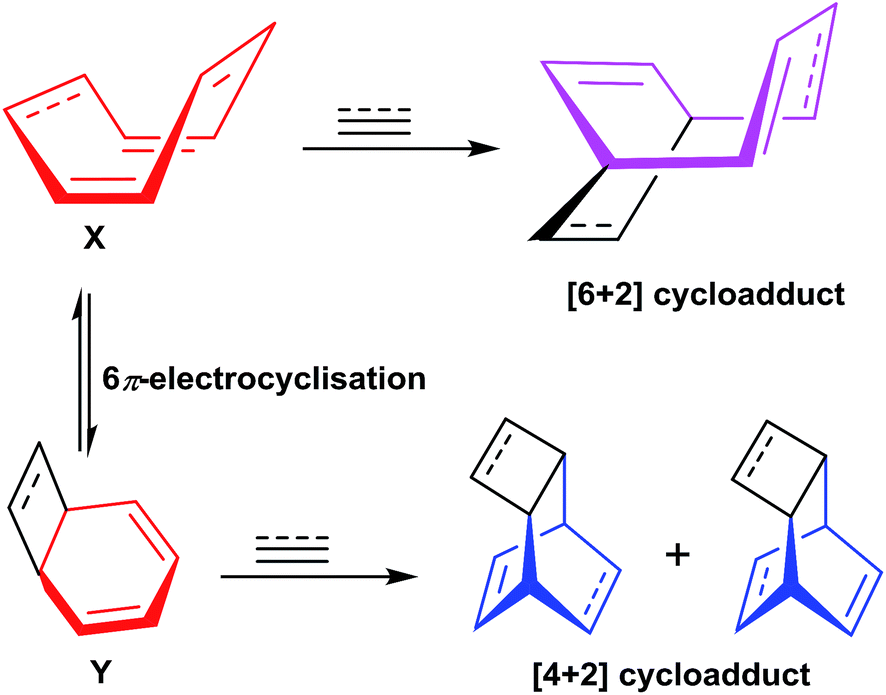
The COT 32/COTT 26 can exist in two isomeric forms namely monocyclic form X and 6π bicyclic isomeric electrocycle Y. The [6 + 2] cycloaddition of terminal alkyne 30 to isomeric form X have resulted in the formation of desired cycloadduct 33, whereas, [4 + 2] cycloaddition of 30 to bicyclic isomeric form Y have led to the isolation of cycloadduct 34. The direct non participation of X in [4 + 2] cycloaddition with terminal alkyne 30 have resulted in non isolation of hypothetical products 35 and 36 (Fig. 7).
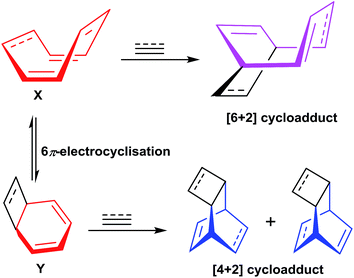 |
| | Fig. 7 Valance tautomeric forms of COT/COTT and their participation in cycloadditions with alkynes. | |
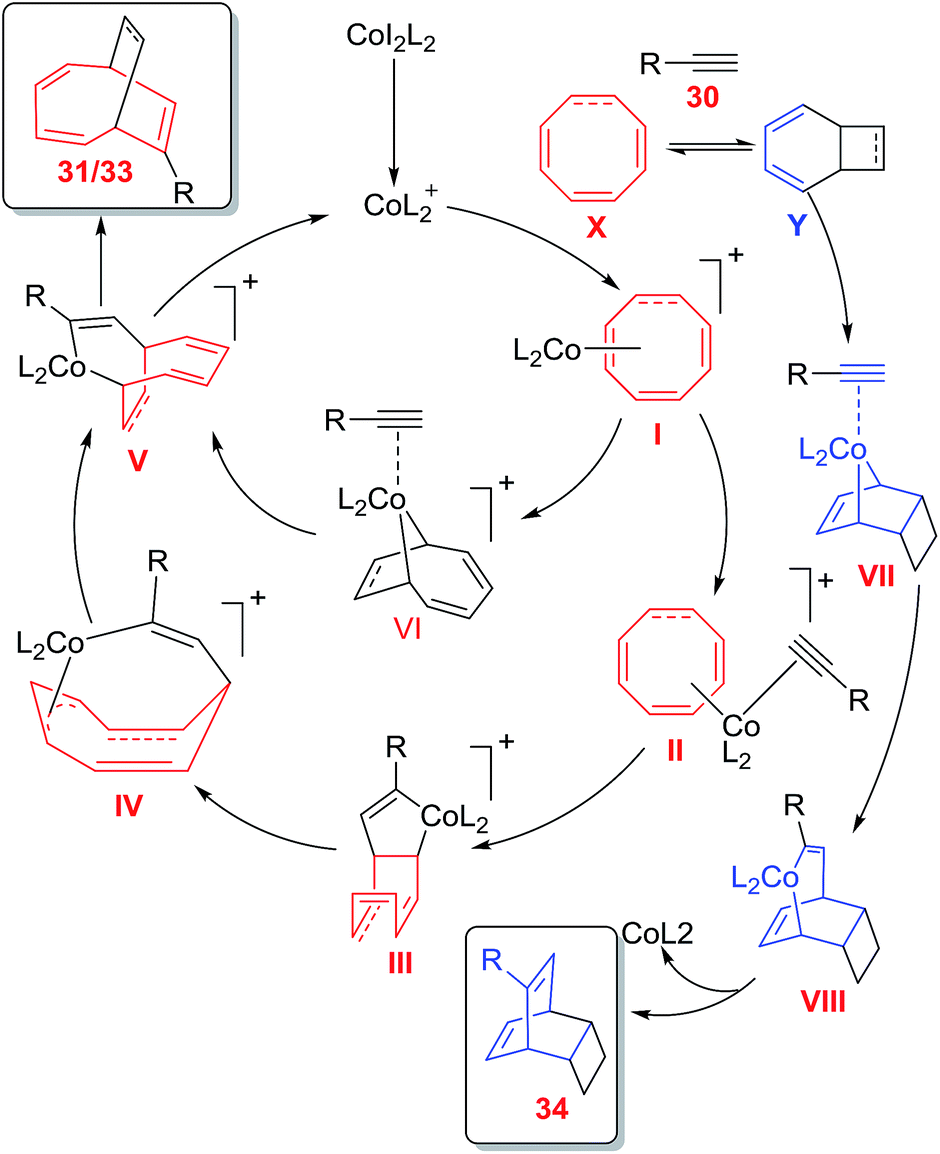
Mechanistically, the reaction is expected to proceed through two pathways. Oxidative cyclometallation of I followed by its coordination to alkyne 30 generates cobaltacycle VI in the first path. Alkyne insertion to Co–C bond leads to cobaltabicycle V, which on reductive elimination of cobalt affords cycloadduct 31/33 and regenerates active [CoL2]+ to renter the catalytic cycle. In the second path, oxidative cyclometallation of II leads to complex III which on 1,3-migration of Co–C(sp3) bond through σ-π-allyl complex forms cobaltabicycle adduct V. Formation of [4 + 2] cycloadduct proceeds via sequential oxidative cyclometallation, alkyne insertion and reductive elimination of intermediate VII and VIII leading to product 34 (Fig. 8).42
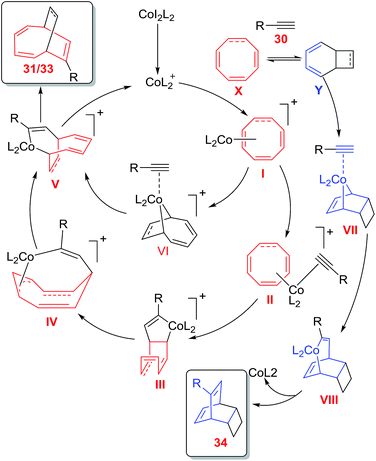 |
| | Fig. 8 Proposed mechanism for cobalt catalysed cycloadditions of COTT 26/COT 32 with terminal alkynes 30. | |
[6 + 2] cycloadditions of dicobalt acetylene complex with enol silyl ethers
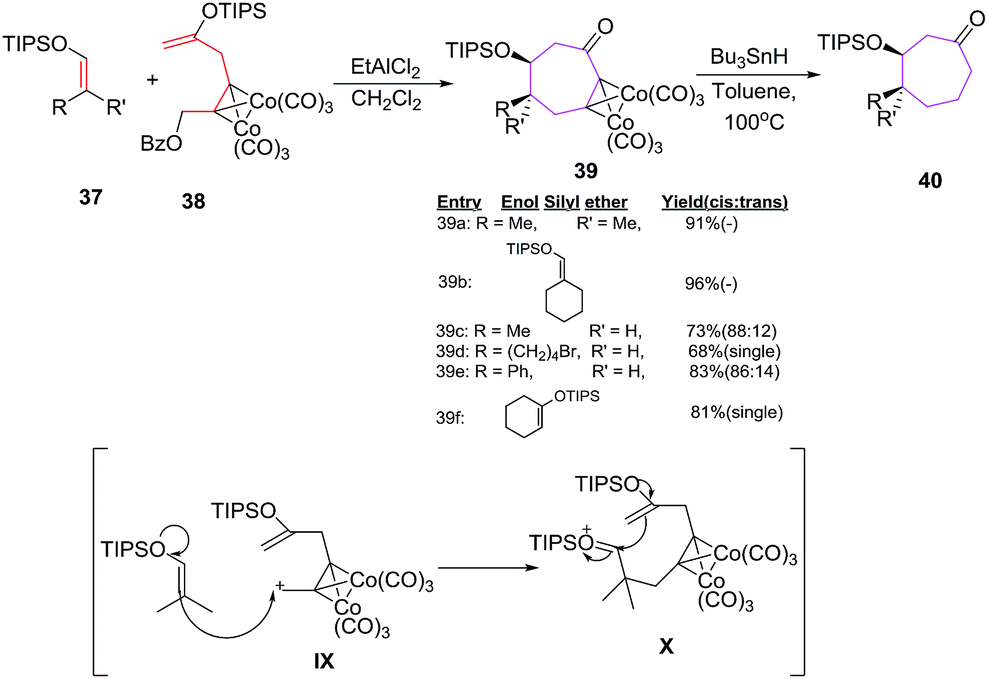
Tanino et al. have developed methodology for the stereoselective synthesis of cyclooctanones 40 by formal [6 + 2] cycloadditions of a hexa carbonyl dicobalt acetylene complex 38 (6π component) with several enol silyl ethers 2π component 37 under the influence of EtAlCl2.
The methodology holds special importance as no side product arising from intramolecular cyclisation/homo coupling of the cobalt complex is noticed which allowed equivalent mixing of reactants without employing high dilution synthesis (Scheme 17).43
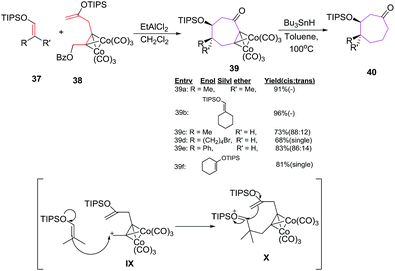 |
| | Scheme 17 [6 + 2] cycloadditions of various enol silyl ether with dicobalt acetylene complex. | |
The reaction proceeds with the stereoselective formation of dicobalt acetylene complex 39 in good yields. Acyclic enol silyl ethers 37c, 37d, 37e produce cycloadducts 39c, 39d and 39e in 73% (88:12), 68% (cis) and 83% (86:14) yields respectively with the major product having cis arrangement of alkyl and silyloxy group. However, trans fused cyclooctanone 39f is obtained as a single cycloadduct (81%) by employing cyclic enol silyl ether 37f.43,44
The [6 + 2] cycloadditions can be utilized to generate a model compound of taxane diterpenoid. The B–C ring system of taxane is constructed by employing [6 + 2] cycloaddition reaction of diacetylene dicobalt complex 38 with cyclic enol silyl ether 37f.45
Titanium(II) catalyzed [6 + 2] cycloadditions
Titanium complexes has successfully tested in [6 + 2] cycloadditions of variety of cycloheptatrienes, cyclooctatetraenes, bis(1,3,5-cycloheptatriene-7-yl)lkanes as 6π-components with alkyne, 1,2-dienes, as 2π-components to yield variety of monocyclic or condensed eight membered carbocycles. All the reported studies involve only intermolecular variants of [6 + 2] cycloadditions.
Titanium(II) catalyzed [6 + 2] cycloadditions of cycloheptatrienes with alkynes
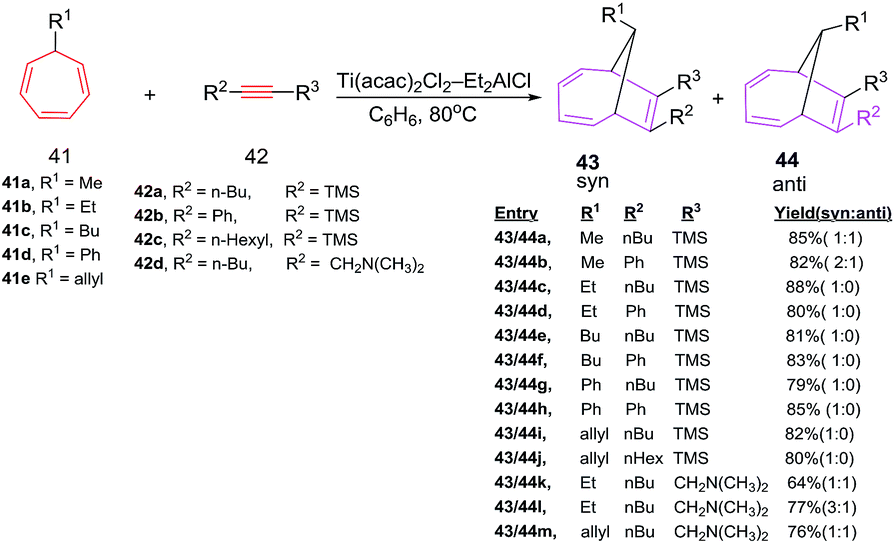
D'yakonov et al. have explored the [6 + 2] cycloadditions of 7-alkyl(allyl,phenyl)-1,3,5-cycloheptatrienes 41 with alkynes 42 catalyzed by the two-component Ti(acac)2Cl2–Et2AlCl system, resulting in the formation of substituted bicyclo[4.2.1]-nona-2,4,7-trienes 43/44 in excellent yields (up to 90%). The addition of alkyne 42 to the cycloheptatriene 41 (1:1) occurs in a stereoselective manner to afford predominantly syn-isomer. The [6 + 2] cycloadditions using alkyne 42 having ethyl-, butyl-, phenyl-, and allyl-substituted cycloheptatrienes 41b, 41c, 41d and 41e lead to cycloadducts 43c–j syn-specificity (Scheme 18).46
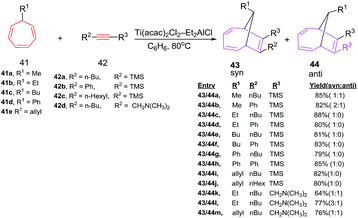 |
| | Scheme 18 Ti-catalyzed [6 + 2] cycloadditions of alkynes to 7-substituted 1,3,5-cycloheptatrienes. | |
Symmetrical alkynes such as hex-3-yne, oct-4-yne and dec-5-yne are incompetent 2π components in [6 + 2] cycloadditions and have shown competitive homo-cyclotrimerization. The [6 + 2] reactions afford bicyclic systems 43/44 using (i-PrO)2TiCl2 and (t-BuO)2TiCl2 or TiCl4 as catalysts.46 The [6 + 2] reactions of 7-alkyl(allyl,phenyl)-1,3,5-cycloheptatrienes 41 with Si-containing alkynes 45 catalyzed by the Ti(acac)2Cl2/Et2AlCl system, furnish substituted bicyclo[4.2.1]nona-2,4,7-trienes 46 in good yields (79–88%; Scheme 19).
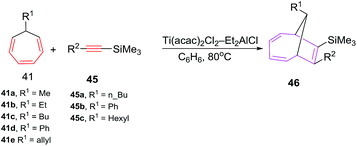 |
| | Scheme 19 Ti-catalyzed [6 + 2] cycloadditions of silicon containing alkynes to 7-substituted 1,3,5-cycloheptatrienes (CHT). | |
[6 + 2] cycloadditions of cycloheptatrienes with 1,2-dienes
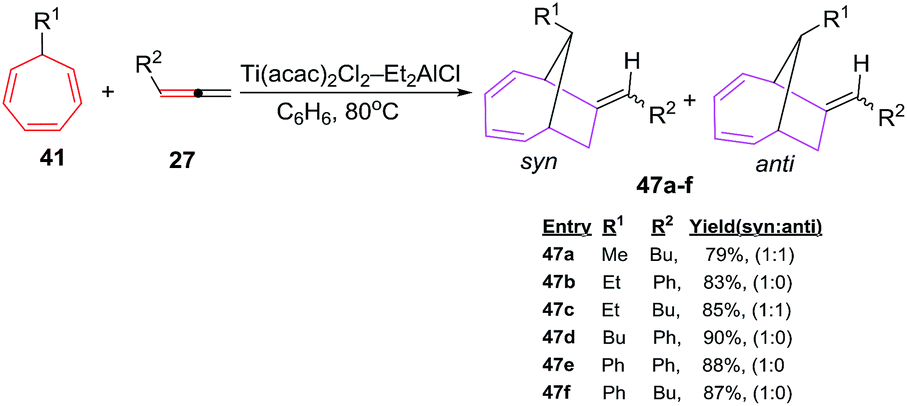
D'yakonov et al. have also envisaged [6 + 2] cycloadditions of functionalized cycloheptatrienes 41 with allenes(1,2-dienes) 27 using Ti(acac)2Cl2–Et2AlCl system as catalyst to generate predominantly endobicyclo[4.2.1]-nona-2,4-dienes 47a–f in good yields.47 The enhanced syn selectivity is largely attributed to the steric bulk at 7-position of CHT nucleus (Scheme 20).
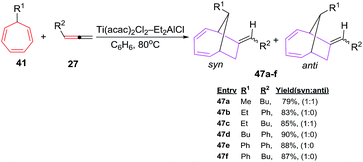 |
| | Scheme 20 Ti-catalyzed [6 + 2] cycloadditions of 1,2-dienes to 7-substituted 1,3,5-cycloheptatrienes (CHT). | |
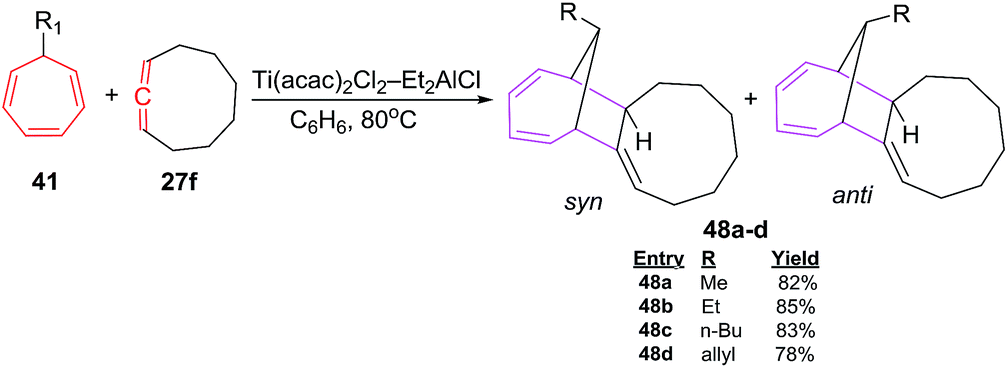
D'yakonov et al. have also studied [6 + 2] cycloadditions reactions using functionalized cycloheptatrienes 41 and 1,2 cyclononadiene 27f to yield a mixture of syn and anti of isomeric tricycle[9.4.1.0]-hexadeca-2,12,14-trienes 48a–d (78–85%; Scheme 21).47
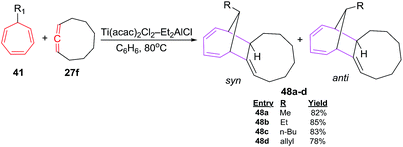 |
| | Scheme 21 Ti-catalyzed [6 + 2] cycloadditions of 1,2 cyclononadiene 7-substituted 1,3,5-cycloheptatrienes (CHT). | |
[6 + 2] cycloadditions of bis(1,3,5-cycloheptatriene-7-yl)lkanes 47 and alkynes
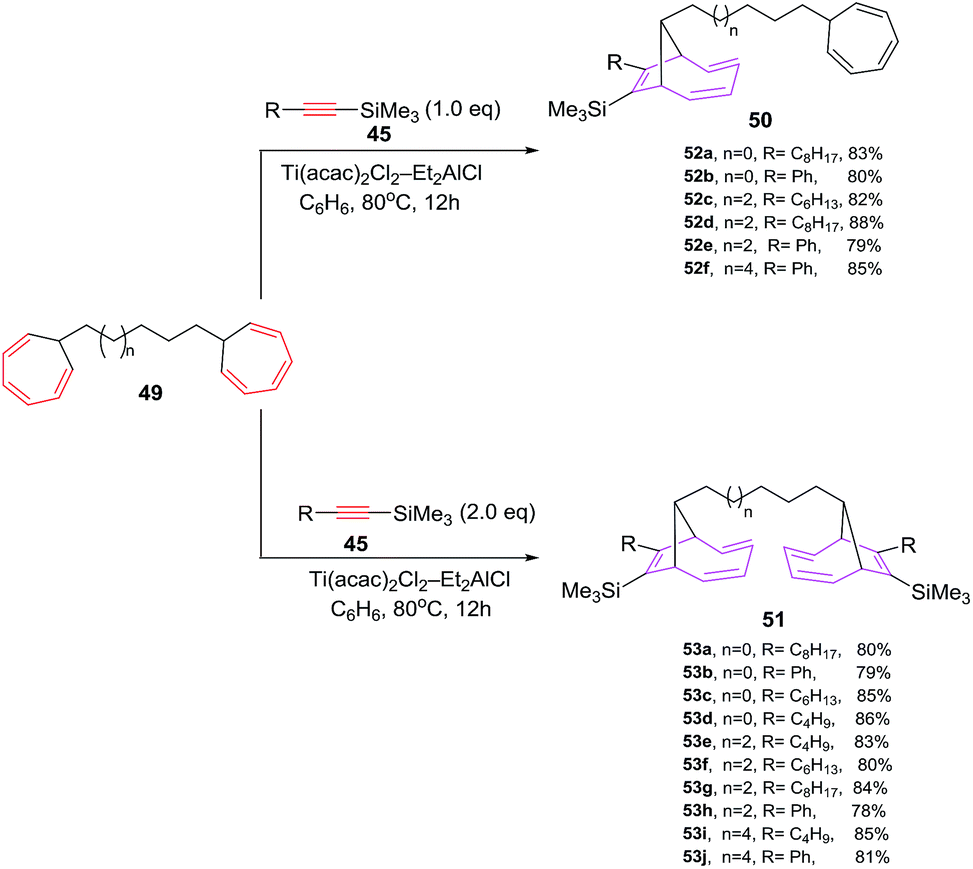
D'yakonov et al. have explored [6 + 2] cycloadditions of Si-containing alkynes 45 and bis(1,3,5-cycloheptatriene-7-yl)alkanes 49 using Ti(acac)2Cl2–Et2AlCl as catalytic system, leading to selective formation of mono- and bis-adducts [9-[4-(2,4,6-cycloheptatrienyl)alkyl]-8-alkyl(phenyl)bicyclo[4.2.1]nona-2,4,7-triene-7-yl](trimethyl)silanes 50a–f and bis(7-trimethylsilyl-8-alkyl(phenyl)bicyclo[4.2.1]nona-2,4,7-triene-7-yl)alkanes 51a–f in 78–88% yields respectively. The cycloaddition reactions of alkynes 45 and bis(1,3,5-cycloheptatriene-7-yl)alkanes 49 are stereoselective, resulting in the isolation of syn-isomer. The high stereoselectivity is presumably attributable to steric factors associated with the bulky substituent at the bridging carbon atom in the initial bis(1,3,5-cycloheptatriene-7-yl)alkane 4948 (Scheme 22).
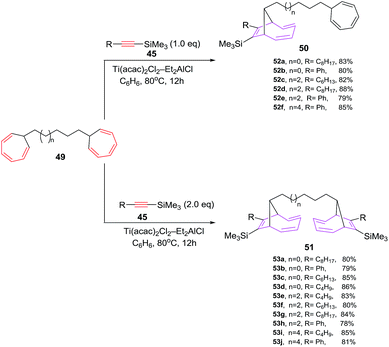 |
| | Scheme 22 Ti-catalyzed [6 + 2] cycloadditions of silicon containing alkynes with bis-1,3,5-cycloheptatrienes (CHT). | |
Platinum(II) catalyzed intramolecular [6 + 2] cycloadditions
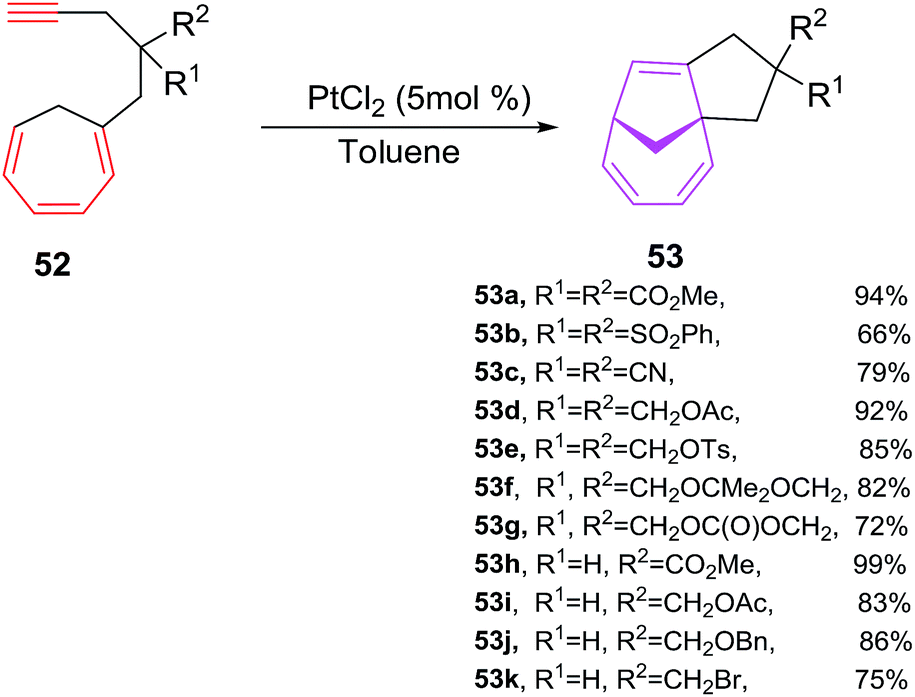
Platinum(II) complexes has efficiently been explored in relatively less explored intramolecular variant of [6 + 2] cycloadditions to afford bicyclic cyclopentane fused bicyclo[4.2.1]nona-2,4,7-trienes. Tenaglia et al. have developed a facile, chemoselective and high yielding platinum-catalyzed (5 mol%) en route for intramolecular [6 + 2] cycloadditions of alkynes tethered to cycloheptatrienes 52a–k at room-temperature to afford cyclopentane fused bicyclo[4.2.1]nona-2,4,7-trienes 53a–k in good yields (66–99%). The cycloisomerizations of 52 with other metal complexes such as [{RuCl2(CO)3}2] and AuCl3 provide satisfactory results, whereas rhodium complexes are poor in catalysing these reactions. The best results are observed using PtCl2 (5 mol%) as catalysts. The reactions are also analyzed in different solvents, such as THF (85%), acetone (91%), toluene (94%) are observed most promising solvent and cycloadditions in methanol, acetonitrile and DMF are unsuccessful. The [6 + 2] cycloaddition can tolerate variety of substitutions at cycloheptatrienes such as CHT carrying acetal 52f, ether 52d–e, ester 52h–j, sulfonate 52b, cyclic carbonate 52g, halide 52k or nitrile substituents 52c afford [6 + 2] cycloadducts in good yields. However, lengthening the tether by an additional carbon unit in 52 has a detrimental effect and no [6 + 2] cycloadduct is realised49 (Scheme 23).
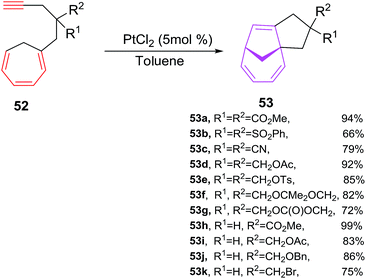 |
| | Scheme 23 PtCl2-catalyzed [6 + 2] cycloadditions of alkynes tethered to cycloheptatrienes 54. | |
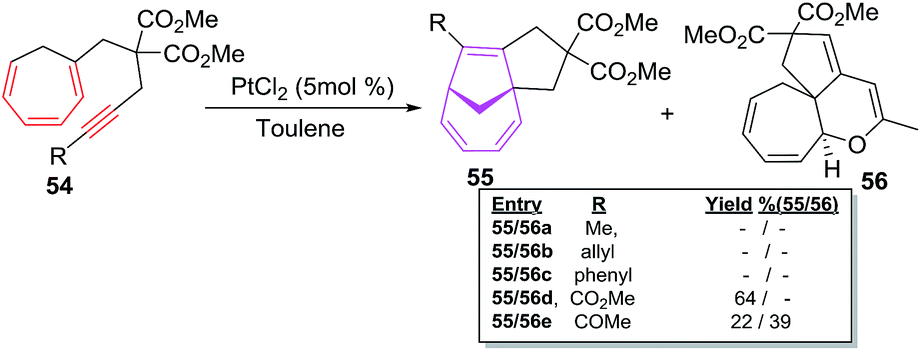
The CHT bearing internal alkynes 54a–e have been observed reluctant participants in intramolecular [6 + 2] cycloadditions even at elevated reaction temperature (110 °C; Scheme 24).
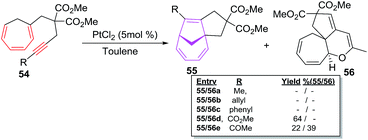 |
| | Scheme 24 PtCl2-catalyzed [6 + 2] cycloadditions of trienynes 54a–e. | |
Alkyl 54a–b and phenyl 54c substituted trienynes are unreactive whereas, electron deficient 54d–e afford [6 + 2] adducts 55d–e albeit in low yields of 64% and 22% respectively.49
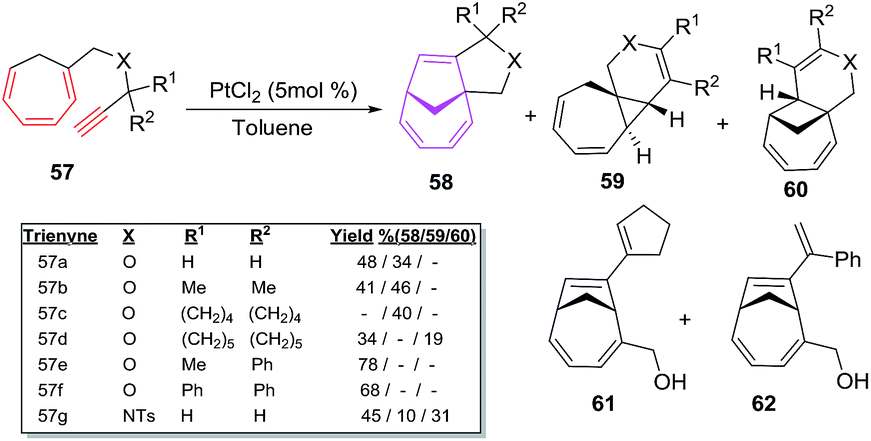
The intramolecular [6 + 2] cycloisomerisations of trienynes 57a–g bearing heteroatom in the tether yield [6 + 2] cycloadducts 58 along with the formation of side products such as dihydropyranes or tetrahydropyranes 59 via 1,2-hydrogen or alkyl migration, along with other products 60, 61 and 62 as depicted in Scheme 25.49
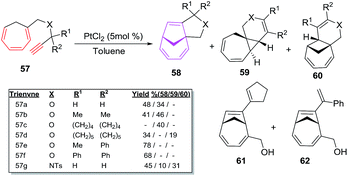 |
| | Scheme 25 PtCl2-catalyzed [6 + 2] cycloadditions of heteroatom tethered trienynes 57a–g. | |
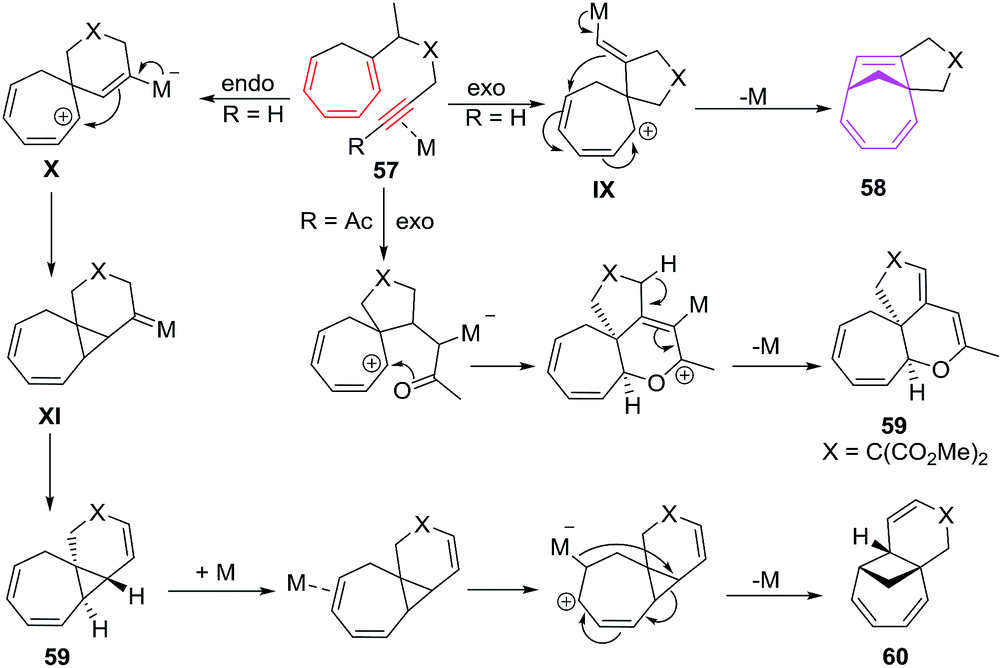
The proposed mechanism involves the exo cyclisations of 57 leading to pentadienyl cationic intermediate IX which through electronic redistribution affords [6 + 2] cycloadducts 58. With trienyne bearing an acyl functionality, carbonyl group trigger the cyclisation of 57, leading to dihydropyran 59. Endocyclisation of 57 through zwitter ion X and platinacarbene intermediate XI leads to product 59 presumably through a 1,2-hydrogen/alkyl shift followed by the elimination of metal ion. Formation of 60 is owed to the coordination of Pt2+ species to distant double bond of 59 followed by ring cleavage of the cyclopropane moiety. The tetrahydropyran ring opening of 58c/58e driven by HCl (generated from PtCl2) affords tertiary allylic carbocation yielding products 61 and 62 respectively49 (Fig. 9).
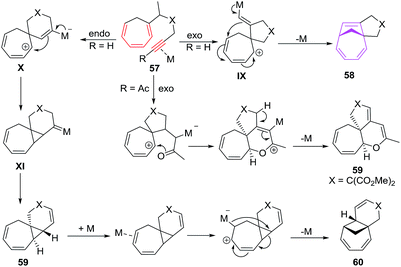 |
| | Fig. 9 Proposed mechanism for cycloisomerisations of trienynes. | |
Ruthenium(0) catalyzed [6 + 2] cycloadditions of cyclooctatriene (COT) with alkenes
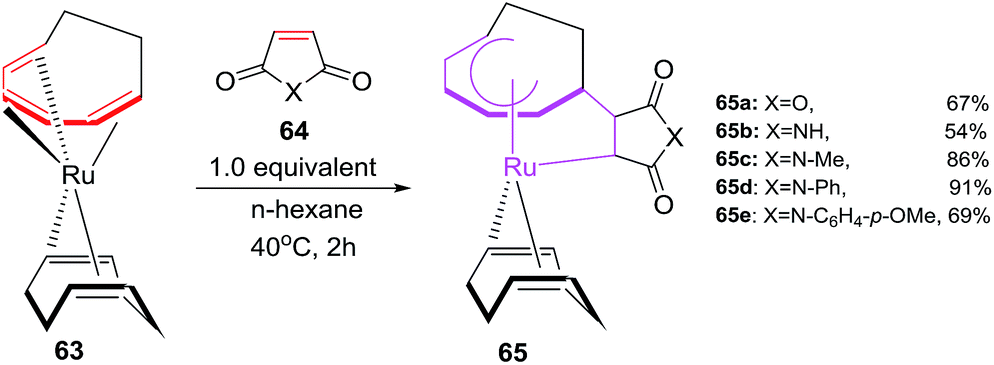
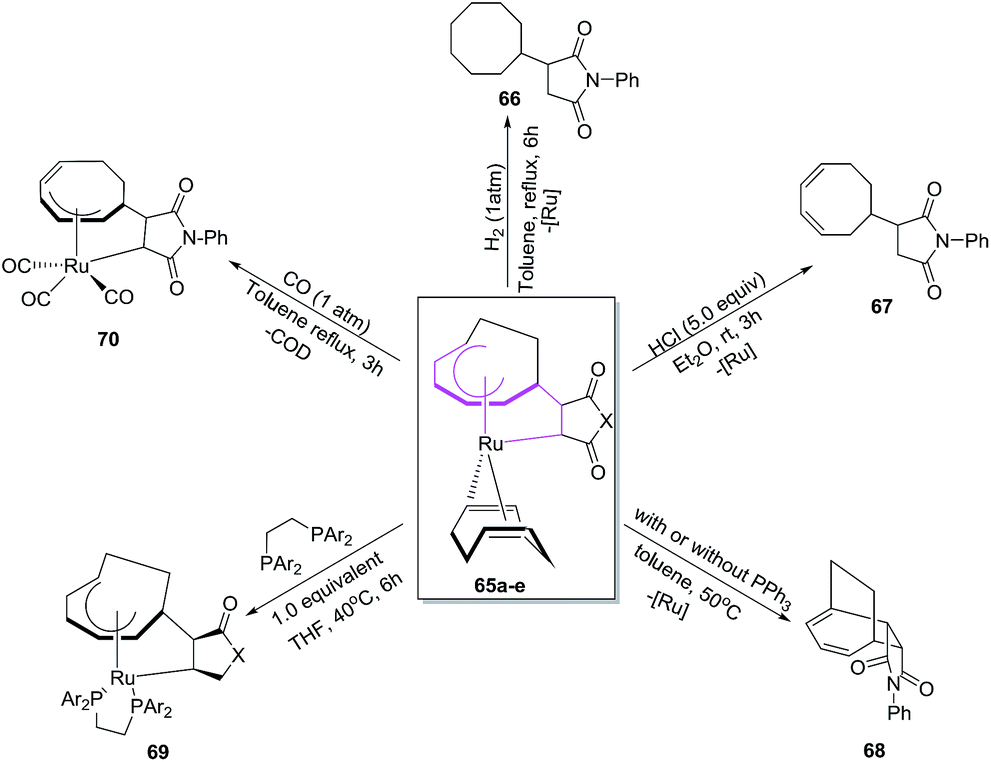
Mitsudo et al. have envisaged [6 + 2]cycloaddition reactions of ruthenium(0) complex such as Ru(η4-cod)(η6-cot) 63 with maleic anhydride 64a or maleimides 64b–e to generate a series of novel divalent ruthenacycles 65a–e. The reaction proceeds by employing equivalent amounts of both reactants in n-hexane as solvent for 2 h to afford ruthenacycles 65a–e50 (Scheme 26).
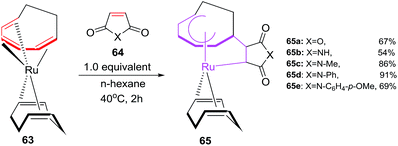 |
| | Scheme 26 Formation of ruthenacycles 67 via oxidative cyclisations. | |
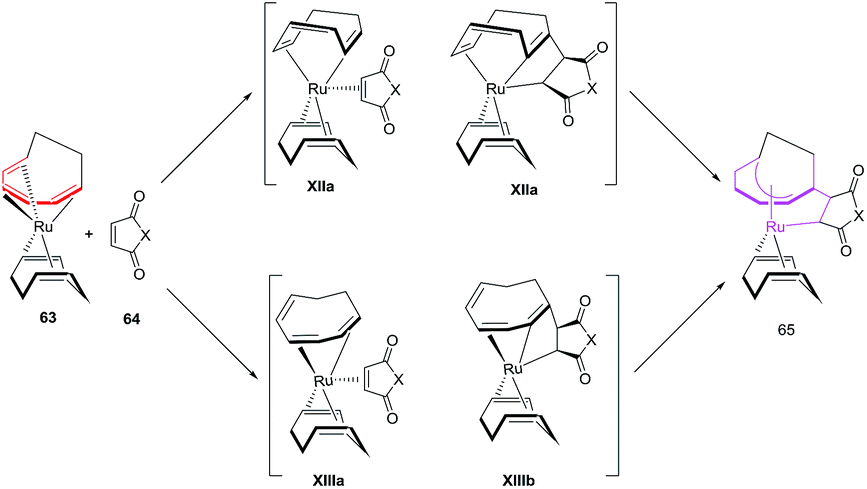
Mitsudo et al. have also studied electron deficient alkenes such as dimethyl fumarate and dimethyl maleate in [6 + 2] cycloadditions to afford Ru(η5-cot)(η2-dimethyl fumarate).51 Employing p-benzoquinone in place of maleic anhydride produces an uncharacterizable mixture of products. N,N-Dimethylacrylamide and methyl vinyl ketone are ineffective in conceiving these reactions. Terminal alkynes on the other hand have afforded a zerovalent η4,η2-bicyclodecatriene product.52 The proposed mechanism for the production of ruthenacycles 65 involves the change of coordination mode of COT ligand of 63 from 1-6-η6 to 1-2:5-6-η4 or 1-4-η4 followed by 64 occupying its vacant coordination site to either produce intermediate XIIa or XIIIa. The oxidative cyclisation of COT and 64 generates ruthenacycles intermediates XIIb and XIIIb, which re-coordinates of the CC bond affording desired ruthenacycles 65 (Fig. 10).50
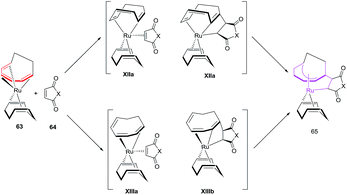 |
| | Fig. 10 Proposed mechanism for formation of ruthenacycles 65. | |
The ruthenacycles 65a–e react in a number of ways to create cyclooctyl complexes 66–70 respectively. The ruthenacycles 65d, for example, on hydrogenation at 1 atm pressure affords N-phenylcyclooctylsucciniimide 66 in overall yields of 56%. The treatment of 65d with HCl in Et2O, however, have resulted in the formation of N-phenyl(3,5-cyclooctadien-1-yl)succiniimide 67 in 62% yield using mild reaction conditions. The ruthenacycle 65d undergoes ring closure via reductive elimination at high temperature to afford tricyclic [6 + 2] cycloadduct 68. Introduction additives such as PPh3 promotes the production of 68 from 65d, while 1,2-bis(diphenylphosphino)ethane (dppe) does not yield 68, instead stable ruthenacycle 69 is formed (71%) presumably via ligand displacement between COD and dppe. The reaction of 65d with CO in toluene at 1 atm pressure for 3 h have resulted in the isolation of ticarbonyl ruthenacycle complex 70 in an overall yield of 76%. The reaction proceeds with full dissociation of COD ligand from 65d followed with the insertion of three molecules of CO coordinated to Ru along with cycloheptadienyl hapicity change from the change η5 to η3. The theoretical studies have also been carried to support the experimental results (Scheme 27).50
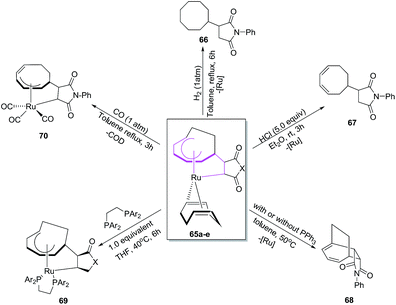 |
| | Scheme 27 Reactivity of ruthenacycle 65. | |
Cu(II) catalysed [6 + 2] cycloaddition reactions
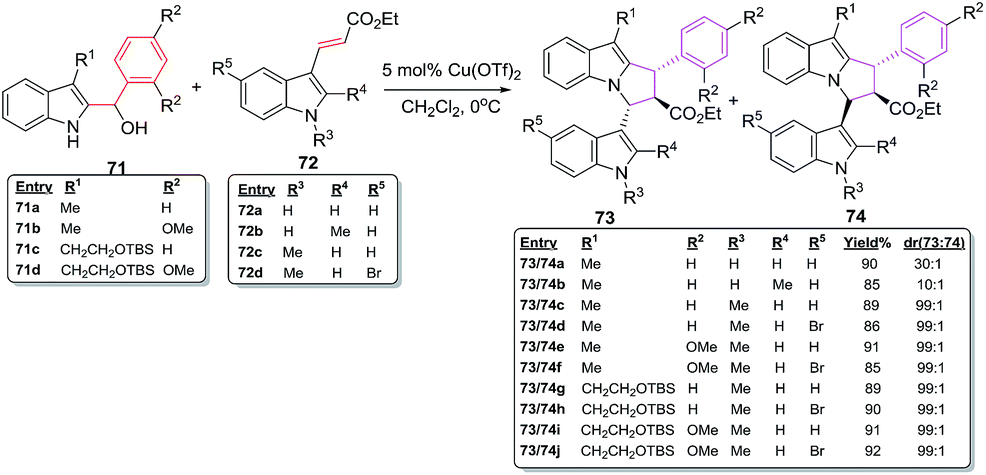
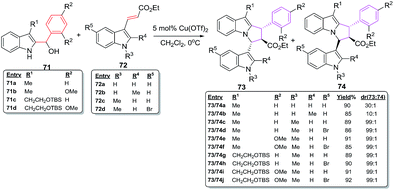
Dethe et al. have recently studied highly regio- and diastereoselective facile, one pot Lewis acid catalysed [6 + 2] cycloaddition reactions for the synthesis of highly substituted pyrrolo[1,2-a]indoles having three contiguous stereocenters. The reactions of indole alcohol 71a–d with conjugated indole ester 72a–e in the presence of 5 mol% Cu(OTf)2 afford pyrroloindoles 73a–j and 74a–j in excellent overall yields of 85–92% with outstanding regio- and diastereoselectivity53 (Scheme 28).
 |
| | Scheme 28 Cu(OTf)2 catalysed [6 + 2] cycloadditions reactions of 71 and 72. | |
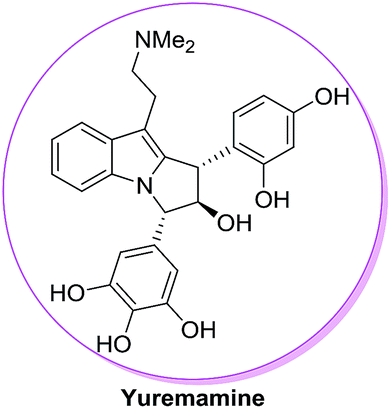
The ester moiety in case of 72 is crucial in controlling diastereoselectivity in these cycloadditions. Other Lewis acid catalysts such as TiCl4, BF3–Et2O, FeCl3, AlCl3, Sn(OTf)2 and Fe(OTf)3 have resulted in poor diastereoselectivity of 11:1, 14:1, 4:1, 3:1, 7:1 and 6:1 for products 73/74. The methodology has also been used for the construction of the fully functionalized tricyclic core of bioactive natural product yuremamine which has been isolated from the stem bark of Mimosa hostilis having hallucinogenic and psychoactive effects54 (Fig. 11).
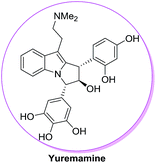 |
| | Fig. 11 Structure of yuremamine. | |
Chromium catalysed [6 + 2] cycloaddition reactions
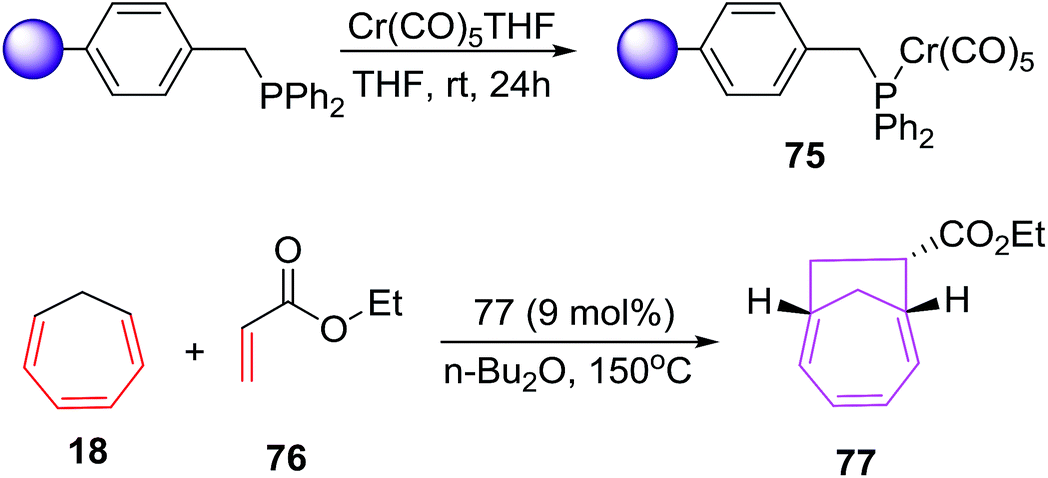
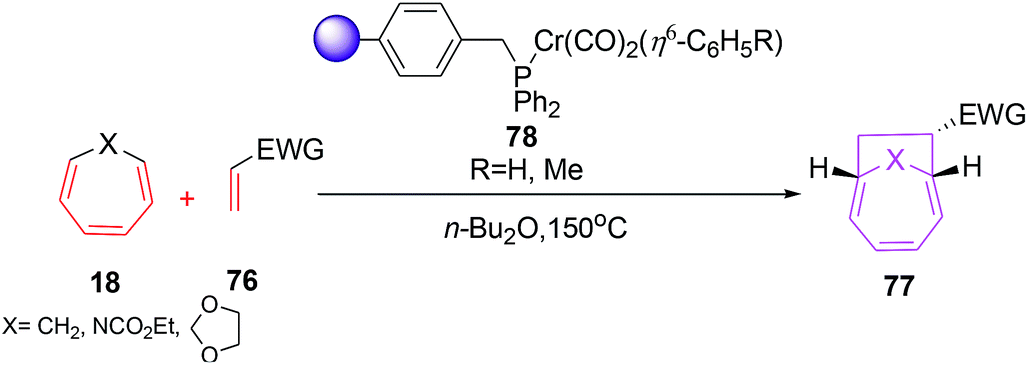
Chromium(0) complexes are also been tested in [6 + 2] cycloadditions and a single report on Cr(0) catalysed [6 + 2] cycloadditions is disclosed by Rigby et al. It involves the development of resin supported chromium(0) catalytic system and their use as catalyst in [6 + 2] cycloadditions of cycloheptatriene (CHT) with variety of alkenes. The resin based chromium catalyst prepared from chloromethylated polystyrene (PS-DVB) (Merrifield resin, 2% cross linked) 75 (9 mol%) catalyses the [6 + 2] cycloadditions of CHT 18 with excess ethyl acrylate 76 to afford bicyclo[4.2.1]nona-2,4-diene 77 in 59% yield. However, the catalytic system has a serious drawback of recyclability owing to thermal denaturation of the catalytic systems (Scheme 29).
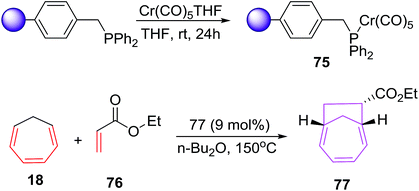 |
| | Scheme 29 [6 + 2] cycloadditions of CHT with ethyl acrylate catalysed by solid supported chromium(0) complex. | |
Replacing chromium coordination sphere to η6-C6H5 has marked effect on its multiple recyclability by simple filtration followed by washing with non polar solvents (Scheme 30).55 The catalyst 78 affords [6 + 2] cycloadditions in excellent yield (64–92%) in the reactions of CHT 18 with various electron rich trienophiles.56
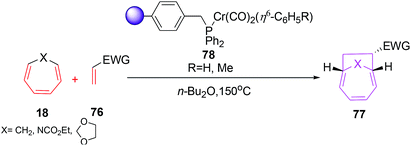 |
| | Scheme 30 [6 + 2] cycloadditions of CHT with ethyl acrylate catalysed by solid supported chromium(0) complex. | |
Conclusion
In conclusion, the present review summarises transition metal catalysed [6 + 2] cycloaddition reactions that have appeared in literature since 2000. Various inter and intramolecular versions have been explored employing various substrates contributing as 6π component promoted by different transition metal catalysts. The developed methodologies have provided a facile access to mono- and bicyclic carbo-/heterocyclic eight membered core structures which are otherwise difficult to achieve. Although, the developed protocols have provided an informal to functionalised ring systems, but, the true potential of [6 + 2] cycloadditions still need to be scrutinized so as to access its regio and stereselective versions under milder reaction conditions. Copper, ruthenium, chromium and platinum based complexes have made entry to the list of metals catalysed [6 + 2] cycloadditions, but the detailed studies on such metals catalysed [6 + 2] cycloaddition involving other prevalent substrates as 6π components still need to be explored. Moreover, the semicyclic variants of 6π components in transition metal catalysed still need to be studied. The studies on [6 + 2] cycloadditions involving different heterodienophiles also need to be explored. Hence, [6 + 2] cycloadditions have an enormous potential for the synthesis of eight membered monocyclic or fused heterocyclic systems.
The authors have tried to recapitulate most of the reports appearing within the stipulated time frame, nonetheless there will always be some experts missing for which we deeply apologize.
Conflicts of interest
There are no conflicts to declare.
Notes and references
-
(a) Advances in Cycloaddition, ed. D. P. Curran, JAI Press, Greenwich, 1994, vol. 1–3 Search PubMed;
(b) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS;
(c) M. Lautens, W. Klute and W. Tam, Chem. Rev., 1996, 96, 49 CrossRef CAS PubMed;
(d) N. E. Schore, Chem. Rev., 1988, 88, 1081 CrossRef CAS.
-
(a) B. M. Trost, Acc. Chem. Res., 2002, 35, 695 CrossRef CAS PubMed;
(b) M. Dai, D. Sarlah, M. Yu, S. J. Danishefsky, G. O. Jones and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 645 CrossRef CAS PubMed.
- E. J. Corey, N. M. Weinshnker, T. K. Schaaf and W. J. Huber, J. Am. Chem. Soc., 1969, 91, 5675 CrossRef CAS PubMed.
- [5+2] cycloaddition references see: H. Pellissiera, Adv. Synth. Catal., 2011, 353, 189 CrossRef and reference cited therein.
-
(a) For [6+2] cycloadditions see: X. Z. Yu, Y. Wang and Y. Wang, Chem.–Asian J., 2010, 5, 1072 CrossRef PubMed and reference cited therein;
(b) For a review on [8+2] cycloadditions, see: V. Nair and K. G. Abhilash, Synlett, 2008, 301 CrossRef CAS.
-
(a) D. J. Faulkner, Nat. Prod. Rep., 1988, 5, 613 RSC;
(b) D. G. I. Kingston, J. Org. Chem., 2008, 73, 3975 CrossRef CAS PubMed;
(c) R. S. Daum, S. Kar and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2007, 6, 865 CrossRef CAS.
- For pleuromutilin, see: P. Brown and M. Dawson, Prog. Med. Chem., 2015, 54, 135 Search PubMed and reference cited therein.
- Z. -K. Yao, J. Li and Z.-X. Yu, Org. Lett., 2011, 13, 134 CrossRef CAS PubMed.
- For Rh(I)-catalysed intramolecular [6+2] cycloadditions, see:
(a) P. A. Wender, A. G. Correa, Y. Sato and R. Sun, J. Am. Chem. Soc., 2000, 122, 7815 CrossRef CAS;
(b) F. Inagaki, K. Sugikubo, Y. Oura and C. Mukai, Chem.–Eur. J., 2011, 17, 9062 CrossRef CAS PubMed;
(c) Y. Oonishi, A. Hosotani and Y. Sato, J. Am. Chem. Soc., 2011, 133, 10386 CrossRef CAS PubMed.
- For Rh(I)-catalyzed [5+2+1] cycloadditions, see:
(a) P. A. Wender, G. G. Gamber, R. D. Hubbard and L. Zhang, J. Am. Chem. Soc., 2002, 124, 2876 CrossRef CAS PubMed;
(b) H. A. Wegner, A. de Meijere and P. A. Wender, J. Am. Chem. Soc., 2005, 127, 6530 CrossRef CAS PubMed;
(c) Y. Wang, J. Wang, J. Su, F. Huang, L. Jiao, Y. Liang, D. Yang, S. Zhang, P. A. Wender and Z.-X. Yu, J. Am. Chem. Soc., 2007, 129, 10060 CrossRef CAS PubMed;
(d) F. Huang, Z.-K. Yao, Y. Wang, J. Zhang and Z.-X. Yu, Chem.–Asian J., 2010, 5, 1555 CrossRef CAS PubMed.
- For Rh(I)-catalyzed [4+2+2] cycloadditions, see
(a) P. A. Evans, J. E. Robinson, E. W. Baum and A. N. Fazal, J. Am. Chem. Soc., 2002, 124, 8782 CrossRef CAS PubMed;
(b) S. R. Gilbertson and B. J. DeBoef, J. Am. Chem. Soc., 2002, 124, 8784 CrossRef CAS PubMed;
(c) P. A. Evans and E. W. Baum, J. Am. Chem. Soc., 2004, 126, 11150 CrossRef CAS PubMed;
(d) P. A. Evans, E. W. Baum, A. N. Fazal and M. Pink, Chem. Commun., 2005, 63 RSC;
(e) P. A. Wender and J. P. Christy, J. Am. Chem. Soc., 2006, 128, 5354 CrossRef CAS PubMed;
(f) B. DeBoef, W. R. Counts and S. R. Gilbertson, J. Org. Chem., 2007, 72, 799 CrossRef CAS PubMed.
- For the synthesis of a monocyclic eight-membered ring by Rh(I) catalyzed hydroacylation, see
(a) A. D. Aloise, M. E. Layton and M. D. Shair, J. Am. Chem. Soc., 2000, 122, 12610 CrossRef CAS;
(b) D. Crepin, J. Dawick and C. Aissa, Angew. Chem., 2010, 122, 630 (Angew. Chem. Int. Ed., 2010, 49, 620) CrossRef.
-
(a) M. E. Maier, Angew. Chem., 2000, 112, 2153 (Angew. Chem., Int. Ed., 2000, 39, 2073) CrossRef;
(b) A. Michaut and J. Rodriguez, Angew. Chem., 2006, 118, 5870 (Angew. Chem., Int. Ed., 2006, 45, 5740) CrossRef;
(c) For an exampleof applying Cope rearrangement in total synthesis of natural product asteriscanolide, see: J. Limanto and M. L. Snapper, J. Am. Chem. Soc., 2000, 122, 8071 CrossRef CAS.
- For uncatalysed [6+2] cycloadditions since year 2000, which are not covered in the current article, see:
(a) O. Yahiaoui, L. F. Pasteka, B. Judeel and T. Fallon, Angew. Chem., Int. Ed., 2018, 57, 2570 CrossRef CAS PubMed;
(b) Z. Wang, Y. Addepalli and Y. He, Org. Lett., 2018, 20, 644 CrossRef CAS PubMed;
(c) R. Hanada, K. Mitachi and K. Tanino, Tetrahedron Lett., 2014, 55, 1097 CrossRef CAS;
(d) N. Coskun, J. Maa, A. Saeed, C. Gartner and I. Erden, Org. Lett., 2011, 13, 5952 CrossRef CAS PubMed;
(e) S. Koya, K. Yamanoi, R. Yamasaki, I. Azumaya, H. Masu and S. Saito, Org. Lett., 2009, 11, 5438 CrossRef CAS PubMed;
(f) T. Sugimura, C.-Y. Im and T. Okuyama, Tetrahedron Lett., 2004, 45, 1519 CrossRef CAS;
(g) B.-C. Hong, Y.-J. Shr, J.-L. Wu, A. K. Gupta and K.-J. Lin, Org. Lett., 2002, 4, 2249 CrossRef CAS PubMed;
(h) K. Takaoka, T. Aoyama and T. Shioiri, Heterocycles, 2001, 54, 209 CrossRef CAS.
- For selected examples:
(a) J. J. Wang and J. H. Wang, Angew. Chem., Int. Ed., 2012, 51, 12334 CrossRef CAS PubMed;
(b) P. Metza, Adv. Synth. Catal., 2009, 351, 3128 CrossRef;
(c) J. R. Kong, C. W. Cho and M. J. Krische, J. Am. Chem. Soc., 2005, 127, 11269 CrossRef CAS PubMed;
(d) M. E. Vander Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS PubMed;
(e) C. H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC;
(f) D. Nečas and M. Kotora, Curr. Org. Chem., 2007, 11, 1566 CrossRef;
(g) M. Tobisu and N. Chatani, Chem. Soc. Rev., 2008, 37, 300 RSC;
(h) S. M. Bonesi and M. Fagnoni, Chem.–Eur. J., 2010, 16, 13572 CrossRef CAS PubMed.
- For selected examples:
(a) S. Hashimotoa, Tetrahedron Lett., 2009, 50, 3675 CrossRef;
(b) S. Shin, A. K. Gupta and C. Y. Rhim, Chem. Commun., 2005, 35, 4429 RSC;
(c) A. J. Padwa, J. Org. Chem., 1995, 60, 6258 CrossRef CAS.
-
(a) C. S. Bryan and M. Lautens, Org. Lett., 2008, 10, 4633 CrossRef CAS PubMed;
(b) M. Lautens and J. Mancuso, Org. Lett., 2002, 4, 2105 CrossRef CAS PubMed;
(c) M. Lautens and M. Yoshida, Org. Lett., 2002, 4, 123 CrossRef CAS PubMed;
(d) J. L. Mascareñas and G. Saya, Angew. Chem., 2010, 122, 10082 CrossRef;
(e) J. L. Mascareñas, G. Bhargava, B. Trillo and M. Araya, Chem. Commun., 2010, 46, 270 RSC;
(f) K. Teruyuki, Molecules, 2010, 15, 4189 CrossRef PubMed;
(g) A. Padwa and D. B. England, J. Org. Chem., 2008, 73, 2792 CrossRef PubMed;
(h) J. M. Mejia-Oneto and A. Padwa, Org. Lett., 2006, 8, 3275 CrossRef CAS PubMed;
(i) A. Padwa, J. Boonsombat, P. Rashatasakhon and J. Willis, Org. Lett., 2005, 7, 3725 CrossRef CAS PubMed;
(j) P. A. Evans, J. E. Robinson, E. W. Baum and A. N. Fazal, J. Am. Chem. Soc., 2002, 124, 8782 CrossRef CAS PubMed;
(k) S. R. Gilbertson and B. DeBoef, J. Am. Chem. Soc., 2002, 124, 8784 CrossRef CAS PubMed;
(l) T. Fukuyama, Y. Ohta, C. Brancour, K. Miyagawa, I. Ryu, A. L. Dhimane, L. Fensterbank and M. Malacria, Chem.–Eur. J., 2012, 18, 7243 CrossRef CAS PubMed;
(m) Z. X. Yu, Y. A. Guo, G. Y. Kang and M. Lin, J. Am. Chem. Soc., 2012, 134, 398 CrossRef PubMed.
- P. A. Wender, A. G. Correa, Y. Sato and R. Sun, J. Am. Chem. Soc., 2000, 122, 7815 CrossRef CAS.
- Y. Oonishi, A. Hosotani and Y. Sato, J. Am. Chem. Soc., 2011, 133, 10386 CrossRef CAS PubMed.
- Y. Oonishi, A. Hosotani and Y. Sato, Angew. Chem., Int. Ed., 2012, 51, 11548 CrossRef CAS PubMed.
- F. Inagaki, K. Sugikubo, Y. Miyashita and C. Mukai, Angew. Chem., 2010, 122, 2252 (Angew. Chem., Int. Ed., 2010, 49, 2206) CrossRef.
- F. Inagaki, K. Sugikubo, Y. Oura and C. Mukai, Chem.–Eur. J., 2011, 17, 9062 CrossRef CAS PubMed.
-
(a) Z. X. Yu, P. A. Wender and K. N. Houk, J. Am. Chem. Soc., 2004, 126, 9154 CrossRef CAS PubMed;
(b) Z. X. Yu, P. H. Y. Cheong, P. Liu, C. Y. Legault, P. A. Wender and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 2378 CrossRef CAS PubMed;
(c) P. Liu, H. Y. Cheong, Z. X. Yu, P. A. Wender and K. N. Houk, Angew. Chem., 2008, 120, 4003 (Angew. Chem., Int. Ed., 2008, 47, 3939) CrossRef;
(d) P. Liu, L. E. Siros, P. H. Y. Cheong, Z. X. Yu, I. V. Hartung, H. Rieck, P. A. Wender and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 10127 CrossRef CAS PubMed.
- P. A. Wender, N. M. Deschamps and R. Sun, Angew. Chem., 2006, 118, 4061 (Angew. Chem., Int. Ed., 2006, 45, 3957) CrossRef.
- M. R. Siebert, J. M. Osbourn, K. M. Brummond and D. J. Tantillo, J. Am. Chem. Soc., 2010, 132, 11952 CrossRef CAS PubMed.
- Ti catalyzed reactions:
(a) K. Mach, H. Antropiusová, P. Sedmera, V. Hanuš and F. Tureček, J. Chem. Soc., Chem. Commun., 1983, 805 RSC;
(b) K. Mach, H. Antropiusová, L. Petrusová, V. Hanuš, F. Tureček and P. Sedmera, Tetrahedron, 1984, 40, 3295 CrossRef CAS;
(c) R. Klein, P. Sedmera, J. Cÿejka and K. Mach, J. Organomet. Chem., 1992, 436, 143 CrossRef CAS.
- Ru catalyzed reactions:
(a) M. Green, S. M. Heathcock, T. W. Turney and D. M. P. Mingos, J. Chem. Soc., Dalton Trans., 1977, 204 RSC;
(b) K. Itoh, K. Mukai, H. Nagashima and H. Nishiyama, Chem. Lett., 1983, 499 CrossRef CAS;
(c) H. Nagashima, H. Matsuda and K. Itoh, J. Organomet. Chem., 1983, 258, C15 CrossRef CAS.
- Mo mediated reactions:
(a) D. G. Bourner, L. Brammer, M. Green, G. Moran, A. G. Orpen, C. Reeve and C. J. Schaverien, J. Chem. Soc., Chem. Commun., 1985, 1409 RSC;
(b) T. Schmidt, F. Bienewald and R. Goddard, J. Chem. Soc., Chem. Commun., 1994, 1857 RSC.
- Co catalysed cyclotrimerizations of alkynes:
(a) A. Geny, N. Agenet, L. annazzo, M. Malacria, C. Aubert and V. Gandon, Angew. Chem., Int. Ed., 2009, 48, 1810 CrossRef CAS PubMed;
(b) G. Hilt, C. Hengst and W. Hess, Eur. J. Org. Chem., 2008, 2293 CrossRef CAS;
(c) G. Hilt, T. Vogler, W. Hess and F. Galbiati, Chem. Commun., 2005, 1474 RSC;
(d) L. Doszczak, P. Fey and R. Tacke, Synlett, 2007, 753 CAS;
(e) H. T. Chang, M. Jeganmohan and C. H. Cheng, Org. Lett., 2007, 9, 505 CrossRef CAS PubMed;
(f) N. Saino, F. Amemiya, E. Tanabe, K. Kase and S. Okamoto, Org. Lett., 2006, 8, 1439 CrossRef CAS PubMed;
(g) M. S. Wu, M. Shanmugasundaram and C. H. Cheng, Chem. Commun., 2003, 718 RSC;
(h) L. Yong and H. Butenschön, Chem. Commun., 2002, 2852 RSC;
(i) T. A. Sugihara, Wakabayashi, Y. Nagai, H. Takao, H. Imagawa and M. Nishizawa, Chem. Commun., 2002, 576 RSC;
(j) J. H. Rigby, M. S. Laxmisha, A. R. Hudson, C. H. Heap and M. J. Heeg, J. Org. Chem., 2004, 69, 6751 CrossRef CAS PubMed.
-
(a) J. H. Rigby, K. M. Short, H. S. Ateeq and J. A. Henshilwood, J. Org. Chem., 1992, 57, 5290 CrossRef CAS;
(b) J. H. Rigby, H. S. Ateeq, N. R. Charles, J. A. Henshilwood, K. M. Short and P. M. Sugathapala, Tetrahedron, 1993, 49, 5495 CrossRef CAS;
(c) J. H. Rigby, M. A. Kondratenko and C. Fiedler, Org. Lett., 2000, 2, 3917 CrossRef CAS PubMed;
(d) J. H. Rigby, L. M. Mann and B. J. Myers, Tetrahedron Lett., 2001, 42, 8773 CrossRef CAS;
(e) C. G. Kreiter, E. Michels and H. Kurz, J. Organomet. Chem., 1982, 232, 249 CAS.
-
(a) J. S. Ward and R. Pettit, J. Am. Chem. Soc., 1971, 93, 262 CrossRef;
(b) M. Green, S. M. Heathcock, T. W. Turney and D. M. P. Mingos, J. Chem. Soc., Dalton Trans., 1977, 204 RSC;
(c) A. J. Pearson and X. Wang, Tetrahedron Lett., 2005, 46, 3123 CrossRef CAS.
- See ref. 26a and b J. F. Kaagman, M. Rep, M. Horacek, P. Sedmera, J. Cejka, V. Varga and K. Mach, Collect. Czech. Chem. Commun., 1996, 61, 1722 CrossRef CAS . and references therein.
-
(a) N. Toselli, D. Martin, M. Achard, A. Tenaglia, T. Bürgi and G. Buono, Adv. Synth. Catal., 2008, 350, 280 CrossRef CAS;
(b) M. Achard, M. Mosrin, A. Tenaglia and G. Buono, J. Org. Chem., 2006, 71, 2907 CrossRef CAS PubMed;
(c) M. Achard, A. Tenaglia and G. Buono, Org. Lett., 2005, 7, 2353 CrossRef CAS PubMed.
- M. Achard, M. Mosrin, A. Tenaglia and G. Buono, J. Org. Chem., 2006, 71, 2907 CrossRef CAS PubMed.
- H. Clavier, K. L. Jeune and G. Buono, Org. Lett., 2011, 13, 308 CrossRef CAS PubMed.
- X. Zhang, J. Wang, H. Zhao, H. Zhao and J. Wang, Organometallics, 2013, 32, 3529–3536 CrossRef CAS.
- D. B. Ramachary and V. V. Narayana, Eur. J. Org. Chem., 2011, 3514 CrossRef CAS.
- M. Achard, A. Tenaglia and G. Buono, Org. Lett., 2005, 7, 2353 CrossRef CAS PubMed.
-
(a) O. Pardigon and G. Buono, Tetrahedron: Asymmetry, 1993, 4, 1977 CrossRef CAS;
(b) O. Pardigon, A. Tenaglia and G. Buono, J. Org. Chem., 1995, 60, 1868 CrossRef CAS;
(c) A. Tenaglia, O. Pardigon and G. Buono, J. Org. Chem., 1996, 61, 1129 CrossRef CAS;
(d) O. Pardigon, A. Tenaglia and G. Buono, J. Mol. Catal. A: Chem., 2003, 196, 157 CrossRef CAS;
(e) G. Hilt and K. I. Smolko, Synthesis, 2002, 5, 686 CrossRef;
(f) I. F. Duan, C. H. Cheng, J. S. Shaw, S. S. Cheng and K. F. Liou, J. Chem. Soc., Chem. Commun., 1991, 1347 RSC;
(g) M. S. Wu, M. Shanmugasundaram and C. H. Cheng, J. Chem. Soc., Chem. Commun., 2003, 718 RSC;
(h) P. Binger and S. J. Albus, Organomet. Chem., 1995, 493, C6–C8 CrossRef CAS;
(i) [4 + 2 + 2] cycloaddition of norbornadiene with 1,3-dienes, see: ;
(j) Y. Chen, R. Kiattansakul, B. Ma and J. Snyder, J. Org. Chem., 2001, 66, 6932 CrossRef CAS PubMed;
(k) B. Ma and J. K. Snyder, Organometallics, 2002, 21, 4688 CrossRef CAS;
(l) G. Hilt and K. I. Smolko, Angew. Chem., Int. Ed., 2003, 42, 2795 CrossRef CAS PubMed;
(m) G. Hilt, K. I. Smolko and B. V. Lotsch, Synlett, 2002, 7, 1081 CrossRef.
- H. Clavier, K. L. Jeune, I. D. Riggi, A. Tenaglia and G. Buono, Org. Lett., 2011, 13, 308–311 CrossRef CAS PubMed.
- V. A. D'yakonov, G. N. Kadikova, G. F. Gazizullina, L. M. Khalilov and U. M. Dzhemilev, Tetrahedron Lett., 2015, 56, 2005 CrossRef.
- M. Achard, M. Mosrin, A. Tenaglia and G. Buono, J. Org. Chem., 2006, 71, 2907 CrossRef CAS PubMed.
- K. Mitachi, T. Shimizu, M. Miyashita and K. Tanino, Tetrahedron Lett., 2010, 51, 3983 CrossRef CAS.
- T. Jeffery, Tetrahedron Lett., 1989, 30, 2225 CrossRef CAS.
- R. Hanada, K. Mitachi and K. Tanino, Tetrahedron Lett., 2014, 55, 1097 CrossRef CAS.
- V. A. D'yakonov, G. N. Kadikova, D. I. Kolokol'tsev, I. R. Ramazanov and U. M. Dzhemilev, J. Organomet. Chem., 2015, 794, 23 CrossRef.
- V. A. D'yakonov, G. N. Kadikova, D. I. Kolokol'tsev, I. R. Ramazanov and U. M. Dzhemilev, Eur. J. Org. Chem., 2015, 4464 CrossRef.
- V. A. D'yakonov, G. N. Kadikova, R. N. Nasretdinov, D. I. Kolokol'tsev and U. M. Dzhemilev, Tetrahedron Lett., 2017, 58, 1839 CrossRef.
- S. Tenaglia and S. Gaillard, Angew. Chem., Int. Ed., 2008, 47, 2454 CrossRef PubMed.
- Y. Ura, T. Utsumi, H. Tsujita, K. Weda, T. Kondo and T. Mitsudo, Organometallics, 2006, 25, 2934–2942 CrossRef CAS.
- T. Mitsudo, T. Suzuki, S. W. Zhang, D. Imai, K. Fujita, T. Manabe, M. Shiotsuki, Y. Wantanbe, K. Wada and T. Kondo, J. Am. Chem. Soc., 1999, 121, 1839 CrossRef CAS.
- K. Itoh, K. Mukai, H. Nagashima and H. Nishiyama, Chem. Lett., 1983, 499 CrossRef CAS.
- D. Dethe, R. Boda and S. Das, Chem. Commun., 2013, 49, 3260 RSC.
- J. J. Vepsalainen, S. Auriola, M. Tukianen, N. ropponen and J. C. Callaway, Planta Med., 2005, 71, 1053 CrossRef CAS PubMed.
- J. H. Rigby, M. A. Kondratenko and C. Fiedler, Org. Lett., 2000, 24, 3917 CrossRef.
- J. A. S. Howell, M. G. Palin, P. McArdle, D. Cunningham, Z. Goldschmidt, H. E. Gottlieb and D. Herzoni-Langerman, Organometallics, 1993, 12, 1694 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Vipan Kumar
b,
Vipan Kumar