DOI:
10.1039/C9RA02805F
(Paper)
RSC Adv., 2019,
9, 23425-23434
Specific purification of a single protein from a cell broth mixture using molecularly imprinted membranes for the biopharmaceutical industry†
Received
14th April 2019
, Accepted 11th July 2019
First published on 29th July 2019
Abstract
A surface imprinting method is presented herein for the development of a highly selective yet highly permeable molecularly imprinted membrane for protein separation and purification. The resultant protein imprinted membrane was shown to exhibit great potential for the efficient separation of the template protein from a binary mixture and a cell lysate solution, while maintaining high transport flux for complementary molecules. Bovine Serum Albumin (BSA) and Lysozyme (Lys) were individually immobilized on a cellulose acetate membrane as template molecules. In situ surface crosslinking polymerization was then used for protein imprinting on the membrane for a controlled duration. Both membranes showed high adsorption capacity towards template proteins in the competitive batch rebinding tests. In addition, the adsorption capacity could be greatly enhanced in a continuous permeation procedure, where the resultant membrane specifically adsorbed the template protein for more than 40 h. Moreover, this is the first report of purification of a specific protein from the cell broth mixture using a molecularly imprinted membrane. The protein imprinted membrane enables the transport of multiple non-template proteins with high permeation rate in a complex system, thus opening the way for high efficiency protein separation at a low cost for the industry.
Introduction
Molecularly imprinted polymers (MIPs) have a wide range of applications, such as in the field of separation,1 catalysis,2 drug design,3 analytical chemistry,4,5 biosensing6 and other materials-related areas.7,8 MIPs display molecular selectivity that is comparable to their biological counterparts such as enzymes and antibodies while possessing intrinsic strengths which are absent in biological molecules, such as robustness, reusability, and being inexpensive to prepare.9 As such, MIPs could potentially be used to mimic and replace their biological equivalents. In recent years, the molecular imprinting technique (MIT) has achieved great progress, especially in the separation of small molecules. Challenges still remain in the separation of biomacromolecules due to their flexible, complex and large-sized structures.10 A major challenge lies in the limited mass transfer of the biomolecules in the bulk polymer, which hinders the templates' removal and targets' rebinding. The surface imprinting technique is one of the most efficient strategies to overcome this obstacle.11 Surface imprinted thin films could be employed as shell layers of various kinds of substrates, for instance, nanoparticles,12–14 nanowires15–17 nanofibers,18 nanotubes19 and microspheres.20,21 However, further separation steps might be required, such as dispersion and recollection of core–shell particles in the protein solution.
Membrane separation has been widely used in industrial purification due to its single-step operation procedure, low energy consumption and ease of scaling-up. Besides, it has great potential in protein separation and purification based on the size, charge and other properties of proteins.22–24 However, most of membranes were not tailored specially for a particular protein. Besides, membrane properties were adapted specifically for each separation system.25
In contrast, molecularly imprinted membranes (MIMs) offer researchers opportunities to design membranes that are targeted to specific molecules.26,27 Thereby, a generic MIM can be employed for separating different protein mixtures and systems as long as the desired target molecules are the same. MIMs combine advantages of both the molecular imprinting technology (i.e. high intrinsic selectivity) and the membrane technology (i.e. low energy consumption, ease of scale-up and the potential for continuous operation).26 Recently, MIMs for protein separation have been reported with several strategies, including electrospinning,28 surface grafting29–31 and nanopores incorporation.32 High specific recognition of the template proteins has been achieved. However, drawbacks such as weak binding force and low perm-selectivity, are still challenging the comprehensive membrane performance.33
Consequently, finding new approaches to construct the surface imprinted layer is of great importance to achieve new MIMs with excellent perm-selectivity for protein purification. Furthermore, the separation mechanism of MIMs in the continuous membrane separation process, which differs greatly from that of the static adsorption process, needs to be investigated systematically.
To address the above problems, we herein report a novel protein surface imprinting technique for the preparation of a protein imprinted membrane with high binding capacity to the template protein and high permeability to the non-template protein. Following a similar molecularly imprinting technique based on our previous works,34–36 the template protein was first covalently immobilized on a porous cellulose acetate membrane. Subsequently, redox initiators were spread onto the porous substrate by spin-coating, and then in situ crosslinking polymerization was carried out. Finally, the immobilized template protein was removed by hydrolysis to leave imprinted sites on the membrane (as shown in Fig. 1). In this study, two molecularly imprinted membranes, MIMBSA and MIMLys, were prepared by using bovine serum albumin (BSA) and lysozyme (Lys) as template proteins respectively. The effectiveness of this method was examined through the separation of template proteins from the BSA–Lys binary mixture. In addition, membrane performance was tested in a continuous separation setup, with a complex mixture to simulate an industrial system, consisting of BSA, lysozyme and bacterial cell lysate. Furthermore, membrane separation mechanism of MIMs was investigated.
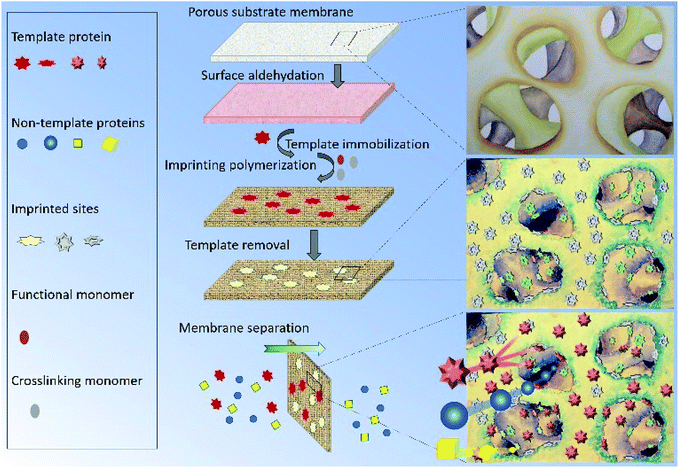 |
| | Fig. 1 Schematic diagram of preparation and separation of the protein-imprinted membrane. | |
Experimental section
Materials
Cellulose acetate (CA) (CA-389-30) was purchased from Eastman Chemical Company, USA. Phosphate buffered saline (PBS, 0.1 M), sodium periodate, bovine serum albumin (BSA), lysozyme (Lys), fluorescein isothiocyanate (FITC), methyl methacrylate (MMA), ethylene glycol dimethacrylate (EGDMA), ammonium persulfate (APS), sodium bisulfite (SBS), sodium dodecyl sulfate (SDS) and trifluoroacetic acid (TFA) were purchased from Sigma Aldrich, USA. N-methyl-2-pyrrolidone (NMP, 99.5%) was purchased from Merck KGaA, Darmstadt, Germany. Tryptone, yeast extract and agar were purchased from BactoTM. Escherichia coli ATCC®19853 was purchased from the American Type Culture Collection, USA.
Preparation of MIMs and NIMs
Surface imprinting for MIMBSA and MIMLys. The substrate porous membrane of cellulose acetate was prepared through phase inversion method. Aldehyde group was then chemically modified onto the membrane surface. After that, the template protein was covalently immobilized onto the porous substrate membrane. The preparation and optimization details of the protein immobilized membrane (BSA-immobilized membrane and lysozyme-immobilized membrane) can be found in the ESI.† Afterwards, methyl methacrylate (MMA, Sigma Aldrich) (3.2 ml) and ethylene glycol dimethacrylate (EGDMA, Sigma Aldrich) (22.4 ml) were mixed at 40 °C under nitrogen. Ammonium persulfate (APS, Sigma Aldrich) and sodium bisulfite (SBS, Sigma Aldrich) (4 mg ml−1) solutions were prepared separately and filtered through a poly(ether sulfone) filter with the pore size of 0.45 μm. The initiators were then coated onto the surface of the-BSA-immobilized membranes or the-lysozyme-immobilized membranes using a spin-coater (Laurel MSC-650) under nitrogen. 4 ml of the initiator solution was used for coating of each membrane. And the resultant membrane was immediately dipped into the monomer solution for the redox polymerization in a water bath (40 °C). The duration of the polymerization reaction was adjusted to control the thickness of the molecularly imprinted polymer layer. Upon completion, the membranes were washed twice with 10% sodium dodecyl sulfate (SDS, Sigma Aldrich) to remove any remaining monomers and several times with de-ionized water to remove the remaining SDS.
Template removal. The immobilized template BSA or lysozyme molecules were removed by acid hydrolysis. Membranes prepared above were shaken in 1 M sulfuric acid. A sample of the acid was analyzed hourly using UV-vis spectrophotometry (Biochrom Libra S32) until no change in the absorption was detected, indicating that all of the template protein molecules had been removed. Upon completion, the membrane was washed with de-ionized water until the pH of the washing water was close to neutral.
Preparation of non-imprinted membrane (NIM). NIMs were prepared as control. Preparation of NIMs follows that of MIMs described above except that no template protein was used. The polymerization duration of NIMBSA was the same with MIMBSA, and polymerization duration of NIMLys was the same with MIMLys.
Structural characterization of membranes
Membrane structures before and after aldehyde functionalization were monitored using a FTIR Spectrum 2000 (PerkinElmer) with an attenuated total reflection (ATR-FTIR) technique. The surface elemental composition of the membranes after each surface modification step were determined through X-ray photoelectron spectroscopy (XPS) (AXIS HIS, 165 Ultra, Shimadzu). Surface roughness changes of membranes after each modification step were analyzed using a Nanoscope IIIa AFM (Veeco) in tapping mode. Morphological observation of the membranes was conducted using a field emission scanning electron microscopy (FESEM) (JSM-6700F, JEOL). Membranes were first freeze-dried and then fractured in liquid nitrogen. The fractured membranes were then mounted onto the sample holder using conductive carbon adhesive tape. The samples were sputtered with platinum before FESEM observation. The nitrogen distribution of the protein-adsorbed membranes was measured using an energy dispersive X-ray (EDX, Oxford Instruments, Aztec X-MaxN). All EDX analyses were performed using the line scan mode for 150 s along the cross-section of the MIMs. The pore size of the original CA membrane, MIMs and NIMs were characterized via neutral solute rejection by using a dead-end permeation cell, and detailed technical descriptions can be found in the ESI.†
Membrane performance characterization
Protein rebinding tests. In the membrane adsorption experiment of MIMBSA, 10 ml of 0.1 M PBS (phosphate buffered saline, Sigma Aldrich) solution was used to prepare the BSA solution with an initial concentration of 2.5 mg ml−1 (36 nmol ml−1). The MIMBSA was shaken in the BSA solution at room temperature for 24 h. The amount of BSA adsorbed by the tested membrane after 24 h was determined via eqn (S1) (see ESI†). BSA concentrations in the solution before and after rebinding were determined using Agilent 1200 series HPLC with a C18, 4.6 × 250 mm, reversed phase column (Grace Vydac, Alltech). Samples were prefiltered with a 0.2 μm filter before analyzed by HPLC. Two mobile phases, (A) ultrapure water with 0.1 vol% trifluoroacetic acid (TFA, Sigma Aldrich) and (B) 80 vol% acetonitrile (Fisher Scientific) and 20 vol% water with 0.09 vol% TFA, were used for linear gradient elution. The flow rate of the mobile phase was set at 1 ml min−1 with solvent B increasing from 25 to 36% (v/v) in 10 min. The analyte injection volume was 40 μl and the column temperature was set at 40 °C. Samples were analyzed by UV-vis absorption at a wavelength of 220 nm. The NIMBSA were also subjected to the same rebinding test for control purposes. The protein rebinding test of MIMLys is the same with that of MIMBSA except that 0.54 mg ml−1 (36 nmol ml−1) lysozyme solution was used instead of 2.5 mg ml−1 (36 nmol ml−1) BSA solution. The NIMLys were subjected to the same rebinding test for control purpose. All tests were conducted in triplicate.
Adsorption kinetics study. Adsorption kinetics of MIMs and NIMs were studied using an experimental procedure similar to that of the single rebinding test. The initial BSA concentration was 2.5 mg ml−1 for MIMIBSA and NIMBSA, and the initial lysozyme concentration was 0.54 mg ml−1 for MIMLys and NIMLys. Analytes were drawn from the systems at regular intervals for HPLC analysis. The protein concentration obtained with HPLC were used to determine the adsorption profiles of the membranes over time. The tests were conducted in triplicate.
Adsorption isotherm study. 0.1 M PBS (phosphate buffered saline, Sigma Aldrich) solution was used to prepare the BSA solutions with various initial concentrations. The MIMBSA/NIMBSA with a membrane area of 1.77 cm2 was incubated in 10 ml of the BSA solution with a certain protein concentration at room temperature for 24 h to reach equilibrium. The protein adsorption capacity at equilibrium (Qe) was estimated by measuring the variation of the protein concentration before and after adsorption through UV-vis Absorption Spectroscopy (Biochrom Libra S32). Same experiments were conducted with MIMlys and NIMlys using lysozyme solutions. Data was fitted to the Langmuir isotherm with eqn (1) and to the Freundlich isotherm with eqn (2)37| |
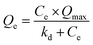 | (1) |
where Qe is the protein adsorption capacity at equilibrium in nmol cm−2, Ce is the free protein concentration in the equilibrium solution in nmol ml−1, Qmax is the maximum adsorption capacity of the template protein in nmol cm−2, kd is the dissociation constant in nmol ml−1| |
lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Qe = lgA + m × lgCe Qe = lgA + m × lgCe
| (2) |
where Qe and Ce are the same with that in eqn (1), A (nmol cm−2) and m are Freundlich parameters.
Competitive batch rebinding tests. In the competitive batch rebinding test, separation factors of the MIMs were studied in a BSA–Lysozyme binary solution in a 0.1 M PBS solution with initial BSA concentration of 2.5 mg ml−1 and Lys concentration of 0.54 mg ml−1 respectively. The MIMBSA or MIMLys with a surface area of 1.77 cm2 was shaken for 24 h in 10 ml of the binary protein solution. Afterwards, the binary solution was analyzed by HPLC using the same procedure described for the batch rebinding experiment. NIMs were tested with the same procedure. All the competitive batch rebinding tests were conducted in triplicate.The separation factor of MIMBSA and NIMBSA was expressed as eqn (3)38 and the separation factor of MIMLys and NIMLys was expressed as eqn (4)39
| |
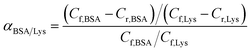 | (3) |
| |
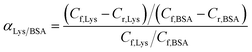 | (4) |
where
Cf,BSA (nmol ml
−1) is the BSA concentration in the feed solution,
Cr,BSA (nmol ml
−1) is the concentration of BSA in the solution after 24 h of competitive batch rebinding,
Cf,Lys (nmol ml
−1) is the Lysozyme concentration in the feed solution, and
Cr,Lys (nmol ml
−1) is the concentration of Lysozyme in the solution after 24 h of competitive batch rebinding.
Permeation study with BSA–Lys binary solution. The membrane performance in the protein permeation experiment was studied with a dialysis permeation cell consisting of one feed chamber and one strip chamber (Fig. S1, see ESI†). The membrane sample was clamped between these two chambers.40 The feed solution was a BSA–Lys binary mixture in 0.1 M PBS solution with initial concentration of 36 nmol ml−1 for BSA and Lys respectively. The strip solution was a 0.1 M PBS solution. Both solutions were stirred at 80 rpm using Teflon impellers. 0.5 ml of samples from the feed and the strip chambers were collected periodically for HPLC analysis separately. The tests were conducted in triplicate. Protein permeation rate J (nmol cm−2 h−1) of the membranes was defined as:| |
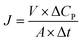 | (5) |
where ΔCP (nmol ml−1) is the concentration change of the solute in the strip chamber, Δt (h) is the permeation time, V (ml) is the stripping volume, and A (cm2) is the effective membrane area, which is 1.77 cm2 in this experiment.
Preparation of bacterial lysate solution. Medium including solid media and liquid media were prepared as follows. Bottom agar solid media was prepared by firstly mixing 1 g of tryptone (BactoTM), 0.5 g of yeast extract (BactoTM), 1 g of sodium chloride (Merck KGaA, Darmstadt, Germany) and 1.5 g of agar (BactoTM) with 100 ml DI water. The mixture was then autoclaved for the purpose of sterilization. At a temperature of 50 °C, the medium was poured into sterile disposable Petri dishes and allowed to solidify. The same procedure was repeated for preparing the top agar medium having 0.5% of agar. The liquid broth medium was also prepared in a similar fashion without the addition of the agar for propagating the host bacterium. The bacterium used in this study was Escherichia coli ATCC®19853. A starter culture of the bacteria was prepared by picking an isolated colony from stock agar plates and grown for 6 h. A 50 ml broth culture was prepared by inoculating with 0.5 ml of the 6 h culture and grown for 18 h. All the broth cultures were grown at 37 °C while shaking at 300 rpm. The cells were then collected by centrifuging at 9000 rpm. 10 ml broth media was added for harvesting of the cells. Then, mixture of the cell and media was put into a 4 °C refrigerator followed by 1 h thawing at room temperature. The freeze–thaw cycles were repeated 3 times. Disruption of cells was performed using an ultrasonic processor (VCX 130, Sonics and Materials) at 20 kHz equipped with a needle titanium alloy Ti-6Al-4V probe of 6 mm diameter and 113 mm length (Model CV18 6085). The disruption period was 3 s with 7 s intervals in an ice bath for 10 min and the acoustic power was 91 W. Subsequently, the mixture of bacterial lysate and the culture medium was filtered using a membrane with pore size of 0.22 μm and stored at 4 °C for future use. The concentration of the resultant lysate solution (in mg ml−1) was tested through weighting the residue after drying the solution.
Permeation study with a complex protein mixture. The permeation study with a complex protein mixture was carried out with the dialysis permeation cell. 1 ml of bacterial lysate solution prepared above was added into 34 ml of BSA–Lys binary solution with an initial BSA and Lys concentration of 36 nmol ml−1 respectively to prepare the feed solution. The strip solution was a 0.1 M PBS solution. Both solutions were stirred at 80 rpm using Teflon impellers. 0.5 ml samples of both strip and feed solutions were collected periodically for HPLC analysis. The tests were conducted in triplicate. The concentration of BSA, lysozyme and bacterial lysate in each sample were tested by HPLC and calculated by Peak Fitting Module of Origin software. Permeation rates of BSA and Lys were defined in eqn (5). Permeation rate of bacterial lysate JBac (mg cm−2 h−1) were defined in eqn (6):| |
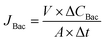 | (6) |
where ΔCBac (mg ml−1) is the bacterial lysate concentration change of the solute, Δt (h) is the permeation time, V (ml) is the stripping volume and A (cm2) is the effective membrane area, which is 1.77 cm2 in this experiment.
Results and discussion
Synthesis of membranes
Cellulose acetate (CA) membrane was used as the substrate for surface chemical modification. The fabrication of the MIM began by immobilizing the template protein onto the CA membrane substrate. The appearance of the characteristic aldehyde peak at ∼1720 cm−1 in the FTIR spectrum proved the successful modification of the membrane using NaIO4-generated aldehyde groups from the hydroxyl groups of the CA (Fig. S2†). The maximum number of aldehyde groups on the CA membrane surface was generated to maximize the density of the immobilized proteins on the CA substrates (Fig. S3, see ESI† for details). Subsequently, an in situ surface crosslinking polymerization on the protein-immobilized CA membrane was applied and followed by hydrolysis to remove the template protein. The duration of the polymerization reaction was used to control the thickness of the imprinting polymer layer. Specifically, if the polymerization time was too short, the thickness of the polymer film may not be sufficient to imprint the shape of the template protein. On the contrary, an excessive polymerization imprinting time would form denser polymer monoliths that could completely cover and permanently entrap the template proteins. The AFM images of original CA membrane and a series of non-imprinted membranes (NIMs) in Fig. 2 indicate average roughness values of 1.7 nm before polymerization and 2.4, 3.2, 5.9 and 13.3 nm after polymerization for 5, 8, 14 and 18 h respectively. The thickness of the imprinted polymer layer after 14 h of polymerization was estimated to be around 4.2 nm by comparing with the average roughness of the original CA membrane, which suggests a high possibility of fully covering the BSA template as the hydrodynamic radius of BSA is 4.5 nm.41 Therefore, the 8 h polymerization time was chosen in subsequent studies, to balance between efficiency of the imprinting process and the surface imprinting coverage for fabrication of the MIMBSA. By using a similar estimation, the 5 h polymerization provided a 0.7 nm imprinting polymer layer which was used to imprint the Lys template molecules with a hydrodynamic radius of 1.9 nm.42 The reduction in nitrogen content detected after protein removal indicated that 83% and 75% of the immobilized template protein molecules was removed via acidic hydrolysis for both MIMBSA and MIMLys respectively (Table S1†).
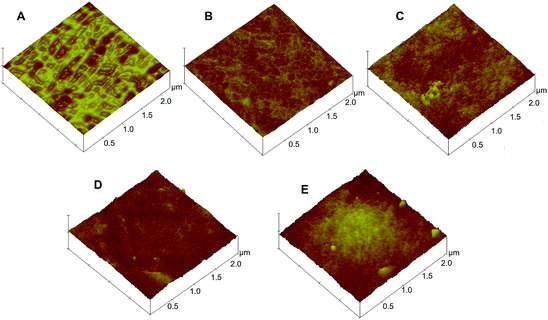 |
| | Fig. 2 Surface morphology of membranes by atomic force microscopy (AFM): (A) original CA membrane, and NIMs with various polymerization duration of (B) 5 h; (C) 8 h; (D) 14 h; (E) 18 h. | |
Structural characterization
FESEM images in Fig. 3 show the aldehyde functionalized membranes and of those after in situ crosslinking polymerization. The comparison among the more detailed morphologies in Fig. 3B, E and H suggests that both the substrate underlayer and the top skin layer of the post-imprinted membrane are slightly thicker and denser than those before imprinting. Such morphological change is more pronounced in MIMBSA, indicating that the imprinting layer on MIMBSA is thicker than MIMLys. The comparison among Fig. 3C, F and I shows higher roughness on the surfaces of the membranes after imprinting. These morphological changes observed in the post-imprinted membrane suggests that in situ crosslinking polymerization might have taken place on both the skin layer and the substrate under layer due to the porous structural nature of the membrane.
 |
| | Fig. 3 Membranes morphology by FESEM. The cross-sections (A and B) and the top surface (C) of the aldehyde functionalized membrane; the cross-sections (D and E) and the top surface (F) of the MIMLys; the cross-sections (G and H) and the top surface (I) of the MIMBSA. | |
Membrane performance
Protein batch rebinding tests. Protein batch rebinding tests of MIMs and NIMs were performed at an initial template protein concentration of 36 nmol ml−1. More template proteins were observed to be adsorbed on MIMs than on NIMs (Fig. S4†). The saturated adsorption amount of BSA on MIMBSA was 5.8 nmol cm−2, which was about 6-fold higher than that of the NIMBSA (0.9 nmol cm−2). Similarly, the saturated adsorption amount of lysozyme on MIMLys was 4.0 nmol cm−2, which was about 3.6-fold higher than that of NIMLys (0.7 nmol cm−2). Thus, both of the protein imprinted membranes exhibit high affinities towards the template molecules. Based on the saturated adsorption amounts of MIMs and NIMs, the imprinting efficiency can be calculated as eqn (7):43| |
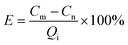 | (7) |
where Qi is the theoretical maximum number of imprinted sites on MIM, which can be calculated from the maximum immobilized template protein and the removal efficiency during membrane synthesis, and Cm and Cn are the apparent maximum number of binding sites from the rebinding test on the MIMs and the NIMs, respectively. The imprinting efficiencies were thus calculated to be 98% and 76% for MIMBSA and MIMLys, respectively. The slightly lower imprinting efficiency of MIMLys can be attributed to the smaller size of Lysozyme as compared with BSA. In order to remove the template, the optimum thickness of the MIP layer should be controlled to be less than the size of Lysozyme. Due to the thin imprinting layer of only 0.7 nm, it is highly possible that some parts of the coated layer may be too thin to suitably imprint the shape of lysozyme.
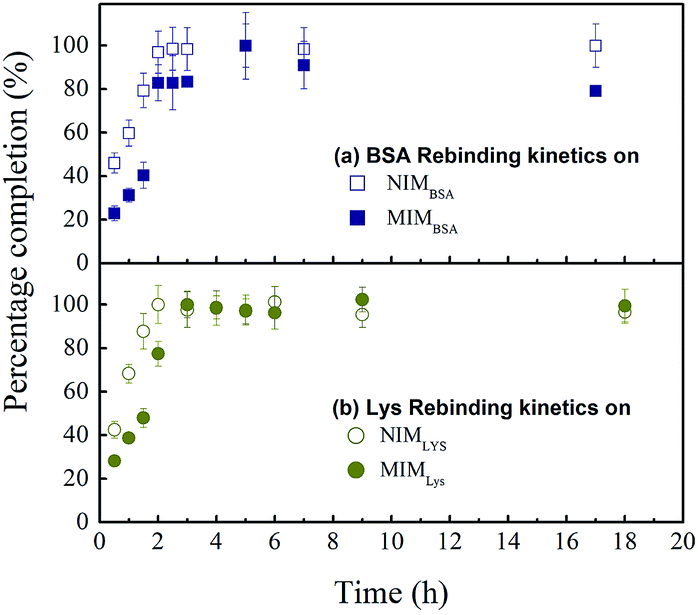
Rebinding kinetic. Rebinding kinetic is an important index for practical MIM application. Adsorption processes are often accompanied by other processes like conformational changes or lateral interaction changes, resulting in a delay in reaching adsorption equilibrium for the system. As observed in Fig. 4, adsorption equilibrium was reached quickly, within 2 h for two non-imprinted membranes NIMBSA and NIMLys, but 5 h for the MIMBSA and 4 h for the MIMLys respectively, suggesting that adsorption might have occurred mainly on the surfaces of the membranes. In this process, the most available binding sites were first occupied followed by a decreasing rate until the system reaches a pseudo-steady state. Interestingly, both MIMBSA and MIMLys underwent a sudden increase in adsorption speed between 1.5 and 2 h. It had been pointed out by van Tassel44 that protein adsorption kinetics was history dependent, i.e. the protein molecules that were first adsorbed might act as templates or nucleating sites to accelerate the adsorption of more protein molecules. On the other hand, the adsorption equilibriums of the NIMs were reached faster than those of the MIMs, which could be attributed to the much lower and non-specific adsorption capacity of the NIMs as well as the time required for the recognition of template protein on the MIMs.38
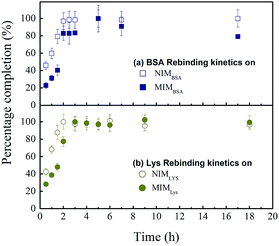 |
| | Fig. 4 Protein rebinding kinetic behaviour of MIMs and NIMs: (a) BSA rebinding on NIMBSA ( ), and MIMBSA( ), and MIMBSA( ); (b) lysozyme rebinding on NIMLys ( ); (b) lysozyme rebinding on NIMLys ( ) and MIMLys ( ) and MIMLys ( ). Values represent mean ± standard deviation (error bars) with n = 3. ). Values represent mean ± standard deviation (error bars) with n = 3. | |
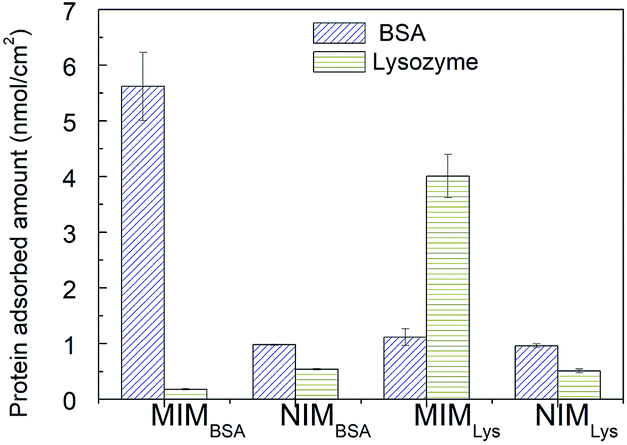
Competitive rebinding tests. Competitive rebinding tests were conducted in a binary BSA–Lys protein solution, where a non-template protein was employed as an adsorption competitor. As, shown in Fig. 6, the difference in protein adsorption between the NIMBSA and the NIMLys was insignificant; however, the template protein adsorption on the MIMs was significantly higher than the non-template protein as well as both proteins on the NIMs. The separation factor of MIMBSA was calculated to be 32 (BSA to Lys), while the separation factor of MIMLys was 3.6 (Lys to BSA), indicating that both MIMBSA and MIMLys possess high recognition ability towards their template molecules in the presence of competitors.
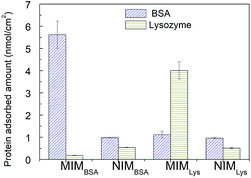 |
| | Fig. 6 Proteins competitive adsorption amount on MIMs and NIMs. Values represent mean ± standard deviation (error bars) with n = 3. | |
Permeation performance and mechanism
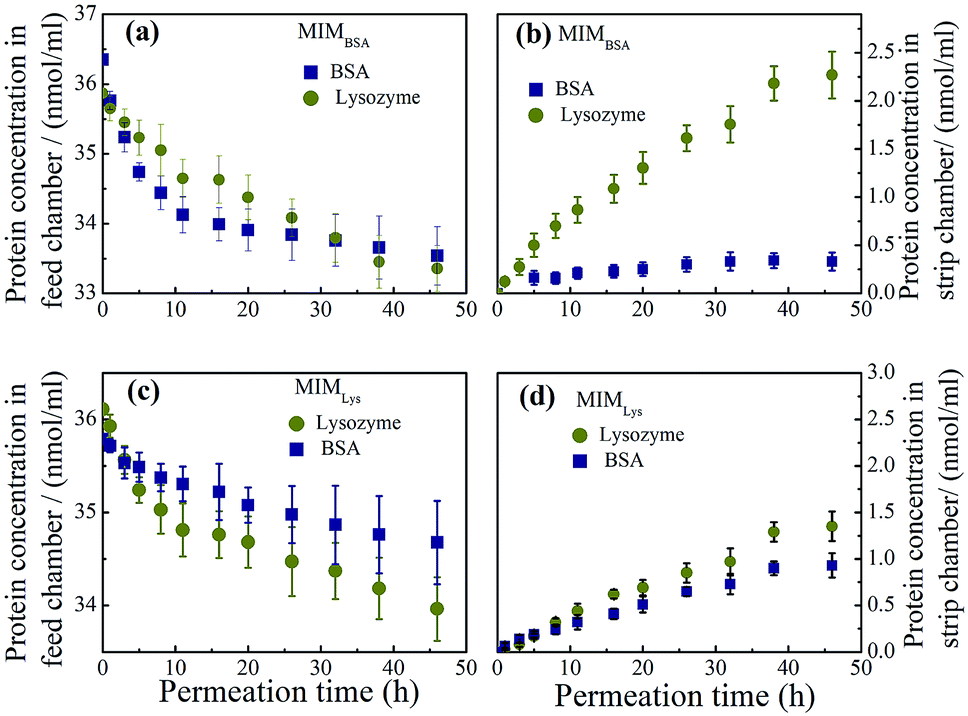
Permeation experiments. Permeation experiments were conducted to further investigate the specificity of MIM for the template molecule, using the binary mixture of BSA and lysozyme at the same molar concentration of 36 nmol ml−1. The mean pore diameters on membrane skin layer for MIMBSA, MIMlys, NIMBSA and NIMlys are 14.5, 19.6, 18.0 and 22.9 nm respectively (Table S2†). As was expected, both BSA and lysozyme molecules were detected in the strip chamber when NIMs were used (Fig. S5†), because NIMBSA and NIMLys had mean pore diameter of ∼18 nm and ∼23 nm respectively, which was much larger than the Stokes radius of both BSA (4.5 nm) and Lys (1.9 nm). The MIMBSA exhibited surprisingly excellent competence to separate a BSA–Lys binary mixture with a negligible permeation of BSA and a high permeation rate of lysozyme. Both the BSA concentration and the Lysozyme concentration in the feed chamber decreased with time (Fig. 7a). In the strip chamber, only trace amount of BSA could be found even after 48 h, although the MIMBSA has a mean pore size of 14.5 nm which should also be large enough for both proteins to pass through. This could partially be explained by the separation mechanism of MIMs,26 where the high selectivity of membranes would be achieved by retarding of the permeation of the template molecules (i.e. BSA) until saturation of the imprinted sites was reached. However, the adsorption equilibrium time for BSA was found to be about 8 h (Fig. 7a), which was 3 h longer than that in the competitive batch adsorption. Thus, it is hypothesized that another reason for the long and sustained separation performance of the MIMBSA is that the BSA molecules accumulate both on the skin layer and in the pores of the membrane during permeation causing the pores to become smaller. This phenomenon is similar to membrane fouling which is common in membrane separation processes and may play a positive role in prolonging the saturation time of BSA adsorption and maintaining a high purity of lysozyme in the other chamber. It was found that, in Fig. 7b, the permeation rate of Lys dropped from 1.7 nmol cm−2 h−1 to 0.8 nmol cm−2 h−1 after ∼8 h permeation when the BSA-adsorption equilibrium was reached. Nonetheless, even after the imprinted sites on MIMBSA were saturated, BSA molecules might still not be able to freely diffuse through the membrane as the pore sizes decreased due to the adsorbed BSA layers.
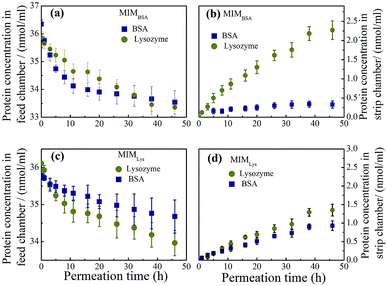 |
| | Fig. 7 Protein concentration profiles in (a) the feed chamber and (b) the strip chamber of the permeation device with MIMBSA during permeation test ( Lys, Lys,  BSA); protein concentration profiles in (c) the feed chamber and (d) the strip chamber with MIMLys during permeation test ( BSA); protein concentration profiles in (c) the feed chamber and (d) the strip chamber with MIMLys during permeation test ( Lys, Lys,  BSA). Values represent mean ± standard deviation (error bars) with n = 3. BSA). Values represent mean ± standard deviation (error bars) with n = 3. | |
On the other hand, the separation performance of MIMLys was not as good as MIMBSA. Intrinsically, due to the lower imprinting efficiency of the MIMLys, the adsorption of lysozyme was not significantly higher than BSA as also found in the batch competitive rebinding test.
The other possible factor was that positive charges on lysozyme under a neutral pH were unfavorable for adsorption. Comparing the two permeation systems, the electrical charge density of lysozyme was much higher than that of BSA at the neutral permeation condition considering the huge differences of the isoelectric point (pI) and the molecular weight (MW) between BSA (pI: 5.1, MW: 67000)45 and Lys (pI: 11, MW: 14400).39 Consequently, a higher electron repulsion could be expected to exist among the lysozyme molecules in solution and those adsorbed on the MIMLys compared with that of the BSA and MIMBSA.46
Permeation mechanism. It is worth noting that the adsorption capacity of the template proteins on MIMs in the permeation test was much higher compared with that in the competitive rebinding test; i.e. MIMBSA was about 9 times higher, and MIMLys was about 4 times higher. The possible reason might be that the imprinted polymer was formed not just on the surface but also beneath the skin layer in the porous structure of CA membranes, as previously observed in FESEM images, thus forming more imprinted binding sites for the template proteins. The distributions of protein adsorption sites along the cross section of the membrane after permeation were investigated through the nitrogen distribution profiles obtained by EDX. A higher quantity of protein was found just beneath the skin layer in the MIMs after permeation than after only adsorption (see ESI Fig. S6†).To further verify the above hypothesis of existence of the imprinted sites beneath the skin layer of MIMs, two pieces of MIMBSA bound with BSA-FITC conjugates were imaged with a confocal laser scanning microscope. One MIMBSA was after 48 h in the competitive rebinding test; and the other MIMBSA was after 48 h in the permeation test with BSA–Lys binary solution. From Fig. 8, it can be observed that more BSA-FITC conjugates were adsorbed on the top layer of MIMBSA after permeation than after rebinding. In addition, BSA-FITC conjugates adsorbed onto the inside layer of the porous substrates after permeation test. These sites were less accessible to BSA-FITC conjugates during the rebinding test.
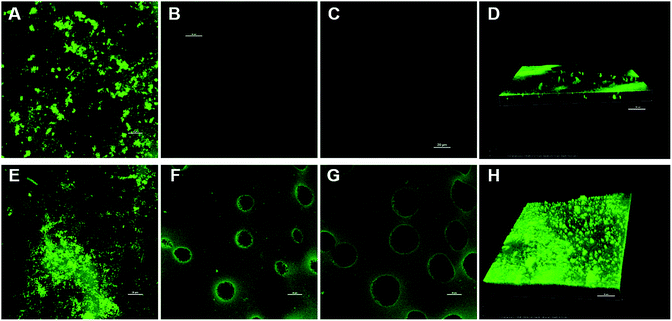 |
| | Fig. 8 Confocal laser scanning microscope images of MIMBSA adsorbed with BSA-FITC. (A) Top surface, (B) inside layer I, (C) inside layer II, and (D) volume view of the MIMBSA adsorbed with BSA-FITC after 48 h of rebinding test; (E) top surface, (F) inside layer I, (G) inside layer II, and (H) volume view of MIMBSA adsorbed with BSA-FITC after 48 h of continuous permeation test. Scale bars on all images denote 20 μm. | |
Permeation purification from bacterial lysate
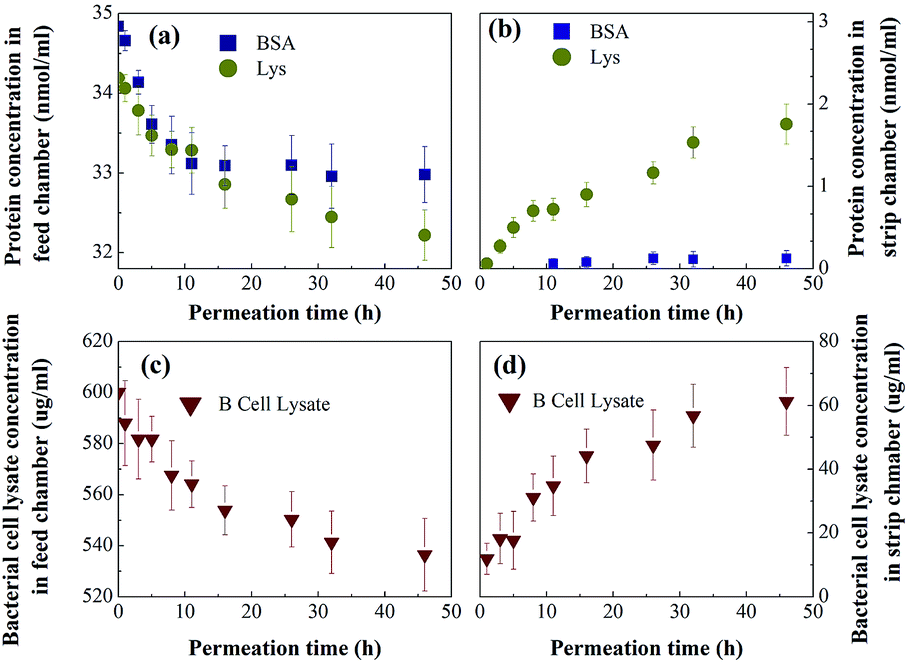
To better mimic the complexity and diversity of a real industrial protein purification, MIMBSA was applied to a complex mixture consisting of BSA, lysozyme and bacterial lysate which was composed of proteins, lipids and nucleic acids. From Fig. 9, both the concentration of lysozyme and bacterial lysate increased in the strip chamber with a permeation rate of 0.85 nmol cm−2 h−1 and 27 μg cm−2 h−1 respectively, and only negligible amount of BSA could be found in the strip chamber even after 48 h. These results illustrated that template BSA was successfully separated from a multicomponent mixture with a high flux of other components. Thus, the protein imprinted membrane possesses a wide potential in the application of protein purification due to its high specification and selectivity in a complex system.
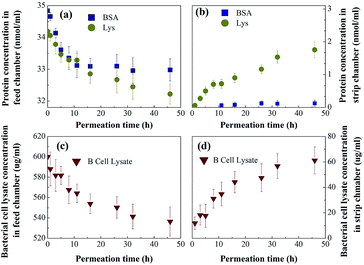 |
| | Fig. 9 Protein ( Lys, Lys,  BSA) concentration profiles in (a) the feed chamber and (b) the strip chamber, and the bacterial cell lysate ( BSA) concentration profiles in (a) the feed chamber and (b) the strip chamber, and the bacterial cell lysate ( ) concentration profile in the (c) feed chamber and the (d) strip chamber with MIMBSA during the permeation tests. Values represent the mean ± the standard deviation (error bars) with n = 3. ) concentration profile in the (c) feed chamber and the (d) strip chamber with MIMBSA during the permeation tests. Values represent the mean ± the standard deviation (error bars) with n = 3. | |
Reusability of the protein imprinted membranes was also tested. The adsorbed protein after the rebinding tests and the protein residue after protein removal experiments were characterized by XPS. As shown in Fig. S7,† the adsorbed amounts of proteins remained the same even after the third rebinding cycle. Specifically, after the third rebinding or removal, the nitrogen element contents on the surface of MIM can still go up to or decrease to almost the same amount as the first rebinding, indicating good reusability of the MIM.
Conclusions
We have shown a well-controlled molecular imprinting method for MIM fabrication. It is based on a simple yet elegant modification of a conventional cellulose acetate membrane, thus enabling common membranes to be used for highly effective protein separation. The above results showed that the protein separation was characterized in detail using adsorption and also continuous assays to elucidate the mechanism of this membrane imprinting method. This work documents that a combination of the specific recognition ability of a molecular imprinted polymer and the size-exclusion effect of a membrane during permeation synergistically enhance protein separation performance in terms of purity. Finally, we have shown that this novel membrane has the ability to separate protein from a complex protein mixture with a high flux thus making it suitable for industrial applications in protein purification. Although only BSA and lysozyme are used as target template in this study, this surface imprinting technique has demonstrated the great potential and viability for the preparation of protein-imprinted membranes using a wide range of other target template molecules.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
The authors appreciate the National Natural Science Foundation of China (NSFC No. 51803176) and NUS Foundation (R279000249646) for financially supporting this research. Special thanks are given to Dr Jingnan Luo and Dr Shirlaine Koh for their suggestions and help in this work.
References
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- C. Alvarez-Lorenzo and A. Concheiro, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 804, 231–245 CrossRef CAS PubMed.
- R. Kecili and C. M. Hussain, Int. J. Anal. Chem., 2018, 8503853 Search PubMed.
- J. Wang, Q. Xu, W. W. Xia, Y. Shu, D. Jin, Y. Zang and X. Hu, Sens. Actuators, B, 2018, 271, 215–224 CrossRef CAS.
- W. S. P. Carvalho, M. Wei, N. Ikpo, Y. Gao and M. J. Serpe, Anal. Chem., 2018, 90, 459–479 CrossRef CAS.
- J. Wackerlig and R. Schirhagl, Anal. Chem., 2016, 88, 250–261 CrossRef CAS PubMed.
- H. Wang, Q. Xu, J. Wang, W. Du, F. Liu and X. Hu, Biosens. Bioelectron., 2018, 100, 105–114 CrossRef CAS PubMed.
- A. Katz and M. E. Davis, Nature, 2000, 403, 286–289 CrossRef CAS PubMed.
- M. J. Whitcombe, I. Chianella, L. Larcombe, S. A. Piletsky, J. Noble, R. Porter and A. Horgan, Chem. Soc. Rev., 2011, 40, 1547–1571 RSC.
- G. De Middeleer, P. Dubruel and S. De Saeger, TrAC, Trends Anal. Chem., 2016, 76, 71–85 CrossRef CAS.
- C. Yang, X. Yan, H. Guo and G. Fu, Biosens. Bioelectron., 2016, 75, 129–135 CrossRef CAS PubMed.
- Y. Hoshino, T. Miyoshi, M. Nakamoto and Y. Miura, J. Mater. Chem. B, 2017, 5, 9204–9210 RSC.
- J. Wackerlig and P. A. Lieberzeit, Sens. Actuators, B, 2015, 207, 144–157 CrossRef CAS.
- H.-H. Yang, S.-Q. Zhang, F. Tan, Z.-X. Zhuang and X.-R. Wang, J. Am. Chem. Soc., 2005, 127, 1378–1379 CrossRef CAS PubMed.
- T. Chen, M. Shao, H. Xu, S. Zhuo, S. Liu and S.-T. Lee, J. Mater. Chem., 2012, 22, 3990–3996 RSC.
- X. Xu, P. Guo, Z. Luo, Y. Ge, Y. Zhou, R. Chang, W. Du, C. Chang and Q. Fu, RSC Adv., 2017, 7, 18765–18774 RSC.
- Y. Li, Q. Bin, Z. Lin, Y. Chen, H. Yang, Z. Cai and G. Chen, Chem. Commun., 2015, 51, 202–205 RSC.
- Z. Luo, W. Du, P. Guo, P. Zheng, R. Chang, J. Wang, A. Zeng, C. Chang and Q. Fu, RSC Adv., 2015, 5, 72610–72620 RSC.
- L. Qin, X.-W. He, X. Yuan, W.-Y. Li and Y.-K. Zhang, Anal. Bioanal. Chem., 2011, 399, 3375–3385 CrossRef CAS PubMed.
- J. Wang, L. Tian, Y. Yan, Y. Liu, Y. Zhang and C. Yang, J. Braz. Chem. Soc., 2018, 29, 2–10 CAS.
- X. Q. Cheng, Z. X. Wang, X. Jiang, T. Li, C. H. Lau, Z. Guo, J. Ma and L. Shao, Prog. Mater. Sci., 2018, 92, 258–283 CrossRef CAS.
- D. Xu, S. Hein and K. Wang, Mater. Sci. Technol., 2008, 24, 1076–1087 CrossRef CAS.
- D. Y. Xing, S. Y. Chan and T.-S. Chung, Green Chem., 2012, 14, 1405–1412 RSC.
- N. F. Ishak, N. A. Hashim, M. H. D. Othman, P. Monash and F. M. Zuki, Ceram. Int., 2017, 43, 915–925 CrossRef CAS.
- M. Yoshikawa, K. Tharpa and Ş.-O. Dima, Chem. Rev., 2016, 116, 11500–11528 CrossRef CAS PubMed.
- R. I. Boysen, L. J. Schwarz, D. V. Nicolau and M. T. W. Hearn, J. Sep. Sci., 2017, 40, 314–335 CrossRef CAS PubMed.
- T. Zhu, D. Xu, Y. Wu, J. Li, M. Zhou, T. Tian, Y. Jiang, F. Li and G. Li, J. Mater. Chem. B, 2013, 1, 6449–6458 RSC.
- D. Liu and M. Ulbricht, RSC Adv., 2017, 7, 11012–11019 RSC.
- Y. Wu, M. Yan, J. Cui, Y. Yan and C. Li, Adv. Funct. Mater., 2015, 25, 5823–5832 CrossRef CAS.
- R.-R. Chen, L. Qin, M. Jia, X.-W. He and W.-Y. Li, J. Membr. Sci., 2010, 363, 212–220 CrossRef CAS.
- Z. Luo, W. Du, P. Guo, P. Zheng, R. Chang, J. Wang, A. Zeng, C. Chang and Q. Fu, RSC Adv., 2015, 5, 72610–72620 RSC.
- W. Li and J. Y. Walz, Sci. Rep., 2014, 4, 4418 CrossRef PubMed.
- W. Xie, F. He, B. Wang, T.-S. Chung, K. Jeyaseelan, A. Armugam and Y. W. Tong, J. Mater. Chem. A, 2013, 1, 7592–7600 RSC.
- N. Sankarakumar and Y. W. Tong, J. Mater. Chem. B, 2013, 1, 2031–2037 RSC.
- N. Sankarakumar and Y. W. Tong, RSC Adv., 2013, 3, 1519–1527 RSC.
- R. J. Ansell, in Molecularly Imprinted Polymers in Biotechnology, ed. B. Mattiasson and L. Ye, Springer International Publishing, Cham, 2015, vol. 150, pp. 51–93 Search PubMed.
- C. J. Tan, H. G. Chua, K. H. Ker and Y. W. Tong, Anal. Chem., 2008, 80, 683–692 CrossRef CAS PubMed.
- K. P. Wilson, B. A. Malcolm and B. W. Matthews, J. Biol. Chem., 1992, 267, 10842–10849 CAS.
- Y. Xiao and T.-S. Chung, J. Membr. Sci., 2007, 290, 78–85 CrossRef CAS.
- U. Böhme and U. Scheler, Chem. Phys. Lett., 2007, 435, 342–345 CrossRef.
- A. Bonincontro, V. Calandrini and G. Onori, Colloids Surf., B, 2001, 21, 311–316 CrossRef CAS PubMed.
- Z. Zhang and B. Wang, J. Appl. Polym. Sci., 2009, 113, 1050–1062 CrossRef CAS.
- P. R. Van Tassel, L. Guemouri, J. J. Ramsden, G. Tarjus, P. Viot and J. Talbot, J. Colloid Interface Sci., 1998, 207, 317–323 CrossRef CAS PubMed.
- B. Jachimska, M. Wasilewska and Z. Adamczyk, Langmuir, 2008, 24, 6866–6872 CrossRef CAS PubMed.
- A. Vinu, M. Miyahara and K. Ariga, J. Phys. Chem. B, 2005, 109, 6436–6441 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02805f |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ad,
Honglei Wangb,
Yen Wah Tong*bc,
Niranjani Sankarakumarb,
Ming Yinc,
Defeng Wu
*ad,
Honglei Wangb,
Yen Wah Tong*bc,
Niranjani Sankarakumarb,
Ming Yinc,
Defeng Wu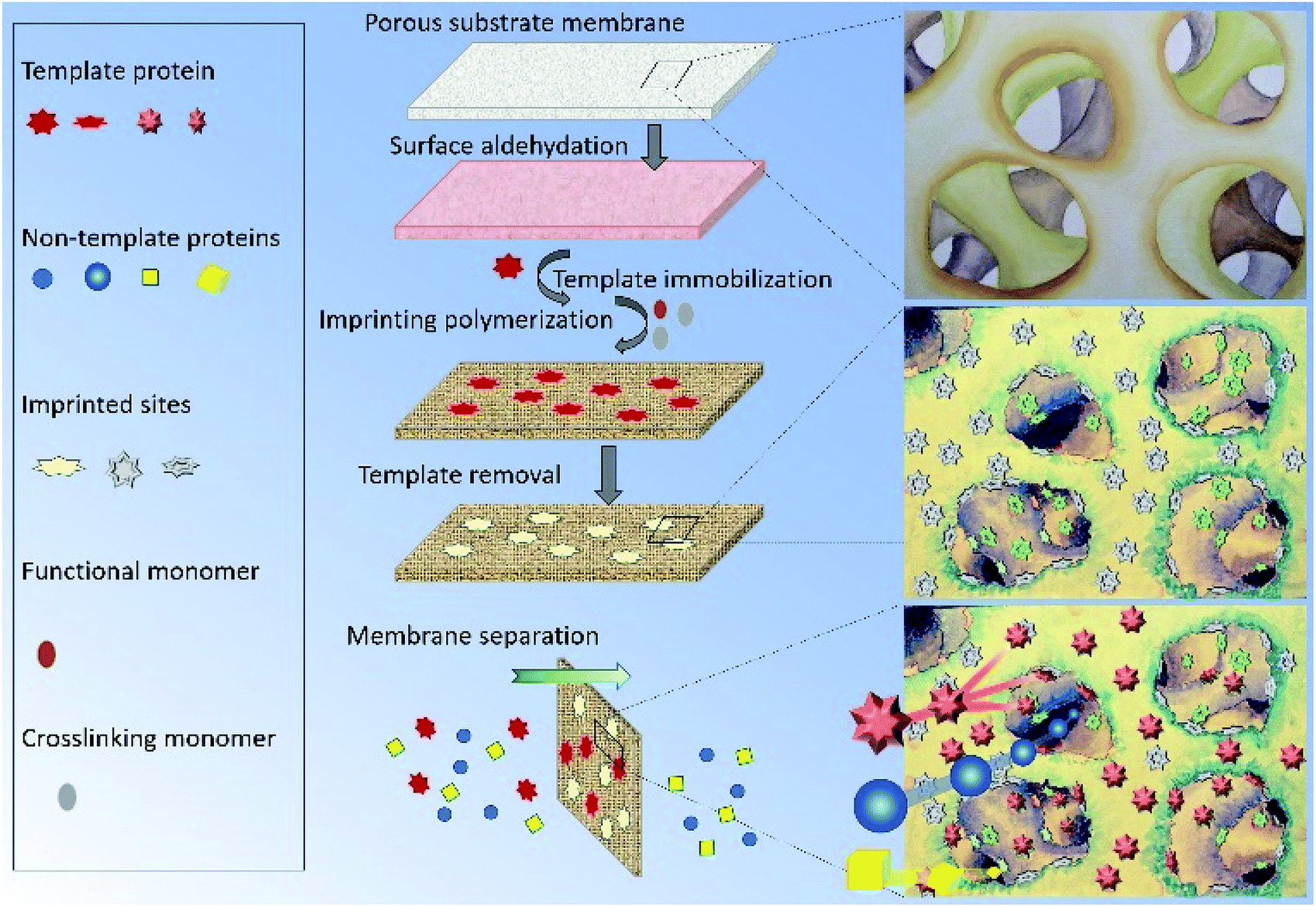
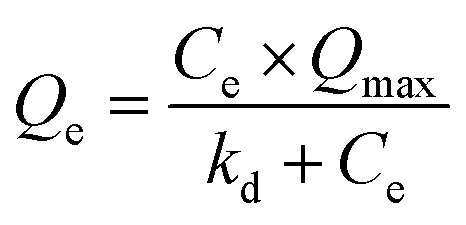
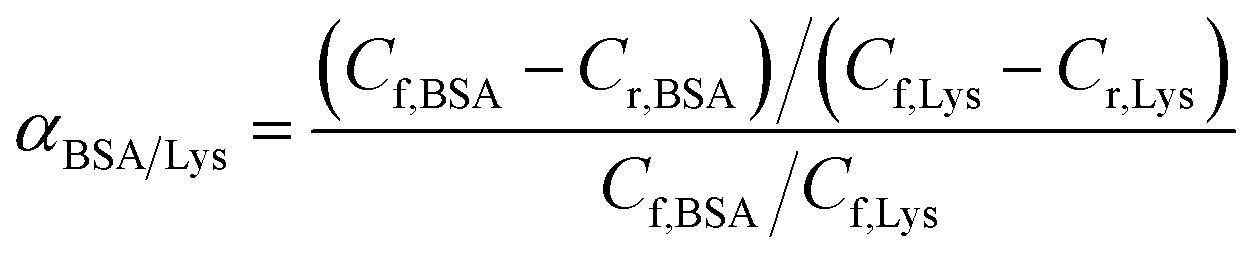
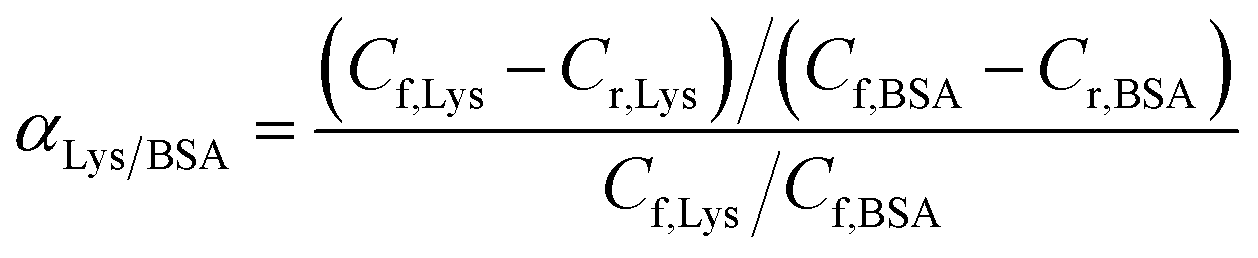
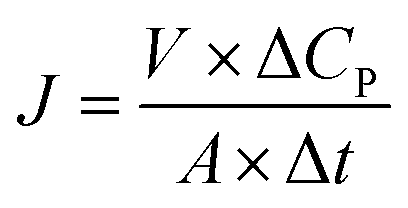
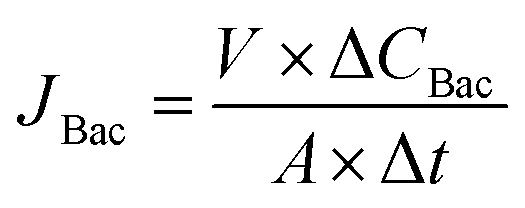



 ), and MIMBSA(
), and MIMBSA( ); (b) lysozyme rebinding on NIMLys (
); (b) lysozyme rebinding on NIMLys ( ) and MIMLys (
) and MIMLys ( ). Values represent mean ± standard deviation (error bars) with n = 3.
). Values represent mean ± standard deviation (error bars) with n = 3.
 Lys,
Lys,  BSA); protein concentration profiles in (c) the feed chamber and (d) the strip chamber with MIMLys during permeation test (
BSA); protein concentration profiles in (c) the feed chamber and (d) the strip chamber with MIMLys during permeation test ( Lys,
Lys,  BSA). Values represent mean ± standard deviation (error bars) with n = 3.
BSA). Values represent mean ± standard deviation (error bars) with n = 3.
 Lys,
Lys,  BSA) concentration profiles in (a) the feed chamber and (b) the strip chamber, and the bacterial cell lysate (
BSA) concentration profiles in (a) the feed chamber and (b) the strip chamber, and the bacterial cell lysate ( ) concentration profile in the (c) feed chamber and the (d) strip chamber with MIMBSA during the permeation tests. Values represent the mean ± the standard deviation (error bars) with n = 3.
) concentration profile in the (c) feed chamber and the (d) strip chamber with MIMBSA during the permeation tests. Values represent the mean ± the standard deviation (error bars) with n = 3.