
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring biomimetic polymer networks towards an unprecedented combination of versatile mechanical characteristics†
Eun Jung Cha‡
,
Dong Soo Lee‡,
Hyohye Kim,
Yun Ho Kim ,
Byoung Gak Kim,
Youngjae Yoo,
Yong Seok Kim and
Dong-Gyun Kim*
,
Byoung Gak Kim,
Youngjae Yoo,
Yong Seok Kim and
Dong-Gyun Kim*
Advanced Materials Division, Korea Research Institute of Chemical Technology, 141 Gajeong-ro, Yuseong-gu, Daejeon 34114, Republic of Korea. E-mail: dgkim@krict.re.kr
First published on 21st May 2019
Abstract
Biomimetic polymeric materials, adopting the basic molecular design principles of biological materials, have been extensively studied in recent years but it is still challenging to combine assorted mechanical characteristics in a single material. Here, we present a simple and effective strategy to prepare mechanically robust yet resilient biomimetic polymer networks by utilizing dual noncovalent and covalent cross-linkings. Tailoring the dual cross-links consisting of thiourea noncovalent interactions and epoxy–amine covalent linkages in the biomimetic polymer networks enables a rare combination of excellent elastic modulus (1.1 GPa), yield stress (39 MPa), extensibility (320%), as well as complete strain and performance recovery after deformation at room temperature. The biomimetic polymer networks also exhibit highly adaptive mechanical properties in response to multiple-stimuli including strain rate, temperature, light, and solvent.
Biological polymeric materials are known for their remarkable combinations of seemingly mutually exclusive mechanical properties, such as stiffness, strength, toughness, resilience, and adaptability.1 However, achieving such combinations of diverse mechanical properties in a single synthetic material remains a challenge despite the great progress in polymeric materials science and technology.2 The understanding and application of the underlying physical/chemical design principles of biological materials are thus of increasing interest to develop biomimetic polymeric materials meeting the disparate physical requirements of emerging applications.1,3 The unique mechanical properties of biological materials originate from their hierarchical structures constructed with weak noncovalent interactions as well as strong covalent bonds.1 A representative example is the skeletal muscle protein titin, which contributes to the mechanical strength, toughness, and resilience of the muscle.1a,4 Titin is a giant protein (3 MDa, ≈1 mm long), composed of ≈300 repeating units of hard immunoglobulin (Ig) tertiary structure domains interconnected with soft amorphous fibronectin type III domains.4 The high molecular weight and hard Ig domains of titin provide the mechanical strength to sustain a mechanical load. The Ig domains also unfold via rupturing noncovalent interactions, releasing hidden lengths, and dissipating energy, when the load reaches a certain level, thereby contributing to the toughness. Without the load, the unfolded domains refold due to the reversibility of the noncovalent interactions, thus exhibiting the resilience.1a,4
Based on the molecular design principles of biological polymeric materials, the sacrificial noncovalent interactions have been employed extensively in recent years for the preparation of mechanically robust hydrogel materials.3b,c While the hydrogel networks utilizing the noncovalent interactions, such as hydrogen (H) bonds,5 host–guest interactions,6 ionic bonds,7 and metal–ligand interactions,8 have much improved multifaceted mechanical properties over the conventional ones, their elastic modulus and strength except for the high extensibility and toughness are limited intrinsically due to the large content of water in the loosely networked structures.3b,c A seminal work by Guan et al. described that a non-hydrated biomimetic polymer based on a quadruple H-bonding 2-ureido-4[1H]-pyrimidone (UPy) motif, as the modular domain-forming mimic of the Ig domains in titin, exhibited relatively high elastic modulus (E ≈ 200 MPa) and yield stress (σy ≈ 5 MPa) as well as the extensibility (strain at break, εb ≈ 100%, without crazing or necking) and resilience (full recovery of shape and properties with time or upon heat treatment).9 Moreover, adaptive property, such as shape memory behavior, was observed from the polymer. After which the application of different noncovalent interactions to non-hydrated polymeric materials has been investigated,3c,10 yet imparting high mechanical stiffness and strength to the polymeric materials while maintaining the toughness and resilience still remains elusive.
Recently, Aida et al. reported amorphous poly(ether-thiourea)s with dense H-bondings, which were mechanically robust yet readily repairable at room temperature.11 The study focused on elucidating the interesting healing behaviors of the polymers in glassy state by investigating the effect of structural elements, such as thiourea/urea H-bonding units and oligoether/alkylene spacers connecting the thiourea units. Although not the main focus in that work, the mechanical properties of TUEG are also notable for potential applications other than the healable materials. Despite its low molecular weight (Mn = 9.5 kg mol−1), the poly(ether-thiourea) with triethylene glycol spacer (TUEG) exhibited high elastic modulus (E ≈ 1.4 GPa), yield stress (σy ≈ 40 MPa), and extensibility (εb ≈ 350%) at 21 °C (≈6 °C lower than its glass transition temperature, Tg). Thus, based on the robust mechanical properties of TUEG, we sought to explore its use, as a sacrificial noncovalent cross-linking building block, in the preparation of non-hydrated biomimetic polymer networks.
Herein, we report a new class of biomimetic polymer networks (BMPNs), consisting of densely H-bonding thiourea units, randomly coiled TUEG chains, and covalently cross-linked epoxy–amine linkages (Fig. 1). By tailoring the chemical composition of the networks, we have controlled their mechanical properties, consequently achieving the intriguing combination of resilience (complete strain and performance recovery within 48 h) as well as high stiffness (E ≈ 1 GPa), strength (σy ≈ 40 MPa), and toughness (UT ≈ 80 MJ m−3, with εb ≈ 300%) at room temperature (26 ± 1 °C). In addition, the BMPNs are found to present adaptive mechanical properties in response to stimuli such as strain rate, temperature, light, and solvent.
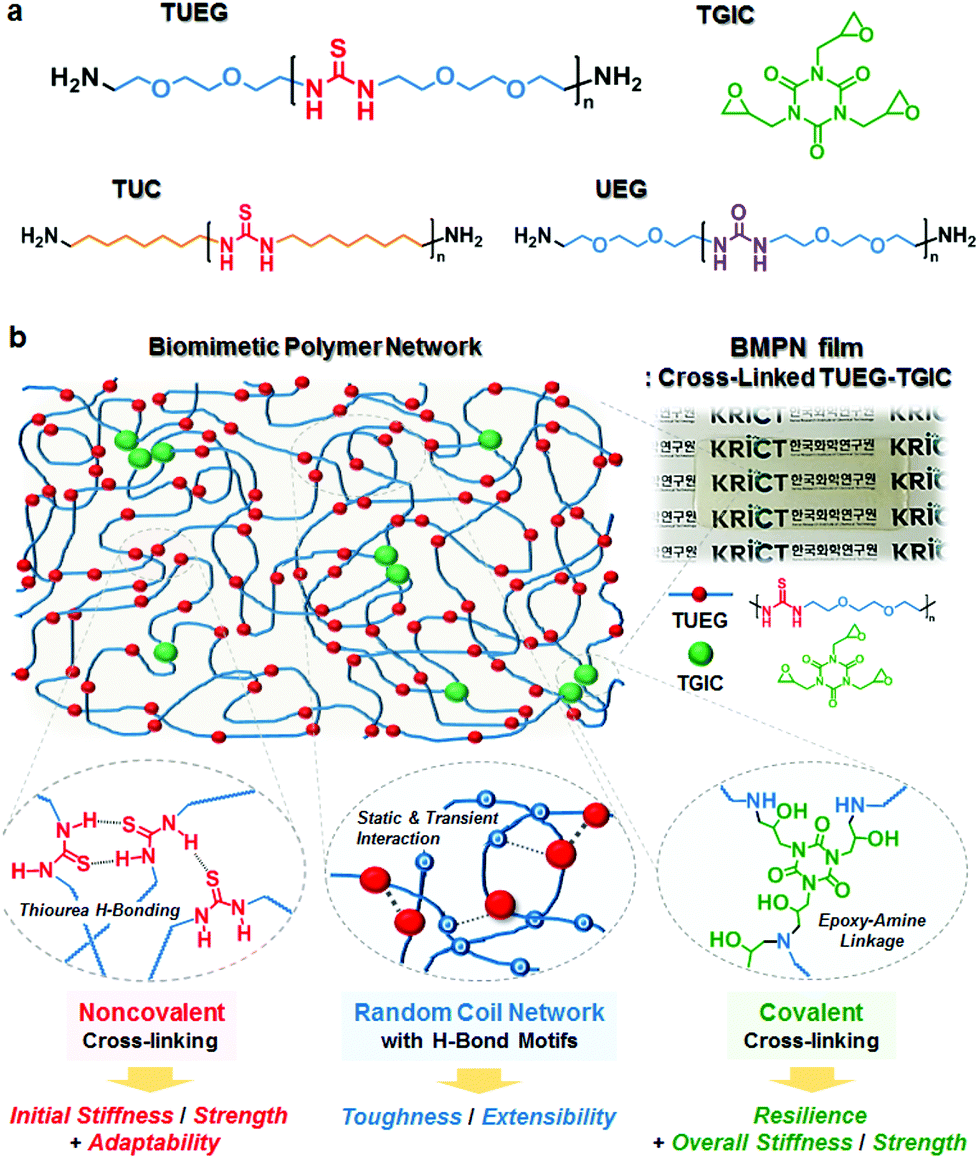 | ||
| Fig. 1 (a) Chemical structures of poly(ether-thiourea) (TUEG), poly(alkylene-thiourea) (TUC), poly(ether-urea) (UEG), and triglycidyl isocyanurate (TGIC). (b) Schematic illustration of biomimetic polymer networks (BMPNs) based on TUEG and TGIC. The smaller red circle in the schematic network represents thiourea unit connected by blue-colored triethylene glycol spacers and the larger green circle represents TGIC cross-linker. | ||
A series of poly(ether-thiourea)s with triethylene glycol spacer (TUEG#, where # indicates the average degree of polymerization) were synthesized by polycondensation of 1,1′-thiocarbonyldiimidazole (TCDI) and 1,2-bis(2-aminoethoxy) ethane (EGDA) (Fig. S1a†).11 1H/13C nuclear magnetic resonance (NMR) and gel permeation chromatography (GPC) revealed that the polymers were successfully synthesized (Fig. S2–S4 and Table S1†). The number-average molecular weights (Mn,NMR, estimated by 1H NMR end group analysis) of TUEGs were controlled to be in the range of 1500–6700 g mol−1, varying the EGDA to TCDI feed ratio. A poly(alkylene-thiourea) with octamethylene spacer (TUC46) and a poly(ether-urea) with triethylene glycol spacer (UEG107) were also prepared for comparison purposes (Fig. S1 and Table S1†).
The presence of both thiourea unit and triethylene glycol spacer in the TUEGs is important for exhibiting the robust mechanical properties. Under an applied tensile load, the TUEG34 undergoes a sequence of deformation mechanisms at progressively higher strains, displaying initial elastic stretching, yielding at ≈10% strain, and large deformation up to ≈500% before rupture at 26 ± 1 °C, whereas both TUC46 and UEG107 are highly brittle and fracture within 10% strain (Fig. S6 and Table S2†). Urea units in UEG107 are known to form H-bonded linear arrays adopting only a trans/trans conformation (Fig. S7a†),11,12 which results in the brittle, semicrystalline structure (Fig. S5†). In contrast, the amorphous nature of TUEGs originates from the non-linear arrays of H-bonded thiourea units adopting cis/trans and strained trans/trans conformations.11,12 Meanwhile, compared to the octamethylene spacer in TUC46, the triethylene glycol spacer in TUEGs also contributes to their large stretch behavior, possibly by working as temporal H-bond acceptors under chain slip motions (Fig. S7b†).11 Therefore, the TUEG chains can serve as the noncovalently cross-linked random coil network in the BMPNs for providing initial stiffness and strength as well as toughness and extensibility.
The biomimetic polymer networks (BMPN#s) in this study are composed of covalently cross-linked TUEG and triglycidyl isocyanurate (TGIC) moieties, wherein the weight percentage of TGIC cross-linker (#) in BMPNs was controlled to be 4, 8, and 17 wt% in order to investigate the effect of the cross-linking density on the mechanical properties (Table 1). BMPN films were prepared via simple solution casting of the TUEG and TGIC mixture onto a Teflon plate, followed by drying on a hot plate at 50 °C overnight and subsequent thermal treatment (at 80 °C for 1 h, 120 °C for 2 h, and 150 °C for 2 h) under vacuum. The molar feed ratio of amine end group of TUEG to epoxy group of TGIC ([amine]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[epoxy]) was fixed at 1:1.5 for all the films, so TUEG7, TUEG18, and TUEG34 with different molecular weights were used for the preparation of BMPN17, BMPN8, and BMPN4, respectively. The completion of curing reaction between epoxy groups of TGIC and terminal amine groups of TUEG could be confirmed by Fourier transform infrared (FT-IR) spectroscopy, where the characteristic peaks of epoxy at 929 and 843 cm−1 disappeared after the thermal treatment (Fig. S9a†).13 The characteristic peak at around 3060 cm−1, corresponding to the NH deformation vibration of nonlinearly H-bonded thiourea units, is still observed in the BMPNs, thus attesting the presence of the amorphous H-bonded TUEG chains in the networks (Fig. S9b†).11 Although the BMPNs exhibit some irregular surface topologies with root-mean-square roughness of 1–3 nm (Fig. S11†), possibly originating from subtle phase separation of the TUEG/TGIC and/or different degree of shrinkage in the networks during the curing reaction, there is no distinct microphase separation or crystallinity as confirmed by X-ray scattering profiles (Fig. S12 and S13†).
:[epoxy]) was fixed at 1:1.5 for all the films, so TUEG7, TUEG18, and TUEG34 with different molecular weights were used for the preparation of BMPN17, BMPN8, and BMPN4, respectively. The completion of curing reaction between epoxy groups of TGIC and terminal amine groups of TUEG could be confirmed by Fourier transform infrared (FT-IR) spectroscopy, where the characteristic peaks of epoxy at 929 and 843 cm−1 disappeared after the thermal treatment (Fig. S9a†).13 The characteristic peak at around 3060 cm−1, corresponding to the NH deformation vibration of nonlinearly H-bonded thiourea units, is still observed in the BMPNs, thus attesting the presence of the amorphous H-bonded TUEG chains in the networks (Fig. S9b†).11 Although the BMPNs exhibit some irregular surface topologies with root-mean-square roughness of 1–3 nm (Fig. S11†), possibly originating from subtle phase separation of the TUEG/TGIC and/or different degree of shrinkage in the networks during the curing reaction, there is no distinct microphase separation or crystallinity as confirmed by X-ray scattering profiles (Fig. S12 and S13†).
| Sample | WTGICa | νeb (mol m−3) | fgc | Ed (GPa) | σyd (MPa) | εbd (%) | UTd (MJ m−3) |
|---|---|---|---|---|---|---|---|
| a Weight fraction of TGIC in the BMPNs.b Cross-linking density, calculated by νe = E′/3RT, where E′, R, and T are the storage modulus, universal gas constant, and absolute temperature in the rubbery region (ca. 353.15 K), respectively.c Gel fraction, obtained by fg = Wa/Wd, where Wd and Wa are the weights of dried film before and after N,N-dimethylformamide (DMF) solvent extraction.d Determined from tensile testing at room temperature and humidity (26 ± 1 °C, RH 45 ± 8%) and a strain rate of 0.013 s−1, where E, σy, εb, and UT are elastic modulus, yield stress, strain at break, and toughness, respectively. | |||||||
| BMPN4 | 0.04 | 53 | 0.63 | 0.9 ± 0.0 | 29 ± 2 | 370 ± 40 | 79 ± 7 |
| BMPN8 | 0.08 | 110 | 0.77 | 1.1 ± 0.1 | 39 ± 2 | 320 ± 30 | 83 ± 9 |
| BMPN17 | 0.17 | 637 | 0.91 | 1.4 ± 0.2 | 61 ± 2 | 12 ± 2 | 4 ± 1 |
The integrity of the networks, in terms of cross-linking density and gel fraction, monotonically increases with increasing the TGIC content in the BMPNs (Table 1), which results in gradual increase of Tg of the BMPNs (Fig. S14b†).14 In comparison to the TUEG34 (Table S2 and Fig. S6†), the BMPNs with covalent cross-links exhibit similar deformation behaviors under the tensile load, but have much improved elastic modulus (E), yield stress (σy), and toughness (UT) (Table 1 and Fig. S15†). Moreover, the mechanical properties of BMPNs could be controlled by the cross-linking density. The elastic modulus and yield stress tend to increase as the cross-linking density increases, whereas the strain at break decreases due to the decrease of the random coiled TUEG chain length in the networks. When the TGIC content is 17 wt% (i.e., BMPN17), the elastic modulus and yield stress reach 1.4 GPa and 61 MPa, respectively, with much decreased extensibility of 12% at 26 ± 1 °C. It should be noted that the use of TGIC as the cross-linker is important for constructing mechanically robust polymer networks. The TGIC contains a rigid isocyanurate ring structure with H-bond accepting carbonyl groups (Fig. S9a†).13 Accordingly, when compared to BMPN17 cross-linked by 17 wt% of trimethylolpropane triglycidyl ether (TMPTGE) without the ring structure and H-bond accepting moieties (Table S3 and Fig. S16†), the BMPN8 with 8 wt% of TGIC cross-linker exhibits comparable or even better mechanical properties in terms of modulus, strength, extensibility, and toughness.
The BMPNs exhibit combined mechanical properties including resilience in addition to the high stiffness, strength, and toughness at room temperature. For example, as shown in Fig. 2a, the initially stiff BMPN8 film (ca. 45 mm (L) × 20 mm (W) × 0.4 mm (T)) supports a mechanical load of 75 g, which is deformed without breaking under larger load or tensile stress. Interestingly, the plastic deformation of BMPN8 is found to be temporary, and recover gradually with time or promptly upon heat treatment above its Tg. The strain recovery behaviors of BMPNs are further characterized by cyclic tensile tests. After the first loading–unloading cycle within a strain limit of 200%, BMPN8 shows a residual, temporary strain of ≈140%, which increases slightly up to ≈160% by the five consecutive cycles (Fig. 2b). When allowed to rest for 48 h at 25 °C, the pre-stretched BMPN8 fully recovers its original shape and mechanical properties (Fig. 2c). In comparison to the BMPN8, the BMPN4 also exhibits similar recovery behavior but shorter time is required for the strain recovery after deformation (Fig. S17†), likely due to the larger polymer chain mobility as expected by the smaller Tg value (Fig. S14b†). By contrast, the complete recovery behavior is not observed for the TUEG34 without the covalent cross-linking (Fig. S8†), confirming that the resilience of the BMPNs is provided by the TGIC cross-linker through establishing the permanent covalent networks.5c,6 Although showing somewhat fast strain recovery behavior during 1 h of recovery after the first deformation cycle, the TUEG34 tends to break without complete recovery of strain and mechanical properties during the repetitive cyclic tensile testing even with longer recovery time.
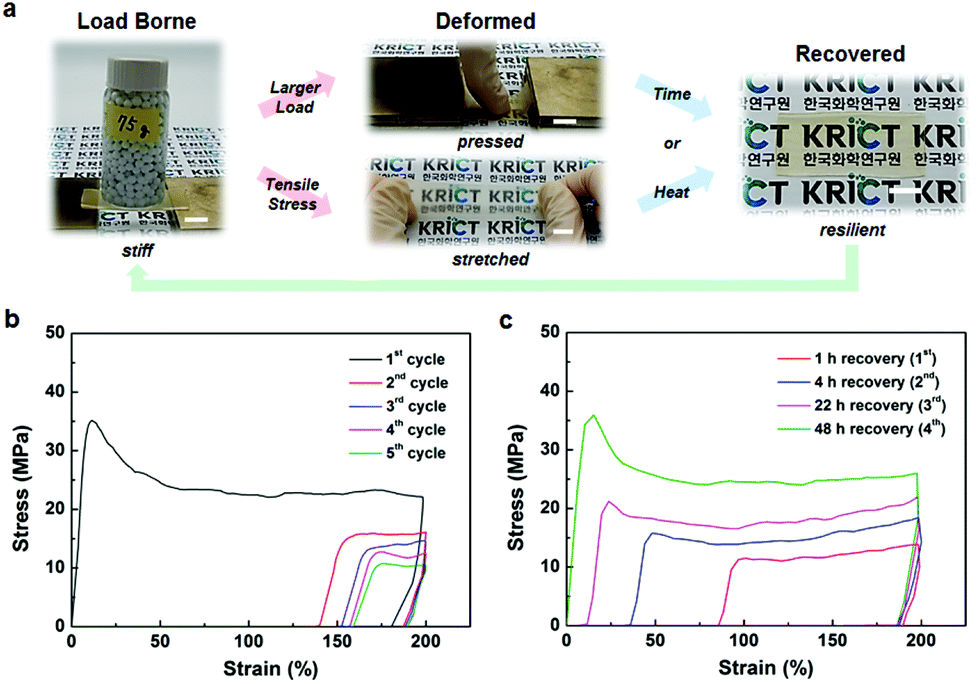 | ||
| Fig. 2 Stiff, strong, tough, and resilient properties of BMPN8. (a) Photographs of load borne, deformed, and recovered BMPN8 (scare bar = 1 cm). (b) Cyclic stress–strain curves of pristine BMPN8. (c) Cyclic stress–strain curves of the pre-stretched BMPN8 (in (b)) after recovered for 1, 4, 22, and 48 h at 25 °C. The cyclic tensile testing was performed at strain rate and limit of 0.0067 s−1 and 200%, respectively. | ||
The BMPNs are also found to possess adaptability, presenting shape memory behaviors responsive to diverse stimuli as well as dynamic mechanical properties depending on the strain rate and temperature. The BMPN8, which has optimum mechanical properties among the BMPNs in terms of the balanced combination of elastic modulus, yield stress, toughness, and resilience, was subjected to consecutive shape memory cycles for quantitative analysis (Fig. 3a). A typical thermally triggered one-way dual-shape memory behavior is observed from the BMPN8, constructed with covalent cross-links (TGIC) for the permanent shape formation and switch segments (Tg = 31.5 °C) for the temporary shape fixation.15 Once the extended shape (≈28% strain, at 50 °C and 0.2 MPa) is fixed at 0 °C with released stress, the BMPN8 quickly recovers its original shape upon heating to 50 °C. The consecutive shape memory cycles are stable, with the shape fixity (Rf) and recovery (Rr) ratios approaching 100% and 98%, repectively. The shape recovery can be induced by solvent as well as by the heat (Fig. 3b), in which the solvent molecules plastisize the amorphous phase of BMPN8, thereby leading to the decrease in Tg to a level below the room temperature to trigger the recovery.15 It is shown that different solvents affect the shape recovery time significantly (Fig. S18†), because the BMPN8 is a polar, hydrophilic network with dense thiourea H-bonds. Besides, upon irradiated with an 808 nm near-infrared (NIR) laser (0.2 W mm−2), the BMPN8 experiences a temperature rise up to about 32 °C (Fig. S19†), which is above its Tg. Thus, this enables remotely and regionally controlled shape recovery of the BMPN8 via NIR laser irradiation (Fig. 3b). Meanwhile, BMPN4 and BMPN17 are also found to exhibit similarly stable shape memory behaviors, although the strains at the same stress value tend to decrease with increasing cross-linking density of the BMPNs (Fig. 3a and S20†).
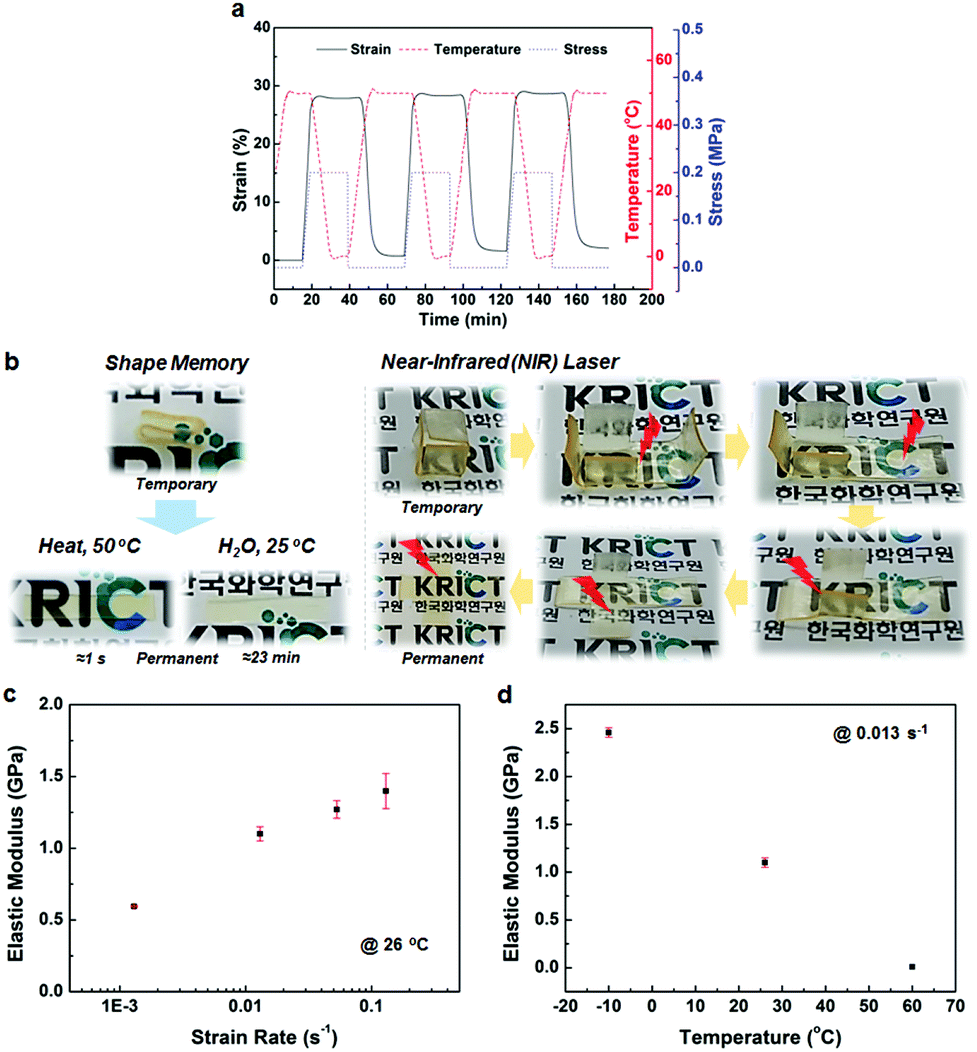 | ||
| Fig. 3 Adaptive and dynamic mechanical properties of BMPN8. (a) Quantitative shape memory cycles. (b) Photographs of heat-, solvent-, and NIR-induced shape recovery behaviours. (c) Strain rate and (d) temperature dependent elastic modulus. | ||
Although the BMPN8 shows high elastic modulus and yield stress at a certain strain rate (0.013 s−1) and room temperature (26 ± 1 °C), its mechanical properties are found to be affected by different strain rates and temperatures (Fig. 3c and d). Strain rate dependence of mechanical properties for solid polymers is considered more challenging to realize compared to strain rate dependent fluids.16 It is important to utilize a large amount of dynamic noncovalent interactions in polymer networks for the strain rate dependent mechanical properties.16 H-bonding polyurea-based elastomers have shown strain rate dependence of modulus under tensile tests, although the variance was not large (18 MPa to 27 MPa, ≈1.5× increase).17 In a more recent study, a solid polymer network with high content of the quadruple H-bonding UPy groups showed 14 times increase in elastic modulus (≈0.3 MPa to ≈4.1 MPa), when the strain rate varied from 1.7 × 10−3 to 1.7 s−1.16 For the BMPN8 with a large amount of thiourea H-bonds, as the strain rate increases from 1.3 × 10−3 to 1.3 × 10−1 s−1, the elastic modulus increases from 0.6 GPa to 1.4 GPa (Fig. 3c and S21a†). This increase in modulus (2.3 times) is notable, because the BMPN8 possesses much higher mechanical modulus (≈1000×) than the UPy-based soft polymer networks. The BMPN8 also shows the temperature dependence of mechanical properties (Fig. 3d, S21b and S22b†). The high stiffness and strength dominate among the mechanical properties of BMPN8 at temperatures below its Tg (e.g., at −10 °C), whereas the large extensibility up to ≈375%, with much decreased stiffness and strength, is clearly observed at temperatures above its Tg (e.g., at 60 °C), exhibiting the rubbery behavior.2
While the polymer chemistry has been progressed considerably over the past century, it is still difficult to achieve diverse mechanical properties, especially such as stiffness, strength, toughness, and resilience, in a synthetic polymer.2 These properties are usually considered mutually exclusive in conventional synthetic polymers (Fig. S23†). For example, engineering thermoplastics and thermosets are known for their excellent mechanical stiffness and strength, but generally have poor toughness and resilience.2 Whereas elastomers possess high resilience, their stiffness and strength are often restricted.2 In contrast to the conventional synthetic polymers, the BMPNs in this study have combined the seemingly mutually exclusive mechanical properties; they exhibit high elastic modulus (≈1 GPa) and yield stress (≈40 MPa), comparable to polyethylene (PE) or polypropylene (PP), yet large extensibility (≈300%), intriguing resilience (complete recovery of shape and performance), and adaptability for various stimuli as well. To our knowledge, this can be considered an unprecedented combination of multifaceted, robust mechanical properties even compared with other recent reports, concerning the use of noncovalent sacrificial bonds in polymeric materials (Table S4†). Despite the application of the biomimetic molecular design principles, most polymeric materials in the literature have outstanding extensibility and resilience, but lack high mechanical stiffness and strength.3,5–10 It should be also noted that the strong epoxy–amine covalent bonding, combined with the weak thiourea hydrogen bonding, in this study is still important for the significant improvements of versatile mechanical properties. The linear TUEG34 without any covalent cross-linking cannot provide complete recovery of shape and mechanical properties after large deformation over the yield point (Fig. 2 and S8†). In addition, the robust mechanical properties of BMPNs, approaching to those of PE or PP in terms of elastic modulus, yield stress, and toughness, at room temperature (26 °C) are achieved by the covalent cross-linking (Tables 1 and S2†).
In summary, we have demonstrated that dual cross-linkings based on thiourea noncovalent interactions and epoxy–amine covalent linkages can bestow the biomimetic polymer networks with a remarkable combination of versatile mechanical characteristics. By the covalent cross-linking of the noncovalent cross-linking building block (TUEG), the polymer networks (e.g., BMPN8) exhibit greatly enhanced overall mechanical properties, including elastic modulus (1.1 GPa), yield stress (39 MPa), and toughness (83 MJ m−3) while slightly compromising extensibility (320%). Furthermore, the complete recovery of shape and mechanical performance of the BMPNs, even after large deformation over the yield point, is provided by the covalent cross-linking. We believe that the present work provides insight into the design and preparation of biomimetic polymer networks with advanced properties for various applications, by taking advantage of the non-crystalline, dense thiourea H-bondings. In particular, the multifaceted mechanical properties of BMPNs herein, in combination with their adaptability responsive to multi-stimuli, hold great promise for emerging applications, such as 4D printing, shape-morphing devices, and soft robotics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Korea Research Institute of Chemical Technology (KRICT) core project (KK1961-06, SI1803-03) and the Center for Advanced Soft Electronics funded by the Ministry of Science and ICT as a Global Frontier Project (2015M3A6A5065315).References
- (a) A. M. Kushner and Z. Guan, Angew. Chem., Int. Ed., 2011, 50, 9026 CrossRef CAS PubMed; (b) M. J. Buehler and Y. C. Yung, HFSP J., 2010, 4, 26 CrossRef CAS PubMed.
- (a) J. W. S. Hearle, Polymers and Their Properties, vol. 1: Fundamentals of Structure and Mechanics, Ellis Horwood, 1982 Search PubMed; (b) T. R. Crompton, Physical Testing of Plastics, Smithers Rapra, 2012 Search PubMed.
- (a) L. M. de Espinosa, W. Meesorn, D. Moatsou and C. Weder, Chem. Rev., 2017, 117, 12851 CrossRef PubMed; (b) W. Wang, Y. Zhang and W. Liu, Prog. Polym. Sci., 2017, 71, 1 CrossRef; (c) X. Zhou, B. Guo, L. Zhang and G.-H. Hu, Chem. Soc. Rev., 2017, 46, 6301 RSC.
- (a) S. Labeit and B. Kolmerer, Science, 1995, 270, 293 CrossRef CAS PubMed; (b) M. Rief, M. Gautel, F. Oesterhelt, J. M. Fernandez and H. E. Gaub, Science, 1997, 276, 1109 CrossRef CAS; (c) M. S. Z. Kellermayer, S. B. Smith, H. L. Granzier and C. Bustamante, Science, 1997, 276, 1112 CrossRef CAS PubMed.
- (a) Q. Chen, L. Zhu, C. Zhao, Q. Wang and J. Zheng, Adv. Mater., 2013, 25, 4171 CrossRef CAS PubMed; (b) X. Dai, Y. Zhang, L. Gao, T. Bai, W. Wang, Y. Cui and W. Liu, Adv. Mater., 2015, 27, 3566 CrossRef CAS PubMed; (c) X. Hu, M. Vatankhah-Varnoosfaderani, J. Zhou, Q. Li and S. S. Sheiko, Adv. Mater., 2015, 27, 6899 CrossRef CAS PubMed.
- J. Liu, C. S. Y. Tan, Z. Yu, Y. Lan, C. Abell and O. A. Scherman, Adv. Mater., 2017, 29, 1604951 CrossRef PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, Md. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932 CrossRef CAS PubMed.
- (a) J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133 CrossRef CAS; (b) P. Lin, S. Ma, X. Wang and F. Zhou, Adv. Mater., 2015, 27, 2054 CrossRef CAS PubMed.
- A. M. Kushner, J. D. Vossler, G. A. Williams and Z. Guan, J. Am. Chem. Soc., 2009, 131, 8766 CrossRef CAS PubMed.
- (a) C.-H. Li, C. Wang, C. Keplinger, J.-L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X.-Z. You and Z. Bao, Nat. Chem., 2016, 8, 618 CrossRef CAS PubMed; (b) S. Yoshida, H. Ejima and N. Yoshie, Adv. Funct. Mater., 2017, 27, 1701670 CrossRef; (c) Q. Zhang, C.-Y. Shi, D.-H. Qu, Y.-T. Long, B. L. Feringa and H. Tian, Sci. Adv., 2018, 4, eaat8192 CrossRef PubMed.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Science, 2018, 359, 72 CrossRef CAS PubMed.
- (a) R. Custelcean, M. G. Gorbunova and P. V. Bonnesen, Chem. –Eur. J., 2005, 11, 1459 CrossRef CAS PubMed; (b) R. Custelcean, Chem. Commun., 2008, 295 RSC.
- (a) X. Wang, Y. Hu, L. Song, W. Xing and H. Lu, Polym. Adv. Technol., 2012, 23, 190 CrossRef CAS; (b) P. Sun, Z. Li, S. Wang and X. Yin, J. Membr. Sci., 2018, 549, 660 CrossRef CAS.
- J. Shim, K. Y. Bae, H. J. Kim, J. H. Lee, D.-G. Kim, W. Y. Yoon and J.-C. Lee, ChemSusChem, 2015, 8, 4133 CrossRef CAS PubMed.
- Q. Zhao, H. J. Qi and T. Xie, Prog. Polym. Sci., 2015, 49–50, 79 CrossRef CAS.
- G. Zhang, Q. Zhao, W. Zou, Y. Luo and T. Xie, Adv. Funct. Mater., 2016, 26, 931 CrossRef CAS.
- D. Mohotti, M. Ali, T. Ngo, J. Lu and P. Mendis, Mater. Des., 2014, 53, 830 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02760b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |