
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceToward heterostructured transition metal hybrids with highly promoted electrochemical hydrogen evolution†
Guanhui Gao‡
 *ab,
Xiaobin Xie‡c,
Shendong Kangd,
Yanhua Leie,
Achim Trampertb and
Lintao Cai*d
*ab,
Xiaobin Xie‡c,
Shendong Kangd,
Yanhua Leie,
Achim Trampertb and
Lintao Cai*d
aMaterials Science and NanoEngineering Department, Rice University, Houston, 77005, USA. E-mail: gg13@rice.edu
bPaulPaul-Drude Institut Für Festkörperelektronik, Leibniz-Institut Im Forschungsverbund Berlin, Berlin, 10117, Germany
cSoft Condensed Matter, Debye Institute for Nanomaterials Science, Utrecht University, Utrecht, 3584CC, Netherlands
dGuangdong Key Laboratory of Nanomedicine, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, 518055, China. E-mail: lt.cai@siat.ac.cn
eCollege of Ocean Environment and Engineering, Shanghai Maritime University, 1550 Haigang Ave, Shanghai, 201306, China
First published on 25th June 2019
Abstract
It is essential to precisely develop low-cost and sustainable electrocatalysts for the hydrogen evolution reaction. Herein, we explore a robust and controllable hydrothermal approach to synthesize defect-rich MoS2 nanoflakes on exfoliated MoS2 and WS2. Such well-designed hetero-structural hybrids of MoS2/exfoliated MoS2 and MoS2/exfoliated WS2 exhibit dramatically promoted electrochemical activity and high stability. The as-grown MoS2 nanoflakes hybridized on exfoliated MoS2 and WS2 generate abundant active edge sites (rich in basal defects) and unsaturated sulfur atoms, resulting in highly enhanced electrocatalytic performance. This is expected to pave the way towards a significant improvement in transition metal dichalcogenide heterostructures as electrocatalysts.
1 Introduction
Two-dimensional graphene-based atomic crystals and transition metal dichalcogenides (TMDs) have been attracting intense interest owing to their extraordinary chemical and physical properties,1–6 and they are expected to have a prosperous future in catalysis, water splitting, optic-electronic performance and energy storage devices.7–12 Very recently, TMD heterostructures with highly enhanced electrochemical performance have been suggested as efficient catalysts to substitute the scarce and expensive noble metals such as platinum-based catalysts, which could supply a promising strategy to generate sustainable hydrogen by electrochemical water splitting. In particular, layered molybdenum disulfide (MoS2) and tungsten disulfide (WS2) have been put forward as robust and efficient catalysts for the hydrogen evolution reaction (HER), attributed to their abundant catalytically active sites in the atomic superlattice structure.13–15 To date, anchoring massive sulfide nano-flakes onto multi-layered TMD surfaces has been explored for building heterogeneous structural hybrids, which demonstrate promoted electrochemical performance and long-term stability.16,17 Hinnemann et al. simulated the Gibbs free energy (ΔGH) at the edge sites of MoS2 via density functional theory, which can be applied as a reasonable descriptor of HER activity for general catalysts. An optimal HER performance is considered to be achieved at a value of ΔGH ≈ 0. A lower ΔGH will lead to very high surface coverage of Hads, while a higher ΔGH will make the proton bonding on the catalyst surface tremendously fragile, resulting in slow HER kinetics.18 Prof. Song Jin's research group has instead engaged in efforts toward enriching defects on the basal planes and edges of TMD heterostructures to expose abundant active sites, triggering catalytic enhancement.19–24 Tuning 1T (octahedral) structural TMDs could result in the restoration of the 2H (trigonal prismatic) phase with a band gap ∼2.0 eV, ideal for photocatalysis.13,25 Tailoring the electronic arrangement of TMD crystals by doping could significantly promote their catalytic activity.26–32Herein, defect-rich MoS2 nanoflakes are synthesized via a chemical hydrothermal approach of loading onto the surface of exfoliated MoS2/WS2 layers to prepare extraordinary heterostructures. The exfoliated MoS2/WS2 layers act as scaffolds. The Mo source is generated from (NH4)6Mo7O24·4H2O, and the S source is supplied by excessive thiourea triggering to obtain defect-rich MoS2, as shown as Scheme 1 (details of the experiments are given in ESI†), which is expected to enhance their catalytic performance and stability for HER due to the abundant edges and defect active sites.33 The aim is to supply a platform for designing TMD-based heterostructures as alternatives for noble metal catalysts with increased catalytic properties.
 | ||
| Scheme 1 Schematic illustration of the fabrication of defect-rich MoS2 nanoflakes grown on exfoliated MoS2 and WS2 hybrids, respectively. | ||
2 Experiment
2.1 Exfoliation of layered MoS2 and WS2
Pristine MoS2 (WS2) powder is dissolved into NMP solvent (initial concentration of 1.0 mg mL−1) sonicated in a low-power sonication bath for 1 h with the bath temperature kept at 50 °C. Then the above mixture is transferred to a higher power sonicator and continually sonicated for 6 h. Finally, the mixture is centrifuged at 8000 rpm for 15 minutes. The supernatant is collected with a pipette and filtered with a filtration system (PTFE membrane, diameter 47 mm, pore diameter 0.22 μm, Millipore filter). The above exfoliated MoS2 nanosheets are then uniformly dispersed into IPA, and dried at 60 °C in a vacuum. Finally, the exfoliated MoS2 layers containing single, double and multi-layers are stored under vacuum for their characteristics to be investigated further.2.2 Preparation of hybrids of MoS2 grown on exfoliated MoS2 or WS2 via a hydrothermal approach
Exfoliated MoS2 (WS2) dispersion: 20.00 mg of as-exfoliated MoS2 (WS2) is dissolved into 20.00 mL of IPA, sonicated for 30 min to obtain a homogeneous MoS2 (WS2)/IPA mixture (1 mg mL−1). Precursor: prepare 0.03 mM mL−1 (NH4)6Mo7O24·4H2O solution and 0.1 mM mL−1 thiourea solution. The synthesis of MoS2 hybridized MoS2 (WS2) heterostructures: add 20 mL of ultrapure water into 5 mL of the above MoS2 (WS2)/IPA mixture (1 mg mL−1), 0.5 mL of 0.03 mM mL−1 (NH4)6Mo7O24·4H2O solution is mixed with 0.5 mL of 0.1 mM mL−1 thiourea solution, sonicated for 10 minutes, transferred to a 45 mL PTFE reactor and reacted for 12 h at 210 °C. Then the reactant is centrifuged (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm) at room temperature for 10 min, the supernatant solution removed, and the precipitate continuously washed three times using ultrapure water to remove precursor residues and surface adsorption contamination. Finally, the obtained products are freeze-dried for investigation.
000 rpm) at room temperature for 10 min, the supernatant solution removed, and the precipitate continuously washed three times using ultrapure water to remove precursor residues and surface adsorption contamination. Finally, the obtained products are freeze-dried for investigation.
2.3 Electrocatalytic performance
Electrochemical measurements are conducted with a three-electrode system on a CHI 660D electrochemical workstation (Shanghai Chenhua Instruments). The working electrode is a glassy-carbon electrode (GCE, CHI104, diameter: 3 mm, area: 0.071 cm2); Pt wires and an Ag/AgCl are used as counter and reference electrodes, respectively. The preparation of working electrodes: 4 mg catalyst with 30 μL Nafion solution are dispersed into 1 mL of DI water/ethanol (1:3 in volume) mixture, and ultrasonicated for 30 minutes to obtain a homogeneous dispersion. Then 5 μL of the above dispersion (containing 20 μg of catalyst) is transferred onto the glassy-carbon electrode, and dried naturally for later investigation.
| ERHE = EAg/AgCl + 0.213 V |
The electrochemically active surface area (ECSA) measurements are determined by integrating the hydrogen adsorption charge on the cyclic voltammetry (CV) at room temperature in argon-saturated 0.5 M H2SO4 solution. The potential scan rate is 50 mV s−1 for the CV measurement. The Tafel plots are investigated by replotting the polarization curves as overpotential (η) vs. log current (logj) to assess the HER kinetics of the obtained catalysts. The durability tests are evaluated at room temperature in 0.5 M H2SO4 solution by applying a cyclic potential between −0.6 and 0.2 V versus Ag/AgCl electrode at a sweeping rate of 50 mV s−1 for 3000 cycles.
2.4 Characterization
Transmission electron microscopy (TEM), high-resolution TEM (HRTEM) images and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and STEM-EDS mapping images are performed with a FEI Tecnai G2 F20 S-Twin microscope at an acceleration voltage of 200 kV. Cross-sectional method preparing TEM sample: Cross-sections of exfoliated MoS2 and WS2 sheets transferred onto Si substrates are prepared by using the standard strategy of mechanical grinding and dimpling (dimple grinder II Model 657, Gatan) the specimen followed by Ar-ion beam milling with the Precision Ion Polishing System (PIPS, model 691, Gatan) down to electron transparency. TEM imaging measurements are performed with a JEOL 2100F field emission microscope operated at 200 kV. The microscope is equipped with a Gatan Ultra Scan 4000 CCD camera for image recording.X-ray diffraction (XRD) analysis is conducted with a Bruker D8 Advance with Cu Kα radiation (λ = 1.54178 Å). X-ray Photoelectron Spectrometry (XPS) is carried out with a Thermo Fisher ESCALAB 250Xi. The sonication equipment is adapted with a sonicator, Fisherbrand FB15061, 750 W. The higher power sonicator is supplied with a Cole-Parmer 1200 W.
3 Results and discussion
Exfoliated MoS2 layers are observed by TEM measurement, as shown in Fig. 1A. The size distribution has a rather large range, mostly with multi-layers. The lattice structure of MoS2 nano-sheets is obtained (Fig. 1B) and hardly any defects are found. The inset indicates the MoS2 simulated lattice structure with the yellow standing for S atoms and the blue being Mo atoms. And the typical layered structure with an interlayer spacing distance of 0.635 nm is shown in Fig. 1C, which is evaluated via a cross-sectional method with the sample prepared by using the standard strategy of mechanical grinding and dimpling the specimen followed by Ar-ion beam milling down to electron transparency. The high-resolution TEM (HRTEM) image with fast Fourier transform (FFT) pattern given in ESI (Fig. S1†) illustrates the complete hexagonal atomic arrangement with 2H phase.31 From the TEM and high-angle annular dark-field (HAADF) STEM images, a large quantity of defect-rich MoS2 nanoflakes grown on the exfoliated MoS2 layers can clearly be observed (Fig. 1D and E). The hexagonal lattice of this well-designed 3D hybrid is characterized by selected area electron diffraction (SAED), as shown as Fig. 1F, indicating its polycrystalline structure. The HRTEM images provide more obvious proof of the lattice structures of both synthesized MoS2 nanoflakes and exfoliated MoS2 layers (Fig. 1G, H and S2†). The lattice structure of exfoliated MoS2 layers uncovered at the edge is complete with a spacing distance of 0.270 nm, (100) facet. In comparison, the atomic arrangement of synthesized MoS2 seems irregularly disordered, inducing surface defects, and is expected to supply more activate sites for improving catalytic function. | ||
| Fig. 1 (A) TEM image of the exfoliated MoS2 nano-sheets produced by a chemical liquid method. (B) Lattice structure of the exfoliated MoS2 sheets. (C) Cross-sectional HRTEM image of exfoliated MoS2 sheets. (D) The TEM image of defect-rich MoS2 hybridized exfoliated MoS2 layers. (E) High-angle annular dark-field (HAADF)-STEM image of (D). (F) SAED pattern of (D). (G and H) HRTEM images of local defect-rich MoS2 hybridized exfoliated MoS2 layers. | ||
The energy dispersive X-ray spectrometry (EDS) mapping results indicate that the synthesized MoS2 nanoflakes are uniformly deposited onto the exfoliated MoS2 surface (Fig. 2A–D). Furthermore, X-ray diffraction (XRD) is conducted to explore the structural information between exfoliated MoS2 layers and hybrids of defect-rich MoS2 nanoflakes grown on exfoliated MoS2 layers (MoS2/exfoliated MoS2), as shown as Fig. 3E. This reveals that the exfoliated MoS2 layers are 2H phase, corresponding to JCPDS 65-0160.
 | ||
| Fig. 2 (A) Bright-field TEM image of hybrids of defect-rich MoS2 nanoflakes grown on exfoliated MoS2. (B) HAADF-STEM image of (A). (C and D) S and Mo elemental mappings. (E) XRD spectra of exfoliated MoS2 layers and MoS2 nanoflakes grown on exfoliated MoS2 hybrids. (F) High-resolution XPS spectra of MoS2 nanoflakes grown on exfoliated MoS2 hybrids, Mo 3d, S 2s and S 2p core level, respectively. | ||
 | ||
| Fig. 3 (A) TEM image of the exfoliated WS2 nano-sheets produced by a chemical liquid method. (B) Lattice structure of the exfoliated WS2 layers. (C) Cross-sectional HRTEM image of exfoliated WS2 nano-sheets. (D) The TEM image of the hybrids of defect-rich MoS2 nanoflakes grown on exfoliated WS2 via a hydrothermal strategy. (E) HAADF-STEM image of (D). (F) SAED pattern of (D). (G and H) HRTEM image of local defect-rich MoS2 hybridized exfoliated WS2 layers. | ||
Obviously, the crystallinity of the MoS2/exfoliated MoS2 hybrids is not as high as that of pure exfoliated MoS2. Hybrids of MoS2/exfoliated MoS2 are produced to further evaluate the elements' binding mechanism using X-ray photoelectron spectroscopy (XPS). The main peaks of Mo 3d are at 227 eV and 232 eV, corresponding to Mo 3d5/2 and Mo 3d3/2, respectively, which indicates the valence of Mo4+. The binding energies of 161 eV and 163 eV contribute to S 2p3/2 and S 2p1/2, illustrating that the valence of S is −2 (Fig. 2F).
The exfoliated WS2 nano-sheets, as shown in Fig. 3A and B, are obtained by a chemical liquid method which is the same as for the fabrication of layered MoS2. The lattice structure of WS2 nano-sheets is obtained (Fig. 3C). The inset shows the WS2 simulated lattice structure where blue stands for a W atom, and yellow represents an S atom. Similarly, the cross-sectional lamellar structure of exfoliated WS2 is investigated, and the number of WS2 layers can clearly be seen. And the FFT mode exhibits the hexagonal structure of layered WS2 with 2H phase (Fig. S3†). It is observed that the synthesized MoS2 nanoflakes hybridize on layered WS2 nano-sheets, as shown in Fig. 3D and E. The hexagonal lattice with the polycrystalline structure of the hybrids is investigated by SAED, as shown as Fig. 3F. The high-resolution TEM images indicate defect-rich MoS2 with a multilayered morphology grown on exfoliated WS2 (Fig. 3G). The clear boundary between as-grown MoS2 and exfoliated WS2 is demonstrated in Fig. 3H and S4† due to the obvious lattice mismatch.
A morphological image of MoS2 grown on exfoliated WS2 hybrids is shown in Fig. 4A, indicating that the branch-like grafted MoS2 grows in situ on the surface of exfoliated WS2. This typical hetero-structure might be advantageous for enhancing electro-catalytical performance. STEM and EDS elemental mappings illustrate the spatial distribution of these S, Mo and W elements of the obtained hybrids (Fig. 4B–E). In comparison, the pixel density of W is more compact than that of Mo, revealing that the W content is locally higher than the Mo content. Moreover, the distribution area of Mo is larger than that of W, suggesting that Mo element is dominant in the external surface of the hybrids. XRD spectra are conducted to explore the structural information between exfoliated MoS2 layers, exfoliated WS2 layers and hybrids of MoS2 grown on exfoliated WS2 layers (MoS2/WS2), as shown as Fig. 4F. This indicates that the exfoliated WS2 layer is 2H phase, corresponding to JCPDS 65-0160, but the crystallinity is not particularly high in comparison. In order to further investigate the variation in surface electron states of hetero-structural MoS2/WS2 hybrids, XPS measurement is conducted, as shown as Fig. 4G. The peaks located at 33 eV and 38 eV are assigned to W 4f5/2 and W 4f7/2, respectively, indicating the valence state of W with +4. The binding energies of S 2p3/2 and S 2p1/2 negatively shift to 161.9 eV and 163.1 eV (compared to pure MoS2 layers with their binding energies of 162.7 eV and 163.9 eV for S 2p3/2 and S 2p1/2), illustrating the electron transfer from WS2 to MoS2 in MoS2/WS2 hybrids. And the typical peaks are located at 227 eV and 232 eV corresponding to Mo 3d5/2 and Mo 3d3/2, indicating the valence state of Mo is +4.
 | ||
| Fig. 4 (A) Morphology of the hybrids of MoS2 grown on exfoliated WS2 (MoS2/exfoliated WS2). (B–E) HAADF-STEM and STEM-EDS elemental mapping images of MoS2/exfoliated WS2 hybrids. (F) XRD pattern of exfoliated WS2, exfoliated MoS2 layers and hybrids of defect-rich MoS2 nanoflakes grown on exfoliated WS2. (G) XPS spectra of the hybrids of MoS2 grown on exfoliated WS2, W 5p, W 4f, Mo 3d, S 2s and S 2p core level. | ||
Electrocatalytic measurements were investigated at room temperature using a working electrode made of glassy-carbon electron (GCE, CHI104, ∅ = 3 mm) in argon-saturated 0.5 M H2SO4 electrolyte (see ESI† for the experimental methods in detail). The polarization curves are acquired at a scan rate of 5 mV s−1, sweeping the potential from −0.6 to 0.2 V (vs. Ag/AgCl) at room temperature. Subsequently, we evaluate the electrocatalytic performance for the hydrogen evolution reaction of the well-designed MoS2/WS2 hybrids, MoS2/MoS2 hybrids, exfoliated MoS2, and exfoliated WS2 as investigated catalysts. Fig. 5A exhibits that the initial overpotentials of both MoS2/WS2 hybrids and MoS2/MoS2 hybrids are much lower than those of exfoliated MoS2, or exfoliated WS2, indicating that the electrocatalytic activities of MoS2 hybridized layered MoS2 and WS2 heterostructures are promoted significantly. Comparably, the catalytic performance of the hetero-structural MoS2/WS2 hybrids is superior to that of the MoS2/MoS2 hybrids, a possible reason being that the structure of MoS2 is not infected by MoS2 hybridization, the lattice structure and constants of synthesized MoS2 remain consistent with exfoliated MoS2 layers, and thus the electronic effect would not be varied. But in terms of MoS2/WS2 hybrids, the banding energy and electronic property of exfoliated WS2 are influenced dramatically after MoS2 hybridization.32
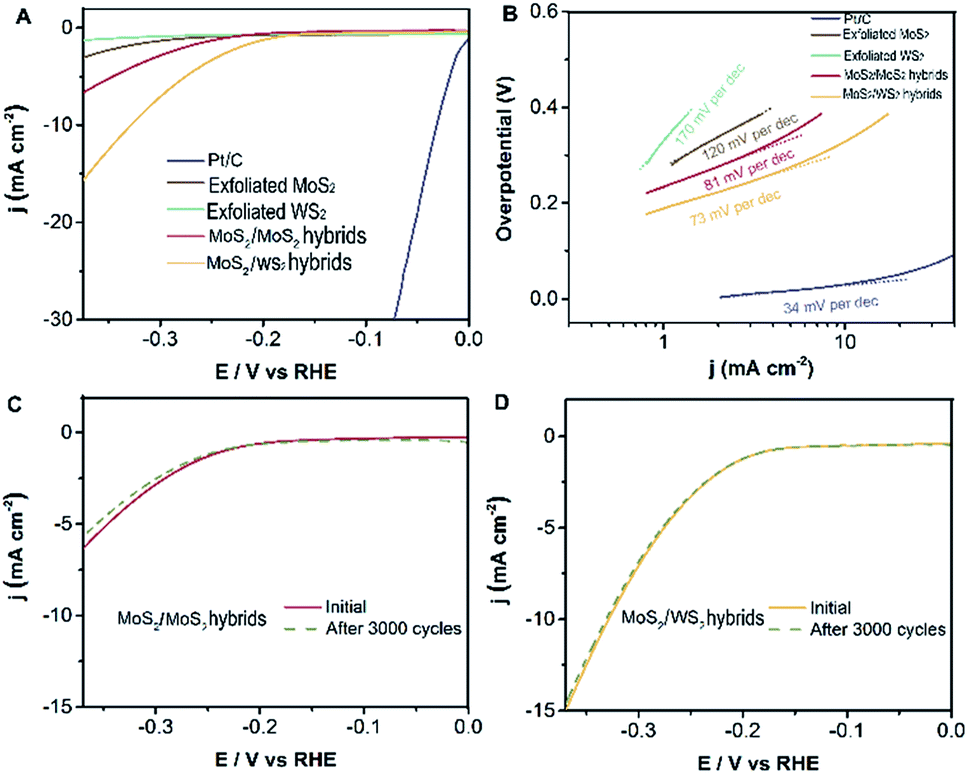 | ||
| Fig. 5 Electrocatalytic hydrogen evolution of different catalysts. (A) Polarization curves for HER on pure exfoliated MoS2, exfoliated WS2, obtained MoS2/WS2 hybrids, MoS2/MoS2 hybrids and a high-quality commercial Pt/C catalyst for comparison. (B) Tafel plot for the different catalysts derived from (A). (C) Comparison of polarization curves for HER on MoS2/MoS2 hybrids and after 3000 cycles. (D) Comparison of polarization curves for HER on MoS2/WS2 hybrids and after 3000 cycles. | ||
In principle, Tafel slopes demonstrate the hydrogen evolution reaction (HER) kinetics of the as-synthesized catalysts. In an acidic system, there are generally three stages to describe the procedure of H+ converting to H2, famously the reactions of Volmer, Heyrovsky, Tafel, shown as follows:33,34
| Volmer: M + H+ + e− → M⋯Had |
| Heyrovsky: M⋯Had + H+ + e− → M + H2 |
| Tafel: M⋯2Had → M + H2 |
The Tafel slope (b = 2.3RT/αT) is a convenient and reasonable way to analyze electrode properties and the reaction mechanism. It is can be derived to be reversely proportional to the charge transfer coefficient (α), where T is temperature and F is the Faraday constant.
The charge transfer coefficient is a significant quantity in electrochemistry, reflecting the nature of electron transfer in an elementary reaction. While it is commonly assumed that the charge transfer coefficient of an elementary reaction is either 0.5 (single electron transfer) or 0 (nonelectron transfer). For instance, HER kinetic models suggest that a Tafel slope of about 120, 40 or 30 mV per decade will be obtained if the Volmer, Heyrovsky or Tafel reaction, respectively, is the rate-determining step. Likewise, the Tafel slope of Pt/C is 34 mV dec−1, suggesting a Tafel-step-determined Volmer–Tafel mechanism at work in the Pt/C catalyst. Based on this analysis and the Butler–Volmer kinetics, we show that the Volmer–Heyrovsky mechanism is dominant for HER in these MoS2 hybridized MoS2 and MoS2 hybridized WS2 nanostructures due to their Tafel slopes of 81 mV dec−1 and 73 mV dec−1 (Fig. 5B) respectively. (The Tafel slope is higher than 30 mV dec−1, indicating there is no electron transfer, which is attributed to the reaction mechanism mainly being dominated by the Volmer–Heyrovsky mechanism.) However, this value of the Tafel slope is still higher than that of a Pt/C catalyst, the main possible reason for which is that the electrodes are extremely small in our electrochemical workstation system, resulting in an inefficient evaluation. Nevertheless, the building blocks of MoS2 hybridized MoS2 (WS2) demonstrate superior catalytic performance compared with exfoliated MoS2 (WS2) layers. In order to further evaluate the durability of as-catalysts, the electrochemical activity of MoS2 hybridized MoS2 (WS2) heterostructures are retained in the current after 3000 cycles, as shown in Fig. 5C and D. There is almost no variation in the polarization curves for HER before and after 3000 cycles. This confirms the outstanding stability and durability of MoS2/exfoliated MoS2 hybrids and MoS2/exfoliated WS2 hybrids.
4 Conclusions
In summary, we explored heterogeneous hybrids of MoS2/exfoliated MoS2 (WS2) via a facile and high-yielding hydrothermal strategy. These well-designed heterostructures possess abundant edges and basal plane defects which could supply rather more active sites, triggering significant promotion of electrochemical performance and stability for HER as catalysts. This is expected to pave the way for the development of innovative efficient catalysts in transition metal dichalcogenides instead of rare noble catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The presented research was financially supported by Key International Science and Technology Cooperation Project (2015DFH50230), the Natural Science Foundation of China (51602195, 51502333), the Guangdong Natural Science Foundation of Research Team (2016A030312006), and the Science and Technology Program of Shenzhen (JCYJ20120615124830232, JCYJ20160429191503002).Notes and references
- Y. Yin, J. Han, Y. Zhang, X. Zhang, P. Xu, Q. Yuan, L. Samad, X. Wang, Y. Wang, Z. Zhang, P. Zhang, X. Cao, B. Song and S. Jin, J. Am. Chem. Soc., 2016, 138, 7965–7972 CrossRef CAS PubMed.
- H. S. Matte, A. Gomathi, A. K. Manna, D. J. Late, R. Datta, S. K. Pati and C. N. Rao, Angew. Chem., Int. Ed., 2010, 49, 4059–4062 CrossRef CAS PubMed.
- Z. Y. Zeng, Z. Yin, X. Huang, H. Li, Q. He, G. Lu, F. Boey and H. Zhang, Angew. Chem., Int. Ed., 2011, 50, 11093–11097 CrossRef CAS PubMed.
- A. J. Mannix, X. F. Zhou, B. Kiraly, J. D. Wood, D. Alducin, B. D. Myers, X. Liu, B. L. Fisher, U. Santiago, J. R. Guest, M. J. Yacaman, A. Ponce, A. R. Oganov, M. C. Hersam and N. P. Guisinger, Science, 2015, 350, 1513–1516 CrossRef CAS PubMed.
- X. Huang, Z. Zeng and H. Zhang, Chem. Soc. Rev., 2013, 42, 1934–1946 RSC.
- C. Zhang, Y. Shi, Y. Yu, Y. Du and B. Zhang, ACS Catal., 2018, 8, 8077–8083 CrossRef CAS.
- Y. Yu, Y. Shi and B. Zhang, Acc. Chem. Res., 2018, 51, 1711–1721 CrossRef CAS PubMed.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H. Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS PubMed.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Nørskov and X. Zheng, Nat. Mater., 2016, 15, 48–53 CrossRef CAS PubMed.
- S. F. Zhuo, Y. Xu, W. W. Zhao, J. Zhang and B. Zhang, Angew. Chem., Int. Ed., 2013, 52, 8602–8606 CrossRef CAS PubMed.
- H. Li, G. Lu, Z. Yin, Q. He, H. Li, Q. Zhang and H. Zhang, Small, 2012, 8, 682–686 CrossRef CAS PubMed.
- J. Duan, S. Chen, B. A. Chambers, G. G. Andersson and S. Z. Qiao, Adv. Mater., 2015, 27, 4234–4241 CrossRef CAS PubMed.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850–855 CrossRef CAS PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- D. Voiry, M. Salehi, R. Silva, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nano Lett., 2013, 13, 6222–6227 CrossRef CAS PubMed.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- H. T. Wang, Z. Lu, S. Xu, D. Kong, J. J. Cha, G. Zheng, P. C. Hsu, K. Yan, D. Bradshaw, F. B. Prinz and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19701–19706 CrossRef CAS PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- L. Cheng, W. Huang, Q. Gong, C. Liu, Z. Liu, Y. Li and H. Dai, Angew. Chem., Int. Ed., 2014, 53, 7860–7863 CrossRef CAS PubMed.
- Y. Yin, Y. Zhang, T. Gao, T. Yao, X. Zhang, J. Han, X. Wang, Z. Zhang, P. Xu, P. Zhang, X. Cao, B. Song and S. Jin, Adv. Mater., 2017, 29, 1700311 CrossRef PubMed.
- Q. Ding, B. Song, P. Xu and S. Jin, Chem, 2016, 1, 699–726 CAS.
- H. Vrubel, D. Merki and X. Hu, Energy Environ. Sci., 2012, 5, 6136–6144 RSC.
- J. D. Benck, Z. Chen, L. Y. Kuritzky, A. J. Forman and T. F. Jaramillo, ACS Catal., 2012, 2, 1916–1923 CrossRef CAS.
- D. Merki and X. Hu, Energy Environ. Sci., 2011, 4, 3878–3888 RSC.
- M. A. Lukowski, A. S. Daniel, C. R. English, F. Meng, A. Forticaux, R. J. Hamersa and S. Jin, Energy Environ. Sci., 2014, 7, 2608–2613 RSC.
- J. L. Zhang, M. B. Vukmirovic, Y. Xu, M. Mavrikakis and R. R. Adzic, Angew. Chem., Int. Ed., 2005, 44, 2132–2135 CrossRef CAS PubMed.
- W. F. Chen, K. Sasaki, C. Ma, A. I. Frenkel, N. Marinkovic, J. T. Muckerman, Y. Zhu and R. R. Adzic, Angew. Chem., Int. Ed., 2012, 51, 6131–6135 CrossRef CAS PubMed.
- L. T. Qu, Y. Liu, J. B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- R. Silva, D. Voiry, M. Chhowalla and T. Asefa, J. Am. Chem. Soc., 2013, 135, 7823–7826 CrossRef CAS PubMed.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. Chen and S. Huang, ACS Nano, 2012, 6, 205–211 CrossRef CAS PubMed.
- R. Kappera, D. Voiry, S. E. Yalcin, B. Branch, G. Gupta, A. D. Mohite and M. Chhowalla, Nat. Mater., 2014, 13, 1128–1134 CrossRef CAS PubMed.
- X. Chen and A. R. McDonald, Adv. Mater., 2016, 28, 5738–5746 CrossRef CAS PubMed.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- M. R. Gao, J. X. Liang, Y. R. Zheng, Y. F. Xu, J. Jiang, Q. Gao, J. Li and S. H. Yu, Nat. Commun., 2014, 6, 1–7 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02673h |
| ‡ Dr Guanhui Gao and Xiaobin Xie contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |